Ion channel trafficking implications in heart failure
 Jean-Baptiste Reisqs
Jean-Baptiste Reisqs Yongxia Sarah Qu1,2
Yongxia Sarah Qu1,2  Mohamed Boutjdir
Mohamed Boutjdir- 1Cardiovascular Research Program, VA New York Harbor Healthcare System, New York, NY, United States
- 2Department of Cardiology, New York Presbyterian Brooklyn Methodist Hospital, New York, NY, United States
- 3Department of Medicine, Cell Biology and Pharmacology, State University of New York Downstate Health Sciences University, New York, NY, United States
- 4Department of Medicine, New York University Grossman School of Medicine, New York, NY, United States
Heart failure (HF) is recognized as an epidemic in the contemporary world, impacting around 1%–2% of the adult population and affecting around 6 million Americans. HF remains a major cause of mortality, morbidity, and poor quality of life. Several therapies are used to treat HF and improve the survival of patients; however, despite these substantial improvements in treating HF, the incidence of HF is increasing rapidly, posing a significant burden to human health. The total cost of care for HF is USD 69.8 billion in 2023, warranting a better understanding of the mechanisms involved in HF. Among the most serious manifestations associated with HF is arrhythmia due to the electrophysiological changes within the cardiomyocyte. Among these electrophysiological changes, disruptions in sodium and potassium currents’ function and trafficking, as well as calcium handling, all of which impact arrhythmia in HF. The mechanisms responsible for the trafficking, anchoring, organization, and recycling of ion channels at the plasma membrane seem to be significant contributors to ion channels dysfunction in HF. Variants, microtubule alterations, or disturbances of anchoring proteins lead to ion channel trafficking defects and the alteration of the cardiomyocyte's electrophysiology. Understanding the mechanisms of ion channels trafficking could provide new therapeutic approaches for the treatment of HF. This review provides an overview of the recent advances in ion channel trafficking in HF.
1 Clinical presentation
Heart failure (HF) is a chronic disease depicted by the inability of the myocardium to propel blood and oxygen effectively to comply with the body's needs and remains a major cause of death. It occurs when the heart muscles weaken or stiffen, leading to a reduced ability to pump blood. This cardiac pathology is characterized by dyspnea, peripheral edema, elevated jugular venous pressure, and tachycardia (1, 2). HF is a multifactorial disease mostly caused by various factors such as valvular diseases, coronary artery disease, endocardial or pericardial abnormalities, and high blood pressure (3, 4). In the US, HF affects 5.7 million individuals and is responsible for approximately 300,000 deaths per year (5). The incidence of HF is 5–10 per 1,000 individuals and the prevalence is 1%–2% annually (3). HF is classified based on the left ventricular ejection fraction (EF) into three EF categories: HF with reduced (HFrEF, ≤40%), mildly reduced (HFmrEF, 41%–49%), and preserved EF (HFpEF, ≥50%) (6). Given the scarce literature on ion channel trafficking in HFmrEF and HFpEF, the focus of this review is on HFrEF.
Pharmacotherapy is used for the treatment of patients with HFrEF and for improving their survival. This includes, β-blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and mineralocorticoid receptor antagonists (MRAs). Two new classes of drugs, angiotensin receptor-neprilysin inhibitor, sacubitril/valsartan (ENTRESTO®), and sodium-glucose cotransporter 2 (SGLT2) inhibitors are also used (7, 8). Adding MRAs, including spironolactone and eplerenone to the standard therapy in HF, reduced morbidity and mortality (9). Aldosterone, an end product of the upregulated renin-angiotensin-aldosterone system in HF, has been implicated as playing a major role in the progression of HF (10). In addition to the promotion of inflammation and fibrosis, aldosterone has been responsible for electrical remodeling (11). The chronotropic action of aldosterone was averted by spironolactone, indicating the role of MRA in this response (12, 13). HF is also associated with increased activity of the sympathetic nervous system (14). This chronic elevated plasma catecholamines lead to desensitization of the β-adrenergic signaling pathway. This includes downregulation of the number of receptor proteins from enhanced degradation and loss of receptor function (15, 16). Increased sympathetic activity leads to the upregulation of β-adrenergic receptor kinase, which phosphorylates the β-adrenergic and uncouples it from the second messenger triggered downstream pathway (17, 18). The result is a modification in the signaling pathway from a mainly β1-receptor in the healthy myocardium to a combined β1/β2 receptors in HF. These changes lead to a reduction of 50%–60% in β-adrenergic signaling capacity in advanced HF (19). β-blockers are efficient in restoring the adrenergic signaling pathway. Despite substantial advances in treating HF, the incidence of HF continues to rapidly increase posing a significant burden on human health. The total cost of healthcare for HF is projected to be USD 69.8 billion in 2023 (20).
Changes in the functional activity of ion channels play an important role in HF (21). Nevertheless, the mechanisms responsible for these alterations remain not fully understood. This incomplete knowledge can be attributed to the complexity of ion channels trafficking, which involves several proteins and serves as a significant contributor to the arrhythmogenic process. This review presents an update on the current understanding of ion channels trafficking physiology and, subsequently, provides the evidence related to trafficking defects observed in HF.
2 Electrical remodeling
2.1 Action potential
The cardiac action potential (AP) is important for the generation and the propagation of excitation leading to contraction and is the result of ion channels composition in each cardiomyocyte (22–24). AP corresponds to a transient depolarization due to the sequential activation and inactivation of voltage-dependent ion channels, ensuring a flow of ions between the intracellular and extracellular fluid according to the electrochemical gradient. AP is composed of five phases in the non-pacemaker myocytes that determine its duration and amplitude (Figure 1). It is carried by a sequential activation and inactivation of different currents (25, 26). The depolarization phase (Phase 0) is generated by the inward sodium (Na+) current (INa) encoded by the Na+ voltage-gated channel alpha subunit 5 (SCN5A). This phase is followed by a rapid repolarization (Phase 1), carried by the transient outward potassium (K+) current (Ito) encoded by the K+ voltage-gated channel subfamily D 2 (KCND2). This repolarization is very fast and followed by a plateau phase (Phase 2), which is a balance between the ion calcium (Ca2+) inflow through L-type Ca2+ current (ICaL), encoded by the Ca2+ voltage-gated channel subunit alpha 1C (CACNA1C) and the outward flow of K+ ions. The rapid (IKr) and the slow (IKs) K+ currents, conduct this outward flow of K+, and are encoded by human ether-a-go-go–related gene (hERG) and by the K+ voltage-gated subfamily Q member1 (KCNQ1), respectively. These two currents terminate the AP resulting in the final repolarization (Phase 3). Finally, Phase 4 corresponds to the resting state around −80 mV in non-pacemaker cells, mainly held by the inwardly rectifying K+ currents (IK1) encoded by the K+ inwardly rectifying channel subfamily J 12/14 and 4 (KCNJ12/14/4) (27, 28) and also to the diastolic depolarization in the pacemaker cells carried by the funny current (If) encoded by the hyperpolarization activated cyclic nucleotide gated K+ channel 4 (HCN4) (29, 30). Under pathological conditions such as HF, these ion channels undergo a remodeling process impacting the AP shape and AP duration (APD), part of which is due to trafficking disorders (31, 32). The APD of both atrial and ventricular cells is prolonged in HFrEF and HFpEF (33). However, Ca2+ handling is not impaired in the same way in HFrEF and HFpEF. In HFrEF, the systolic Ca2+ is decreased and the diastolic Ca2+ is increased, because of a decrease of T-tubule density and an increase of Na+/Ca2+ exchanger (NCX) expression. In HFpEF, there is a high cytoplasmic Ca2+ concentration associated with an unchanged NCX expression and an increased density of T-tubules (34).
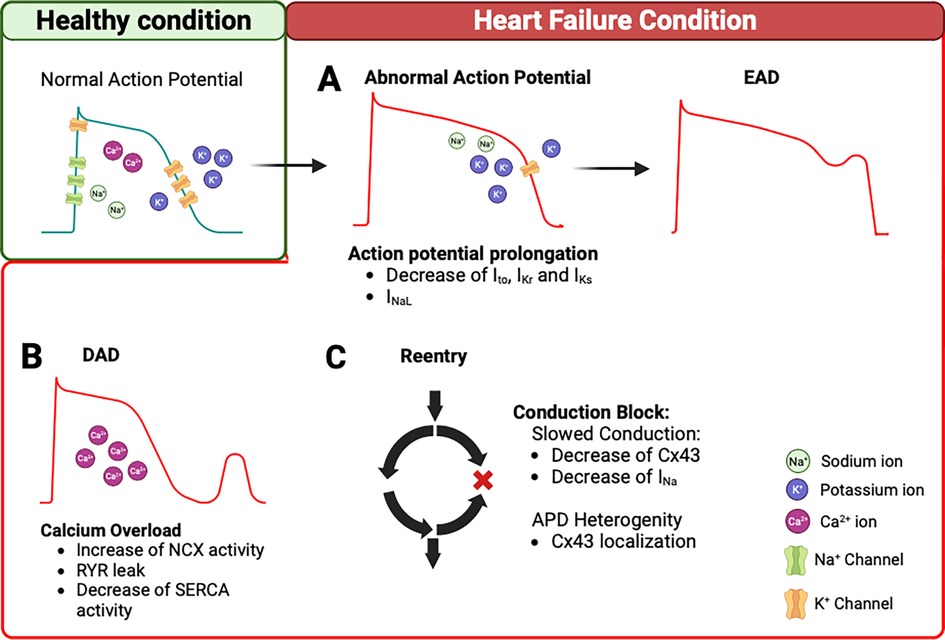
Figure 1. Arrhythmogenesis mechanisms in heart failure. (A) In HF condition, decrease of K+ current (Ito, IKr, IKs) and/or INaL can prolong the AP. This remodeling triggers EADs. (B) In addition, intracellular Ca2+ overload during the diastole can trigger DADs. This is due to an increase in NCX reverse mode, Ca2+ leak from the RYR2, and a decrease of SERCA function. (C) The remodeling of Cx43 and/or decrease of INa contribute to slowing the conduction velocity and APD heterogeneity, which creates conduction block and promotes re-entry pathways. AP, action potential; EADs, early aftredepolarizations; DADs, delayed aftredepolarizations; NCX, Na+/Ca2+ exchanger; RYR2, ryanodine receptor 2; SERCA, Sarcoendoplasmic Reticulum Calcium ATPase; Cx43, connexin 43; APD, action potential duration.
2.2 Sodium current
INa alterations have been implicated in HF. A HF study conducted on mice missing the expression of the Muscle Lim Protein (MLP) demonstrated a decreased of INa density by post-translational reduction (35, 36). This INa reduction decreased the electrical conduction, favored re-entry, and contributed to inefficient synchronized conduction. Furthermore, studies conducted on dog and human HF led to alterations in the inactivation of INa causing a late persistent current (37, 38). Most of INa is rapidly inactivated, but a small fraction remains and contributes to generate a late INa (INaL), thus contributing to increase in the concentration of Na+ ions in the cardiomyocyte and consequently increasing the APD (39). However, the molecular mechanisms which explain this INaL in HF are still unclear. One study showed that the heterologous expression of Nav1.5 produced a late opening in gating modes (40). Furthermore, another explanation for this late current would be the contribution of the neuronal isoform Nav1.1. One study, on a dog model of HF, demonstrated the participation of this neuronal isoform in inducing the INaL (41). Targeting the neuronal type of Na+ channel by riluzole was reported to reduce this INaL (42). Riluzole could thus offer a therapeutic target to prevent arrhythmia and slow the progression of HF (43). The importance of this INaL abnormality has been well demonstrated to contribute to arrhythmias in HF, and medications targeting the INa could prevent arrhythmia (44). In fact, some studies used ranolazine, an INaL inhibitor, to reduce and prevent arrhythmias in HF (45, 46). To this end, a medication empagliflozin, an SGLT2 inhibitor, was described to interact directly with the Na+ channel and reduce the INaL in a mouse model (47). Recently, another study used empagliflozin, a SGLT2 inhibitor, to revert the INaL upregulation and arrhythmogenic AP in an HFpEF mice model, induced by high-fat diet and nitric oxide synthases inhibition (48). These studies showed that inhibiting INaL may be a therapeutic target in the prevention of arrhythmia onset in HF (49). The accumulation of Na+ concentration can alter the Ca2+ homeostasis because the Ca2+ efflux is via the NCX, which will compensate by extruding excessive intracellular Na+ resulting in intracellular Ca2+ overload and subsequent triggered arrhythmic events (50).
2.3 Potassium current
In HF, alterations in IKr, IKs, and Ito can be present. IK plays a crucial role in the cardiac AP shape, and its remodeling is an important contributor to repolarization abnormalities in HF (51, 52). First, the Ito is known to be reduced in HF and to participate in APD prolongation (53, 54). The reduction of Ito participates in increasing the Ca2+ influx and Ca2+ concentration in the cells, which triggered the activation of hypertrophic pathway in the mice model (55, 56). Hypertrophy is a compensatory response to HF, but sustained cardiac hypertrophy contributes to the development of HF. Remodeling in IKr and IKs in HF have also been reported (57–59). A study conducted in rabbits with atrioventricular block showed a reduction of both IKr and IKs, using the patch-clamp technique (60, 61). IK1 was also shown to be downregulated in HF (59). The small-conductance Ca2+-activated K+ currents (SK) are upregulated in HF (62). This current participates in the repolarization of healthy atria but not in the ventricle (63). Other studies have shown an upregulation in this channel in ventricular myocytes, which prolongs the APD and promotes the arrhythmogenesis in HF (64, 65). Finally, a downregulation of the TREK channel was observed in patients with atrial fibrillation complicated by HF (66). In all, the remodeling of IK in HF underlies, in part, the electrical abnormality and associated arrhythmogenesis (67).
2.4 Calcium handling
During the development of HF, important changes in Ca2+ handling occur (68). Key players in Ca2+ homeostasis include L-type Ca2+ channels, NCX, ryanodine receptor type 2 (RYR2), sarcoplasmic reticulum Ca2+-ATPase (SERCA), and Na+/H+ exchangers (NHE).
2.4.1 Calcium current
Dysregulation of ICaL can impair the ability of the heart to contract affectively, leading to a decreased contractility and pumping function. The alteration of Ca2+ channels, which allow Ca2+ entry in failing cardiomyocytes, is still debatable in HF. In fact, the existing data on Ca2+ channel gene expression during HF show either a decrease or small changes in transcript levels of the gene CACNA1C (69–71). In addition, the increased phosphorylation of the Ca2+ channel leads to an increase of its open probability in human failing ventricle, suggesting a compensatory mechanism (72, 73). Furthermore, our group has demonstrated the re-expression of alpha1D L-type Ca2+ channels encoded by the CACNA1D gene in failing human ventricles, which is normally not expressed in the healthy ventricles (74). Their unique activation at a more negative voltage allows to play a larger role in Phase 4 depolarization and spontaneous beating. Furthermore, the density of ICaL is influenced by the severity of failure (75). In HF, adult ventricular myocytes reactivate the fetal gene expression program and acquire spontaneous beating (76, 77). In addition, some studies reported the re-expression of T-type Ca2+ channels in post-infarcted hearts, in hypertrophied adult cat hearts and in rat models of HF (78–80). Despite these modifications, the L-type Ca2+ channels are the main sources of extracellular Ca2+.
2.4.2 Sodium-calcium exchanger
NCX works in two modes: forward mode (Ca2+ efflux and Na+ influx) to reduce diastolic Ca2+ levels and reverse mode (Ca2+ influx/Na+ efflux) depends on the sarcolemma Na+ gradient (81). In HF, with an aberrant accumulation of Na+, NCX changes from forward to reversed mode, resulting in an increase of Ca2+ concentration in the cell (82, 83). The implication of NCX dysregulation has been demonstrated in human HF for triggering arrhythmias (84, 85). Studies have also demonstrated an upregulation of both mRNA and protein expression of NCX in human and animal models, contributing to the arrhythmogenicity of HF (86). These changes are associated with arrhythmogenic mechanisms, such as early (EADs) and delayed afterdepolarizations (DADs) in HF (87, 88). Therefore, the block of NCX may represent a potential therapy for the prevention of arrhythmia in HF (85). The selective NCX blocker, SEA-0400, has already demonstrated a potential benefit in pig and mice models of HF (89, 90). This NCX blocker restores the sarcoplasmic reticulum (SR) Ca2+ load and releases it, improving Ca2+ handling (91). Recently, new NCX blockers were developed, ORM-11035 and SAR296968, which have significantly attenuated Ca2+ handling remodeling and diastolic dysfunction in a rat model of HF (85, 92).
2.4.3 Ryanodine receptor
Dysregulation of Ca2+ homeostasis can impair the capacity of the myocardium to contract efficiently, leading to a decreased contractility and pumping function. These modifications in Ca2+ handling result in higher diastolic Ca2+ and enhanced diastolic Ca2+ loss from the SR, together impacting the Ca2+ induced Ca2+ release mechanism and excitation–contraction coupling culminating in reduced contractile force. The RYR2 is a Ca2+-release channel primarily found in the SR of cardiomyocytes. Its function is to regulate the release of Ca2+ during the cardiac contraction. RYR2 is directly or indirectly linked to the pathological cellular mechanisms of HF (93, 94). It interacts with a multitude of proteins regulating its activity, such as protein kinase A (PKA) and calmodulin-dependent kinase II (CamKII) (95). When one of these elements is perturbed, the RYR2 undergoes a pathological remodeling, which can initiate or amplify the cellular mechanisms contributing to HF. PKA hyperphosphorylation of RYR2 in HF results in the depletion of its stabilizing FK506 binding protein, FKBP12.6 (96). RYR2 are hyperphosphorylated by PKA and/or CAMKII, causing aberrant Ca2+ leaks from the SR (97). The diastolic releases of SR Ca2+ have been linked to DADs triggering fatal ventricular arrhythmias (Figure 1). In a canine model of rapid pacing-induced HF, administration of a β-adrenergic blocker, metoprolol, reverses PKA hyperphosphorylation of RYR2, restoring the stoichiometry of RYR2 macrocomplex, and normalizes single-channel function (98–100). Recently it has been demonstrated that Entresto® diminished diastolic Ca2+ spark frequency and SR leaks in mouse myocytes subjected to catecholaminergic stress, as well as in human left ventricular cardiomyocytes with end stage HF (101). The findings suggest that Entresto® ameliorates cardiac function in part by improving myocardial Ca2+ homeostasis.
The impairment of SR function has proven to be predominantly induced by a SR Ca2+ load reduction due to decreased activity of the SERCA and also from the RYR2 leak (102–104). It is associated with a Ca2+ leak from the SR through the RYR2 and a higher RYR2 activity (105, 106). Other studies have highlighted that the SR Ca2+ leak triggers mitochondrial dysfunction and leads to an increased production of free radicals, which in turn leads to pathological RYR2 remodeling (107). A further SR Ca2+ leak through RYR2 is then observed with HF progression exacerbation (108). Further results published show that silencing the expression of junctophilin-2 in the mice provokes SR Ca2+ leak through RYR2, and the development of HF (109). Conversely, a study demonstrated that maintaining the level of junctophilin-2 protein at a high level can prevent the progression of HF (110). This study underlies a key role of junctophilin-2 in HF and could be an interesting therapeutic target for HF. One of the largest gene transfer clinical studies (CUPID2), targeting the SERCA activity, was initiated in patients with HFrEF (111). However, the preliminary results did not show any improvement of the heart function.
Collectively, the Ca2+ mishandling observed in HF contributes to reduced contractility of the myocardium triggering pump failure or arrhythmias. The Ca2+ cycling disruption is one of the central elements initiating and progressing the cardiac function deterioration in HF (72).
2.4.4 Sodium/proton exchanger
The NHE simultaneously transports 1Na+ ion into the cells and extrudes 1H+ ion (112). NHE type 1 is ubiquitous in all tissue plasma membranes, including the heart (113). In cardiac cells, NHE is localized at intercalated disks and T-tubules, and may influence local pH (114). NHE activity is significantly increased in both patients with HF and animal models of HF (82, 115). The subsequent rise in intracellular Na+ stimulates the NCX and results in an increase in intracellular Ca2+, which promotes myocardial damage. The mechanism of SGLT2 inhibitor-mediated cardioprotection in HF is possibly through its effect on NHE. Specifically, SGLT2 inhibitors (empagliflozin, dapagliflozin, and canagliflozin) act directly on NHE, by cross reacting with the Na+-binding site of NHE, inhibiting the activity of NHE, thereby lowering the intracellular Na+ and thus producing cardioprotective effects (116). It was suggested that diminished NHE activity in the heart by SGLT2 inhibitors is due to the reduction in reactive oxygen species (ROS), oxidative stress production, cardiac inflammation, and fibrosis (117). Altogether, inhibition of NHE could open a new therapeutic avenue for HF (115).
2.5 Pacemaker current
Clinical and experimental studies have demonstrated that the sinus node function is impaired in HF (118, 119). These observations are correlated with a downregulation of the If in the sinus node pacemaker in a rabbit HF model (120). Another study in dogs with HF reported a decrease of the transcript and protein level of both HCN4 and HCN2, which explains the decrease of If and the impairment of the sinus node function indicating that the pacemaker current is remodeled in HF (119). Unlike these studies, the study conducted on human HF demonstrated an upregulation of both atrial and ventricular HCN2 and HCN4, accounts for the observed increase in If, which could contribute to atrial and ventricular arrhythmias (121). A healthy adult ventricle normally has low levels of HCN2 and HCN4. However, the expressions of these genes increases as a result of HF (122). In support of this study, clinical trials have demonstrated the beneficial effects of ivabradine, an If inhibitor, in HF (123–125). Additional studies are warranted to establish the exact role of If and its genes HCN2/HCN4 in HF.
2.6 Connexins
The electrical propagation in cardiomyocytes occurs through the connexins and forms an electrical cell-to-cell coupling (126). The connexin-43 (Cx43) protein is highly expressed in the ventricles (127). In diseased hearts such as in the case of hypertrophy and HF, Cx43 is localized at the lateral side of the ventricular myocytes (128). While Cx43 remodeling has been associated with spontaneous ventricular arrhythmia, the exact mechanisms for arrhythmogenesis are just emerging. Interestingly, the expression of Cx43 has been described to be downregulated in HF condition (129, 130). In addition, a decreased phosphorylation of Cx43 was detected in HF cardiomyocytes, which impairs the function and the localization from the intercalated disc to the lateral membrane (131, 132). Abnormalities in function and expression of Cx43 result in slow conduction in HF, which increases the propensity for arrhythmic events. Lahnwong et al. showed that pretreatment with dapagliflozin, a SGLT2 inhibitor, in a rat model of HF had lower arrhythmia score (133). The anti-arrhythmic effects of SGLT2 inhibitor are likely explained by its effect on the phosphorylation level of Cx43.
These ion channels, exchangers, and connexins remodeling in HF increase the propensity of arrhythmias. EADs and DADs represent triggers of arrhythmias. EADs are due to AP prolongation caused by a reduction of repolarizing currents, such as Ito, IKr, and IKs, and an increase the ICaL and INaL (Figure 1A). The DADs are caused by Ca2+ handling abnormalities during the diastole, due to an increased activity of NCX, decreased SERCA function, and Ca2+ leak from the RYR (Figure 1B). Furthermore, the re-entry mechanisms are the result of the remodeling process that leads to slower conduction velocities and the heterogeneity of APD, which can create conduction blocks. A reduction, observed in the HF model, of INa, If, and Cx43 favors this phenomenon and contributes to sustained arrhythmic events (Figure 1C). These ion channel changes are summarized in Table 1.
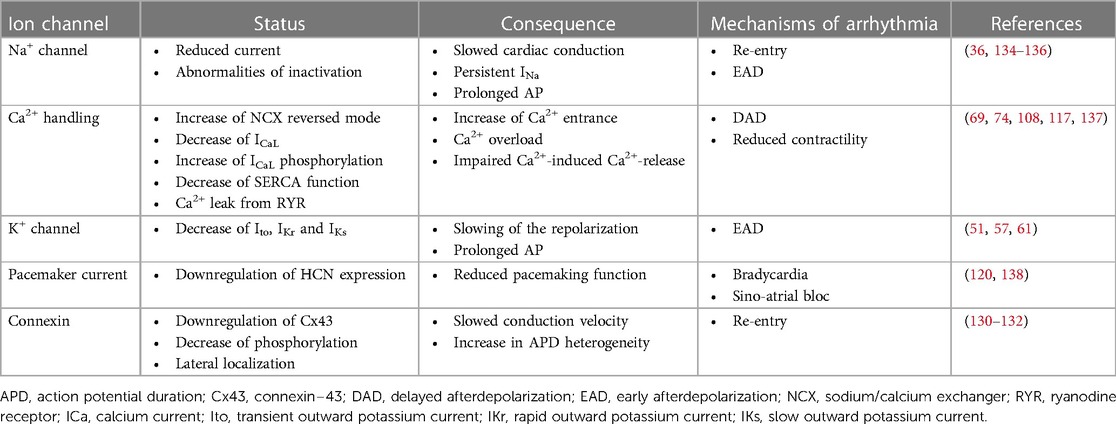
Table 1. Ion channels involvement and heart failure arrhythmogenesis.
3 Ion channel trafficking
The newly synthesized proteins go through the process of trafficking to reach their destination. The two major components are the endoplasmic reticulum (ER) and the Golgi apparatus and allow the synthesis of functional proteins. The ion channels trafficking into microdomains of the plasma membrane is critical for the anisotropic transmission of the electromechanical signal at the tissue level (139).
The starting point of ion channels trafficking is in the ER, where the ion channels are synthetized by the ribosomes. A quality control is carried out in the ER before the protein is transported to the Golgi (140). The newly formed proteins are supported by chaperone proteins (calnexin, calreticulin) which will facilitate their maturation and their oligomerization (Figure 2A) (141). However, misassembled ion channels can exhibit the ER-retention signals. Among these signals, dual bi-arginine (RXR) and C-terminal Lys-Asp-Glu-Leu (KDEL) motifs are well known (142, 143). A misfolded protein in the ER triggers the unfolded protein response (UPR) using three pathways such as inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase R-like ER kinase (PERK) (11, 144). These pathways will activate the genes involved in the ER-associated degradation system (ERAD) and induce a cytosolic degradation by the proteasome (Figure 2B) (145). In the Golgi, the ion channels undergo post-translational modifications, such as the glycosylation before being trafficked to the plasma membrane (146, 147). Many ion channels and their subunits are glycosylated. N-glycosylation will regulate the stability and the expression level of the membrane channels, their transport, and also their biophysical properties (148, 149).
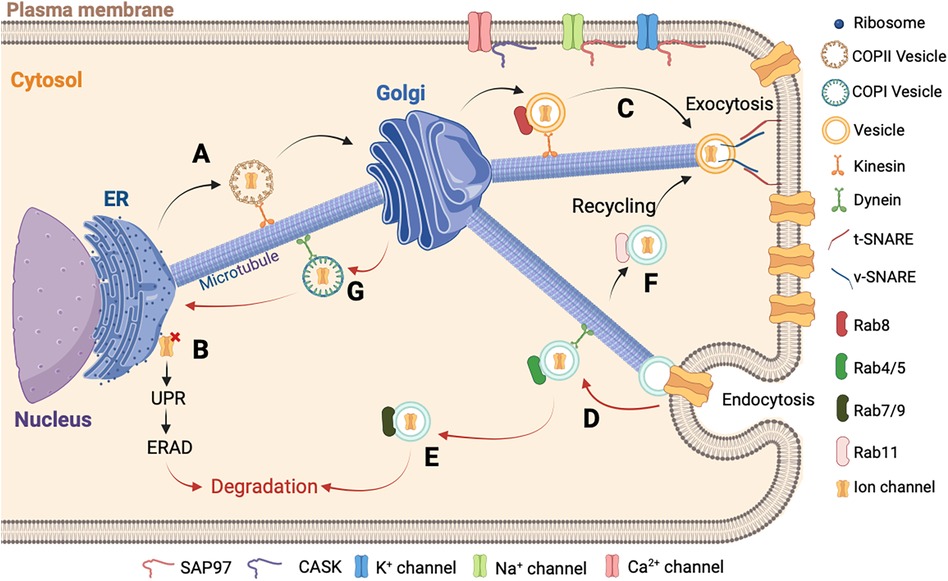
Figure 2. Schematic representation of ion channels trafficking. (A) The newly synthesized proteins in the ER are carried by the COPII vesicles until the Golgi apparatus in the anterograde direction. (B) In cases where the assembly of the newly synthesized protein is misfolded, it is retained in the ER followed by the activation of the UPR and ERAD to allow its degradation in the cytosol. (C) After the final maturation process, the new ion channel protein is directed to the plasma membrane by a vesicle. At the final localization, the v-SNARE and t-SNARE proteins interact to allow the fusion between the vesicle and the plasma membrane. (D) Then the ion channel is recaptured into the cell by endocytosis and carried out by a vesicle with Rab4/5 protein and dynein to allow the retrograde direction. (E) The ion channel is transported by a late vesicle with Rab7/9 protein and degraded in the cytosol, or (F) is recycled by another vesicle with the Rab11 protein. (G) The final step of the ion channel trafficking is carried out by the COPI vesicle for the retrograde direction to be recycled into the ER. It is important to note that the accessory molecules, such as CASK and SAP97, stabilize the ion channel at the destination.
Proteins will leave the ER through a region without ribosomes called ER Exist Site (ERES) (150, 151). In this region, the ion channels are packaged in coatomer protein II (COPII) vesicles. These COPII vesicles ensure anterograde transport from the ER to the Golgi, whereas the coatomer protein I (COPI) vesicles mediate the retrograde transport (Figure 2) (152, 153).
In cardiomyocytes, the transport of ion channels needs molecular motors and regulators. In the cytoplasm, the ion channel trafficking is coordinated by various Rab GTPases (154). Rab proteins belong to the largest family of small Ras-like GTPases. Active in the presence of GTP and inactive in the presence of GDP, they are associated with practically all stages of vesicular transport, including the Golgi stages. Rab8 is involved in the anterograde direction for the delivery of newly synthetized ion channels (155). Rab4 and Rab5 are associated with vesicles following ion channel internalization (156, 157). Rab7 and Rab9 are associated with the late endosome and Rab11 is associated with the recycling endosome (156, 158). In the anterograde direction, the transportation is provided by the action of different kinesins and thereafter along the actin cytoskeleton by the action of myosin V. In the retrograde direction, the dynein associated with the microtubules and actin carry out the transportation (Figure 2) (159, 160).
Another important aspect of ion channels regulation is their anchoring and aggregation in different areas of the sarcolemma. Many adapter proteins capable of interacting with the channels and regulating their membrane localizations have been identified, such as the membrane-associated guanylate kinase (MAGUK) protein family (161). Most MAGUKs have one or more PDZ domains, an SH3 domain, and a GUK domain. The presence of these many protein interaction domains allows MAGUK to interact with multiple partners simultaneously and explains their multiple roles such as anchoring and aggregation of ion channels in specialized areas of cellular communication, scaffolding of macromolecular complexes, functional interaction of channels with signaling elements, and intracellular trafficking (162). For example, cardiomyocytes express several MAGUK proteins, where the most important is SAP97 (synapse-associated protein 97), which interacts with the major ion currents, IKr, IK1, and INa (Figure 2) (163–165). Another protein of the MAGUK family participates in the trafficking and anchoring of ion channels, the Ca2+/calmodulin-dependent serine protein kinase (CASK) protein. This proteins has a domain homologous to CAMKII. The CASK protein is composed of the combination of typical MAGUK domains plus two L27 domains and an N-terminal CAMKII domain (166). Studies have demonstrated that the CASK protein is involved in the Ca2+ channel trafficking and its stabilization at the membrane level (Figure 2) (167). In addition, the expression and phosphorylation levels of hERG are dependent on the presence of CASK protein (168). Furthermore, CASK protein interacts with the Nav1.5 channel and regulates its trafficking and its surface expression at the lateral membrane (169).
The end stage of the ion channel trafficking corresponds to the attachment of vesicles to the membrane. This attachment is carried out by the N-ethylmaleimide sensitive fusion protein (NSF), the NSF attachment proteins (SNAP), and the SNAP REceptor (SNARE) proteins (170, 171). Each transport vesicle contains one or more v-SNAREs (v for vesicle), which interacts with a t-SNARE (t for target) on the target membrane. The assembly of the pair of SNARE proteins is regulated by different types of proteins including the GTPases of the Ypt/Rab family and the Sec1 family (170, 172). This process is catalyzed by NSF and SNAPs. This assembly will destabilize the lipid bilayers and permits the ion channel to be directed to the plasma membrane (Figure 2C) (173). Another contributor to the ion channel trafficking is ankyrin. Ankyrin constitutes a group of intracellular proteins responsible for arranging, transporting, and securing membrane protein complexes to the actin/spectrin cytoskeleton, thereby establishing microdomains within membranes that exhibit functional properties (174). The isoform G and B are present in the heart tissue and are implicated in the stabilization of ion channels at the membrane level (175). The ankyrin-G was identified to stabilize the Nav1.5 channel in the heart and is important for the localization at the intercalated discs as described in the ankyrin-G knock-out mice (176). The ankyrin-B interacts with the NCX, the Cav1.3 channel, and stabilizes the SERCA, three important Ca2+ transporters (177, 178). A defect of ankyrin-B was previously described to induce Ca2+ abnormalities, increase injury, and reduce contractility in a mouse heart that is ankyrin-B deficient (179).
It is important to note that the level of ion channels in the plasma membrane is also dependent on a balance between the phenomena of exocytosis and endocytosis, recycling, and degradation. This balance is important to allow a satisfactory regulation of ion channel activity.
4 Ion channel trafficking defect contributes to the development of heart failure
Cardiomyocytes have distinct microdomains, such as t-tubules, lateral membranes, and intercalated discs. These microdomains have precise physiological roles in ion channels trafficking to microdomains and hence it is crucial to maintain these specific roles. However, in HF, a mislocalization or redistribution of ion channels disrupts the normal electrical gradients and alters the propagation of electrical signals. The different aspects of the ion channel trafficking in HF are summarized in Table 2.

Table 2. Summary of ion channel trafficking alterations and their consequences in heart failure.
Since the 1990s, it was discovered that variants in ion channels are responsible for trafficking defects, leading to the development of HF and arrhythmias (Figure 3A) (199). Also, several abnormal post-translational modifications, which affect many cellular pathways, have been reported in patients with HF (183). Among these modifications, alterations in phosphorylation, glycosylation, ubiquitin, acetylation, and succinylation were reported (168, 200, 201). Several enzymes control these post-translational modifications and are summarized in Table 3. Alterations in these processes can lead to the degradation of channel proteins by the proteasome leading to electrical abnormalities such as the long QT syndrome or arrhythmias associated with HF, revealing its importance in the functional expression of ion channels (Figure 3B) (184). In recent years, the studies conducted on post-translational modifications have gradually increased and become a new field of medical research for the HF (207–209).
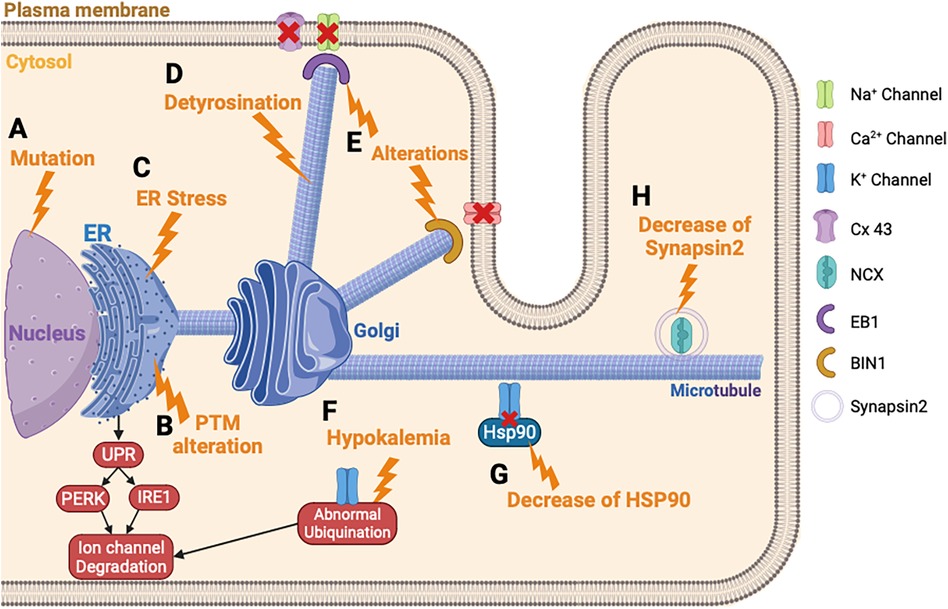
Figure 3. Ion channels trafficking perturbation in heart failure. (A) Ion channel variant leads to ER retention of the ion channel. (B) PTM alteration triggers the ion channel retention in the ER and the degradation of the ion channel. (C) ER stress leads to an abnormal degradation of the ion channel protein by the UPR response. (D) The detyrosination of the microtubules alters the ion channel trafficking. (E) Alteration of EB1 or BIN1 inhibits the ion channel incorporation into the membrane. (F) Hypokalemia triggers an abnormal ubiquitination of hERG and its degradation. (G) Decrease of HSP90 chaperone disturbs the K+ channel trafficking. (H) Decrease of synapsin2 vesicle decreases the trafficking of NCX. PTM, post-translational modification.
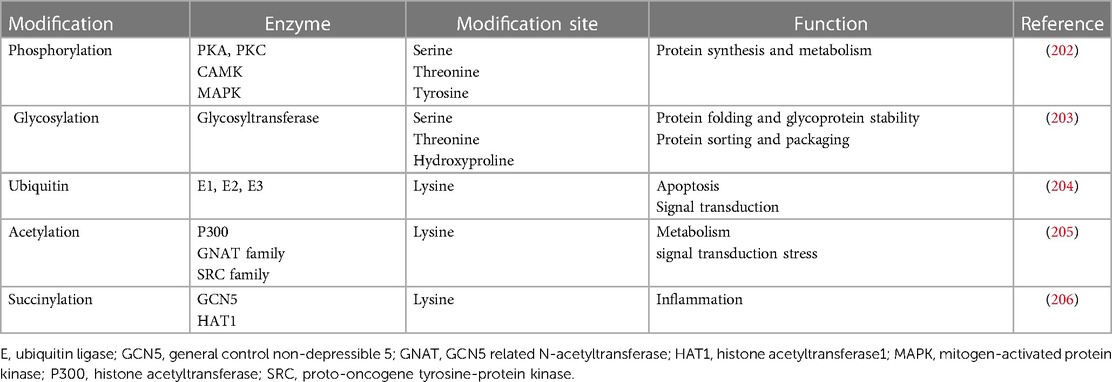
Table 3. Post-translational modifications in heart failure.
The ER, which is the first step of the maturation and trafficking of ion channels, is impacted by HF, which induces a stress of this cellular compartment (210, 211). In mice injected with tunicamycin to induce HF, a prolonged activation of UPR and PERK is provoked, which activates the degradation pathway of the newly formed proteins by ERAD (Figure 3C) (185). This study showed that INa, IKr, and IK1 are downregulated by this mechanism, in response to ER stress induced by HF. Each ion channel has specific trafficking defects, detailed as follows.
4.1 Sodium current
Most trafficking defects come from a direct variant on ion channels, which induces a misfolded protein and the retention in ER. Variants in the Na+ channel were discovered to be linked with a trafficking defect, leading to HF (212). A recent study demonstrated that two different variants of Nav1.5 can interact with each other to rescue the INa (181). Specifically, this study showed that one variant of Na+ channel, trafficking-deficient but gating-competent, was able to restore a fraction of the INa by interacting with another Na+ channel variant that was trafficking-competent but gating-deficient. This study supports the notion that Na+ channel α-subunits interact and cooperate with each other. The interaction between Na+ channels will allow to rescue the trafficking and could lead to a new approach to treat patients.
The post-translational modifications are important for the trafficking of Na+ channels (213). Specifically, the phosphorylation of Na+ channel protein process is well known to be a key factor in the trafficking (214, 215). The PKA, protein kinase C (PKC), and CAMKII are among the most abundant kinases expressed in the heart (216, 217). More recently, a study conducted on rabbit cardiomyocyte showed that the ubiquitination of Na+ channel by the ubiquitin ligase NEDD4 could regulate the endosomal trafficking (218). Targeting these enzymes could lead to enhancing the ion channel trafficking of Na+ channel and rescue the INa in HF. Interestingly, empagliflozin was recently described to reduce the INaL by inhibition of CAMKII activity (219). This medication could be used to restore a normal INa in patients with HF.
Changes in microtubule dynamics are known to alter ion channel trafficking. In fact, an upregulation of the detyrosination of microtubules can induce HF (Figure 3D) (187). The detyrosination is a post-translational modification of tubulin, the protein building block of microtubules, which are essential components of the cytoskeleton. In this process, the C-terminal tyrosine residue of tubulin is removed enzymatically, resulting in a stable and long-lived form of the microtubules. It was previously described that the detyrosination reduces the density and the peak INa in mice cardiomyocytes (220). It will be interesting to suppress the detyrosination to restore normal ion channel trafficking of the Na+ channel. Chen et al. showed that a pharmacological suppression of detyrosinated microtubules restores 50% of the lost contractile function in the left ventricular myocardium of a failing human heart (221).
Furthermore, Marchal et al. demonstrated in human-induced pluripotent stem cells-derived cardiomyocytes (hiPSC-CM) that the plus-end binding protein (EB1) improves the trafficking and the function of Na+ channel trafficking (190). In mammals, the EB protein family consists of three proteins: EB1, EB2, and EB3, which bind directly to the end of microtubules and facilitate the anchoring of proteins at the plasma membrane (222, 223). In addition, other microtubule plus-end tracking proteins, also called +TIP, have been found to be associated with the Na+ channel and potentially involved in its trafficking. For example, alterations in EB1, EB2, CLASP, CLIP1 CENP-F, MACF1, or iASPP are linked to a decrease in conduction or abnormal contraction, thereby emphasizing the impact of +TIP function on cardiac electrophysiology and function (Figure 3E) (189, 192, 224). One of the consequences of HF is the dysregulation of microtubule homeostasis, which is necessary for guiding ion channels to the membrane and this process explains the reduced INa reported in patients with HF.
4.2 Potassium current
Variants in K+ channels were the first described and represent the most important factor in the development of HF (52, 225). Indeed, a study described that 90% of hERG variants inhibited the trafficking from the ER to the Golgi (226). These variants cause hERG transport disorders by stimulating the UPR and leading the degradation of the ion channel protein (182). Variants on KCNQ1 were also revealed to play a role in the abnormal KCNQ1 trafficking, leading to HF and long QT syndrome (227, 228). Furthermore, another study described that a variant of KCNQ1 impaired the hERG trafficking (229). Indeed, this variant of KCNQ1 interacts in the cytosol with hERG and impairs the trafficking. The alteration of hERG trafficking, caused by the KCNQ1 variant, leads to HF and long QT syndrome.
Hypokalemia or hyperkaliemia disturbs the trafficking of hERG (198). Guo et al. were the first to reveal how the density of hERG channels is regulated under hypokalemic conditions (230). This situation triggered an ubiquitination of hERG, internalization, and finally degradation (231) (Figure 3F).
The trafficking of K+ channels, especially hERG protein, involves a chaperone protein from the family of heat shock protein (HSP) (232, 233). HSP90 and HSP70 interact with hERG and play a crucial role for the maturation. In HF, the role of these chaperone proteins was not well described, but it was suggested as playing a potential role for therapeutic modulation of HSP in HF (196). Indeed, an inhibition of HSP90 in mice model of HF demonstrated a preserved left ventricular pressure (234). More recently, this team showed that simvastatin also inhibits HSP90 and reduces cardiac remodeling in HF rat model (235). For hERG protein trafficking, a study showed that the reduced expression level of hERG protein is linked with impaired trafficking because of the diminished interaction between hERG and HSP90 (Figure 3G) (197). This observation could explain the decrease of IK observed in HF, and showed the potential therapeutic role of HSP90 in HF (236). There is limited knowledge regarding the trafficking defects for the Ito and IK1 in HF. However, the Kv channel-interacting protein (KChIP) is downregulated in HF and this contributes to the inhibition of the trafficking of Ito (237). Furthermore, one study mentioned the N-glycosylation defect of TREK channel, which contributes to reducing the IK in HF (238).
4.3 Calcium handling
Ca2+ channel trafficking and function are regulated by auxiliary protein subunits, including the β-subunits, which help in their trafficking (239, 240). These β-subunits are important in facilitating the Ca2+ channel exiting from the ER (241), and different β-subunit variants have been involved in HF (180, 241). ER stress also induced a repression of Ca2+ channel trafficking. Indeed, a study conducted on hiPSC-CM showed that the activation IRE1, a pathway activated by the UPR response, decreases the protein level of the Ca2+ channel (Figure 3B) (186). In failing human hearts a decrease of Myc box-dependent-interacting protein 1 (BIN1) protein has been demonstrated. In myocytes, BIN1 facilitates microtubule-based delivery of Cav1.2 channels directly to T-tubules for normal Ca2+ transient development (242). BIN1 acts like a membrane scaffolding protein to anchor the Ca2+ channel at the plasma membrane and could explain the decrease of ICaL observed in HF (193) (Figure 3E). In case of this channel, a recent study conducted in mice knock-out for CASK demonstrated that the loss of ICaL accelerates HF development (194). This latest study demonstrated the importance of the actors involved in the trafficking of ion channels and that their destabilization can lead to aggravating the development of HF.
Furthermore, studies have shown that the post-translational modifications could affect the other components of the Ca2+ handling like the RYR2. PKA hyperphosphorylation, CAMKII, oxidation, and nitrosylation trigger the destabilization and dissociation of FKBP12.6 from the channel and contribute to SR Ca2+ leak that is thought to induce arrhythmias and decrease myocardium contractile strength in HF (243). These post-translational modifications can cause defects of RYR2 trafficking and contribute to development of HF.
Few studies attempted to understand the mechanisms of NCX trafficking in HF. In fact, the NCX activity in HF occurs more in response to a persistent INa that increases its reverse mode or by a Ca2+ leak coming from the SR, thus increasing its activity and its arrhythmic role. However, a recent study highlighted the role of the synapsin-2 on the NCX regulation of trafficking in HF (195). The synapsin-2 is a protein associated with the vesicle transport during protein trafficking. First, the authors showed a decrease of synapsin-2 expression in the left ventricle in mice model of HF and demonstrated a co-localization of synapsin-2 and NCX. These data suggest that reduced synapsin-2 in HF alters the NCX trafficking and leads to ventricular arrhythmias (Figure 3H) (195).
4.4 Connexin
For the Cx43, the localization of this hemichannel is at the intercalated discs. The trafficking of Cx43 involves a major plus-end EB1 protein (244, 245). EB1 allows anterograde movement to the intercalated discs by linking the microtubules to the desmosomes via the interaction with another plus-end protein (p150) (246). In case of HF, oxidative stress causes the microtubule plus-end EB1 dissociation from the end of the microtubules (Figure 3E). Consequently, it impairs the microtubule attachment to adherent junction structures and the delivery of Cx43 to the plasma membrane at the intercalated discs (191, 247). This observation could explain the decrease of Cx43 observed in patients with HF (248).
Collectively, ion channel trafficking disturbances lead to HF. A better understanding of these mechanisms allows to develop new therapeutic strategies or to better understand certain therapies used. First, targeting HSP chaperone proteins, suppressing microtubule detyrosination, or even targeting Rab recycling proteins could be interesting new strategies to restore ion channels trafficking. Therapy using an adeno-associated virus (AAV), as in the CUPID2 study, does not seem to be the best solution as of yet in the treatment of HF (111). However, therapies carried by SGLT2 inhibitors seem the most promising for trafficking restoration, specifically empagliflozin, which makes it possible to restore normal ion channels trafficking through its chaperone-like effect by increasing the forward trafficking of ion channels (249). Finally, Entresto® is a new therapeutic option to reduce the occurrence of arrhythmias in patients with HF. Its precise role in the treatment of these arrhythmias is still being explored, but it appears that its role affects the three pathways of B-type natriuretic peptide, angiotensin II, and bradykinin (250). Its role in ion channel trafficking is yet to be discovered.
5 Summary
Ion channel trafficking is a central regulator of cardiac electrophysiology, contractility, and alterations in the cell homeostasis in HF, thereby contributing to arrhythmias. Several actors are involved in this process, from the formation of new synthesized proteins to their assembly at the membrane. HF can induce an intracellular stress, which also disturbs the ion channels trafficking and accelerates the development of the electrical activity and function of the failing heart. However, few studies demonstrate the direct link between the HF and protein trafficking. A better understanding of the mechanisms leading to ion channels trafficking disorders in HF could open new therapeutic targets.
Author contributions
JR: Conceptualization, Investigation, Validation, Writing – original draft, Writing – review & editing. YQ: Investigation, Validation, Writing – original draft. MB: Funding acquisition, Supervision, Validation, Writing – review & editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by a Merit Review Grant from Biomedical Laboratory Research & Development Service of Veterans Affairs Office of Research and Development (I01 BX002137 to MB), from the National Heart, Lung, and Blood Institute (1R01HL164415 to MB), and from the US Department of Defense (W81XWH-21-1-0424 to MB).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med. (1971) 285:1441–6. doi: 10.1056/NEJM197112232852601
2. Sapna F, Raveena F, Chandio M, Bai K, Sayyar M, Varrassi G, et al. Advancements in heart failure management: a comprehensive narrative review of emerging therapies. Cureus. (2023) 15:e46486. doi: 10.7759/cureus.46486
3. Abovich A, Matasic DS, Cardoso R, Ndumele CE, Blumenthal RS, Blankstein R, et al. The AHA/ACC/HFSA 2022 heart failure guidelines: changing the focus to heart failure prevention. Am J Prev Cardiol. (2023) 15:100527. doi: 10.1016/j.ajpc.2023.100527
4. Nicholson C. Chronic heart failure: pathophysiology, diagnosis and treatment. Nurs Older People. (2014) 26:29–38. doi: 10.7748/nop.26.7.29.e584
5. Clark KAA, Reinhardt SW, Chouairi F, Miller PE, Kay B, Fuery M, et al. Trends in heart failure hospitalizations in the US from 2008 to 2018. J Card Fail. (2022) 28:171–80. doi: 10.1016/j.cardfail.2021.08.020
6. Lam CSP, Solomon SD. Classification of heart failure according to ejection fraction: JACC review topic of the week. J Am Coll Cardiol. (2021) 77:3217–25. doi: 10.1016/j.jacc.2021.04.070
7. Balan I, Khayo T, Sultanova S, Lomakina Y. Overview of sodium-glucose co-transporter 2 (SGLT2) inhibitors for the treatment of non-diabetic heart failure patients. Cureus. (2021) 13:e17118. doi: 10.7759/cureus.17118
8. Burnett H, Earley A, Voors AA, Senni M, McMurray JJV, Deschaseaux C, et al. Thirty years of evidence on the efficacy of drug treatments for chronic heart failure with reduced ejection fraction. Circ Heart Fail. (2017) 10:e003529. doi: 10.1161/CIRCHEARTFAILURE.116.003529
9. Jadhav U, Nair T, Mohanan P, Chopra V, Kerkar P, Das Biswas A, et al. Impact of mineralocorticoid receptor antagonists in the treatment of heart failure: targeting the heart failure cascade. Cureus. (2023) 15:e45241. doi: 10.7759/cureus.45241
10. Buffolo F, Tetti M, Mulatero P, Monticone S. Aldosterone as a mediator of cardiovascular damage. Hypertension. (2022) 79:1899–911. doi: 10.1161/HYPERTENSIONAHA.122.17964
11. Fan R, Wang S, Wu Y, Feng Y, Gao M, Cao Y, et al. Activation of endoplasmic reticulum stress by harmine suppresses the growth of esophageal squamous cell carcinoma. Phytother Res. (2023) 37:4655–73. doi: 10.1002/ptr.7933
12. Beldhuis IE, Myhre PL, Bristow M, Claggett B, Damman K, Fang JC, et al. Spironolactone in patients with heart failure, preserved ejection fraction, and worsening renal function. J Am Coll Cardiol. (2021) 77:1211–21. doi: 10.1016/j.jacc.2020.12.057
13. Rossier MF, Lenglet S, Vetterli L, Python M, Maturana A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertens Dallas Tex. (2008) 52:721–8. doi: 10.1161/HYPERTENSIONAHA.108.114223
14. Triposkiadis F, Briasoulis A, Sarafidis P, Magouliotis D, Athanasiou T, Paraskevaidis I, et al. The sympathetic nervous system in hypertensive heart failure with preserved LVEF. J Clin Med. (2023) 12:6486. doi: 10.3390/jcm12206486
15. Bencivenga L, Liccardo D, Napolitano C, Visaggi L, Rengo G, Leosco D. β-adrenergic receptor signaling and heart failure: from bench to bedside. Heart Fail Clin. (2019) 15:409–19. doi: 10.1016/j.hfc.2019.02.009
16. Hall SA, Cigarroa CG, Marcoux L, Risser RC, Grayburn PA, Eichhorn EJ. Time course of improvement in left ventricular function, mass and geometry in patients with congestive heart failure treated with beta-adrenergic blockade. J Am Coll Cardiol. (1995) 25:1154–61. doi: 10.1016/0735-1097(94)00543-y
17. Bristow MR. Mechanism of action of beta-blocking agents in heart failure. Am J Cardiol. (1997) 80:26l–40l. doi: 10.1016/s0002-9149(97)00846-1
18. Garnier A, Leroy J, Deloménie C, Mateo P, Viollet B, Veksler V, et al. Modulation of cardiac CAMP signaling by AMPK and its adjustments in pressure overload-induced myocardial dysfunction in rat and mouse. PLoS One. (2023) 18:e0292015. doi: 10.1371/journal.pone.0292015
19. Prijic S, Buchhorn R. Mechanisms of beta-blockers action in patients with heart failure. Rev Recent Clin Trials. (2014) 9:58–60. doi: 10.2174/1574887109666140908125402
20. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. (2023) 118:3272–87. doi: 10.1093/cvr/cvac013
21. Ravens U, Peyronnet R. Electrical remodelling in cardiac disease. Cells. (2023) 12:230. doi: 10.3390/cells12020230
22. Bers DM. Cardiac excitation-contraction coupling. Nature. (2002) 415:198–205. doi: 10.1038/415198a
23. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. (2005) 85:1205–53. doi: 10.1152/physrev.00002.2005
24. Varró A, Tomek J, Nagy N, Virág L, Passini E, Rodriguez B, et al. Cardiac transmembrane ion channels and action potentials: cellular physiology and arrhythmogenic behavior. Physiol Rev. (2021) 101:1083–176. doi: 10.1152/physrev.00024.2019
25. Garg P, Garg V, Shrestha R, Sanguinetti MC, Kamp TJ, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes as models for cardiac channelopathies: a primer for non-electrophysiologists. Circ Res. (2018) 123:224–43. doi: 10.1161/CIRCRESAHA.118.311209
26. Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. (2002) 90:939–50. doi: 10.1161/01.res.0000018627.89528.6f
27. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. (2010) 90:291–366. doi: 10.1152/physrev.00021.2009
28. Klein MG, Shou M, Stohlman J, Solhjoo S, Haigney M, Tidwell RR, et al. Role of suppression of the inward rectifier current in terminal action potential repolarization in the failing heart. Heart Rhythm. (2017) 14:1217–23. doi: 10.1016/j.hrthm.2017.04.001
29. Chen K, Zuo D, Wang S-Y, Chen H. Kir2 inward rectification-controlled precise and dynamic balances between Kir2 and HCN currents initiate pacemaking activity. FASEB J. (2018) 32:3047–57. doi: 10.1096/fj.201701260R
30. DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol. (1993) 55:455–72. doi: 10.1146/annurev.ph.55.030193.002323
31. Husti Z, Varró A, Baczkó I. Arrhythmogenic remodeling in the failing heart. Cells. (2021) 10:3203. doi: 10.3390/cells10113203
32. Rahm A-K, Lugenbiel P, Schweizer PA, Katus HA, Thomas D. Role of ion channels in heart failure and channelopathies. Biophys Rev. (2018) 10:1097–106. doi: 10.1007/s12551-018-0442-3
33. Brette F, Dos Santos P, Hulot J-S. Editorial: heart failure with preserved ejection fraction: basic, translational, and clinical research. Front Physiol. (2022) 13:1092009. doi: 10.3389/fphys.2022.1092009
34. Shou J, Huo Y. Changes of calcium cycling in HFrEF and HFpEF. Mechanobiol Med. (2023) 1:100001. doi: 10.1016/j.mbm.2023.100001
35. Hou J, Wen X, Long P, Xiong S, Liu H, Cai L, et al. The role of post-translational modifications in driving abnormal cardiovascular complications at high altitude. Front Cardiovasc Med. (2022) 9:886300. doi: 10.3389/fcvm.2022.886300
36. Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quiñones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. (2001) 276:28197–203. doi: 10.1074/jbc.M102548200
37. Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. (2007) 9:219–27. doi: 10.1016/j.ejheart.2006.08.007
38. Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. (2005) 38:475–83. doi: 10.1016/j.yjmcc.2004.12.012
39. Chadda KR, Jeevaratnam K, Lei M, Huang CL-H. Sodium channel biophysics, late sodium current and genetic arrhythmic syndromes. Pflugers Arch. (2017) 469:629–41. doi: 10.1007/s00424-017-1959-1
40. Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc Res. (2006) 69:116–27. doi: 10.1016/j.cardiores.2005.08.015
41. Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN, Undrovinas A. Contribution of sodium channel neuronal isoform Nav1.1 to late sodium current in ventricular myocytes from failing hearts. J Physiol. (2015) 593:1409–27. doi: 10.1113/jphysiol.2014.278259
42. Munger MA, Olğar Y, Koleske ML, Struckman HL, Mandrioli J, Lou Q, et al. Tetrodotoxin-sensitive neuronal-type Na+ channels: a novel and druggable target for prevention of atrial fibrillation. J Am Heart Assoc. (2020) 9:e015119. doi: 10.1161/JAHA.119.015119
43. Kim K, Kim S, Katana M, Terentyev D, Radwański PB, Munger M. Riluzole is associated with reduced risk of heart failure. medRxiv [preprint]. (2023). Available at: 10.1101/2023.12.28.23300552
44. Bai J, Wang K, Bai X, Yuan Y, Zhang H. Pro-arrhythmic effects of increased late sodium current in failing human heart. Computing in Cardiology 2014 (2014). pp. 857–60.
45. Fukaya H, Plummer BN, Piktel JS, Wan X, Rosenbaum DS, Laurita KR, et al. Arrhythmogenic cardiac alternans in heart failure is suppressed by late sodium current blockade by ranolazine. Heart Rhythm. (2019) 16:281–9. doi: 10.1016/j.hrthm.2018.08.033
46. Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, et al. Altered Na(+) currents in atrial fibrillation: effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. (2010) 55:2330–42. doi: 10.1016/j.jacc.2009.12.055
47. Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, et al. Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation. (2021) 143:2188–204. doi: 10.1161/CIRCULATIONAHA.121.053350
48. Hegyi B, Mira Hernandez J, Shen EY, Habibi NR, Bossuyt J, Bers DM. Empagliflozin reverses late Na + current enhancement and cardiomyocyte proarrhythmia in a translational murine model of heart failure with preserved ejection fraction. Circulation. (2022) 145:1029–31. doi: 10.1161/CIRCULATIONAHA.121.057237
49. Horváth B, Hézső T, Kiss D, Kistamás K, Magyar J, Nánási PP, et al. Late sodium current inhibitors as potential antiarrhythmic agents. Front Pharmacol. (2020) 11:413. doi: 10.3389/fphar.2020.00413
50. Wagner S, Maier LS, Bers DM. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res. (2015) 116:1956–70. doi: 10.1161/CIRCRESAHA.116.304678
51. Hegyi B, Bossuyt J, Ginsburg KS, Mendoza LM, Talken L, Ferrier WT, et al. Altered repolarization reserve in failing rabbit ventricular myocytes: calcium and β-adrenergic effects on delayed- and inward-rectifier potassium currents. Circ Arrhythm Electrophysiol. (2018) 11:e005852. doi: 10.1161/CIRCEP.117.005852
52. Näbauer M, Kääb S. Potassium channel down-regulation in heart failure. Cardiovasc Res. (1998) 37:324–34. doi: 10.1016/s0008-6363(97)00274-5
53. Rahm A-K, Müller ME, Gramlich D, Lugenbiel P, Uludag E, Rivinius R, et al. Inhibition of cardiac Kv4.3 (Ito) channel isoforms by class I antiarrhythmic drugs lidocaine and mexiletine. Eur J Pharmacol. (2020) 880:173159. doi: 10.1016/j.ejphar.2020.173159
54. Zicha S, Xiao L, Stafford S, Cha TJ, Han W, Varro A, et al. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. (2004) 561:735–48. doi: 10.1113/jphysiol.2004.075861
55. Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. (2011) 51:468–73. doi: 10.1016/j.yjmcc.2011.01.012
56. Wei X, Jin J, Wu J, He Y, Guo J, Yang Z, et al. Cardiac-specific BACH1 ablation attenuates pathological cardiac hypertrophy by inhibiting the Ang II type 1 receptor expression and the Ca2+/CaMKII pathway. Cardiovasc Res. (2023) 119:1842. doi: 10.1093/cvr/cvad086
57. Bossuyt J, Borst JM, Verberckmoes M, Bailey LRJ, Bers DM, Hegyi B. Protein kinase D1 regulates cardiac hypertrophy, potassium channel remodeling, and arrhythmias in heart failure. J Am Heart Assoc. (2022) 11:e027573. doi: 10.1161/JAHA.122.027573
58. Li G-R, Lau C-P, Ducharme A, Tardif J-C, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. (2002) 283:H1031–41. doi: 10.1152/ajpheart.00105.2002
59. Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, et al. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. (2005) 288:H2077–87. doi: 10.1152/ajpheart.00526.2003
60. Ning B, Zhang F, Song X, Hao Q, Li Y, Li R, et al. Cardiac contractility modulation attenuates structural and electrical remodeling in a chronic heart failure rabbit model. J Int Med Res. (2020) 48:300060520962910. doi: 10.1177/0300060520962910
61. Tsuji Y, Zicha S, Qi X-Y, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. (2006) 113:345–55. doi: 10.1161/CIRCULATIONAHA.105.552968
62. Chang P-C, Chen P-S. SK Channels and ventricular arrhythmias in heart failure. Trends Cardiovasc Med. (2015) 25:508–14. doi: 10.1016/j.tcm.2015.01.010
63. Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, et al. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. (2005) 289:H2714–23. doi: 10.1152/ajpheart.00534.2005
64. Bonilla IM, Long VP, Vargas-Pinto P, Wright P, Belevych A, Lou Q, et al. Calcium-activated potassium current modulates ventricular repolarization in chronic heart failure. PLoS One. (2014) 9:e108824. doi: 10.1371/journal.pone.0108824
65. Chang P-C, Turker I, Lopshire JC, Masroor S, Nguyen B-L, Tao W, et al. Heterogeneous upregulation of apamin-sensitive potassium currents in failing human ventricles. J Am Heart Assoc. (2013) 2:e004713. doi: 10.1161/JAHA.112.004713
66. Lugenbiel P, Wenz F, Syren P, Geschwill P, Govorov K, Seyler C, et al. TREK-1 (K2P2.1) K+ channels are suppressed in patients with atrial fibrillation and heart failure and provide therapeutic targets for rhythm control. Basic Res Cardiol. (2017) 112:8. doi: 10.1007/s00395-016-0597-7
67. Shugg T, Somberg JC, Molnar J, Overholser BR. Inhibition of human ether-a-go-go-related gene (hERG) potassium current by the novel sotalol analogue, soestalol. JACC Clin Electrophysiol. (2020) 6:756–9. doi: 10.1016/j.jacep.2020.05.037
68. Phadumdeo VM, Mallare BL, Hund TJ, Weinberg SH. Long-term changes in heart rate and electrical remodeling contribute to alternans formation in heart failure: a patient-specific in silico study. Am J Physiol Heart Circ Physiol. (2023) 325:H414–31. doi: 10.1152/ajpheart.00220.2023
69. Hersel J, Jung S, Mohacsi P, Hullin R. Expression of the L-type calcium channel in human heart failure. Basic Res Cardiol. (2002) 97:1–1. doi: 10.1007/s003950200022
70. Kepenek ES, Ozcinar E, Tuncay E, Akcali KC, Akar AR, Turan B. Differential expression of genes participating in cardiomyocyte electrophysiological remodeling via membrane ionic mechanisms and Ca2+-handling in human heart failure. Mol Cell Biochem. (2020) 463:33–44. doi: 10.1007/s11010-019-03626-4
71. Takahashi T, Allen PD, Lacro RV, Marks AR, Dennis AR, Schoen FJ, et al. Expression of dihydropyridine receptor (Ca2+channel) and calsequestrin genes in the myocardium of patients with end-stage heart failure. J Clin Invest. (1992) 90:927–35. doi: 10.1172/JCI115969
72. Benitah J-P, Perrier R, Mercadier J-J, Pereira L, Gómez AM. RyR2 and calcium release in heart failure. Front Physiol. (2021) 12:734210. doi: 10.3389/fphys.2021.734210
73. Schröder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, et al. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. (1998) 98:969–76. doi: 10.1161/01.cir.98.10.969
74. Srivastava U, Jennings-Charles R, Qu YS, Sossalla S, Chahine M, Boutjdir M. Novel re-expression of L-type calcium channel Cav1.3 in left ventricles of failing human heart. Heart Rhythm. (2020) 17:1193–7. doi: 10.1016/j.hrthm.2020.02.025
75. Sanchez-Alonso JL, Fedele L, Copier JS, Lucarelli C, Mansfield C, Judina A, et al. Functional LTCC-β2AR complex needs caveolin-3 and is disrupted in heart failure. Circ Res. (2023) 133:120–37. doi: 10.1161/CIRCRESAHA.123.322508
76. Schwartz K, Mercadier JJ. Molecular and cellular biology of heart failure. Curr Opin Cardiol. (1996) 11:227–36. doi: 10.1097/00001573-199605000-00002
77. Zaveri S, Srivastava U, Qu YS, Chahine M, Boutjdir M. Pathophysiology of Cav1.3 L-type calcium channels in the heart. Front Physiol. (2023) 14:1144069. doi: 10.3389/fphys.2023.1144069
78. Huang B, Qin D, Deng L, Boutjdir M, El-Sherif N. Reexpression of T-type Ca2+channel gene and current in post-infarction remodeled rat left ventricle. Cardiovasc Res. (2000) 46:442–9Z. doi: 10.1016/s0008-6363(00)00017-1
79. Izumi T, Kihara Y, Sarai N, Yoneda T, Iwanaga Y, Inagaki K, et al. Reinduction of T-type calcium channels by endothelin-1 in failing hearts in vivo and in adult rat ventricular myocytes in vitro. Circulation. (2003) 108:2530–5. doi: 10.1161/01.CIR.0000096484.03318.AB
80. Nuss HB, Houser SR. T-type Ca2+ current is expressed in hypertrophied adult feline left ventricular myocytes. Circ Res. (1993) 73:777–82. doi: 10.1161/01.res.73.4.777
81. Bögeholz N, Knappe V, Pauls P, Schulte JS, Goldhaber JI, Müller FU, et al. Increased in vivo perpetuation of whole-heart ventricular arrhythmia in heterozygous Na+/Ca2+exchanger knockout mice. IJC Heart Vasc. (2023) 44:101168. doi: 10.1016/j.ijcha.2022.101168
82. Baartscheer A, Van Borren M. Sodium ion transporters as new therapeutic targets in heart failure. Cardiovasc Hematol Agents Med Chem. (2008) 6:229–36. doi: 10.2174/187152508785909546
83. Hutchings DC, Madders GWP, Niort BC, Bode EF, Waddell CA, Woods LS, et al. Interaction of background Ca2+ influx, sarcoplasmic reticulum threshold and heart failure in determining propensity for Ca2+ waves in sheep heart. J Physiol. (2022) 600:2637–50. doi: 10.1113/JP282168
84. Flesch M, Schwinger RH, Schiffer F, Frank K, Südkamp M, Kuhn-Regnier F, et al. Evidence for functional relevance of an enhanced expression of the Na(+)-Ca2+exchanger in failing human myocardium. Circulation. (1996) 94:992–1002. doi: 10.1161/01.cir.94.5.992
85. Primessnig U, Bracic T, Levijoki J, Otsomaa L, Pollesello P, Falcke M, et al. Long-term effects of Na+/Ca2+ exchanger inhibition with ORM-11035 improves cardiac function and remodelling without lowering blood pressure in a model of heart failure with preserved ejection fraction. Eur J Heart Fail. (2019) 21:1543–52. doi: 10.1002/ejhf.1619
86. Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. (1999) 85:1009–19. doi: 10.1161/01.RES.85.11.1009
87. Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. (2006) 1080:165–77. doi: 10.1196/annals.1380.015
88. Kawakami S, Takada K, Aimoto M, Nagasawa Y, Kusakabe T, Kato K, et al. The antiarrhythmic action of the Na+/Ca2+ exchanger inhibitor SEA0400 on drug-induced long QT syndrome depends on the severity of proarrhythmic conditions in anesthetized atrioventricular block rabbits. Biol Pharm Bull. (2023) 46:1120–7. doi: 10.1248/bpb.b23-00202
89. Antoons G, Willems R, Sipido KR. Alternative strategies in arrhythmia therapy: evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol Ther. (2012) 134:26–42. doi: 10.1016/j.pharmthera.2011.12.001
90. Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier J-J, Varro A, et al. Pharmacological inhibition of Na/Ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res. (2008) 102:1398–405. doi: 10.1161/CIRCRESAHA.108.173922
91. Oravecz K, Kormos A, Gruber A, Márton Z, Kohajda Z, Mirzaei L, et al. Inotropic effect of NCX inhibition depends on the relative activity of the reverse NCX assessed by a novel inhibitor ORM-10962 on canine ventricular myocytes. Eur J Pharmacol. (2018) 818:278–86. doi: 10.1016/j.ejphar.2017.10.039
92. Hegner P, Drzymalski M, Biedermann A, Memmel B, Durczok M, Wester M, et al. SAR296968, a novel selective Na+/Ca2+ exchanger inhibitor, improves Ca2+ handling and contractile function in human atrial cardiomyocytes. Biomedicines. (2022) 10:1932. doi: 10.3390/biomedicines10081932
93. Joshi P, Estes S, DeMazumder D, Knollmann BC, Dey S. Ryanodine receptor 2 inhibition reduces dispersion of cardiac repolarization, improves contractile function, and prevents sudden arrhythmic death in failing hearts. eLife. (2023) 12:RP88638. doi: 10.7554/eLife.88638
94. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. (2000) 101:365–76. doi: 10.1016/s0092-8674(00)80847-8
95. Sleiman Y, Lacampagne A, Meli AC. “Ryanopathies” and RyR2 dysfunctions: can we further decipher them using in vitro human disease models? Cell Death Dis. (2021) 12:1041. doi: 10.1038/s41419-021-04337-9
96. Richardson SJ, Thekkedam CG, Casarotto MG, Beard NA, Dulhunty AF. FKBP12 binds to the cardiac ryanodine receptor with negative cooperativity: implications for heart muscle physiology in health and disease. Philos Trans R Soc Lond B Biol Sci. (2023) 378:20220169. doi: 10.1098/rstb.2022.0169
97. McCauley MD, Wehrens XHT. Ryanodine receptor phosphorylation, calcium/calmodulin-dependent protein kinase II, and life-threatening ventricular arrhythmias. Trends Cardiovasc Med. (2011) 21:48–51. doi: 10.1016/j.tcm.2012.02.004
98. Fischer TH, Eiringhaus J, Dybkova N, Saadatmand A, Pabel S, Weber S, et al. Activation of protein phosphatase 1 by a selective phosphatase disrupting peptide reduces sarcoplasmic reticulum Ca2+ leak in human heart failure. Eur J Heart Fail. (2018) 20:1673–85. doi: 10.1002/ejhf.1297
99. Reiken S, Gaburjakova M, Gaburjakova J, He KL, Prieto A, Becker E, et al. Beta-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation. (2001) 104:2843–8. doi: 10.1161/hc4701.099578
100. Zhang S, Liu Z, Zou C, Wang L. Effect of metoprolol on sarcoplasmic reticulum Ca2+ leak in a rabbit model of heart failure. Chin Med J (Engl). (2012) 125:815–22. doi: 10.3760/cma.j.issn.0366-6999.2012.05.016
101. Eiringhaus J, Wünsche CM, Tirilomis P, Herting J, Bork N, Nikolaev VO, et al. Sacubitrilat reduces pro-arrhythmogenic sarcoplasmic reticulum Ca2+ leak in human ventricular cardiomyocytes of patients with end-stage heart failure. ESC Heart Fail. (2020) 7:2992–3002. doi: 10.1002/ehf2.12918
102. Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Györke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. (2007) 93:4083–92. doi: 10.1529/biophysj.107.114546
103. Denniss AL, Dashwood AM, Molenaar P, Beard NA. Sarcoplasmic reticulum calcium mishandling: central tenet in heart failure? Biophys Rev. (2020) 12:865–78. doi: 10.1007/s12551-020-00736-y
104. Pieske B, Maier LS, Bers DM, Hasenfuss G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res. (1999) 85:38–46. doi: 10.1161/01.res.85.1.38
105. Ruiz-Hurtado G, Li L, Fernández-Velasco M, Rueda A, Lefebvre F, Wang Y, et al. Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. J Gen Physiol. (2015) 146:295–306. doi: 10.1085/jgp.201511366
106. Sleiman Y, Reiken S, Charrabi A, Jaffré F, Sittenfeld LR, Pasquié J-L, et al. Personalized medicine in the dish to prevent calcium leak associated with short-coupled polymorphic ventricular tachycardia in patient-derived cardiomyocytes. Stem Cell Res Ther. (2023) 14:266. doi: 10.1186/s13287-023-03502-5
107. Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. (2020) 115:38. doi: 10.1007/s00395-020-0797-z
108. Val-Blasco A, Gil-Fernández M, Rueda A, Pereira L, Delgado C, Smani T, et al. Ca2+ mishandling in heart failure: potential targets. Acta Physiol (Oxf). (2021) 232:e13691. doi: 10.1111/apha.13691
109. Wang W, Landstrom AP, Wang Q, Munro ML, Beavers D, Ackerman MJ, et al. Reduced junctional Na+/Ca2+-exchanger activity contributes to sarcoplasmic reticulum Ca2+ leak in junctophilin-2-deficient mice. Am J Physiol Heart Circ Physiol. (2014) 307:H1317–26. doi: 10.1152/ajpheart.00413.2014
110. Guo A, Zhang X, Iyer VR, Chen B, Zhang C, Kutschke WJ, et al. Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc Natl Acad Sci U S A. (2014) 111:12240–5. doi: 10.1073/pnas.1412729111
111. Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet Lond Engl. (2016) 387:1178–86. doi: 10.1016/S0140-6736(16)00082-9
112. Murphy E, Eisner DA. Does the cardioprotective effect of empagliflozin involve inhibition of the sodium-proton exchanger? Cardiovasc Res. (2021) 117:2696–8. doi: 10.1093/cvr/cvab137
113. Fliegel L. Structural and functional changes in the Na+/H+ exchanger isoform 1, induced by Erk1/2 phosphorylation. Int J Mol Sci. (2019) 20:2378. doi: 10.3390/ijms20102378
114. Fliegel L. Role of genetic mutations of the Na+/H+ exchanger isoform 1, in human disease and protein targeting and activity. Mol Cell Biochem. (2021) 476:1221–32. doi: 10.1007/s11010-020-03984-4
115. Pérez-Carrillo L, Aragón-Herrera A, Giménez-Escamilla I, Delgado-Arija M, García-Manzanares M, Anido-Varela L, et al. Cardiac sodium/hydrogen exchanger (NHE11) as a novel potential target for SGLT2i in heart failure: a preliminary study. Pharmaceutics. (2022) 14:1996. doi: 10.3390/pharmaceutics14101996
116. Despa S. Myocyte [Na+]i dysregulation in heart failure and diabetic cardiomyopathy. Front. Physiol. (2018) 9:1303. doi: 10.3389/fphys.2018.01303
117. Chen S, Coronel R, Hollmann MW, Weber NC, Zuurbier CJ. Direct cardiac effects of SGLT2 inhibitors. Cardiovasc Diabetol. (2022) 21:45. doi: 10.1186/s12933-022-01480-1
118. Yampolsky P, Koenen M, Mosqueira M, Geschwill P, Nauck S, Witzenberger M, et al. Augmentation of myocardial If dysregulates calcium homeostasis and causes adverse cardiac remodeling. Nat Commun. (2019) 10:3295. doi: 10.1038/s41467-019-11261-2
119. Zicha S, Fernández-Velasco M, Lonardo G, L’Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. (2005) 66:472–81. doi: 10.1016/j.cardiores.2005.02.011
120. Verkerk AO, Wilders R, Coronel R, Ravesloot JH, Verheijck EE. Ionic remodeling of sinoatrial node cells by heart failure. Circulation. (2003) 108:760–6. doi: 10.1161/01.CIR.0000083719.51661.B9
121. Stillitano F, Lonardo G, Zicha S, Varro A, Cerbai E, Mugelli A, et al. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. (2008) 45:289–99. doi: 10.1016/j.yjmcc.2008.04.013
122. Dias P, Terracciano CM. Hyperpolarization-activated cyclic nucleotide-gated channels and ventricular arrhythmias in heart failure: a novel target for therapy? J Am Heart Assoc. (2013) 2:e000287. doi: 10.1161/JAHA.113.000287
123. Fox K, Ford I, Steg PG, Tendera M, Ferrari R, BEAUTIFUL Investigators. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet Lond Engl. (2008) 372:807–16. doi: 10.1016/S0140-6736(08)61170-8
124. Iness AN, Shah KM, Kukreja RC. Physiological effects of ivabradine in heart failure and beyond. Mol Cell Biochem. (2023). doi: 10.1007/s11010-023-04862-5. [Epub ahead of print]37768496
125. Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet Lond Engl. (2010) 376:875–85. doi: 10.1016/S0140-6736(10)61198-1
126. Rodríguez-Sinovas A, Sánchez JA, Valls-Lacalle L, Consegal M, Ferreira-González I. Connexins in the heart: regulation, function and involvement in cardiac disease. Int J Mol Sci. (2021) 22:4413. doi: 10.3390/ijms22094413
127. Boengler K, Rohrbach S, Weissmann N, Schulz R. Importance of Cx43 for right ventricular function. Int J Mol Sci. (2021) 22:987. doi: 10.3390/ijms22030987
128. Liu S, Lan Y, Zhao Y, Zhang Q, Lin T, Lin K, et al. Expression of connexin 43 protein in cardiomyocytes of heart failure mouse model. Front Cardiovasc Med. (2022) 9:1028558. doi: 10.3389/fcvm.2022.1028558
129. Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res. (2005) 96:54–63. doi: 10.1161/01.RES.0000152325.07495.5a
130. Epifantseva I, Shaw RM. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim Biophys Acta Biomembr. (2018) 1860:40–7. doi: 10.1016/j.bbamem.2017.05.018
131. Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. (2004) 95:717–25. doi: 10.1161/01.RES.0000144125.61927.1c
132. Yan J, Killingsworth C, Walcott G, Zhu Y, Litovsky S, Huang J, et al. Molecular remodeling of Cx43, but not structural remodeling, promotes arrhythmias in an arrhythmogenic canine model of nonischemic heart failure. J Mol Cell Cardiol. (2021) 158:72–81. doi: 10.1016/j.yjmcc.2021.05.012
133. Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T, et al. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc Diabetol. (2020) 19:91. doi: 10.1186/s12933-020-01066-9
134. Bengel P, Ahmad S, Sossalla S. Inhibition of late sodium current as an innovative antiarrhythmic strategy. Curr Heart Fail Rep. (2017) 14:179–86. doi: 10.1007/s11897-017-0333-0
135. Cumberland MJ, Riebel LL, Roy A, O’Shea C, Holmes AP, Denning C, et al. Basic research approaches to evaluate cardiac arrhythmia in heart failure and beyond. Front Physiol. (2022) 13:806366. doi: 10.3389/fphys.2022.806366
136. Naveed M, Mohammed ASA, Topal L, Kovács ZM, Dienes C, Ovári J, et al. Selective inhibition of cardiac late Na+ current is based on fast offset kinetics of the inhibitor. Biomedicines. (2023) 11:2383. doi: 10.3390/biomedicines11092383
137. George SA, Brennan-McLean JA, Trampel KA, Rytkin E, Faye NR, Knollmann BC, et al. Ryanodine receptor inhibition with acute dantrolene treatment reduces arrhythmia susceptibility in human hearts. Am J Physiol Heart Circ Physiol. (2023) 325:H720–8. doi: 10.1152/ajpheart.00103.2023
138. Xue J-B, Val-Blasco A, Davoodi M, Gómez S, Yaniv Y, Benitah J-P, et al. Heart failure in mice induces a dysfunction of the sinus node associated with reduced CAMKII signaling. J Gen Physiol. (2022) 154:e202112895. doi: 10.1085/jgp.202112895
139. van der Heyden MAG, Delisle BP, Abriel H. Editorial: ion channel trafficking and cardiac arrhythmias. Front Physiol. (2018) 9:1254. doi: 10.3389/fphys.2018.01254
140. Arrieta A, Blackwood EA, Glembotski CC. ER protein quality control and the unfolded protein response in the heart. Curr Top Microbiol Immunol. (2018) 414:193–213. doi: 10.1007/82_2017_54
141. Paskevicius T, Farraj RA, Michalak M, Agellon LB. Calnexin, more than just a molecular chaperone. Cells. (2023) 12:403. doi: 10.3390/cells12030403
142. Cela I, Dufrusine B, Rossi C, Luini A, De Laurenzi V, Federici L, et al. KDEL receptors: pathophysiological functions, therapeutic options, and biotechnological opportunities. Biomedicines. (2022) 10:1234. doi: 10.3390/biomedicines10061234
143. Shiwarski DJ, Crilly SE, Dates A, Puthenveedu MA. Dual RXR motifs regulate nerve growth factor-mediated intracellular retention of the delta opioid receptor. Mol Biol Cell. (2019) 30:680–90. doi: 10.1091/mbc.E18-05-0292
144. Hu L, Gao D, Lv H, Lian L, Wang M, Wang Y, et al. Finding new targets for the treatment of heart failure: endoplasmic reticulum stress and autophagy. J Cardiovasc Transl Res. (2023) 16:1349–56. doi: 10.1007/s12265-023-10410-9
145. Christianson JC, Jarosch E, Sommer T. Mechanisms of substrate processing during ER-associated protein degradation. Nat Rev Mol Cell Biol. (2023) 24:777–96. doi: 10.1038/s41580-023-00633-8
146. Baycin-Hizal D, Gottschalk A, Jacobson E, Mai S, Wolozny D, Zhang H, et al. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem Biophys Res Commun. (2014) 453:243–53. doi: 10.1016/j.bbrc.2014.06.067
147. Nguyen T-T-D, Le N-Q-K, Tran T-A, Pham D-M, Ou Y-Y. Incorporating a transfer learning technique with amino acid embeddings to efficiently predict N-linked glycosylation sites in ion channels. Comput Biol Med. (2021) 130:104212. doi: 10.1016/j.compbiomed.2021.104212
148. Cortada E, Brugada R, Verges M. N-glycosylation of the voltage-gated sodium channel β2 subunit is required for efficient trafficking of NaV1.5/β2 to the plasma membrane. J Biol Chem. (2019) 294:16123–40. doi: 10.1074/jbc.RA119.007903
149. Kim H, Yang H, Ednie AR, Bennett ES. Simulation modeling of reduced glycosylation effects on potassium channels of mouse cardiomyocytes. Front Physiol. (2022) 13:816651. doi: 10.3389/fphys.2022.816651
150. Pagano A, Letourneur F, Garcia-Estefania D, Carpentier J-L, Orci L, Paccaud J-P. Sec24 proteins and sorting at the endoplasmic reticulum. J Biol Chem. (1999) 274:7833–40. doi: 10.1074/jbc.274.12.7833
151. Wilfling F, Kaksonen M, Stachowiak J. Protein condensates as flexible platforms for membrane traffic. Curr Opin Cell Biol. (2023) 85:102258. doi: 10.1016/j.ceb.2023.102258
152. Hutchings J, Stancheva VG, Brown NR, Cheung ACM, Miller EA, Zanetti G. Structure of the complete, membrane-assembled COPII coat reveals a complex interaction network. Nat Commun. (2021) 12:2034. doi: 10.1038/s41467-021-22110-6
153. Pietrosemoli N, Pancsa R, Tompa P. Structural disorder provides increased adaptability for vesicle trafficking pathways. PLoS Comput Biol. (2013) 9:e1003144. doi: 10.1371/journal.pcbi.1003144
154. Liu J, Zheng X, Wu X. The Rab GTPase in the heart: pivotal roles in development and disease. Life Sci. (2022) 306:120806. doi: 10.1016/j.lfs.2022.120806
155. Takahashi T, Shirai J, Matsuda M, Nakanaga S, Matsushita S, Wakita K, et al. Protein quality control machinery supports primary ciliogenesis by eliminating GDP-bound Rab8-family GTPases. iScience. (2023) 26:106652. doi: 10.1016/j.isci.2023.106652
156. Martínez-Morales JC, Romero-Ávila MT, Reyes-Cruz G, García-Sáinz JA. Roles of receptor phosphorylation and Rab proteins in G protein-coupled receptor function and trafficking. Mol Pharmacol. (2022) 101:144–53. doi: 10.1124/molpharm.121.000429
157. Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. (2000) 149:901–14. doi: 10.1083/jcb.149.4.901
158. Satoh AK, O’Tousa JE, Ozaki K, Ready DF. Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila photoreceptors. Dev Camb Engl. (2005) 132:1487–97. doi: 10.1242/dev.01704
159. Hummel JJA, Hoogenraad CC. Inducible manipulation of motor-cargo interaction using engineered kinesin motors. J Cell Sci. (2021) 134:jcs258776. doi: 10.1242/jcs.258776
160. Karcher RL, Deacon SW, Gelfand VI. Motor-cargo interactions: the key to transport specificity. Trends Cell Biol. (2002) 12:21–7. doi: 10.1016/s0962-8924(01)02184-5
161. Bahouth SW, Nooh MM, Mancarella S. Involvement of SAP97 anchored multiprotein complexes in regulating cardiorenal signaling and trafficking networks. Biochem Pharmacol. (2023) 208:115406. doi: 10.1016/j.bcp.2022.115406
162. Beuriot A, Eichel CA, Dilanian G, Louault F, Melgari D, Doisne N, et al. Distinct calcium/calmodulin-dependent serine protein kinase domains control cardiac sodium channel membrane expression and focal adhesion anchoring. Heart Rhythm. (2020) 17:786–94. doi: 10.1016/j.hrthm.2019.12.019
163. Fourie C, Li D, Montgomery JM. The anchoring protein SAP97 influences the trafficking and localisation of multiple membrane channels. Biochim Biophys Acta. (2014) 1838:589–94. doi: 10.1016/j.bbamem.2013.03.015
164. Gillet L, Rougier J-S, Shy D, Sonntag S, Mougenot N, Essers M, et al. Cardiac-specific ablation of synapse-associated protein SAP97 in mice decreases potassium currents but not sodium current. Heart Rhythm. (2015) 12:181–92. doi: 10.1016/j.hrthm.2014.09.057
165. Matamoros M, Pérez-Hernández M, Guerrero-Serna G, Amorós I, Barana A, Núñez M, et al. Nav1.5 N-terminal domain binding to α1-syntrophin increases membrane density of human Kir2.1, Kir2.2 and Nav1.5 channels. Cardiovasc Res. (2016) 110:279–90. doi: 10.1093/cvr/cvw009
166. Mori T, Zhou M, Tabuchi K. Diverse clinical phenotypes of CASK-related disorders and multiple functional domains of CASK protein. Genes (Basel). (2023) 14:1656. doi: 10.3390/genes14081656
167. Nafzger S, Rougier J-S. Calcium/calmodulin-dependent serine protein kinase CASK modulates the L-type calcium current. Cell Calcium. (2017) 61:10–21. doi: 10.1016/j.ceca.2016.10.001
168. Capera J, Serrano-Novillo C, Navarro-Pérez M, Cassinelli S, Felipe A. The potassium channel odyssey: mechanisms of traffic and membrane arrangement. Int J Mol Sci. (2019) 20:734. doi: 10.3390/ijms20030734
169. Eichel CA, Beuriot A, Chevalier MYE, Rougier J-S, Louault F, Dilanian G, et al. Lateral membrane-specific MAGUK CASK down-regulates NaV1.5 channel in cardiac myocytes. Circ Res. (2016) 119:544–56. doi: 10.1161/CIRCRESAHA.116.309254
170. Jahn R, Cafiso DC, Tamm LK. Mechanisms of SNARE proteins in membrane fusion. Nat Rev Mol Cell Biol. (2023) 25:101–18. doi: 10.1038/s41580-023-00668-x
171. Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. (1993) 75:409–18. doi: 10.1016/0092-8674(93)90376-2
172. Søgaard M, Tani K, Ye RR, Geromanos S, Tempst P, Kirchhausen T, et al. A Rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell. (1994) 78:937–48. doi: 10.1016/0092-8674(94)90270-4
173. Bowman PRT, Smith GL, Gould GW. Cardiac SNARE expression in health and disease. Front Endocrinol. (2019) 10:881. doi: 10.3389/fendo.2019.00881
174. Cunha SR, Mohler PJ. Cardiac ankyrins: essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc Res. (2006) 71:22–9. doi: 10.1016/j.cardiores.2006.03.018
175. Stevens SR, Rasband MN. Pleiotropic ankyrins: scaffolds for Ion channels and transporters. Channels Austin Tex. (2022) 16:216–29. doi: 10.1080/19336950.2022.2120467
176. Makara MA, Curran J, Little SC, Musa H, Polina I, Smith SA, et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. (2014) 115:929–38. doi: 10.1161/CIRCRESAHA.115.305154
177. Skogestad J, Aronsen JM, Tovsrud N, Wanichawan P, Hougen K, Stokke MK, et al. Coupling of the Na+/K+-ATPase to ankyrin B controls Na+/Ca2+exchanger activity in cardiomyocytes. Cardiovasc Res. (2020) 116:78–90. doi: 10.1093/cvr/cvz087
178. Vandecaetsbeek I, Raeymaekers L, Wuytack F, Vangheluwe P. Factors controlling the activity of the SERCA2a pump in the normal and failing heart. Biofactors. (2009) 35:484–99. doi: 10.1002/biof.63
179. Kashef F, Li J, Wright P, Snyder J, Suliman F, Kilic A, et al. Ankyrin-B protein in heart failure: identification of a new component of metazoan cardioprotection. J Biol Chem. (2012) 287:30268–81. doi: 10.1074/jbc.M112.368415
180. Buraei Z, Yang J. The ß subunit of voltage-gated Ca2+channels. Physiol Rev. (2010) 90:1461–506. doi: 10.1152/physrev.00057.2009
181. Clatot J, Coulombe A, Deschênes I, Guicheney P, Neyroud N. Trafficking and gating cooperation between deficient Nav1.5-mutant channels to rescue INA. Front Biosci Landmark Ed. (2022) 27:209. doi: 10.31083/j.fbl2707209
182. Tang W, Cai D, Fu Y, Zheng Z, Huang X, Khouzam RN, et al. 4-phenylbutyric acid re-trafficking hERG/G572R channel protein by modulating the endoplasmic reticulum stress-associated chaperones and endoplasmic reticulum-associated degradation gene. J Thorac Dis. (2023) 15:4472–85. doi: 10.21037/jtd-23-1252
183. Cheng X, Wang K, Zhao Y, Wang K. Research progress on post-translational modification of proteins and cardiovascular diseases. Cell Death Discov. (2023) 9:275. doi: 10.1038/s41420-023-01560-5
184. Ednie AR, Parrish AR, Sonner MJ, Bennett ES. Reduced hybrid/complex N-glycosylation disrupts cardiac electrical signaling and calcium handling in a model of dilated cardiomyopathy. J Mol Cell Cardiol. (2019) 132:13–23. doi: 10.1016/j.yjmcc.2019.05.001
185. Liu M, Shi G, Dudley SC. Cardiac ion channel changes in response to ER stress. Heart Rhythm. (2014) 11:2129. doi: 10.1016/j.hrthm.2014.09.012
186. Liu M, Shi G, Zhou A, Rupert CE, Coulombe KLK, Dudley SC. Activation of the unfolded protein response downregulates cardiac ion channels in human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol. (2018) 117:62–71. doi: 10.1016/j.yjmcc.2018.02.011
187. Liu C, Chen Y, Xie Y, Xiang M. Tubulin post-translational modifications: potential therapeutic approaches to heart failure. Front Cell Dev Biol. (2022) 10:872058. doi: 10.3389/fcell.2022.872058
188. Sanyal C, Pietsch N, Ramirez Rios S, Peris L, Carrier L, Moutin M-J. The detyrosination/re-tyrosination cycle of tubulin and its role and dysfunction in neurons and cardiomyocytes. Semin Cell Dev Biol. (2023) 137:46–62 (Special Issue: The tubulin code by Maria M. Magiera). doi: 10.1016/j.semcdb.2021.12.006
189. Manalo A, Schroer AK, Fenix AM, Shancer Z, Coogan J, Brolsma T, et al. Loss of CENP-F results in dilated cardiomyopathy with severe disruption of cardiac myocyte architecture. Sci Rep. (2018) 8:7546. doi: 10.1038/s41598-018-25774-1
190. Marchal GA, Jouni M, Chiang DY, Pérez-Hernández M, Podliesna S, Yu N, et al. Targeting the microtubule EB1-CLASP2 complex modulates NaV1.5 at intercalated discs. Circ Res. (2021) 129:349–65. doi: 10.1161/CIRCRESAHA.120.318643
191. Smyth JW, Hong T-T, Gao D, Vogan JM, Jensen BC, Fong TS, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. (2010) 120:266–79. doi: 10.1172/JCI39740
192. Yashirogi S, Nagao T, Nishida Y, Takahashi Y, Qaqorh T, Yazawa I, et al. AMPK regulates cell shape of cardiomyocytes by modulating turnover of microtubules through CLIP-170. EMBO Rep. (2021) 22:e50949. doi: 10.15252/embr.202050949
193. Hong T-T, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, et al. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. (2012) 9:812–20. doi: 10.1016/j.hrthm.2011.11.055
194. Mustroph J, Sag CM, Bähr F, Schmidtmann A-L, Gupta SN, Dietz A, et al. Loss of CASK accelerates heart failure development. Circ Res. (2021) 128:1139–55. doi: 10.1161/CIRCRESAHA.120.318170
195. Ottesen AH, Raiborg C, Melleby AO, Hansen MSH, Hafver TL, Sandbu RA, et al. Synapsin 2 regulates NCX1 trafficking and is down-regulated in the failing myocardium, which increases the risk of ventricular arrhythmia and heart failure mortality. Eur Heart J. (2022) 43:ehac544.2974. doi: 10.1093/eurheartj/ehac544.2974
196. Ranek MJ, Stachowski MJ, Kirk JA, Willis MS. The role of heat shock proteins and co-chaperones in heart failure. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20160530. doi: 10.1098/rstb.2016.0530
197. Shi Y-Q, Yan M, Liu L-R, Zhang X, Wang X, Geng H-Z, et al. High glucose represses hERG K+ channel expression through trafficking inhibition. Cell Physiol Biochem. (2015) 37:284–96. doi: 10.1159/000430353
198. Meier S, Grundland A, Dobrev D, Volders PGA, Heijman J. In silico analysis of the dynamic regulation of cardiac electrophysiology by Kv 11.1 ion-channel trafficking. J Physiol. (2023) 601:2711–31. doi: 10.1113/JP283976
199. Blandin CE, Gravez BJ, Hatem SN, Balse E. Remodeling of ion channel trafficking and cardiac arrhythmias. Cells. (2021) 10:2417. doi: 10.3390/cells10092417
200. Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CAMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. (2013) 305:H431–45. doi: 10.1152/ajpheart.00306.2013
201. Schulz DJ, Temporal S, Barry DM, Garcia ML. Mechanisms of voltage-gated ion channel regulation: from gene expression to localization. Cell Mol Life Sci. (2008) 65:2215–31. doi: 10.1007/s00018-008-8060-z
202. Zhuang L, Jia K, Chen C, Li Z, Zhao J, Hu J, et al. DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation. (2022) 145:829–46. doi: 10.1161/CIRCULATIONAHA.121.055727
203. Yang S, Chatterjee S, Cipollo J. The glycoproteomics-MS for studying glycosylation in cardiac hypertrophy and heart failure. Proteomics Clin Appl. (2018) 12:e1700075. doi: 10.1002/prca.201700075
204. Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun. (2021) 12:1684. doi: 10.1038/s41467-021-21931-9
205. Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca2+-ATPase in heart failure. Circ Res. (2019) 124:e63–80. doi: 10.1161/CIRCRESAHA.118.313865
206. Byrne NJ, Soni S, Takahara S, Ferdaoussi M, Al Batran R, Darwesh AM, et al. Chronically elevating circulating ketones can reduce cardiac inflammation and blunt the development of heart failure. Circ Heart Fail. (2020) 13:e006573. doi: 10.1161/CIRCHEARTFAILURE.119.006573
207. Bruyneel AAN, McKeithan WL, Feyen DAM, Mercola M. Using iPSC models to probe regulation of cardiac ion channel function. Curr Cardiol Rep. (2018) 20:57. doi: 10.1007/s11886-018-1000-0
208. Kanner SA, Shuja Z, Choudhury P, Jain A, Colecraft HM. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat Methods. (2020) 17:1245–53. doi: 10.1038/s41592-020-00992-6
209. Loh KWZ, Liang MC, Soong TW, Hu Z. Regulation of cardiovascular calcium channel activity by post-translational modifications or interacting proteins. Pflugers Arch. (2020) 472:653–67. doi: 10.1007/s00424-020-02398-x
210. Dickhout JG, Krepinsky JC. Endoplasmic reticulum stress and renal disease. Antioxid Redox Signal. (2009) 11:2341–52. doi: 10.1089/ars.2009.2705
211. Nakamura S, Numata G, Yamaguchi T, Tokiwa H, Higashikuni Y, Nomura S, et al. Endoplasmic reticulum stress-activated nuclear factor-kappa B signaling pathway induces the upregulation of cardiomyocyte dopamine D1 receptor in heart failure. Biochem Biophys Res Commun. (2022) 637:247–53. doi: 10.1016/j.bbrc.2022.11.031
212. Li S, Fraser S, Ranpura G, Lim S, Singer ES, Parker JDK, et al. Generation of an induced pluripotent stem cell line from a patient with conduction disease and recurrent ventricular fibrillation with a sodium voltage-gated channel alpha subunit 5 (SCN5A) gene c.392+3A >G splice-site variant. Stem Cell Res. (2023) 71:103153. doi: 10.1016/j.scr.2023.103153
213. Laedermann CJ, Abriel H, Decosterd I. Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front Pharmacol. (2015) 6:263. doi: 10.3389/fphar.2015.00263
214. Chen X, Zhu C, Zhou H, Zhang Y, Cai Z, Wu H, et al. Key role of the membrane trafficking of Nav1.5 channel protein in antidepressant-induced Brugada syndrome. Front Physiol. (2018) 9:1230. doi: 10.3389/fphys.2018.01230
215. Shao D, Okuse K, Djamgoz MBA. Protein–protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. Int J Biochem Cell Biol. (2009) 41:1471–81. doi: 10.1016/j.biocel.2009.01.016
216. Aromolaran AS, Chahine M, Boutjdir M. Regulation of cardiac voltage-gated sodium channel by kinases: roles of protein kinases A and C. Handb Exp Pharmacol. (2018) 246:161–84. doi: 10.1007/164_2017_53
217. Ferreira JCB, Mochly-Rosen D, Boutjdir M. Regulation of cardiac excitability by protein kinase C isozymes. Front Biosci Sch Ed. (2012) 4:532–46. doi: 10.2741/s283
218. Turan NN, Moshal KS, Roder K, Baggett BC, Kabakov AY, Dhakal S, et al. The endosomal trafficking regulator LITAF controls the cardiac Nav1.5 channel via the ubiquitin ligase NEDD4-2. J Biol Chem. (2020) 295:18148–59. doi: 10.1074/jbc.RA120.015216
219. Mustroph J, Baier MJ, Pabel S, Stehle T, Trum M, Provaznik Z, et al. Empagliflozin inhibits cardiac late sodium current by Ca/calmodulin-dependent kinase II. Circulation. (2022) 146:1259–61. doi: 10.1161/CIRCULATIONAHA.122.057364
220. Casini S, Nasilli G, De Waal T, Dekkers Y, Van Amersfoorth S, Marchal GA, et al. Decreasing microtubule detyrosination by parthenolide restores sodium current in mdx cardiomyocytes. Europace. (2023) 25:euad122.592. doi: 10.1093/europace/euad122.592
221. Chen CY, Caporizzo MA, Bedi K, Vite A, Bogush AI, Robison P, et al. Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat Med. (2018) 24:1225–33. doi: 10.1038/s41591-018-0046-2
222. Hayashi I, Ikura M. Crystal structure of the amino-terminal microtubule-binding domain of end-binding protein 1 (EB1). J Biol Chem. (2003) 278:36430–4. doi: 10.1074/jbc.M305773200
223. Song X, Yang F, Yang T, Wang Y, Ding M, Li L, et al. Phase separation of EB1 guides microtubule plus-end dynamics. Nat Cell Biol. (2023) 25:79–91. doi: 10.1038/s41556-022-01033-4
224. Barc J, Tadros R, Glinge C, Chiang DY, Jouni M, Simonet F, et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat Genet. (2022) 54:232–9. doi: 10.1038/s41588-021-01007-6
225. Severino P, D’Amato A, Pucci M, Infusino F, Birtolo LI, Mariani MV, et al. Ischemic heart disease and heart failure: role of coronary ion channels. Int J Mol Sci. (2020) 21:3167. doi: 10.3390/ijms21093167
226. Ono M, Burgess DE, Schroder EA, Elayi CS, Anderson CL, January CT, et al. Long QT syndrome type 2: emerging strategies for correcting class 2 KCNH2 (hERG) mutations and identifying new patients. Biomolecules. (2020) 10:1144. doi: 10.3390/biom10081144
227. Lu H, Ding W, Xiao H, Dai M, Xue Y, Jia Z, et al. Association of the P441l KCNQ1 variant with severity of long QT syndrome and risk of cardiac events. Front Cardiovasc Med. (2022) 9:922335. doi: 10.3389/fcvm.2022.922335
228. Wilson A, Quinn K, Graves F, Bitnerglindzicz M, Tinker A. Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1). Cardiovasc Res. (2005) 67:476–86. doi: 10.1016/j.cardiores.2005.04.036
229. Wu J, Sakaguchi T, Takenaka K, Toyoda F, Tsuji K, Matsuura H, et al. A trafficking-deficient KCNQ1 mutation, T587M, causes a severe phenotype of long QT syndrome by interfering with intracellular hERG transport. J Cardiol. (2019) 73:343–50. doi: 10.1016/j.jjcc.2018.10.011
230. Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, et al. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. (2009) 119:2745–57. doi: 10.1172/JCI39027
231. Melgari D, Barbier C, Dilanian G, Rücker-Martin C, Doisne N, Coulombe A, et al. Microtubule polymerization state and clathrin-dependent internalization regulate dynamics of cardiac potassium channel: microtubule and clathrin control of KV1.5 channel. J Mol Cell Cardiol. (2020) 144:127–39. doi: 10.1016/j.yjmcc.2020.05.004
232. Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. (2003) 92:e87–100. doi: 10.1161/01.RES.0000079028.31393.15
233. Nair SP, Sharma RK. Heat shock proteins and their expression in primary murine cardiac cell populations during ischemia and reperfusion. Mol Cell Biochem. (2020) 464:21–6. doi: 10.1007/s11010-019-03645-1
234. Marunouchi T, Nishiumi C, Iinuma S, Yano E, Tanonaka K. Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice. Eur J Pharmacol. (2021) 898:173987. doi: 10.1016/j.ejphar.2021.173987
235. Marunouchi T, Fujita K, Takahashi K, Namiki S, Kyo L, Uchida M, et al. Simvastatin attenuates the c-Raf/Erk and calcineurin-NFATc2 pathways via inhibition of Hsp90 activity during the development of heart failure. J Pharmacol Sci. (2023) 151:17–27. doi: 10.1016/j.jphs.2022.11.002
236. Roberts RJ, Hallee L, Lam CK. The potential of Hsp90 in targeting pathological pathways in cardiac diseases. J Pers Med. (2021) 11:1373. doi: 10.3390/jpm11121373
237. Wu L-Y, Song Y-J, Zhang C-L, Liu J. KV channel-interacting proteins in the neurological and cardiovascular systems: an updated review. Cells. (2023) 12:1894. doi: 10.3390/cells12141894
238. Wiedmann F, Schlund D, Faustino F, Kraft M, Ratte A, Thomas D, et al. N-Glycosylation of TREK-1/hK2P2.1 two-pore-domain potassium (K2P) channels. Int J Mol Sci. (2019) 20:5193. doi: 10.3390/ijms20205193
239. Bogdanov V, Soltisz AM, Moise N, Sakuta G, Orengo BH, Janssen PML, et al. Distributed synthesis of sarcolemmal and sarcoplasmic reticulum membrane proteins in cardiac myocytes. Basic Res Cardiol. (2021) 116:63. doi: 10.1007/s00395-021-00895-3
240. Smyth JW, Shaw RM. Forward trafficking of ion channels: what the clinician needs to know. Heart Rhythm. (2010) 7:1135–40. doi: 10.1016/j.hrthm.2010.05.030
241. Loh KWZ, Liu C, Soong TW, Hu Z. Β subunits of voltage-gated calcium channels in cardiovascular diseases. Front Cardiovasc Med. (2023) 10:1119729. doi: 10.3389/fcvm.2023.1119729
242. Guo J, Tian Q, Barth M, Xian W, Ruppenthal S, Schaefers H-J, et al. Human BIN1 isoforms grow, maintain, and regenerate excitation-contraction couplons in adult rat and human stem cell-derived cardiomyocytes. Cardiovasc Res. (2022) 118:1479–91. doi: 10.1093/cvr/cvab195
243. Dridi H, Liu Y, Reiken S, Liu X, Argyrousi EK, Yuan Q, et al. Heart failure-induced cognitive dysfunction is mediated by intracellular Ca2+ leak through ryanodine receptor type 2. Nat Neurosci. (2023) 26:1365–78. doi: 10.1038/s41593-023-01377-6
244. Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan Y-N, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. (2007) 128:547–60. doi: 10.1016/j.cell.2006.12.037
245. Wang Q, Xiaoyan L, Shang S, Fan Y, Lv H, Tang B, et al. Desmosomal junctions and connexin-43 remodeling in high-pacing-induced heart failure dogs. Anatol J Cardiol. (2023) 27:462–71. doi: 10.14744/AnatolJCardiol.2023.2823
246. Basheer WA, Shaw RM. Connexin 43 and CaV1.2 ion channel trafficking in healthy and diseased myocardium. Circ Arrhythm Electrophysiol. (2016) 9:e001357. doi: 10.1161/CIRCEP.115.001357
247. Caporizzo MA, Prosser BL. The microtubule cytoskeleton in cardiac mechanics and heart failure. Nat Rev Cardiol. (2022) 19:364–78. doi: 10.1038/s41569-022-00692-y
248. Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. (2001) 33:359–71. doi: 10.1006/jmcc.2000.1308
249. Dago M, Crespo-García T, Cámara-Checa A, Rapún J, Rubio-Alarcón M, Marín M, et al. Empagliflozin and dapagliflozin increase Na+ and inward rectifier K+ current densities in human cardiomyocytes derived from induced pluripotent stem cells (hiPSC-CMs). Cells. (2022) 11:3707. doi: 10.3390/cells11233707
Keywords: ion channels, trafficking, heart failure, electrophysiology, arrhythmia, treatment
Citation: Reisqs J-B, Qu YS and Boutjdir M (2024) Ion channel trafficking implications in heart failure. Front. Cardiovasc. Med. 11:1351496. doi: 10.3389/fcvm.2024.1351496
Received: 6 December 2023; Accepted: 25 January 2024;
Published: 14 February 2024.
Edited by:
Hua-Qian Yang, Soochow University, ChinaReviewed by:
Florentina Pluteanu, University of Bucharest, RomaniaPrzemyslaw Radwanski, The Ohio State University, United States
© 2024 Reisqs, Qu and Boutjdir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Boutjdir mohamed.boutjdir@va.gov
Abbreviations AAV, adeno-associated virus; AP, action potential; APD, action potential duration; ATF6, activating transcription factor 6; Ca2+, calcium; CAMKII, calmodulin-dependent kinase II; CASK, calcium/calmodulin-dependent serine protein kinase; COP, coatomer protein; Cx43, connexin-43; DAD, delayed afterdepolarization; EAD, early afterdepolarization; EB1, plus-end binding protein; EF, ejection fraction; ER, endoplasmic reticulum; ERAD, ER-associated degradation system; ERES, endoplasmic reticulum exit site; hERG, human ether-a-go-go–related gene; HF, heart failure; HFrEF, heart failure with reduced ejection fraction; HFmrEF, heart failure with mildly reduced ejection fraction; HFpEF, heart failure with preserved ejection fraction; hiPSC, human-induced pluripotent stem cells; HSP, heat shock protein; ICaL, L-type calcium current; If, funny current; IK1, inwardly rectifying potassium current; IKr, rapid outward potassium current; IKs, slow outward potassium current; INa, sodium current; INaL, late sodium current; Ito, transient outward potassium current; IRE1, inositol-requiring enzyme 1; KCNQ1, potassium voltage-gated subfamily Q member 1; MAGUK, membrane-associated guanylate kinase; MLP, Muscle Lim Protein; MRA, mineralocorticoid receptor antagonist; Na+, sodium; NCX, Na+/Ca2+ exchanger; NHE, Na+/H+ exchanger; NSF, N-ethylmaleimide sensitive fusion protein; PERK, protein kinase R-like ER kinase; PKA, protein kinase A; PKC, protein kinase C; ROS, reactive oxygen species; RYR, ryanodine receptor; SAP97, synapse-associated protein 97; SERCA, sarcoplasmic reticulum Ca2+-ATPase; SGLT2, sodium-glucose cotransporter 2; SNAP, NSF attachment protein; SNARE, SNAP receptor; SR, sarcoplasmic reticulum; UPR, unfolded protein response.