Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism
 Mark C. Blaser
Mark C. Blaser Elena Aikawa
Elena Aikawa- 1Division of Cardiovascular Medicine, Department of Medicine, Center for Interdisciplinary Cardiovascular Sciences, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, United States
- 2Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women's Hospital, Center of Excellence in Cardiovascular Biology, Harvard Medical School, Boston, MA, United States
Cardiovascular calcification is a multifaceted disease that is a leading independent predictor of cardiovascular morbidity and mortality. Recent studies have identified a calcification-prone population of extracellular vesicles as the putative elementary units of vascular microcalcification in diseased heart valves and vessels. Their action is highly context-dependent; extracellular vesicles released by smooth muscle cells, valvular interstitial cells, endothelial cells, and macrophages may promote or inhibit mineralization, depending on the phenotype of their originating cells and/or the extracellular environment to which they are released. In particular, emerging roles for vesicular microRNAs, bioactive lipids, metabolites, and protein cargoes in driving this pro-calcific switch underpin the necessity of innovative strategies to employ next-generation sequencing and omics technologies in order to better understand the pathobiology of these nano-sized entities. Furthermore, a recent body of work has emerged that centers on the novel re-purposing of extracellular vesicles and exosomes as potential therapeutic avenues for cardiovascular calcification. This review aims to highlight the role of extracellular vesicles as constituents of cardiovascular calcification and summarizes recent advances in our understanding of the biophysical nature of vesicle accumulation, aggregation, and mineralization. We also comprehensively discuss the latest evidence that extracellular vesicles act as key mediators and regulators of cell/cell communication, osteoblastic/osteoclastic differentiation, and cell/matrix interactions in cardiovascular tissues. Lastly, we highlight the importance of robust vesicle isolation and characterization when studying these phenomena, and offer a brief primer on working with cardiovascular applications of extracellular vesicles.
Introduction
Cardiovascular calcification is both a strong predictor of morbidity and mortality (1, 2) as well as a direct driver of cardiovascular events (3). In a mineralization-promoting milieu (e.g., atherosclerosis, diabetes, chronic kidney disease (CKD)], such calcification occurs when vascular smooth muscle cells (SMCs) or circulating cells differentiate to an osteogenic-like phenotype that actively synthesizes an extracellular matrix (ECM) and regulates the deposition of hydroxyapatite [Figure 1, reviewed in (5)]. This mineral nucleation often begins as microcalcifications, which can potentiate atherosclerotic plaque rupture by concentrating mechanical forces within the fibrous cap (6, 7). Such calcification is not limited to the vasculature: in the cardiac valves, resident valvular interstitial cells (VICs) undergo transformation into myofibroblast-like and osteoblast-like cells, leading to thickening, stiffening, fibrosis, and calcification of the valve leaflets. In turn, this calcific burden impairs leaflet opening/closure, resulting in stenosis or regurgitation and a deadly load on the cardiac muscle: aortic stenosis conveys a 75% 5-year risk of heart failure, valve replacement surgery, or mortality (8, 9).
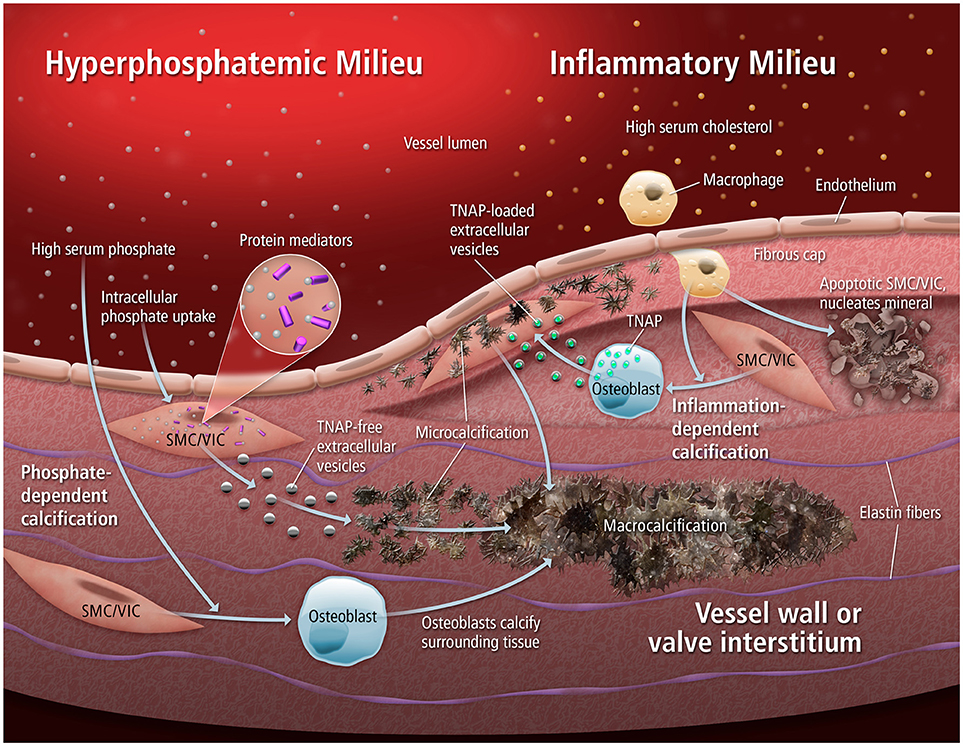
Figure 1. Mechanisms of calcification. Extracellular vesicles can mediate multiple forms of cardiovascular calcification, in both hyperphosphatemic and inflammatory milieus. When an abundance of phosphate is present, rapid tissue calcification can occur through the nucleation of mineral by EVs low in TNAP. Alternatively, inflammatory stimuli can drive osteogenic differentiation of resident SMCs/VICs that leads to the release of TNAP-loaded EVs that progressively aggregate into microcalcifications. From (4).
Expressed ubiquitously by all human cell types, extracellular vesicles (EVs) are found throughout the body's tissues and biofluids (10). EVs are membrane-bound, nanometer-scale structures that contain cargo (RNA, proteins, and metabolites) and whose secretion is highly conserved between forms of eukaryotic life [reviewed in (11)]. The release of these particles was originally thought to be a mode of waste elimination (12), but they are now understood to play an active role in the regulation of cellular processes. For example, EVs are known to mediate the adaptive immune response (13), modulate tumor metastasis (14), and control coagulation cascades (15). For some time, there has been a growing body of evidence that calcifying EVs released from SMCs are strongly implicated in mineralization of the vasculature (16). Cell types known to be involved in cardiovascular calcification release membrane-bound EVs that have the potential to nucleate minerals, aggregate into microcalcifications, and drive the pathological differentiation of neighboring tissues (17–19). It appears likely then that the release of these calcifying EVs initiates and drives the progression of vascular calcification. Besides taking part in homeostasis and pathogenesis, attempts are being made to exploit the unique properties of these structures [low toxicity, stable phenotypes, targeted delivery, extended half-life; reviewed in (20)] for other purposes. Recent studies by our lab and others have identified many putative anti-calcific and anti-fibrotic proteins that may be amenable to arresting both the origins and advancement of vascular calcification (21–23); therapeutic transport of these or other compounds by EVs could allow for a leap in the treatment of cardiovascular diseases. Treatment efforts directed at vascular/valvular calcification have to date been largely ineffective, due in part to low delivery efficiency and non-specific targeting in the complex multi-cell type calcifying vascular milieu [reviewed in (24)]. An acellular EV treatment approach would reduce regulatory burdens and is amenable to multiple forms of delivery (injection, impregnated scaffolds, etc).
In this review, we clarify the historically-confusing nomenclature and cellular origins of calcifying EVs, and discuss the latest work on roles for EV accumulation in the initiation of microcalcification as well as their putative roles as mediators of intercellular communication in the vasculature. When aspects of this rapidly-progressing area are short on information from cardiovascular sources, we supplement with what is known from bone EV metabolism, and comment on the relevance of these findings to the vasculature. Finally, we discuss current gold-standard and forthcoming methods for the isolation, characterization, and study of EVs, and provide practical guidance for investigators interested in the study of EV-mediated vascular calcification.
Release, Structure, and Composition of Calcifying Extracellular Vesicles
According to the guidelines of the International Society of Extracellular Vesicles (ISEV), the term “extracellular vesicles” collectively refers to secreted membrane-enclosed particles whose subsets have historically been referred to by field-dependent terms such as exosomes, ectosomes, microvesicles, microparticles, multivesicular bodies, matrix vesicles, and apoptotic bodies (25). Subset classification is based primarily on the route of biogenesis, as well as EV size, density, and protein markers. Cells release EVs through multiple processes (Figure 2): generally, exosomes refer to EVs released through the endosomal-sorting complex, where intraluminal vesicles formed by the inward budding of endosomal membrane are packaged within multivesicular bodies (MVBs). MVBs then fuse with the plasma membrane to release their enclosed exosomes (~50–150 nm in diameter) outside of the cell in a Rab GTPase-dependent manner [reviewed in (11, 26, 27)]. In contrast, microvesicles (or microparticles or “platelet dust”) are formed by direct budding and fission from the plasma membrane (~100–500 nm) (28). Other forms of secreted particles exist as well: during apoptosis, breakdown of the cytoskeleton induces bulges in the plasma membrane, which then separate as large apoptotic bodies (1,000–5,000 nm) (29). Even with the classification of these subsets, there is clearly substantial overlap between exosomes and microvesicles. Further complicating this scenario is the fact that these two populations are typically shed concurrently (30), a spectrum of heterogeneity exists within these subtypes (31), and there are currently no techniques that can identify the route of biogenesis beyond an estimate of EV size and subsequent correlation of size with known routes of release (see section Methods and Guidelines for Isolation of and Experimentation With Extracellular Vesicles).
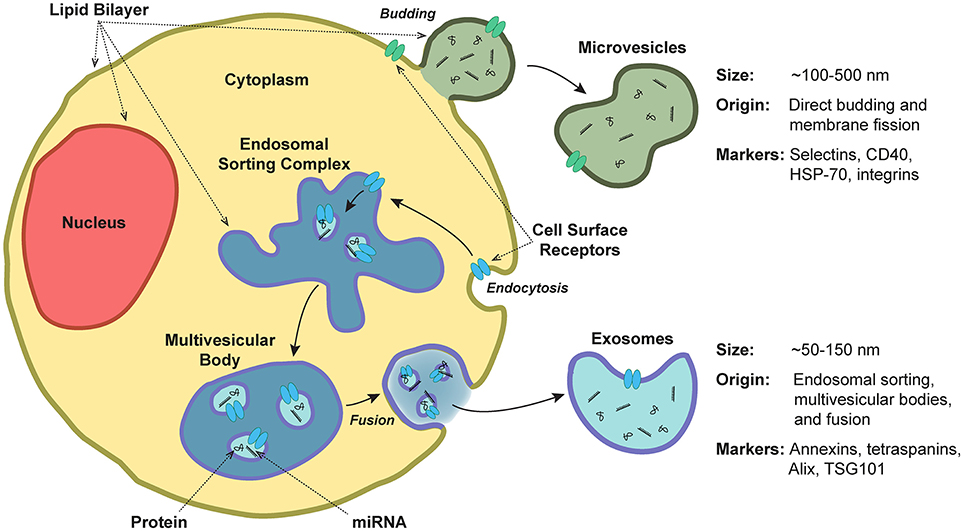
Figure 2. Extracellular vesicle release, structure, and composition. Extracellular vesicles are a heterogeneous population of secreted lipid bilayer-enclosed particles. Microvesicles are formed by direct budding and fission from the plasma membrane (~100–500 nm in diameter). In contrast, exosomes are released via the endosomal-sorting complex, where intraluminal vesicles formed by the inward budding of endosomal membrane are packaged within multivesicular bodies. These bodies then fuse with the plasma membrane to release their enclosed exosomes (~50–150 nm). Both exosomes and microvesicles contain miRNA and protein cargoes.
Since 1967, the release of small vesicular bodies from cartilaginous epiphyseal plates has been implicated in the initiation of calcification (32–34). Epiphyseal chondrocytes, bone-derived osteoblasts, odontoblasts, etc. are known to release EVs that were originally termed matrix vesicles (MVs, not to be confused with microvesicles or multivesicular bodies), with diameters in the range of 100–400 nm (35, 36), and bud from specific locations on the plasma membrane [apical microvilli (36)]—indicating that at least a subset of MVs have a microvesicle-like origin (37). Macrophage-derived calcifying EVs are 30–300 nm in diameter (mean of ~50 nm), and originate as membranous protrusions (17). Electron microscopy demonstrates that MV-like vesicles are present in vascular calcifications of patients with chronic kidney disease (38) or atherosclerotic lesions (39). However, SMCs also release calcifying EVs that are typically on the order of 100–150 nm in diameter (19, 40), and recent work has shown that SMC-derived calcifying EVs originate from an exosomal pathway, are processed through multivesicular bodies, and are enriched in members of the Rab GTPase family (18). The structure of these calcifying EVs resembles that of exosomes, and inhibition of exosomal release in vascular SMCs inhibits their ability to mineralize (18). Importantly, MVs are also reported to be released from growth plate chondrocytes in such a way as to transit across intact plasma membranes (41), from epiphyseal cartilage as dense lysosomal bodies (42), and from osteoblasts as aggregates contained in a membrane-like sac that ruptures after release from the cell (43), indicating the presence of a MV subpopulation with exosomal origins. In short: (i) not all EVs are exosomes, (ii) all exosomes are EVs, (iii) osteoblasts/chondrocytes release mineralizing matrix vesicles that may be exosomes or microvesicles, (iv) macrophages release calcifying microvesicles, and (v) vascular SMCs release calcifying exosomes. Circulating vesicles from a myriad of other cell types (e.g., platelets, erythrocytes, leukocytes, MSCs, etc.) also likely contribute to the calcification of vascular plaques [reviewed in (44, 45)].
The intracellular origins of these calcifying EVs remain understudied, and are not always well-defined. Calcifying EVs can be produced by both exosomal and microvesicular modes of biogenesis, and vesicle release is clearly a highly dynamic process—cells can produce both forms of EVs (11). Importantly, many calcifying EV populations are described as having sizes that overlap with the 100–150 nm diameter range shared between prototypical exosomes and microvesicles. As noted above, recent work has clearly demonstrated that under high calcium/inorganic phosphate culture conditions (reminiscent of a CKD-like state), SMCs produce calcifying vesicles that are exosomal in origin (though their average diameter of ~140 nm places them on the high end of exosomal size) (18, 46). MVBs and exosomes have also been identified in the calcified arteries of dialysis patients, and appear to modulate mineralization in a fetuin-A- and sphingomyelin phosphodiesterase 3-dependent manner (18). In contrast, SMCs cultured under osteogenic conditions (rich in organic phosphate sources) produce larger calcifying EVs with a diameter of >150 nm. In 3D culture, these EVs mimic the collagen-dependent aggregation and calcifying behavior of microcalcifications found in mouse and human lipid/inflammation-rich atherosclerotic plaques. Consistent with this work, there is some evidence that macrophage-derived calcifying EVs have a microvesicular origin, as electron microscopy and time-lapse imaging has shown that they are released directly by budding from the plasma membrane (17). However, these calcifying macrophage-derived EVs have diameters of ~50 nm—more in line with that of exosomes. Regardless, these directly-budding EVs drive calcification through externalization of phosphatidylserine and the subsequent production of a phosphatidylserine-annexin V-S100A9 complex. The relative importance, contributions of, and mechanisms of actions for calcifying EVs derived from SMCs vs. macrophages remains to be determined. The limited experimental evidence available could indicate that microvesicular EVs may be the more dominant form of calcifying EVs produced in an inflammatory/atherosclerotic milieu, while exosomal calcifying EVs could play a larger role in CKD-associated calcification. In the future, rigorous identification and separation of putative EV sub-populations followed by proteomics-based phenotyping for biogenesis markers could enable a better understanding of the relative contributions of these differing routes of EV biogenesis to cardiovascular calcification. Specific inhibition of exosomal and, separately, microvesicle release pathways in model systems of disease would also ensure confirmation of the origins of the EVs of interest.
Despite their varied routes of biogenesis, EV subsets are all characterized by the presence of a membranous phospholipid bilayer. EV and cellular plasma membrane compositions differ: calcifying vesicle membranes are enriched in annexins and phosphatidylserines, which enhance their mineralizing potential (see section Biophysical Vesicle Aggregation and Mineralization) (47, 48). Internally and externally, exosomes and microvesicles carry protein, miRNA, and mRNA cargoes while apoptotic bodies are large enough to encapsulate nuclear fractions and entire organelle structures. As reviewed by van Niel et al. (11), exosomes/EVs are enriched in markers of their endosomal origins such as the members of the annexin family, tetraspanins (e.g., CD9, CD81, CD63), and proteins including Alix and TSG101 that are involved in exosomal biogenesis. They are also characterized by the presence of MHC complexes, lactadherin, RAB GTPases, and flotillin-1. Microvesicles, on the other hand, tend to be enriched in selectins (e.g., CD62), CD40, isoforms of heat shock protein-70, and various integrins. MVs are enriched in matrix metalloproteinases (MMPs, e.g., MMP-2,−3,−9,−13) that presumably assist with the remodeling of the extracellular matrix in order to expose charged sites on collagen fibers which are prone to initiation of mineral nucleation (49, 50).
In SMCs, there are large differences in cellular vs. vesicular miRNA populations, and a substantial number of miRNAs that are enriched in SMCs target genes involved in osteogenesis (51–54): miR-30,−125-b,−143,−145, and−155 modulate expression of proteins such as Smad1, Runx2, alkaline phosphatase (ALP, also known as tissue non-specific alkaline phosphatase or TNAP), and Osterix, resulting in altered MAPK signaling and calcium metabolism that drive vascular calcification (55). Next-generation sequencing reveals that osteoblast EVs have depleted levels of messenger RNAs (mRNAs) encoding proteins associated with basic homeostatic processes such as cytoskeletal function, cell survival, and apoptosis, and are enriched in mRNAs associated with protein translation, RNA processing, and stromal cell proliferation (56). There appears to be a disease-dependent localization of circulating miRNA: in plasma from healthy donors 90% of circulating miRNA is non-membrane-bound (57), whereas in patients with coronary artery disease the majority of plasma miRNAs are associated with EVs (58). In a CKD-mimicking environment (e.g., high-calcium, high-phosphate culture), EV loading with both inhibitors of calcification such as fetuin-A or matrix Gla protein (MGP) and drivers of mineral formation such as ALP is attenuated (18, 48). The nascent field of metabolomics has also begun to shed light on the small-molecule contents of EVs, and the potential for these cargoes to affect cellular function (59). Recent work has found that EVs can function as independent metabolic units with asparginase activity (60), with enrichment of a selected subset of cytosol-derived nucleotide and spermidine (autophagy-linked) pathway-derived metabolites (61).
Biophysical Vesicle Aggregation and Mineralization
Extracellular accumulation and aggregation of calcifying EVs occurs via several interrelated mechanisms, many of which involve EV interactions with the ECM. In both atherosclerotic plaque formation and fibrocalcific aortic valve disease, there is a robust fibrotic response and resultant accumulation of large amounts of disorganized collagen prior to the onset of calcification. Calcifying EVs contain high levels of collagen binding proteins [e.g., ALP, proteoglycan link proteins, hyaluronic-acid binding regions, annexins (62, 63)]. Exosomes also associate with other ECM components such as fibronectin by integrin binding (64), and expression of the collagen-binding receptor DDR-1 by SMCs modulates EV release and mineral deposition (21). As a counterpart to these collagen-binding sites, charged regions on the proteoglycans and collagen fibers of the ECM likely interact with charged EV membranes to promote accumulation via charge-charge interactions. Thus, in early-stage diseases characterized by tissue fibrosis, altered ECM turnover and degradation may expose charged structures that act as templates for EV binding and mineral nucleation (65). Local microenvironmental cues such as hypoxia may exacerbate EV trapping by collagenous ECM: circulating EV populations are enriched in an isoform of the collagen-crosslinking enzyme lysyl oxidase under hypoxic conditions (66). Using a novel 3D collagen hydrogel system paired with high-resolution optical and electron microscopy, Hutcheson and colleagues were able to directly observe the progressive aggregation of individual EVs to coalesce and then form microcalcifications (19). Notably, these microcalcifications are comparable to those of human atherosclerotic plaques (Figure 3) when detected by Fourier-transformed infrared spectroscopy (FTIR), indicating similar mineral maturation induced by the 3D model system. The most interesting findings of this study revolve around new evidence that EV aggregation kinetics may impact the stability of the fibrous cap. In silico modeling of human plaques has found that microcalcifications cause large increases in local tissue stresses, thereby potentiating plaque rupture (6, 7). In both 3D hydrogels and human plaques, there is an inverse relationship between collagen content and microcalcification size: as collagen degrades, EVs can accumulate, aggregate, nucleate hydroxyapatite, and form microcalcifications (19). Recent work from Hodroge and colleagues has identified specific regions on collagen fibers to which EVs bind: isoforms of 8-degree-polymerized oligogalacturonic acid mask the GFOGER sequence, inhibit vesicle-collagen binding, and impair the development of calcification (68).
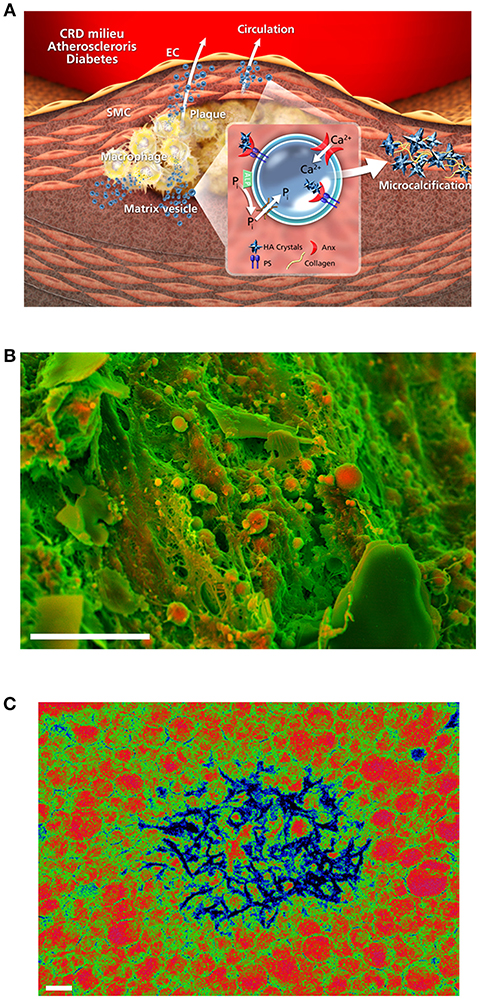
Figure 3. Accumulation and mineralization of calcifying EVs. (A) ALP produces inorganic phosphate (Pi) in the extravesicular space, and a mineral concentration gradient between the intra- and extra-vesicular spaces drives an influx of phosphate and calcium into EVs via suitable transporters. Hydroxyapatite nucleation is modulated by annexins and phosphatidylserines, leading to the production of calcified EVs and microcalcification; adapted from New and Aikawa (67). (B) Density-dependent color scanning electron microscopy (DDC-SEM) of calcified human atheroma (scale bar = 10 μm) reveals the accumulation of EVs as key building blocks of vascular calcification. (C) Pseudocolor transmission electron microscopy of macrophage-derived EVs reveals membrane-associated and intravesicular hydroxyapatite nucleation after calcium/phosphate treatment; adapted from New et al. (17) (scale bar = 200 nm).
How, then, does mineral first begin to nucleate and crystalize in EVs? While the specific location of nucleation remains unclear, under inflammatory or hyperlipidemic stimuli calcification is typically dependent on the increased expression and activity of ALP to free phosphate from a biological source (e.g., ATP) for nucleation. During cellular osteogenic transition, the multiligand sorting receptor sortilin regulates the release of EVs (69) and explicitly drives loading of activated ALP into calcifying EVs in a Rab11-dependent manner (70). Dimerization of this receptor appears necessary for the trafficking of sortilin, and thus ALP, into EVs (71). Notably, serum sortilin levels associate with increased risk of major adverse cerebrovascular and cardiovascular events, as well as with the severity of abdominal aortic calcification (72). In contrast, it appears likely that under hyperphosphatemic conditions (e.g., CKD or Monckeberg's syndrome), endocytosed phosphate binds with intracellular protein mediators of mineralization. Subsequently, exosomal EVs act to shuttle this mineral outside of the cell (18) in an ALP-independent manner. In bone, a precursor phase of amorphous calcium phosphate that acts as prenucleation clusters precedes hydroxyapatite crystallization (73). When compared with the parental cells (74), calcifying EVs are enriched in annexins, sphingomyelins, and phosphatidylserine (PS), a key mineral nucleation site on the EV membrane (75). Multiscale molecular dynamics simulations of calcium triphosphate Ca2(HPO4 formation show that palmitoyloleoylphosphatidylserine has the greatest affinity for binding Ca2+ ions, with a ~4-fold greater contact with Ca2+ ions than palmitoyloleoylphosphatidylethanolamine (followed by palmitoyloleoylphosphatidylinositol, palmitoylsphingomyelin, and palmitoyloleoylphosphatidylcholine having progressively fewer calcium ion interactions) (76). In addition, coarse-grained simulations have found that ion:water concentrations of >0.26 are necessary for EV mineral nucleation to occur within reasonable time periods at physiological temperatures (76).
In addition to PS, there is ample indication that several members of the annexin family may also be direct drivers of EV mineralization, though it is notable that specific mechanisms of action for the regulation of calcification by this protein family remain unclear. The 14 annexins act as voltage-gated Ca2+ ion channels and/or as Ca2+-dependent anionic phospholipid binding proteins [reviewed in (77)], and are intimately involved in endo/exocytosis, membrane structure, and lipid raft organization [reviewed in (78)]. Typically, EVs are enriched in annexins A2, A5, and A6, and annexin-enriched EVs derived from osteoblasts drive mineralization of cultured mesenchymal stem cells (MSCs) (79). Calcium and phosphate stimulation of macrophages induces externalization of PS and a resultant interaction of PS, annexin A5, and the calcium-binding protein S100A9 that leads to hydroxyapatite nucleation and which likely contributes to plaque calcification in CKD (17). Annexin A6 is preferentially increased in exosomes derived from calcifying vascular SMCs, where overexpression/knockdown studies demonstrate that it appears to function as an active mediator of ECM mineralization (18, 48). In calcified aortic valve leaflets, annexin A6 co-localizes with EV and is elevated in rat VIC-derived EVs under high calcium/phosphate conditions (80). Such high-phosphate conditions also increase shedding of calcifying EVs from both odontoblasts (81) and vascular SMCs (82) while driving the incorporation of annexin family members into EVs derived from the latter. Annexin A2 also prevents loading of the mineralization inhibitor fetuin-A into vascular SMC-derived EVs by directly binding to and inducing its endocytosis (63). Indeed, in both vascular SMC and osteoblast-derived EVs, increased annexin A2 levels result in a concomitant activation of EV ALP and calcification potential (63, 83).
Beyond annexins and phosphatidylserines, there is a large body of evidence that homeostatic loading of EVs with inhibitors of calcification such as fetuin-A and MGP (16) is disrupted in calcifying EVs (48), leading to a loss of inhibition of mineralization. Recent work by Viegas and colleagues found that serum from patients with advanced CKD contains EVs with reduced levels of fetuin-A, and Gla-rich protein (GRP) (84). Notably, circulating GRP-rich EVs appear to be produced by monocyte/macrophage populations and have additional anti-inflammatory functionality through the down-regulation of proinflammatory cytokine expression (85). Other recent proteomics studies have identified roles for the deposition of EV-derived prothrombin in modulating calcific burden and thrombogenesis in the vasculature (46). These EVs drive vascular SMC calcification via inflammatory induction of an osteochondrogenic phenotype, and this study determined that production of calcium phosphate crystals can be reduced specifically by the addition of γ-carboxylated GRP in vitro (84). In concert, EV levels of proteins that drive intracellular Ca2+ concentration (e.g., NADPH oxidase) and modulate oxidative stress (e.g., SOD2) are enriched (86). Proteomics of an ultracentrifugation-isolated calcifying EV population derived from human coronary artery SMCs demonstrates significant enrichment in proteins associated with glycosaminoglycan binding, calcium ion binding, ECM binding, and nitric oxide synthase regulation, among others (40). Most recently, the importance of cellular senescence to atherosclerotic calcification has come to light—senescent macrophages accumulate in the subendothelial space at disease onset, SMCs in advanced atherosclerotic plaques are highly senescent, and the deletion of senescent cells in mouse models of atherosclerosis and vascular calcification drives substantial lesion regression (87, 88). EVs isolated from the plasma of elderly donors or from cultures of senescent endothelial cells promote the calcification of human aortic SMCs; as subjects age, these EVs are more prevalent and contain greater amounts of calcium, annexin A2, annexin A6, and bone morphogenic protein-2 (BMP2) (89).
Roles in Intercellular Communication
As noted above, EVs are selectively enriched in specific miRNA, mRNA, and protein cargoes, and are utilized by cells as a means of local or long-distance intercellular paracrine/endocrine communication [Figure 4; (90)]. Functionally, the EV lipid bilayer serves to protect miRNAs/mRNAs and proteins from degradation by extracellular ribonucleases and proteases, respectively (91, 92)—thereby dramatically increasing the plasma/tissue half-life of these compounds in comparison to their un-packaged forms. As an example, preserved phosphoproteins have been recovered from exosomal samples even after 5 years of cryopreservation (92). While a not inconsequential portion of extracellular RNA is associated with Argonaute 2 protein complexes and not vesicles, the composition of these two pools is significantly different (57) and both differ from that of their donor cells (58). It is therefore likely that cells preferentially and actively select miRNAs for packaging into EVs. Substantially less is known about whether or how EV protein cargoes are processed or packaged. The advent of inexpensive, higher-throughput proteomics and low-input peptide isolation techniques (93) may help to tackle the question of whether protein pools are similarly regulated.
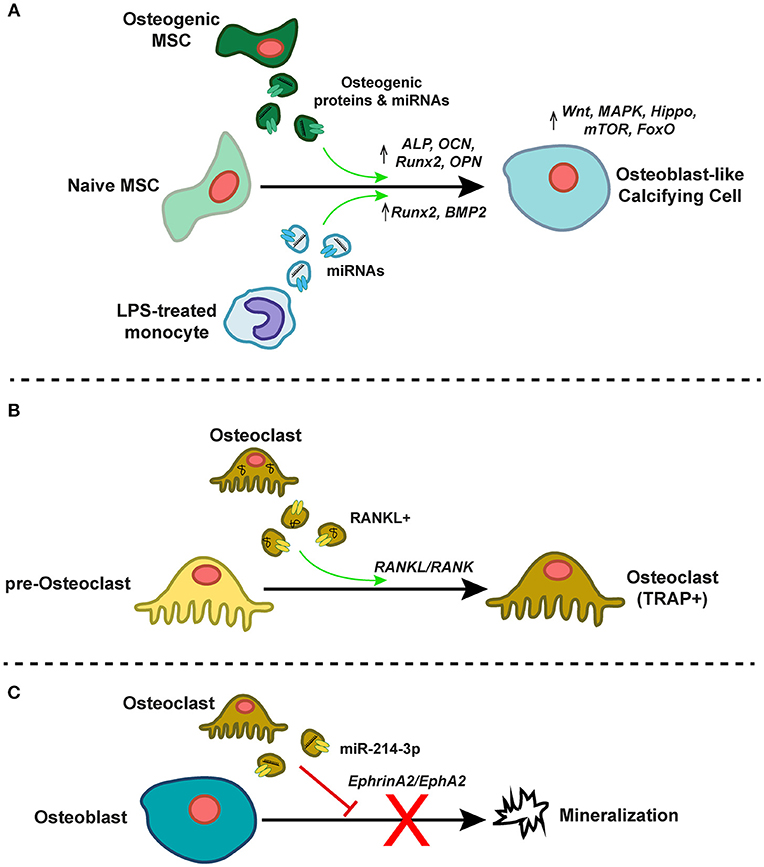
Figure 4. EVs as mediators of intercellular communication in the calcifying milieu. (A) EVs from osteogenic mesenchymal stem cells (MSCs) or lipopolysaccharide (LPS)-treated inflammatory monocytes contain pro-osteogenic proteins and miRNAs that induce pro-osteogenic gene expression and signaling programs in recipient naïve MSCs, thereby driving differentiation toward an osteoblast-like calcifying cell type. (B) TRAP- pre-osteoclasts differentiate to mature TRAP+ osteoclasts after treatment with RANKL-containing EVs derived from mature osteoclasts. This EV-associated RANKL induces RANK signaling in recipient pre-osteoclasts. (C) Osteoclast-derived EVs enriched in miR-214-3p inhibit mineralization by mature osteoblasts, perhaps via ephrinA2/EphA2 signaling.
Calcific and inflammatory burdens in both the arteries and aortic valve correlate with paradoxical osteoporotic bone remodeling (94), and so the well-described phenomena of EV-mediated control of osteoblasts- and/or osteoclastogenesis is of particular relevance to vascular calcification. EVs released by actively mineralizing pre-osteoblastic cells promote differentiation of recipient bone marrow stromal cells (BMSCs) to an osteogenic phenotype (95), and the proteome of these vesicles is enriched in pathways associated with exosome biogenesis, formation, uptake, and osteogenesis (96). This EV-induced osteogenic differentiation appears to be sensitive to extracellular magnesium levels, likely due to induction of autophagic processes (97). In concert, miRNA profiles in recipient pre-osteoblastic cells change significantly due to exosomal miRNA transfer, and result in activation of Wnt/β-catenin signaling via inhibition of Axin1 (95). Furthermore, treatment of human fetal pre-osteoblasts with EVs isolated from BMSCs induces osteogenic gene expression (ALP, OCN, Runx2, OPN), osteoblastic differentiation, and mineralization. These BMSC-derived EVs are enriched in the pro-osteogenic miRs−27a,−196a, and−206, and pharmacological abolishment of EV miR-196a partially inhibits EV-induced osteogenic gene expression (98). In vivo, hydrogel-based delivery of BMSC-derived EVs to rat critical-sized calvarial bone defects promotes bone regeneration. Interestingly, these EVs were derived from BMSCs under normal culture conditions, thus implying that even basal state EVs from these cell types have the ability to induce an osteogenic phenotypic switch. There is also evidence that EV-induced osteoblastogenesis is sensitive to the temporal stage of osteogenesis of the EV “donor” cell population. Vesicles collected from human MSCs at both early and late stages of osteogenic differentiation can induce ALP expression in naïve homotypic cells, but only those derived from late-stage osteogenic MSCs produce mineralization of the ECM in recipient cell cultures. This effect is mediated, at least in part, by significant temporal alterations in 16 EV miRNAs whose functions are associated with the modulation of the Wnt, MAPK, Hippo, mTOR, and FoxO signaling pathways—all of which are involved in regulation of osteogenesis (99). The pro-osteoblastogenic effect of EVs is not confined to those derived from osteoblast-like cells. An inflammatory stimulus of miRNA-containing EVs from LPS-treated monocytes drives increases in osteogenic gene programs (e.g., Runx2, BMP2) in human MSCs within 72 h of EVs being taken up (100, 101).
Understanding osteoblast-osteoclast interplay and EV-induced osteoclastogenesis in the cardiovascular system is paramount. Vesicles shed from osteoblasts contain the receptor activator of nuclear factor kappa-β ligand (RANKL), and LAMP1-positive EVs shed by both osteoblasts and osteoclasts also contain tartrate-resistant acid phosphatase (TRAP) and osteoprotegerin (OPG) (102). When administered to RAW264.7 osteoclast precursors, these EVs deliver RANKL, causing stimulation of RANKL-RANK signaling, and differentiation of the recipient osteoclast precursors to become TRAP-positive osteoclasts (103). In contrast, while EVs from murine pre-osteoclasts drive mouse marrow hematopoietic precursors to differentiate to TRAP-positive osteoclasts, those from mature osteoclasts do not induce osteoclastogenesis in recipient cultures, but rather inhibited it (104). Mature osteoclast-derived EVs are enriched in RANK, and subsequent depletion of RANK in these EVs restored their ability to induce osteoclast differentiation. RANK-positive EVs may competitively bind to RANKL (in a similar manner to RANKL/OPG binding), thus inhibiting osteoclastogenesis. Alternatively, mature osteoclastic EVs may deliver other regulatory cargoes (104). Osteoclasts may also regulate osteoblastic bone formation via EVs (105, 106). Levels of exosomal miR-214-3p are highly associated with inhibition of bone formation in elderly women with fractures (107), and overexpression of miR-214-3p in osteoclasts inhibits osteoblast-mediated mineralization in co-culture (107). This phenomenon may be mediated by ephrinA2/EphA2 signaling (106). Together, these findings indicate the presence of a complex, EV-dependent paracrine regulatory system of osteoblast/osteoclastogenesis [reviewed in (108)]. Despite this multitude of studies, exact mechanisms and the specific miRNAs/proteins responsible for this cross-talk remain elusive, as does confirmatory evidence of in vivo EV crosstalk in vascular calcification.
Endothelial-derived EVs from both patients with acute coronary syndrome and senescent endothelial cell (EC) EC cultures can induce early senescence in receptor ECs (the former of which occurs in a shear-sensitive manner at low atheroprone shear stresses) and are associated with increased levels of receptor cell oxidative stress, MAPK and Akt signaling, and upregulation of p53 (109). These findings demonstrate that EVs released from senescent cells can also drive senescence of neighboring cells in a paracrine manner, and EV levels/composition may serve as important diagnostic biomarkers of vascular calcification. For example, urinary exosomal fetuin-A levels are a strong predictive biomarker of acute renal injury (110). Other studies have shown that SMC phenotypes in the vessel wall are regulated by EC-derived EVs that are selectively loaded with miR-126,−143, and−145 (111, 112).
There is also evidence of EV-driven cell-cell transfer of mRNA: in co-culture, next generation sequencing shows that a multitude of mRNAs are transferred through EVs between differing cell types. The function of these mRNAs ranges from transcriptional regulation to protein coding, and their biological roles are associated with ECM regulation, cell adhesion, glycoproteins, and signal peptides (113). One long-time mystery in valvular biology has been the question of how valvular endothelial cell (VEC)/VIC paracrine signaling occurs—how do VEC-detected changes in shear stress, stretch, circulating growth factor, and/or lipid levels modulate VIC function, and vice-versa (114). In VIC/VEC co-cultures, VIC-derived EVs are taken up into the perinuclear space by VECs (115) through the endosomal pathway, and EV transfer of miRNA, mRNA, and protein may therefore be a key mode of VEC/VIC communication.
Therapeutic Potential
Along with their clear implication in the pathogenesis of cardiovascular calcification, EVs may also hold the key to successfully treating these pathologies in the clinic. Recently, a number of studies have begun to explore the notion that EVs could themselves have a unique ability to act as therapeutic agents [reviewed in (116)]. Indeed, the fact that they are so centrally positioned in the progression of mineralization means that delivery of therapeutic forms of EVs or disruption of the contents, synthesis, release, or extracellular accumulation of endogenous pro-calcifying EV populations could efficaciously prevent or regress calcification. MSC-derived exosomes have shown positive results in a number of disease models, such as reduction of myocardial infarction size and improvements to post-MI inflammation (117, 118), resolution of pulmonary hypertension (119), amelioration of kidney fibrosis (120), and restoration of neurovascular function and plasticity (121). Importantly, EVs derived from immunocompetent MSC populations may themselves also be immunocompetent—thus providing a non-means of delivering complex biological cargoes [reviewed in (122)]. One popular approach that can maintain tissue homeostasis or reverse cardiovascular pathogenesis is to deliver EVs collected from the conditioned media of cultured “producer” cells [reviewed in (123)]. Cargo loading generally occurs either during biogenesis (by overexpression of cargo in the producer cells and relying on increased intracellular levels to enrich EV levels), or by post-biogenesis, by collecting EVs then subsequently loading them by transfection or hydrophobic tagging. miRNAs and siRNAs have been successfully loaded into EVs by electroporation for nearly a decade (124, 125). EVs can deliver miRNAs to difficult-to-transfect cells such as MH-S lines and bone marrow-derived macrophages (126), and more contemporary approaches that leverage conjugation of RNA with hydrophobic cholesterols have increased loading efficiencies to nearly 50% (127). While RNA cargoes have been most frequently employed, protein or small molecule loading is frequently a more attractive proposition (obviates the need for transcription/translation in treated cells, along with the advantage of inherently higher cargo stability). By tagging with a WW domain, protein of interest can be forced to interact with ubiquitination-promoting Ndfip1, that then drives loading of the target protein into exosomes (128). As an alternative light-inducible and reversible approach (i.e., spatially/temporally controlled loading and controlled release), cargo proteins can be tagged with the photoreceptor CRY2 while CD9 is fused with a truncated CRY-interacting domain. Upon exposure to blue light, these domains interact and the complex is loaded into EVs (129). In addition, small molecules such as doxorubicin and porphyrins have been loaded into EVs by electroporation, saponin treatment, and extrusion (130, 131), though encapsulation efficiency is often <20%. Another option is to use fusogenic liposomes and a packaging cell line: the small molecule of interest is incorporated into liposomes, which efficiently fuse with the cellular plasma membrane, delivering the small molecule into the packaging cells. EVs produced by those cells (and naturally enriched in the molecule of interest due to mass action) can then be collected (132).
Though the advantages of active EV loading are clear, they do come with additional regulatory requirements centered on demonstrations of purity. As an alternative, vesicular secretome fractions (VSFs) or naïve, unmanipulated EVs derived from cell types relevant to the pathology of interest can be employed. EVs from clonal cardiac stem cells mitigate ventricular damage in animal models of acute myocardial infarction (133). Oxidative stress (frequently coincident with vascular calcification) causes uncoupling of endothelial nitric oxide synthase (eNOS) and endothelial dysfunction. EVs derived from cultured human umbilican vein endothelial cells (HUVECs) carry functional eNOS and protect the vasculature against oxidative stress by Akt-mediated upregulation of superoxide dismutase under ex vivo culture conditions. Despite these promising initial efforts having become a key focus of the drug discovery field, the impact of exosomal therapeutics on vascular calcification remains unstudied. A degree of care will be required when such studies are undertaken, as MSC-derived EVs can—under the proper conditions—robustly drive osteogenic differentiation, endochondral ossification, and bone regeneration (95, 98, 134). Besides using exosomes as a delivery vehicle, the contents of calcifying EVs are proving to be an important source of drug targets in and of themselves. Treatment with the inhibitor of acid sphingomyelinase imipramine reduces calcifying EV release from osteoblasts (135), and the Ca2+ channel blocker verapamil reduces SMC calcifying EV biogenesis, vesicle ALP activity, and ECM mineralization, while also impairing atheroma formation in the rat aorta (136). Dimerization of the aforementioned sortilin protein also appears to regulate its packaging in EVs—inhibition of sortilin homodimer formation is therefore another promising EV-associated therapeutic strategy (71). EVs thus offer the promise of a biological, acellular, targeted, and immunocompetent means of delivering complex therapeutic cocktails to calcifying cardiovascular tissue (24).
Methods and Guidelines for Isolation of and Experimentation With Extracellular Vesicles
There are several important methodological considerations when performing experiments involving exosomes or EVs. As this field has matured, a number of vesicle isolation techniques have become popular (Table 1), however EV yield, integrity, biodistribution, clearance, and breakdown are all affected by the choice of isolation method (137). Ultracentrifugation is one of the most simple, widely-available, and cost-effective strategies for vesicle collection, though it will promote vesicle clumping and may cause soluble factors and ECM-rich protein aggregates to pellet along with EVs themselves (138). Recovery is typically variable (10–80%), and is capable of concentrating a sample ~8x (139). Fortunately, calcifying EVs have an increased density over other EVs (due perhaps to elevated mineral content) and pellet more quickly under ultracentrifugation (40). Alternative methods include density gradient isolation/flotation, where ultracentrifugation is paired with sucrose or ioxidanol gradients to obtain fractions enriched in EVs based on mass density and can obtain EV recovery in the range of 5–50% (140). Care must be taken when using sucrose, as there is evidence that non-isosmotic sucrose concentrations cause leaching of vesicle cargoes during isolation [summarized in (141)]. Size exclusion chromatography can be performed on a single column, removes 99% of soluble plasma proteins and does not induce EV aggregation, though it is prone to co-isolation of viruses, protein aggregates, and very large proteins (142–144). Recovery rates are typically 30–90% of EVs (145). Ultrafiltration's ability to concentrate from soluble components EVs is currently unmatched, with an ability to do so up to 250x with nearly an 80% recovery (146). To isolate specific subpopulations of EVs, immunocapture assays using immobilized monoclonal antibodies can be utilized (147). Non-EV proteins are often recovered in these assays, and care must be taken to account for cross-reactivity and non-specific binding (31, 148). Recently, commercially-available kit-based precipitation approaches have become popular due to their simplicity and ease of use. These kits leverage the volume-excluding properties of polyethylene glycol polymers (149) to reduce EV solubility (150). Recovery of ~90% and a 50x concentration performance can be expected by these kits (151). Caution is necessary when attempting to quantify precipitation-isolated EVs by nanoparticle tracking analysis, as the polymer components of the precipitation buffer appear to frequently be detected as false-positive EV particles. Besides these standard approaches, a number of innovative techniques are also under development. By using ultrasonic standing waves, acoustic separation can exert differential force on exosomes in a scalable manner (152). Flow field fractionation exploits perpendicular flow fields to separate exosomes by their size-based diffusivity (153). Lastly, microfabricated micropillar-based nano-traps can be tuned to capture EVs in a particular size range of interest, while washing out cellular debris, other EV size fractions, and cellular debris (154). It is important to note that each of the methods described above has its own unique sets of tradeoffs (on ease of use, availability, cost, recovery, concentration, etc.), none is perfect, and there exists no consensus on a single gold-standard isolation approach. Instead, investigators must make an effort to comprehensively report the method that was selected, and to rigorously assess and describe the phenotype of their isolated EV population (e.g., purity, concentration, size range, morphology).
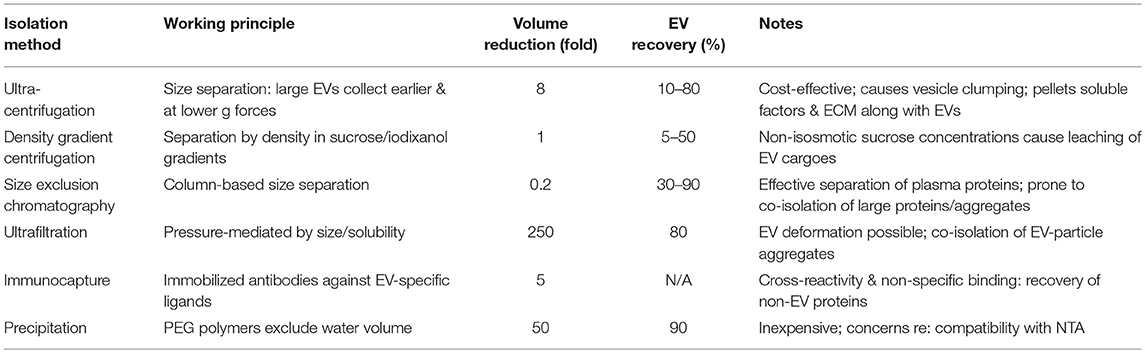
Table 1. Methods of extracellular vesicle isolation.
Though researchers have several options available in order to characterize EV populations of interest, electron microscopy (EM) is the gold standard for exosome imaging. The resolution of this technique is ~1–5 nm (139), allows for multiplexed immunogold labeling to phenotype EVs, and can reveal EV population size distributions, presence of aggregation, and inclusion of debris contaminants. EM is not, however, able to measure EV concentrations. The recent advent of density-dependent scanning electron microscopy (SEM) allows for in-situ imaging of calcifying EVs and assessment of their size, density, and biophysical accumulation in calcified tissues, hydrogels, or matrices (19). In situ imaging of vesicles in concert with (immuno)fluorescence/confocal light-based approaches is now feasible with the advent of super-resolution microscopy (e.g., SIM, TIRF, PALM, or STORM), which overcomes the traditional resolution limitations that diffraction places on light microscopy (155). These imaging techniques enable resolutions as low as 20 nm, sufficient to visualize individual exosomes within a physiological ECM (19).
Though typically used to count, phenotype, and/or separate cells, flow cytometry can also be applied to EVs—with the essential caveat that because EVs are orders of magnitude smaller than cells, light scattering from these exosomes may be close to, or below, the background noise detected on current-generation flow cytometers (156). Combinatorial counting that leverages tags, membrane dyes, and/or antibodies against vesicle markers (e.g., annexins) can improve this performance (157). Instrument settings must be tuned for EV analysis, and one must remember that for the purposes of calibration, the size of EVs cannot be related directly to the size of calibrator polystyrene/silica beads due to differences in refractive index (158), though models do exist that attempt to account for these elements (159). By passing EVs through a pore and measuring a resultant change in electrical impedance, resistive pulse sensing (RPS) can quantify exosome size and electrophoretic mobility (160), though it cannot differentiate between EVs and contaminating particles (139) and pores can be prone to clogging if samples are not pre-filtered and/or have already undergone size exclusion chromatography (161). One of the most commonly used and cost-effective means of measuring EV size distributions and concentrations is nanoparticle tracking analysis (NTA), which optically tracks vesicle Brownian motion (162). Many NTA instruments also incorporate measurement of membrane charge state, or zeta potential. A sufficiently-large number of particles (typically >4,000) must be measured to ensure the statistically-relevant accuracy of the size distribution, and reference particles should be utilized for concentration calibration (139).
Once EVs have been isolated, quantified, and characterized, their contents can then be measured. Next-generation sequencing has been used to examine EV RNA populations derived from a number of different cell types/microenvironmental milieus (163, 164), and rigorous guidelines have been developed for the use of NGS techniques on exosomes (165). qPCR and microarray techniques can be used for EV RNA analysis, but both lack an ability to identify novel sequences and do not perform as well as NGS when quantifying transcript numbers (139). Both miRNA and mRNA expression profiles differ based on isolation technique [e.g., precipitation vs. ultracentrifugation (166)], with iodixanol density gradients having the best purity performance (167). At the next stop on the central dogma, EV protein levels are frequently assessed by Western blotting or ELISA. However, the advent of proteomic approaches to quantify thousands of proteins has revolutionized this approach—nearly 10,000 vesicle-associated proteins have been annotated (168, 169). While care must be taken to remove or account for co-isolating non-EV proteins, recent advantages in density-dependent ultracentrifugation-based isolation of SMC-derived calcifying EV populations enabled LC-MS/MS identification and quantification of over 400 EV proteins, with subsequent pathway analysis finding calcification-associated functionalities (40). Lastly, EVs also carry cellular metabolites that may provide insight into the biochemical status of their originating cells, and mass spectrometry-based metabolomics techniques to assess the EV metabolome have recently begun to be presented (60, 61, 170, 171). Importantly, label-free and non-targeted metabolite identification is a massively more complex undertaking than global proteomics, and metabolite libraries must be carefully and rigorously employed to confirm target IDs (172).
One additional area of caution should be noted when working with EVs: when cell types relevant to vascular calcification are cultured (e.g., endothelial, SMCs, VICs, monocyte/macrophage, osteoblast, osteoclasts, etc.), the culture media is typically supplemented with ~1–10% fetal bovine serum (FBS) in order to provide essential growth factors and adsorbed ECM components that aid in cell attachment. Importantly, even commercially-available lots of FBS are replete with EVs and exosomes (often somewhere on the order of ~10e10 exosomes per ml). Besides acting as a contaminating source of extracellular RNA or protein when co-isolated with endogenously-produced EVs (173), this undefined and highly variable component of the culture conditions can have an outsized effect on cell behavior (174, 175). Various strategies are used to account for this: exosome-depleted serum can be purchased, though (i) it is costly, (ii) there is currently no exosome-depleted human serum on the market, (iii) producing it in-lab typically requires access to an ultracentrifuge, (iv) such serum is only exosome-depleted (of ≥90% of endogenous serum exosomes), not exosome-free, and (v) there is evidence that exosome-depleted serum has a reduced capacity to support cell growth (176). Alternatively, cells can be cultured in normal concentrations of FBS to support growth, attachment, and phenotype, then switched low concentrations of FBS (e.g., incomplete media or 0.1% FBS) for the final portion of an experiment. This solution is not ideal either; as differing FBS levels alter endogenous cellular exosome production, release, and contents. Furthermore, if EV-ECM interactions are relevant to the study aims or the milieu being modeled [e.g., experiments performed on 3D hydrogels, ex vivo tissue culture, or matrix-bound vesicles (63)], then a short-term switch to incomplete media will have a limited capacity to flush/remove FBS-derived EVs that have built up in the ECM throughout the remainder of the experimental timeline. One solution: in proteomics experiments, the species of detected/sequenced proteins can be determined with a reasonable degree of accuracy (177). If the cells being studied are non-bovine, then the entire extracellular EV proteome can be isolated, detected, and sequenced at once, followed by filtering out all proteins identified as bovine in origin. Unfortunately, this is much more difficult with miRNAs due to their high degree of conservation across species (178). Regardless, great care should be taken to minimize and account for any exogenous vesicle contamination from culture media.
Conclusions and Future Directions
The discovery of calcifying EVs has led to a growing appreciation for the key role that these bodies play as both active and passive constituents of vascular calcification. One appropriate analogy may be that of masonry: by inherently regulating their own aggregation and mineralization, EVs act as both the bricks and the mortar of microcalcifications. Importantly, they also actively control the work of the bricklayers themselves (e.g., osteoblasts, osteoclasts). In short, EVs seem to be intimately connected to all stages of the pathogenesis of vascular calcification. As this burgeoning field matures, a number of emerging questions remain unanswered: Can we identify specific roles for EV microRNAs in preventing or promoting vascular calcification? In vivo, how does osteoblast/osteoclast vesicle cross-talk in vivo modulate mineralization of vascular tissue in both homeostasis and disease? By what specific mechanisms do annexins mediate calcifying vesicle aggregation and mineralization, and how do annexins interact with sphingomyelin and phosphatidylserine to promote nucleation? Can EVs be therapeutically administered locally/systemically without off-target effects (e.g., unintended promotion of vascular osteogenesis and/or skeletal osteoporosis). In the future, the benefits of better understanding the mechanistic basis of vascular EV calcification may be two-fold: first, robust study of calcifying EVs could yield a novel set of anti-calcification drug targets for small molecule high-throughput screening that are largely distinct from existing (intra)cellular targets due to the selective loading of EV cargoes. Second, purposeful introduction of exogenous engineered EVs to the calcifying vascular milieu might be a viable and revolutionary therapeutic delivery strategy, whereby inhibitors of osteogenesis and mineralization or inducers of osteoclast differentiation could be efficiently conveyed into atherosclerotic plaques or fibrocalcific valve leaflets. Indeed, such an approach could circumvent many of the issues associated with intracellular delivery of small-molecules and/or nucleic acids. In the long run, it may even be feasible to develop artificially-functionalized EVs that leverage the physicochemical mechanisms of mineral formation in order to locally arrest mineral nucleation, prevent further aggregation of endogenous calcifying EVs, and even stabilize vulnerable atheromata.
Author Contributions
MB performed an extensive literature review, drafted the manuscript, prepared figures and tables. EA proposed the subject of the review, and critically revised and edited the manuscript.
Funding
This work was funded by the National Institutes of Health (NIH) R01 grants R01HL114805, R01HL136431, and R01HL109889 (EA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALP, alkaline phosphatase; BMSC, bone marrow stromal cell; CKD, Chronic kidney disease; ECM, extracellular matrix; EC, endothelial cell; EV, extracellular vesicle; LPS, lipopolysaccharide; MGP, matrix Gla protein; MSC, mesenchymal stem cell; MV, matrix vesicle; MVB, multivesicular bodies; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor kappa-β; RANKL, receptor activator of nuclear factor kappa-β ligand; SMC, smooth muscle cell; TNAP, tissue non-specific alkaline phosphatase; TRAP, tartrate-resistant acid phosphatase; VEC, valvular endothelial cell; VIC, valvular interstitial cell.
References
1. Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension (2001) 38:938–42. doi: 10.1161/hy1001.096358
2. Chen J, Budoff MJ, Reilly MP, Yang W, Rosas SE, Rahman M, et al. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. (2017) 2:635–43. doi: 10.1001/jamacardio.2017.0363
3. Dweck MR, Chow MW, Joshi NV, Williams MC, Jones C, Fletcher AM, et al. Coronary arterial 18F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol. (2012) 59:1539–48. doi: 10.1016/j.jacc.2011.12.037
4. Hutcheson JD, Blaser MC, Aikawa E. Giving calcification its due: recognition of a diverse disease: A first attempt to standardize the field. Circ Res. (2017) 120:270–3. doi: 10.1161/circresaha.116.310060.
5. Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. (2006) 99:1044–59. doi: 10.1161/01.RES.0000249379.55535.21
6. Kelly-Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L, Weinbaum S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci USA. (2013) 110:10741–6. doi: 10.1073/pnas.1308814110
7. Maldonado N, Kelly-Arnold A, Cardoso L, Weinbaum S. The explosive growth of small voids in vulnerable cap rupture; cavitation and interfacial debonding. J Biomech. (2013) 46:396–401. doi: 10.1016/j.jbiomech.2012.10.040
8. Otto CM, Burwash IG, Legget ME, Munt BI, Fujioka M, Healy NL, et al. Prospective study of asymptomatic valvular aortic stenosis. Clin Echocardiogr Exerc Predict Outcome Circul. (1997) 95:2262–70. doi: 10.1161/01.CIR.95.9.2262
9. Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, et al. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol. (2014) 34:2387–93. doi: 10.1161/ATVBAHA.114.302523
10. van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. (2012) 64:676–705. doi: 10.1124/pr.112.005983
11. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
12. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes) J Biol Chem. (1987) 262:9412–20.
13. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. (1996) 183:1161–72. doi: 10.1084/jem.183.3.1161
14. Pucci F, Garris C, Lai CP, Newton A, Pfirschke C, Engblom C, et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science (2016) 352:242–6. doi: 10.1126/science.aaf1328
15. Wei H, Malcor JM, Harper MT. Lipid rafts are essential for release of phosphatidylserine-exposing extracellular vesicles from platelets. Sci Rep. (2018) 8:9987. doi: 10.1038/s41598-018-28363-4
16. Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. (2004) 15:2857–67. doi: 10.1097/01.ASN.0000141960.01035.28
17. New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. (2013) 113:72–7. doi: 10.1161/CIRCRESAHA.113.301036
18. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. (2015) 116:1312–23. doi: 10.1161/CIRCRESAHA.116.305012
19. Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. (2016) 15:335–43. doi: 10.1038/nmat4519
20. Delcayre A, Le Pecq JB. Exosomes as novel therapeutic nanodevices. Curr Opin Mol Ther. (2006) 8:31–8.
21. Krohn JB, Hutcheson JD, Martinez-Martinez E, Irvin WS, Bouten CV, Bertazzo S, et al. Discoidin domain receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell fibrocalcific response via transforming growth factor-beta signaling. Arterioscler Thromb Vasc Biol. (2016) 36:525–33. doi: 10.1161/ATVBAHA.115.307009
22. Blaser MC, Wei K, Adams RL, Zhou YQ, Caruso LL, Mirzaei Z, et al. Deficiency of natriuretic peptide receptor 2 promotes bicuspid aortic valves, aortic valve disease, left ventricular dysfunction, and ascending aortic dilatations in mice. Circ Res. (2017) 122:405–16. doi: 10.1161/CIRCRESAHA.117.311194
23. Schlotter F, Halu A, Goto S, Blaser MC, Body SC, Lee LH, et al. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation (2018) 138:377–393. doi: 10.1161/CIRCULATIONAHA.117.032291
24. Azoidis I, Cox SC, Davies OG. The role of extracellular vesicles in biomineralisation: current perspective and application in regenerative medicine. J Tissue Eng. (2018) 9:2041731418810130. doi: 10.1177/2041731418810130
25. Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles (2014) 3:26913. doi: 10.3402/jev.v3.26913
26. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
27. Blanc L, Vidal M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases (2018) 9:95–106. doi: 10.1080/21541248.2016.1264352
28. Al-Nedawi K, Meehan B, Rak J. Microvesicles: messengers and mediators of tumor progression. Cell Cycle (2009) 8:2014–8. doi: 10.4161/cc.8.13.8988
29. Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, et al. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles (2013) 2:20677. doi: 10.3402/jev.v2i0.20677
30. Hutcheson JD, Aikawa E. Extracellular vesicles in cardiovascular homeostasis and disease. Curr Opin Cardiol. (2018) 33:290–297. doi: 10.1097/HCO.0000000000000510
31. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. (2016) 113:E968–77. doi: 10.1073/pnas.1521230113
32. Anderson HC. Electron microscopic studies of induced cartilage development and calcification. J Cell Biol. (1967) 35:81–101. doi: 10.1083/jcb.35.1.81
33. Bonucci E. Fine structure of early cartilage calcification. J Ultrastruct Res. (1967) 20:33–50. doi: 10.1016/S0022-5320(67)80034-0
34. Anderson HC. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J Cell Biol. (1969) 41:59–72. doi: 10.1083/jcb.41.1.59
35. Hale JE, Wuthier RE. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J Biol Chem. (1987) 262:1916–25.
36. Thouverey C, Strzelecka-Kiliszek A, Balcerzak M, Buchet R, Pikula S. Matrix vesicles originate from apical membrane microvilli of mineralizing osteoblast-like Saos-2 cells. J Cell Biochem. (2009) 106:127–38. doi: 10.1002/jcb.21992
37. Leach RJ, Schwartz Z, Johnson-Pais TL, Dean DD, Luna M, Boyan BD. Osteosarcoma hybrids can preferentially target alkaline phosphatase activity to matrix vesicles: evidence for independent membrane biogenesis. J Bone Miner Res. (1995) 10:1614–24. doi: 10.1002/jbmr.5650101103
38. Kim KM. Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc. (1976) 35:156–62.
39. Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ, Valenzuela SM. Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic Res Cardiol. (2007) 102:133–43. doi: 10.1007/s00395-006-0637-9
40. Hutcheson JD, Goettsch C, Pham T, Iwashita M, Aikawa M, Singh SA, et al. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. J Extracell Vesicles (2014) 3:25129. doi: 10.3402/jev.v3.25129
41. Akisaka T, Gay CV. The plasma membrane and matrix vesicles of mouse growth plate chondrocytes during differentiation as revealed in freeze-fracture replicas. Am J Anat. (1985) 173:269–86. doi: 10.1002/aja.1001730404
42. Thyberg J, Nilsson S, Friberg U. Electron microscopie and enzyme cytochemical studies on the guinia pig metaphysis with special reference to the lysosomal system of different cell types. Cell Tissue Res. (1975) 156:273–99.
43. Xiao Z, Camalier CE, Nagashima K, Chan KC, Lucas DA, de la Cruz MJ, et al. Analysis of the extracellular matrix vesicle proteome in mineralizing osteoblasts. J Cell Physiol. (2007) 210:325–35. doi: 10.1002/jcp.20826
44. Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. (2017) 14:259–72. doi: 10.1038/nrcardio.2017.7
45. Schurgers LJ, Akbulut AC, Kaczor DM, Halder M, Koenen RR, Kramann R. Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Front Cardiovasc Med (2018) 5:36. doi: 10.3389/fcvm.2018.00036
46. Kapustin AN, Schoppet M, Schurgers LJ, Reynolds JL, McNair R, Heiss A, et al. Prothrombin loading of vascular smooth muscle cell-derived exosomes regulates coagulation and calcification. Arterioscler Thromb Vasc Biol. (2017) 37:e22–32. doi: 10.1161/ATVBAHA.116.308886
47. Majeska RJ, Wuthier RE. Studies on matrix vesicles isolated from chick epiphyseal cartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Biochim Biophys Acta (1975) 391:51–60. doi: 10.1016/0005-2744(75)90151-5
48. Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. (2011) 109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808
49. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. (2002) 160:673–80. doi: 10.1016/S0002-9440(10)64887-0
50. Schmidt JR, Kliemt S, Preissler C, Moeller S, von Bergen M, Hempel U, et al. Osteoblast-released matrix vesicles, regulation of activity and composition by sulfated and non-sulfated glycosaminoglycans. Mol Cell Proteomics (2016) 15:558–72. doi: 10.1074/mcp.M115.049718
51. Mizuno Y, Yagi K, Tokuzawa Y, Kanesaki-Yatsuka Y, Suda T, Katagiri T, et al. miR-125b inhibits osteoblastic differentiation by down-regulation of cell proliferation. Biochem Biophys Res Commun. (2008) 368:267–72. doi: 10.1016/j.bbrc.2008.01.073
52. Goettsch C, Rauner M, Pacyna N, Hempel U, Bornstein SR, Hofbauer LC. miR-125b regulates calcification of vascular smooth muscle cells. Am J Pathol. (2011) 179:1594–600. doi: 10.1016/j.ajpath.2011.06.016
53. Wu T, Zhou H, Hong Y, Li J, Jiang X, Huang H. miR-30 family members negatively regulate osteoblast differentiation. J Biol Chem. (2012) 287:7503–11. doi: 10.1074/jbc.M111.292722
54. Chen NX, Kiattisunthorn K, O'Neill KD, Chen X, Moorthi RN, Gattone VH II, et al. Decreased microRNA is involved in the vascular remodeling abnormalities in chronic kidney disease (CKD). PLoS ONE (2013) 8:e64558. doi: 10.1371/journal.pone.0064558
55. Chaturvedi P, Chen NX, O'Neill K, McClintick JN, Moe SM, Janga SC. Differential miRNA Expression in Cells and Matrix Vesicles in Vascular Smooth Muscle Cells from Rats with Kidney Disease. PLoS ONE (2015) 10:e0131589. doi: 10.1371/journal.pone.0131589
56. Morhayim J, van de Peppel J, Dudakovic A, Chiba H, van Wijnen AJ, van Leeuwen JP. Molecular characterization of human osteoblast-derived extracellular vesicle mRNA using next-generation sequencing. Biochim Biophys Acta (2017) 1864:1133–41. doi: 10.1016/j.bbamcr.2017.03.011
57. Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. (2011) 108:5003–8. doi: 10.1073/pnas.1019055108
58. Diehl P, Fricke A, Sander L, Stamm J, Bassler N, Htun N, et al. Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovasc Res. (2012) 93:633–44. doi: 10.1093/cvr/cvs007
59. Mastrangelo A, Armitage EG, Garcia A, Barbas C. Metabolomics as a tool for drug discovery and personalised medicine. A review. Curr Top Med Chem. (2014) 14:2627–36. doi: 10.2174/1568026614666141215124956
60. Iraci N, Gaude E, Leonardi T, Costa ASH, Cossetti C, Peruzzotti-Jametti L, et al. Extracellular vesicles are independent metabolic units with asparaginase activity. Nat Chem Biol. (2017) 13:951–5. doi: 10.1038/nchembio.2422
61. Puhka M, Takatalo M, Nordberg ME, Valkonen S, Nandania J, Aatonen M, et al. Metabolomic profiling of extracellular vesicles and alternative normalization methods reveal enriched metabolites and strategies to study prostate cancer-related changes. Theranostics (2017) 7:3824–41. doi: 10.7150/thno.19890
62. Wu LN, Genge BR, Lloyd GC, Wuthier RE. Collagen-binding proteins in collagenase-released matrix vesicles from cartilage. Interaction between matrix vesicle proteins and different types of collagen. J Biol Chem. (1991) 266:1195–203.
63. Chen NX, O'Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res. (2008) 23:1798–805. doi: 10.1359/jbmr.080604
64. Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. (2015) 6:7164. doi: 10.1038/ncomms8164
65. Hsu HH, Tawfik O, Sun F. Mechanism of dystrophic calcification in rabbit aortas: temporal and spatial distributions of calcifying vesicles and calcification-related structural proteins. Cardiovasc Pathol. (2004) 13:3–10. doi: 10.1016/S1054-8807(03)00093-0
66. de Jong OG, van Balkom BW, Gremmels H, Verhaar MC. Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2. J Cell Mol Med. (2016) 20:342–50. doi: 10.1111/jcmm.12730
67. New SE, Aikawa E. Role of extracellular vesicles in de novo mineralization: an additional novel mechanism of cardiovascular calcification. Arterioscler Thromb Vasc Biol. (2013) 33:1753–8. doi: 10.1161/ATVBAHA.112.300128
68. Hodroge A, Trecherel E, Cornu M, Darwiche W, Mansour A, Ait-Mohand K, et al. Oligogalacturonic acid inhibits vascular calcification by two mechanisms: inhibition of vascular smooth muscle cell osteogenic conversion and interaction with collagen. Arterioscler Thromb Vasc Biol. (2017) 37:1391–401. doi: 10.1161/ATVBAHA.117.309513
69. Wilson CM, Naves T, Vincent F, Melloni B, Bonnaud F, Lalloue F, et al. Sortilin mediates the release and transfer of exosomes in concert with two tyrosine kinase receptors. J Cell Sci. (2014) 127(Pt 18):3983–97. doi: 10.1242/jcs.149336
70. Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. (2016) 126:1323–36. doi: 10.1172/JCI80851
71. Itoh S, Mizuno K, Aikawa M, Aikawa E. Dimerization of sortilin regulates its trafficking to extracellular vesicles. J Biol Chem. (2018) 293:4532–44. doi: 10.1074/jbc.RA117.000732
72. Goettsch C, Iwata H, Hutcheson JD, O'Donnell CJ, Chapurlat R, Cook NR, et al. Serum sortilin associates with aortic calcification and cardiovascular risk in men. Arterioscler Thromb Vasc Biol. (2017) 37:1005–11. doi: 10.1161/ATVBAHA.116.308932
73. Mahamid J, Aichmayer B, Shimoni E, Ziblat R, Li C, Siegel S, et al. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc Natl Acad Sci USA. (2010) 107:6316–21. doi: 10.1073/pnas.0914218107
74. Wuthier RE, Lipscomb GF. Matrix vesicles: structure, composition, formation and function in calcification. Front Biosci. (2011) 16:2812–902. doi: 10.2741/3887
75. Genge BR, Wu LN, Wuthier RE. In vitro modeling of matrix vesicle nucleation: synergistic stimulation of mineral formation by annexin A5 and phosphatidylserine. J Biol Chem. (2007) 282:26035–45. doi: 10.1074/jbc.M701057200
76. Pokhrel R, Gerstman BS, Hutcheson JD, Chapagain PP. In silico investigations of calcium phosphate mineralization in extracellular vesicles. J Phys Chem B (2018) 122:3782–9. doi: 10.1021/acs.jpcb.8b00169
77. Lizarbe MA, Barrasa JI, Olmo N, Gavilanes F, Turnay J. Annexin-phospholipid interactions. Functional implications. Int J Mol Sci. (2013) 14:2652–83. doi: 10.3390/ijms14022652
78. Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. (2005) 6:449–61. doi: 10.1038/nrm1661
79. Davies OG, Cox SC, Williams RL, Tsaroucha D, Dorrepaal RM, Lewis MP, et al. Annexin-enriched osteoblast-derived vesicles act as an extracellular site of mineral nucleation within developing stem cell cultures. Sci Rep. (2017) 7:12639. doi: 10.1038/s41598-017-13027-6
80. Cui L, Rashdan NA, Zhu D, Milne EM, Ajuh P, Milne G, et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J Cell Physiol. (2017) 232:2985–95. doi: 10.1002/jcp.25935
81. Chaudhary SC, Kuzynski M, Bottini M, Beniash E, Dokland T, Mobley CG, et al. Phosphate induces formation of matrix vesicles during odontoblast-initiated mineralization in vitro. Matrix Biol. (2016) 52–54:284–300. doi: 10.1016/j.matbio.2016.02.003
82. Chaudhary SC, Khalid S, Smethurst V, Monier D, Mobley J, Huet A, et al. Proteomic profiling of extracellular vesicles released from vascular smooth muscle cells during initiation of phosphate-induced mineralization. Connect Tissue Res. (2018) 59:55–61. doi: 10.1080/03008207.2018.1444759
83. Gillette JM, Nielsen-Preiss SM. The role of annexin 2 in osteoblastic mineralization. J Cell Sci. (2004) 117(Pt 3):441–9. doi: 10.1242/jcs.00909
84. Viegas CSB, Santos L, Macedo AL, Matos AA, Silva AP, Neves PL, et al. Chronic kidney disease circulating calciprotein particles and extracellular vesicles promote vascular calcification: a role for GRP (Gla-Rich Protein). Arterioscler Thromb Vasc Biol. (2018) 38:575–87. doi: 10.1161/ATVBAHA.117.310578
85. Viegas CSB, Costa RM, Santos L, Videira PA, Silva Z, Araujo N, et al. Gla-rich protein function as an anti-inflammatory agent in monocytes/macrophages: implications for calcification-related chronic inflammatory diseases. PLoS ONE (2017) 12:e0177829. doi: 10.1371/journal.pone.0177829
86. Chen NX, O'Neill KD, Moe SM. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. (2018) 93:343–54. doi: 10.1016/j.kint.2017.07.019
87. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation (2015) 132:1909–19. doi: 10.1161/CIRCULATIONAHA.115.016457
88. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (2016) 354:472–7. doi: 10.1126/science.aaf6659
89. Alique M, Ruiz-Torres MP, Bodega G, Noci MV, Troyano N, Bohorquez L, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging (Albany NY) (2017) 9:778–89. doi: 10.18632/aging.101191
90. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
91. Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles (2014) 3:23743. doi: 10.3402/jev.v3.23743
92. Li A, Zhang T, Zheng M, Liu Y, Chen Z. Exosomal proteins as potential markers of tumor diagnosis. J Hematol Oncol. (2017) 10:175. doi: 10.1186/s13045-017-0542-8
93. Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods (2014) 11:319–24. doi: 10.1038/nmeth.2834
94. Hjortnaes J, Butcher J, Figueiredo JL, Riccio M, Kohler RH, Kozloff KM, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J. (2010) 31:1975–84. doi: 10.1093/eurheartj/ehq237
95. Cui Y, Luan J, Li H, Zhou X, Han J. Exosomes derived from mineralizing osteoblasts promote ST2 cell osteogenic differentiation by alteration of microRNA expression. FEBS Lett. (2016) 590:185–92. doi: 10.1002/1873-3468.12024
96. Ge M, Ke R, Cai T, Yang J, Mu X. Identification and proteomic analysis of osteoblast-derived exosomes. Biochem Biophys Res Commun. (2015) 467:27–32. doi: 10.1016/j.bbrc.2015.09.135
97. Li Y, Wang J, Yue J, Wang Y, Yang C, Cui Q. High magnesium prevents matrix vesicle-mediated mineralization in human bone marrow-derived mesenchymal stem cells via mitochondrial pathway and autophagy. Cell Biol Int. (2018) 42:205–15. doi: 10.1002/cbin.10888
98. Qin Y, Wang L, Gao Z, Chen G, Zhang C. Bone marrow stromal/stem cell-derived extracellular vesicles regulate osteoblast activity and differentiation in vitro and promote bone regeneration in vivo. Sci Rep. (2016) 6:21961. doi: 10.1038/srep21961
99. Wang X, Omar O, Vazirisani F, Thomsen P, Ekstrom K. Mesenchymal stem cell-derived exosomes have altered microRNA profiles and induce osteogenic differentiation depending on the stage of differentiation. PLoS ONE (2018) 13:e0193059. doi: 10.1371/journal.pone.0193059
100. Omar OM, Graneli C, Ekstrom K, Karlsson C, Johansson A, Lausmaa J, et al. The stimulation of an osteogenic response by classical monocyte activation. Biomaterials (2011) 32:8190–204. doi: 10.1016/j.biomaterials.2011.07.055
101. Ekstrom K, Omar O, Graneli C, Wang X, Vazirisani F, Thomsen P. Monocyte exosomes stimulate the osteogenic gene expression of mesenchymal stem cells. PLoS ONE (2013) 8:e75227. doi: 10.1371/journal.pone.0075227
102. Solberg LB, Stang E, Brorson SH, Andersson G, Reinholt FP. Tartrate-resistant acid phosphatase (TRAP) co-localizes with receptor activator of NF-KB ligand (RANKL) and osteoprotegerin (OPG) in lysosomal-associated membrane protein 1 (LAMP1)-positive vesicles in rat osteoblasts and osteocytes. Histochem Cell Biol. (2015) 143:195–207. doi: 10.1007/s00418-014-1272-4
103. Deng L, Wang Y, Peng Y, Wu Y, Ding Y, Jiang Y, et al. Osteoblast-derived microvesicles: a novel mechanism for communication between osteoblasts and osteoclasts. Bone (2015) 79:37–42. doi: 10.1016/j.bone.2015.05.022
104. Huynh N, VonMoss L, Smith D, Rahman I, Felemban MF, Zuo J, et al. Characterization of regulatory extracellular vesicles from osteoclasts. J Dent Res. (2016) 95:673–9. doi: 10.1177/0022034516633189
105. Ciardiello C, Cavallini L, Spinelli C, Yang J, Reis-Sobreiro M, de Candia P, et al. Focus on extracellular vesicles: new frontiers of cell-to-cell communication in cancer. Int J Mol Sci. (2016) 17:175. doi: 10.3390/ijms17020175
106. Sun W, Zhao C, Li Y, Wang L, Nie G, Peng J, et al. Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discov. (2016) 2:16015. doi: 10.1038/celldisc.2016.15
107. Li D, Liu J, Guo B, Liang C, Dang L, Lu C, et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun. (2016) 7:10872. doi: 10.1038/ncomms10872
108. Yuan FL, Wu QY, Miao ZN, Xu MH, Xu RS, Jiang DL, et al. Osteoclast-derived extracellular vesicles: novel regulators of osteoclastogenesis and osteoclast-osteoblasts communication in bone remodeling. Front Physiol. (2018) 9:628. doi: 10.3389/fphys.2018.00628
109. Abbas M, Jesel L, Auger C, Amoura L, Messas N, Manin G, et al. Endothelial microparticles from acute coronary syndrome patients induce premature coronary artery endothelial cell aging and thrombogenicity: role of the Ang II/AT1 receptor/NADPH oxidase-mediated activation of MAPKs and PI3-kinase pathways. Circulation (2017) 135:280–96. doi: 10.1161/CIRCULATIONAHA.116.017513
110. Zhou H, Pisitkun T, Aponte A, Yuen PS, Hoffert JD, Yasuda H, et al. Exosomal Fetuin-A identified by proteomics: a novel urinary biomarker for detecting acute kidney injury. Kidney Int. (2006) 70:1847–57. doi: 10.1038/sj.ki.5001874
111. Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. (2009) 2:ra81. doi: 10.1126/scisignal.2000610
112. Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56. doi: 10.1038/ncb2441
113. Yang J, Hagen J, Guntur KV, Allette K, Schuyler S, Ranjan J, et al. A next generation sequencing based approach to identify extracellular vesicle mediated mRNA transfers between cells. BMC Genomics (2017) 18:987. doi: 10.1186/s12864-017-4359-1
114. Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis (2015) 242:251–60. doi: 10.1016/j.atherosclerosis.2015.07.008
115. Krohn JB, Hutcheson JD, Martinez-Martinez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. (2016) 594:2895–903. doi: 10.1113/JP271338
116. Stranford DM, Leonard JN. Delivery of biomolecules via extracellular vesicles: a budding therapeutic strategy. Adv Genet. (2017) 98:155–75. doi: 10.1016/bs.adgen.2017.08.002
117. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. (2010) 4:214–22. doi: 10.1016/j.scr.2009.12.003
118. Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. (2013) 10:301–12. doi: 10.1016/j.scr.2013.01.002
119. Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, et al. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation (2012) 126:2601–11. doi: 10.1161/CIRCULATIONAHA.112.114173
120. Dorronsoro A, Robbins PD. Regenerating the injured kidney with human umbilical cord mesenchymal stem cell-derived exosomes. Stem Cell Res Ther. (2013) 4:39. doi: 10.1186/scrt187
121. Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, et al. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J Neurosurg. (2015) 122:856–67. doi: 10.3171/2014.11.JNS14770
122. Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem Pharmacol. (2011) 81:1171–82. doi: 10.1016/j.bcp.2011.02.011
123. Willis GR, Kourembanas S, Mitsialis SA. Toward exosome-based therapeutics: isolation, heterogeneity, and fit-for-purpose potency. Front Cardiovasc Med. (2017) 4:63. doi: 10.3389/fcvm.2017.00063
124. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. (2011) 29:341–5. doi: 10.1038/nbt.1807
125. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. (2013) 21:185–91. doi: 10.1038/mt.2012.180
126. Zhang D, Lee H, Zhu Z, Minhas JK, Jin Y. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L110–21. doi: 10.1152/ajplung.00423.2016
127. Didiot MC, Hall LM, Coles AH, Haraszti RA, Godinho BM, Chase K, et al. Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol Ther. (2016) 24:1836–47. doi: 10.1038/mt.2016.126
128. Sterzenbach U, Putz U, Low LH, Silke J, Tan SS, Howitt J. Engineered exosomes as vehicles for biologically active proteins. Mol Ther. (2017) 25:1269–78. doi: 10.1016/j.ymthe.2017.03.030
129. Yim N, Ryu SW, Choi K, Lee KR, Lee S, Choi H, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun. (2016) 7:12277. doi: 10.1038/ncomms12277
130. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials (2014) 35:2383–90. doi: 10.1016/j.biomaterials.2013.11.083
131. Fuhrmann G, Serio A, Mazo M, Nair R, Stevens MM. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J Control Release (2015) 205:35–44. doi: 10.1016/j.jconrel.2014.11.029
132. Lee J, Lee H, Goh U, Kim J, Jeong M, Lee J, et al. Cellular engineering with membrane fusogenic liposomes to produce functionalized extracellular vesicles. ACS Appl Mater Interfaces (2016) 8:6790–5. doi: 10.1021/acsami.6b01315
133. de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, et al. Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation (2017) 136:200–14. doi: 10.1161/CIRCULATIONAHA.116.024590
134. Zhang S, Chu WC, Lai RC, Lim SK, Hui JH, Toh WS. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr Cartil. (2016) 24:2135–40. doi: 10.1016/j.joca.2016.06.022
135. Deng L, Peng Y, Jiang Y, Wu Y, Ding Y, Wang Y, et al. Imipramine protects against bone loss by inhibition of osteoblast-derived microvesicles. Int J Mol Sci. (2017) 18:1013. doi: 10.3390/ijms18051013
136. Chen NX, Kircelli F, O'Neill KD, Chen X, Moe SM. Verapamil inhibits calcification and matrix vesicle activity of bovine vascular smooth muscle cells. Kidney Int. (2010) 77:436–42. doi: 10.1038/ki.2009.481
137. Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G, et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Vesicles (2015) 4:30087. doi: 10.3402/jev.v4.30087
138. Linares R, Tan S, Gounou C, Arraud N, Brisson AR. High-speed centrifugation induces aggregation of extracellular vesicles. J Extracell Vesicles (2015) 4:29509. doi: 10.3402/jev.v4.29509
139. Coumans FAW, Brisson AR, Buzas EI, Dignat-George F, Drees EEE, El-Andaloussi S, et al. Methodological guidelines to study extracellular vesicles. Circ Res. (2017) 120:1632–48. doi: 10.1161/CIRCRESAHA.117.309417
140. Cantin R, Diou J, Belanger D, Tremblay AM, Gilbert C. Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J Immunol Methods (2008) 338:21–30. doi: 10.1016/j.jim.2008.07.007
141. Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles (2013) 2:20360. doi: 10.3402/jev.v2i0.20360
142. Baranyai T, Herczeg K, Onodi Z, Voszka I, Modos K, Marton N, et al. Isolation of exosomes from blood plasma: qualitative and quantitative comparison of ultracentrifugation and size exclusion chromatography methods. PLoS ONE (2015) 10:e0145686. doi: 10.1371/journal.pone.0145686
143. Nordin JZ, Lee Y, Vader P, Mager I, Johansson HJ, Heusermann W, et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine (2015) 11:879–83. doi: 10.1016/j.nano.2015.01.003
144. Xu R, Greening DW, Zhu HJ, Takahashi N, Simpson RJ. Extracellular vesicle isolation and characterization: toward clinical application. J Clin Invest. (2016) 126:1152–62. doi: 10.1172/JCI81129
145. Welton JL, Webber JP, Botos LA, Jones M, Clayton A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J Extracell Vesicles (2015) 4:27269. doi: 10.3402/jev.v4.27269
146. Lobb RJ, Becker M, Wen SW, Wong CS, Wiegmans AP, Leimgruber A, et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles (2015) 4:27031. doi: 10.3402/jev.v4.27031
147. Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, et al. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods (2012) 56:293–304. doi: 10.1016/j.ymeth.2012.01.002
148. Juncker D, Bergeron S, Laforte V, Li H. Cross-reactivity in antibody microarrays and multiplexed sandwich assays: shedding light on the dark side of multiplexing. Curr Opin Chem Biol. (2014) 18:29–37. doi: 10.1016/j.cbpa.2013.11.012
149. Ingham KC. Protein precipitation with polyethylene glycol. Methods Enzymol. (1984) 104:351–6. doi: 10.1016/S0076-6879(84)04101-X
150. Brown PN, Yin H. Polymer-based purification of extracellular vesicles. Methods Mol Biol. (2017) 1660:91–103. doi: 10.1007/978-1-4939-7253-1_8
151. Kim J, Shin H, Kim J, Kim J, Park J. Isolation of high-purity extracellular vesicles by extracting proteins using aqueous two-phase system. PLoS ONE (2015) 10:e0129760. doi: 10.1371/journal.pone.0129760
152. Lee K, Shao H, Weissleder R, Lee H. Acoustic purification of extracellular microvesicles. ACS Nano (2015) 9:2321–7. doi: 10.1021/nn506538f
153. Kang D, Oh S, Ahn SM, Lee BH, Moon MH. Proteomic analysis of exosomes from human neural stem cells by flow field-flow fractionation and nanoflow liquid chromatography-tandem mass spectrometry. J Proteome Res. (2008) 7:3475–80. doi: 10.1021/pr800225z
154. Wang Z, Wu HJ, Fine D, Schmulen J, Hu Y, Godin B, et al. Ciliated micropillars for the microfluidic-based isolation of nanoscale lipid vesicles. Lab Chip (2013) 13:2879–82. doi: 10.1039/c3lc41343h
155. Sydor AM, Czymmek KJ, Puchner EM, Mennella V. Super-resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol. (2015) 25:730–48. doi: 10.1016/j.tcb.2015.10.004
156. Harrison P, Gardiner C. Invisible vesicles swarm within the iceberg. J Thromb Haemost. (2012) 10:916–8. doi: 10.1111/j.1538-7836.2012.04711.x
157. Stoner SA, Duggan E, Condello D, Guerrero A, Turk JR, Narayanan PK, et al. High sensitivity flow cytometry of membrane vesicles. Cytometry A (2016) 89:196–206. doi: 10.1002/cyto.a.22787
158. Chylek P. Absorption and scattering of light by small particles. By C F Bohren and d R Huffman. Appl Opt. (1986) 25:3166.
159. van der Pol E, Coumans FA, Sturk A, Nieuwland R, van Leeuwen TG. Refractive index determination of nanoparticles in suspension using nanoparticle tracking analysis. Nano Lett. (2014) 14:6195–201. doi: 10.1021/nl503371p
160. Coumans FA, van der Pol E, Boing AN, Hajji N, Sturk G, van Leeuwen TG, et al. Reproducible extracellular vesicle size and concentration determination with tunable resistive pulse sensing. J Extracell Vesicles (2014) 3:25922. doi: 10.3402/jev.v3.25922
161. Vogel R, Coumans FA, Maltesen RG, Boing AN, Bonnington KE, Broekman ML, et al. A standardized method to determine the concentration of extracellular vesicles using tunable resistive pulse sensing. J Extracell Vesicles (2016) 5:31242. doi: 10.3402/jev.v5.31242
162. Gardiner C, Shaw M, Hole P, Smith J, Tannetta D, Redman CW, et al. Measurement of refractive index by nanoparticle tracking analysis reveals heterogeneity in extracellular vesicles. J Extracell Vesicles (2014) 3:25361. doi: 10.3402/jev.v3.25361
163. Huang X, Yuan T, Tschannen M, Sun Z, Jacob H, Du M, et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics (2013) 14:319. doi: 10.1186/1471-2164-14-319
164. van Balkom BW, Eisele AS, Pegtel DM, Bervoets S, Verhaar MC. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J Extracell Vesicles (2015) 4:26760. doi: 10.3402/jev.v4.26760
165. Hill AF, Pegtel DM, Lambertz U, Leonardi T, O'Driscoll L, Pluchino S, et al. ISEV position paper: extracellular vesicle RNA analysis and bioinformatics. J Extracell Vesicles (2013) 2:22859. doi: 10.3402/jev.v2i0.22859
166. Rekker K, Saare M, Roost AM, Kubo AL, Zarovni N, Chiesi A, et al. Comparison of serum exosome isolation methods for microRNA profiling. Clin Biochem. (2014) 47:135–8. doi: 10.1016/j.clinbiochem.2013.10.020
167. Van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, et al. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J Extracell Vesicles (2014) 3. doi: 10.3402/jev.v3.24858
168. Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics (2009) 9:4997–5000. doi: 10.1002/pmic.200900351
169. Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. (2012) 10:e1001450. doi: 10.1371/journal.pbio.1001450
170. Altadill T, Campoy I, Lanau L, Gill K, Rigau M, Gil-Moreno A, et al. Enabling metabolomics based biomarker discovery studies using molecular phenotyping of exosome-like vesicles. PLoS ONE (2016) 11:e0151339. doi: 10.1371/journal.pone.0151339
171. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife (2016) 5:e10250. doi: 10.7554/eLife.10250
172. Shahaf N, Rogachev I, Heinig U, Meir S, Malitsky S, Battat M, et al. The WEIZMASS spectral library for high-confidence metabolite identification. Nat Commun. (2016) 7:12423. doi: 10.1038/ncomms12423
173. Wei Z, Batagov AO, Carter DR, Krichevsky AM. Fetal bovine serum RNA interferes with the cell culture derived extracellular RNA. Sci Rep. (2016) 6:31175. doi: 10.1038/srep31175
174. Shelke GV, Lasser C, Gho YS, Lotvall J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J Extracell Vesicles (2014) 3:24783. doi: 10.3402/jev.v3.24783
175. Angelini F, Ionta V, Rossi F, Miraldi F, Messina E, Giacomello A. Foetal bovine serum-derived exosomes affect yield and phenotype of human cardiac progenitor cell culture. Bioimpacts (2016) 6:15–24. doi: 10.15171/bi.2016.03
176. Eitan E, Zhang S, Witwer KW, Mattson MP. Extracellular vesicle-depleted fetal bovine and human sera have reduced capacity to support cell growth. J Extracell Vesicles (2015) 4:26373. doi: 10.3402/jev.v4.26373
177. Hummel J, Keshvari N, Weckwerth W, Selbig J. Species-specific analysis of protein sequence motifs using mutual information. BMC Bioinformatics (2005) 6:164. doi: 10.1186/1471-2105-6-164
Keywords: extracellular vesicles, exosomes, calcification, mineralization, atherosclerosis, calcific aortic valve disease
Citation: Blaser MC and Aikawa E (2018) Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 5:187. doi: 10.3389/fcvm.2018.00187
Received: 03 October 2018; Accepted: 10 December 2018;
Published: 21 December 2018.
Edited by:
Dwight A. Towler, University of Texas Southwestern Medical Center, United StatesReviewed by:
Adriana Georgescu, Institute of Cellular Biology and Pathology (ICBP), RomaniaTeresa Padro, Sant Pau Institute for Biomedical Research, Spain
Copyright © 2018 Blaser and Aikawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Aikawa, eaikawa@bwh.harvard.edu