Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition
 Machteld Sillen
Machteld Sillen Paul J. Declerck
Paul J. Declerck- Laboratory for Therapeutic and Diagnostic Antibodies, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, Leuven, Belgium
Plasminogen activator inhibitor-1 (PAI-1), a member of the serine protease inhibitor (serpin) superfamily with antiprotease activity, is the main physiological inhibitor of tissue-type (tPA) and urokinase-type (uPA) plasminogen activators (PAs). Apart from being crucially involved in fibrinolysis and wound healing, PAI-1 plays a pivotal role in various acute and chronic pathophysiological processes, including cardiovascular disease, tissue fibrosis, cancer, and age-related diseases. In the prospect of treating the broad range of PAI-1-related pathologies, many efforts have been devoted to developing PAI-1 inhibitors. The use of these inhibitors, including low molecular weight molecules, peptides, antibodies, and antibody fragments, in various animal disease models has provided ample evidence of their beneficial effect in vivo and moved forward some of these inhibitors in clinical trials. However, none of these inhibitors is currently approved for therapeutic use in humans, mainly due to selectivity and toxicity issues. Furthermore, the conformational plasticity of PAI-1, which is unique among serpins, poses a real challenge in the identification and development of PAI-1 inhibitors. This review will provide an overview of the structural insights into PAI-1 functionality and modulation thereof and will highlight diverse approaches to inhibit PAI-1 activity.
Introduction
Hemostasis is an essential physiological process that preserves the integrity of the vascular system and secures sufficient blood flow throughout the circulatory system. The balance between clot formation (coagulation) and clot dissolution (fibrinolysis) is very tightly regulated in a spatiotemporal manner and requires a dynamic interplay with other systems involved, such as the vascular system and platelets (1). Briefly, upon vascular injury, a sequence of cellular and molecular events is triggered that can be characterized by three distinct but overlapping phases of initiation, amplification, and propagation (coagulation) (2, 3). The end result of the coagulation cascade is the conversion of fibrinogen, a soluble plasma protein, into an insoluble fibrin meshwork that constitutes blood clots. To limit the coagulatory response to the site of injury and prevent vascular occlusion, the prothrombotic response is balanced by the fibrinolytic system. Fibrinolysis revolves around the enzymatic activation of plasminogen into the key fibrinolytic enzyme plasmin through tissue-type (tPA) and urokinase-type (uPA) plasminogen activators (PAs) (4). Tissue-type PA is produced by vascular endothelial cells and released in response to thrombin and venous occlusion. It is primarily involved in the activation of plasminogen that is required for fibrin dissolution in the circulation (5, 6). In contrast, uPA is expressed by a variety of cells, including renal epithelial cells, inflammatory cells, and cancer cells. It is considered more important in pericellular proteolysis during tissue remodeling and cell migration through the activation of cell-bound plasminogen (7, 8). Plasminogen activator inhibitor-1, a member of the serpin superfamily, is a key component of the plasminogen/plasmin system as it is the primary inhibitor of tPA and uPA.
Synthesis, Distribution, and Biochemical Properties of PAI-1
PAI-1 was first detected almost four decades ago as an inhibitor of the fibrinolytic system associated with cultured bovine endothelial cells (9). Not much later, several research groups demonstrated its presence in human plasma (10–12), as well as various other cell types throughout the body, including the spleen, liver, kidney, lung, and adipocytes, albeit at different concentrations and with variable functional activities (13, 14). Furthermore, PAI-1 expression and release are strongly regulated by various factors, including growth factors (e.g., transforming growth factor-β, epidermal growth factor), inflammatory cytokines (e.g., tumor necrosis factor-α and interleukin-1β), hormones (e.g., insulin, glucocorticoid, and angiotensin II), glucose, and endotoxin of Gram-negative bacteria (15, 16). In the blood, PAI-1 occurs in two distinct pools, free in plasma or retained in platelets (17). Plasma PAI-1 circulates mainly in the active conformation at relatively low levels (5–50 ng/mL) (17) showing a large interpersonal variability caused by factors including race/ethnicity (18), gender (19), and body composition (20). In contrast, platelet PAI-1 serves as the main blood pool of PAI-1 with concentrations up to ~300 ng/mL (17). Initially, several studies showed that platelet-derived PAI-1 is less active compared to plasma PAI-1, considered being only 2–5% functionally active (21, 22). However, the pre-analytical methods used in these studies, such as sonication or freeze-thawing, may have reduced the activity of platelet-derived PAI-1 since more recent studies were able to demonstrate a substantially higher activity for PAI-1 (23, 24). Even though platelets do not contain a nucleus, they retain the ability for de novo PAI-1 synthesis through translationally active PAI-1 messenger RNA, of which the synthesis rate is importantly increased by platelet activation (23). As a result, at least 50% of platelet-derived PAI-1 was shown to be in the biologically active form and capable of forming an irreversible PAI-1/tPA complex. Importantly, platelet-derived PAI-1 has a substantial role in conferring thrombolysis resistance to the clot through local accumulation caused by its release from activated platelets and subsequent partial retention of functional PAI-1 on the platelet membrane (24–26).
The 12.3 kb human PAI-1 gene (SERPINE1) was mapped to chromosome 7 (7q21.3-q22) and contains nine exons and eight introns (27, 28). The exons encode for a 23 amino acid long signal peptide and the 379 amino acid long mature PAI-1 protein (29). Additionally, a mature form comprising 381 amino acids, including two extra amino-terminal (N-terminal) residues, has been identified and is most likely the result of cleavage at an alternative cleavage site for signal peptidases (30). Native PAI-1 is a 45-kDa single-chain glycoprotein that lacks cysteines. Based on the amino acid sequence, three potential sites for N-linked glycosylation were identified of which Asn209 and Asn165 display a heterogeneous tissue-type specific glycosylation pattern while Asn329 is not utilized in vivo (31, 32). Even though glycosylation often has a critical role in determining protein structure, function, and stability for many proteins, glycosylation of PAI-1 is not a prerequisite for its ability to inactivate PAs or to interact with its cofactor vitronectin (33). In contrast, several studies demonstrated that glycosylation can have a tremendous effect on the neutralizing activity of PAI-1 inhibitors and therefore emphasizes the significance of the source of PAI-1 used in the development of PAI-1 inhibitors (31, 34, 35).
Structural and Functional Properties
PAI-1 Is an Inhibitory Serpin
The serpin superfamily comprises over 1,500 inhibitory and non-inhibitory proteins that are broadly distributed among several species, including humans, animals, viruses, bacteria, and plants (36). Despite their profound structural similarity, serpins are functionally very diverse. Whereas, their biological function often requires inhibition of proteases, some non-inhibitory serpins function as, for example, hormone transporters (37), tumor repressors (38), or molecular chaperones (39). Based on their evolutionary relatedness, eukaryotic serpins have been divided into 16 clades (termed A-P), with clades A-I representing human serpins. PAI-1 is categorized as a clade E serpin and is considered to be the main physiological inhibitor of tPA and uPA. However, other serpins with inhibitory activity toward PAs have been identified and include plasminogen activator inhibitor-2 (clade B), protease nexin I (clade E), and neuroserpin (clade I) (40).
PAI-1 displays the well-conserved structure of serpins (Figure 1), characterized by three β-sheets [termed A–C, with strand numbers indicated as s(#)A, s(#)B, and s(#)C] and nine α-helices (termed hA-hI) (42, 43). As the primary inhibitor of PAs, PAI-1 rapidly inactivates both tPA and uPA with second-order rate constants between 106 and 107 M−1 s−1 following the basic mechanism applied to all serpin/serine proteinase reactions (43, 44). The key to this reaction is that the PA recognizes PAI-1 as a (pseudo)substrate. Therefore, PAI-1 carries a flexible surface-exposed reactive center loop (RCL) of 26 residues long (331-SGTVASSSTAVIVSARMAPEEIIMDR-356, designated P16-P10′) that presents a substrate-mimicking peptide sequence (Arg346-Met347, designated as P1-P1′). PAI-1 is synthesized in a metastable active conformation, i.e., with the RCL protruding from the top of the molecule, which is essential for the kinetic trapping of PAs in a thermodynamically favorable complex. Several regions–the hinge region (P15-P9 of the RCL), the breach region (the top of β-sheet A), the shutter domain (the central part of s3A and s5A and the N-terminal part of hB), the gate region (s3C and s4C), and the flexible joint region (hD, hE, hF, and s1A)–have been shown to be important in controlling and modulating PAI-1 functionality through conformational changes (Figure 1).
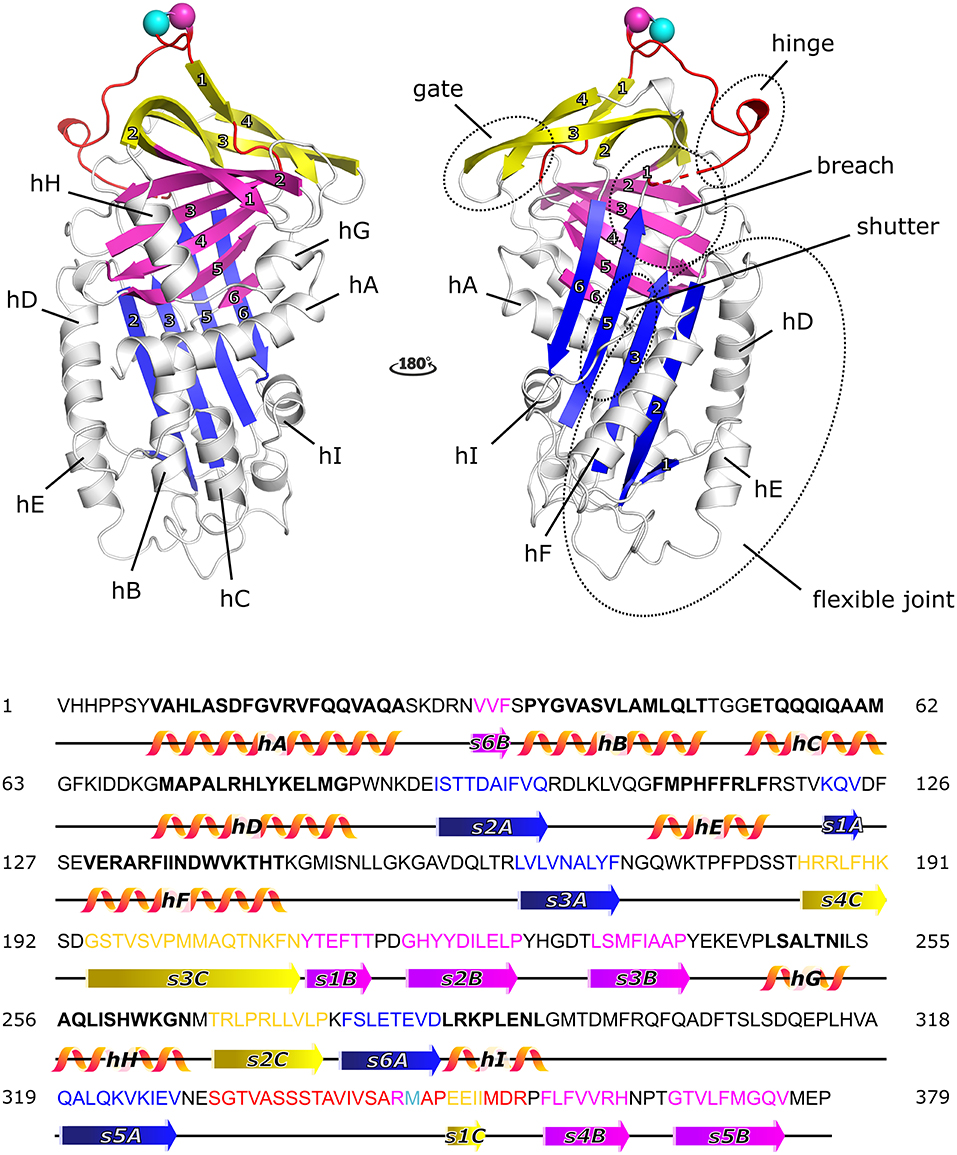
Figure 1. Cartoon representation of the crystal structure of the active form of plasminogen activator inhibitor-1 (PAI-1) [PDB ID 6ZRV (41)] and the amino acid sequence of native PAI-1. PAI-1 shows the evolutionarily conserved topology of serpins, consisting of three β-sheets (A–C) and nine α-helices (hA-hI). β-sheet A, B, and C are shown in blue, magenta, and yellow, respectively, with numbers labeling the individual strands. The α-helices are indicated in the figure. The reactive center loop (RCL) of PAI-1 connects strand 5 of β-sheet A (s5A) to strand 4 of β-sheet B (s4B) and comprises strand 1 of β-sheet C (s1C). The RCL is shown in red, with the reactive center Arg346 (P1) and Met347 (P1′) represented by a magenta and cyan sphere, respectively. Other important domains that control and modulate PAI-1 conformational changes (the gate, hinge, breach, shutter, and flexible joint regions) are also indicated. Residues missing in the crystal structure are indicated by a dashed line. The amino acid sequence is presented and secondary structures (α-helices and β-strands) are indicated in the colors corresponding to the cartoon representation.
Mechanism of Protease Inhibition
The PAI-1/PA reaction is initiated by the formation of a non-covalent 1:1 stoichiometric Michaelis complex (EI) between PAI-1 (inhibitor, I) and the PA (enzyme, E) (Figure 2). Initially, PAs bind to PAI-1 through several exosite interactions, defined as secondary interactions between regions outside of the PA active site and the PAI-1 P1-P1′ reactive center (47, 53). The nature of this Michaelis complex is now well-understood from the X-ray structure determination of PAI-1 in complex with active-site mutants of tPA (tPA-S478A) and uPA (uPA-S195A) (47, 53). Through flexible loops on their surface, PAs contact several exosites adjacent to the RCL to facilitate the initial docking step and, in addition to P1 and nearby residues in the RCL, confer proteinase specificity. By forming tight interactions, exosites stabilize the Michaelis complex and lock the PA into a particular orientation to warrant optimal positioning of the P1 residue in the active site of the PA. Furthermore, these additional interactions slow down the dissociation of the PA from its initial docking site, allowing the PA active site serine to attack the P1-P1′ bond to form a tetrahedral intermediate with PAI-1 (54). Successful cleavage of this bond yields the acyl-enzyme intermediate (E~I) in which the PA is covalently linked to the main chain carbonyl of the P1 residue in PAI-1. Following a branched pathway mechanism, the PAI-1/PA reaction is directed either into the inhibitory or into the substrate pathway.
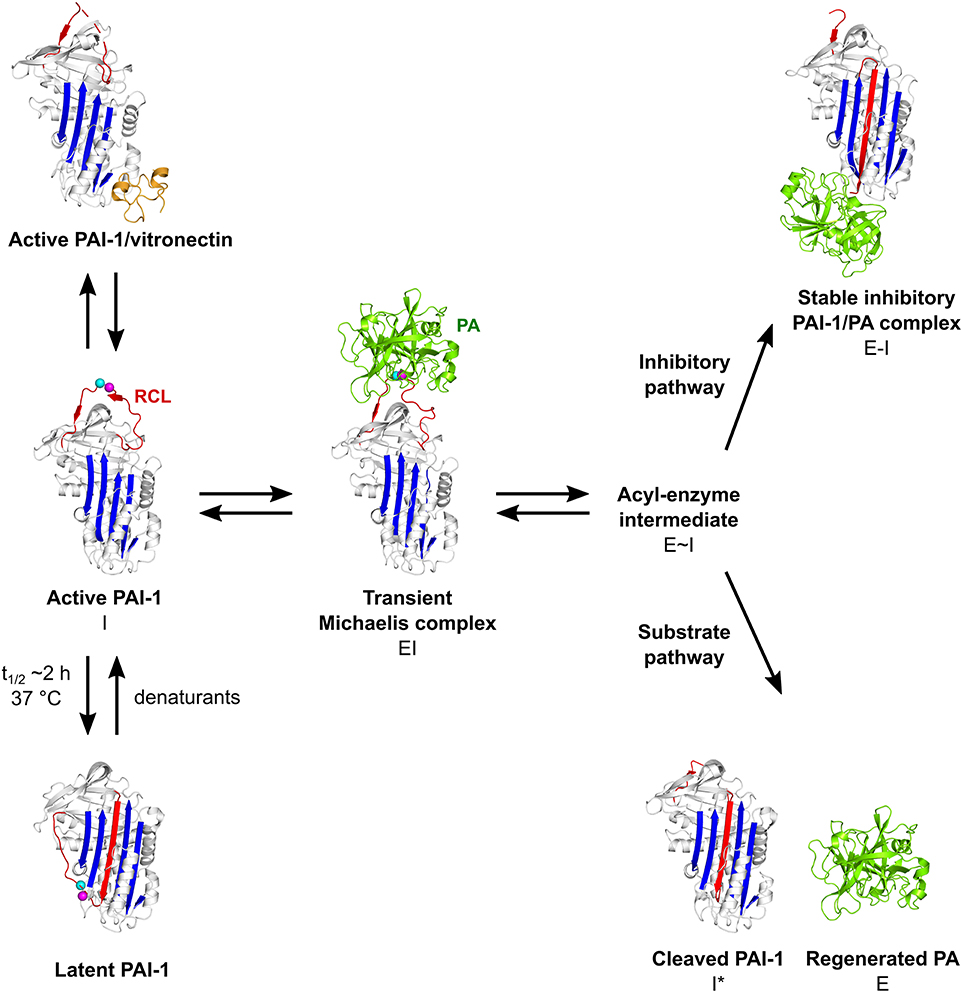
Figure 2. Schematic overview of the PAI-1 (I) conformations as well as its interactions with plasminogen activators (PAs, E) and cofactor vitronectin. Following the formation of a non-covalent PAI-1/PA Michaelis complex (EI), the P1-P1′ bond is cleaved to generate an acyl-enzyme intermediate (E~I). From here on, the reaction proceeds through a branched pathway, resulting in either the formation of an irreversible inhibitory complex (E-I) or the generation of cleaved PAI-1 (I*) due to the hydrolysis of the acyl-enzyme intermediate. PAI-1 is shown in white; the central β-sheet A of the PAI-1 molecule in blue; the flexible reactive center loop (RCL) in red, and Arg346 and Met347 (P1-P1′) of the reactive center are indicated by magenta and cyan spheres, respectively. The PA is shown in green. Vitronectin is shown in orange. PDB structures 1DVN (45), 1DB2 (46), 5BRR (47), 3EOX (48), 1EZX (49), 1H4W (50), and 1OC0 (51) were used to generate this figure. Figure adapted from Sillen et al. (52).
In the inhibitory pathway (Figure 2), the formation of the acyl-enzyme intermediate is coupled to a rapid and full insertion of the N-terminal part of the RCL (P16-P1) as strand 4 into the central β-sheet A (s4A) (54). This major conformational change coincides with a 70 Å translocation of the bound PA to the opposite side of the PAI-1 molecule. There, a large part of the PA, including the active site, is deformed by compression against the body of PAI-1. As a result, hydrolysis of the acyl-enzyme intermediate is prevented and the PA remains trapped as a stable PAI-1/PA inhibitory complex (E-I) (49, 55). This mechanism of inhibition was demonstrated by the crystallographic structure of the α1-antitrypsin/trypsin complex (49), which is in line with the results from studies that investigated serpin exosite distortion by using nuclear magnetic resonance (56, 57) and circular dichroism (58) studies. In this serpin-protease complex (49), trypsin shows a high degree of conformational disorder as compared to its native form, i.e., a loss of structure for ~37% of the protease. Furthermore, the active site of trypsin is disrupted as Ser195 of the catalytic triad is moved away from its catalytic partners. Several regions in PAI-1 are crucial for the orchestration of loop insertion and are furthermore involved in the energetical coupling of this favorable conformational change to the energy-demanding process of PA distortion (Figure 1) (43). Upon cleavage of the P1-P1′ bond, the PA dissociates from its initial docking site on PAI-1 while releasing the distal P′ side of the cleaved RCL from the PA active site cleft. Simultaneously, the breach region at the top and the shutter region near the center of β-sheet A open up to accommodate the RCL as s4A. The hinge region that contains a conserved series of small hydrophobic residues (P15-P9) initially inserts into the breach region and is a prerequisite for rapid loop insertion. Whereas, the surface-exposed RCL only makes a few contacts with the serpin body, it now becomes an integral part of the central β-sheet A. Further insertion of the RCL, however, is obstructed by a steric clash with hF that is located across β-sheet A. Experimental data favor the hypothesis that hF plays an essential role in PA inhibition by (I) being actively displaced until the loop is fully inserted and the PA has passed to the very end of β-sheet A and (II) by temporarily storing the energy derived from loop insertion in order to distort the PA upon return of hF to its original position, ultimately leading to the formation of the irreversible inhibitory complex (59). Through basic residues in hD and hE in the flexible joint region of PAI-1, these PAI-1/PA complexes bind certain receptors of the low-density lipoprotein receptor (LDLR) family, including low-density lipoprotein receptor-related protein-1 (LRP1), leading to endocytosis and degradation of the complex (60, 61).
In the substrate pathway (Figure 2), the acyl-enzyme intermediate is hydrolyzed prior to PA distortion, resulting in the release of regenerated PA (E) from cleaved RCL-inserted PAI-1 (I*) (48, 62). This substrate behavior has been associated with the pre-existence of a conformational distinct substrate-like subset of PAI-1 (63, 64), or results from a change in the kinetic parameters that define the partitioning between both branches of the PAI-1/PA reaction (65, 66).
Factors Influencing the PAI-1/PA Reaction
Several factors have been established that determine target proteinase specificity or influence the partitioning between the inhibitory and substrate branch of the PAI-1/PA reaction pathway. The first region to determine target specificity of PAI-1, and serpins in general, consists of residue at the P1 position and the immediately adjacent residues in the RCL. Indeed, by replacing residues P3-P3′ of the PAI-1 RCL with the corresponding residues of another serpin antithrombin III, this PAI-1 mutant acquired thrombin inhibitory properties (67). Interestingly, vitronectin was shown to alter PAI-1 specificity by also enhancing PAI-1 reactivity toward thrombin in a dose-dependent manner (66, 67). Importantly, studies using PAI-1/serpin chimeras, in which the RCL was replaced with that of other serpins, showed that all chimeras were still effective inhibitors of both PAs, and thus strongly suggested a major contribution of regions outside of the RCL to differences in specificity (68). Based on the crystal structure of the PAI-1/PA Michaelis complexes, interactions between several complementary electrostatic surfaces on PAI-1 and the PAs, referred to as exosites, have been identified (47, 53). One particular region of PAs has been shown to make strong and extensive interactions with PAI-1. The positively charged 37-loop of PAs contacts (I) a negatively charged patch comprising residues in s1B, s2B, and the s3B-hG loop, and (II) the P4′ (Glu350) and P2′ (Ala348) residues in the RCL. This 37-loop/exosite interaction has been proven necessary to ensure the rapid and high-affinity association between PAI-1 and PAs in studies using 37-loop mutants or antibodies specifically binding to this region on the PA molecule (69–71). Furthermore, the residues in the 37-loop of tPA that are responsible for the direct interaction with PAI-1 are less charged in the 37-loop of uPA, resulting in a stronger exosite interaction of PAI-1 to tPA (72). These stronger exosite interactions result in a twice as large contact area of tPA with PAI-1 when compared to uPA, and provide a rationale for the difference in second-order inhibitory rate constants between the two PAs (47, 53).
As mentioned, the importance of several regions within the PAI-1 molecule for RCL insertion as well as for its interaction with the target PAs has been extensively studied. Ample evidence suggested that the nature of the amino acids and the flexibility of these segments are crucial for PAI-1 functionality and that changes made within these regions result in an altered reaction mechanism. Mutations within the hinge region of the RCL (residues P14-P8) altered the specificity toward tPA or uPA and moreover caused PAI-1 to behave predominantly as a substrate toward PAs (65, 73, 74). Stable substrate behavior without any detectable inhibitory activity was also conferred upon a PAI-1 deletion mutant lacking hF and the hF-s3A loop (75). Since both regions are involved in RCL insertion, i.e., the hinge region is initially inserted and hF coordinates the final insertion step, mutations in these regions may change the initial conformation of PAI-1 or impair the kinetics of RCL insertion. Therefore, these changes may ultimately result in the hydrolysis of the acyl-enzyme intermediate (76). Apart from these PAI-1 mutants, the behavior of PAI-1 as an inhibitor or as a substrate has also been reported to be influenced by external conditions, such as low temperature and non-ionic detergents, and ligands (77–79).
Functional Stability of PAI-1
PAI-1 Spontaneously Converts Into an Unreactive Latent Form
Unlike other serpins, PAI-1 has the unique ability to spontaneously convert into a thermodynamically stable latent form with a half-life of ~2 h at 37 °C in vitro. This active-to-latent transition occurs by slowly self-inserting the N-terminal part of the RCL into the core of the protein, thereby making the P1-P1′ bond inaccessible for PAs (80). Spontaneous latency transition is rather exceptional and is reported in only a few other serpins (81–83). Several lines of evidence indicate that latency transition in PAI-1 is evolutionarily conserved (84, 85). Therefore, it suggests an important role in auto-regulation of PAI-1 activity to reduce the risk of thrombosis due to the otherwise prolonged antifibrinolytic action of PAI-1. Based on the structures of active and latent PAI-1, the dynamical mechanisms involved in the active-to-latent transition were simulated using a computational approach, and could later be supported by experimental evidence (72, 86, 87). In a concerted manner, strand 1 (s1C) is peeled away from β-sheet C and allows the RCL to move around the gate region while it partially inserts up to P11 (Ser336) in the central β-sheet A that opened up at the breach and shutter region to form s4A. To reach this prelatent state, for which experimental evidence indicates that it co-exists with active PAI-1 in solution (88–91), a change in the bend in hA is required. After being held for an extended period in the prelatent form, full insertion of the final P6-P4 residues of the RCL is blocked by steric clashes between the RCL and the hF-s3A loop that overlies β-sheet A, posing a high-energy barrier. Similar to the conformational changes required for PA translocation, the favorable energy that is released upon partial insertion of the RCL is temporarily stored by an outward movement or unfolding of hF to enable full RCL insertion. Finally, hF returns to its native position across β-sheet A and irreversibly locks PAI-1 in its unreactive latent state. Whereas, during the inhibitory reaction with PAs energy can be recovered to distort the active site of the PA, latency transition is an energetically silent process. Therefore, it has been hypothesized that the energy gain from the favorable insertion of the RCL is used to extract s1C from β-sheet C and to position it alongside the PAI-1 molecule (92). Even though this transition is generally considered to be irreversible, limited reactivation by an unknown mechanism may occur in vivo (93). In vitro, the inhibitory properties can be restored by treating latent PAI-1 with denaturants followed by refolding (94).
In vivo Stabilization
In vivo, the active form of PAI-1 is stabilized at least 2-fold by the high-affinity association (KD ~ 0.1–1 nM) with the glycoprotein vitronectin that is abundant in plasma and the extracellular matrix (Figure 2) (95–99). The interaction between PAI-1 and vitronectin has been extensively characterized by mutagenesis and competition experiments using monoclonal antibodies, PAI-1/PAI-2 chimeras, and (synthetic) peptides (100–108). Based on these results, the N-terminal somatomedin B (SMB) domain within vitronectin and the flexible joint region, defined by hE, hF, and s1A, within PAI-1 were identified as the primary regions to engage in the interaction. Later, the crystal structure of PAI-1 in complex with the SMB domain of vitronectin (PDB ID 1OC0) provided additional details on the interaction interface, restricting their respective binding sites to the central region of the SMB domain (residues 10–30) and residues in the hE-s2A loop (Arg101), in hE (Pro111 and Phe114), hF (Asp138, Ile135, and Trp139) and s1A (Thr120, Lys122, Gln123, Val124, and Asp125) of PAI-1 (51). Through allosteric modulation of several regions remote from the SMB binding site, vitronectin causes a strong and widespread stabilization of the lower half of the PAI-1 molecule, including hB, hC, hD, hI, and the hI-s5A loop, and induces conformational changes in the RCL without compromising the ability of PAI-1 to associate with PAs (99, 109, 110). By reducing the structural flexibility, binding of vitronectin interferes with the sliding movement that is required to open up the shutter region, and consequently decreases the rate of RCL insertion that ultimately slows down the transition to latent PAI-1. Alternatively, expansion of β-sheet A due to loop insertion during latency transition or during the interaction with PAs results in the dissociation of vitronectin from inactive PAI-1 (95, 111, 112). Apart from the primary high-affinity PAI-1 binding site in the SMB domain, there is experimental evidence for additional PAI-1 binding sites in vitronectin. These sites, comprising a cluster of basic amino acids (residues 348–370 of vitronectin) in the C-terminal region of vitronectin (111–115) as well as the region connecting the SMB domain to the remainder of the vitronectin molecule (residues 111–121 of vitronectin) (116), have been shown to bind PAI-1 with a lower affinity and promote the assembly of higher-order PAI-1/vitronectin complexes (114, 115, 117).
One of the major acute-phase proteins, α1-acid glycoprotein, has also been shown to bind and stabilize the active form of PAI-1. Extensive binding studies allowed to identify a binding region that is distinct from that of vitronectin. This α1-acid glycoprotein binding region resides in the hI-s5A loop, comprising residues Arg300–Asp305 located at the bottom of PAI-1 β-sheet A (118). Even though this interaction occurs at a slower rate and is less stable as compared to the interaction with vitronectin, it might contribute to the PAI-1-mediated effects during inflammation or acute phase reactions (119).
PAI-1 Mutants With Increased Stability
Since its discovery, a vast amount of PAI-1 variants has been generated by both site-directed and random mutagenesis (120). These mutants have been employed in order to gain insights into the structure/function relationship in PAI-1, to identify regions that are important for its biological interactions, and to investigate its pleiotropic functions in various pathological processes. Due to its conformational flexibility, structural studies have benefited in particular from the generation of PAI-1 variants of which the functionally active form is stabilized. While single substitutions cause only a moderate stabilization of PAI-1, the combination of multiple mutations often results in a markedly enhanced stability with half-lives up to 450 h (85). Alternatively, the active conformation can also be maintained by introducing Cys-residues to crosslink flexible regions in PAI-1 that are crucially involved in latency transition (121, 122). This way, PAI-1 variants have been generated in which hD of the flexible joint region is connected to the N-terminal part of hA (engineered disulfide bridge between Val8Cys and Ala74Cys), in which s3A and s5A in the shutter region are covalently linked (Gln169Cys–Gly324Cys), in which the N-terminal part of the RCL is connected to the top of s3A in the breach region (Gln174Cys–Gly332Cys), or in which a combination is used. However, the introduction of only one single disulfide bond at the breach region is sufficient to most effectively preclude latency transition (very long half-life PAI-1, t1/2 > 700 h) without affecting its structure (122).
As mentioned, several crystal structures of PAI-1 in its alternative conformations (active, latent, and cleaved PAI-1) or of PAI-1 in complex with biological ligands have been determined by employing these stabilized active mutants (Figure 2 and Table 1). The first stable mutants to be successfully crystallized in the active conformation were the quadruple mutant PAI-1-N150H-K154T-Q319L-M354I, commonly referred to as PAI-1 14-1B (t1/2 ~ 145 h) (123, 133) and a variant harboring a fifth mutation, PAI-1-N150H-K154T-Q301P-Q319L-M354I, referred to as PAI-1-stab (46, 134). Later, the structure of active PAI-1-W175F (t1/2 ~ 7 h) was resolved as well (124). Apart from its prolonged half-life, PAI-1-W175F behaves similarly to wild-type PAI-1 and is therefore a more valid representative of wild-type PAI-1. Comparison of the available PAI-1 14-1B and PAI-1-W175F structures revealed numerous structural differences, with the most prominent one located in the region containing hF and the hF-s3A loop. Three of the mutations in PAI-1 14-1B and PAI-1-stab are clustered in and below the hF-s3A loop (Asn150His, Lys154Thr, and Gln319Leu) and induce a 310-like helix covering residues 151–157 that connects hF to the underlying β-sheet A through a hydrogen-bonding network. As a consequence, the energy barrier for hF displacement during the final step in RCL insertion is raised, explaining both the stabilization of the active conformation as well as the increased substrate behavior upon interaction with PAs that is observed for PAI-1 14-1B and PAI-1-stab. In contrast, the stabilization caused by the single amino acid substitution of the conserved tryptophan in PAI-1-W175F appears to be the result of local effects in the breach region that restrict initial loop insertion (124, 135).
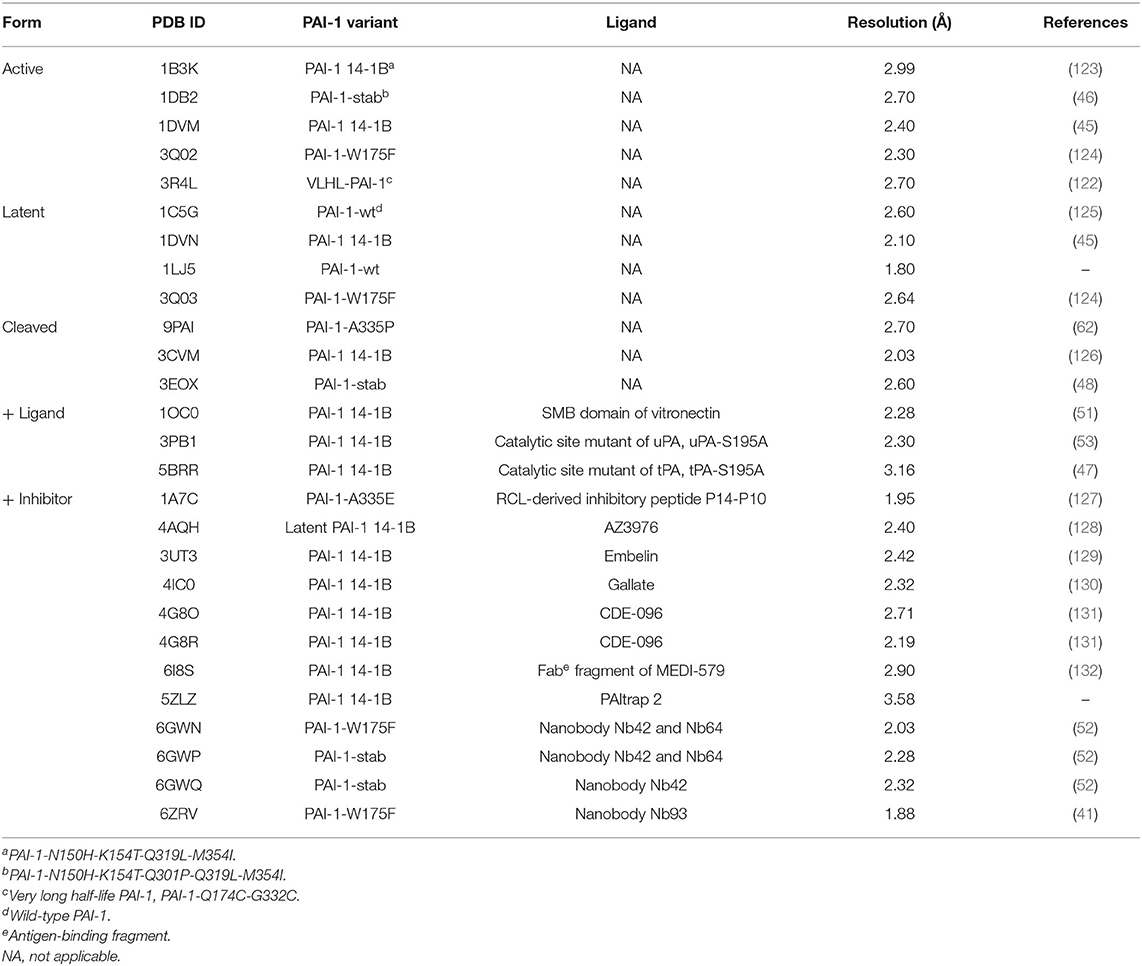
Table 1. List of X-ray crystallographic structures containing human PAI-1 available in the Protein Data Bank (PDB).
External Conditions Affecting PAI-1 Stability
Apart from being stabilized through interactions with its physiological ligands, several external conditions have been shown to affect the rate of latency transition in PAI-1 in vitro. During the search for the optimal purification conditions of recombinant PAI-1, a low temperature (4°C), a low pH (~5.5), and a high salt concentration (1 M NaCl) contributed to increased PAI-1 stability (98). Since a decrease in pH causes protonation of imidazole groups, it was suggested that one or more histidine residues might be directly responsible for the pH-dependent stability of PAI-1. It was first speculated that His143, localized at the top of hF, might be responsible for this effect (136). However, site-directed mutagenesis studies could only demonstrate a direct role for His364, situated on the C-terminal end of s4B in close vicinity to hD in the flexible joint region and to the W86-loop (137). The salt stabilization was further investigated based on the observation of an anion-binding site in a crystal structure of PAI-1 14-1B in the active conformation (45). It was suggested that by forming close interactions with partially positive nitrogen residues on each side of the anion-binding site, i.e., with Lys323 and Lys325 in β-sheet A and Ser149 and His150 in the hF-s3A loop, chloride binding increases the energy barrier of the final stage in latency transition. Also, a more pronounced stabilization was correlated with an increased electronegativity of the anion (F− ≥ Cl− > Br− > I−), resulting in tighter interactions. Notably, the proposed anion-binding site is located in the hF-s3A loop that is structurally different in PAI-1 14-1B as compared to “wild-type” PAI-1-W175F. Indeed, anion-binding could not be observed within this region in the more recent structure of active PAI-1-W175F (124) and is thus likely to be an artifact resulting from the induced conformational changes in the hF-s3A loop region in PAI-1 14-1B. However, a previously unknown chloride-binding site centered in the gate region could be unambiguously identified. This led to a revised hypothesis of the salt stabilizing effect on PAI-1 inhibitory activity, i.e., delaying latency transition by blocking the gate through bridging of several structural elements located between the s3C/s4C loop and the hG-s3B loop. Also, the preferential stabilization of other halide salts could not be extended to PAI-1-W175F or wild-type PAI-1, which were most dramatically stabilized by sodium chloride yielding half-lives well above 30 h at a 1 M salt concentration (124). Interestingly, two zinc-binding sites could clearly be identified within the same crystal structure of PAI-1-W175F. Since the metals appeared at the interface between two PAI-1 molecules inside the crystal, it remained debatable whether one or both binding sites are physiologically relevant. However, one zinc ion was strongly coordinated by N-terminal His2 and His3 (124). Almost simultaneously it was shown that type I metal ions (calcium, magnesium, and manganese) have modest stabilizing effects on PAI-1 activity, whereas type II metals (cobalt, copper, and nickel) had a more pronounced effect, either destabilizing PAI-1 in the absence of vitronectin or adding up onto the stabilization caused by simultaneous binding of vitronectin (138). Even though competitive binding experiments suggested that these effects were mediated through a single metal-binding site (139), a copper-binding site involving N-terminal His2 and His3 was identified that only accounted for the stabilizing, and not the destabilizing, effect of copper (140). The existence of a second copper-binding site has been further confirmed by the observation that copper facilitates an early step in PAI-1 latency transition by increasing protein dynamics in the flexible joint region and the helices underlying the shutter region, which could also be observed when copper bound to a mutant lacking His2 and His3 (141). Apart from salts and metals, high concentrations of arginine have been shown to elute subendothelial matrix-bound PAI-1 and to specifically stabilize the PAI-1 active conformation (142, 143). Since arginine residues are clustered in the stretch of basic residues in the C-terminal region of vitronectin, free arginine might contribute to an enhanced PAI-1 stability in a similar way as the arginine-rich C-terminal region of vitronectin.
(PATHO)Physiological Roles of PAI-1
As the major physiological inhibitor of plasminogen activators tPA and uPA, PAI-1 plays a regulatory role in the fibrinolytic system by controlling plasmin formation. Not only is the plasminogen activator/plasmin system involved in fibrinolysis, it has also a profound role in multiple physiological processes, including the degradation of extracellular matrix (ECM), tissue remodeling, wound healing, angiogenesis, cell migration, and inflammation (144). Upon uPA-mediated activation of plasminogen, either by two-chain uPA or single-chain uPA bound to the uPA receptor (uPAR), plasmin can degrade several ECM components either directly or indirectly through the activation of matrix metalloproteases (MMPs). Degradation of the ECM may then facilitate cell invasion into the surrounding tissue. Furthermore, by increasing the availability of growth factors, such as vascular endothelial growth factor, fibroblast growth factor, and transforming growth factor-β, the role of plasmin further extends to the control of angiogenesis, cell growth, and cell differentiation. Importantly, independent of its effect on plasmin formation, PAI-1 directly interacts with specific matrix components, including vitronectin, LRP1, and the uPA/uPAR complex to affect cell migration and intracellular signaling.
The (patho)physiological role of PAI-1 has been extensively studied by comparing the phenotype observed in human PAI-1 deficiency with that of mice engineered to be completely PAI-1 deficient by gene targeting (PAI-1−/− mice). In humans, PAI-1 deficiency is an uncommon disorder that can be caused by mutations in the SERPINE1 gene leading to the production of non-functional PAI-1 protein (145) or by a complete absence of PAI-1 plasma antigen (146–148). Typically, this disorder is characterized by mild to moderate bleeding in response to injury, trauma, or surgery. In women, PAI-1 deficiency may cause severe blood loss during menstruation and pregnancy-related complications, such as prepartum bleeding, preterm labor, or miscarriage (149–151). PAI-1−/− mice were shown to be viable, fertile, and developed normally (152). Furthermore, disruption of the PAI-1 gene did not appear to impair hemostasis, but was associated with increased resistance to thrombosis and with a milder hyperfibrinolytic state as compared to humans (153). In contrast to PAI-1 deficient mice, several lines of transgenic mice overexpressing native or stabilized PAI-1 of human and murine origin have been established. These lines have been generated to explore the effects of elevated PAI-1 levels on, e.g., the progress of thrombosis (154, 155), pulmonary fibrosis (156), and obesity (157). Furthermore, these transgenic mice often display hair loss and skin abnormalities. Importantly, transgenic mice expressing a reactive site inactive PAI-1 mutant exhibit complete phenotypic rescue, while transgenic mice expressing PAI-1 with reduced affinity for vitronectin manifest all of the phenotypic abnormalities, underscoring the fact that PAI-1 affects physiological processes by acting through multiple pathways (158). In humans, two variations in the promoter region of the PAI-1 gene occur frequently and have been shown to affect PAI-1 levels (159–161). Firstly, the 4G/5G polymorphism refers to a single guanosine insertion/deletion at position 675 upstream of the transcription site (159). It has been suggested that the 4G allele is associated with higher PAI-1 activity since the 5G allele harbors an additional binding site for a transcriptional repressor. Secondly, the G/A polymorphism is characterized by a single nucleotide substitution of guanine for adenine at position 844 upstream of the transcription site, generating a consensus binding sequence for transcription factor Ets-1 which increases the transcription rate (161). Taken together, the association of these PAI-1 gene polymorphisms and/or elevated PAI-1 levels with several pathologies have been extensively studied in both humans and in PAI-1 deficient or transgenic mice.
PAI-1 in Cardiovascular Disease
Elevated levels of PAI-1 downregulate tPA and uPA activity and create a prothrombotic or hypofibrinolytic state that was suggested to contribute to the pathogenesis of cardiovascular diseases (CVD) (162). As mentioned, several lines of transgenic mice that overexpress PAI-1 have been developed and support a contribution of elevated PAI-1 levels to thrombosis and CVD. The first line of transgenic PAI-1 mice overexpressed native human PAI-1 and was shown to develop transient venous thrombosis in the tail and hind limbs and subcutaneous hemorrhage (154). Later, human PAI-1-stab mice were generated that displayed age-dependent coronary arterial thrombosis and myocardial infarction (155). In contrast, spontaneous thrombosis could not be observed in transgenic mice that overexpress stabilized murine PAI-1 (163). However, it should be noted that (I) the choice of the promoter that drives PAI-1 expression, (II) the nature of the stable variant, and (III) a cross-species difference in PAI-1 function may have contributed to the observed phenotypic differences. In humans, many studies have suggested that PAI-1 gene polymorphisms, possibly leading to higher PAI-1 levels, are an independent risk factor for major adverse cardiovascular events (MACE) including myocardial infarction (MI) (164–167) and ischemic stroke (168), as well as coronary heart disease (CHD) (169), venous thrombosis (170–172), and atherosclerosis (173). However, despite the observed link in these studies, these findings are in contradiction with other available data (174–178). Similarly, independent of the contribution from PAI-1 gene polymorphisms, ample evidence has been provided of a link between elevated PAI-1 levels and MI or stroke (179–181), CHD (182), venous thrombosis (183), and atherosclerosis (184, 185). In a recent systematic review of all studies published between 1991 and 2016 that examined the association of PAI-1 with MACE (defined as death, MI, and stroke) or restenosis (the recurrence of treated coronary artery stenosis), Jung et al. substantiated a link between elevated plasma PAI-1 antigen levels, but not PAI-1 activity levels, and MACE in both incident and secondary event populations (181). MI is most often a result of CHD and is caused by the disruption of an atherosclerotic plaque, thereby exposing a procoagulatory surface of the coronary vessel that gives rise to occlusive thrombus formation (186). Several studies have reported elevated PAI-1 levels in several cell types associated with atherosclerotic plaques in human coronary arteries, including endothelial cells, vascular smooth muscle cells (SMCs), and macrophages (184, 187, 188). In mice, PAI-1 deficiency has been shown to be protective (189) or promoting (190) in the development of atherosclerosis, however, no effect of PAI-1 on atherosclerosis has been observed as well (191). Indeed, overproduction of PAI-1 in a diseased vessel wall may contribute to the progression of atherosclerosis by reducing local plasmin production which is physiologically required for the removal of fibrin, ECM remodeling, and SMC proliferation. However, when the controlling effect of PAI-1 on plasmin formation is abolished, it may contribute to the atherogenic role of plasmin, as plasmin is also involved in lipoprotein modification, macrophage cholesterol accumulation, inflammation, and foam cell formation (192, 193). Furthermore, PAI-1 has been shown to have an ambiguous role in neointima formation (194–198). In this respect, in the same systematic review by Jung et al., low PAI-1 antigen and activity levels were associated with increased restenosis, highlighting the complex role of PAI-1 in vascular remodeling (181). Despite the links provided between PAI-1 and CVD, certain studies could not confirm these associations or the significance was lost after adjustment for other risk factors (199–201). A positive correlation has been demonstrated for plasma PAI-1 levels and known risk factors for developing CVD, including age, sex, obesity, hyperlipidemia, insulin resistance, and diabetes (162, 181, 202, 203). Age is an important risk factor for most chronic diseases including cardiovascular disease, type 2 diabetes, and metabolic syndrome. Furthermore, PAI-1 levels have been reported to increase with age in various tissues. More recently, PAI-1 has been identified not only as a marker but also as a mediator of cellular senescence associated with aging and aging-related pathologies (204).
Diverse Approaches To Inhibit PAI-1
As PAI-1 is considered a risk factor in various pathological conditions, many efforts have been devoted to the development of PAI-1 inhibitors, including small molecules, synthetic peptides, RNA aptamers, monoclonal antibodies (mAbs), and antibody derivatives. Whereas, some marketed drugs have been shown to attenuate PAI-1 synthesis or secretion (205), the majority of PAI-1 inhibitors currently in development can influence PAI-1 functionality in at least three possible ways: (I) by directly blocking the initial formation of the Michaelis complex between PAI-1 and PAs, (II) by preventing the formation of the final inhibitory complex, resulting in substrate behavior of PAI-1, or (III) by accelerating the active-to-latent transition of the PAI-1 molecule or an otherwise inert form. Despite extensive in vitro and in vivo characterization, no PAI-1 inhibitor is currently approved for therapeutic use in humans. This is mainly due to affinity and specificity issues, which are especially observed for small molecules. Furthermore, the structural plasticity of PAI-1 and the possible counteraction of PAI-1 binding partners, such as vitronectin, pose a real challenge to develop PAI-1 inhibitors that retain their capacity to modulate PAI-1 activity in vivo. To improve their properties or to guide the rational design of novel PAI-1 inhibitors it is essential to get a deeper understanding of PAI-1 inhibition at the molecular level. In addition to the several crystal structures of PAI-1 in active, latent, or cleaved conformation (45, 48, 124), a handful of structures containing PAI-1 in complex with inhibitory peptides, small molecules, and antibody fragments have been described (Table 1). Furthermore, by using a broad range of biophysical and biochemical methods, including competitive binding experiments, mutagenesis, and computational docking, the presumptive binding regions of the majority of PAI-1 inhibitors have been mapped and can be related to the mechanisms by which they interfere with PAI-1 functionality.
Synthetic Peptides
RCL-Mimicking Peptides
Insertion of the RCL into the central β-sheet A is a crucial step in the inhibitory mechanism of serpins in order to translocate and irreversibly trap the target proteinase. In this regard, synthetic peptides derived from the RCL-sequences of antithrombin III and α1-antitrypsin were shown to convert the respective serpins from an inhibitor to a substrate. By binding between s3A and s5A in β-sheet A and thus becoming s4A, these peptides prevent endogenous RCL insertion upon interaction with the target proteinase, resulting in cleavage of the serpin and release of regenerated proteinase (206). Taking a similar approach, several peptides that mimic different fragments of the RCL of PAI-1, such as P14-P1 (207), P14-P10 (127), P14-P9 (208), P14-P7 (136), and P8-P3 (208), were designed and evaluated for their PAI-1 modulating properties. The first peptide, corresponding to residues P14-P1 of the RCL, was shown to rapidly inhibit PAI-1 activity by accelerating the conversion to a non-reactive PAI-1 form and effectively enhanced in vitro fibrinolysis in both platelet-poor and platelet-rich clots (207). However, when PAI-1 was bound to its biological cofactor vitronectin, the PAI-1 neutralizing effects of this peptide were considerably reduced. A comparable mechanism was observed for the P8-P3 peptide that mimics the C-terminal part of the RCL that inserts at the bottom of the β-sheet A in latent PAI-1. Unlike peptides P14-P1 and P8-P3, peptides P14-P7, P14-P10, or P14-P9 that correspond to the N-terminal part of the RCL converted PAI-1 from an inhibitor to a substrate for tPA (127, 136, 208). By showing that binding of P14-P7 and the formation of latent PAI-1 are competitive events, the first evidence was provided for a binding site in the central β-sheet A cleft (136). The high-resolution crystal structure of PAI-1 mutant PAI-1-Ala335Glu in complex with two P14-P10 peptides (PDB ID 1A7C) further confirmed this presumption (127). The structure revealed that both peptide molecules bound inside the cleft between s3A and s5A, with the first molecule occupying the same space as RCL residues P14-P10 in latent and cleaved PAI-1, and the second one occupying the same space as residues P6-P2 in cleaved PAI-1. Since the different effects of RCL-derived peptides on the outcome of the PAI-1/PA reaction, i.e., conversion to either inert or substrate PAI-1, did not seem compatible with one common binding position inside the β-sheet A cleft, it was finally suggested that peptides mimicking the C-terminal part of the RCL (P8-P3) act by accelerating the irreversible transition to an inert form of PAI-1, whereas peptides that mimic the N-terminal part of the RCL (P14-P9) primarily induce PAI-1 substrate behavior (208).
Other Peptides
A few other peptides have been isolated from a phage-display peptide library including paionin-1 (209), which does not impair PAI-1 activity, and paionin-4 (210), which accelerates the active-to-latent conversion of PAI-1. In silico docking of paionin-1 into the crystal structure of PAI-1 suggested a binding site in the flexible joint region that was supported by site-directed mutagenesis and competitive binding of other molecules targeting the same region, such as XR-5118 and bis-ANS. As paionin-1 is able to prevent binding of the PAI-1/uPA complex to LRP1, paionin-1 or other compounds binding in the same region may be of benefit in cases where disruption of the signaling function of uPA/uPAR/LRP1 is desired. Paionin-4 presumably binds PAI-1 at the loop between hD and s2A and is suggested to induce a conformational change that facilitates loop insertion.
RNA Aptamers
RNA-aptamers are 8–15 kDa single-stranded nucleic acid ligands that tend to bind to highly positive regions on proteins and block protein-protein interactions. In this respect, a few RNA aptamers have been developed in order to interfere with the interactions between PAI-1 and its binding partners. WT-15 and SM-20 are able to disrupt the functional interaction between vitronectin and PAI-1 without compromising the PA-inhibitory function of PAI-1 (211). Expression of these aptamers in human breast cancer cells decreased cell migration and invasion and additionally decreased PAI-1 and uPA levels while increasing the stable PAI-1/uPA complex (212). Other aptamers, R10-4 and R10-2, were able to interfere with the formation of the stable PAI-1/tPA complex but not with the PAI-1/uPA complex, suggesting a binding site not directly involved in PAI-1/PA interactions, without disrupting the PAI-1/vitronectin interaction (213).
Small Molecules
Since the mid-90s, several low molecular weight (LMW) inhibitors possessing a large structural diversity have been described and grouped based on their chemical composition [extensively reviewed by Fortenberry (214) and Rouch et al. (215)]. Lead molecules have been discovered using various approaches, including through isolation from bacterial biomass or a library of natural products, by high-throughput screening (HTS) of synthetic libraries, and by structure-based virtual screening. Subsequently, many of these compounds have been modified based on structure-activity relationship (SAR) studies in order to improve their selectivity and specificity and their inhibitory and physicochemical properties. Even though only a few structures of PAI-1 complexed with small molecules were determined, they provided evidence for a common compound-binding pocket within the flexible joint area of PAI-1 (Figure 3).
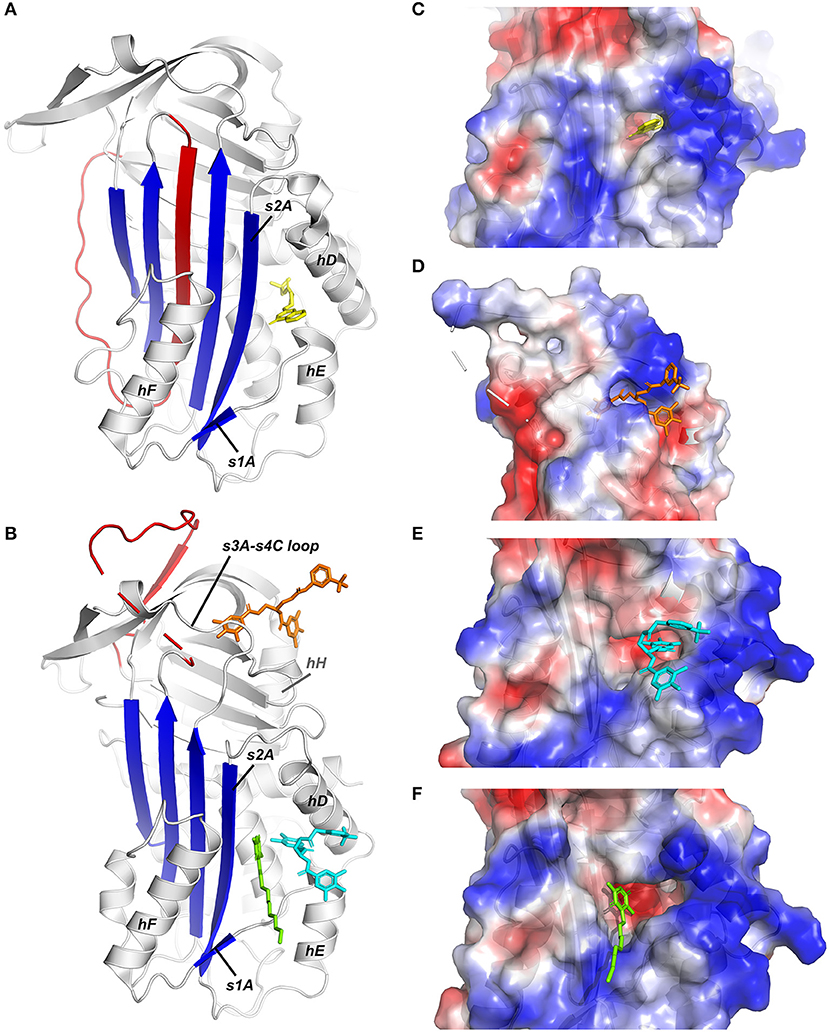
Figure 3. X-ray crystallographic structures of small molecule inhibitors bound to PAI-1. (A) Structure of latent PAI-1 in complex with AZ3976 (128). (B) Superimposition of the structures of active PAI-1 in complex with embelin (129) and CDE-096 (131). PAI-1 is colored white with the central β-sheet A colored blue and the RCL colored red. Secondary structure elements involved in binding to the compounds are indicated. AZ3976 is colored yellow, and embelin is colored green. CDE-096 in the structures obtained by cocrystallization or crystal soaking is colored orange or cyan, respectively. (C) AZ3976 bound to a deep pocket aligned by hD and s2A in latent PAI-1 [PDB ID 4AQH (128)]. (D) CDE-096 bound to active PAI-1 obtained by cocrystallization [PDB ID 4G8O (131)]. (E) Detail of the structure of CDE-096 bound to active PAI-1 obtained by crystal soaking [PDB ID 4G8R (131)]. (F) Detail of the structure of embelin bound a groove aligned by hD, hF, s2A, and the hE-s1A loop in active PAI-1 [PDB ID 3UT3 (129)].
The first published crystal structure of a PAI-1/inhibitor complex involved compound AZ3976 (PDB ID 4AQH) (Figure 3C) (128). AZ3976 was identified by HTS of the AstraZeneca compound collection and shown to inhibit PAI-1 activity in in vitro chromogenic and clot lysis assays. Titration of PAI-1 with AZ3976 revealed that the compound was only bound to 30% of the total PAI-1 present, corresponding to the non-reactive subpopulation (latent or cleaved) in the preparation of active PAI-1. This was confirmed by affinity data, showing a high binding affinity toward latent PAI-1 whereas no binding was observed toward the active form. Based on the structure of latent PAI-1 complexed with AZ3976, a deep ligand-binding pocket within the flexible joint region was identified with the entry located between hD and s2A. Importantly, this binding site appeared to be more accessible in latent PAI-1, however, tight binding of AZ3976 requires small conformational changes. Since AZ3976 has been shown to accelerate the active to latent transition of PAI-1, it was therefore suggested to bind to a latent-like prelatent form from which latent PAI-1 is then generated more rapidly.
Not much later, a second crystal structure of PAI-1 containing a small molecule inhibitor, embelin, was published (PDB ID 3UT3) (Figure 3F) (129). Embelin was identified as a PAI-1 antagonist by screening a library of natural products. Structural and site-directed mutagenesis results have shown that embelin binds to a small and charged groove aligned by hD, hF, s2A, and the hE-s1A loop in active PAI-1 (129), located adjacent to the larger and deeper pocket in (pre)latent PAI-1 that can be occupied by AZ3976 (128). It was proposed that embelin fixes s2A to the neighboring hD and hE, and thereby interferes with the sliding movement of s2A and s3A into the flexible joint region in order to accommodate RCL insertion. Indeed, consistent with this theory, embelin has been shown to act by a two-step mechanism including a fast reversible step of inducing PAI-1 substrate behavior, followed by a slow irreversible induction of an inactive form. Despite the ability of AZ3976 and embelin to inhibit PAI-1 activity in vitro, the presence of vitronectin abolished the capacity of both compounds to bind and modulate PAI-1 activity. Since the binding sites for AZ9376 and embelin partially overlap with the binding site for vitronectin in active PAI-1, the protective effects of vitronectin might be caused by sterically blocking their binding sites or, in the case of AZ3976, by preventing the formation of the prelatent structural state to which AZ3976 preferably binds. Importantly, the binding pocket within the flexible joint region, which has been observed in the crystal structures of PAI-1 complexed with AZ3976 and embelin, is consistent with the binding sites for other small molecules that were determined mainly through competitive binding studies, mutagenesis studies, and molecular modeling. Several negatively [AR-H029953XX (216), ANS, Bis-ANS] and positively [XR5118 (217, 218)] charged amphipathic inhibitors have been shown to bind overlapping but non-identical binding sites within this hydrophobic pocket, resulting in variable induced molecular changes in PAI-1 and in a differential susceptibility to vitronectin-bound PAI-1 (219). First, it was demonstrated that both groups inhibit PAI-1 via a two-step mechanism, involving a rapid reversible conversion into a PAI-1 form exhibiting substrate behavior, followed by a slower irreversible conversion into a non-reactive form. However, a different study showed that both AR-H029953XX and XR5118 induce a direct conversion of PAI-1 into a non-reactive form, possibly due to the differences in compound concentrations that were used to conduct experiments. A concentration-dependent effect could also be observed for tiplaxtinin (PAI-039), which induces PAI-1 substrate behavior at lower concentrations and converts PAI-1 to a non-reactive form at high concentrations (88). Interestingly, several studies reported that PAI-1 polymerization could be induced by negatively charged organochemical inhibitors following conversion to a non-reactive form (219–221).
Apart from this common compound-binding pocket, a third crystal structure of the cocrystallized PAI-1/CDE-096 complex (PDB ID 4G8O) elucidated the binding mode of CDE-096 which is, in contrast to the aforementioned compounds, active against both free and vitronectin-bound PAI-1 (Figure 3D) (131). CDE-096 was synthesized based on a SAR study of a high-affinity polyphenolic PAI-1 inhibitor (222). Structural studies, substantiated with site-directed mutagenesis results, revealed a binding site at the interface composed of residues from the s3A/s4C loop, β-sheets B and C, and hH, referred to as the sB/sC pocket. Using a combination of biochemical experiments, a mechanism of action was proposed that involves reversible allosteric modulation of RCL conformation to block initial PAI-1/PA Michaelis complex formation. Furthermore, although CDE-096 and vitronectin reduce PAI-1's affinity for one another, their binding is not strictly mutually exclusive, suggesting allosteric modulation through reciprocal communication between the high-affinity compound- and vitronectin-binding sites. In crystal soaking studies that require high concentrations of CDE-096 to be incubated with preformed PAI-1 14-1B crystals, a second possible binding site was observed and shows overlap with the binding site for AZ3976 in latent PAI-1 (PDB ID 4G8R) (Figure 3E). However, based on the mutagenesis results and the capacity of CDE-096 to bind both active and latent PAI-1 with similar affinity, it was argued to be an artifact induced by the high concentrations used for crystal soaking.
Another class of small molecules was discovered by virtual screening of a library of commercially available chemicals. To address the lack of efficacy when translated into in vivo settings often encountered in high-throughput screening of large compound libraries, two filters were applied representing (I) the general drug-likeliness based on clinically used drug molecules, and (II) the specific lead-likeliness based on the RCL peptide P14-P10 as well as reference inhibitors that bind in the region of the vitronectin binding site in PAI-1 (219, 223). Next, docking simulations for the remaining compounds focused on the cleft in β-sheet A that is occupied by the RCL following loop insertion. TM5007, the most effective compound, exhibited high specificity for the PAI-1/PA system and was furthermore effective in in vivo models of thrombosis and fibrosis (223). SAR studies on TM5007 resulted in the selection of TM5275 with an improved inhibitory profile and better oral bioavailability (224). A similar docking simulation suggested that TM5275 binds within the cleft between the strands of β-sheet A, albeit at a different position compared to its precursor TM5007. Whereas, TM5007 docked into the space occupied by P8-P3 in the latent form, TM5275 docked at the P14-P9 position. The differences in their presumed binding sites within the cleft also seemed to correlate with their mechanisms of action, i.e., by either preventing PAI-1/PA complex formation (TM5007) or inducing substrate behavior of PAI-1 (TM5275) (223, 224). Further structure-optimization by substituting the lipophilic moiety and varying the acyl-type linker length led to the discovery of smaller derivatives, including TM5441 and TM5484 (225). Although these compounds have originally been designed to bind the central β-sheet A cleft, there is no experimental evidence that confirms their binding site or their mechanism of action.
Even though many small molecules have been shown to be potent PAI-1 inhibitors in vitro or in vivo, several other factors, such as the lack of information regarding their exact inhibitory mechanism and/or binding area in PAI-1, the inability to modulate the activity of vitronectin-bound PAI-1, and the possibility to induce PAI-1 polymerization may partially hamper the future rational design of novel effective small molecules.
Antibodies and Antibody Derivatives
Conventional antibodies are Y-shaped heterotetrameric glycoproteins (150 kDa) composed of two light and two heavy chains that are linked together by multiple disulfide bonds (Figure 4A) (228). Each light chain comprises one variable (VL) and one constant domain (CL), whereas each heavy chain comprises one variable domain (VH) and three constant domains (CH1, CH2, and CH3). The Ig unit can be divided into three functional components, namely two identical antigen-binding fragments (Fabs) and one crystallizable fragment (Fc). In each Fab fragment, the variable fragment (Fv) is responsible for the recognition of and high-affinity binding to a specific antigen and is composed of the variable domains of both chains (VL and VH). The amino acid residues of the V-regions that are in direct contact with the antigen are referred to as the paratope, whereas the binding site for the antibody on the surface of the antigen is referred to as the epitope. By connecting the VH and VL domain of a conventional antibody through a flexible polypeptide linker consisting of serines and glycines, a ~25 kDa single-chain variable fragment (scFv) can be created that usually retains the antigen-binding capacity of the parental mAb (Figure 4B). Subsequently, two scFvs can be combined in order to generate a ~55 kDa diabody that can either target the same epitope on another molecule of the same antigen (bivalent), another epitope on the same antigen (biparatopic), or another antigen (bispecific) (Figure 4B).
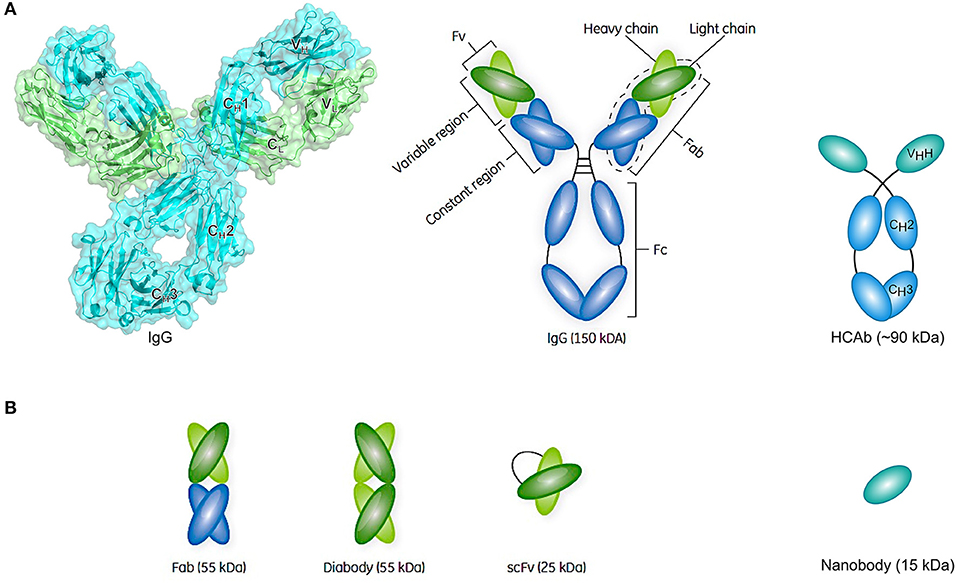
Figure 4. Schematic representation of an antibody structure and different antibody fragment formats. (A) The left panel shows the cartoon and biological surface representation of the full-size crystal structure of Pembrolizumab, a human anti-PD1 immunoglobulin G4 (IgG4) antibody [PDB ID 5DK3 (226)]. A conventional antibody is a Y-shaped heterotetrameric glycoprotein consisting of two identical heavy (cyan) and two identical light chains (green). The heavy chain comprises one variable domain (VH) and three constant domains (CH1, CH2, and CH3), whereas the light chain comprises one variable (VL) and one constant (CL) domain. The panel on the right shows a simplified schematic representation of a conventional antibody and a heavy-chain-only antibody (HCAb). Each arm of the conventional antibody represents the antigen-binding fragment (Fab) that comprises the constant region (CH1 and CL domains) and the variable fragment (Fv) containing VL and VH. The stem of the antibody comprises two copies of the CH2-CH3 domains that form the crystallizable fragment (Fc). The HCAb comprises two heavy chains, each combining one variable VHH domain, referred to as nanobody, and two constant domains (CH2 and CH3). (B) Schematic representation of a selection of antibody fragments, including Fab, diabody, single-chain variable fragment (scFv), and nanobody. Figure adapted from Rodrigo et al. (227).
Later it was discovered that, besides conventional antibodies, the sera of camelids (such as camels and llamas) (229) as well as cartilaginous fish (such as nurse sharks) (230) naturally contain functional heavy-chain-only antibodies (HCAbs) (231). In contrast to conventional antibodies, camel HCAbs (~90 kDa) are devoid of the light chains and the heavy chain CH1 domains that normally serve to anchor the light chains (Figure 4A). The VHH domain can be isolated from the HCAb and be produced as such at a large scale in bacterial expression systems, ultimately yielding a recombinant single-domain antibody referred to as nanobody (Nb) (Figure 4B). In addition to having high binding affinities toward antigens in the nano- or even picomolar range, Nbs express a favorable stable behavior in harsh conditions, including high temperatures, high protein concentrations, high pressure, and the presence of detergents or denaturants (232, 233). Furthermore, resistance to pepsin or chymotrypsin can be conferred upon nanobodies by introducing an additional disulfide bond, suggesting the possibility of oral administration (234). Furthermore, their small size makes them good candidates when it is required to penetrate dense tissues in order to bind hard to reach targets or to deliver functional molecules to the cytoplasm. As they can easily be linked, nanobodies can serve as “building blocks” to construct multispecific, multivalent, or multiparatopic molecules.
Antibody-Based PAI-1 Inhibitors
Due to the efforts of many research groups, a large panel of mAbs that interfere with PAI-1 activity is readily available. More recently, nanobody libraries have been constructed as well (35). In contrast to inhibitory peptides and small molecules, the epitopes of antibody-based PAI-1 inhibitors have been mapped to different regions of the PAI-1 molecule (Figure 5). Antibodies have been shown to affect PAI-1 functionality at distinct levels during the PAI-1/PA reaction, i.e., by preventing Michaelis complex formation, by inducing substrate behavior of PAI-1, or by accelerating the conversion to latent PAI-1 (243).
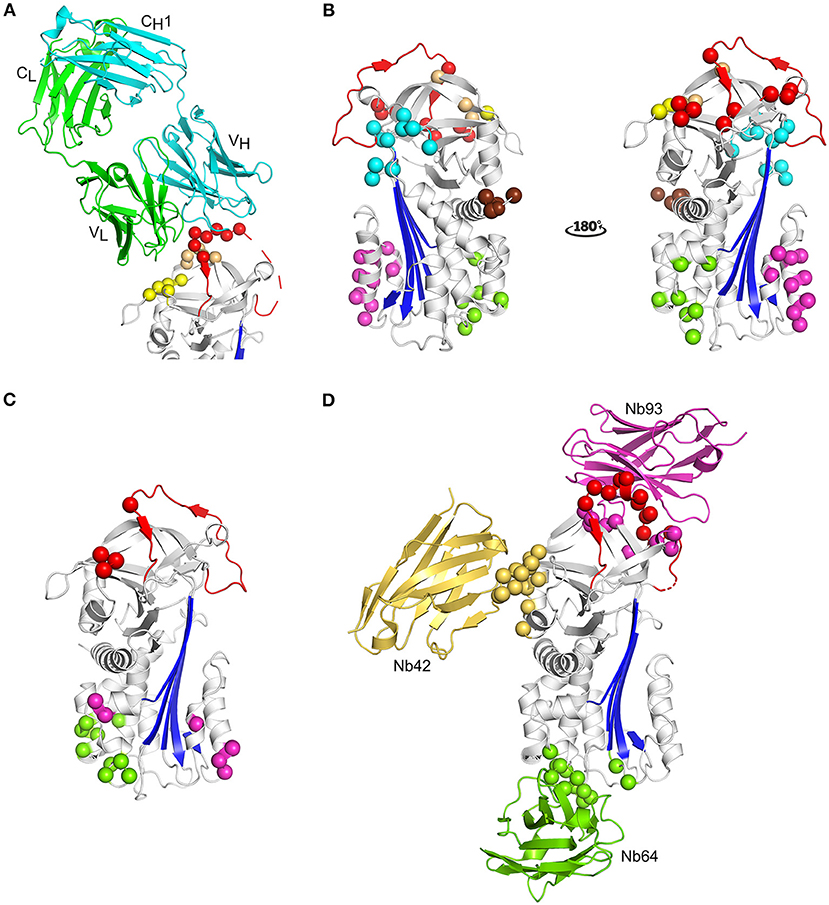
Figure 5. Localization of different epitopes in the structure of active PAI-1. (A) Cartoon representation of the crystal structure of PAI-1 in complex with the Fab fragment of MEDI-579 (PDB ID 6I8S) (132). The heavy and the light chain of the Fab fragment are colored cyan and green, respectively. The constant and variable heavy (CH1 and VH) and light (CL and VL) domains are indicated in the figure. Residues closer than 4 Å to MEDI-579 are indicated as spheres. Residues located in the RCL are colored red, residues residing in the exosite regions for the 37-loop and 60-loop of PAs are colored yellow and orange, respectively. (B) Localization of different epitopes of monoclonal antibodies (mAbs) as determined by mutagenesis studies. The epitopes of mAbs that prevent the interaction between PAI-1 and PAs are indicated as red (MA-42A2F6, MA-56A7C10, and MA-44E4) (235) and yellow (MA-124K1) (236) spheres. The epitopes of switching mAbs that bind to hF or the hF-s3A loop (MA-33H1F7, MA-55F4C12, and Mab2) (237, 238) are indicated as magenta spheres. The epitope of switching mAb MA-8H9D4 (239) that binds to the hI-s5A loop is indicated as green spheres. The epitopes of latency-inducing antibody MA-33B8 (240, 241), MA-H4B3 (90), and MA-159M12 (242) are indicated as cyan, orange, and brown spheres, respectively. (C) Localization of different epitopes of nanobodies as determined by mutagenesis studies (35). The epitope of substrate-inducing nanobody Nb98 is indicated by green and magenta spheres, whereas only the magenta spheres indicate the epitope of Nb64. The epitope of Nb93 that interferes with PAI-1/PA complex formation is indicated as red spheres. (D) Cartoon representation of the superimposed crystal structures of PAI-1 in complex with Nb42 [PDB ID 6GWN, 6GWP, and 6GWQ (52)], Nb64 [PDB ID 6GWN and 6GWP (52)], and Nb93 [PDB ID 6ZRV (41)]. Nb42, Nb64, and Nb93 are colored yellow, green, and magenta, respectively. Residues closer than 4 Å to Nb42 are indicated as yellow spheres. Residues closer than 4 Å to Nb64 are indicated as green spheres. Residues closer than 4 Å to Nb93 are indicated as red and magenta spheres. Red spheres represent residues located in the RCL of PAI-1, whereas magenta spheres represent residues located in the exosite binding regions for PAs.
Only recently, the first crystal structure of PAI-1 complexed with an inhibitory antibody fragment belonging to the first class was reported (Table 1) (132). This structure containing the Fab fragment of neutralizing antibody MEDI-579 revealed that its epitope is concentrated around the C-terminal region of the RCL (residues Ala345–Glu350 or P2–P4′) and the neighboring exosites for the 37-loop (Tyr210, Glu212, Tyr220, and Tyr241) and 60-loop (Leu269, Pro270, and Arg271) of PAs (Figure 5A) (132). As a consequence, by simultaneously interfering with exosite interactions and shielding the P1-P1′ reactive center, MEDI-579 prevents the initial interaction between PAI-1 and PAs. A similar mechanism has been described for nanobody VHH-s-a93 (Nb93) that binds an epitope slightly overlapping with that for MEDI-579 (41). The X-ray crystallographic structure of the PAI-1/Nb93 complex revealed that Nb93 binds PAI-1 in a PA-like manner through interactions with the almost full-length RCL and adjacent exosites for PAs other than that for their 37-loop (Table 1 and Figure 5D) (41). In addition, Nb93 was shown to selectively bind and stabilize the active conformation of PAI-1 by anchoring the RCL to the top of the PAI-1 molecule. Notably, similar binding sites including the RCL of PAI-1 and/or neighboring exosites for PAs have been described for other mAbs (Figure 5B), including ESPI-12 (between residues 342–349), MAI-12 (between residues 320–379), MA-42A2F6 (Lys243 and Glu350), MA-56A7C10 (Glu242, Lys243, Glu244, Glu350, Asp355, and Arg356), and MA-44E4 (His185, His186, and Arg187), suggesting that they directly interfere with Michaelis complex formation as well (132, 235, 244, 245). The epitopes of two other nanobodies, VHH-s-a27 (Nb27) and VHH-2g-42 (Nb42), could not be deduced by mutational studies (35). However, it was hypothesized that they might interfere with the PAI-1/PA reaction by directly preventing PAI-1/PA complex formation as well. Indeed, using a hybrid approach employing structural and biochemical methods, Nb42 was shown to destabilize the initial Michaelis complex by binding to the exosite region for the 37-loop of PAs (Table 1 and Figure 5B) (52). Interestingly, MA-124K1 that inhibits rat PAI-1 was found to bind the exosite region for the 37-loop of PAs (Glu212 and Glu220) and thereby inhibits PAI-1 activity while simultaneously enhancing the binding of PAI-1 to vitronectin (236).
The second class of mAbs, referred to as “switching antibodies,” redirect the inhibitory PAI-1/PA reaction toward the substrate branch. Within the category of substrate-inducing antibodies, two different subclasses have been identified. Even though both subclasses ultimately increase the relative fraction of cleaved PAI-1, each class acts through a distinct mechanism by binding different epitope regions localized in the lower half of the PAI-1 molecule (Figure 5B). Several mAbs, including MA-33H1F7 (Glu130, Arg131, and Lys154), MA-55F4C12 (Glu128, Val129, Glu130, Arg131, and Lys154), and Mab2 (Arg131, Arg133, Phe134, Asn137, Asp138, Leu152, and Lys154) were shown to have overlapping epitopes located in hF and the loop connecting hF with s3A of the central PAI-1 β-sheet (237, 238). These mAbs have been shown to slow down the rate of cleaved RCL insertion, possibly by restricting the structural rearrangements within this region during RCL insertion. A different epitope was identified for switching antibody MA-8H9D4 that binds the loop between hI and s5A at the bottom of the PAI-1 molecule (Arg300, Gln303, and Asp305) and possibly residues in hC (Glu53) and hI (Arg287, Glu297, and Asp297) (239). A similar epitope, i.e., comprising residues in hB, hC, and the hI-s5A loop, was identified for nanobody VHH-2w-64 (Nb64). The crystallographic structures of the PAI-1/Nb64 complex later confirmed the crucial involvement of the latter loop (Table 1) (52). Based on structures of other serpin/serine proteinase complexes, the binding site of Nb64 in all probability overlaps with the position of the PA in the final inhibitory complex (Figure 1, Figure 5D). In contrast to the first subclass binding hF, MA-8H9D4 (246), and Nb64 (52) neither affected the formation of the initial PAI-1/PA complex, nor the kinetics of RCL insertion for the PAI-1/tPA reaction. It was therefore suggested that MA-8H9D4 and Nb64 interfere with the final step of inhibitory complex formation by hindering the PA to come close enough to the PAI-1 surface for PA distortion. Furthermore, strong functional additivity has been observed for the MA-33H1F7/MA-8H9D4 and MA-55F4C12/MA-8H9D4 antibody pairs which demonstrates that these mAbs bind structurally distinct epitopes and affect different steps of the PAI-1/PA reaction (246). Importantly, the effects of Mab2, MA-8H9D4, and Nb64 are potentiated by simultaneous binding of vitronectin to the opposite side of hF in PAI-1, which further increases the rigidity within this region (99). Another substrate-inducing nanobody VHH-s-a98 (Nb98) was suggested to bind a cavity aligned by hB and hC (Gln47, Glu53, and Gln55-Gln56-Gln57), the hF-s3A loop (Glu128-Val129-Glu130-Arg131 and Lys154), hI (Glu291 and Asn292), and the hI-s5A loop (Gln303 and Asp305). Since this region harbors binding sites for both subclasses of switching Abs (MA-33H1F7 and MA-55F4C12 vs. MA-8H9D4 and Nb64), the exact mechanism by which Nb98 induces substrate behavior remains unclear.
The third class of mAbs, including MA-35A5, MA-33B8, M5, and MA-H4B3, have the ability to accelerate the active to latent transition of PAI-1 and bind epitopes that are spread more across the PAI-1 surface (Figure 5B). The major determinants of the MA-33B8 epitope were simultaneously reported by two research groups, and are comprised in the same region that covers the turn connecting hD with s2A (Asn87, Lys88, and Asp89), the top of s3A (Gln174 and Lys176), the loop connecting s2B with s3B (His229, Gly230, and Thr232), and the loop connecting s5A with the RCL in the breach region (Asn329 and Ser331) (240, 241). Interestingly, this epitope is relatively less accessible in the active form of PAI-1 and undergoes a structural rearrangement to become more compact in the loop-inserted forms of PAI-1. Furthermore, since the putative epitope contains residues located on both sides of the RCL insertion site, i.e., on s5A and s3A, and MA-33B8 promotes loop insertion, the binding must occur to a prelatent form of PAI-1 in which the RCL is already partially inserted. Additional evidence for a prelatent form that can be accelerated into latency transition has been provided by the binding site and inhibitory mechanisms of M-5 and MA-H4B3. The dominant epitope residue for M-5 was mapped to Asp181 located directly below the RCL at the loop connecting s3A with s4C (247). Since this residue was shown to be more accessible in the latent form, it was hypothesized that M-5 displaces and forces insertion of the RCL into the central β-sheet A upon binding. The epitope of MA-H4B3 includes residues on s1B, s2B, and s3B (Tyr210, Glu212, and Tyr241) as well as s2C (Arg271) and thus partially overlaps with the epitope for MEDI-579 that prevents Michaelis complex formation (90). However, since the epitope of MA-H4B3 was shown to be occluded by s1C in active PAI-1, it was suggested that the epitope only becomes available upon s1C detachment during latency transition. Thus, by binding to a prelatent form that exists in equilibrium with active PAI-1, MA-H4B3 accelerates the rate of RCL insertion, resulting in an enhanced latency transition. Another mAb, MA-159M12, binds to the N-terminal part of hA (Pro2, Leu3, Pro4, and Glu5) and accelerates the active to latent transition in rat PAI-1 (242). However, MA-31C9 that targets a similar region in human PAI-1 (His3, Ser6, Tyr7, and His10) has been shown to be non-inhibitory. Furthermore, introduction of the epitope of MA-159M12 in human PAI-1 only caused MA-159M12 to bind PAI-1 with low affinity. This observation emphasized that two mAbs generated toward the same region in different orthologs can display very divergent functional effects, either caused by subtle structural differences between human and rat PAI-1 or by subtle differences in the binding mode of these mAbs (35, 235–238, 248).
Alternatively, single-chain variable fragments have been derived from several of the aforementioned mAbs (249–251). Since crystallization attempts with these scFvs and their full-size mAbs remained unsuccessful thus far, a mutagenesis approach was used to identify epitope (235, 239) and paratope (251, 252) residues involved in the interaction between PAI-1 and the scFvs. Subsequently, this information was used to drive protein-protein docking in order to predict the structures of the respective PAI-1/scFv complexes (253). Notably, due to the in vitro and in vivo potency and cross-reactivity toward rodent PAI-1, one scFv (scFv-33H1F7) was developed into a bispecific diabody format together with scFv-TCK26D6 that inhibits the antifibrinolytic enzyme thrombin activatable fibrinolysis inhibitor (TAFI) (254). Further in vivo evaluation and comparison with the standard thrombolytic therapy (tPA) showed that the simultaneous administration of MA-33H1F7 and MA-TCK26D6 or the use of diabody Db-TCK26D6x33H1F7 holds a great promise in both prophylaxis and treatment of thrombotic disease (255, 256).
Conclusions
Over the last four decades, the role of PAI-1 in various pathophysiological processes including cardiovascular disease has been extensively studied (257, 258). As the main physiological inhibitor of PAs, PAI-1 exerts an antifibrinolytic activity mainly by attenuating the plasmin-mediated fibrin degradation and thereby contributes to the pathogenesis of thrombotic cardiovascular diseases. Apart from being a regulator of the plasminogen activation system, PAI-1 has a pleiotropic biological function stemming from its ability to interact with ligands, such as the extracellular matrix protein vitronectin and cellular low-density lipoprotein receptors including LRP1. As a consequence, PAI-1 is also involved in the (patho)physiological processes associated with tissue remodeling, cell migration, and inflammation. Even though the precise role of PAI-1 in these diverse pathological processes is not always fully understood, elevated levels of PAI-1 have been shown to be related to the incidence, severity, and prognosis of various diseases. Therefore, significant clinical interest has been tied to PAI-1 as a putative drug target for the treatment of PAI-1-related pathologies. A very diverse collection of PAI-1 inhibitors has already been developed, including peptides, RNA aptamers, small organochemical molecules, antibodies, and antibody fragments. Even though several antagonists have been extensively characterized in vitro and in vivo, no PAI-1 inhibitors were approved to date for therapeutic use in humans. However, it should be noted that a few PAI-1 antagonists are currently proceeding through clinical trials, underscoring the persistent clinical interest in safe and efficient modulators of PAI-1 activity. The growing number of available structures from PAI-1 in complex with biological ligands and inhibitors may provide access to useful information for guiding the development of the continuously growing segment of PAI-1 antagonists.
Author Contributions
MS and PJD contributed to the conception and the design of the manuscript. MS did the literature research, wrote the first draft of the review, and generated the figures and tables. PJD reviewed the manuscript and provided the critical feedback. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
MS was a recipient of a Ph.D. fellowship Fundamental Research of the Research Foundation–Flanders (FWO), grant number 11ZQ918N.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Brzoska T, Suzuki Y, Sano H, Suzuki S, Tomczyk M, Tanaka H, et al. Imaging analyses of coagulation-dependent initiation of fibrinolysis on activated platelets and its modification by thrombin-activatable fibrinolysis inhibitor. Thromb Haemost. (2017) 117:682–90. doi: 10.1160/TH16-09-0722
2. Hoffman M, Monroe DM. A cell-based model of hemostasis. Thromb Haemost. (2001) 85:958–65. doi: 10.1055/s-0037-1615947
3. Monroe Dougald M, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol. (2006) 26:41–8. doi: 10.1161/01.ATV.0000193624.28251.83
4. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. (2015) 29:17–24. doi: 10.1016/j.blre.2014.09.003
5. Levin EG, Marzec U, Anderson J, Harker LA. Thrombin stimulates tissue plasminogen activator release from cultured human endothelial cells. J Clin Invest. (1984) 74:1988–95. doi: 10.1172/JCI111620
6. Oliver JJ, Webb DJ, Newby DE. Stimulated tissue plasminogen activator release as a marker of endothelial function in humans. Arterioscler Thromb Vasc Biol. (2005) 25:2470–9. doi: 10.1161/01.ATV.0000189309.05924.88
7. Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. (2005) 129:307–21. doi: 10.1111/j.1365-2141.2005.05444.x
8. Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost. (2009) 7:4–13. doi: 10.1111/j.1538-7836.2008.03220.x
9. Loskutoff DJ, van Mourik JA, Erickson LA, Lawrence D. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc Natl Acad Sci USA. (1983) 80:2956–60. doi: 10.1073/pnas.80.10.2956
10. Chmielewska J, Rånby M, Wiman B. Evidence for a rapid inhibitor to tissue plasminogen activator in plasma. Thromb Res. (1983) 31:427–36. doi: 10.1016/0049-3848(83)90407-3
11. Kruithof EK, Tran-Thang C, Ransijn A, Bachmann F. Demonstration of a fast-acting inhibitor of plasminogen activators in human plasma. Blood. (1984) 64:907–13. doi: 10.1182/blood.V64.4.907.907
12. Verheijen JH, Chang GT, Kluft C. Evidence for the occurrence of a fast-acting inhibitor for tissue-type plasminogen activator in human plasma. Thromb Haemost. (1984) 51:392–5. doi: 10.1055/s-0038-1661109
13. Simpson AJ, Booth NA, Moore NR, Bennett B. Distribution of plasminogen activator inhibitor (PAI-1) in tissues. J Clin Pathol. (1991) 44:139. doi: 10.1136/jcp.44.2.139
14. Crandall DL, Quinet EM, Morgan GA, Busler DE, McHendry-Rinde B, Kral JG. Synthesis and secretion of plasminogen activator inhibitor-1 by human preadipocytes. J Clin Endocrinol Metab. (1999) 84:3222–7. doi: 10.1210/jcem.84.9.5987
15. Rabieian R, Boshtam M, Zareei M, Kouhpayeh S, Masoudifar A, Mirzaei H. Plasminogen activator inhibitor type-1 as a regulator of fibrosis. J Cell Biochem. (2018) 119:17–27. doi: 10.1002/jcb.26146
16. Yamamoto K, Takeshita K, Shimokawa T, Yi H, Isobe K-i, Loskutoff DJ, et al. Plasminogen activator inhibitor-1 is a major stress-regulated gene: Implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci USA. (2002) 99:890. doi: 10.1073/pnas.022608799
17. Booth NA, Simpson AJ, Croll A, Bennett B, MacGregor IR. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. (1988) 70:327–33. doi: 10.1111/j.1365-2141.1988.tb02490.x
18. Lutsey PL, Cushman M, Steffen LM, Green D, Barr RG, Herrington D, et al. Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: the MESA study. J Thromb Haemost. (2006) 4:2629–35. doi: 10.1111/j.1538-7836.2006.02237.x
19. Asselbergs FW, Williams SM, Hebert PR, Coffey CS, Hillege HL, Navis G, et al. Gender-specific correlations of plasminogen activator inhibitor-1 and tissue plasminogen activator levels with cardiovascular disease-related traits. J Thromb Haemost. (2007) 5:313–20. doi: 10.1111/j.1538-7836.2007.02311.x
20. Sundell IB, Nilsson TK, Rånby M, Hallmans G, Hellsten G. Fibrinolytic variables are related to age, sex, blood pressure, and body build measurements: a cross-sectional study in Norsjö, Sweden. J Clin Epidemiol. (1989) 42:719–23. doi: 10.1016/0895-4356(89)90067-X
21. Booth NA, Croll A, Bennett B. The activity of plasminogen activator inhibitor-1 (PAI-1) of human platelet. Fibrinolysis. (1990) 4:138–40. doi: 10.1016/0268-9499(90)90391-V
22. Declerck PJ, Alessi MC, Verstreken M, Kruithof EK, Juhan-Vague I, Collen D. Measurement of plasminogen activator inhibitor 1 in biologic fluids with a murine monoclonal antibody-based enzyme-linked immunosorbent assay. Blood. (1988) 71:220–5. doi: 10.1182/blood.V71.1.220.220
23. Brogren H, Wallmark K, Deinum J, Karlsson L, Jern S. Platelets retain high levels of active plasminogen activator inhibitor 1. PLoS ONE. (2011) 6:e26762. doi: 10.1371/journal.pone.0026762
24. Morrow GB, Whyte CS, Mutch NJ. Functional plasminogen activator inhibitor 1 is retained on the activated platelet membrane following platelet activation. Haematologica. (2019) 105:2824–33. doi: 10.3324/haematol.2019.230367
25. Torr-Brown SR, Sobel BE. Attenuation of thrombolysis by release of plasminogen activator inhibitor type-1 from platelets. Thromb Res. (1993) 72:413–21. doi: 10.1016/0049-3848(93)90241-F
26. Stringer HA, van Swieten P, Heijnen HF, Sixma JJ, Pannekoek H. Plasminogen activator inhibitor-1 released from activated platelets plays a key role in thrombolysis resistance. Studies with thrombi generated in the Chandler loop. Arterioscler Thromb. (1994) 14:1452–8. doi: 10.1161/01.ATV.14.9.1452
27. Strandberg L, Lawrence D, Ny T. The organization of the human-plasminogen-activator-inhibitor-1 gene. Implications on the evolution of the serine-protease inhibitor family. Eur J Biochem. (1988) 176:609–16. doi: 10.1111/j.1432-1033.1988.tb14320.x
28. Klinger KW, Winqvist R, Riccio A, Andreasen PA, Sartorio R, Nielsen LS, et al. Plasminogen activator inhibitor type 1 gene is located at region q21.3-q22 of chromosome 7 and genetically linked with cystic fibrosis. Proc Natl Acad Sci USA. (1987) 84:8548–52. doi: 10.1073/pnas.84.23.8548
29. Bosma PJ, van den Berg EA, Kooistra T, Siemieniak DR, Slightom JL. Human plasminogen activator inhibitor-1 gene. Promoter and structural gene nucleotide sequences. J Biol Chem. (1988) 263:9129–41.
30. Andreasen PA, Riccio A, Welinder KG, Douglas R, Sartorio R, Nielsen LS, et al. Plasminogen activator inhibitor type-1: reactive center and amino-terminal heterogeneity determined by protein and cDNA sequencing. FEBS Lett. (1986) 209:213–8. doi: 10.1016/0014-5793(86)81113-9
31. Gils A, Pedersen KE, Skottrup P, Christensen A, Naessens D, Deinum J, et al. Biochemical importance of glycosylation of plasminogen activator inhibitor-1. Thromb Haemost. (2003) 90:206–17. doi: 10.1160/TH03-01-0034
32. Brogren H, Sihlbom C, Wallmark K, Lönn M, Deinum J, Karlsson L, et al. Heterogeneous glycosylation patterns of human PAI-1 may reveal its cellular origin. Thromb Res. (2008) 122:271–81. doi: 10.1016/j.thromres.2008.04.008
33. Lawrence D, Strandberg L, Grundström T, Ny T. Purification of active human plasminogen activator inhibitor 1 from Escherichia coli. Comparison with natural and recombinant forms purified from eucaryotic cells. Eur J Biochem. (1989) 186:523–33. doi: 10.1111/j.1432-1033.1989.tb15238.x
34. Van De Craen B, Scroyen I, Vranckx C, Compernolle G, Lijnen HR, Declerck PJ, et al. Maximal PAI-1 inhibition in vivo requires neutralizing antibodies that recognize and inhibit glycosylated PAI-1. Thromb Res. (2012) 129:e126–33. doi: 10.1016/j.thromres.2011.11.038
35. Zhou X, Hendrickx ML, Hassanzadeh-Ghassabeh G, Muyldermans S, Declerck PJ. Generation and in vitro characterisation of inhibitory nanobodies towards plasminogen activator inhibitor 1. Thromb Haemost. (2016) 116:1032–40. doi: 10.1160/TH16-04-0306
36. Heit C, Jackson BC, McAndrews M, Wright MW, Thompson DC, Silverman GA, et al. Update of the human and mouse SERPIN gene superfamily. Hum Genomics. (2013) 7:22. doi: 10.1186/1479-7364-7-22
37. Pemberton PA, Stein PE, Pepys MB, Potter JM, Carrell RW. Hormone binding globulins undergo serpin conformational change in inflammation. Nature. (1988) 336:257–8. doi: 10.1038/336257a0
38. Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S, et al. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. (1994) 263:526–9. doi: 10.1126/science.8290962
39. Nagata K. Hsp47: a collagen-specific molecular chaperone. Trends Biochem Sci. (1996) 21:22–6. doi: 10.1016/S0968-0004(06)80023-X
40. Law RHP, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. (2006) 7:216. doi: 10.1186/gb-2006-7-5-216
41. Sillen M, Weeks SD, Strelkov SV, Declerck PJ. Structural insights into the mechanism of a nanobody that stabilizes PAI-1 and modulates its activity. Int J Mol Sci. (2020) 21:5859. doi: 10.3390/ijms21165859
42. Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. (2000) 10:1845–64. doi: 10.1101/gr.GR-1478R
43. Gettins PGW, Olson ST. Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochemical J. (2016) 473:2273–93. doi: 10.1042/BCJ20160014
44. Thorsen S, Philips M, Selmer J, Lecander I, Astedt B. Kinetics of inhibition of tissue-type and urokinase-type plasminogen activator by plasminogen-activator inhibitor type 1 and type 2. Eur J Biochem. (1988) 175:33–9. doi: 10.1111/j.1432-1033.1988.tb14162.x
45. Stout TJ, Graham H, Buckley DI, Matthews DJ. Structures of active and latent PAI-1: a possible stabilizing role for chloride ions. Biochemistry. (2000) 39:8460–9. doi: 10.1021/bi000290w
46. Nar H, Bauer M, Stassen JM, Lang D, Gils A, Declerck PJ. Plasminogen activator inhibitor 1. Structure of the native serpin, comparison to its other conformers and implications for serpin inactivation. J Mol Biol. (2000) 297:683–95. doi: 10.1006/jmbi.2000.3604
47. Gong L, Liu M, Zeng T, Shi X, Yuan C, Andreasen PA, et al. Crystal structure of the Michaelis complex between tissue-type plasminogen activator and plasminogen activators inhibitor-1. J Biol Chem. (2015) 290:25795–804. doi: 10.1074/jbc.M115.677567
48. Dewilde M, Strelkov SV, Rabijns A, Declerck PJ. High quality structure of cleaved PAI-1-stab. J Struct Biol. (2009) 165:126–32. doi: 10.1016/j.jsb.2008.11.001
49. Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. (2000) 407:923–6. doi: 10.1038/35038119
50. Katona G, Berglund GI, Hajdu J, Gráf L, Szilágyi L. Crystal structure reveals basis for the inhibitor resistance of human brain trypsin. J Mol Biol. (2002) 315:1209–18. doi: 10.1006/jmbi.2001.5305
51. Zhou A, Huntington JA, Pannu NS, Carrell RW, Read RJ. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol. (2003) 10:541–4. doi: 10.1038/nsb943
52. Sillen M, Weeks SD, Zhou X, Komissarov AA, Florova G, Idell S, et al. Molecular mechanism of two nanobodies that inhibit PAI-1 activity reveals a modulation at distinct stages of the PAI-1/plasminogen activator interaction. J Thromb Haemost. (2020) 18:681–92. doi: 10.1111/jth.14716
53. Lin Z, Jiang L, Yuan C, Jensen JK, Zhang X, Luo Z, et al. Structural basis for recognition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1. J Biol Chem. (2011) 286:7027–32. doi: 10.1074/jbc.M110.204537
54. Lawrence DA, Ginsburg D, Day DE, Berkenpas MB, Verhamme IM, Kvassman JO, et al. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J Biol Chem. (1995) 270:25309–12. doi: 10.1074/jbc.270.43.25309
55. Perron MJ, Blouse GE, Shore JD. Distortion of the catalytic domain of tissue-type plasminogen activator by plasminogen activator inhibitor-1 coincides with the formation of stable serpin-proteinase complexes. J Biol Chem. (2003) 278:48197–203. doi: 10.1074/jbc.M306184200
56. Peterson FC, Gettins PG. Insight into the mechanism of serpin-proteinase inhibition from 2D [1H-15N] NMR studies of the 69 kDa alpha 1-proteinase inhibitor Pittsburgh-trypsin covalent complex. Biochemistry. (2001) 40:6284–92. doi: 10.1021/bi010100x
57. Kaslik G, Kardos J, Szabó E, Szilágyi L, Závodszky P, Westler WM, et al. Effects of serpin binding on the target proteinase: global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry. (1997) 36:5455–64. doi: 10.1021/bi962931m
58. Bousquet JA, Duranton J, Mély Y, Bieth JG. Conformational change in elastase following complexation with alpha1-proteinase inhibitor: a CD investigation. Biochem J. (2003) 370:345–9. doi: 10.1042/bj20020890
59. Gettins PGW. The F-helix of serpins plays an essential, active role in the proteinase inhibition mechanism. FEBS Lett. (2002) 523:2–6. doi: 10.1016/S0014-5793(02)02924-1
60. Horn IR, van den Berg BM, Moestrup SK, Pannekoek H, van Zonneveld AJ. Plasminogen activator inhibitor 1 contains a cryptic high affinity receptor binding site that is exposed upon complex formation with tissue-type plasminogen activator. Thromb Haemost. (1998) 80:822–8. doi: 10.1055/s-0037-1615365
61. Stefansson S, Muhammad S, Cheng XF, Battey FD, Strickland DK, Lawrence DA. Plasminogen activator inhibitor-1 contains a cryptic high affinity binding site for the low density lipoprotein receptor-related protein. J Biol Chem. (1998) 273:6358–66. doi: 10.1074/jbc.273.11.6358
62. Aertgeerts K, De Bondt HL, De Ranter CJ, Declerck PJ. Mechanisms contributing to the conformational and functional flexibility of plasminogen activator inhibitor-1. Nat Struct Biol. (1995) 2:891–7. doi: 10.1038/nsb1095-891
63. Declerck PJ, De Mol M, Vaughan DE, Collen D. Identification of a conformationally distinct form of plasminogen activator inhibitor-1, acting as a noninhibitory substrate for tissue-type plasminogen activator. J Biol Chem. (1992) 267:11693–6.
64. Urano T, Strandberg L, Johansson LB, Ny T. A substrate-like form of plasminogen-activator-inhibitor type 1. Conversions between different forms by sodium dodecyl sulphate. Eur J Biochem. (1992) 209:985–92. doi: 10.1111/j.1432-1033.1992.tb17372.x
65. Audenaert AM, Knockaert I, Collen D, Declerck PJ. Conversion of plasminogen activator inhibitor-1 from inhibitor to substrate by point mutations in the reactive-site loop. J Biol Chem. (1994) 269:19559–64.
66. van Meijer M, Smilde A, Tans G, Nesheim ME, Pannekoek H, Horrevoets AJ. The suicide substrate reaction between plasminogen activator inhibitor 1 and thrombin is regulated by the cofactors vitronectin and heparin. Blood. (1997) 90:1874–82. doi: 10.1182/blood.V90.5.1874
67. Ehrlich HJ, Gebbink RK, Keijer J, Linders M, Preissner KT, Pannekoek H. Alteration of serpin specificity by a protein cofactor. Vitronectin endows plasminogen activator inhibitor 1 with thrombin inhibitory properties. J Biol Chem. (1990) 265:13029–35.
68. Lawrence DA, Strandberg L, Ericson J, Ny T. Structure-function studies of the SERPIN plasminogen activator inhibitor type 1. Analysis of chimeric strained loop mutants. J Biol Chem. (1990) 265:20293–301.
69. Tachias K, Madison EL. Variants of tissue-type plasminogen activator that display extraordinary resistance to inhibition by the serpin plasminogen activator inhibitor type 1. J Biol Chem. (1997) 272:14580–5. doi: 10.1074/jbc.272.23.14580
70. Florova G, Azghani AO, Karandashova S, Schaefer C, Yarovoi SV, Declerck PJ, et al. Targeting plasminogen activator inhibitor-1 in tetracycline-induced pleural injury in rabbits. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L54–L68. doi: 10.1152/ajplung.00579.2016
71. Petersen HH, Hansen M, Schousboe SL, Andreasen PA. Localization of epitopes for monoclonal antibodies to urokinase-type plasminogen activator: relationship between epitope localization and effects of antibodies on molecular interactions of the enzyme. Eur J Biochem. (2001) 268:4430–9. doi: 10.1046/j.1432-1327.2001.02365.x
72. Qureshi T, Goswami S, McClintock CS, Ramsey MT, Peterson CB. Distinct encounter complexes of PAI-1 with plasminogen activators and vitronectin revealed by changes in the conformation and dynamics of the reactive center loop. Protein Sci. (2016) 25:499–510. doi: 10.1002/pro.2841
73. Gils A, Knockaert I, Declerck PJ. Substrate behavior of plasminogen activator inhibitor-1 is not associated with a lack of insertion of the reactive site loop. Biochemistry. (1996) 35:7474–81. doi: 10.1021/bi960079d
74. Gils A, Vleugels N, Tobback K, Knockaert I, Declerck PJ. Characterization of plasminogen activator inhibitor 1 mutants containing the P13 to P10 region of ovalbumin or antithrombin III: evidence that the P13 residue contributes significantly to the active to substrate transition. Biochim Biophys Acta. (1998) 1387:291–7. doi: 10.1016/S0167-4838(98)00139-3
75. Vleugels N, Gils A, Bijnens AP, Knockaert I, Declerck PJ. The importance of helix F in plasminogen activator inhibitor-1. Biochim Biophys Acta. (2000) 1476:20–6. doi: 10.1016/S0167-4838(99)00224-1
76. Lawrence DA, Olson ST, Muhammad S, Day DE, Kvassman JO, Ginsburg D, et al. Partitioning of serpin-proteinase reactions between stable inhibition and substrate cleavage is regulated by the rate of serpin reactive center loop insertion into beta-sheet A. J Biol Chem. (2000) 275:5839–44. doi: 10.1074/jbc.275.8.5839
77. Gils A, Declerck PJ. Modulation of plasminogen activator inhibitor 1 by Triton X-100–identification of two consecutive conformational transitions. Thromb Haemost. (1998) 80:286–91. doi: 10.1055/s-0037-1615189
78. Andreasen PA, Egelund R, Jensen S, Rodenburg KW. Solvent effects on activity and conformation of plasminogen activator inhibitor-1. Thromb Haemost. (1999) 81:407–14. doi: 10.1055/s-0037-1614487
79. Kjøller L, Martensen PM, Sottrup-Jensen L, Justesen J, Rodenburg KW, Andreasen PA. Conformational changes of the reactive-centre loop and beta-strand 5A accompany temperature-dependent inhibitor-substrate transition of plasminogen-activator inhibitor 1. Eur J Biochem. (1996) 241:38–46. doi: 10.1111/j.1432-1033.1996.0038t.x
80. Mottonen J, Strand A, Symersky J, Sweet RM, Danley DE, Geoghegan KF, et al. Structural basis of latency in plasminogen activator inhibitor-1. Nature. (1992) 355:270–3. doi: 10.1038/355270a0
81. Ricagno S, Pezzullo M, Barbiroli A, Manno M, Levantino M, Santangelo MG, et al. Two latent and two hyperstable polymeric forms of human neuroserpin. Biophysical J. (2010) 99:3402–11. doi: 10.1016/j.bpj.2010.09.021
82. Lomas DA, Elliott PR, Chang WS, Wardell MR, Carrell RW. Preparation and characterization of latent alpha 1-antitrypsin. J Biol Chem. (1995) 270:5282–8. doi: 10.1074/jbc.270.10.5282
83. Chang WS, Lomas DA. Latent alpha1-antichymotrypsin. A molecular explanation for the inactivation of alpha1-antichymotrypsin in chronic bronchitis and emphysema. J Biol Chem. (1998) 273:3695–701. doi: 10.1074/jbc.273.6.3695
84. Jendroszek A, Sønnichsen MS, Muñoz AC, Leyman K, Christensen A, Petersen SV, et al. Latency transition of plasminogen activator inhibitor type 1 is evolutionarily conserved. Thromb Haemost. (2017) 117:1688–99. doi: 10.1160/TH17-02-0102
85. Stoop AA, Eldering E, Dafforn TR, Read RJ, Pannekoek H. Different structural requirements for plasminogen activator inhibitor 1 (PAI-1) during latency transition and proteinase inhibition as evidenced by phage-displayed hypermutated PAI-1 libraries. J Mol Biol. (2001) 305:773–83. doi: 10.1006/jmbi.2000.4356
86. Krüger P, Verheyden S, Declerck PJ, Engelborghs Y. Extending the capabilities of targeted molecular dynamics: simulation of a large conformational transition in plasminogen activator inhibitor 1. Protein Sci. (2001) 10:798–808. doi: 10.1110/ps.40401
87. Cazzolli G, Wang F, a Beccara S, Gershenson A, Faccioli P, Wintrode PL. Serpin latency transition at atomic resolution. Proc Natl Acad Sci USA. (2014) 111:15414–9. doi: 10.1073/pnas.1407528111
88. Gorlatova NV, Cale JM, Elokdah H, Li D, Fan K, Warnock M, et al. Mechanism of inactivation of plasminogen activator inhibitor-1 by a small molecule inhibitor. J Biol Chem. (2007) 282:9288–96. doi: 10.1074/jbc.M611642200
89. Verhamme I, Kvassman JO, Day D, Debrock S, Vleugels N, Declerck PJ, et al. Accelerated conversion of human plasminogen activator inhibitor-1 to its latent form by antibody binding. J Biol Chem. (1999) 274:17511–7. doi: 10.1074/jbc.274.25.17511
90. Dupont DM, Blouse GE, Hansen M, Mathiasen L, Kjelgaard S, Jensen JK, et al. Evidence for a pre-latent form of the serpin plasminogen activator inhibitor-1 with a detached beta-strand 1C. J Biol Chem. (2006) 281:36071–81. doi: 10.1074/jbc.M606851200
91. Hägglöf P, Bergström F, Wilczynska M, Johansson LBÅ, Ny T. The reactive-center loop of active PAI-1 is folded close to the protein core and can be partially inserted. J Mol Biol. (2004) 335:823–32. doi: 10.1016/j.jmb.2003.11.005
92. Boudier C, Gils A, Declerck PJ, Bieth JG. The conversion of active to latent plasminogen activator inhibitor-1 is an energetically silent event. Biophys J. (2005) 88:2848–54. doi: 10.1529/biophysj.104.053306
93. Vaughan DE, Declerck PJ, Van Houtte E, De Mol M, Collen D. Studies of recombinant plasminogen activator inhibitor-1 in rabbits. Pharmacokinetics and evidence for reactivation of latent plasminogen activator inhibitor-1 in vivo. Circ Res. (1990) 67:1281–6. doi: 10.1161/01.RES.67.5.1281
94. Hekman CM, Loskutoff DJ. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J Biol Chem. (1985) 260:11581–7.
95. Lawrence DA, Palaniappan S, Stefansson S, Olson ST, Francis-Chmura AM, Shore JD, et al. Characterization of the binding of different conformational forms of plasminogen activator inhibitor-1 to vitronectin. Implications for the regulation of pericellular proteolysis. J Biol Chem. (1997) 272:7676–80. doi: 10.1074/jbc.272.12.7676
96. Declerck PJ, De Mol M, Alessi MC, Baudner S, Pâques EP, Preissner KT, et al. Purification and characterization of a plasminogen activator inhibitor 1 binding protein from human plasma. Identification as a multimeric form of S protein (vitronectin). J Biol Chem. (1988) 263:15454–61.
97. Preissner KT, Grulich-Henn J, Ehrlich HJ, Declerck P, Justus C, Collen D, et al. Structural requirements for the extracellular interaction of plasminogen activator inhibitor 1 with endothelial cell matrix-associated vitronectin. J Biol Chem. (1990) 265:18490–8.
98. Lindahl TL, Sigurdardottir O, Wiman B. Stability of plasminogen activator inhibitor 1 (PAI-1). Thromb Haemost. (1989) 62:748–51. doi: 10.1055/s-0038-1646895
99. Komissarov AA, Andreasen PA, Bødker JS, Declerck PJ, Anagli JY, Shore JD. Additivity in effects of vitronectin and monoclonal antibodies against alpha-helix F of plasminogen activator inhibitor-1 on its reactions with target proteinases. J Biol Chem. (2005) 280:1482–9. doi: 10.1074/jbc.M408608200
100. Seiffert D, Ciambrone G, Wagner NV, Binder BR, Loskutoff DJ. The somatomedin B domain of vitronectin. Structural requirements for the binding and stabilization of active type 1 plasminogen activator inhibitor. J Biol Chem. (1994) 269:2659–66.
101. Sigurdardottir O, Wiman B. Identification of a PAI-1 binding site in vitronectin. Biochim Biophys Acta. (1994) 1208:104–10. doi: 10.1016/0167-4838(94)90166-X
102. Deng G, Royle G, Wang S, Crain K, Loskutoff DJ. Structural and functional analysis of the plasminogen activator inhibitor-1 binding motif in the somatomedin B domain of vitronectin. J Biol Chem. (1996) 271:12716–23. doi: 10.1074/jbc.271.22.12716
103. Royle G, Deng G, Seiffert D, Loskutoff DJ. A method for defining binding sites involved in protein-protein interactions: analysis of the binding of plasminogen activator inhibitor 1 to the somatomedin domain of vitronectin. Anal Biochem. (2001) 296:245–53. doi: 10.1006/abio.2001.5316
104. Lawrence DA, Berkenpas MB, Palaniappan S, Ginsburg D. Localization of vitronectin binding domain in plasminogen activator inhibitor-1. J Biol Chem. (1994) 269:15223–8.
105. van Meijer M, Gebbink RK, Preissner KT, Pannekoek H. Determination of the vitronectin binding site on plasminogen activator inhibitor 1 (PAI-1). FEBS Lett. (1994) 352:342–6. doi: 10.1016/0014-5793(94)00990-2
106. Padmanabhan J, Sane DC. Localization of a vitronectin binding region of plasminogen activator inhibitor-1. Thromb Haemost. (1995) 73:829–34. doi: 10.1055/s-0038-1653876
107. Jensen JK, Wind T, Andreasen PA. The vitronectin binding area of plasminogen activator inhibitor-1, mapped by mutagenesis and protection against an inactivating organochemical ligand. FEBS Lett. (2002) 521:91–4. doi: 10.1016/S0014-5793(02)02830-2
108. Jensen JK, Durand MK, Skeldal S, Dupont DM, Bødker JS, Wind T, et al. Construction of a plasminogen activator inhibitor-1 variant without measurable affinity to vitronectin but otherwise normal. FEBS Lett. (2004) 556:175–9. doi: 10.1016/S0014-5793(03)01405-4
109. Trelle MB, Hirschberg D, Jansson A, Ploug M, Roepstorff P, Andreasen PA, et al. Hydrogen/deuterium exchange mass spectrometry reveals specific changes in the local flexibility of plasminogen activator inhibitor 1 upon binding to the somatomedin B domain of vitronectin. Biochemistry. (2012) 51:8256–66. doi: 10.1021/bi3008998
110. Fa M, Karolin J, Aleshkov S, Strandberg L, Johansson LB, Ny T. Time-resolved polarized fluorescence spectroscopy studies of plasminogen activator inhibitor type 1: conformational changes of the reactive center upon interactions with target proteases, vitronectin and heparin. Biochemistry. (1995) 34:13833–40. doi: 10.1021/bi00042a015
111. Gechtman Z, Sharma R, Kreizman T, Fridkin M, Shaltiel S. Synthetic peptides derived from the sequence around the plasmin cleavage site in vitronectin. Use in mapping the PAI-1 binding site. FEBS Lett. (1993) 315:293–7. doi: 10.1016/0014-5793(93)81181-X
112. Gechtman Z, Belleli A, Lechpammer S, Shaltiel S. The cluster of basic amino acids in vitronectin contributes to its binding of plasminogen activator inhibitor-1: evidence from thrombin-, elastase- and plasmin-cleaved vitronectins and anti-peptide antibodies. Biochem J. (1997) 325:339–49. doi: 10.1042/bj3250339
113. Chain D, Kreizman T, Shapira H, Shaltiel S. Plasmin cleavage of vitronectin Identification of the site and consequent attenuation in binding plasminogen activator inhibitor-1. FEBS Lett. (1991) 285:251–6. doi: 10.1016/0014-5793(91)80810-P
114. Schar CR, Blouse GE, Minor KH, Peterson CB. A deletion mutant of vitronectin lacking the somatomedin B domain exhibits residual plasminogen activator inhibitor-1-binding activity. J Biol Chem. (2008) 283:10297–309. doi: 10.1074/jbc.M708017200
115. Schar CR, Jensen JK, Christensen A, Blouse GE, Andreasen PA, Peterson CB. Characterization of a site on PAI-1 that binds to vitronectin outside of the somatomedin B domain. J Biol Chem. (2008) 283:28487–96. doi: 10.1074/jbc.M804257200
116. Mimuro J, Muramatsu S, Kurano Y, Uchida Y, Ikadai H, Watanabe S, et al. Identification of the plasminogen activator inhibitor-1 binding heptapeptide in vitronectin. Biochemistry. (1993) 32:2314–20. doi: 10.1021/bi00060a024
117. Podor TJ, Shaughnessy SG, Blackburn MN, Peterson CB. New insights into the size and stoichiometry of the plasminogen activator inhibitor type-1.vitronectin complex. J Biol Chem. (2000) 275:25402–10. doi: 10.1074/jbc.M000362200
118. Smolarczyk K, Gils A, Boncela J, Declerck PJ, Cierniewski CS. Function-stabilizing mechanism of plasminogen activator inhibitor type 1 induced upon binding to alpha1-acid glycoprotein. Biochemistry. (2005) 44:12384–90. doi: 10.1021/bi050690p
119. Boncela J, Papiewska I, Fijalkowska I, Walkowiak B, Cierniewski CS. Acute phase protein α1-acid glycoprotein interacts with plasminogen activator inhibitor type 1 and stabilizes its inhibitory activity. J Biol Chem. (2001) 276:35305–11. doi: 10.1074/jbc.M104028200
120. De Taeye B, Gils A, Declerck PJ. The story of the serpin plasminogen activator inhibitor 1: is there any need for another mutant? Thromb Haemost. (2004) 92:898–924. doi: 10.1160/TH04-05-0269
121. Chorostowska-Wynimko J, Swiercz R, Skrzypczak-Jankun E, Wojtowicz A, Selman SH, Jankun J. A novel form of the plasminogen activator inhibitor created by cysteine mutations extends its half-life: relevance to cancer and angiogenesis. Mol Cancer Ther. (2003) 2:19.
122. Jankun J, Yang J, Zheng H, Han FQ, Al-Senaidy A, Skrzypczak-Jankun E. Remarkable extension of PAI-1 half-life surprisingly brings no changes to its structure. Int J Mol Med. (2012) 29:61–4. doi: 10.3892/ijmm.2011.798
123. Sharp AM, Stein PE, Pannu NS, Carrell RW, Berkenpas MB, Ginsburg D, et al. The active conformation of plasminogen activator inhibitor 1, a target for drugs to control fibrinolysis and cell adhesion. Structure. (1999) 7:111–8. doi: 10.1016/S0969-2126(99)80018-5
124. Jensen JK, Thompson LC, Bucci JC, Nissen P, Gettins PG, Peterson CB, et al. Crystal structure of plasminogen activator inhibitor-1 in an active conformation with normal thermodynamic stability. J Biol Chem. (2011) 286:29709–17. doi: 10.1074/jbc.M111.236554
125. Tucker HM, Mottonen J, Goldsmith EJ, Gerard RD. Engineering of plasminogen activator inhibitor-1 to reduce the rate of latency transition. Nat Struct Biol. (1995) 2:442–5. doi: 10.1038/nsb0695-442
126. Jensen JK, Gettins PGW. High-resolution structure of the stable plasminogen activator inhibitor type-1 variant 14–1B in its proteinase-cleaved form: a new tool for detailed interaction studies and modeling. Protein Sci. (2008) 17:1844–9. doi: 10.1110/ps.036707.108
127. Xue Y, Björquist P, Inghardt T, Linschoten M, Musil D, Sjölin L, et al. Interfering with the inhibitory mechanism of serpins: crystal structure of a complex formed between cleaved plasminogen activator inhibitor type 1 and a reactive-centre loop peptide. Structure. (1998) 6:627–36. doi: 10.1016/S0969-2126(98)00064-1
128. Fjellström O, Deinum J, Sjögren T, Johansson C, Geschwindner S, Nerme V, et al. Characterization of a small molecule inhibitor of plasminogen activator inhibitor type 1 that accelerates the transition into the latent conformation. J Biol Chem. (2013) 288:873–85. doi: 10.1074/jbc.M112.371732
129. Lin Z, Jensen JK, Hong Z, Shi X, Hu L, Andreasen PA, et al. Structural insight into inactivation of plasminogen activator inhibitor-1 by a small-molecule antagonist. Chem Biol. (2013) 20:253–61. doi: 10.1016/j.chembiol.2013.01.002
130. Hong Z-B, Lin Z-H, Li-Hu G, Ming-Dong H. Crystal structure of PAI-1 in complex with gallate. Chinese J Struct Chem. (2013) 32:1005–12. doi: 10.2210/pdb4ic0/pdb
131. Li SH, Reinke AA, Sanders KL, Emal CD, Whisstock JC, Stuckey JA, et al. Mechanistic characterization and crystal structure of a small molecule inactivator bound to plasminogen activator inhibitor-1. Proc Natl Acad Sci USA. (2013) 110:E4941–9. doi: 10.1073/pnas.1216499110
132. Vousden KA, Lundqvist T, Popovic B, Naiman B, Carruthers AM, Newton P, et al. Discovery and characterisation of an antibody that selectively modulates the inhibitory activity of plasminogen activator inhibitor-1. Sci Rep. (2019) 9:1605. doi: 10.1038/s41598-019-38842-x
133. Berkenpas MB, Lawrence DA, Ginsburg D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. (1995) 14:2969–77. doi: 10.1002/j.1460-2075.1995.tb07299.x
134. Vleugels N, Gils A, Mannaerts S, Knockaert I, Declerck PJ. Evaluation of the mechanism of inactivation of plasminogen activator inhibitor-1 by monoclonal antibodies using a stable variant. Fibrinol Proteol. (1998) 12:277–82. doi: 10.1016/S0268-9499(98)80020-9
135. Blouse GE, Perron MJ, Kvassman J-O, Yunus S, Thompson JH, Betts RL, et al. Mutation of the highly conserved tryptophan in the serpin breach region alters the inhibitory mechanism of plasminogen activator inhibitor-1. Biochemistry. (2003) 42:12260–72. doi: 10.1021/bi034737n
136. Kvassman J-O, Lawrence DA, Shore JD. The acid stabilization of plasminogen activator inhibitor-1 depends on protonation of a single group that affects loop insertion into β-sheet A. J Biol Chem. (1995) 270:27942–7. doi: 10.1074/jbc.270.46.27942
137. Mångs H, Sui G-C, Wiman B. PAI-1 stability: the role of histidine residues. FEBS Lett. (2000) 475:192–6. doi: 10.1016/S0014-5793(00)01656-2
138. Thompson LC, Goswami S, Ginsberg DS, Day DE, Verhamme IM, Peterson CB. Metals affect the structure and activity of human plasminogen activator inhibitor-1. I. Modulation of stability and protease inhibition. Protein Sci. (2011) 20:353–65. doi: 10.1002/pro.568
139. Thompson LC, Goswami S, Peterson CB. Metals affect the structure and activity of human plasminogen activator inhibitor-1. II. Binding affinity and conformational changes. Protein Sci. (2011) 20:366–78. doi: 10.1002/pro.567
140. Bucci JC, McClintock CS, Chu Y, Ware GL, McConnell KD, Emerson JP, et al. Resolving distinct molecular origins for copper effects on PAI-1. J Biol Inorg Chem. (2017) 22:1123–35. doi: 10.1007/s00775-017-1489-5
141. Bucci JC, Trelle MB, McClintock CS, Qureshi T, Jørgensen TJ, Peterson CB. Copper(II) ions increase plasminogen activator inhibitor type 1 dynamics in key structural regions that govern stability. Biochemistry. (2016) 55:4386–98. doi: 10.1021/acs.biochem.6b00256
142. Keijer J, Linders M, Ehrlich H, Gebbink RK, Pannekoek H. Stabilisation of plasminogen activator inhibitor type 1 (PAI-1) activity by arginine: possible implications for the interaction of PAI-1 with vitronectin. Fibrinolysis. (1990) 4:153–9. doi: 10.1016/S0268-9499(05)80047-5
143. Mimuro J, Schleef RR, Loskutoff DJ. Extracellular matrix of cultured bovine aortic endothelial cells contains functionally active type 1 plasminogen activator inhibitor. Blood. (1987) 70:721–8. doi: 10.1182/blood.V70.3.721.721
144. Aisina RB, Mukhametova LI. Structure and function of plasminogen/plasmin system. Russ J Bioorg Chem. (2014) 40:590–605. doi: 10.1134/S1068162014060028
145. Schleef RR, Higgins DL, Pillemer E, Levitt LJ. Bleeding diathesis due to decreased functional activity of type 1 plasminogen activator inhibitor. J Clin Invest. (1989) 83:1747–52. doi: 10.1172/JCI114076
146. Lee MH, Vosburgh E, Anderson K, McDonagh J. Deficiency of plasma plasminogen activator inhibitor 1 results in hyperfibrinolytic bleeding. Blood. (1993) 81:2357–62. doi: 10.1182/blood.V81.9.2357.2357
147. Dieval J, Nguyen G, Gross S, Delobel J, Kruithof EK. A lifelong bleeding disorder associated with a deficiency of plasminogen activator inhibitor type 1. Blood. (1991) 77:528–32. doi: 10.1182/blood.V77.3.528.528
148. Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. (1997) 90:204–8. doi: 10.1182/blood.V90.1.204
149. Heiman M, Gupta S, Shapiro AD. The obstetric, gynaecological and fertility implications of homozygous PAI-1 deficiency: single-centre experience. Haemophilia. (2014) 20:407–12. doi: 10.1111/hae.12313
150. Iwaki T, Nagahashi K, Kobayashi T, Umemura K, Terao T, Kanayama N. The first report of uncontrollable subchorionic and retroplacental haemorrhage inducing preterm labour in complete PAI-1 deficiency in a human. Thromb Res. (2012) 129:e161–3. doi: 10.1016/j.thromres.2011.10.008
151. Iwaki T, Nagahashi K, Takano K, Suzuki-Inoue K, Kanayama N, Umemura K, et al. Mutation in a highly conserved glycine residue in strand 5B of plasminogen activator inhibitor 1 causes polymerisation. Thromb Haemost. (2017) 117:860–9. doi: 10.1160/TH16-07-0572
152. Carmeliet P, Kieckens L, Schoonjans L, Ream B, van Nuffelen A, Prendergast G, et al. Plasminogen activator inhibitor-1 gene-deficient mice. I. Generation by homologous recombination and characterization. J Clin Invest. (1993) 92:2746–55. doi: 10.1172/JCI116892
153. Carmeliet P, Stassen JM, Schoonjans L, Ream B, van den Oord JJ, De Mol M, et al. Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis, and thrombolysis. J Clin Invest. (1993) 92:2756–60. doi: 10.1172/JCI116893
154. Erickson LA, Fici GJ, Lund JE, Boyle TP, Polites HG, Marotti KR. Development of venous occlusions in mice transgenic for the plasminogen activator inhibitor-1 gene. Nature. (1990) 346:74–6. doi: 10.1038/346074a0
155. Eren M, Painter Corrie A, Atkinson James B, Declerck Paul J, Vaughan Douglas E. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. (2002) 106:491–6. doi: 10.1161/01.CIR.0000023186.60090.FB
156. Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, et al. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. (1996) 97:232–7. doi: 10.1172/JCI118396
157. Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, et al. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. (2003) 23:78–84. doi: 10.1161/01.ATV.0000044457.60665.DD
158. Eren M, Gleaves LA, Atkinson JB, King LE, Declerck PJ, Vaughan DE. Reactive site-dependent phenotypic alterations in plasminogen activator inhibitor-1 transgenic mice. J Thromb Haemost. (2007) 5:1500–8. doi: 10.1111/j.1538-7836.2007.02587.x
159. Dawson S, Hamsten A, Wiman B, Henney A, Humphries S. Genetic variation at the plasminogen activator inhibitor-1 locus is associated with altered levels of plasma plasminogen activator inhibitor-1 activity. Arterioscler Thromb. (1991) 11:183–90. doi: 10.1161/01.ATV.11.1.183
160. Kathiresan S, Gabriel SB, Yang Q, Lochner AL, Larson MG, Levy D, et al. Comprehensive survey of common genetic variation at the plasminogen activator inhibitor-1 locus and relations to circulating plasminogen activator inhibitor-1 levels. Circulation. (2005) 112:1728–35. doi: 10.1161/CIRCULATIONAHA.105.547836
161. Grubic N, Stegnar M, Peternel P, Kaider A, Binder BR. A novel G/A and the 4G/5G polymorphism within the promoter of the plasminogen activator inhibitor-1 gene in patients with deep vein thrombosis. Thromb Res. (1996) 84:431–43. doi: 10.1016/S0049-3848(96)00211-3
162. Ploplis VA. Effects of altered plasminogen activator inhibitor-1 expression on cardiovascular disease. Curr Drug Targets. (2011) 12:1782–9. doi: 10.2174/138945011797635803
163. Fahim AT, Wang H, Feng J, Ginsburg D. Transgenic overexpression of a stable plasminogen activator inhibitor-1 variant. Thromb Res. (2009) 123:785–92. doi: 10.1016/j.thromres.2008.07.004
164. Abboud N, Ghazouani L, Saidi S, Ben-Hadj-Khalifa S, Addad F, Almawi WY, et al. Association of PAI-1 4G/5G and−844G/A gene polymorphisms and changes in PAI-1/tissue plasminogen activator levels in myocardial infarction: a case–control study. Genet Test Mol Biomarkers. (2009) 14:23–7. doi: 10.1089/gtmb.2009.0039
165. Fu L, Jin H, Song K, Zhang C, Shen J, Huang Y. Relationship between gene polymorphism of the PAI-1 promoter and myocardial infarction. Chin Med J. (2001) 114:266–9.
166. García-González IJ, Valle Y, Sandoval-Pinto E, Valdés-Alvarado E, Valdez-Haro A, Muñoz-Valle JF, et al. The−844 G>A PAI-1 polymorphism is associated with acute coronary syndrome in Mexican population. Dis Markers. (2015) 2015:460974. doi: 10.1155/2015/460974
167. Osmak GJ, Sidko AR, Kiselev IS, Favorova OO. Age-dependent approach to search for genetic variants associated with myocardial infarction. Mol Biol. (2020) 54:618–22. doi: 10.1134/S0026893320040123
168. Wiklund P-G, Nilsson L, Ardnor Sofie N, Eriksson P, Johansson L, Stegmayr B, et al. Plasminogen activator inhibitor-1 4G/5G polymorphism and risk of stroke. Stroke. (2005) 36:1661–5. doi: 10.1161/01.STR.0000174485.10277.24
169. Liang Z, Jiang W, Ouyang M, Yang K. PAI-1 4G/5G polymorphism and coronary artery disease risk: a meta-analysis. Int J of Clin Exp Med. (2015) 8:2097–107.
170. Seguí R, Estellés A, Mira Y, España F, Villa P, Falcó C, et al. PAI-1 promoter 4G/5G genotype as an additional risk factor for venous thrombosis in subjects with genetic thrombophilic defects. Br J Haematol. (2000) 111:122–8. doi: 10.1046/j.1365-2141.2000.02321.x
171. Tang J, Zhu W, Mei X, Zhang Z. Plasminogen activator inhibitor-1: a risk factor for deep vein thrombosis after total hip arthroplasty. J Orthop Surg Res. (2018) 13:8. doi: 10.1186/s13018-018-0716-2
172. Huang G, Wang P, Li T, Deng X. Genetic association between plasminogen activator inhibitor-1 rs1799889 polymorphism and venous thromboembolism: Evidence from a comprehensive meta-analysis. Clin Cardiol. (2019) 42:1232–8. doi: 10.1002/clc.23282
173. Liu Y, Cheng J, Guo X, Mo J, Gao B, Zhou H, et al. The roles of PAI-1 gene polymorphisms in atherosclerotic diseases: a systematic review and meta-analysis involving 149,908 subjects. Gene. (2018) 673:167–73. doi: 10.1016/j.gene.2018.06.040
174. Ye S, Green FR, Scarabin PY, Nicaud V, Bara L, Dawson SJ, et al. The 4G/5G genetic polymorphism in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene is associated with differences in plasma PAI-1 activity but not with risk of myocardial infarction in the ECTIM study. Etude CasTemoins de I'nfarctus du Mycocarde. Thromb Haemost. (1995) 74:837–41. doi: 10.1055/s-0038-1649833
175. Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Miletich JP. Arterial and venous thrombosis is not associated with the 4G/5G polymorphism in the promoter of the plasminogen activator inhibitor gene in a large cohort of US men. Circulation. (1997) 95:59–62. doi: 10.1161/01.CIR.95.1.59
176. Hindorff LA, Schwartz SM, Siscovick DS, Psaty BM, Longstreth WT Jr., et al. The association of PAI-1 promoter 4G/5G insertion/deletion polymorphism with myocardial infarction and stroke in young women. J Cardiovasc Risk. (2002) 9:131–7. doi: 10.1177/174182670200900209
177. Mansilha A, Araújo F, Severo M, Sampaio SM, Toledo T, Albuquerque R. Genetic polymorphisms and risk of recurrent deep venous thrombosis in young people: prospective cohort study. Eur J Vasc Endovasc Surg. (2005) 30:545–9. doi: 10.1016/j.ejvs.2005.05.038
178. Morange PE, Saut N, Alessi MC, Yudkin JS, Margaglione M, Di Minno G, et al. Association of plasminogen activator inhibitor (PAI)-1 (SERPINE1) SNPs with myocardial infarction, plasma PAI-1, and metabolic parameters. Arterioscler Thromb Vasc Biol. (2007) 27:2250–7. doi: 10.1161/ATVBAHA.107.149468
179. Hamsten A, Wiman B, de Faire U, Blombäck M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med. (1985) 313:1557–63. doi: 10.1056/NEJM198512193132501
180. Tofler GH, Massaro J, O'Donnell CJ, Wilson PWF, Vasan RS, Sutherland PA, et al. Plasminogen activator inhibitor and the risk of cardiovascular disease: the Framingham Heart Study. Thromb Res. (2016) 140:30–5. doi: 10.1016/j.thromres.2016.02.002
181. Jung RG, Motazedian P, Ramirez FD, Simard T, Di Santo P, Visintini S, et al. Association between plasminogen activator inhibitor-1 and cardiovascular events: a systematic review and meta-analysis. Thromb J. (2018) 16:12. doi: 10.1186/s12959-018-0166-4
182. Song C, Burgess S, Eicher John D, null n, O'Donnell Christopher J, Johnson Andrew D, et al. Causal effect of plasminogen activator inhibitor type 1 on coronary heart disease. J Am Heart Association. 6:e004918.
183. Meltzer ME, Lisman T, de Groot PG, Meijers JC, le Cessie S, Doggen CJ, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. (2010) 116:113–21. doi: 10.1182/blood-2010-02-267740
184. Schneiderman J, Sawdey MS, Keeton MR, Bordin GM, Bernstein EF, Dilley RB, et al. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci USA. (1992) 89:6998–7002. doi: 10.1073/pnas.89.15.6998
185. Chomiki N, Henry M, Alessi MC, Anfosso F, Juhan-Vague I. Plasminogen activator inhibitor-1 expression in human liver and healthy or atherosclerotic vessel walls. Thromb Haemost. (1994) 72:44–53. doi: 10.1055/s-0038-1648809
186. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
187. Padró T, Steins M, Li C-X, Mesters RM, Hammel D, Scheld HH, et al. Comparative analysis of plasminogen activator inhibitor-1 expression in different types of atherosclerotic lesions in coronary arteries from human heart explants. Cardiovasc Res. (1997) 36:28–36. doi: 10.1016/S0008-6363(97)00144-2
188. Lupu F, Bergonzelli GE, Heim DA, Cousin E, Genton CY, Bachmann F, et al. Localization and production of plasminogen activator inhibitor-1 in human healthy and atherosclerotic arteries. Arterioscler Thromb. (1993) 13:1090–100. doi: 10.1161/01.ATV.13.7.1090
189. Eitzman DT, Westrick RJ, Xu Z, Tyson J, Ginsburg D. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood. (2000) 96:4212–5. doi: 10.1182/blood.V96.13.4212
190. Luttun A, Lupu F, Storkebaum E, Hoylaerts MF, Moons L, Crawley J, et al. Lack of plasminogen activator inhibitor-1 promotes growth and abnormal matrix remodeling of advanced atherosclerotic plaques in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2002) 22:499–505. doi: 10.1161/hq0302.104529
191. Sjöland H, Eitzman DT, Gordon D, Westrick R, Nabel EG, Ginsburg D. Atherosclerosis progression in LDL receptor-deficient and apolipoprotein E-deficient mice is independent of genetic alterations in plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. (2000) 20:846–52. doi: 10.1161/01.ATV.20.3.846
192. Haka AS, Grosheva I, Singh RK, Maxfield FR. Plasmin promotes foam cell formation by increasing macrophage catabolism of aggregated low-density lipoprotein. Arterioscler Thromb Vasc Biol. (2013) 33:1768–78. doi: 10.1161/ATVBAHA.112.301109
193. Kremen M, Krishnan R, Emery I, Hu JH, Slezicki KI, Wu A, et al. Plasminogen mediates the atherogenic effects of macrophage-expressed urokinase and accelerates atherosclerosis in apoE-knockout mice. Proc Natl Acad Sci USA. (2008) 105:17109. doi: 10.1073/pnas.0808650105
194. Suzuki J-i, Ogawa M, Muto S, Yamaguchi Y, Itai A, Isobe M. The effects of pharmacological PAI-1 inhibition on thrombus formation and neointima formation after arterial injury. Expert Opin Ther Targets. (2008) 12:783–94. doi: 10.1517/14728222.12.7.783
195. Ji Y, Weng Z, Fish P, Goyal N, Luo M, Myears SP, et al. Pharmacological targeting of plasminogen activator inhibitor-1 decreases vascular smooth muscle cell migration and neointima formation. Arterioscler Thromb Vasc Biol. (2016) 36:2167–75. doi: 10.1161/ATVBAHA.116.308344
196. Lijnen HR, Van Hoef B, Umans K, Collen D. Neointima formation and thrombosis after vascular injury in transgenic mice overexpressing plasminogen activator inhibitor-1 (PAI-1). J Thromb Haemost. (2004) 2:16–22. doi: 10.1111/j.1538-7836.2003.00533.x
197. Carmeliet P, Moons L, Lijnen R, Janssens S, Lupu F, Collen D, et al. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation. Circulation. (1997) 96:3180–91. doi: 10.1161/01.CIR.96.9.3180
198. Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. (2004) 279:22595–604. doi: 10.1074/jbc.M313004200
199. Juhan-Vague I, Pyke SD, Alessi MC, Jespersen J, Haverkate F, Thompson SG. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European Concerted Action on Thrombosis and Disabilities. Circulation. (1996) 94:2057–63. doi: 10.1161/01.CIR.94.9.2057
200. Johansson L, Jansson JH, Boman K, Nilsson TK, Stegmayr B, Hallmans G. Tissue plasminogen activator, plasminogen activator inhibitor-1, and tissue plasminogen activator/plasminogen activator inhibitor-1 complex as risk factors for the development of a first stroke. Stroke. (2000) 31:26–32. doi: 10.1161/01.STR.31.1.26
201. Folsom AR, Cushman M, Heckbert SR, Rosamond WD, Aleksic N. Prospective study of fibrinolytic markers and venous thromboembolism. J Clin Epidemiol. (2003) 56:598–603. doi: 10.1016/S0895-4356(03)00052-0
202. Juhan-Vague I, Alessi MC, Vague P. Increased plasma plasminogen activator inhibitor 1 levels. A possible link between insulin resistance and atherothrombosis. Diabetologia. (1991) 34:457–62. doi: 10.1007/BF00403280
203. Carratala A, Martinez-Hervas S, Rodriguez-Borja E, Benito E, Real JT, Saez GT, et al. PAI-1 levels are related to insulin resistance and carotid atherosclerosis in subjects with familial combined hyperlipidemia. J Investig Med. (2018) 66:17–21. doi: 10.1136/jim-2017-000468
204. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol. (2017) 37:1446–52. doi: 10.1161/ATVBAHA.117.309451
205. Baluta MM, Vintila MM. PAI-1 inhibition–another therapeutic option for cardiovascular protection. Maedica. (2015) 10:147–52.
206. Björk I, Nordling K, Larsson I, Olson ST. Kinetic characterization of the substrate reaction between a complex of antithrombin with a synthetic reactive-bond loop tetradecapeptide and four target proteinases of the inhibitor. J Biol Chem. (1992) 267:19047–50.
207. Eitzman DT, Fay WP, Lawrence DA, Francis-Chmura AM, Shore JD, Olson ST, et al. Peptide-mediated inactivation of recombinant and platelet plasminogen activator inhibitor-1 in vitro. J Clin Invest. (1995) 95:2416–20. doi: 10.1172/JCI117937
208. D'Amico S, Martial JA, Struman I. A peptide mimicking the C-terminal part of the reactive center loop induces the transition to the latent form of plasminogen activator inhibitor type-1. FEBS Lett. (2012) 586:686–92. doi: 10.1016/j.febslet.2012.02.013
209. Jensen JK, Malmendal A, Schiøtt B, Skeldal S, Pedersen KE, Celik L, et al. Inhibition of plasminogen activator inhibitor-1 binding to endocytosis receptors of the low-density-lipoprotein receptor family by a peptide isolated from a phage display library. Biochem J. (2006) 399:387–96. doi: 10.1042/BJ20060533
210. Mathiasen L, Dupont DM, Christensen A, Blouse GE, Jensen JK, Gils A, et al. A peptide accelerating the conversion of plasminogen activator inhibitor-1 to an inactive latent state. Mol Pharmacol. (2008) 74:641–53. doi: 10.1124/mol.108.046417
211. Blake CM, Sullenger BA, Lawrence DA, Fortenberry YM. Antimetastatic potential of PAI-1-specific RNA aptamers. Oligonucleotides. (2009) 19:117–28. doi: 10.1089/oli.2008.0177
212. Fortenberry YM, Brandal SM, Carpentier G, Hemani M, Pathak AP. Intracellular expression of PAI-1 specific aptamers alters breast cancer cell migration, invasion and angiogenesis. PLoS ONE. (2016) 11:e0164288. doi: 10.1371/journal.pone.0164288
213. Damare J, Brandal S, Fortenberry YM. Inhibition of PAI-1 antiproteolytic activity against tPA by RNA aptamers. Nucleic Acid Ther. (2014) 24:239–49. doi: 10.1089/nat.2013.0475
214. Fortenberry YM. Plasminogen activator inhibitor-1 inhibitors: a patent review (2006–present). Expert Opin Ther Pat. (2013) 23:801–15. doi: 10.1517/13543776.2013.782393
215. Rouch A, Vanucci-Bacqué C, Bedos-Belval F, Baltas M. Small molecules inhibitors of plasminogen activator inhibitor-1–an overview. Eur J Med Chem. (2015) 92:619–36. doi: 10.1016/j.ejmech.2015.01.010
216. Björquist P, Ehnebom J, Inghardt T, Hansson L, Lindberg M, Linschoten M, et al. Identification of the binding site for a low-molecular-weight inhibitor of plasminogen activator inhibitor type 1 by site-directed mutagenesis. Biochemistry. (1998) 37:1227–34. doi: 10.1021/bi971554q
217. Einholm AP, Pedersen KE, Wind T, Kulig P, Overgaard MT, Jensen JK, et al. Biochemical mechanism of action of a diketopiperazine inactivator of plasminogen activator inhibitor-1. Biochemical J. (2003) 373:723–32. doi: 10.1042/bj20021880
218. Charlton P, Faint R, Barnes C, Bent F, Folkes A, Templeton D, et al. XR5118, a novel modulator of plasminogen activator inhibitor-1 (PAI-1), increases endogenous tPA activity in the rat. Fibrinol Proteol. (1997) 11:51–6. doi: 10.1016/S0268-9499(97)80009-4
219. Egelund R, Einholm AP, Pedersen KE, Nielsen RW, Christensen A, Deinum J, et al. A regulatory hydrophobic area in the flexible joint region of plasminogen activator inhibitor-1, defined with fluorescent activity-neutralizing ligands: ligand-unduced serpin polymerization. J Biol Chem. (2001) 276:13077–86. doi: 10.1074/jbc.M009024200
220. Gils A, Stassen JM, Nar H, Kley JT, Wienen W, Ries UJ, et al. Characterization and comparative evaluation of a novel PAI-1 inhibitor. Thromb Haemost. (2002) 88:137–43. doi: 10.1055/s-0037-1613166
221. Pedersen KE, Einholm AP, Christensen A, Schack L, Wind T, Kenney JM, et al. Plasminogen activator inhibitor-1 polymers, induced by inactivating amphipathic organochemical ligands. Biochem J. (2003) 372:747–55. doi: 10.1042/bj20021868
222. Cale JM, Li S-H, Warnock M, Su EJ, North PR, Sanders KL, et al. Characterization of a novel class of polyphenolic inhibitors of plasminogen activator inhibitor-1. J Biol Chem. (2010) 285:7892–902. doi: 10.1074/jbc.M109.067967
223. Izuhara Y, Takahashi S, Nangaku M, Takizawa S, Ishida H, Kurokawa K, et al. Inhibition of plasminogen activator inhibitor-1: its mechanism and effectiveness on coagulation and fibrosis. Arterioscler Thromb Vasc Biol. (2008) 28:672–7. doi: 10.1161/ATVBAHA.107.157479
224. Yamaoka N, Kawano Y, Izuhara Y, Miyata T, Meguro K. Structure-activity relationships of new 2-acylamino-3-thiophenecarboxylic acid dimers as plasminogen activator inhibitor-1 inhibitors. Chem Pharm Bull. (2010) 58:615–9. doi: 10.1248/cpb.58.615
225. Yamaoka N, Murano K, Kodama H, Maeda A, Dan T, Nakabayashi T, et al. Identification of novel plasminogen activator inhibitor-1 inhibitors with improved oral bioavailability: Structure optimization of N-acylanthranilic acid derivatives. Bioorg Med Chem Lett. (2018) 28:809–13. doi: 10.1016/j.bmcl.2017.11.016
226. Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, et al. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. (2015) 22:953–8. doi: 10.1038/nsmb.3129
227. Rodrigo G, Gruvegård M, Van Alstine JM. Antibody fragments and their purification by protein L affinity chromatography. Antibodies. (2015) 4:259–77. doi: 10.3390/antib4030259
228. Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies. (2019) 8:55. doi: 10.3390/antib8040055
229. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, et al. Naturally occurring antibodies devoid of light chains. Nature. (1993) 363:446–8. doi: 10.1038/363446a0
230. Greenberg AS, Avila D, Hughes M, Hughes A, McKinney EC, Flajnik MF. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature. (1995) 374:168–73. doi: 10.1038/374168a0
231. Flajnik MF, Deschacht N, Muyldermans S. A case of convergence: why did a simple alternative to canonical antibodies arise in sharks and camels? PLoS Biol. (2011) 9:e1001120. doi: 10.1371/journal.pbio.1001120
232. Ewert S, Cambillau C, Conrath K, Plückthun A. Biophysical properties of camelid V(HH) domains compared to those of human V(H)3 domains. Biochemistry. (2002) 41:3628–36. doi: 10.1021/bi011239a
233. Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LGJ, et al. Single-domain antibody fragments with high conformational stability. Protein Sci. (2002) 11:500–15. doi: 10.1110/ps.34602
234. Harmsen MM, van Solt CB, van Zijderveld-van Bemmel AM, Niewold TA, van Zijderveld FG. Selection and optimization of proteolytically stable llama single-domain antibody fragments for oral immunotherapy. Appl Microbiol Biotechnol. (2006) 72:544–51. doi: 10.1007/s00253-005-0300-7
235. Bijnens AP, Gils A, Stassen JM, Komissarov AA, Knockaert I, Brouwers E, et al. The distal hinge of the reactive site loop and its proximity: a target to modulate plasminogen activator inhibitor-1 activity. J Biol Chem. (2001) 276:44912–8. doi: 10.1074/jbc.M103077200
236. Ngo TH, Hoylaerts MF, Knockaert I, Brouwers E, Declerck PJ. Identification of a target site in plasminogen activator inhibitor-1 that allows neutralization of its inhibitor properties concomitant with an allosteric up-regulation of its antiadhesive properties. J Biol Chem. (2001) 276:26243–8. doi: 10.1074/jbc.M008241200
237. Bijnens AP, Gils A, Knockaert I, Stassen JM, Declerck PJ. Importance of the hinge region between alpha-helix F and the main part of serpins, based upon identification of the epitope of plasminogen activator inhibitor type 1 neutralizing antibodies. J Biol Chem. (2000) 275:6375–80. doi: 10.1074/jbc.275.9.6375
238. Wind T, Jensen MA, Andreasen PA. Epitope mapping for four monoclonal antibodies against human plasminogen activator inhibitor type-1: implications for antibody-mediated PAI-1-neutralization and vitronectin-binding. Eur J Biochem. (2001) 268:1095–106. doi: 10.1046/j.1432-1327.2001.2680041095.x
239. Naessens D, Gils A, Compernolle G, Declerck PJ. Elucidation of a novel epitope of a substrate-inducing monoclonal antibody against the serpin PAI-1. J Thromb Haemost. (2003) 1:1028–33. doi: 10.1046/j.1538-7836.2003.00206.x
240. Naessens D, Gils A, Compernolle G, Declerck PJ. Elucidation of the epitope of a latency-inducing antibody: identification of a new molecular target for PAI-1 inhibition. Thromb Haemost. (2003) 90:52–8. doi: 10.1055/s-0037-1613598
241. Gorlatova NV, Elokdah H, Fan K, Crandall DL, Lawrence DA. Mapping of a conformational epitope on plasminogen activator inhibitor-1 by random mutagenesis: Implications for serpin function. J Biol Chem. (2003) 278:16329–35. doi: 10.1074/jbc.M208420200
242. Ngo TH, Zhou Y, Stassen JM, Declerck PJ. Importance of N-terminal residues in plasminogen activator inhibitor 1 on its antibody induced latency transition. Thromb Haemost. (2002) 88:288–93. doi: 10.1055/s-0037-1613200
243. Bijnens AP, Ngo TH, Gils A, Dewaele J, Knockaert I, Stassen JM, et al. Elucidation of the binding regions of PAI-1 neutralizing antibodies using chimeric variants of human and rat PAI-1. Thromb Haemost. (2001) 85:866–74. doi: 10.1055/s-0037-1615761
244. Perrie AM, MacGregor IR, Booth NA. Definition of epitopes within plasminogen activator inhibitor type-1 (PAI-1) using multiple peptide synthesis. Fibrinolysis. (1993) 7:257–63. doi: 10.1016/0268-9499(93)90134-H
245. Keijer J, Linders M, van Zonneveld AJ, Ehrlich HJ, de Boer JP, Pannekoek H. The interaction of plasminogen activator inhibitor 1 with plasminogen activators (tissue-type and urokinase-type) and fibrin: localization of interaction sites and physiologic relevance. Blood. (1991) 78:401–9. doi: 10.1182/blood.V78.2.401.401
246. Komissarov AA, Declerck PJ, Shore JD. Mechanisms of conversion of plasminogen activator inhibitor 1 from a suicide inhibitor to a substrate by monoclonal antibodies. J Biol Chem. (2002) 277:43858–65. doi: 10.1074/jbc.M204110200
247. Stoop AA, Jespers L, Lasters I, Eldering E, Pannekoek H. High-density mutagenesis by combined DNA shuffling and phage display to assign essential amino acid residues in protein-protein interactions: application to study structure-function of plasminogen activation inhibitor 1 (PAI-I). J Mol Biol. (2000) 301:1135–47. doi: 10.1006/jmbi.2000.4035
248. Gils A, Meissenheimer LM, Compernolle G, Declerck PJ. Species-dependent molecular drug targets in plasminogen activator inhibitor-1 (PAI-1). Thromb Haemost. (2009) 102:609–10. doi: 10.1160/TH09-04-0257
249. Debrock S, Sironi L, Declerck PJ. Cloning of a single-chain variable fragment (scFv) switching active plasminogen activator inhibitor-1 to substrate. Gene. (1997) 189:83–8. doi: 10.1016/S0378-1119(96)00838-4
250. Verbeke K, Gils A, Declerck PJ. Inhibition of plasminogen activator inhibitor-1: antibody fragments and their unique sequences as a tool for the development of profibrinolytic drugs. J Thromb Haemost. (2004) 2:298–305. doi: 10.1111/j.1538-7933.2004.00583.x
251. Verbeke K, Gils A, Declerck PJ. Cloning and paratope analysis of an antibody fragment, a rational approach for the design of a PAI-1 inhibitor. J Thromb Haemost. (2004) 2:289–97. doi: 10.1111/j.1538-7933.2004.00582.x
252. Verbeke K, Gils A, Stassen JM, Declerck PJ. Elucidation of the paratope of scFv-8H9D4, a PAI-1 neutralizing antibody derivative. Thromb Haemost. (2003) 89:74–82. doi: 10.1055/s-0037-1613545
253. Novoa de Armas H, Dewilde M, Verbeke K, De Maeyer M, Declerck PJ. Study of recombinant antibody fragments and PAI-1 complexes combining protein-protein docking and results from site-directed mutagenesis. Structure. (2007) 15:1105–16. doi: 10.1016/j.str.2007.07.009
254. Wyseure T, Gils A, Declerck PJ. Evaluation of the profibrinolytic properties of a bispecific antibody-based inhibitor against human and mouse thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1. J Thromb Haemost. (2013) 11:2069–71. doi: 10.1111/jth.12399
255. Wyseure T, Rubio M, Denorme F, Martinez de Lizarrondo S, Peeters M, Gils A, et al. Innovative thrombolytic strategy using a heterodimer diabody against TAFI and PAI-1 in mouse models of thrombosis and stroke. Blood. (2015) 125:1325–32. doi: 10.1182/blood-2014-07-588319
256. Denorme F, Wyseure T, Peeters M, Vandeputte N, Gils A, Deckmyn H, et al. Inhibition of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 reduces ischemic brain damage in mice. Stroke. (2016) 47:2419–22. doi: 10.1161/STROKEAHA.116.014091
257. Declerck PJ, Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Semin Thromb Hemost. (2013) 39:356–64. doi: 10.1055/s-0033-1334487
Keywords: plasminogen activator inhibitor 1 (PAI-1), PAI-1 inhibitors, serpin (serine proteinase inhibitor), fibrinolyisis, cardiovascular disease
Citation: Sillen M and Declerck PJ (2020) Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 7:622473. doi: 10.3389/fcvm.2020.622473
Received: 28 October 2020; Accepted: 03 December 2020;
Published: 22 December 2020.
Edited by:
Alexandra R. Lucas, Arizona State University, United StatesReviewed by:
Katsuya Hirano, Kagawa University, JapanMargarethe Geiger, Medical University of Vienna, Austria
Copyright © 2020 Sillen and Declerck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul J. Declerck, paul.declerck@kuleuven.be