Antibodies and Immunity During Kawasaki Disease
 Mark Daniel Hicar1,2,3*
Mark Daniel Hicar1,2,3*- 1University at Buffalo, Buffalo, NY, United States
- 2John R. Oishei Children's Hospital, Buffalo, NY, United States
- 3Department of Pediatrics, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, United States
The cause of Kawasaki disease (KD), the leading cause of acquired heart disease in children, is currently unknown. Epidemiology studies support that an infectious disease is involved in at least starting the inflammatory cascade set off during KD. Clues from epidemiology support that humoral immunity can have a protective effect. However, the role of the immune system, particularly of B cells and antibodies, in pathogenesis of KD is still unclear. Intravenous immunoglobulin (IVIG) and other therapies targeted at modulating inflammation can prevent development of coronary aneurysms. A number of autoantibody responses have been reported in children with KD and antibodies have been generated from aneurysmal plasma cell infiltrates. Recent reports show that children with KD have similar plasmablast responses as other children with infectious diseases, further supporting an infectious starting point. As ongoing studies are attempting to identify the etiology of KD through study of antibody responses, we sought to review the role of humoral immunity in KD pathogenesis, treatment, and recovery.
Introduction
The cause of Kawasaki disease (KD) continues to perplex clinicians and researchers. Also known as Kawasaki syndrome or mucocutaneous lymph node syndrome, KD is the leading cause of acquired cardiac disease in children. Recent murine and human clinical trials are enhancing our understanding of this disorder (1). The mainstay of treatment has been intravenous immunoglobulin (IVIG), initially implying a major role for humoral immunity. However, newer therapies that also have broad immunomodulatory effects have become widely used for refractory cases (2). Studies from different fields, from maternal immunity to genome-wide association studies, also imply a role of humoral immunity (3–6). This review will give an overview of the present knowledge of the field directed toward the etiology of KD and what role B cells and antibodies may play in treatment, pathogenesis, and diagnosis.
Review With a Focus on B Cells and Antibodies
Clinical Presentation
Diagnosis is purely clinical, as there are no adequately specific or sensitive tests available. The “classic” diagnosis involves 5 days of fever and having four of the five following criteria: mucous membrane inflammation, rash, hands and feet swelling, conjunctivitis, and a solitary inflamed lymph node mass (6–9). If left untreated, roughly one-quarter of the children meeting clinical criteria will go on to have coronary artery inflammation, including aneurysms. Incomplete cases, those which do not fulfill four of five of the classic criteria, have similar risk of coronary aneurysms (10). Treating affected patients with IVIG reduces the rates of coronary aneurysms, with a minority seemingly resistant to treatment (6–8, 11–14). Although most aneurysms resolve, some defects are retained. Initial studies done on adults with a history of KD implies there is a greater lifetime risk of cardiac issues and early mortality (15–18). To add to the diagnostic confusion, several infectious etiologies have also been independently associated with aneurysms (19). It remains a frustrating diagnosis because of the unknown etiology, clinical variability, lack of specific testing, and unclear pathogenesis.
Epidemiology
There appears to be a genetic influence in exhibiting KD. Incidence is higher in some genetic backgrounds and consistently appears in males greater than females within those backgrounds (20). By age five in the United States, 1 in 1,000 African-American children and 1 in 2,000 Caucasian children will have been affected (21–23). In general, Asians have a much higher rate of KD; which is especially evident in Japanese children, whose lifetime incidence rate is near 1% (24). This predisposition holds even for those persons of Japanese heritage raised in foreign lands, such as the United States (20). Sibling can have a 10–30-fold higher risk of KD compared to the general population (25).
The etiology of KD is unknown (6, 26, 27). However, there is a proposed relation to an infectious agent. Epidemiological evidence for this comes from the fact that there are seasonal peaks of KD during winter and spring months and outbreaks have been described (27–34). Siblings have a 10–30-fold higher rate, with most occurring within 1 year of each other (35), and up to 50% of sibling cases are within 10 days of each other (25).
Recent studies support a protective role for humoral immunity. There is a lower incidence in breastfed infants (4) and KD is rare in both newborns and individuals over 5 years of age. This implies a maternally derived protective immunity to a ubiquitous infectious agent (36). Trans-placental passage of maternal antibodies is thought to be protective and explain the paucity of cases in infancy (3).
Proposed Etiologies
It is possible that there is not one cause of KD, but multiple etiologies that result in similar pathogenesis. This may explain the clinical variability and lack of discovery of a definitive agent, however, the low recurrence rate even in high prevalent areas speaks against a large number of infectious causes (37).
Previously proposed infectious agents include Herpesviridae (HHV-6, Epstein Barr Virus, Cytomegalovirus), human coronavirus, retroviruses, Parvovirus B19, bocavirus, and bacterial infections such as staphylococci, streptococci, Bartonella, and Yersinia infections (15, 20). Some of these agents have been independently associated with aneurysm formation (19), with the Epstein Barr Virus most commonly associated (38). Several non-infectious agents have also been proposed such as carpet shampoos, mercury exposure and living near bodies of water (15, 20). Additionally, the recent report of tropospheric wind patterns correlating with outbreaks in Japan would not be consistent with many of the viruses that have been proposed (26, 34, 39). These reports imply a relationship to an environmental antigen, as either a priming or inciting event. This “two-hit” hypothesis is also suggested by similar data from Canada (40).
If a ubiquitous childhood pathogen is the cause of KD, the mode of entry would likely be a common mode of infection such as fecal-oral or respiratory spread. Outbreaks in the United States have been associated with preceding viral illness (41). To note, mild upper respiratory symptoms and gastrointestinal complaints have been described in up to 35 and 61% of cases, respectively (42). Rare but more significant pulmonary disease has also been reported (43). Notably, however, concomitant respiratory viruses are near 10% of cases (44, 45). A persistent infection has been theorized (46). Although numerous viruses that can reactivate during stress (Herpesviridae family) or are considered “slow” viral infections (47), the failure of numerous attempts to identify a specific infectious agent argues against a prolonged infection. There are difficult to culture viruses, such as coronavirus which had also enjoyed a short-lived consideration as the cause of KD (48). An abnormal response to normal flora has been proposed (49, 50) and studies on a relationship to the emerging field of microbiome research have recently been reviewed (51).
Human Biomarkers
Currently, diagnosis is aided by utilizing sensitive but not specific biomarkers such as C-reactive protein, sedimentation rate, liver function tests, urine leukocytes, platelets, leukocyte count, and hemoglobin (2). As highlighted by recommendations for diagnosis of incomplete cases, many biomarkers do not reveal the nature of the underlying illness. A number of traditional laboratory and clinical findings have been built into scoring systems to predict IVIG resistance that are used in Japanese populations (52). These scoring systems (Sano, Kobayashi, Egami) all have relatively high sensitivity, near 80%, in Asian populations; however, they have poor predictive ability in heterogenous lower pretest probability populations of North America (53).
Numerous other biomarkers have been proposed to aid in diagnosis of KD and have been recently reviewed (54). Although some of the transcriptomic approaches mirror genomic findings, intriguingly, there has been a disconnect between proteomics and genomic associations. A recent study using serum mass spectrometry to look at differentially expressed proteins showed lipopolysaccharide binding protein (LBP), leucine rich alpha-2 glycoprotein (LRG1), and angiotensinogen were higher in acute phase, and retinol binding protein was diminished (55). The marker of tenascin, involved in tissue remodeling, is promising as a marker of coronary involvement during acute KD (56, 57).
Numerous cytokine associations in KD have been shown, and those with unique B cell associations will be reviewed. IL-10 has long been shown to be elevated during acute KD (58, 59). This is likely a natural anti-inflammatory response, as recent studies show supplementing IL-10 via an AAV vector protected against aneurysms in the Candida albicans murine model (60). IL-10 is produced by myeloid dendritic cells and regulatory B cells, and recently has been shown to drive plasmablast responses (discussed later) (61). IP-10, an activator of B cells and macrophages, has also been associated with clinical KD. Notably, this group did not see peripheral IL-1B elevation. (62). IL-21, produced mainly by T cells and Natural Killer cells (63, 64), has recently been proposed as a specific marker in KD in a Korean cohort of children when compared to prolonged fevers from mononucleosis (65). IL-21 modulates immunoglobulin isotype switching and is involved in the differentiation of both naïve and memory B cells into mature plasma cells (66). However, in a study of IL-21 levels in children presenting to a North American emergency room with fever, KD and febrile children could not be distinguished by IL-21 levels (67).
Biomarkers Supporting Innate Immunity
A number of transcriptomic approaches show some promise in distinguishing KD from viral infections. Initial studies that look at IVIG response in PBMCs and monocytes suggested monocyte regulation was a main role of IVIG (68) FCGR1a, FCGR3A, CCR2, S100A9, S100A12, and adrenomedullin were notably effected. FCGR2A transcripts were reduced, but surface expression on monocytes was variable. The S100A9 and S100A12 are involved in monocyte adhesion and chemotaxis. Adrenomedullin, important for vascular integrity, was shown in monocytes by gene array as well (69) and these were both supported in other studies (70). Transcriptome analysis of PBMCs showed upregulation of NAIP, IPAF, S100A9, FCGR1A, and GCA which is also supportive of a role for the innate immune system (71).
Transcriptomics Supportive of a Role for B Cells in KD
Pathway significance analysis of blood lymphocyte-specific gene markers revealed that PI3K signaling in B lymphocytes was the most significant finding; however, T cell receptor signaling, B cell receptor signaling, T helper cell differentiation, and natural killer cell signaling were also significantly down-regulated in KD compared to febrile control patients (72). On whole blood expression analysis, comparing acute vs. convalescent samples, numerous upregulated pathways involved in innate immunity (particularly IL-1 and the Nlrp3 inflammasome) were shown in the acute phase. IL-10 was notably the highest association, and will be reviewed later. For pathways downregulated, outside of EIF2/p70S6K/MTOR pathways, “B cell development” was the most significant. As mTOR is essential in signaling through PI3K/Akt activity after B cell receptor (BCR) engagement, this association also supports that B cells may be playing a role. A number of these associations; however, were also seen in their controls. There was a surprising similarity with influenza and virally infected individuals. Of the pathways completely unique to KD, most of the specific associations related to myofibroblast migration (paxillin signaling, G-protein coupled receptor signaling, triacylglycerol and relaxin signaling) but PI3K/AKT signaling pathway showed a minor association. Lastly, there was surprisingly few transcript differences noted in those with and without aneurysms (73). A recent similar study comparing KD to adenovirus did not show much specific overlap with previous studies, but was also consistent with relation to a number of inflammatory pathways (74).
Overall, transcriptomics and cytokine profiling suggest a significant, but not exclusive, role for B cells in the pathogenesis of KD.
Models Systems of KD (See Table 1)
The first KD model system developed depended on intraperitoneal Candida albicans alkaline extract injections in susceptible mouse strains (75). Injection of the water soluble fraction of this, (CAWS) had increased incidence of arteritis which can be partially blocked by IVIG. IL-1 inhibition also diminishes the coronary inflammation (76). Notably this vasculitis is a panvasculitis predominantly and also effects the aortic root, which has not been described in KD cases (77).
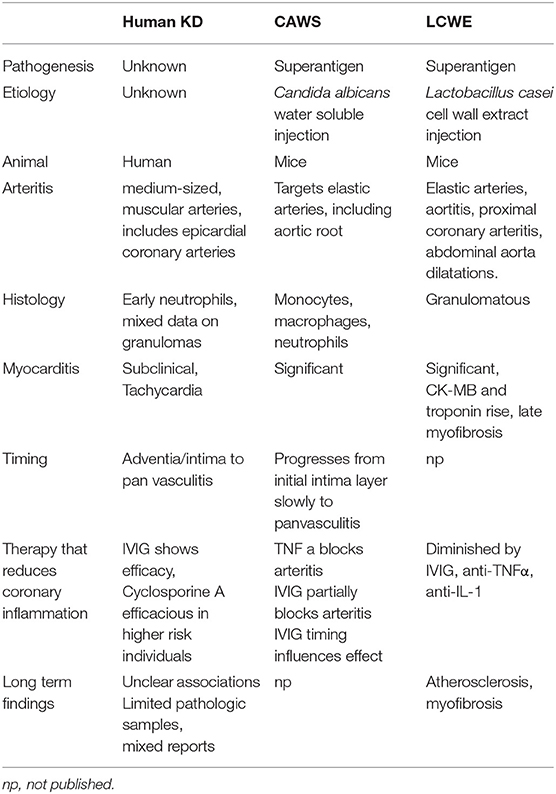
Table 1. Comparison of human KD to common murine models of KD.
Similarly, mice develop coronary artery inflammation after intraperitoneal injection with Lactobacillus casei cell wall extract (LCWE) (78, 79). Pathogenesis in this model parallels KD in that younger mice are more predisposed to develop arteritis and there is a favorable response to IVIG treatment. This disease exhibits mostly a T-cell infiltrate in coronary arterial specimens (79). In fact, in both RAG-1 (80) and TCR-α (81) deficient mice, this arteritis is diminished (82). Etanercept completely blocked these lesions and this was apparently related to signaling through TNFRI (83). Blocking IL-1 can also prevent progression of coronary pathology (84, 85). TNFα can drive metalloproteinase mmp9 activity to cause elastin breakdown. Doxycycline has been shown to prevent this (86), and human data will come from the currently open trial (87). A number of studies show marked myocarditis in these models with late fibrosis (88); however, following troponins is generally not clinically relevant. One group did associate serum troponins and ck-mb (89) but others have not seen that association. A number of the model systems have granulomatous changes, which have variably been seen in human specimens (90, 91). Other models depend on immune complex deposition. This was observed after bovine serum albumin injection into rabbits, which exhibited a disease similar to serum sickness (92). Presently, there is not a model system consistent with direct infectious coronary artery invasion.
No model system exactly replicates the pathologic changes seen in humans, and the utility of these models have been called into question (93). Although most data from model systems are supportive of superantigen involvement, studies from human peripheral lymphocyte responses as reviewed are variable and inconsistent (94). These models and other data are driving clinical trials, but results seen in the models aren't equitably transferable to treatment modalities (reviewed later). Since the cause in humans is unknown, it is still unclear if any of these models of arteritis are truly applicable.
The Superantigen Theory
A superantigen response was considered by numerous groups and is supported by murine models (79, 95–99). Certain bacterial infections contain proteins that non-specifically bind effector cell receptors causing a more generalized polyclonal expansion and inflammation, termed a superantigen effect. Polyclonality of T cell receptor usage has been shown in KD (100, 101); however, the reports are variable as to which subset of T cell receptors are effected (102). Other studies support a traditional oligoclonal response consistent with an immune response against a specific etiologic agent. Oligoclonal expansion of CD8+ T cells (103) and peripheral IgM+ B cell responses have been shown (9, 104). Numerous other studies have not shown superantigen associated expansions of cell subsets (103, 105, 106). Overall, there is not a clear role for a superantigen response during KD.
Genetic Implications
Inflammation during KD is universally present, so it is no surprise that a recent network and pathway analysis was consistent with global activation of the immune response (107). Large dataset and related human genomic studies have been performed to look for associated genes with KD incidence and treatment response. These study co-segregation of single nucleotide polymorphisms (SNPs) with the disease state to define significance. These are excellent hypothesis generating techniques, as each dataset generated has numerous implicated genes, but assigning biological significance can at times be difficult. These have been more successful in more homogenous populations and, for KD, have been recently reviewed (108–110).
Genome wide association studies (GWAS) offer unbiased approaches to explore genetic associations. A SNP (rs28493229) near inositol 1, 4, 5-trisphosphate 3-kinase (ITPKC, 19q13) was implicated first by a linkage disequilibrium study in siblings (111). ITPKC modifies the IP3 pathway leading to activation of NFAT transcription factor. A number of GWAS studies were negative or needed metanalysis to confirm this finding (112, 113) or showed a different SNP (rs2290692) association in the area (114). This may be an effect of differing genetic backgrounds in these studies. Other studies associate this gene area with coronary artery risk (115) and a number of SNPs associated with ITPKC were also seen in a study that initially identified FCRG2a (116). Caspase 3 (rs113420705) has been proposed to act downstream of ITPKC, and a genetic variance in the enhancer has been associated (rs72689236) (117, 118). Single-nucleotide polymorphisms (SNPs) in ITPKC (rs28493229) and caspase-3 (CASP3, rs113420705), when analyzed together, associate with increased risk for aneurysms (119). Separately they show a trend of overrepresentation in IVIG non-responders and together with coronary risk (120). These two genes could work on a similar calcium influx pathway. The polymorphism rs28493229 has been shown to effect ITPKC protein levels and thereby IL-1β and inflammasome NLRP3 expression, which was shown by studying murine models, KD subject samples, and EBV transformed B cells (121). There is a general lack of studies in non-Asian populations and disparate results can be seen depending on the genetic background. One of the first studies of this nature showed NAALADL2 and ZFHX3 highly associated with susceptibility in Dutch Caucasians. Reanalysis implicated other genes as well (DGKB, PPP1R14C, LNX1, CAMK2D, CSMD1, and TCP1) and network analysis could associate a number of these with CAMK2D (122).
The most significant and repeatedly associated susceptibility genes are FCGR2A, BLK and CD40, as reviewed in (110). The rs1801274 SNP is a functional polymorphism associated in the IgG receptor gene FCGR2A, which is a cell surface receptor found on phagocytic cells such as macrophages and neutrophils (116, 123). This receptor is a low-affinity receptor for immunoglobulin and is involved in the process of phagocytosis and clearing of immune complexes. Intriguingly, male gender influences the association of the FCRG2a (124). Genome-wide genetic marker association studies show that specific polymorphisms in CD40 and in the B lymphoid tyrosine kinase genes associate with KD (5, 6). One of the more consistent findings from GWAS data has been identifying SNPs in the region of the B lymphocyte kinase (BLK) (123, 125). BLK encodes a non-receptor protein tyrosine kinase and of the Src family of tyrosine kinases that acts downstream of the B cell receptor. Functional validation shows BLK is induced during acute KD (from patient samples) and the protective genotype of this SNP correlates with lower BLK expression on induction of EBV transformed B cells. Intriguingly, on a recent meta-analysis rs2736340 of BLK is also associated with systemic lupus erythematosus and rheumatoid arthritis-associated (126). Hypothesizing the importance of B cells based solely on BLK association is tenuous, as Blk knockout mice also suggest a role for Blk in the development of γδ TCR+ T cells (127) and marginal zone B cells (128). The association of CD40 would also potentially point to a more specific B cell function (5, 123, 125), although CD40 is expressed on a number of cell types and functional inference of this association is lacking. The association of BLK and FCGR2a were recently confirmed; however, did not seem to correlate with incomplete or cases from children older than 5 years of age (129).
A recent meta-analysis did see nominal associations with four previously noted genes (FCGR2A rs1801274, TCP1 rs3818298, BLK rs2736340, and CD40 rs4813003) but did not replicate a number of these associations (ZFHX3 rs9937546, NAALADL2 rs1870740, CAMK2D rs4834340, LNX1 rs6554112, MIA-RAB4B rs2233152, HLA-DQB-HLA-DOB rs2857151) (130). When specifically studying susceptibility to cardiac aneurysms, a number of intergenic associations were also shown, highlighting the complex organization and influence of chromatin structure (131). The most review and meta-analysis of available data supports a role for in susceptibility for genetic variations of ACE, BLK, CASP3, CD40, FCGR2A, FGb, HLA-E, IL1A, IL6, ITPKC, LTA, MPO, PD1, SMAD3, TARC/CCL17, and TNF. Also, genetic variations in BTNL2, CASP3, FCGR2A, FGF23, FGb, GRIN3A, HLA-E, IL10, ITPKC, and TGFBR2 may serve as biomarkers of CALs in KD (109).
Overall, association with a variety of pathways is not surprising for a disease with a likely complex pathogenesis. However, most of the major repetitively associated and/or validated genomic findings could support a role for B cells and antibodies influencing susceptibility (110).
Clues From Treatment (See Table 2)
Intravenous Immunoglobulin (IVIG)
The first implication of humoral immunity possibly being involved in KD was the response to immunoglobulin infusions. IVIG was started after success with preparations in Immune thrombocytopenic purpura and was shown to protect against cardiac involvement (142). After discovery that aneurysms are associated with KD, studies, including randomized controlled trials, supported a protective effect of IVIG (136). IVIG is known to broadly effect numerous mechanisms that have been outlined in recent reviews, including: direct neutralization of organism or toxin, inhibition of autoantibodies, inhibition of the complement cascade, inhibition of adhesion molecules on vascular endothelium, modulation of cytokine response, expansion of T regulatory cells, modulation of dendritic cell responses, and decreasing pro-inflammatory effects of monocytes by FC interaction (143–146). Possible mechanisms are detailed in Figure 1. The exact mechanism that correlates with clinical improvement seems to depend on the underlying disorder. On the most simplistic level, supplementation of a specific necessary immune response is still theorized for KD (147).
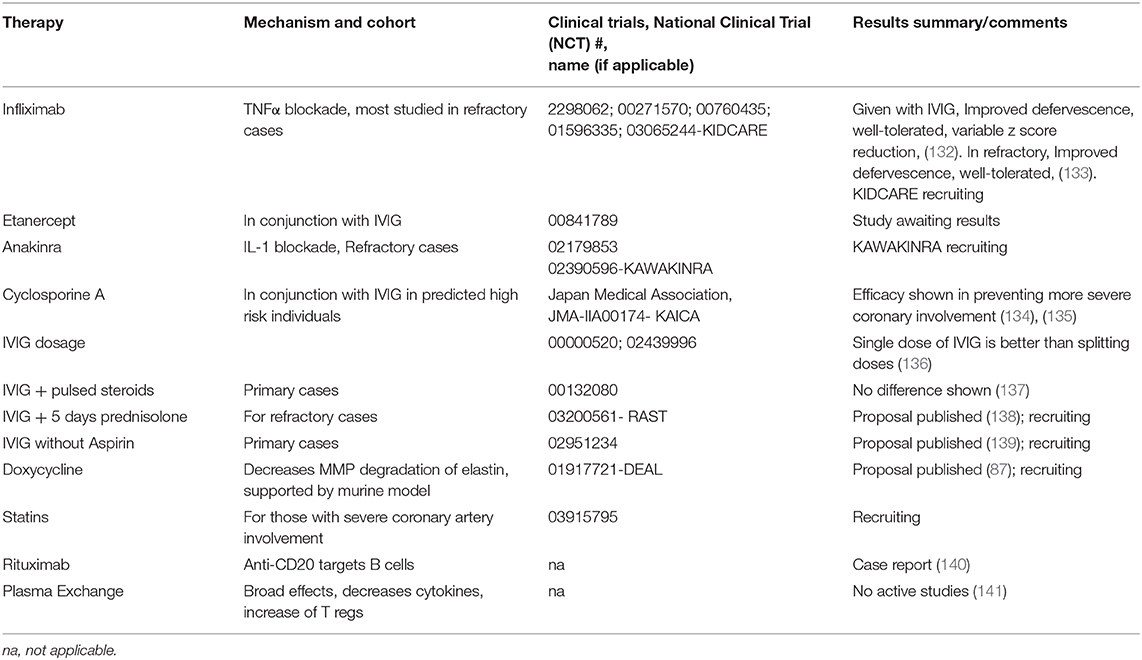
Table 2. Notable therapies and trials for treatment to prevent coronary aneurysms in Kawasaki Disease.
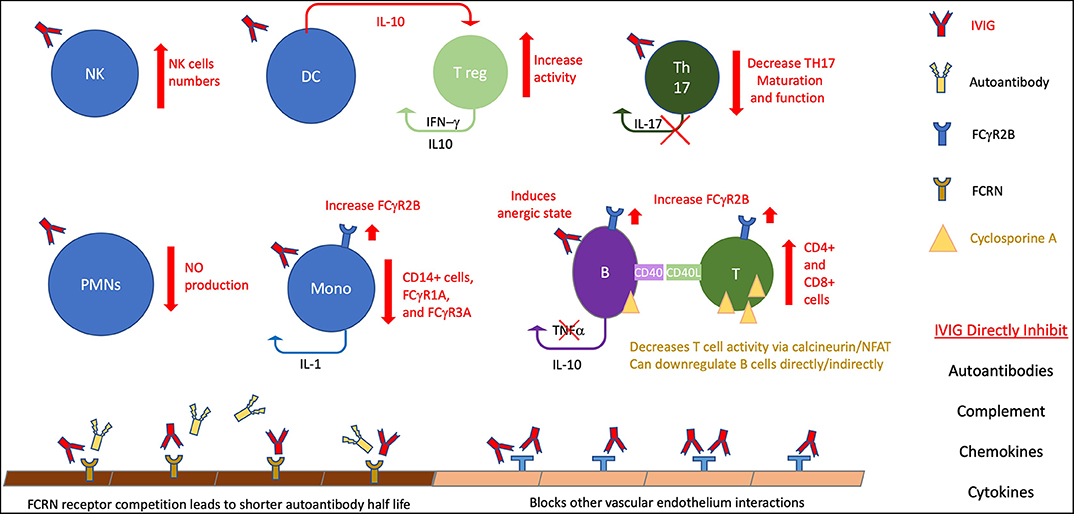
Figure 1. The many and varied potential activities in KD. This figure highlight potential therapeutic effects on B cell activity, particularly for IVIG. IVIG effects are noted by red elements, cyclosporin A noted by gold. B, B cells; T, T cells; T reg, T regulatory cells, DC, dendritic cells; nk, natural killer; PMNs, polymorphonuclear cells; Mono, monocytes, NO, Nitric Oxide.
However, it is unclear how IVIG actually functions during KD and if specific antibody responses are responsible for pathogenesis. Early studies focused on dosing regimens and showed single large infusions (2 grams/kg) are superior to smaller more frequent doses (136). It is possible that a high level of antibody is needed to clear a pathogen, or to improve diffusion to certain areas; however, why this difference is unclear. The mechanism of loading of the neonatal FcRn receptor may require such high volume administration. Other Fc receptor modulation, particularly upregulation of FcγRIIB has also been proposed (148, 149). Without knowledge of the cause of KD, the mechanism of action will remain difficult to prove. The anti-inflammatory properties of IVIG also treat many infectious agents so a clinical response to IVIG is not definitive proof of the diagnosis of KD (147).
Recent studies show specific immune changes induced by treatment with IVIG. TH17 cells and IL-17 levels are elevated during acute KD, which is seen in autoimmune disorders, and this can be improved after IVIG (150, 151). T regulatory cells (T regs) are reduced during acute KD, and this is improved with IVIG (152). These cells, as well as myeloid dendritic cells are expanded by IVIG (145). In a study comparing IVIG to aspirin treatment alone group, IVIG treatment showed increase CD4, restoration of CD8 to normal control levels, and a significant suppression of CD19 cells below that of normal controls (153). Other studies also have explored diminished CD8 T cells in acute KD (154). Abnormal elevations of BAFF, which is in involved in survival of and proliferation of B cells, are decreased after treatment with IVIG (155). In what authors termed, “functional silencing,” IVIG suppressed phosphoinositide 3-kinase signaling (via NFAT) which would affect calcium signaling, coreceptor activation, and BCR activation (156). This finding supportive of B cell signaling involvement in KD could potentially link some of the genomic and transcriptomic associations.
Other Treatments
Adjunct steroids showed efficacy in certain populations (157). Meta-analysis of this and other clinical trials, mostly from Japan, support the use of steroids along with IVIG in high-risk patients (158). Since it is difficult to predict risk in heterogenous populations outside of Japan, and historical studies have shown worse outcomes with steroid use (159), there is not a universal recommendation to give adjunct steroids (2). Even in the revised 2012 guidelines from the Japanese Society of Pediatric Cardiology, “controversy remains” on universal initial steroid use (160). Aspirin is begun at anti-inflammatory doses initially, then is changed to a lower dose to prevent complications of coronary involvement (2). As this has not been shown to effect development of aneurysms, other platelet- aggregation inhibitors such as Clopidogrel or dipyridamole, can be considered in special situations (2). The main treatment modalities hoped to target prevention of coronary aneurysms used for refractory treatment are second doses of IVIG, steroids, calcineurin inhibitors, anti-IL-1 monoclonal antibodies, and anti-TNF monoclonal antibodies; all of which have broad immunological effects (53, 161).
Success with anti-TNFα treatment seemingly argues against a significant role of B cells, as this would affectively release a suppressive action of TNFα on B cell proliferation. The main treatments studied have been infliximab, a humanized mouse monoclonal antibody, and etanercept, a human recombinant mimic of the TNFα receptor (162). TNFα produced from B cells is implicated in atherosclerosis, so perhaps B cell derived TNFα is targeted (163). Notably, IVIG decreases TNFα production. The status of clinical trials have been recently reviewed (164). Although tolerance and improved defervescence are clear in trials with infliximab, there is less clarity of efficacy for improvement of aneurysms (132, 133). The KIDCARE trial, which is focused on recalcitrant cases may prove to be more insightful than previous trials. Etanercept has also undergone recent phase II randomized trial, but publications of results have not been done to date. A recent meta-analysis of twelve studies showed that both IV methylprednisolone and infliximab were not superior to a second dose of IVIG for prevention of aneurysms (165). The other main class of biologics used are Interluekin-1 (IL 1) inhibitors. These obviously have a broad inflammatory response (145) with notable effects on B cell activity (166) There is support in the Lactobacillus casei mouse model for IL-1 playing a role (167).
Genetic associations and data from murine models have supported targeting of NFAT with calcineurin inhibitors, and treatment protocols for KD have been developed (168). A recent Japanese clinical trial (135) show that addition of Cyclosporine A to IVIG in high risk patients is efficacious in prevention of the largest of aneurysms. Cyclosporine A mainly targets T cell activity by inhibition of calcineurin and its downstream transcription factor NFAT. This downregulation of T cell activity has been shown to decrease B cell immunoglobulin production (169). Although encouraging, the applicability of this treatment outside of the highest risk individuals is unclear. Limited reports of successful treatment with anti-B cell monoclonal antibodies (anti-CD20) also support a role for B cell activation in KD pathogenesis (140). Based on data regarding MMP in murine models, doxycycline is also being used (87). Supplementation with statins also has a currently recruiting trial ongoing. Plasma exchange, which also has a broad range of activity, is used in some centers, particularly in Japan (2). Recent data suggests restitution of circulating T regulatory cells occurs which aligns with other mechanisms similar to IVIG (141).
The broad effects of most treatments make drawing specific conclusions about correlation with pathogenesis (see Figure 1); however, a number of treatments and reported success with anti-CD20 monoclonal antibodies supports a significant role for B cells.
Autoimmune Antibodies in KD
A role for B cells could imply a significant autoimmune component in KD (170). Self-antigen responses to a variety of targets have actually been well-described in KD. These include recent reports of antibody responses to type III collagen, myosin (171), cardiolipin (172), alpha-enolase (173), and anti-endothelial antibodies. Anti-endothelial antibodies are particularly interesting as these are seen in other disorders, such as SLE and renal allograft rejection (174). Other vasculitides have also been associated with anti-endothelial antibodies. These have been shown to cause upregulation of E-selectin, VCAM-1, ICAM-1, and NFκB (175). Responses to these antibodies include upregulation of inflammatory cytokines and apoptosis of the endothelial cells.
In KD subjects, a polyclonal response against endothelial cells has been described (176); however, not universally (177). Particularly during generalized inflammation, cytokines such as IFN-γ, IL-1, and TNF can reveal this IgG and IgM anti-endothelial response (178, 179). In cell lysis assays, pathogenesis was eliminated by clearing the serum through anti-IgG and anti-IgM sepharose columns arguing against a role of peripheral anti-IgA responses. This does not eliminate the potential role of intra-tissue IgA+ plasma cell development in pathogenesis as has been postulated (180, 181). Other studies support significant IgM mediated cytotoxicity against endothelial cells in KD patients (182). Prevalent IgM anti-endothelial responses in KD have also been shown without cytokine stimulation (182, 183). In a mouse model system, anti-endothelial antibody responses were replicated, but these did not demonstrate cardiac vascular involvement (184).
Recently, the autoimmune considerations in KD have been reviewed (170). The case report of anti-B cell monoclonal antibody success was proposed by the authors to be due to the downregulation of such an anti-endothelial invasive effect (140).
Although intriguing, it remains unknown if these anti-endothelial responses actually contribute to the vasculitis in KD and other vasculitides (175).
Human Pathologic Studies
A small number of studies, due to the necessary reliance on autopsy specimens, support limited B cell infiltration into the coronary arteries (185). These limited studies have shown that coronary infiltrates develop over time with late fibrosis occurring in the intima and adventitial layers and can be divided into an acute, subacute and chronic state formation (90). In the acute phase, neutrophils are the predominant initial cell infiltrate (186). This can result in necrotizing arteritis that destroys the adventitia leading to aneurysm formation (90).
A number of studies have noted lymphocytic infiltrates in samples from later timepoints. In a study of eight specimens, two of which were within the first 2 weeks of illness, showed predominant monocytes on day 6 and no CD3+ (T cell) nor CD20+ (B cells) cells. T and B cells were shown in the one sample from day 10 but both monocytes and neutrophils were more predominant (186). B cell responses were highest on day 10 and 17 overall, but one of the two samples from day 17 had very few B cells. Another small study of seven subjects, all from over 13 days of illness, showed IgA+ plasma cell infiltrates. These were seemingly specific to KD; however, the fourteen control specimens were from autopsies that succumbed generally from non-inflammatory and non-cardiac syndromes. Notably, mature memory and immature B cells (CD20+ cells) were lacking (180). If the previous day 10 B cell infiltrates became plasma cells as has been postulated (187), these studies would be consistent. Also consistent, in a series of six KD specimens who were not diagnosed with aneurysms compared to 21 non-inflamed controls, neutrophil infiltration in adventia and intima layers was quickly followed by lymphocyte infiltrates, then mixed lymphocyte, eosinophils, and plasma cell infiltrates were demonstrated later, near day 19 of illness (186, 188). Other reports support monocyte infiltration with more significant CD8 T cell responses, however there were few early samples here as well (187). Overall, innate immune cells are the predominant in early infiltrates.
Prominent nodular infiltrates, similar to atherosclerotic plaque formation, have also been described, but these appear to occur at later timepoints (>3 weeks). These infiltrates consisted of T cells, macrophages, B cells and prevalent IgM+ plasma cells, with less frequent IgA+ plasma cells. The authors compare these to similar B cell rich lesions driven by both superantigens and specific infectious antigens (189).
The largest study, relying on electron microscopic studies, suggests that there is an early necrotizing arteritis suggestive of an acute viral infection, followed by a vasculitis, then luminal myofibroblast proliferation (90). There were a number of differences from previous reported pathological specimens, including lack of granulomatous inflammation, lack of small vessel pathology, minimal medial hyperplasia or scarring, and lack of atherosclerosis. Overall most studies are consistent with an early phase with neutrophil infiltration when severe is termed necrotizing arteritis, with later lymphocyte and plasma cell infiltration still in the background of significant monocytes.
Only few studies on long-term sequelae have been published. There seems to be an increased risk of cardiac death in survivors of KD. Limited human pathology specimens show no long-term inflammatory infiltrates (186, 188). The limited data on adult follow-up cases of KD implies there is a greater lifetime risk of cardiac issues and early mortality (15–18). Even those who had KD without noticeable aneurysms may have endothelial dysfunction and increased arterial stiffness (158). A clinical trial on long-term consequences is actively recruiting (NCT03750123-CAVASAKI trial). As established clinical cohorts age, the lifetime risk of childhood KD should be revealed.
Unfortunately, disparity of reports, lack of early timepoints, and lack of control tissue sample from cases experiencing ongoing inflammation make firm conclusions difficult. Collection and study of these types rare samples should continue.
Studies on Circulating Immune Cells
In the few published studies on peripheral cell subsets during acute KD, circulating B cells are generally elevated (6, 9, 45, 71, 190). Lack of changes in acute and convalescent B cells subgroups and increases in CD69+ natural killer and γδ T cells supported a role for the innate immune system (71). B cells did seem to be in an “activated” phenotype, being positive for CD86 (190). After stimulation of the TLR-9 receptor there was a global increase in the ability of B cells to secrete IgM, IgG, and IgA with a notable expansion in IgA+ B cell numbers (190). One study showed a paucity of IgA + peripheral B cells from acute KD samples compared to controls continuing through convalescence (8) that was not replicated in other studies. Reports do show increased IgA immune complexes and levels (13), although immune complexes do not necessarily portend worse prognosis (191). Overall, in the small number of studies relating to the peripheral blood B cell compartment, overall B cells numbers are increased and B cells are more reactive.
Although, total numbers of cells do not show consistent results, clonal expansion within the B cell compartment can be studied. A specific immune response to an agent typically has an initial inherent immune component that leads to antigen presentation to effector cells. Receptors on the effector cell surface (T-cell receptors in T cells and Immunoglobulin, or antibody, in B cells) bind specific targeted areas of the agent, termed epitopes. Specific recognition by T and B lymphocytes leads to stimulation, lymphocyte replication and clonal expansion; what is termed an oligoclonal response. Oligoclonal expansion is shown in peripheral IgM+ B cells in KD (9). Oligoclonal plasma cell infiltrates, predominantly IgA+, have been shown in KD arterial specimens (192). Heavy and light chain sequences were obtained and cloned into full length expression vectors from these cardiac vessel samples. These chimeric antibodies identified intracellular inclusions (ICI) in bronchial epithelium samples from children with KD (46, 104). However, similarly cloned antibodies revealed self-antigen targeting (181). Additionally, 26% of the control group, composed primarily of adult patients, had similar inclusion bodies, and next generation sequencing from these ICIs did not reveal an etiology (193). Although plasma cell infiltration outlined above is intriguing, a similar pathological response is seen in a number of inflammatory conditions such as NMDAR encephalitis (194), primary sclerosing cholangitis, (195) multiple sclerosis, (196) and responses to tumors (197). ICI in cells can be any number of structures (aggresomes, stress granules, p-bodies, prion-aggregates, aggresome-like induced structures (ALIS) and autophagosomes) and this has been recently reviewed (164). Currently, it is unclear of the significance of this work.
Similar Plasmablast (PB) Responses in KD Compared to Infections
Plasmablasts (PBs), derived from naïve and memory responses, are B cells transitioning to plasma cells that circulate in the peripheral blood cell compartment. They are characterized by surface expression of CD19, with CD20 downregulation, and high levels of CD27 and CD38. After antigenic challenge (vaccination and natural infections), CD19+, CD20lo, CD27+ and CD38 + PBs can be seen in the peripheral blood (8, 198–200). Immunization studies in adults, with pathogens such as tetanus, influenza and rabies, show PBs are enriched for specific antibodies against the challenge antigen, temporally peak 5–10 days after immunization, and are predictive of later sero-immunity (198, 201–203). In comparison to the general circulating B cell population, PBs are enriched for B cells that produce infection-specific antibodies (204–209) Although certain infections, such as dengue virus, may set off exceedingly high PB levels (210–212), excessive PB responses are usually an indicator of autoimmune flares (213). This excessive circulating PB response seen specifically correlates with CRP level in studies on ulcerative colitis (212, 214) and IGG4 related disease (215, 216). Besides correlation of an inflammatory state consistent with infection and inflammation, little is known regarding regulatory or pathologic consequences of PB elevation.
We postulated if KD is caused by an infection, we should see a predictable rise of PBs in the peripheral blood. On single timepoint collection, we showed 15 of 18 KD samples had elevation of their PBs, and overall this response was similar to the range of data shown in our 69 infected controls (45). Results of this study are consistent with the majority of the literature that show B cell stimulation and increasing peripheral B cell numbers during KD (6, 9, 71, 190). Importantly, the levels were not consistent with a pure autoimmune response as they did not correlate with CRPs and were not overly excessive. Unfortunately, only five children had repeat samples, but from these it did not appear that IVIG has an effect on PB numbers. As PBs rise and fall over time, we have ongoing studies collecting samples over time to confirm a dynamic PB response similar to infections occurs in KD. Other groups have recently shown similar PB elevation during KD, but this was compared to healthy controls (217).
Ongoing studies are exploring heavy and light chain usage in B cells and PBs during KD with next generation sequencing techniques. From our initial evaluation, a number of these clones have markers of affinity maturation (multiple clonal members, isotype switching, increased nucleotide substitutions from predicted germlines, and increases in the subgroup replacement to silent nucleotide mutation (R/S) ratios) (218). Our group and other are expressing cloned sequences as monoclonal antibodies in the attempt to find their antigenic target.
Because of reported oligoclonal responses and characteristics suggesting response to infection seen in PBs and antibodies during KD, we hypothesize the antibodies derived from PBs during KD are specific against the etiology that led to KD within that child.
Discussion
As reviewed, a number of studies support a roll for B cells, plasma cells, and antibodies in the pathogenesis of KD. Although B cell and plasma cell infiltration in pathology specimens is intriguing, whether they are bystanders activated by a superantigen effect, are responding to a self-antigen revealed by inflammation, or specific against an infectious etiology is currently unknown. An autoimmune component to this disease has long been postulated, and overlapping genomic risk associations with lupus and rheumatoid arthritis suggest this to play a role. Genomic and proteomic approaches strongly suggest a role for the innate immune system in pathogenesis, but associations with BLK, and CD40 also support a potential role for B cells. The increasingly appreciated role of B cells as immune system modulators may explain these associations and align the disparate data.
Humoral immune responses continue to be worth exploration in children with KD. Like the mouse models and attempts at developing new therapeutics, it is hard to be confident in any one approach without knowledge of the etiology. Compelling recent data suggests that exploration of the specific B cell responses is an encouraging path to discovering improved diagnostics and potentially the pathogen that sets off this immune cascade.
This is a rich opportunity for clinical investigators. Rigorous studies are needed on those children who present with KD. If any pulmonary findings are found, bronchial washings should be obtained and stored for potential molecular diagnostics. Other samples, such as PBMCs and serum, should be taken and banked for future studies. Thorough autopsy evaluation should be pursued on any subjects who succumb during the acute or convalescent phases of KD. Improved reporting and national registries would go a long way in establishing a representative pool of patients. Studies currently ongoing on peripheral cytokine profiles, B cells and PBs may show a consistent marker to help define who has KD. A correlative diagnostic marker, possibly even antibody derived, would be a highly desirable first step in future studies.
As outlined herein, evidence from genomics, transcriptomics, and proteomics support a role for B cells in the pathogenesis of KD, and continued studies on B cell responses may assist in identifying the etiology of KD.
Author Contributions
MH conceived and was the sole contributor to this manuscript.
Funding
Relative funding includes a generous grant from the Wildermuth Foundation provided through the Variety Club of Buffalo.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kawasaki T. Kawasaki disease. Acta Paediatr. (1995) 84:713–5. doi: 10.1111/j.1651-2227.1995.tb13742.x
2. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. (2017) 135:e927–99. doi: 10.1161/CIR.0000000000000484
3. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis and kawasaki disease, council on cardiovascular disease in the young, American Heart Association. Circulation. (2004) 110:2747–71. doi: 10.1161/01.CIR.0000145143.19711.78
4. Yorifuji T, Tsukahara H, Doi H. Breastfeeding and risk of kawasaki disease: a nationwide longitudinal survey in Japan. Pediatrics. (2016) 137:e20153919. doi: 10.1542/peds.2015-3919
5. Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat Genet. (2012) 44:522–5. doi: 10.1038/ng.2227
6. Chang CJ, Kuo HC, Chang JS, Lee JK, Tsai FJ, Khor CC, et al. Replication and meta-analysis of GWAS identified susceptibility loci in Kawasaki disease confirm the importance of B lymphoid tyrosine kinase (BLK) in disease susceptibility. PLoS ONE. (2013) 8:e72037. doi: 10.1371/journal.pone.0072037
7. Kuo HC, Lo MH, Hsieh KS, Guo MM, Huang YH. High-dose aspirin is associated with anemia and does not confer benefit to disease outcomes in Kawasaki disease. PLoS ONE. (2015) 10:e0144603. doi: 10.1371/journal.pone.0144603
8. Shingadia D, O'Gorman M, Rowley AH, Shulman ST. Surface and cytoplasmic immunoglobulin expression in circulating B-lymphocytes in acute Kawasaki disease. Pediatr Res. (2001) 50:538–43. doi: 10.1203/00006450-200110000-00019
9. Lee HH, Park IH, Shin JS, Kim DS. Immunoglobulin V(H) chain gene analysis of peripheral blood IgM-producing B cells in patients with Kawasaki disease. Yonsei Med J. (2009) 50:493–504. doi: 10.3349/ymj.2009.50.4.493
10. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis, and Kawasaki disease, council on cardiovascular disease in the young, American Heart Association. Pediatrics. (2004) 114:1708–33. doi: 10.1542/peds.2004-2182
11. Terai M, Honda T, Yasukawa K, Higashi K, Hamada H, Kohno Y. Prognostic impact of vascular leakage in acute Kawasaki disease. Circulation. (2003) 108:325–30. doi: 10.1161/01.CIR.0000079166.93475.5F
12. Jun HO, Yu JJ, Kang SY, Seo CD, Baek JS, Kim YH, et al. Diagnostic characteristics of supplemental laboratory criteria for incomplete Kawasaki disease in children with complete Kawasaki disease. Korean J Pediatr. (2015) 58:369–73. doi: 10.3345/kjp.2015.58.10.369
13. Ohshio G, Furukawa F, Khine M, Yoshioka H, Kudo H, Hamashima Y. High levels of IgA-containing circulating immune complex and secretory IgA in Kawasaki disease. Microbiol Immunol. (1987) 31:891–8. doi: 10.1111/j.1348-0421.1987.tb03150.x
14. Yanagawa H, Yashiro M, Nakamura Y, Sakata K, Kawasaki T. Iv gamma globulin treatment of Kawasaki disease in Japan: results of a nationwide survey. Acta Paediatr. (1995) 84:765–8. doi: 10.1111/j.1651-2227.1995.tb13752.x
15. Pinna GS, Kafetzis DA, Tselkas OI, Skevaki CL. Kawasaki disease: an overview. Curr Opin Infect Dis. (2008) 21:263–70. doi: 10.1097/QCO.0b013e3282fbf9cd
16. Nakamura Y, Yanagawa H, Harada K, Kato H, Kawasaki T. Mortality among persons with a history of Kawasaki disease in Japan: the fifth look. Arch Pediatr Adolesc Med. (2002) 156:162–5. doi: 10.1001/archpedi.156.2.162
17. Nakamura Y, Aso E, Yashiro M, Uehara R, Watanabe M, Tajimi M, et al. Mortality among persons with a history of Kawasaki disease in Japan: can paediatricians safely discontinue follow-up of children with a history of the disease but without cardiac sequelae? Acta Paediatr. (2005) 94:429–34. doi: 10.1111/j.1651-2227.2005.tb01913.x
18. Gordon JB, Kahn AM, Burns JC. When children with Kawasaki disease grow up: myocardial and vascular complications in adulthood. J Am Coll Cardiol. (2009) 54:1911–20. doi: 10.1016/j.jacc.2009.04.102
19. Chrissoheris MP, Donohue TJ, Young RS, Ghantous A. Coronary artery aneurysms. Cardiol Rev. (2008) 16:116–23. doi: 10.1097/CRD.0b013e31815d0573
20. Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. (2005) 9:185–94. doi: 10.1016/j.ijid.2005.03.002
21. Abuhammour WM, Hasan RA, Eljamal A, Asmar B. Kawasaki disease hospitalizations in a predominantly African-American population. Clin Pediatr. (2005) 44:721–5. doi: 10.1177/000992280504400812
22. Clark DE, Denby KJ, Kaufman LM, Fill MA, Piya B, Krishnaswami S, et al. Predictors of intravenous immunoglobulin non-response and racial disparities in Kawasaki disease. Pediatr Infect Dis J. (2018) 37:1227–34. doi: 10.1097/INF.0000000000002019
23. Porcalla AR, Sable CA, Patel KM, Martin GR, Singh N. The epidemiology of Kawasaki disease in an urban hospital: does African American race protect against coronary artery aneurysms? Pediatr Cardiol. (2005) 26:775–81. doi: 10.1007/s00246-005-0916-5
24. Scuccimarri R. Kawasaki disease. Pediatr Clin North Am. (2012) 59:425–45. doi: 10.1016/j.pcl.2012.03.009
25. Fujita Y, Nakamura Y, Sakata K, Hara N, Kobayashi M, Nagai M, et al. Kawasaki disease in families. Pediatrics. (1989) 84:666–9.
26. Rodo X, Ballester J, Cayan D, Melish ME, Nakamura Y, Uehara R, et al. Association of Kawasaki disease with tropospheric wind patterns. Sci Rep. (2011) 1:152. doi: 10.1038/srep00152
27. Burns JC, Cayan DR, Tong G, Bainto EV, Turner CL, Shike H, et al. Seasonality and temporal clustering of Kawasaki syndrome. Epidemiology. (2005) 16:220–5. doi: 10.1097/01.ede.0000152901.06689.d4
28. Kao AS, Getis A, Brodine S, Burns JC. Spatial and temporal clustering of Kawasaki syndrome cases. Pediatr Infect Dis J. (2008) 27:981–5. doi: 10.1097/INF.0b013e31817acf4f
29. Makino N, Nakamura Y, Yashiro M, Ae R, Tsuboi S, Aoyama Y, et al. Descriptive epidemiology of Kawasaki disease in Japan, 2011–2012: from the results of the 22nd nationwide survey. J Epidemiol. (2015) 25:239–45. doi: 10.2188/jea.JE20140089
30. Mauro A, Fabi M, Da Fre M, Guastaroba P, Corinaldesi E, Calabri GB, et al. Kawasaki disease: an epidemiological study in central Italy. Pediatr Rheumatol Online J. (2016) 14:22. doi: 10.1186/s12969-016-0084-6
31. Chen JJ, Ma XJ, Liu F, Yan WL, Huang MR, Huang M, et al. Epidemiologic features of Kawasaki disease in shanghai from 2008 through 2012. Pediatr Infect Dis J. (2016) 35:7–12. doi: 10.1097/INF.0000000000000914
32. Chang A, Delmerico AM, Hicar MD. Spatiotemporal analysis and epidemiology of Kawasaki disease in Western New York: a sixteen year review of cases presenting to a single tertiary care center. Pediatr Infect Dis J. (2018) 38:582–8. doi: 10.1097/INF.0000000000002239
33. Burns JC, Herzog L, Fabri O, Tremoulet AH, Rodo X, Uehara R, et al. Seasonality of Kawasaki disease: a global perspective. PLoS ONE. (2013) 8:e74529. doi: 10.1371/journal.pone.0074529
34. Manlhiot C, O'Shea S, Bernknopf B, LaBelle M, Chahal N, Dillenburg RF, et al. Epidemiology of Kawasaki disease in Canada 2004 to 2014: comparison of surveillance using administrative data vs periodic medical record review. Can J Cardiol. (2018) 34:303–9. doi: 10.1016/j.cjca.2017.12.009
35. Dergun M, Kao A, Hauger SB, Newburger JW, Burns JC. Familial occurrence of Kawasaki syndrome in North America. Arch Pediatr Adolesc Med. (2005) 159:876–81. doi: 10.1001/archpedi.159.9.876
36. Newburger JW, Taubert KA, Shulman ST, Rowley AH, Gewitz MH, Takahashi M, et al. Summary and abstracts of the seventh international Kawasaki disease symposium: december 4–7, 2001, Hakone, Japan. Pediatr Res. (2003) 53:153–7. doi: 10.1203/00006450-200301000-00026
37. Rowley AH, Shulman ST. The Epidemiology and pathogenesis of Kawasaki disease. Front Pediatr. (2018) 6:374. doi: 10.3389/fped.2018.00374
38. Nakagawa A, Ito M, Iwaki T, Yatabe Y, Asai J, Hayashi K. Chronic active Epstein-Barr virus infection with giant coronary aneurysms. Am J Clin Pathol. (1996) 105:733–6. doi: 10.1093/ajcp/105.6.733
39. Rodo X, Curcoll R, Robinson M, Ballester J, Burns JC, Cayan DR, et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc Natl Acad Sci USA. (2014) 111:7952–7. doi: 10.1073/pnas.1400380111
40. Manlhiot C, Mueller B, O'Shea S, Majeed H, Bernknopf B, Labelle M, et al. Environmental epidemiology of Kawasaki disease: linking disease etiology, pathogenesis and global distribution. PLoS ONE. (2018) 13:e0191087. doi: 10.1371/journal.pone.0191087
41. Bell DM, Brink EW, Nitzkin JL, Hall CB, Wulff H, Berkowitz ID, et al. Kawasaki syndrome: description of two outbreaks in the United States. N Engl J Med. (1981) 304:1568–75. doi: 10.1056/NEJM198106253042603
42. Baker AL, Lu M, Minich LL, Atz AM, Klein GL, Korsin R, et al. Associated symptoms in the ten days before diagnosis of Kawasaki disease. J Pediatr. (2009) 154:592–5.e2. doi: 10.1016/j.jpeds.2008.10.006
43. Freeman AF, Crawford SE, Finn LS, Lopez-Andreu JA, Ferrando-Monleon S, Perez-Tamarit D, et al. Inflammatory pulmonary nodules in Kawasaki disease. Pediatr Pulmonol. (2003) 36:102–6. doi: 10.1002/ppul.10333
44. Jordan-Villegas A, Chang ML, Ramilo O, Mejias A. Concomitant respiratory viral infections in children with Kawasaki disease. Pediatr Infect Dis J. (2010) 29:770–2. doi: 10.1097/INF.0b013e3181dba70b
45. Martin M, Wrotniak BH, Hicar M. Suppressed plasmablast responses in febrile infants, including children with Kawasaki disease. PLoS ONE. (2018) 13:e0193539. doi: 10.1371/journal.pone.0193539
46. Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease–cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. (2008) 6:394–401. doi: 10.1038/nrmicro1853
47. Geme JW Jr. A biological perspective of slow virus infection and chronic disease. West J Med. (1978) 128:382–9.
48. Shimizu C, Shike H, Baker SC, Garcia F, van der Hoek L, Kuijpers TW, et al. Human coronavirus NL63 is not detected in the respiratory tracts of children with acute Kawasaki disease. J Infect Dis. (2005) 192:1767–71. doi: 10.1086/497170
49. Lee KY, Han JW, Lee JS. Kawasaki disease may be a hyperimmune reaction of genetically susceptible children to variants of normal environmental flora. Med Hypotheses. (2007) 69:642–51. doi: 10.1016/j.mehy.2006.12.051
50. Rhim JW, Kang HM, Han JW, Lee KY. A Presumed etiology of Kawasaki disease based on epidemiological comparison with infectious or immune-mediated diseases. Front Pediatr. (2019) 7:202. doi: 10.3389/fped.2019.00202
51. Esposito S, Polinori I, Rigante D. The gut microbiota-host partnership as a potential driver of Kawasaki syndrome. Front Pediatr. (2019) 7:124. doi: 10.3389/fped.2019.00124
52. Sleeper LA, Minich LL, McCrindle BM, Li JS, Mason W, Colan SD, et al. Evaluation of Kawasaki disease risk-scoring systems for intravenous immunoglobulin resistance. J Pediatr. (2011) 158:831–5.e3. doi: 10.1016/j.jpeds.2010.10.031
53. Dominguez SR, Anderson MS. Advances in the treatment of Kawasaki disease. Curr Opin Pediatr. (2013) 25:103–9. doi: 10.1097/MOP.0b013e32835c1122
54. Parthasarathy P, Agarwal A, Chawla K, Tofighi T, Mondal TK. Upcoming biomarkers for the diagnosis of Kawasaki disease: a review. Clin Biochem. (2015) 48:1188–94. doi: 10.1016/j.clinbiochem.2015.02.013
55. Kimura Y, Yanagimachi M, Ino Y, Aketagawa M, Matsuo M, Okayama A, et al. Identification of candidate diagnostic serum biomarkers for Kawasaki disease using proteomic analysis. Sci Rep. (2017) 7:43732. doi: 10.1038/srep43732
56. Okuma Y, Suda K, Nakaoka H, Katsube Y, Mitani Y, Yoshikane Y, et al. Serum Tenascin-C as a novel predictor for risk of coronary artery lesion and resistance to intravenous immunoglobulin in Kawasaki disease- a multicenter retrospective study. Circ J. (2016) 80:2376–81. doi: 10.1253/circj.CJ-16-0563
57. Yokouchi Y, Oharaseki T, Enomoto Y, Sato W, Imanaka-Yoshida K, Takahashi K. Expression of tenascin C in cardiovascular lesions of Kawasaki disease. Cardiovasc Pathol. (2019) 38:25–30. doi: 10.1016/j.carpath.2018.10.005
58. Kim DS, Lee HK, Noh GW, Lee SI, Lee KY. Increased serum interleukin-10 level in Kawasaki disease. Yonsei Med J. (1996) 37:125–30. doi: 10.3349/ymj.1996.37.2.125
59. Hirao J, Hibi S, Andoh T, Ichimura T. High levels of circulating interleukin-4 and interleukin-10 in Kawasaki disease. Int Arch Allergy Immunol. (1997) 112:152–6. doi: 10.1159/000237447
60. Nakamura J, Watanabe S, Kimura H, Kobayashi M, Karasawa T, Kamata R, et al. Adeno-associated virus vector-mediated interleukin-10 induction prevents vascular inflammation in a murine model of Kawasaki disease. Sci Rep. (2018) 8:7601. doi: 10.1038/s41598-018-25856-0
61. Heine G, Drozdenko G, Grun JR, Chang HD, Radbruch A, Worm M. Autocrine IL-10 promotes human B-cell differentiation into IgM- or IgG-secreting plasmablasts. Eur J Immunol. (2014) 44:1615–21. doi: 10.1002/eji.201343822
62. Ko TM, Kuo HC, Chang JS, Chen SP, Liu YM, Chen HW, et al. CXCL10/IP-10 is a biomarker and mediator for Kawasaki disease. Circ Res. (2015) 116:876–83. doi: 10.1161/CIRCRESAHA.116.305834
63. Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. (2002) 16:559–69. doi: 10.1016/S1074-7613(02)00295-9
64. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nature reviews Drug discovery. (2014) 13:379–95. doi: 10.1038/nrd4296
65. Bae YJ, Kim MH, Lee HY, Uh Y, Namgoong MK, Cha BH, et al. Elevated serum levels of IL-21 in Kawasaki disease. Allergy Asthma Immunol Res. (2012) 4:351–6. doi: 10.4168/aair.2012.4.6.351
66. Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. (2005) 175:7867–79. doi: 10.4049/jimmunol.175.12.7867
67. Engelberg R, Martin M, Wrotniak BH, Hicar MD. Observational study of Interleukin-21 (IL-21) does not distinguish Kawasaki disease from other causes of fever in children. Pediatr Rheumatol Online J. (2017) 15:32. doi: 10.1186/s12969-017-0163-3
68. Abe J, Jibiki T, Noma S, Nakajima T, Saito H, Terai M. Gene expression profiling of the effect of high-dose intravenous Ig in patients with Kawasaki disease. J Immunol. (2005) 174:5837–45. doi: 10.4049/jimmunol.174.9.5837
69. Nomura I, Abe J, Noma S, Saito H, Gao B, Wheeler G, et al. Adrenomedullin is highly expressed in blood monocytes associated with acute Kawasaki disease: a microarray gene expression study. Pediatr Res. (2005) 57:49–55. doi: 10.1203/01.PDR.0000147745.52711.DD
70. Nishida K, Watanabe K, Echigo S, Mayumi M, Nishikimi T. Increased plasma adrenomedullin levels in Kawasaki disease with coronary artery involvement. Am J Med. (2001) 111:165–6. doi: 10.1016/S0002-9343(01)00781-1
71. Ikeda K, Yamaguchi K, Tanaka T, Mizuno Y, Hijikata A, Ohara O, et al. Unique activation status of peripheral blood mononuclear cells at acute phase of Kawasaki disease. Clin Exp Immunol. (2010) 160:246–55. doi: 10.1111/j.1365-2249.2009.04073.x
72. Ling XB, Lau K, Kanegaye JT, Pan Z, Peng S, Ji J, et al. A diagnostic algorithm combining clinical and molecular data distinguishes Kawasaki disease from other febrile illnesses. BMC Med. (2011) 9:130. doi: 10.1186/1741-7015-9-130
73. Hoang LT, Shimizu C, Ling L, Naim AN, Khor CC, Tremoulet AH, et al. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. (2014) 6:541. doi: 10.1186/s13073-014-0102-6
74. Jaggi P, Mejias A, Xu Z, Yin H, Moore-Clingenpeel M, Smith B, et al. Whole blood transcriptional profiles as a prognostic tool in complete and incomplete Kawasaki Disease. PLoS ONE. (2018) 13:e0197858. doi: 10.1371/journal.pone.0197858
75. Murata H. Experimental candida-induced arteritis in mice. Relation to arteritis in the mucocutaneous lymph node syndrome. Microbiol Immunol. (1979) 23:825–31. doi: 10.1111/j.1348-0421.1979.tb02815.x
76. Hashimoto Y, Fukazawa R, Nagi-Miura N, Ohno N, Suzuki N, Katsube Y, et al. Interleukin-1beta Inhibition Attenuates Vasculitis in a Mouse Model of Kawasaki Disease. J Nippon Med Sch. (2019) 86:108–16. doi: 10.1272/jnms.JNMS.2019_86-206
77. Takahashi K, Oharaseki T, Yokouchi Y, Miura NN, Ohno N, Okawara AI, et al. Administration of human immunoglobulin suppresses development of murine systemic vasculitis induced with Candida albicans water-soluble fraction: an animal model of Kawasaki disease. Mod Rheumatol. (2010) 20:160–7. doi: 10.3109/s10165-009-0250-5
78. Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. (1985) 28:652–9. doi: 10.1002/art.1780280609
79. Duong TT, Silverman ED, Bissessar MV, Yeung RS. Superantigenic activity is responsible for induction of coronary arteritis in mice: an animal model of Kawasaki disease. Int Immunol. (2003) 15:79–89. doi: 10.1093/intimm/dxg007
80. Schulte DJ, Yilmaz A, Shimada K, Fishbein MC, Lowe EL, Chen S, et al. Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking Kawasaki disease. J Immunol. (2009) 183:5311–8. doi: 10.4049/jimmunol.0901395
81. Chan WC, Duong TT, Yeung RS. Presence of IFN-gamma does not indicate its necessity for induction of coronary arteritis in an animal model of Kawasaki disease. J Immunol. (2004) 173:3492–503. doi: 10.4049/jimmunol.173.5.3492
82. Yeung RS. Kawasaki disease: update on pathogenesis. Curr Opin Rheumatol. (2010) 22:551–60. doi: 10.1097/BOR.0b013e32833cf051
83. Hui-Yuen JS, Duong TT, Yeung RS. TNF-alpha is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J Immunol. (2006) 176:6294–301. doi: 10.4049/jimmunol.176.10.6294
84. Chen S, Lee Y, Crother TR, Fishbein M, Zhang W, Yilmaz A, et al. Marked acceleration of atherosclerosis after Lactobacillus casei-induced coronary arteritis in a mouse model of Kawasaki disease. Arterioscler Thromb Vasc Biol. (2012) 32:e60–71. doi: 10.1161/ATVBAHA.112.249417
85. Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, et al. Role of Interleukin-1 signaling in a mouse model of kawasaki disease-associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. (2016) 36:886–97. doi: 10.1161/ATVBAHA.115.307072
86. Lau AC, Duong TT, Ito S, Wilson GJ, Yeung RS. Inhibition of matrix metalloproteinase-9 activity improves coronary outcome in an animal model of Kawasaki disease. Clin Exp Immunol. (2009) 157:300–9. doi: 10.1111/j.1365-2249.2009.03949.x
87. Bratincsak A, Limm-Chan BN, Nerurkar VR, Ching LL, Reddy VD, Lim E, et al. Study design and rationale to assess Doxycycline Efficacy in preventing coronary Artery Lesions in children with Kawasaki disease (DEAL trial) - A phase II clinical trial. Contemp Clin Trials. (2018) 65:33–8. doi: 10.1016/j.cct.2017.11.014
88. Matundan HH, Sin J, Rivas MN, Fishbein MC, Lehman TJ, Chen S, et al. Myocardial fibrosis after adrenergic stimulation as a long-term sequela in a mouse model of Kawasaki disease vasculitis. JCI Insight. (2019) 4:e126279. doi: 10.1172/jci.insight.126279
89. Kim M, Kim K. Elevation of cardiac troponin I in the acute stage of Kawasaki disease. Pediatr Cardiol. (1999) 20:184–8. doi: 10.1007/s002469900437
90. Orenstein JM, Shulman ST, Fox LM, Baker SC, Takahashi M, Bhatti TR, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS ONE. (2012) 7:e38998. doi: 10.1371/journal.pone.0038998
91. Takahashi K, Oharaseki T, Yokouchi Y. Pathogenesis of Kawasaki disease. Clin Exp Immunol. (2011) 164(Suppl. 1):20–2. doi: 10.1111/j.1365-2249.2011.04361.x
92. Dou J, Li H, Sun L, Yan W, Lv H, Ding Y. Histopathological and ultrastructural examinations of rabbit coronary artery vasculitis caused by bovine serum albumin: an animal model of Kawasaki disease. Ultrastruct Pathol. (2013) 37:139–45. doi: 10.3109/01913123.2012.750409
93. Orenstein JM, Rowley AH. An evaluation of the validity of the animal models of Kawasaki disease vasculopathy. Ultrastruct Pathol. (2014) 38:245–7. doi: 10.3109/01913123.2014.932314
94. De Inocencio J, Hirsch R. Evidence for superantigen mediated process in Kawasaki disease. Arch Dis Child. (1995) 73:275–6. doi: 10.1136/adc.73.3.275-b
95. Curtis N. Kawasaki disease and toxic shock syndrome–at last the etiology is clear? Adv Exp Med Biol. (2004) 549:191–200. doi: 10.1007/978-1-4419-8993-2_26
96. Leung DY, Meissner HC, Fulton DR, Murray DL, Kotzin BL, Schlievert PM. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. (1993) 342:1385–8. doi: 10.1016/0140-6736(93)92752-F
97. Leung DY, Sullivan KE, Brown-Whitehorn TF, Fehringer AP, Allen S, Finkel TH, et al. Association of toxic shock syndrome toxin-secreting and exfoliative toxin-secreting Staphylococcus aureus with Kawasaki syndrome complicated by coronary artery disease. Pediatr Res. (1997) 42:268–72. doi: 10.1203/00006450-199709000-00004
98. Matsubara K, Fukaya T. The role of superantigens of group A Streptococcus and Staphylococcus aureus in Kawasaki disease. Curr Opin Infect Dis. (2007) 20:298–303. doi: 10.1097/QCO.0b013e3280964d8c
99. Uchiyama T, Kato H. The pathogenesis of Kawasaki disease and superantigens. Jpn J Infect Dis. (1999) 52:141–5.
100. Abe J, Kotzin BL, Jujo K, Melish ME, Glode MP, Kohsaka T, et al. Selective expansion of T cells expressing T-cell receptor variable regions V beta 2 and V beta 8 in Kawasaki disease. Proc Natl Acad Sci USA. (1992) 89:4066–70. doi: 10.1073/pnas.89.9.4066
101. Abe J, Kotzin BL, Meissner C, Melish ME, Takahashi M, Fulton D, et al. Characterization of T cell repertoire changes in acute Kawasaki disease. J Exp Med. (1993) 177:791–6. doi: 10.1084/jem.177.3.791
102. Yoshioka T, Matsutani T, Iwagami S, Toyosaki-Maeda T, Yutsudo T, Tsuruta Y, et al. Polyclonal expansion of TCRBV2- and TCRBV6-bearing T cells in patients with Kawasaki disease. Immunology. (1999) 96:465–72. doi: 10.1046/j.1365-2567.1999.00695.x
103. Choi IH, Chwae YJ, Shim WS, Kim DS, Kwon DH, Kim JD, et al. Clonal expansion of CD8+ T cells in Kawasaki disease. J Immunol. (1997) 159:481–6.
104. Rowley AH, Baker SC, Shulman ST, Fox LM, Takahashi K, Garcia FL, et al. Cytoplasmic inclusion bodies are detected by synthetic antibody in ciliated bronchial epithelium during acute Kawasaki disease. J Infect Dis. (2005) 192:1757–66. doi: 10.1086/497171
105. Pietra BA, De Inocencio J, Giannini EH, Hirsch R. TCR V beta family repertoire and T cell activation markers in Kawasaki disease. J Immunol. (1994) 153:1881–8.
106. Sakaguchi M, Kato H, Nishiyori A, Sagawa K, Itoh K. Characterization of CD4+ T helper cells in patients with Kawasaki disease (KD): preferential production of tumour necrosis factor-alpha (TNF-alpha) by V beta 2- or V beta 8- CD4+ T helper cells. Clin Exp Immunol. (1995) 99:276–82. doi: 10.1111/j.1365-2249.1995.tb05545.x
107. Lv YW, Wang J, Sun L, Zhang JM, Cao L, Ding YY, et al. Understanding the pathogenesis of Kawasaki disease by network and pathway analysis. Comput Math Methods Med. (2013) 2013:989307. doi: 10.1155/2013/989307
108. Ozen S, Batu ED. Vasculitis pathogenesis: can we talk about precision medicine? Front Immunol. (2018) 9:1892. doi: 10.3389/fimmu.2018.01892
109. Xie X, Shi X, Liu M. The Roles of Genetic factors in Kawasaki disease: a systematic review and meta-analysis of genetic association studies. Pediatr Cardiol. (2018) 39:207–25. doi: 10.1007/s00246-017-1760-0
110. Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis. (2018) 21:26–30. doi: 10.1111/1756-185X.13218
111. Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. (2008) 40:35–42. doi: 10.1038/ng.2007.59
112. Chi H, Huang FY, Chen MR, Chiu NC, Lee HC, Lin SP, et al. ITPKC gene SNP rs28493229 and Kawasaki disease in Taiwanese children. Hum Mol Genet. (2010) 19:1147–51. doi: 10.1093/hmg/ddp586
113. Lin MT, Wang JK, Yeh JI, Sun LC, Chen PL, Wu JF, et al. Clinical Implication of the C Allele of the ITPKC Gene SNP rs28493229 in Kawasaki disease: association with disease susceptibility and BCG scar reactivation. Pediatr Infect Dis J. (2011) 30:148–52. doi: 10.1097/INF.0b013e3181f43a4e
114. Peng Q, Chen C, Zhang Y, He H, Wu Q, Liao J, et al. Single-nucleotide polymorphism rs2290692 in the 3'UTR of ITPKC associated with susceptibility to Kawasaki disease in a Han Chinese population. Pediatr Cardiol. (2012) 33:1046–53. doi: 10.1007/s00246-012-0223-x
115. Lou J, Xu S, Zou L, Zhong R, Zhang T, Sun Y, et al. A functional polymorphism, rs28493229, in ITPKC and risk of Kawasaki disease: an integrated meta-analysis. Mol Biol Rep. (2012) 39:11137–44. doi: 10.1007/s11033-012-2022-0
116. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
117. Kuo HC, Yu HR, Juo SH, Yang KD, Wang YS, Liang CD, et al. CASP3 gene single-nucleotide polymorphism (rs72689236) and Kawasaki disease in Taiwanese children. J Hum Genet. (2011) 56:161–5. doi: 10.1038/jhg.2010.154
118. Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet. (2010) 19:2898–906. doi: 10.1093/hmg/ddq176
119. Kuo HC, Hsu YW, Wu CM, Chen SH, Hung KS, Chang WP, et al. A replication study for association of ITPKC and CASP3 two-locus analysis in IVIG unresponsiveness and coronary artery lesion in Kawasaki disease. PLoS ONE. (2013) 8:e69685. doi: 10.1371/journal.pone.0069685
120. Onouchi Y, Suzuki Y, Suzuki H, Terai M, Yasukawa K, Hamada H, et al. ITPKC and CASP3 polymorphisms and risks for IVIG unresponsiveness and coronary artery lesion formation in Kawasaki disease. Pharmacogenomics J. (2013) 13:52–9. doi: 10.1038/tpj.2011.45
121. Alphonse MP, Duong TT, Shumitzu C, Hoang TL, McCrindle BW, Franco A, et al. Inositol-Triphosphate 3-Kinase C mediates inflammasome activation and treatment response in Kawasaki disease. J Immunol. (2016) 197:3481–9. doi: 10.4049/jimmunol.1600388
122. Burgner D, Davila S, Breunis WB, Ng SB, Li Y, Bonnard C, et al. A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. (2009) 5:e1000319. doi: 10.1371/journal.pgen.1000319
123. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
124. Kwon YC, Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, et al. Male-specific association of the FCGR2A His167Arg polymorphism with Kawasaki disease. PLoS ONE. (2017) 12:e0184248. doi: 10.1371/journal.pone.0184248
125. Onouchi Y, Onoue S, Tamari M, Wakui K, Fukushima Y, Yashiro M, et al. CD40 ligand gene and Kawasaki disease. Eur J Hum Genet. (2004) 12:1062–8. doi: 10.1038/sj.ejhg.5201266
126. Orozco G, Eyre S, Hinks A, Bowes J, Morgan AW, Wilson AG, et al. Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. (2011) 70:463–8. doi: 10.1136/ard.2010.137174
127. Laird RM, Laky K, Hayes SM. Unexpected role for the B cell-specific Src family kinase B lymphoid kinase in the development of IL-17-producing gammadelta T cells. J Immunol. (2010) 185:6518–27. doi: 10.4049/jimmunol.1002766
128. Samuelson EM, Laird RM, Maue AC, Rochford R, Hayes SM. Blk haploinsufficiency impairs the development, but enhances the functional responses, of MZ B cells. Immunol Cell Biol. (2012) 90:620–9. doi: 10.1038/icb.2011.76
129. Sim BK, Park H, Kim JJ, Yun SW, Yu JJ, Yoon KL, et al. Assessment of the clinical heterogeneity of Kawasaki disease using genetic variants of BLK and FCGR2A. Korean Circ J. (2019) 49:99–108. doi: 10.4070/kcj.2018.0224
130. Lou J, Zhong R, Shen N, Lu XZ, Ke JT, Duan JY, et al. Systematic confirmation study of GWAS-identified genetic variants for Kawasaki disease in a Chinese population. Sci Rep. (2015) 5:8194. doi: 10.1038/srep08194
131. Kuo HC, Li SC, Guo MM, Huang YH, Yu HR, Huang FC, et al. Genome-Wide association study identifies novel susceptibility genes associated with coronary artery aneurysm formation in Kawasaki disease. PLoS ONE. (2016) 11:e0154943. doi: 10.1371/journal.pone.0154943
132. Tremoulet AH, Jain S, Jaggi P, Jimenez-Fernandez S, Pancheri JM, Sun X, et al. Infliximab for intensification of primary therapy for Kawasaki disease: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet. (2014) 383:1731–8. doi: 10.1016/S0140-6736(13)62298-9
133. Mori M, Hara T, Kikuchi M, Shimizu H, Miyamoto T, Iwashima S, et al. Infliximab versus intravenous immunoglobulin for refractory Kawasaki disease: a phase 3, randomized, open-label, active-controlled, parallel-group, multicenter trial. Sci Rep. (2018) 8:1994. doi: 10.1038/s41598-017-18387-7
134. Hamada H, Suzuki H, Onouchi Y, Ebata R, Terai M, Fuse S, et al. Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, phase 3 trial. Lancet. (2019) 393:1128–37. doi: 10.1016/S0140-6736(18)32003-8
135. Aoyagi R, Hamada H, Sato Y, Suzuki H, Onouchi Y, Ebata R, et al. Study protocol for a phase III multicentre, randomised, open-label, blinded-end point trial to evaluate the efficacy and safety of immunoglobulin plus cyclosporin A in patients with severe Kawasaki disease (KAICA Trial). BMJ Open. (2015) 5:e009562. doi: 10.1136/bmjopen-2015-009562
136. Newburger JW, Takahashi M, Beiser AS, Burns JC, Bastian J, Chung KJ, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. (1991) 324:1633–9. doi: 10.1056/NEJM199106063242305
137. Newburger JW, Sleeper LA, McCrindle BW, Minich LL, Gersony W, Vetter VL, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. (2007) 356:663–75. doi: 10.1056/NEJMoa061235
138. Kobayashi T, Kobayashi T, Morikawa A, Ikeda K, Seki M, Shimoyama S, et al. Efficacy of intravenous immunoglobulin combined with prednisolone following resistance to initial intravenous immunoglobulin treatment of acute Kawasaki disease. J Pediatr. (2013) 163:521–6. doi: 10.1016/j.jpeds.2013.01.022
139. Kuo HC, Guo MM, Lo MH, Hsieh KS, Huang YH. Effectiveness of intravenous immunoglobulin alone and intravenous immunoglobulin combined with high-dose aspirin in the acute stage of Kawasaki disease: study protocol for a randomized controlled trial. BMC Pediatr. (2018) 18:200. doi: 10.1186/s12887-018-1180-1
140. Sauvaget E, Bonello B, David M, Chabrol B, Dubus JC, Bosdure E. Resistant Kawasaki disease treated with anti-CD20. J Pediatr. (2012) 160:875–6. doi: 10.1016/j.jpeds.2012.01.018
141. Koizumi K, Hoshiai M, Moriguchi T, Katsumata N, Toda T, Kise H, et al. Plasma exchange downregulates activated monocytes and restores regulatory T cells in Kawasaki disease. Ther Apher Dial. (2019) 23:92–8. doi: 10.1111/1744-9987.12754
142. Lo MS, Newburger JW. Role of intravenous immunoglobulin in the treatment of Kawasaki disease. Int J Rheum Dis. (2018) 21:64–9. doi: 10.1111/1756-185X.13220
143. Galeotti C, Kaveri SV, Bayry J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int Immunol. (2017) 29:491–8. doi: 10.1093/intimm/dxx039
144. Burns JC, Franco A. The immunomodulatory effects of intravenous immunoglobulin therapy in Kawasaki disease. Expert Rev Clin Immunol. (2015) 11:819–25. doi: 10.1586/1744666X.2015.1044980
145. Burns JC, Kone-Paut I, Kuijpers T, Shimizu C, Tremoulet A, Arditi M. Review: found in translation: international initiatives pursuing interleukin-1 blockade for treatment of acute Kawasaki disease. Arthritis Rheumatol. (2017) 69:268–76. doi: 10.1002/art.39975
146. Katz U, Shoenfeld Y, Zandman-Goddard G. Update on intravenous immunoglobulins (IVIg) mechanisms of action and off- label use in autoimmune diseases. Curr Pharm Des. (2011) 17:3166–75. doi: 10.2174/138161211798157540
147. Chaigne B, Mouthon L. Mechanisms of action of intravenous immunoglobulin. Transfus Apher Sci. (2017) 56:45–9. doi: 10.1016/j.transci.2016.12.017
148. Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. (2001) 291:484–6. doi: 10.1126/science.291.5503.484
149. Ballow M. Mechanisms of immune regulation by IVIG. Curr Opin Allergy Clin Immunol. (2014) 14:509–15. doi: 10.1097/ACI.0000000000000116
150. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy. (2015) 70:310–8. doi: 10.1111/all.12558
151. Rasouli M, Heidari B, Kalani M. Downregulation of Th17 cells and the related cytokines with treatment in Kawasaki disease. Immunol Lett. (2014) 162(1 Pt A):269–75. doi: 10.1016/j.imlet.2014.09.017
152. Olivito B, Taddio A, Simonini G, Massai C, Ciullini S, Gambineri E, et al. Defective FOXP3 expression in patients with acute Kawasaki disease and restoration by intravenous immunoglobulin therapy. Clin Exp Rheumatol. (2010) 28(1 Suppl. 57):93–7.
153. Lee HK, Kim DS, Noh GW, Lee KY. Effects of intravenous immune globulin on the peripheral lymphocyte phenotypes in Kawasaki disease. Yonsei Med J. (1996) 37:357–63. doi: 10.3349/ymj.1996.37.5.357
154. Ehara H, Kiyohara K, Izumisawa Y, Ito T. Early activation does not translate into effector differentiation of peripheral CD8T cells during the acute phase of Kawasaki disease. Cell Immunol. (2010) 265:57–64. doi: 10.1016/j.cellimm.2010.07.003
155. Doi M, Takeda T, Sakurai Y, Kato J, Hayashi T, Fukuda K, et al. Altered immunoglobulin A and M levels associated with changes in BAFF and APRIL after administration of intravenous immunoglobulin to treat Kawasaki disease. J Investig Allergol Clin Immunol. (2010) 20:413–8.
156. Seite JF, Goutsmedt C, Youinou P, Pers JO, Hillion S. Intravenous immunoglobulin induces a functional silencing program similar to anergy in human B cells. J Allergy Clin Immunol. (2014) 133:181–8.e1–9. doi: 10.1016/j.jaci.2013.08.042
157. Kobayashi T, Saji T, Otani T, Takeuchi K, Nakamura T, Arakawa H, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. (2012) 379:1613–20. doi: 10.1016/S0140-6736(11)61930-2
158. Chen KY, Curtis N, Dahdah N, Kowalski R, Cheung M, Burgner DP. Kawasaki disease and cardiovascular risk: a comprehensive review of subclinical vascular changes in the longer term. Acta Paediatr. (2016) 105:752–61. doi: 10.1111/apa.13367
159. Kato H, Koike S, Yokoyama T. Kawasaki disease: effect of treatment on coronary artery involvement. Pediatrics. (1979) 63:175–9.
160. Research Committee of the Japanese Society of Pediatric C Cardiac Surgery Committee for Development of Guidelines for Medical Treatment of Acute Kawasaki Disease. Guidelines for medical treatment of acute Kawasaki disease: report of the Research Committee of the Japanese Society of Pediatric Cardiology and Cardiac Surgery (2012 revised version). Pediatr Int. (2014) 56:135–58. doi: 10.1111/ped.12317
161. Tacke CE, Burgner D, Kuipers IM, Kuijpers TW. Management of acute and refractory Kawasaki disease. Expert Rev Anti Infect Ther. (2012) 10:1203–15. doi: 10.1586/eri.12.101
162. Mpofu S, Fatima F, Moots RJ. Anti-TNF-alpha therapies: they are all the same (aren't they?). Rheumatology. (2005) 44:271–3. doi: 10.1093/rheumatology/keh483
163. Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K, et al. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc Res. (2016) 111:385–97. doi: 10.1093/cvr/cvw186
164. Lindquist ME, Hicar MD. B Cells and antibodies in Kawasaki disease. Int J Mol Sci. (2019) 20:1834. doi: 10.3390/ijms20081834
165. Chan H, Chi H, You H, Wang M, Zhang G, Yang H, et al. Indirect-comparison meta-analysis of treatment options for patients with refractory Kawasaki disease. BMC Pediatr. (2019) 19:158. doi: 10.1186/s12887-019-1504-9
166. Lipsky PE, Thompson PA, Rosenwasser LJ, Dinarello CA. The role of interleukin 1 in human B cell activation: inhibition of B cell proliferation and the generation of immunoglobulin-secreting cells by an antibody against human leukocytic pyrogen. J Immunol. (1983) 130:2708–14.
167. Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, et al. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. (2012) 125:1542–50. doi: 10.1161/CIRCULATIONAHA.111.072769
168. Tremoulet AH, Pancoast P, Franco A, Bujold M, Shimizu C, Onouchi Y, et al. Calcineurin inhibitor treatment of intravenous immunoglobulin-resistant Kawasaki disease. J Pediatr. (2012) 161:506–12.e1. doi: 10.1016/j.jpeds.2012.02.048
169. Heidt S, Roelen DL, Eijsink C, Eikmans M, van Kooten C, Claas FH, et al. Calcineurin inhibitors affect B cell antibody responses indirectly by interfering with T cell help. Clin Exp Immunol. (2010) 159:199–207. doi: 10.1111/j.1365-2249.2009.04051.x
170. Sakurai Y. Autoimmune aspects of Kawasaki disease. J Investig Allergol Clin Immunol. (2019) 29:251–61. doi: 10.18176/jiaci.0300
171. Cunningham MW, Meissner HC, Heuser JS, Pietra BA, Kurahara DK, Leung DY. Anti-human cardiac myosin autoantibodies in Kawasaki syndrome. J Immunol. (1999) 163:1060–5.
172. Gupta M, Johann-Liang R, Bussel JB, Gersony WM, Lehman TJ. Elevated IgA and IgM anticardiolipin antibodies in acute Kawasaki disease. Cardiology. (2002) 97:180–2. doi: 10.1159/000063118
173. Chun JK, Lee TJ, Choi KM, Lee KH, Kim DS. Elevated anti-alpha-enolase antibody levels in Kawasaki disease. Scand J Rheumatol. (2008) 37:48–52. doi: 10.1080/03009740701607075
174. Cines DB, Lyss AP, Reeber M, Bina M, DeHoratius RJ. Presence of complement-fixing anti-endothelial cell antibodies in systemic lupus erythematosus. J Clin Invest. (1984) 73:611–25. doi: 10.1172/JCI111251
175. Savage CO, Williams JM. Anti endothelial cell antibodies in vasculitis. J Am Soc Nephrol. (2007) 18:2424–6. doi: 10.1681/ASN.2007070767
176. Barron KS. Kawasaki disease in children. Curr Opin Rheumatol. (1998) 10:29–37. doi: 10.1097/00002281-199801000-00005
177. Nash MC, Shah V, Reader JA, Dillon MJ. Anti-neutrophil cytoplasmic antibodies and anti-endothelial cell antibodies are not increased in Kawasaki disease. Br J Rheumatol. (1995) 34:882–7. doi: 10.1093/rheumatology/34.9.882
178. Leung DY, Collins T, Lapierre LA, Geha RS, Pober JS. Immunoglobulin M antibodies present in the acute phase of Kawasaki syndrome lyse cultured vascular endothelial cells stimulated by gamma interferon. J Clin Invest. (1986) 77:1428–35. doi: 10.1172/JCI112454
179. Leung DY, Geha RS, Newburger JW, Burns JC, Fiers W, Lapierre LA, et al. Two monokines, interleukin 1 and tumor necrosis factor, render cultured vascular endothelial cells susceptible to lysis by antibodies circulating during Kawasaki syndrome. J Exp Med. (1986) 164:1958–72. doi: 10.1084/jem.164.6.1958
180. Rowley AH, Eckerley CA, Jack HM, Shulman ST, Baker SC. IgA plasma cells in vascular tissue of patients with Kawasaki syndrome. J Immunol. (1997) 159:5946–55.
181. Rowley AH, Baker SC, Shulman ST, Garcia FL, Guzman-Cottrill JA, Chou P, et al. Detection of antigen in bronchial epithelium and macrophages in acute Kawasaki disease by use of synthetic antibody. J Infect Dis. (2004) 190:856–65. doi: 10.1086/422648
182. Fujieda M, Oishi N, Kurashige T. Antibodies to endothelial cells in Kawasaki disease lyse endothelial cells without cytokine pretreatment. Clin Exp Immunol. (1997) 107:120–6. doi: 10.1046/j.1365-2249.1997.d01-894.x
183. Kaneko K, Savage CO, Pottinger BE, Shah V, Pearson JD, Dillon MJ. Antiendothelial cell antibodies can be cytotoxic to endothelial cells without cytokine pre-stimulation and correlate with ELISA antibody measurement in Kawasaki disease. Clin Exp Immunol. (1994) 98:264–9. doi: 10.1111/j.1365-2249.1994.tb06136.x
184. Grunebaum E, Blank M, Cohen S, Afek A, Kopolovic J, Meroni PL, et al. The role of anti-endothelial cell antibodies in Kawasaki disease - in vitro and in vivo studies. Clin Exp Immunol. (2002) 130:233–40. doi: 10.1046/j.1365-2249.2002.02000.x
185. Landing BH, Larson EJ. Are infantile periarteritis nodosa with coronary artery involvement and fatal mucocutaneous lymph node syndrome the same? Comparison of 20 patients from North America with patients from Hawaii and Japan. Pediatrics. (1977) 59:651–62.
186. Takahashi K, Oharaseki T, Naoe S, Wakayama M, Yokouchi Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr Int. (2005) 47:305–10. doi: 10.1111/j.1442-200x.2005.02049.x
187. Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. (2001) 184:940–3. doi: 10.1086/323155
188. Fujiwara T, Fujiwara H, Nakano H. Pathological features of coronary arteries in children with Kawasaki disease in which coronary arterial aneurysm was absent at autopsy. Quantitative analysis. Circulation. (1988) 78:345–50. doi: 10.1161/01.CIR.78.2.345
189. Kuijpers TW, Biezeveld M, Achterhuis A, Kuipers I, Lam J, Hack CE, et al. Longstanding obliterative panarteritis in Kawasaki disease: lack of cyclosporin A effect. Pediatrics. (2003) 112:986–92. doi: 10.1542/peds.112.4.986
190. Giordani L, Quaranta MG, Marchesi A, Straface E, Pietraforte D, Villani A, et al. Increased frequency of immunoglobulin (Ig)A-secreting cells following Toll-like receptor (TLR)-9 engagement in patients with Kawasaki disease. Clin Exp Immunol. (2011) 163:346–53. doi: 10.1111/j.1365-2249.2010.04297.x
191. Mason WH, Jordan SC, Sakai R, Takahashi M, Bernstein B. Circulating immune complexes in Kawasaki syndrome. Pediatr Infect Dis. (1985) 4:48–51. doi: 10.1097/00006454-198501000-00012
192. Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. (2001) 166:1334–43. doi: 10.4049/jimmunol.166.2.1334
193. Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, et al. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PLoS ONE. (2008) 3:e1582. doi: 10.1371/journal.pone.0001582
194. Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology. (2011) 77:589–93. doi: 10.1212/WNL.0b013e318228c136
195. Takuma K, Kamisawa T, Igarashi Y. Autoimmune pancreatitis and IgG4-related sclerosing cholangitis. Curr Opin Rheumatol. (2011) 23:80–7. doi: 10.1097/BOR.0b013e3283412f60
196. Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. (2012) 8:613–23. doi: 10.1038/nrneurol.2012.203
197. Wouters MCA, Nelson BH. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin Cancer Res. (2018) 24:6125–35. doi: 10.1158/1078-0432.CCR-18-1481
198. Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. (2008) 453:667–71. doi: 10.1038/nature06890
199. van Zelm MC, van der Burg M, van Dongen JJ. Homeostatic and maturation-associated proliferation in the peripheral B-cell compartment. Cell Cycle. (2007) 6:2890–5. doi: 10.4161/cc.6.23.4952
200. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
201. Fink K. Origin and function of circulating plasmablasts during acute viral infections. Front Immunol. (2012) 3:78. doi: 10.3389/fimmu.2012.00078
202. Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. (2005) 105:1614–21. doi: 10.1182/blood-2004-07-2507
203. Blanchard-Rohner G, Pulickal AS, Jol-van der Zijde CM, Snape MD, Pollard AJ. Appearance of peripheral blood plasma cells and memory B cells in a primary and secondary immune response in humans. Blood. (2009) 114:4998–5002. doi: 10.1182/blood-2009-03-211052
204. Balakrishnan T, Bela-Ong DB, Toh YX, Flamand M, Devi S, Koh MB, et al. Dengue virus activates polyreactive, natural IgG B cells after primary and secondary infection. PLoS ONE. (2011) 6:e29430. doi: 10.1371/journal.pone.0029430
205. Lee FE, Falsey AR, Halliley JL, Sanz I, Walsh EE. Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J Infect Dis. (2010) 202:1659–66. doi: 10.1086/657158
206. Di Niro R, Lee SJ, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, et al. Salmonella infection drives promiscuous B cell activation followed by extrafollicular affinity maturation. Immunity. (2015) 43:120–31. doi: 10.1016/j.immuni.2015.06.013
207. Kauffman RC, Bhuiyan TR, Nakajima R, Mayo-Smith LM, Rashu R, Hoq MR, et al. Single-cell analysis of the plasmablast response to vibrio cholerae demonstrates expansion of cross-reactive memory B cells. MBio. (2016) 7:e02021–16. doi: 10.1128/mBio.02021-16
208. Liao HX, Chen X, Munshaw S, Zhang R, Marshall DJ, Vandergrift N, et al. Initial antibodies binding to HIV-1 gp41 in acutely infected subjects are polyreactive and highly mutated. J Exp Med. (2011) 208:2237–49. doi: 10.1084/jem.20110363
209. Liao H, Yu Y, Li S, Yue Y, Tao C, Su K, et al. Circulating plasmablasts from chronically human immunodeficiency virus-infected individuals predominantly produce polyreactive/autoreactive antibodies. Front Immunol. (2017) 8:1691. doi: 10.3389/fimmu.2017.01691
210. Garcia M, Iglesias A, Landoni VI, Bellomo C, Bruno A, Cordoba MT, et al. Massive plasmablast response elicited in the acute phase of hantavirus pulmonary syndrome. Immunology. (2017) 151:122–35. doi: 10.1111/imm.12713
211. Wrammert J, Onlamoon N, Akondy RS, Perng GC, Polsrila K, Chandele A, et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J Virol. (2012) 86:2911–8. doi: 10.1128/JVI.06075-11
212. Tarlton NJ, Green CM, Lazarus NH, Rott L, Wong AP, Abramson ON, et al. Plasmablast frequency and trafficking receptor expression are altered in pediatric ulcerative colitis. Inflamm Bowel Dis. (2012) 18:2381–91. doi: 10.1002/ibd.22962
213. Rivas JR, Ireland SJ, Chkheidze R, Rounds WH, Lim J, Johnson J, et al. Peripheral VH4+ plasmablasts demonstrate autoreactive B cell expansion toward brain antigens in early multiple sclerosis patients. Acta Neuropathol. (2017) 133:43–60. doi: 10.1007/s00401-016-1627-0
214. Hosomi S, Oshitani N, Kamata N, Sogawa M, Okazaki H, Tanigawa T, et al. Increased numbers of immature plasma cells in peripheral blood specifically overexpress chemokine receptor CXCR3 and CXCR4 in patients with ulcerative colitis. Clin Exp Immunol. (2011) 163:215–24. doi: 10.1111/j.1365-2249.2010.04290.x
215. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. (2014) 134:679–87. doi: 10.1016/j.jaci.2014.03.034
216. Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. (2015) 74:190–5. doi: 10.1136/annrheumdis-2014-205233
217. Xu M, Jiang Y, Wang J, Liu J, Liu C, Liu D, et al. Distinct variations of antibody secreting cells and memory B cells during the course of Kawasaki disease. BMC Immunol. (2019) 20:16. doi: 10.1186/s12865-019-0299-7
Keywords: kawasaki disease, antibodies-monoclonal, B cells, plasmablast, immunoglobulin intravenous
Citation: Hicar MD (2020) Antibodies and Immunity During Kawasaki Disease. Front. Cardiovasc. Med. 7:94. doi: 10.3389/fcvm.2020.00094
Received: 08 July 2019; Accepted: 30 April 2020;
Published: 28 May 2020.
Edited by:
Xupei Huang, Florida Atlantic University, United StatesReviewed by:
Howard Prentice, Florida Atlantic University, United StatesJunjun Quan, Children‘s Hospital of Chongqing Medical University, China
Copyright © 2020 Hicar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Daniel Hicar, markhica@buffalo.edu