Variations in Energy Metabolism Precede Alterations in Cardiac Structure and Function in Hypertrophic Preconditioning
 Jian Wu1†
Jian Wu1†  Jing Lu1†
Jing Lu1†  Jiayuan Huang2†
Jiayuan Huang2†  Jieyun You3* Zhiwen Ding1 Leilei Ma1 Fangjie Dai1 Ran Xu1 Xuan Li1 Peipei Yin1 Gang Zhao1 Shijun Wang1 Jie Yuan1
Jieyun You3* Zhiwen Ding1 Leilei Ma1 Fangjie Dai1 Ran Xu1 Xuan Li1 Peipei Yin1 Gang Zhao1 Shijun Wang1 Jie Yuan1  Xiangdong Yang1
Xiangdong Yang1  Junbo Ge1
Junbo Ge1  Yunzeng Zou1*
Yunzeng Zou1*- 1Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital and Institutes of Biomedical Sciences, Fudan University, Shanghai, China
- 2Key Laboratory of Guangdong Laboratory Animals, Guangdong Laboratory Animals Monitoring Institute, Guangzhou, China
- 3Department of Cardiovascular Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China
Recent studies have unveiled that myocardial hypertrophic preconditioning (HP), which is produced by de-banding (De-TAC) of short-term transverse aortic constriction (TAC), protects the heart against hypertrophic responses caused by subsequent re-constriction (Re-TAC) in mice. Although cardiac substrate metabolism is impaired in heart failure, it remains unclear about the role of HP-driven energetics in the development of cardiac hypertrophy. Here, we investigated energy metabolism, cardiac hypertrophy, and function following variational loading conditions, as well as their relationships in HP. Male C57BL/6J mice (10–12 weeks old) were randomly subjected to Sham, HP [TAC for 3days (TAC 3d), de-banding the aorta for 4 days (De-TAC 4d), and then re-banding the aorta for 4 weeks (Re-TAC 4W)], and TAC (TAC for 4 weeks without de-banding). Cardiac echocardiography, hemodynamics, and histology were utilized to evaluate cardiac remodeling and function. The mRNA expression levels of fetal genes (ANP and BNP), glucose metabolism-related genes (glut4, pdk4), and fatty acid oxidation-related genes (mcad, pgc1α, mcd, pparα) were quantitated by real-time quantitative PCR. Activation of hypertrophy regulators ERK1/2, a metabolic stress kinase AMP-activated protein kinase (AMPK), and its downstream target acetyl-coA carboxylase (ACC) were explored by western blot. Compared with TAC 4W mice, Re-TAC 4W mice showed less impairment in glucose and fatty acid metabolism, as well as less cardiac hypertrophy and dysfunction. Moreover, no significant difference was found in myocardial hypertrophy, fibrosis, and cardiac function in TAC 3d and De-TAC 4d groups compared with Sham group. However, glut4, pdk4, mcad, pgc1α, mcd, and pparα were all decreased, while AMPK and ACC were activated in TAC 3d and returned to Sham level in De-TAC 4d, suggesting that the change in myocardial energy metabolism in HP mice was earlier than that in cardiac structure and function. Collectively, HP improves energy metabolism and delays cardiac remodeling, highlighting that early metabolic improvements drive a potential beneficial effect on structural and functional restoration in cardiac hypertrophy.
Introduction
Heart failure (HF) is a leading cause of death, threatening human health worldwide (1). Left ventricular (LV) hypertrophy is an adaptive alteration of the heart in response to increased mechanical overload, per se serving as an independent risk factor of major cardiac events (2, 3). Clinical studies on hypertensive patients and basic studies using animals with transverse aortic constriction (TAC) indicate that sustained pressure overload causes the transition from compensated to decompensated cardiac hypertrophy with the eventual progression to HF (4). If pressure overload is withdrawn timely, regression of cardiac remodeling is accompanied with functional improvement (5). Intriguingly, similar to ischemic preconditioning whereby a brief episode of ischemia increases cardiac resistance to subsequent prolonged ischemia, hypertrophic preconditioning (HP) also exists in the heart (6, 7). Using a unique in vivo model of HP, we and others sequentially imposed, removed, and re-imposed pressure overload on the mouse heart through TAC, de-banding the aorta (De-TAC), and then re-banding the aorta (Re-TAC). It is found that the heart was protected from the subsequent pressure overload induced by Re-TAC (6, 7). Although the discovery of HP would convincingly explain diverse extent of cardiac hypertrophy shown in hypertensive patients and enlighten us with new strategy to blunt the initiation of cardiac hypertrophy, the underlying mechanism remains largely unknown.
It is well-established that LV hypertrophy and HF are associated with metabolic abnormalities (8). Pressure overload forces a shift of reliance on glucose instead of fatty acids to provide energy for the heart (9, 10). The prominent metabolic changes result in impaired cardiac energetics, which precedes manifestation of cardiac dysfunction and LV hypertrophy (8, 11). Although many of these metabolic changes are due to an aberration of transcriptional expression level of metabolic enzymes, it is only recently found that this abnormality of metabolic flexibility is reversible, for normalization of cardiac energy metabolism and LV hypertrophy precedes recovery of LV function, in HF mice with De-TAC and in HF patients with left ventricular assist device (LVAD)-induced mechanical unloading (1, 5). Moreover, considering HP renders the heart resistant to hypertrophy, it would have clinical implications for early detection and prevention of LV hypertrophy and HF, through examination of correlation between metabolic changes, LV function, and hypertrophy. Therefore, we in this study used a mouse model of HP to determine the temporal and causal relationships of these changes.
Materials and Methods
Experimental Animals
Male C57BL/6J mice (12–13 weeks old, purchased from the Department of Laboratory Animal Sciences, Fudan University, Shanghai, China) were enrolled in this study. The animals were housed under controlled conditions (24 ± 2°C, 12-h dark/12-h light cycle, ad libitum access to normal chow and water). All animal experiments conformed to the Guide for the Care and Use of Laboratory Animals (No. 85-23, revised 1996; National Institutes of Health, Bethesda, MD, USA). The study protocol was approved by the Animal Care and Use Committee of Zhongshan Hospital, Fudan University.
The mice were randomly assigned to Sham (n = 10), TAC (n = 15), HP (n = 40) groups. For HP mice, serial assessments described below were performed at Baseline (1 day before operation), 3 days after TAC (TAC 3d), 4 days after aortic de-banding (De-TAC 4d), and 4 weeks after re-banding (Re-TAC 4W) (Supplementary Figure 1). The similar thoracotomies were also performed in Sham and TAC mice to minimize the possible confounding effects of repeated surgical injuries. At each time point, at least three mice were randomly selected for histology, quantitative real-time PCR, and Western blot analyses.
TAC, De-TAC, and Re-TAC
TAC surgery was performed as we described previously (7, 12). In brief, mice were anesthetized by 2% isoflurane; the aortic arch was isolated and secured against a 27-gauge needle with a 6-0 silk ligature, which was promptly removed to form an aortic constriction. Meloxicam (0.13 mg each) was administered for analgesia. Sham mice underwent similar operation without tying the ligature around the aorta.
Three days after TAC, a second thoracotomy was given through the original incision site to de-band the aorta (De-TAC). An additional 4 days later, mice were subjected to a third thoracotomy to re-band the aorta (Re-TAC).
Echocardiography
Cardiac structure and function under 1.5% isoflurane (7, 13) were evaluated by a Vevo2100 high-frequency ultrasound system with a 30-MHz transducer (VisualSonics, Toronto, ON, Canada). The echocardiographic assessment had good intra- and inter-observer agreement as reported in our previous studies (4, 7, 14, 15). Peak flow velocity at the aortic banding site (PVb, for prediction of pressure gradients, see Supplementary Figure 2), left ventricle posterior wall thickness in diastole (LVPWTd) and systole (LVPWTs), left ventricular end-diastolic (LVEDD) and end-systolic (LVESD) dimensions, and left ventricular ejection fraction (LVEF) and fractional shortening (LVFS) were acquired.
Invasive Hemodynamics
After echocardiography, LV hemodynamics were evaluated as we described previously (4, 7). Briefly, under 2% isoflurane, a micromanometer (Millar 1.4F, SPR 835; Millar Instruments, Houston, TX) was inserted into the right common carotid artery and then advanced into the LV for measurement of end-systolic and end-diastolic pressure (LVESP and LVEDP), maximal contraction, and relaxation velocity (Max dp/dt and Min dp/dt).
Histology
Mice were euthanized after hemodynamic measurements. Hearts were excised, rinsed in saline, and heart weight-to-body weight ratios are compared (HW/BW). The LV tissue was separated into several parts longitudinally and stored at −80°C for further analysis after removing the right ventricular free wall. The hearts were stained with hematoxylin and eosin (H&E) for assessment of cardiomyocyte size or with Masson trichrome for cardiac fibrosis. Digital photographs were taken using an image analysis system (KF-TB-400, Konfoong biotech international, Ningbo, China).
Quantitative Real-Time PCR
LV tissue samples were isolated and homogenized in TRIzol Reagent (ThermoFisher Scientific, Carlsbad, CA, USA). The RNA concentration was assessed by spectrophotometry (NanoDrop, ThermoFisher Scientific, Carlsbad, CA, USA). Equivalent amounts (2 μg) of purified RNA were used as a template to synthesize cDNA by the Hifair III cDNA Synthesis SuperMix (Yeasen, Shanghai, China). Quantitative real-time (qRT)-PCR was performed using QuantStudio 6 Flex Real-Time PCR System with ChamQ Universal SYBR qPCR Master Mix (Vazyme biotech, Nanjing, China). Relative mRNA expression levels of target genes were normalized to the amount of the constitutively expressed housekeeping gene β-actin (Actb). Based on previous studies (4, 5, 7, 8, 16), relative gene expression levels of hypertrophy markers such as atrial natriuretic peptide and brain natriuretic peptide (anp and bnp), fibrosis such as collagen type I alpha 1 (col1α1) and collagen type 3 alpha 1 (col3α1), glucose metabolism such as pyruvate dehydrogenase kinase 4 and glucose transporter type 4 (glut4 and pdk4), and fatty acid metabolism such as medium-chain acyl-CoA dehydrogenase, malonyl-CoA decarboxylase, peroxisome proliferator-activated receptor α, and peroxisome proliferator-activated receptor gamma coactivator 1α (mcad, mcd, pparα, and pgc1α) were analyzed using the 2−ΔΔCt method. The nucleotide sequences of primers used are shown in Supplementary Table 1.
Western Blot Analysis
LV tissue was homogenized in RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China), electrophoresed in 8% or 10% polyacrylamide gels, and transferred to a PVDF membrane (Millipore, Billerica, MA, USA). Membranes were blocked with 5% bovine serum albumin, followed by incubation with appropriate primary antibodies: anti-extracellular signal-regulated kinase (ERK1/2, #9102), anti-phosphorylated (p-)ERK1/2 (p-ERK1/2, #9101A), anti-p-AMP-activated protein kinase α (p-AMPK, #2535), and anti-p-acetyl-coA carboxylase (p-ACC, #3661). Those primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The blotted membranes were incubated with horseradish peroxidase-conjugated rabbit secondary antibody (Kang-Chen Biotechnology, Shanghai, China), detected with chemiluminescence reagents (catalog RPN2106, GE Healthcare, Piscataway, NJ, USA), visualized using a LAS-3000 chemiluminescence detection system (FUJIFILM, Kanagawa, Japan), and analyzed with the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistics
Data are expressed as the means ± SEM. Statistical evaluations were performed using the software SPSS 15.0 (SPSS Inc., Chicago, IL, USA). Multiple comparisons were conducted by one-way ANOVA with the Student-Newman-Keuls (S-N-K) test. P < 0.05 was considered statistically significant.
Results
HP Attenuates LV Hypertrophy in Pressure Overload Hearts
Decompensated LV hypertrophy in response to chronic pressure overload is characterized by thickening of the heart muscle and dilation of LV chamber (7, 17). Compared with Sham mice, TAC 4W mice showed decompensated cardiac hypertrophy evidenced by a striking increase of HW/BW, LV wall thickness, inner dimension, fibrosis, and cardiomyocyte size (Figure 1). Re-TAC 4W mice showed that HP treatment significantly mitigated the morphological changes compared with TAC 4W mice, although still showed moderate cardiac hypertrophy compared with Sham mice (Figure 1).
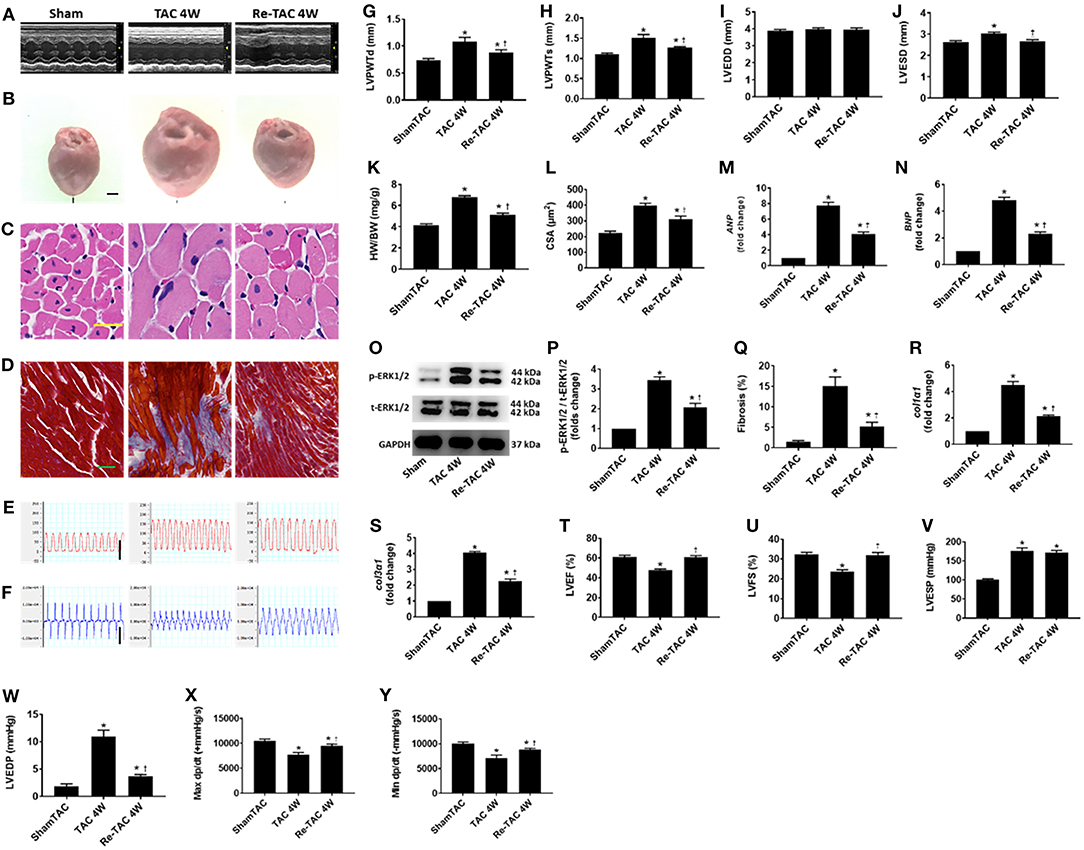
Figure 1. Hypertrophic preconditioning improves cardiac morphological changes and function in pressure overload hearts. (A) M-mode echocardiography from Sham, TAC 4W, and Re-TAC 4W mice. (B) Gross appearance of hearts. Scale bar = 1 mm. (C) Cardiomyocytes stained by H&E. Scale bar = 20 μm. (D) Cardiac fibrosis stained by Masson's trichrome. Scale bar = 50 μm. Representative images of (E) LV pressure and (F) contraction and relaxation velocity (dp/dt). Scale bar = 100 mmHg for pressure, scale bar = 10,000 mmHg/s for dp/dt. (G,H) LV end-diastolic (LVPWTd) and end-systolic (LVPWTs) posterior wall thickness, respectively. (I,J) LV end-diastolic (LVEDD) and end-systolic (LVESD) dimension, respectively. (K) Heart weight-to-body weight ratio (HW/BW). (L) Cross-sectional area (CSA) of cardiomyocytes. (M,N) Real-time quantitative PCR analyses for the gene expression of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), respectively. Representative images (O) and densitometric assessment (P) of Western blot for ERK1/2 activation. (Q) Quantification of collagen staining expressed as % area. Gene expression of makers of cardiac fibrosis (R) collagen type I alpha 1 (col1α1) and (S) collagen type 3 alpha 1 (col3α1). Echocardiographic measurements of (T) LV ejection fraction (LVEF) and (U) LV fractional shortening (LVFS). Invasive hemodynamic assessment of (V,W) LV end-systolic (LVESP) and end-diastolic (LVEDP) pressure, respectively. (X,Y) Maximum (Max dp/dt) and minimum (Min dp/dt) value of LV dp/dt, respectively. *P < 0.05 vs. Sham TAC; †P < 0.05 vs. TAC 4W.
The hypertrophic response to pressure overload are underlined with reprogramming of fetal genes and activation of hypertrophic signaling effectors (12, 17). We therefore measured cardiac mRNA and protein levels of markers of LV hypertrophy and failure. Compared with Sham mice, ANP and BNP in TAC 4W mice are overtly up-regulated, as well as p-ERK1/2 activation (Figure 1). HP in Re-TAC 4W mice notably compromised the hypertrophic response at a molecular level (Figure 1).
HP Improves LV Function in Pressure Overload Hearts
As we and others previously reported, 4 weeks of pressure overload results in cardiac dysfunction (4, 12, 18). To determine whether HP restored cardiac function, mice in this study were subjected to echocardiography and invasive hemodynamic analysis. TAC 4W mice showed a prominent reduction in LVEF, LVFS, Max dp/dt, and Min dp/dt and an increase in LVEDP, compared with Sham mice. Intriguingly, Re-TAC 4W mice presented an improvement in the aforementioned indexes (Figure 1).
HP Improves Substrate Utilization and Alleviates Metabolic Changes in Pressure Overload Hearts
Sustained pressure overload is associated with alterations at the transcriptional level of metabolic enzymes that is detrimental to cardiac energetics (5). To interrogate whether these changes are permanent, the transcript levels of typical genes associated with metabolism of glucose (glut4 and pdk4) and fatty acid (mcad, mcd, pgc1α, and pparα) were measured. They were considerably reduced in TAC 4W hearts but were mostly recovered to the sham-operated level in Re-TAC 4W hearts (Figure 2).
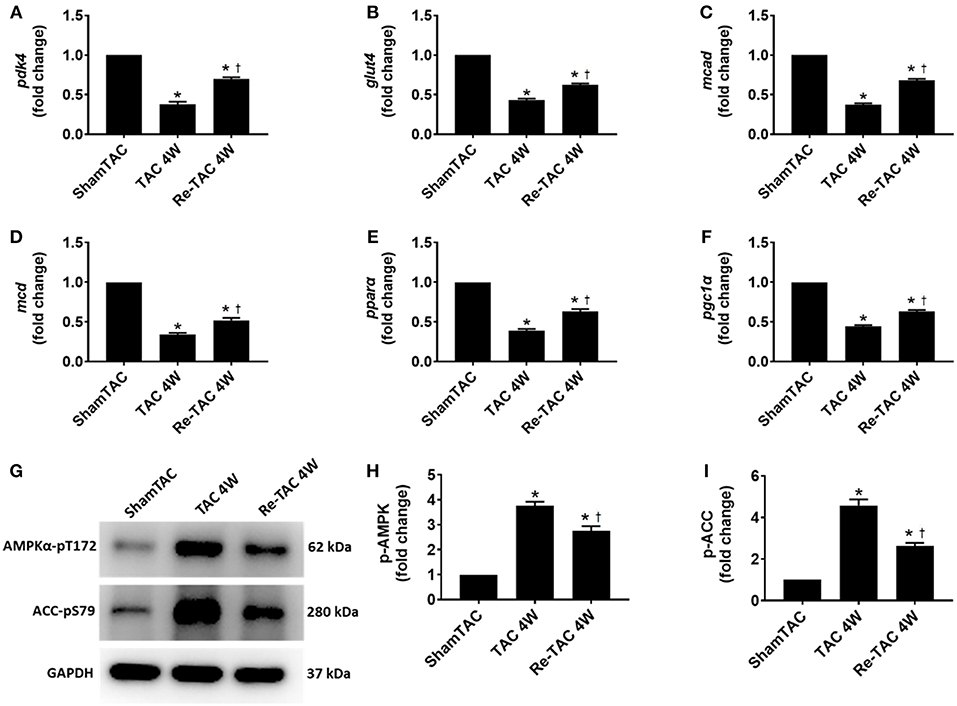
Figure 2. Hypertrophic preconditioning improves substrate utilization in pressure overload hearts. Real-time quantitative PCR analyses for the gene expression of (A) pyruvate dehydrogenase kinase 4 (pdk4), (B) glucose transporter type 4 (glut4), (C) medium-chain acyl-CoA dehydrogenase (mcad), (D) malonyl-CoA decarboxylase (mcd), (E) peroxisome proliferator-activated receptor α (pparα), and (F) peroxisome proliferator-activated receptor gamma coactivator 1α (pgc1α). Representative images (G) and densitometric assessment (H,I) of cardiac phosphorylated AMP-activated protein kinase (p-AMPK) and phosphorylated acetyl-CoA carboxylase (p-ACC) as acquired by Western blot analysis. *P < 0.05 vs. Sham TAC; †P < 0.05 vs. TAC 4W.
To underpin the correlation between mRNA and protein expression levels, we investigated the activation of AMPK, a key metabolic stress kinase, at a post-translational level (19). We found that AMPK was robustly activated in TAC 4W mice, which was recapitulated by its downstream target ACC (Figure 2), whereas Re-TAC 4W mice showed less activation in AMPK and ACC.
Serial Assessment in HP-Treated Mice Reveals Delayed Changes in LV Morphology
To investigate the process of LV hypertrophy in the murine HP model, serial evaluations were performed in Baseline, TAC 3d, De-TAC 4d, and Re-TAC 4W mice. We found that HW/BW, LV wall thickness, inner dimension, fibrosis, and cardiomyocyte size were comparable in Baseline mice, TAC 3d mice, and De-TAC 4d mice. Only Re-TAC 4W mice showed signs of moderate cardiac hypertrophy compared with the above groups (Figure 3).
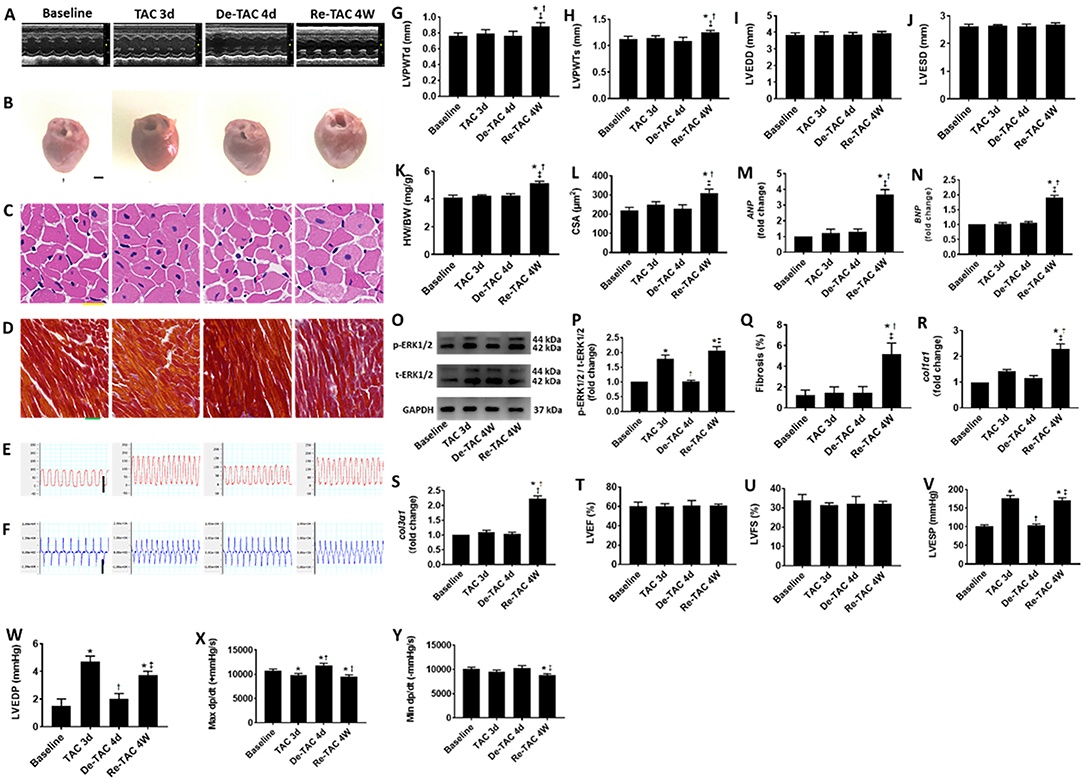
Figure 3. Serial assessment of cardiac morphological and functional changes in HP murine hearts. (A) M-mode echocardiography of LVs at baseline, 3 days after TAC (TAC 3d), 4 days after de-banding (De-TAC 4d), and 4 weeks after re-banding (Re-TAC 4W). (B) Gross appearance of hearts. Scale bar = 1 mm. (C) Cardiomyocytes stained by H&E. Scale bar = 20 μm. (D) Cardiac fibrosis stained by Masson's trichrome. Scale bar = 50 μm. Representative images of (E) LV pressure and (F) dp/dt. Scale bar = 100 mmHg for pressure, scale bar = 10,000 mmHg/s for dp/dt. (G) LVPWTd. (H) LVPWTs. (I) LVEDD. (J) LVESD. (K) HW/BW. (L) CSA of cardiomyocytes. (M,N) Real-time quantitative PCR analyses for the gene expression of ANP and BNP, respectively. Representative images (O) and densitometric assessment (P) of Western blot for ERK1/2 activation. (Q) Quantification of collagen staining expressed as % area. Gene expression of makers of cardiac fibrosis (R) col1α1 and (S) col3α1. Echocardiographic measurements of (T) LVEF and (U) LVFS. Invasive hemodynamic assessment of (V) LVESP, (W) LVEDP, (X) Max dp/dt, (Y) Min dp/dt. *P < 0.05 vs. Baseline; †P < 0.05 vs. TAC 3d; ‡P < 0.05 vs. De-TAC 4d.
At mRNA and protein levels, Baseline mice and De-TAC 4d mice demonstrated similar scenarios without hypertrophic characteristics. Intriguingly, TAC 3d mice displayed increased ERK1/2 phosphorylation, although ANP and BNP showed an increasing trend without statistical significance (Figure 3). Re-TAC 4W mice showed a moderate increase of ANP and BNP expression and p-ERK1/2 activation.
Serial Assessment in HP-Treated Mice Reveals Delayed Changes in Cardiac Function
To explore LV function closely in the murine HP model, serial echocardiography and invasive hemodynamic analyses were performed in Baseline, TAC 3d, De-TAC 4d, and Re-TAC 4W mice. LVEF and LVFS were not significantly different for mice at all time points (Figure 3), whereas invasive hemodynamics showed a transient reduction in cardiac function in TAC 3d hearts especially manifested in LVEDP and Max dp/dt, following a recovery in De-TAC 4d hearts. Cardiac function reflected by LVEDP, Max dp/dt, and Min dp/dt was moderately impaired after Re-TAC 4W (Figure 3). Taken together, in most instances, these data demonstrated that HP delayed the onset of functional decompensation in pressure overload hearts.
Serial Assessment in HP-Treated Mice Reveals Early Changes in Cardiac Energy Metabolism
All genes involved in the metabolism of glucose (glut4 and pdk4) and fatty acid (mcad, mcd, pgc1α, and pparα) were downregulated in TAC 3d hearts when compared to Baseline mice. The gene expression was elevated to Baseline levels in De-TAC 4d hearts, then was suppressed again in Re-TAC 4W hearts, although pdk4 and mcad were downregulated to a lesser extent compared with other metabolism markers (Figure 4).
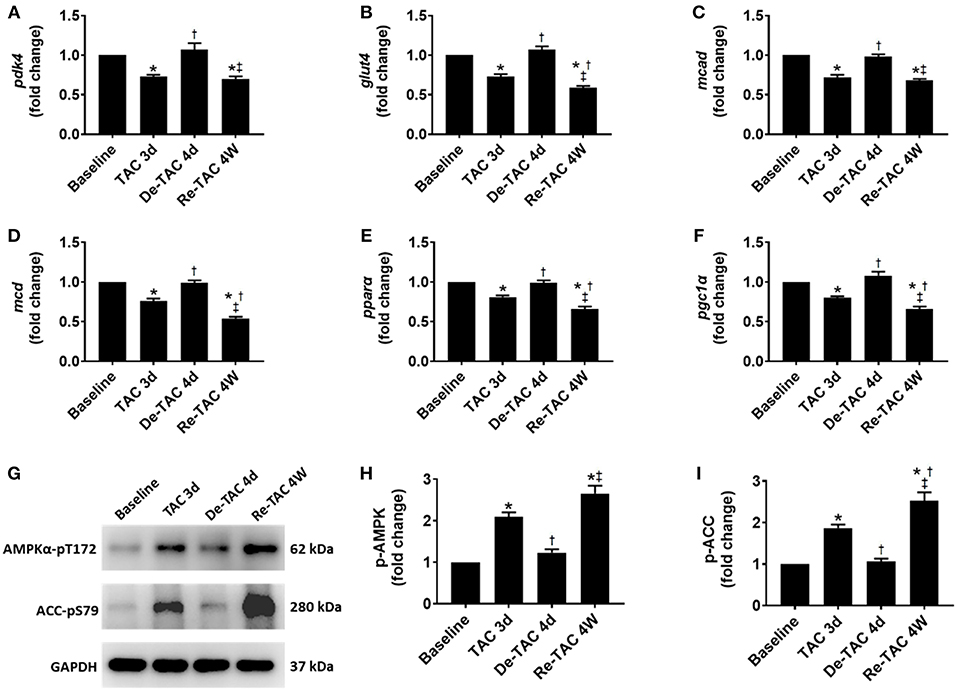
Figure 4. Serial assessment of substrate utilization in HP murine hearts. Real-time quantitative PCR analyses for the gene expression of (A) pdk4, (B) glut4, (C) mcad, (D) mcd, (E) pparα, and (F) pgc1α. Representative images (G) and densitometric assessment (H,I) of cardiac p-AMPK and p-ACC as acquired by Western blot analysis. *P < 0.05 vs. Baseline; †P < 0.05 vs. TAC 3d; ‡P < 0.05 vs. De-TAC 4d.
Consistent with our gene expression data, at a post-translational level, AMPK and ACC were considerably activated in TAC 3d mice, recessed to Baseline level, and fueled again in Re-TAC 4W mice (Figure 4), suggesting improved cardiac energetic status precedes morphological and functional recovery in pressure overload hearts.
Discussion
This study, to the best of our knowledge, for the first time examined the temporal and causal relationships among cardiac morphological, functional, and metabolic changes in a murine model of HP. We observed HP-improved cardiac energetics, morphology, and function effectively in the pressure overload heart. We also identified that short-term TAC treatment induced swift changes in levels of metabolites associated with energy provision and sensing, which varied in accordance with loading conditions. The metabolic changes occurred at early stages before the development of cardiac hypertrophy and dysfunction, which may serve as a sensitive indicator and predisposing factor for cardiac hypertrophy.
LV hypertrophy and HF are tightly associated with metabolic abnormalities (8). Hypertrophied hearts switch from mainly using fatty acids to preferential utilization of glucose for energy production (9). The switch compromises the capacity of cardiac mitochondrial ATP production, contributing to exacerbation of LV hypertrophy and dysfunction (5, 11). On the other hand, preservation of fatty acid oxidation prevents the shift of substrate preference, ameliorates cardiomyocyte hypertrophy, and maintains cardiac function (9). It is thus proposed that maladaptive metabolic remodeling precedes, triggers, and preserves detrimental contractile dysfunction in heart failure progression (20). Nevertheless, only recently, by using pressure overload models and hypertrophy regression models, as well as in patients with LVAD-induced mechanical unloading, it has been evident that metabolic deficit is an early event that contributes to the onset of LV hypertrophy and progression of HF, rather than a consequence of HF (1, 5, 11). In the present study, we with a HP in vivo model further demonstrated that the attenuation of LV hypertrophy and regression of function is associated with early improvements in cardiac metabolism, which corroborates the concept that early interventions in metabolic reprogramming are critical for subsequent morphological and functional recovery.
AMPK, a serine-threonine kinase, coordinates anabolic and catabolic processes as a metabolic stress sensor and thus endogenously reflects cardiac myocardial energetic status (19, 21). AMPK and its downstream target ACC is activated in TAC hearts (22) but normalized in De-TAC hearts (5). We further showed that the activation of AMPK and ACC was attenuated by HP and varied in concert with changes in loading conditions of the heart. Intriguingly, a recent study using 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to activate AMPK transiently before TAC reported decreased maladaptive LV hypertrophy and fibrosis (23). Taken together, these data suggest that AMPK is a nexus linking myocardial energetic with LV remodeling and thus a target for early detection and prevention of LV hypertrophy and HF.
Although it has been well-recognized that HP blunts LV hypertrophy and improves cardiac function (6, 7), the process of LV morphological and functional changes in HP mice has been sparsely investigated. We in this study found that short-term variations of loading conditions (TAC 3d, De-TAC 4d) did not alter morphological appearance. However, ERK1 and ERK2, which are central mediators of LV hypertrophy (24), underwent notable changes during the course, providing evidence that hypertrophic response at a post-translational level is not irreversible. Coincidently, we previously demonstrated that preconditioning with Angiotensin II for a short time prevents the subsequent damage of Angiotensin II in cultured endothelial cells, and the protection was abrogated by an ERK inhibitor PD98059 (25), recapitulating the involvement of ERK1/2 in the cardioprotective role of HP. Moreover, ERK1/2 may contribute to the improvement of cardiac energetics induced by HP, since a recent study reports that AMPK stimulation inhibits cardiac hypertrophy through ERK1/2 (26). Since ERK1/2 and AMPK are hub genes involved in coordinations of cell growth, autophagy, and metabolism, more in-depth studies associated with cardiac apoptosis, autophagy, etc., are warranted.
Limitations
We in this study used β-actin as an internal control for normalization of mRNA expression as described previously (7, 27), for it is stably expressed in murine left ventricles subjected to pressure overload (7, 18, 27, 28). However, as the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines for qPCR suggest, two or multiple validated housekeeping genes should be used to ensure the reliability of qPCR data (29). We thus further performed a stability test across our samples and found that the relative level of GAPDH is almost equal to that of β-actin in our samples (Supplementary Figure 3).
In summary, we for the first time depicted that HP improves energetics, morphology, and function in the pressure overloaded heart. We also demonstrated metabolic changes occurred at early stages before the development of hypertrophy and dysfunction. The time course of events makes it possible that early metabolic abnormalities could serve as antecedent indicators and targets for hypertension-induced LV hypertrophy and HF.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Animal Care and Use Committee of Zhongshan Hospital, Fudan University.
Author Contributions
JW, JL, JH, JYo, and YZ: conception, design, data analysis, and interpretation. JW, JYo, PY, GZ, SW, JYu, XY, JG, and YZ: administrative support. JW, JL, JH, JYo, ZD, LM, FD, RX, XL, and JYu: collection and assembly of data. JW: manuscript writing. All authors wrote first draft, read, amended the draft, and final approval of manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81670228, 81730009, 81941002, and 81770274), Laboratory Animal Science Foundation of Science and Technology Commission of Shanghai Municipality (201409005000), and Health Science and Technology Project of Shanghai Pudong New Area Health Commission (PW2019A-13).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Mr. Jianguo Jia, Mr. Chunjie Yang, Mrs. Bingyu Li, Mrs. Sanli Qian, Mrs. Weiwei Zhang, and Mr. Zhenzhong Zhang, from Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, for their kindness with technical expertise. We also would like to thank Mr. Guoping Zhang for his assistance in lab management.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2020.602100/full#supplementary-material
References
1. Badolia R, Ramadurai DKA, Abel ED, Ferrin P, Taleb I, Shankar TS, et al. The role of non-glycolytic glucose metabolism in myocardial recovery upon mechanical unloading and circulatory support in chronic heart failure. Circulation. (2020) 142:259–74. doi: 10.1161/CIRCULATIONAHA.119.044452
2. Wu J, Dai F, Li C, Zou Y. Gender differences in cardiac hypertrophy. J Cardiovasc Transl Res. (2020) 13:73–84. doi: 10.1007/s12265-019-09907-z
3. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. (2004) 6:499–506. doi: 10.1038/ncb1137
4. You J, Wu J, Zhang Q, Ye Y, Wang S, Huang J, et al. Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am J Physiol Heart Circ Physiol. (2018) 314:H552–62. doi: 10.1152/ajpheart.00212.2017
5. Byrne NJ, Levasseur J, Sung MM, Masson G, Boisvenue J, Young ME, et al. Normalization of cardiac substrate utilization and left ventricular hypertrophy precede functional recovery in heart failure regression. Cardiovasc Res. (2016) 110:249–57. doi: 10.1093/cvr/cvw051
6. Wei X, Wu B, Zhao J, Zeng Z, Xuan W, Cao S, et al. Myocardial hypertrophic preconditioning attenuates cardiomyocyte hypertrophy and slows progression to heart failure through upregulation of S100A8/A9. Circulation. (2015) 131:1506–17. doi: 10.1161/CIRCULATIONAHA.114.013789
7. Huang J, Wu J, Wang S, You J, Ye Y, Ding Z, et al. Asound biomicroscopy validation of a murine model of cardiac hypertrophic preconditioning: comparison with a hemodynamic assessment. Am J Physiol Heart Circ Physiol. (2017) 313:H138–48. doi: 10.1152/ajpheart.00004.2017
8. Li J, Kemp BA, Howell NL, Massey J, Minczuk K, Huang Q, et al. Metabolic changes in spontaneously hypertensive rat hearts precede cardiac dysfunction and left ventricular hypertrophy. J Am Heart Assoc. (2019) 8:e010926. doi: 10.1161/JAHA.118.010926
9. Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res. (2020) 126:182–96. doi: 10.1161/CIRCRESAHA.119.315483
10. Hamirani YS, Kundu BK, Zhong M, McBride A, Li Y, Davogustto GE, et al. Noninvasive detection of early metabolic left ventricular remodeling in systemic hypertension. Cardiology. (2016) 133:157–62. doi: 10.1159/000441276
11. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. (2014) 7:1022–31. doi: 10.1161/CIRCHEARTFAILURE.114.001469
12. You J, Wu J, Jiang G, Guo J, Wang S, Li L, et al. Olmesartan attenuates cardiac remodeling through DLL4/notch1 pathway activation in pressure overload mice. J Cardiovasc Pharmacol. (2013) 61:142–51. doi: 10.1097/FJC.0b013e31827a0278
13. Constantinides C, Murphy K. Molecular and integrative physiological effects of isoflurane anesthesia: the paradigm of cardiovascular studies in rodents using magnetic resonance imaging. Front Cardiovasc Med. (2016) 3:23. doi: 10.3389/fcvm.2016.00023
14. Wu J, You J, Wang X, Wang S, Huang J, Xie Q, et al. Left ventricular response in the transition from hypertrophy to failure recapitulates distinct roles of Akt, β-arrestin-2, and CaMKII in mice with aortic regurgitation. Ann Transl Med. (2020) 8:219–7. doi: 10.21037/atm.2020.01.51
15. Wu J, Bu L, Gong H, Jiang G, Li L, Ma H, et al. Effects of heart rate and anesthetic timing on high-resolution echocardiographic assessment under isoflurane anesthesia in mice. J Ultrasound Med. (2010) 29:1771–8. doi: 10.7863/jum.2010.29.12.1771
16. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. (2001) 104:2923–31. doi: 10.1161/hc4901.100526
17. Satoh M, Nomura S, Harada M, Yamaguchi T, Ko T, Sumida T, et al. High-throughput single-molecule RNA imaging analysis reveals heterogeneous responses of cardiomyocytes to hemodynamic overload. J Mol Cell Cardiol. (2019) 128:77–89. doi: 10.1016/j.yjmcc.2018.12.018
18. Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. (2007) 446:444–8. doi: 10.1038/nature05602
19. Bairwa SC, Parajuli N, Dyck JR. The role of AMPK in cardiomyocyte health and survival. Biochim Biophys Acta. (2016) 1862:2199–210. doi: 10.1016/j.bbadis.2016.07.001
20. Peterzan M, Lygate C, Neubauer S, Rider O. Metabolic remodeling in hypertrophied and failing myocardium: a review. Am J Physiol Heart Circ Physiol. (2017) 313:H597–616. doi: 10.1152/ajpheart.00731.2016
21. Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun. (2018) 9:374. doi: 10.1038/s41467-017-02795-4
22. Shen C, Wang C, Han S, Wang Z, Dong Z, Zhao X, et al. Aldehyde dehydrogenase 2 deficiency negates chronic low-to-moderate alcohol consumption-induced cardioprotecion possibly via ROS-dependent apoptosis and RIP1/RIP3/MLKL-mediated necroptosis. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1912–8. doi: 10.1016/j.bbadis.2016.11.016
23. Nam DH, Kim E, Benham A, Park HK, Soibam B, Taffet GE, et al. Transient activation of AMPK preceding left ventricular pressure overload reduces adverse remodeling and preserves left ventricular function. FASEB J. (2019) 33:711–21. doi: 10.1096/fj.201800602R
24. Zhou N, Li L, Wu J, Gong H, Niu Y, Sun A, et al. Mechanical stress-evoked but angiotensin II-independent activation of angiotensin II type 1 receptor induces cardiac hypertrophy through calcineurin pathway. Biochem Biophys Res Commun. (2010) 397:263–9. doi: 10.1016/j.bbrc.2010.05.097
25. Guan A, Zou Y, Gong H, Niu Y, Ye Y, Jia J, et al. AngiotensinII preconditioning promotes angiogenesis in vitro via ERKs phosphorylation. J Biomed Biotechnol. (2012) 2012:737134. doi: 10.1155/2012/737134
26. Meng R, Pei Z, Zhang A, Zhou Y, Cai X, Chen B, et al. AMPK activation enhances PPARalpha activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway. Arch Biochem Biophys. (2011) 511:1–7. doi: 10.1016/j.abb.2011.04.010
27. Schuman ML, Landa MS, Toblli JE, Peres Diaz LS, Alvarez AL, Finkielman S, et al. Cardiac thyrotropin-releasing hormone mediates left ventricular hypertrophy in spontaneously hypertensive rats. Hypertension. (2011) 57:103–9. doi: 10.1161/HYPERTENSIONAHA.110.161265
28. Fang X, Robinson J, Wang-Hu J, Jiang L, Freeman DA, Rivkees SA, et al. cAMP induces hypertrophy and alters DNA methylation in HL-1 cardiomyocytes. Am J Physiol Cell Physiol. (2015) 309:C425–36. doi: 10.1152/ajpcell.00058.2015
Keywords: cardiac function, cardiac hypertrophy, hypertrophic preconditioning, metabolism, pressure overload
Citation: Wu J, Lu J, Huang J, You J, Ding Z, Ma L, Dai F, Xu R, Li X, Yin P, Zhao G, Wang S, Yuan J, Yang X, Ge J and Zou Y (2020) Variations in Energy Metabolism Precede Alterations in Cardiac Structure and Function in Hypertrophic Preconditioning. Front. Cardiovasc. Med. 7:602100. doi: 10.3389/fcvm.2020.602100
Received: 03 September 2020; Accepted: 09 November 2020;
Published: 11 December 2020.
Edited by:
Brenda Gerull, University Hospital Würzburg, GermanyReviewed by:
Dachun Xu, Tongji University, ChinaRuby C. Y. Lin, Westmead Institute for Medical Research, Australia
Copyright © 2020 Wu, Lu, Huang, You, Ding, Ma, Dai, Xu, Li, Yin, Zhao, Wang, Yuan, Yang, Ge and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunzeng Zou, zou.yunzeng@zs-hospital.sh.cn; Jieyun You, youjieyun1118@hotmail.com
†These authors have contributed equally to this work