Targeting lipid metabolism as a new therapeutic strategy for inherited cardiomyopathies
 Karen R. Gaar-Humphreys1,2 Alyssa van den Brink1,2 Mark Wekking1,2
Karen R. Gaar-Humphreys1,2 Alyssa van den Brink1,2 Mark Wekking1,2  Folkert W. Asselbergs3,4
Folkert W. Asselbergs3,4  Frank G. van Steenbeek1,2,5
Frank G. van Steenbeek1,2,5  Magdalena Harakalova1,2,6
Magdalena Harakalova1,2,6  Jiayi Pei1,2,6*
Jiayi Pei1,2,6*- 1Division Heart and Lungs, Department of Cardiology, Circulatory Health Research Center, University Medical Center Utrecht, Utrecht University, Utrecht, Netherlands
- 2Regenerative Medicine Center Utrecht, University Medical Center Utrecht, Utrecht, Netherlands
- 3Department of Cardiology, Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, Netherlands
- 4Health Data Research United Kingdom and Institute of Health Informatics, University College London, London, United Kingdom
- 5Department of Clinical Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands
- 6Netherlands Heart Institute, Utrecht, Netherlands
Inherited cardiomyopathies caused by pathological genetic variants include multiple subtypes of heart disease. Advances in next-generation sequencing (NGS) techniques have allowed for the identification of numerous genetic variants as pathological variants. However, the disease penetrance varies among mutated genes. Some can be associated with more than one disease subtype, leading to a complex genotype-phenotype relationship in inherited cardiomyopathies. Previous studies have demonstrated disrupted metabolism in inherited cardiomyopathies and the importance of metabolic adaptations in disease onset and progression. In addition, genotype- and phenotype-specific metabolic alterations, especially in lipid metabolism, have been revealed. In this mini-review, we describe the metabolic changes that are associated with dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM), which account for the largest proportion of inherited cardiomyopathies. We also summarize the affected expression of genes involved in fatty acid oxidation (FAO) in DCM and HCM, highlighting the potential of PPARA-targeting drugs as FAO modulators in treating patients with inherited cardiomyopathies.
Introduction
Inherited cardiomyopathies are diseases of the heart muscle due to pathological genetic variants. Based on the clinical presentations, they are often divided into four subtypes, namely, hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic cardiomyopathy (ACM), and restrictive cardiomyopathy (RCM) (1, 2). Among them, the estimated prevalence of DCM is the highest (1:250 individuals), followed by HCM (1:500 individuals) (3). Owing to the advances in next-generation sequencing (NGS) techniques, numerous genetic variants have been identified as disease-causing variants. To date, more than 60 mutated genes are associated with DCM, and they are involved in a wide range of cellular features, including the sarcomere, Z disk, cytoskeleton, sarcoplasmic reticulum and cytoplasm, ion channels, and mitochondria (4). A subset of those mutated genes, such as TTN, LMNA, MYH7, and PLN, exhibits a stronger gene-disease relationship with DCM compared to the rest (5). Unlike the broad range of DCM-causal variants, most pathological variants in HCM affect sarcomeric genes (6). MYH7 and MYBPC3 are the most commonly affected genes in HCM, which account for about 70% of those variants (7). The disease penetrance varies among mutated genes, and the same mutated gene can be associated with more than one subtype (2, 8), leading to a complex genotype-phenotype relationship in inherited cardiomyopathies. Therefore, molecular insights into the affected pathways and biological processes concerning different variants and/or subtypes are needed to characterize the diseases better and provide druggable candidates for novel treatments.
Metabolic changes in inherited cardiomyopathies
The heart has a very high energy demand to fulfill its basic functions. Therefore, sufficient cardiac energy metabolism is crucial. In a healthy adult heart, over 95% of produced ATP is derived from mitochondrial oxidative phosphorylation, and this is predominantly by fatty acid oxidation (FAO) (9). However, a significant metabolic switch toward the less efficient anaerobic glycolytic metabolism occurs in failing hearts, which resembles the energy preference of the fetal heart (10, 11). The inefficiency in utilizing fatty acids results in the accumulation of lipid droplets, which subsequently lead to lipotoxicity and heart failure (12). In addition to lipid accumulation, failing hearts also exhibit impaired metabolic flexibility in switching between different energy substrates, including fatty acids, glucose, ketones, and amino acids (13). The lack of sufficient energy substrates due to prolonged fasting is, in fact, a known trigger for inherited cardiomyopathies (14). Taken together, both internal and external factors could affect cardiac performance and disease progression by disrupting metabolic homeostasis.
Cardiac tissues and plasma samples from patients carrying truncating TTN variants, which account for 15–20% of DCM populations, showed affected genes and metabolites involved in metabolic regulation when compared to DCM patients without TTN variants (15). This suggests a tight relationship between TTN variants and metabolic alterations. In addition, murine and human DCM hearts carrying a PLN variant showed suppressed mitochondrial fatty acid metabolism at mRNA and protein levels (16, 17). Suppressed metabolic genes and mitochondrial enzyme activities were also observed in 2D and 3D human induced pluripotent stem cell-derived cardiomyocytes harboring a mutated PLN gene (18). Multiple omics-based studies showed changes in metabolite levels, such as glutamine, lactate, and acylcarnitines, in DCM patients when compared to healthy individuals and patients with ischemic cardiomyopathy (19–21). Additionally, the metabolic changes correlated with the disease severity (19, 22). Therefore, metabolites involved in metabolic signaling, such as branched-chain amino acid metabolism, glycolysis, and glycolipid metabolism, have been proposed as potential biomarkers for DCM patients (23, 24). In line with these findings, clinical measurements using cardiac magnetic resonance imaging and positron emission tomography scanning also revealed an impaired oxidative metabolism and the subsequent energy starvation mode in DCM (12, 25). Additionally, DCM-related genetic variants, such as LMNA variants, show a direct influence on lipid metabolism (26). Besides the impaired fatty acid metabolism in DCM patients, individuals with FAO disorders also have a higher risk of developing DCM (27). These findings indicate a bi-directional association between DCM and impaired fatty acid metabolism. A recent study showed improved contractility and mitochondrial respiration in cardiomyocytes with various DCM-causing variants, including mutated PLN, TNNT2, TTN, LMNA, TPM1, and LAMA2, by enhancing serine metabolism (28). Serine is a non-essential amino acid and decreased serine availability has been shown to suppress mitochondrial FAO, glucose and glutamine metabolism (29), highlighting the tightly associated metabolic pathways and the promising metabolic-based treatment strategies in DCM.
High energy demand is required in HCM due to the associated hypercontractility (30). Unlike the decreased power cycle (duty ratio) and a lower force-holding capacity in DCM mutations when compared to the wildtype controls, which require much less ATP, HCM mutations exhibit an increased power cycle and a higher force-holding capacity, leading to a higher ATP usage (31). Therefore, alterations in metabolism show a profound impact on HCM pathogenesis. Additionally, in contrast to the decreased Ca2+ sensitivity in DCM, increased Ca2+ sensitivity and cytosolic adenosine diphosphate (ADP) levels are seen in HCM due to sarcomeric variants, resulting in metabolic changes (32). Increased cytosolic ADP increases the oxidation of two metabolic enzymes (NADH and NADPH), which decreases the capacity to attenuate mitochondrial reactive oxygen species (ROS) levels, as NADPH is necessary to detoxify ROS (30). Increased ROS subsequently impairs mitochondrial activation and contributes to HCM development (33). Additionally, reduced phosphocreatine (PCr)/ATP ratios in HCM, both with and without hypertrophy, indicate cardiac energetic impairments are present at an early stage of HCM (34). The switch from FAO to glucose consumption is seen in hypertrophied hearts, along with a decreased expression of CD36, a key lipid transporter (35, 36). Multiple omics-based studies comparing HCM patients to controls further revealed alterations of molecular signatures involved in a wide array of pathways suggesting fatty acid metabolism dysregulation, a reduction of acylcarnitines, and an accumulation of free fatty acids (37–39). A recent study using adult cultured rat cardiomyocytes also demonstrated that increased glucose consumption is necessary for synthesizing aspartate, which directly drives cardiomyocyte hypertrophy (40). Mouse hearts carrying mutated MYH6, one of the HCM-causal genes (41), showed decreased mitochondrial ATP hydrolysis (42). Additionally, a high prevalence of HCM is observed among patients with mitochondrial diseases, and several mutated mitochondrial genes are known to contribute to HCM development, such as HADHB (14, 43). Taken together, impaired mitochondrial lipid metabolism and the switch to glycolysis are important for HCM initiation and progression. Therefore, the potential of various metabolic compounds is currently being studied in HCM, such as perhexiline (44, 45), mavacamten (46, 47), omecamtiv mecarbil (46), and ROS scavengers (48–50). Their efficacy, however, is still to be determined.
Besides DCM and HCM, metabolic disturbances are also observed in ACM and RCM. By comparing the transcriptional landscapes between ACM and control human hearts, affected genes were enriched for several metabolic signaling, including mitochondrial dysfunction and oxidative phosphorylation (51). By comparing the plasma metabolomes between ACM patients and healthy individuals, affected metabolites and lipids further revealed several changed metabolic pathways, including lysine degradation, tryptophan metabolism, and the beta-oxidation of fatty acids (52). A recent paper compared transcriptional changes in RCM, ischemic heart disease, and valvular heart disease to control human hearts and showed that ATP metabolic processes were enriched by altered genes in RCM but not in the other two heart diseases (53). Combined, these findings highlighted the potential benefits of restoring a balanced metabolism in inherited cardiomyopathies.
Shared and unique metabolic alterations between DCM and HCM
As a result of the recent studies indicating impaired metabolism in inherited cardiomyopathies, attention has been drawn to studying and identifying precise metabolic branches and key drivers of disease pathogenesis per subtype. In general, both DCM and HCM exhibit decreased lipid metabolism (30, 54) and increased glucose metabolism (12, 55; Figure 1A). Besides these two major metabolic processes, enhanced ketone body metabolism is also shown in DCM and HCM (56, 57). Interestingly, suppressed oxidative metabolism, amino acid metabolism, pentose phosphate pathway, and nucleotide metabolism are observed in DCM (58–61), whereas they are all elevated in HCM (19, 62–65). A recent study further examined the metabolic alterations in DCM and HCM hearts as compared to non-failing control hearts at the global transcriptional level and demonstrated impaired metabolic signaling of fatty acids, carbohydrates, and amino acids in both DCM and HCM hearts (66). The study also investigated the single-nucleus transcriptome in cardiomyocytes and non-myocyte cell types and showed metabolic pathways were profoundly impaired in DCM cardiomyocytes but not HCM cardiomyocytes, suggesting the disease-specific metabolic alteration. Similarly, another paper also showed HCM- or DCM-specific impaired metabolic processes (67). Disease-specific gene sets, some of which are involved in lipid metabolism, were found to be differentially changed between DCM and HCM, such as the up-regulated APOE and the down-regulated GPT in DCM, as well as the up-regulated APOLD1 and the down-regulated STARD13 and PON3 in HCM. In line with these findings, we also demonstrated that KLF15, an important transcription factor regulating lipid metabolism (68), was significantly up-regulated in HCM hearts carrying mutated MYBPC3 and down-regulated in DCM hearts carrying mutated PLN when compared to non-failing donor hearts (16, 38). Besides the transcriptional level, proteins involved in metabolic pathways, including lipid transfer and fatty acid biosynthetic process, also showed DCM- and HCM-specific changes (69). To conclude, a profoundly impaired metabolism is well-characterized in inherited cardiomyopathies. Meanwhile, DCM- and HCM-specific metabolic alterations, particularly candidate genes in lipid metabolism, have also been cataloged.
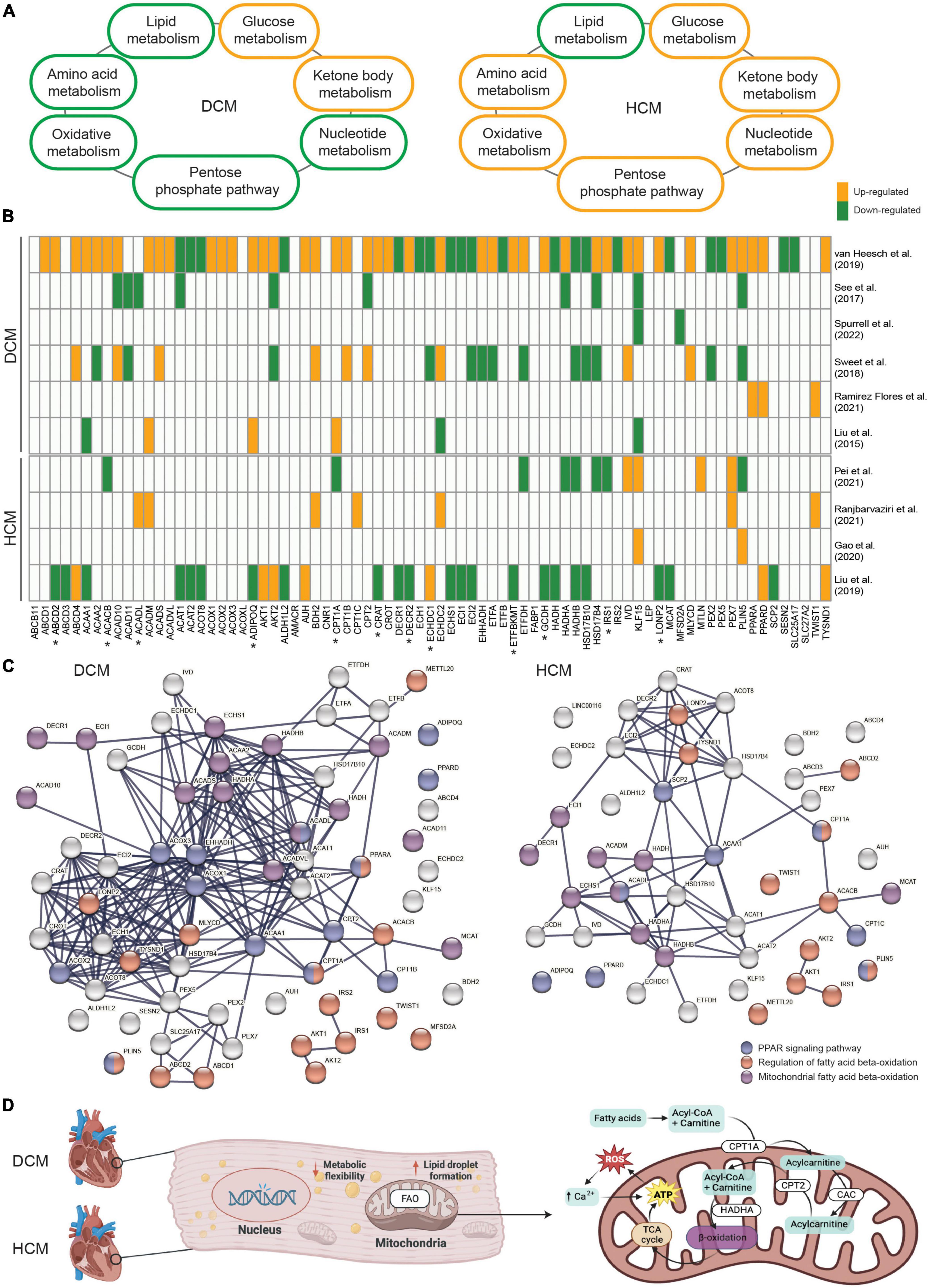
Figure 1. (A) An overview demonstrating the alteration direction of seven main metabolic processes in dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM). (B) The involvement and expression pattern of 77 protein-coding genes associated with fatty acid beta-oxidation (FAO) in DCM and HCM. Up-regulated genes in DCM or HCM vs. the controls are shown in orange, and down-regulated genes are shown in green. Genes that were either not significantly altered or not shown in the related paper were shown in white. Genes with opposite alteration directions between DCM and HCM are indicated by “*”. (C) The enriched protein-protein interaction network in DCM and HCM, respectively. Highlighted genes involved in the regulation of FAO (red), mitochondrial-related FAO (purple), and the peroxisome proliferator-activated receptor (PPAR) signaling pathway (blue). (D) Schematic representation of mitochondrial FAO and its key regulators in inherited cardiomyopathies.
The shared and unique metabolic signaling pathways and candidate genes have opened up new avenues to identify innovative compounds and design novel treatments for inherited cardiomyopathies in general but also for specific cardiomyopathy subtypes. For instance, mavacamten, a selective inhibitor of cardiac myosin ATPase that modulates ATP turnover time, exhibited a promising and beneficial effect on HCM patients (47, 70). FAO inhibitors and lipid-lowering agents have also been studied for treating DCM patients (71). The potential of SGLT2 inhibitors, which improve mitochondrial function, is currently under investigation in treating both DCM and HCM (72–74). Yet, the need for additional drugs targeting metabolism and mitochondrial function as the precision medicine for inherited cardiomyopathies has been urged by the Translational Committee of the Heart Failure Association and the Working Group of Myocardial Function of the European Society of Cardiology (75).
FAO alteration in DCM and HCM
Given FAO is severely impaired in both DCM and HCM, yet a subset of FAO-related genes might be unique for each subset, we searched for relevant studies that presented global transcriptional profiles using either RNA sequencing or microarray in DCM or HCM cohorts. We filtered for studies that were conducted using either human tissue or cells or experiments that were validated in a human model after animal experiments. In total, 10 relevant papers published between 2015 and 2022 were compiled and included for the purpose of this meta-analysis. Next, we collected 76 established genes that are involved in fatty acid beta-oxidation from the gene ontology project (GO:0006635). KLF15 was added to the gene set due to its role in FAO and its unique alteration directions in DCM and HCM. Strikingly, almost all of the 77 genes were significantly differentially expressed between DCM and control hearts in one included paper, confirming the profoundly affected lipid metabolism in DCM (Figure 1B). Interestingly, altered expression patterns for some genes, such as IRS1 and CPT1A, were contrasting between DCM and HCM, suggesting the disease subtype-specific differences in the FAO impairment. Besides, HADHA and HADHB, genes coding for key enzymes in mitochondrial FAO (76), showed generally suppressed mRNA levels in DCM and HCM, whereas ACOX1, ACOX2, and ACOX3, genes coding for key enzymes in peroxisomal FAO (77), showed increased mRNA levels in DCM but not in HCM. This further suggests disease subtype-specific differences in subcellular organelles-related FAO impairment. It is also important to note that some FAO-related genes showed contradicting expression patterns among different DCM-based or HCM-based studies. This could be partially explained by the heterogeneous genetic variants and their mutated genes, the different disease severities of included patients, and the variable group sizes of those studies. Therefore, studies with synchronized patient cohorts and well-characterized genetic information and clinical presentations are needed to address this complex gene-disease relationship. Protein-protein interaction analysis was performed using the genes found to be differentially expressed in DCM and HCM, respectively, to further elucidate their functional networks. Among these affected genes associated with FAO, biological processes, including the regulation of FAO (GO:0031998), mitochondrial FAO (HSA-77289), and peroxisome proliferator-activated receptor (PPAR) signaling pathway (WP3942, Figure 1C), remained significantly enriched in both DCM and HCM. PPARs are important upstream transcription factors in regulating FAO and other facets of lipid metabolism and regulation (78). Notably, PPARA and CPT1A are shown in both the PPAR signaling pathway network and the regulation of FAO network. Both ACADM and ACADL overlap with the PPAR signaling pathway network and are involved with mitochondrial FAO.
PPARA-related FAO modulators as novel candidates for the treatment of inherited cardiomyopathies?
PPARA, PPARD, and PPARG are three isoforms of the peroxisome proliferator-activated receptors (PPARs), which are ligand-activated transcription factors. Since their DNA binding regions are highly similar, they show overlapping biological functions, especially in lipid metabolism (79). PPARA is a key regulator in modulating fatty acid uptake and FAO; PPARD enhances the utilization of lipids and glucose; and PPARY increases fatty acid uptake, triglyceride formation, and lipid storage (80). Notably, multiple studies from us and others have shown suppressed PPARA expressions in DCM and HCM hearts carrying different genetic variants (16, 81, 82). Additionally, the interaction between PPARA and KLF15 showed a significant impact on cardiac lipid metabolism (83).
The cardioprotective effects of ligand-activated PPARA have been reported, including the restored balance between fatty acid uptake and FAO, increased insulin sensitivity, reduced ROS production, and attenuated fibrosis formation (80, 84). Both natural ligands (i.e., omega-3 fatty acids) and synthetic ligands (i.e., fibrates), referred to as PPARA agonists (80), are commonly used to activate PPARA. Previous studies have summarized well-established PPARA agonists and those that are still in development (85–88), some of which are PPARA-specific. Several agonists, such as bezafibrate, ciprofibrate, clofibrate, and fenofibrate, have been FDA-approved for treating type 2 diabetes or dyslipidemia, and many more are under active research (80, 87). However, most research is focused on the application of fibrates as treatments for diseases such as primary biliary cholangitis, COVID-19, and non-alcoholic fatty liver disease (89–91), and limited studies have evaluated fibrates in inherited cardiomyopathies. A recent study using knockout-Dsg2 ACM murine hearts showed that improved myocardial fibrosis was observed after the activation of PPARA by either fenofibrate treatment or adeno-associated virus injections of PPARA (92). Nevertheless, several clinical trials have investigated the effects of bezafibrate on mitochondrial disease, neutral lipid storage disease, muscle/mitochondrial FAO disorders, and Barth syndrome (93–99), which reflect impaired mitochondrial function, lipid accumulation, and heart failure as seen in cardiomyopathies. Therefore, the results obtained from these conducted trials might also shield light on its effect on inherited cardiomyopathies.
Conclusion
Metabolic homeostasis plays an important role in cardiac performance and disrupted metabolism is generally present in inherited cardiomyopathies, regardless of the pathogenic DNA variant and the phenotypes. Despite the shared metabolism alterations among different subtypes of inherited cardiomyopathies, etiology- and phenotype-specific metabolic impairments have been revealed, particularly in relation to FAO (Figure 1D). Those shared and unique metabolic changes provide promising candidate targets for future therapeutic strategies in treating inherited cardiomyopathies. Moreover, due to the importance of PPARA in regulating FAO and the beneficial effects of PPARA agonists observed in cardiomyocytes (100–103), studies have started to specify the pharmacological activities and cardiotoxicity of PPARA agonists (104). However, currently, there is no systematic study on the use of PPARA agonists, even FDA-approved PPARA-targeting fibrates, in patients with inherited cardiomyopathies. In conclusion, the potential of PPARA-activating drugs as FAO modulators to restore a balanced metabolism is worthy of investigation in inherited cardiomyopathies.
Author contributions
KG-H, AB, and JP wrote the manuscript. KG-H, AB, MW, JP, and MH collected and interpreted public datasets. FWA, FvS, and MH edited the text and provided the critical input. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Leducq grant (CURE-PLaN no. 18CVD01 to JP, MH, and FWA), ZonMW Open Competition grant (CONTRACT no. 09120012010018 to KG-H, FWA, and MH), Dutch Cardiovascular Alliance (DCVA) grant (DOUBLE-DOSE no. 2020B005 to MH, FvS, and FWA), and ERA-CVD grant (SCALE no. 2019T109 to JP, FvS, and MH). FWA is supported by UCL Hospitals NIHR Biomedical Research Centre.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
3. McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol. (2020) 18:22–36.
4. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. (2017) 121:731–48.
5. Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation. (2021) 144:7–19.
6. Vander Roest AS, Liu C, Morck MM, Kooiker KB, Jung G, Song D, et al. Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state. Proc Natl Acad Sci U.S.A. (2021) 118:e2025030118. doi: 10.1073/pnas.2025030118
7. Akhtar M, Elliott P. The genetics of hypertrophic cardiomyopathy. Glob Cardiol Sci Pract. (2018) 2018:36. doi: 10.21542/gcsp.2018.36
8. Cahill TJ, Ashrafian H, Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res. (2013) 113:660–75.
9. Ferreira CR, Blau N. Clinical and biochemical footprints of inherited metabolic diseases. IV. Metabolic cardiovascular disease. Mol Genet Metab. (2021) 132:112–8. doi: 10.1016/j.ymgme.2020.12.290
10. Bae J, Paltzer WG, Mahmoud AI. The role of metabolism in heart failure and regeneration. Front Cardiovasc Med. (2021) 8:702920. doi: 10.3389/fcvm.2021.702920
11. Carvajal K, Moreno-Sánchez R. Heart metabolic disturbances in cardiovascular diseases. Arch Med Res. (2003) 34:89–99.
12. Dávila-Román VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. (2002) 40:271–7. doi: 10.1016/s0735-1097(02)01967-8
13. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. (2018) 5:68. doi: 10.3389/fcvm.2018.00068
14. Brailova M, Clerfond G, Trésorier R, Minet-Quinard R, Durif J, Massoullié G, et al. Inherited metabolic diseases and cardiac pathology in adults: diagnosis and prevalence in a CardioMetabo study. J Clin Med Res. (2020) 9:694. doi: 10.3390/jcm9030694
15. Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiarán Aizpurua A, Merken JJ, Wang P, et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J. (2018) 39:864–73. doi: 10.1093/eurheartj/ehx808
16. Pei J, Maas RGC, Nagyova E, Gho JMI, Blok CS, van Adrichem I, et al. Transcriptional regulation profiling reveals disrupted lipid metabolism in failing hearts with a pathogenic phospholamban mutation. bioRxiv [Preprint]. (2020). doi: 10.1101/2020.11.30.402792
17. Eijgenraam TR, Boogerd CJ, Stege NM, Teixeira VON, Dokter MM, Schmidt LE, et al. Protein aggregation is an early manifestation of phospholamban p.(Arg14del)–related cardiomyopathy: development of PLN-R14del–related cardiomyopathy. Circ Heart Fail. (2021) 14:e008532. doi: 10.1161/CIRCHEARTFAILURE.121.008532
18. Cuello F, Knaust AE, Saleem U, Loos M, Raabe J, Mosqueira D, et al. Impairment of the ER/mitochondria compartment in human cardiomyocytes with PLN p.Arg14del mutation. EMBO Mol Med. (2021) 13:e13074. doi: 10.15252/emmm.202013074
19. Ampong I. Metabolic and metabolomics insights into dilated cardiomyopathy. Ann Nutr Metab. (2022) 78:147–55. doi: 10.1159/000524722
20. Haas J, Frese KS, Sedaghat-Hamedani F, Kayvanpour E, Tappu R, Nietsch R, et al. Energy metabolites as biomarkers in ischemic and dilated cardiomyopathy. Int J Mol Sci. (2021) 22:1999. doi: 10.3390/ijms22041999
21. Flam E, Jang C, Murashige D, Yang Y, Morley MP, Jung S, et al. Integrated landscape of cardiac metabolism in end-stage human nonischemic dilated cardiomyopathy. Nat Cardiovasc Res. (2022) 1:817–29.
22. Verdonschot JAJ, Wang P, Van Bilsen M, Hazebroek MR, Merken JJ, Vanhoutte EK, et al. Metabolic profiling associates with disease severity in nonischemic dilated cardiomyopathy. J Card Fail. (2020) 26:212–22. doi: 10.1016/j.cardfail.2019.09.004
23. Zhao J, Yang S, Jing R, Jin H, Hu Y, Wang J, et al. Plasma metabolomic profiles differentiate patients with dilated cardiomyopathy and ischemic cardiomyopathy. Front Cardiovasc Med. (2020) 7:597546. doi: 10.3389/fcvm.2020.597546
24. Liu C, Li R, Liu Y, Li Z, Sun Y, Yin P, et al. Characteristics of blood metabolic profile in coronary heart disease, dilated cardiomyopathy and valvular heart disease induced heart failure. Front Cardiovasc Med. (2021) 7:622236. doi: 10.3389/fcvm.2020.622236
25. Bell SP, Adkisson DW, Ooi H, Sawyer DB, Lawson MA, Kronenberg MW. Impairment of subendocardial perfusion reserve and oxidative metabolism in nonischemic dilated cardiomyopathy. J Card Fail. (2013) 19:802–10. doi: 10.1016/j.cardfail.2013.10.010
26. Boschmann M, Engeli S, Moro C, Luedtke A, Adams F, Gorzelniak K, et al. LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance. J Clin Endocrinol Metab. (2010) 95:1634. doi: 10.1210/jc.2009-1293
27. Merritt JL, MacLeod E, Jurecka A, Hainline B. Clinical manifestations and management of fatty acid oxidation disorders. Rev Endocr Metab Disord. (2020) 21: 479–93.
28. Perea-Gil I, Seeger T, Bruyneel AAN, Termglinchan V, Monte E, Lim EW, et al. Serine biosynthesis as a novel therapeutic target for dilated cardiomyopathy. Eur Heart J. (2022) 43:3477–89.
29. Gao X, Lee K, Reid MA, Sanderson SM, Qiu C, Li S, et al. Serine availability influences mitochondrial dynamics and function through lipid metabolism. Cell Rep. (2018) 22:3507. doi: 10.1016/j.celrep.2018.03.017
30. van der Velden J, Tocchetti CG, Varricchi G, Bianco A, Sequeira V, Hilfiker-Kleiner D, et al. Metabolic changes in hypertrophic cardiomyopathies: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res. (2018) 114:1273. doi: 10.1093/cvr/cvy147
31. Ujfalusi Z, Vera CD, Mijailovich SM, Svicevic M, Yu EC, Kawana M, et al. Dilated cardiomyopathy myosin mutants have reduced force-generating capacity. J Biol Chem. (2018) 293:9017.
32. Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. (2007) 101:1266–73. doi: 10.1161/CIRCRESAHA.107.156380
33. Szyguła-Jurkiewicz B, Szczurek-Wasilewicz W, Osadnik T, Frycz-Kurek AM, Macioł-Skurk K, Małyszek-Tumidajewicz J, et al. Oxidative stress markers in hypertrophic cardiomyopathy. Medicina. (2022) 58:31. doi: 10.3390/medicina58010031
34. Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. (2003) 41:1776–82. doi: 10.1016/s0735-1097(02)03009-7
35. Magida JA, Leinwand LA. Metabolic crosstalk between the heart and liver impacts familial hypertrophic cardiomyopathy. EMBO Mol Med. (2014) 6:482–95. doi: 10.1002/emmm.201302852
36. Glatz JFC, Luiken JJF, Nabben M. CD36 (SR-B2) as a target to treat lipid overload-induced cardiac dysfunction. J Lipid Atheroscler. (2020) 9:66. doi: 10.12997/jla.2020.9.1.66
37. Ranjbarvaziri S, Kooiker KB, Ellenberger M, Fajardo G, Zhao M, Vander Roest AS, et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation. (2021) 144:1714–31.
38. Pei J, Schuldt M, Nagyova E, Gu Z, El Bouhaddani S, Yiangou L, et al. Multi-omics integration identifies key upstream regulators of pathomechanisms in hypertrophic cardiomyopathy due to truncating MYBPC3 mutations. Clin Epigenet. (2021) 13:61. doi: 10.1186/s13148-021-01043-3
39. Schuldt M, Pei J, Harakalova M, Dorsch LM, Schlossarek S, Mokry M, et al. Proteomic and functional studies reveal detyrosinated tubulin as treatment target in sarcomere mutation-induced hypertrophic cardiomyopathy. Circ Heart Fail. (2021) 14:e007022. doi: 10.1161/CIRCHEARTFAILURE.120.007022
40. Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res. (2020) 126:182–96. doi: 10.1161/CIRCRESAHA.119.315483
41. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121:749–70. doi: 10.1161/CIRCRESAHA.117.311059
42. Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, et al. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. (1998) 101:1775–83. doi: 10.1172/JCI1940
43. Chung H, Kim Y, Park C-H, Kim J-Y, Min P-K, Yoon YW, et al. Genetic relevance and determinants of mitral leaflet size in hypertrophic cardiomyopathy. Cardiovasc Ultrasound. (2019) 17:21. doi: 10.1186/s12947-019-0171-1
44. Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. (2010) 122:1562–9. doi: 10.1161/CIRCULATIONAHA.109.934059
45. Gehmlich K, Dodd MS, Allwood JW, Kelly M, Bellahcene M, Lad HV, et al. Changes in the cardiac metabolome caused by perhexiline treatment in a mouse model of hypertrophic cardiomyopathy. Mol Biosyst. (2015) 11:564–73. doi: 10.1039/c4mb00594e
46. Kampourakis T, Zhang X, Sun Y-B, Irving M. Omecamtiv mercabil and blebbistatin modulate cardiac contractility by perturbing the regulatory state of the myosin filament. J Physiol. (2018) 596:31–46. doi: 10.1113/JP275050
47. Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2020) 396:759–69. doi: 10.1016/S0140-6736(20)31792-X
48. Marian AJ, Senthil V, Chen SN, Lombardi R. Antifibrotic effects of antioxidant N-acetylcysteine in a mouse model of human hypertrophic cardiomyopathy mutation. J Am Coll Cardiol. (2006) 47:827–34. doi: 10.1016/j.jacc.2005.10.041
49. Lombardi R, Rodriguez G, Chen SN, Ripplinger CM, Li W, Chen J, et al. Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation. (2009) 119:1398–407. doi: 10.1161/CIRCULATIONAHA.108.790501
50. Wilder T, Ryba DM, Wieczorek DF, Wolska BM, Solaro RJ. N-acetylcysteine reverses diastolic dysfunction and hypertrophy in familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2015) 309:H1720–30. doi: 10.1152/ajpheart.00339.2015
51. Hall CL, Gurha P, Sabater-Molina M, Asimaki A, Futema M, Lovering RC, et al. RNA sequencing-based transcriptome profiling of cardiac tissue implicates novel putative disease mechanisms in FLNC-associated arrhythmogenic cardiomyopathy. Int J Cardiol. (2020) 302:124–30. doi: 10.1016/j.ijcard.2019.12.002
52. Volani C, Rainer J, Hernandes VV, Meraviglia V, Pramstaller PP, Smárason SV, et al. Metabolic signature of arrhythmogenic cardiomyopathy. Metabolites. (2021) 11:195. doi: 10.3390/metabo11040195
53. Zhu M, Zhang C, Zhang Z, Liao X, Ren D, Li R, et al. Changes in transcriptomic landscape in human end-stage heart failure with distinct etiology. iScience. (2022) 25:103935. doi: 10.1016/j.isci.2022.103935
54. Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
55. Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. (2019) 8:e012673. doi: 10.1161/JAHA.119.012673
56. Schugar RC, Moll AR, André d’Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. (2014) 3:754. doi: 10.1016/j.molmet.2014.07.010
57. Selvaraj S, Kelly DP, Margulies KB. Implications of altered ketone metabolism and therapeutic ketosis in heart failure. Circulation. (2020) 141:1800–12. doi: 10.1161/CIRCULATIONAHA.119.045033
58. Naya M, Tamaki N. Imaging of myocardial oxidative metabolism in heart failure. Curr Cardiovasc Imaging Rep. (2014) 7:9244. doi: 10.1007/s12410-013-9244-y
59. Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol. (2019) 18:86. doi: 10.1186/s12933-019-0892-3
60. Dörner A, Schulze K, Rauch U, Schultheiss HP. Adenine nucleotide translocator in dilated cardiomyopathy: pathophysiological alterations in expression and function. Mol Cell Biochem. (1997) 174:261–9.
61. Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. (2011) 90:194.
62. Tuunanen H, Kuusisto J, Toikka J, Jääskeläinen P, Marjamäki P, Peuhkurinen K, et al. Myocardial perfusion, oxidative metabolism, and free fatty acid uptake in patients with hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the α-tropomyosin gene: a positron emission tomography study. J Nucl Cardiol. (2007) 14:354–65. doi: 10.1016/j.nuclcard.2006.12.329
63. Previs MJ, O’Leary TS, Morley MP, Palmer BM, LeWinter M, Yob JM, et al. Defects in the proteome and metabolome in human hypertrophic cardiomyopathy. Circ Heart Fail. (2022) 15:e009521.
64. Rossi A, Olivares J, Aussedat J, Ray A. Increased uracil nucleotide metabolism during the induction of cardiac hypertrophy by β-stimulation in rats. Basic Res Cardiol. (1980) 75:139–42. doi: 10.1007/BF02001405
65. West JA, Beqqali A, Ament Z, Elliott P, Pinto YM, Arbustini E, et al. A targeted metabolomics assay for cardiac metabolism and demonstration using a mouse model of dilated cardiomyopathy. Metabolomics. (2016) 12:1–18. doi: 10.1007/s11306-016-0956-2
66. Chaffin M, Papangeli I, Simonson B, Akkad A-D, Hill MC, Arduini A, et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature. (2022) 608:174–80.
67. Quttainah M, Raveendran VV, Saleh S, Parhar R, Aljoufan M, Moorjani N, et al. Transcriptomal insights of heart failure from normality to recovery. Biomolecules. (2022) 12:731. doi: 10.3390/biom12050731
68. Prosdocimo DA, Anand P, Liao X, Zhu H, Shelkay S, Artero-Calderon P, et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J Biol Chem. (2014) 289:5914.
69. Ghose S, Varshney S, Adlakha K, Bhat A, Naushin S, Seth S, et al. Quantitative proteomics study reveals differential proteomic signature in dilated, restrictive, and hypertrophic cardiomyopathies. J Proteins Proteomics. (2019) 10:33–44.
70. Edelberg JM, Sehnert AJ, Mealiffe ME, del Rio CL, McDowell R. The impact of mavacamten on the pathophysiology of hypertrophic cardiomyopathy: a narrative review. Am J Cardiovasc Drugs. (2022) 22:497–510. doi: 10.1007/s40256-022-00532-x
71. Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation. (2004) 110:894–6.
72. Li N, Zhou H. SGLT2 inhibitors: a novel player in the treatment and prevention of diabetic cardiomyopathy. Drug Des Devel Ther. (2020) 14:4775. doi: 10.2147/DDDT.S269514
73. Paneni F, Costantino S, Hamdani N. Regression of left ventricular hypertrophy with SGLT2 inhibitors. Eur Heart J. (2020) 41:3433–6.
74. Dyck JRB, Sossalla S, Hamdani N, Coronel R, Weber NC, Light PE, et al. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: evidence for potential off-target effects. J Mol Cell Cardiol. (2022) 167:17–31.
75. de Boer RA, Heymans S, Backs J, Carrier L, Coats AJS, Dimmeler S, et al. Targeted therapies in genetic dilated and hypertrophic cardiomyopathies: from molecular mechanisms to therapeutic targets. A position paper from the Heart Failure Association (HFA) and the Working Group on Myocardial Function of the European Society of Cardiology (ESC). Eur J Heart Fail. (2022) 24:406. doi: 10.1002/ejhf.2414
76. Sekine Y, Yamamoto K, Kurata M, Honda A, Onishi I, Kinowaki Y, et al. HADHB, a fatty acid beta-oxidation enzyme, is a potential prognostic predictor in malignant lymphoma. Pathology. (2022) 54:286–93. doi: 10.1016/j.pathol.2021.06.119
77. Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res. (2010) 51:2863.
78. Wagner N, Wagner KD. The role of PPARs in disease. Cells. (2020) 9:2367. doi: 10.3390/cells9112367
79. Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. (2007) 1771:926.
80. Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. (2021) 18:809–23. doi: 10.1038/s41569-021-00569-6
81. Schupp M, Kintscher U, Fielitz J, Thomas J, Pregla R, Hetzer R, et al. Cardiac PPARα expression in patients with dilated cardiomyopathy. Eur J Heart Fail. (2006) 8:290–4. doi: 10.1016/j.ejheart.2005.09.003
82. Burke MA, Chang S, Wakimoto H, Gorham JM, Conner DA, Christodoulou DC, et al. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight. (2016) 1:e86898. doi: 10.1172/jci.insight.86898
83. Prosdocimo DA, John JE, Zhang L, Efraim ES, Zhang R, Liao X, et al. KLF15 and PPARα cooperate to regulate cardiomyocyte lipid gene expression and oxidation. PPAR Res. (2015) 2015:201625. doi: 10.1155/2015/201625
84. Khuchua Z, Glukhov AI, Strauss AW, Javadov S. Elucidating the beneficial role of PPAR agonists in cardiac diseases. Int J Mol Sci. (2018) 19:3464. doi: 10.3390/ijms19113464
85. Takada I, Makishima M. Peroxisome proliferator-activated receptor agonists and antagonists: a patent review (2014-present). Expert Opin Ther Pat. (2020) 30:1–13. doi: 10.1080/13543776.2020.1703952
86. Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr J. (2014) 13:17. doi: 10.1186/1475-2891-13-17
87. Hong F, Xu P, Zhai Y. The opportunities and challenges of peroxisome proliferator-activated receptors ligands in clinical drug discovery and development. Int J Mol Sci. (2018) 19:2189. doi: 10.3390/ijms19082189
88. Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS. Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. Int J Mol Sci. (2019) 20:5055. doi: 10.3390/ijms20205055
89. Chung SW, Lee JH, Kim MA, Leem G, Kim SW, Chang Y, et al. Additional fibrate treatment in UDCA-refractory PBC patients. Liver Int. (2019) 39:1776–85. doi: 10.1111/liv.14165
90. Talasaz AH, Sadeghipour P, Aghakouchakzadeh M, Dreyfus I, Kakavand H, Ariannejad H, et al. Investigating lipid-modulating agents for prevention or treatment of COVID-19: JACC state-of-the-art review. J Am Coll Cardiol. (2021) 78:1635–54. doi: 10.1016/j.jacc.2021.08.021
91. Yoo J, Jeong I-K, Ahn KJ, Chung HY, Hwang Y-C. Fenofibrate, a PPARα agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism. (2021) 120:154798. doi: 10.1016/j.metabol.2021.154798
92. Qiu Z, Zhao Y, Tao T, Guo W, Liu R, Huang J, et al. Activation of PPARα ameliorates cardiac fibrosis in Dsg2-deficient arrhythmogenic cardiomyopathy. Cells. (2022) 11:3184. doi: 10.3390/cells11203184
93. Steele H, Gomez-Duran A, Pyle A, Hopton S, Newman J, Stefanetti RJ, et al. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol Med. (2020) 12:e11589.
94. Garcia MA, Rojas JA, Millán SP, Flórez AA. Neutral lipid storage disease with myopathy and dropped head syndrome. Report of a new variant susceptible of treatment with late diagnosis. J Clin Neurosci. (2018) 58:207–9. doi: 10.1016/j.jocn.2018.10.046
95. Ørngreen MC, Madsen KL, Preisler N, Andersen G, Vissing J, Laforêt P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. (2014) 82:607–13. doi: 10.1212/WNL.0000000000000118
96. Yamada K, Taketani T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet. (2019) 64:73–85. doi: 10.1038/s10038-018-0527-7
97. Suyama T, Shimura M, Fushimi T, Kuranobu N, Ichimoto K, Matsunaga A, et al. Efficacy of bezafibrate in two patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab Rep. (2020) 24:100610. doi: 10.1016/j.ymgmr.2020.100610
98. Bonnefont JP, Bastin J, Laforêt P, Aubey F, Mogenet A, Romano S, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. (2010) 88:101–8. doi: 10.1038/clpt.2010.55
99. Thompson R, Jefferies J, Wang S, Pu WT, Takemoto C, Hornby B, et al. Current and future treatment approaches for Barth syndrome. J Inherit Metab Dis. (2022) 45:17–28. doi: 10.1002/jimd.12453
100. Sharifpanah F, Wartenberg M, Hannig M, Piper HM, Sauer H. Peroxisome proliferator-activated receptor alpha agonists enhance cardiomyogenesis of mouse ES cells by utilization of a reactive oxygen species-dependent mechanism. Stem Cells. (2008) 26:64–71. doi: 10.1634/stemcells.2007-0532
101. Kar D, Bandyopadhyay A. Targeting peroxisome proliferator activated receptor α (PPAR α) for the prevention of mitochondrial impairment and hypertrophy in cardiomyocytes. Cell Physiol Biochem. (2018) 49:245–59. doi: 10.1159/000492875
102. Jen HL, Liu PL, Chen YH, Yin WH, Chen JW, Lin SJ. Peroxisome proliferator-activated receptor α reduces endothelin-1-caused cardiomyocyte hypertrophy by inhibiting nuclear factor- κ B and adiponectin. Mediators Inflamm. (2016) 2016:5609121. doi: 10.1155/2016/5609121
103. Huang Q, Huang J, Zeng Z, Luo J, Liu P, Chen S, et al. Effects of ERK1/2/PPARα/SCAD signal pathways on cardiomyocyte hypertrophy induced by insulin-like growth factor 1 and phenylephrine. Life Sci. (2015) 124:41–9. doi: 10.1016/j.lfs.2015.01.015
Keywords: hypertrophic cardiomyopathy, dilated cardiomyopathy, genetic variants, lipid metabolism, fatty acid oxidation, transcription factor PPARA
Citation: Gaar-Humphreys KR, van den Brink A, Wekking M, Asselbergs FW, van Steenbeek FG, Harakalova M and Pei J (2023) Targeting lipid metabolism as a new therapeutic strategy for inherited cardiomyopathies. Front. Cardiovasc. Med. 10:1114459. doi: 10.3389/fcvm.2023.1114459
Received: 02 December 2022; Accepted: 03 January 2023;
Published: 19 January 2023.
Edited by:
John Lynn Jefferies, The University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Andrew A. Gibb, Temple University, United StatesEmma Louise Robinson, University of Colorado, United States
Kyung Chan Park, University of Oxford, United Kingdom
Copyright © 2023 Gaar-Humphreys, van den Brink, Wekking, Asselbergs, van Steenbeek, Harakalova and Pei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiayi Pei,  j.pei-2@umcutrecht.nl
j.pei-2@umcutrecht.nl