Recent advances of myotubularin-related (MTMR) protein family in cardiovascular diseases
 Jia Wang
Jia Wang Wei Guo3,†
Wei Guo3,†  Xiongshan Sun
Xiongshan Sun- 1Department of Cardiology, The General Hospital of Western Theater Command, Chengdu, Sichuan, China
- 2College of Medicine, Southwest Jiaotong University, Chengdu, Sichuan, China
- 3Clinical Research Center, Stomatological Hospital of Chongqing Medical University, Chongqing, China
Belonging to a lipid phosphatase family containing 16 members, myotubularin-related proteins (MTMRs) are widely expressed in a variety of tissues and organs. MTMRs preferentially hydrolyzes phosphatidylinositol 3-monophosphate and phosphatidylinositol (3,5) bis-phosphate to generate phosphatidylinositol and phosphatidylinositol 5-monophosphate, respectively. These phosphoinositides (PIPs) promote membrane degradation during autophagosome-lysosomal fusion and are also involved in various regulatory signal transduction. Based on the ability of modulating the levels of these PIPs, MTMRs exert physiological functions such as vesicle trafficking, cell proliferation, differentiation, necrosis, cytoskeleton, and cell migration. It has recently been found that MTMRs are also involved in the occurrence and development of several cardiovascular diseases, including cardiomyocyte hypertrophy, proliferation of vascular smooth muscle cell, LQT1, aortic aneurysm, etc. This review summarizes the functions of MTMRs and highlights their pathophysiological roles in cardiovascular diseases.
1 Introduction
As a second messenger within the cell, phosphoinositides (PIPs) participate in a variety of cellular processes, such as protein transport, signal transduction, remodeling of the protein backbone, and fusion of the cell membrane. PIPs can be hydrolyzed into several kinds of substrates, phosphatase and tensin homolog deleted on chromosome ten (PTEN), myotubularin-related (MTMR) protein family, sac1 domain-containing phosphatase, etc. (1, 2). MTMR protein family consists of 16 members, of which 9 members are active phosphatases, while the rest 7 members are inactive phosphatases due to lacking of the conserved cysteine in the catalytic signature (2–4). The most well-known and distinguished effect of MTMRs is the ability to dephosphorylate phosphatidylinositol 3-monophosphate (PI(3)P) and phosphatidylinositol (3, 5) bis-phosphate (PI(3,5)P2), indicating MTMRs are involved in cellular membrane transport and endocytosis (5). MTMRs are widely expressed in different kind of tissue and organs, including the neural system, heart, liver, testicle and gastrointestinal tract (6–8). MTMRs also exert multiple physiological roles such as modulating cell proliferation, differentiation, necrosis and migration (9). Recent studies demonstrate that several MTMRs are also involved in the development of CVDs (10, 11). Thus, illuminating the role of MTMRs in cardiovascular system is of great importance to search novel targets for preventions of CVDs. This review highlights the function of MTMRs in the cardiovascular system and discusses the associated mechanisms.
2 Overview of the canonical physiological mechanisms of MTMRs
2.1 Interactions between MTMRs
MTMRs have been found to have several functional domains that mediate the interactions of protein-protein and protein-lipid, such as the PH-glucosyltransferase, Rab-like GTPase activator and myotubularin (GRAM) domain is involved in the interaction with membranes, Rac-induced recruitment domain which is a membrane-targeted motif, SET interacting domain/PDZ binding domain which mediate protein-protein interactions, and Zinc FYVE domain coupled with phosphatidylinositol (2, 12). Furthermore, the coiled-coil (CC) domain is essential for the homodimerization or heterodimerization between MTMRs (13). The nine members of MTMR with catalytic activity can interact with other members without enzymatic activity, and this interaction between MTMRs plays a crucial role in maintaining its normal function (Table 1). Nandurkar et al. demonstrate that MTMR12, also known as 3-phosphatase adaptor (3-PAP), is a catalytically inactive member of MTMR family and can interact with myotubularin (MTM1) and MTMR2. Co-expression of catalytically inactive MTMR12 with MTM1 can reverse the remodeling of membrane phenotype caused by overexpression of MTM1, translocate MTM1 into cytoplasm, and also attenuate the formation of filamentous pseudopodia caused by overexpression of MTM1 (14). The interaction between MTM1 and MTMR12 is essential for the stability of functional protein complexes in skeletal muscle, which offers novel targets for Mtm1 mutation-induced X-linked myotubular myopathy (XLMTM) (15). MTMR2 is a 73-kDa protein that forms a dimer via its coiled structure, while its interacting partner, MTMR13/SBF2 belongs to the catalytically inactive members. Mutation of either MTMR2 or MTMR13 leads to Charcot-Marie-Tooth type 4B, which is characterized by reduced nerve conductive velocity and folding of myelin within the peripheral nerve (16). MTMR2 binds to MTMR13 and forms a protein complex within Schwann cell, which is critical for the integrity of peripheral nervous system (17). Additionally, Kim et al. demonstrate that MTMR2 interacts with MTMR5 via its CC domain, which enhances the enzymatic activity of MTMR2 and alters its subcellular localization (5).
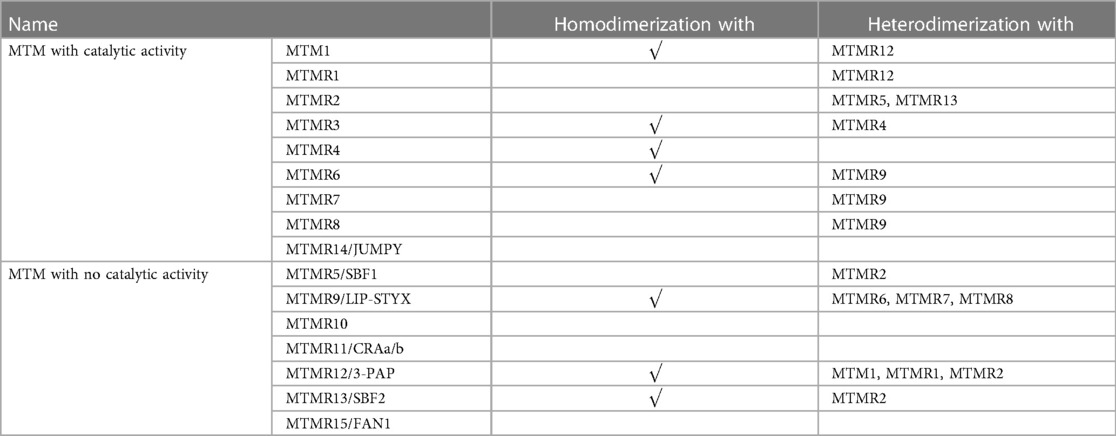
Table 1. Several MTMRs can homodimerize or heterodimerize to form active-active or active-inactive complexes, allowing more precise regulation of phosphoinositide activity.
In addition to heterologous interaction between catalytically active and inactive MTMRs, two catalytically active MTMRs can also interact with each other. The first reported interaction between two catalytically active MTMRs was MTMR3-MTMR4 (18). A subfamily of homologous MTMRs include MTMR6, MTMR7, and MTMR8, all of which can form a heterodimer with MTMR9. The MTMR6/MTMR9 complex has higher activity against, while the MTMR8/MTMR9 complex prefers PI(3)P as substrate (19). Some cellular processes have been implicated in the function of MTMR6/7/8/9 myostuin subsets, such as the heterodimer of MTMR6-MTMR9, which is confirmed to ameliorate cellular apoptosis both in vivo and in vitro (20).
2.2 Modulating the PI3K/AKT pathway
PI3K/AKT pathway plays a vital role in cardiovascular events, including atherosclerosis, cardiac hypertrophy, and vascular remodeling (21). AKT can be activated by a variety of external stimuli in cardiovascular system, such as insulin, vascular endothelial growth factor (VEGF), reactive oxygen species (ROS), and several phosphatase inhibitors (22, 23). These stimuli usually transcriptionally or post-translationally regulate AKT activity. AKT exerts its roles in cardiovascular system by affecting its downstream targets. For example, AKT induces endothelial nitric oxide synthase (eNOS) phosphorylation, vasodilation, and angiogenesis via enhancing VEGF secretion. AKT promotes cellular survival via suppressing FOXOs, caspase 9, and Bcl-2. Additionally, AKT also induces cellular growth and proliferation via increasing mammalian target of rapamycin complex 1 (mTORC1) activity (21).
Razidlo et al. demonstrate that silencing MTM1 significantly inhibits growth factors-induced AKT phosphorylation, which is resulted from abnormal accumulation of PI(3)P, the substrate of MTMR (24). PI(3)P and PI(3,5)P2 generate into phosphatidylinositol (PI) and phosphatidylinositol 5-monophosphate (PI(5)P) when MTM1 is dephosphorylated (Figure 1), while MTM1 knockdown causes a 2-fold rise in total PI(3)P in cell (25). PI(3)P is usually believed to originate from phosphatidylinositol kinase type III. However, increasing evidences suggest that PI3K-C2β can also generates PI(3)P. Overexpression of PI3K-C2β suppresses AKT phosphorylation within mammalian cell, indicating that impaired AKT phosphorylation may be caused by excessive accumulation of PI(3)P (26).
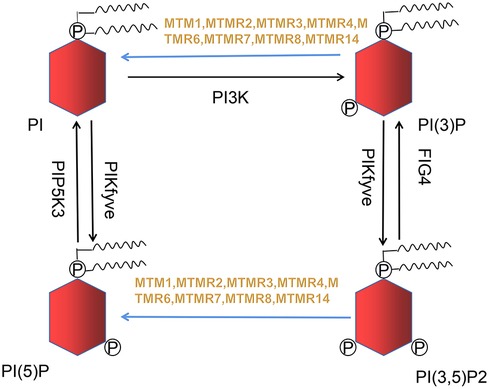
Figure 1. Nine out of sixteen members of the MTMRs family possess catalytic activity, dephosphorylating PI(3)P and PI(3,5)P2 to PI and PI(5)P. This metabolic reaction is indicated in blue arrow.
2.3 Physiological function description of atypical MTMR
The specific pathological and physiological mechanisms of atypical MTMR are as follows. Among the genes abnormally expressed in ankylosing muscular dystrophy type 1 (DM1), myosin associated 1 gene (MTMR1) is associated with impaired muscle differentiation. Maria et al. found that her2 can regulate MTMR11 and promote malignant tumor proliferation. The human heterozygous 15q13.3 microdeletion contains genes FAN1/MTMR15 and MTMR10, which are associated with neuropathological disorders. In addition, MTMR15, as a highly conserved protein, MTMR15/FAN1, interacts with the monoubiquitinated form of FANCD2 and recruits DNA damage sites through FANCD2, promoting repair.
3 Roles of MTMRs in cardiovascular system
There are growing evidences that some MTMRs are differentially expressed in cardiovascular diseases (Table 2). For instance, MTM1 is highly expressed in the membrane of platelets and utilized in the diagnosis of X-linked myotubular myopathy (XMLM). MTMR4 has an effect on the development of LQT1 and aortic aneurysm. As a positive regulator of peroxisome proliferator-activated receptor gamma (PPARγ), MTMR7 has certain diagnostic and therapeutic value for the prevention and treatment of heart failure. MTMR14 regulates cardiomyocyte hypertrophy and proliferation of vascular smooth muscle cell.
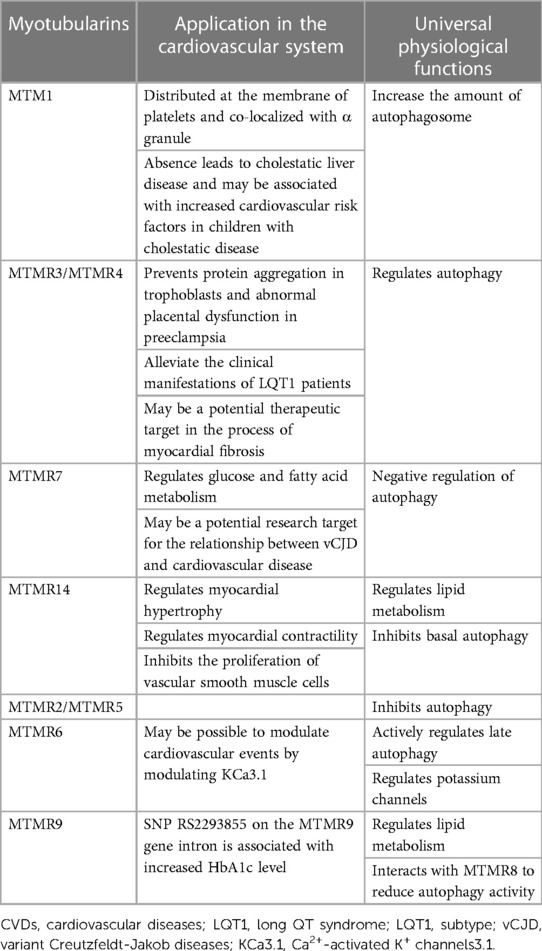
Table 2. Summary of the pathophysiological roles of MTMRs in cardiovascular diseases and common biological functions.
3.1 MTM1
Molecular genetics and histopathology are presently used to diagnose XMLM, which can be resulted from Mtm1 mutation (27). MTM1, a hyperactive 3-phosphatase, is discovered to be abundantly expressed in platelets. MTM1 is mainly distributed at the membrane of platelets and co-localized with α granule. Furthermore, there is no change in aggregation and secretory reaction of platelets after stimulus of thrombin or collagen by using a mouse Mtm1-knocking out model, suggesting that other MTMRs instead of MTM1 play a role in platelets (28). In fact, the mRNA levels of several members of MTMRs are increased during the process of human hemopoietic progenitor cells differentiate into megakaryocyte (29). Whether which kind of MTMR functions in aggregation and secretory reaction of platelet needs further exploration.
MTM1 can interact with a protein which is a striated muscle preferentially expressed protein kinase (SPEG) (30). SPEG plays an important role in the excitation-contraction coupling (31), cytoskeleton organization (32), and other cellular processes (33). The Speg gene is recessively mutated in central nuclear myopathy (CNM) and dilated cardiomyopathy (30). Embryonic Speg-KO mouse shows cardiac enlargement, cardiac fibrosis, decreased cardiac function after birth, and eventually died, which confirms the relationship between SPEG and the occurrence of dilated cardiomyopathy (34). Whether MTM1 plays a role in the pathogenesis of dilated heart disease needs to be further explored. The most recent report was that in 2023, Ka et al. described in a model of zebrafish that loss-of-function mutations in MTM1 lead to severe cholestatic liver disease, while previous studies have reported that there may be increased cardiovascular risk factors in children with cholestatic disease, suggesting that MTM1 may be involved in the occurrence and development of cardiovascular events (35).
3.2 MTMR3/MTMR4
Yeast two-hybrid and co-immunoprecipitation experiments have shown that MTMR3 can bind to MTMR4, so MTMR3 and MTMR4 are introduced together (18). that MTMR3 was found to be a direct target of miR-181a, linking the miRNA to autophagy. In turn, the increase of miR-181a reduced MTMR3, inhibited the occurrence of autophagy, prevented protein aggregation in trophoblasts and abnormal placental dysfunction, and provided a potential therapeutic target for the diagnosis of preeclampsia (36), thereby further reducing the risk of cardiovascular disease, diabetes and other metabolic diseases in women and infants who survived preeclampsia (37).
MTMR4 is a 133-kDa intracellular protein and has two single nucleotide variations (SNVs) within its conserved phosphatase region, which attenuates the degradation of channel proteins and protects ion channels (38). Congenital long QT syndrome (LQTS) is the first reported channelopathy and is associated with mutations of genes encoding ion channels or their regulatory proteins, among which LQT1 is the most common one (39). MTMR4 is confirmed to alleviate the clinical manifestations of LQT1 patients due to the existence of SNVs, which also explain why LQT1 patients has incomplete penetrance and show relatively mild clinical manifestations (38). MTMR4 targets early-stage endosome, regulates TGFβ signaling pathway, and thus participates in cardiovascular diseases via its FEVY domain. Smads protein family act downstream of TGFβ to play critical roles in CVDs. MTMR4 dephosphorylates Smad2/3 within early-stage endosome via binding to the phosphorylated SXS-motif of Smad2/3 and thus stabilizes TGFβ signal (40). Dysregulation of TGF-β signal pathway is involved in the development of aortic aneurysm (41). Three microRNAs (miRNAs) have been identified as diagnostic biomarkers for aortic aneurysm. MTMR4 is the same predictive target of these three miRNAs and shows negative correction with the miRNAs (42). Though MTMR4 may affect aortic aneurysm via affecting TGFβ signaling pathway, there is no direct evidence that MTMR4 has definite curable effects on aortic aneurysm. In addition, Dy et al. (2019) found that MTMR3/MTMR4 regulated interferon gene stimulating factor (STING) trafficking by regulating ptdins3p production, suggesting that MTMR3/MTMR4 may be a potential therapeutic target in the process of myocardial fibrosis, macrophage infiltration and cardiac inflammatory response in patients with diabetes and obesity mediated by STING signaling, which needs to be further experimentally verified (43).
3.3 MTMR7
Peroxisome proliferators-activated receptors (PPARs) belong to the nuclear receptor superfamily, among which a nuclear transcription factor PPARγ inhibits proliferation of cancer cells, exerts lipid lowering and sensitization, and is utilized to prevent against type 2 diabetes (8, 44). RAS-ERK signal transduction has multiple regulatory effects on PPARγ. Weidner et al. demonstrated that downstream effectors of RAS inhibit PPARγ, e.g., by nuclear export and cytosolic sequestration through MEK1. as well as by ERK1/2-dependent phosphorylation (45). Further research found that MTMR7 as a novel interaction partner for PPARγ to counter the inhibitory effect of RAS-ERK on PPARγ (44). MTMR7 is widely expressed in brain, muscle, liver, kidney and cytoplasmic segregation (46). Unlike other MTMRs, MTMR7 is a pro-survival phosphatase and utilizes inositol-1,3 bisphosphate (Ins(1,3)P2) as substrate (47). A synthetic peptide that mimics the CC domain of MTMR7 is able to interact with the steroid receptor coactivator (SRC1) binding site of PPARγ both in vivo and in vitro, indicating that MTMR7 interacts with PPARγ and positively regulates of PPARγ (44). Furthermore, MTMR7 is known to suppress RAS-ERK1/2 and PI3K/AKT/mTOR pathway (8), suggesting that MTMR7 can also indirectly enhance the function of PPARγ. PPARγ is involved in glucose and fatty acid oxidation in cardiac and vascular tissues (48). Pioglitazone, the agonist of PPARγ, ameliorates mitochondrial disorders, reduces lipid deposition during, and thus prevents against severe pulmonary arterial hypertension and vascular remodeling (49). Therefore, further efforts are needed to investigate whether MTMR7 can function in cardiovascular diseases via affecting PPARγ. In addition, a relatively rare SNP variant (rs4921542) in the intron region of MTMR7 is associated with a high risk of variant Creutzfeldt-Jakob disease (vCJD) (50). So, MTMR7 may be a potential research target linking vCJD and cardiovascular disease.
3.4 MTMR14
MTMR14, which takes a variety of phosphates as substrates, is originally identified in human centronuclear myopathy and expressed at kidney, placenta, fat, liver, teste, heart and muscle (51). Recent researches demonstrate that MTMR14 is involved in cardiovascular regulation. The cardiovascular protection mediated by MTMR14 is related to PI3K, which can be activated by G protein-coupled receptors after stress and induces AKT phosphorylation and cardiac hypertrophy (52). MTMR14 can modulate the activity of PI(3,5)P2, which is crucial for maintaining the homeostasis of in muscle (53). MTMR14-mediated regulation of PI(3,5)P2 also exerts in cardiac tissue. Chad et al. demonstrate that PI(3,5)P2 directly binds to RyR2 thus promoting the release of Ca2+ from sarcoplasmic reticulum and improving cardiac contractility (53).
MTMR14 is also discovered to has a specific inhibitory effect on the proliferation of vascular smooth muscle cell (VSMC). Abnormal proliferation and migration of VSMC is the critical step during the development of atherosclerosis and vascular restenosis (54). Kong et al. demonstrate that vascular injury causes neointimal formation and increased expression of MTMR14 in carotid artery (10). Knocking down MTMR14 aggravates neointimal hyperplasia by inducing proliferation of VSMC. Further analysis shows that knocking out MTMR14 (MTMR14-KO) enhances the phosphorylation of polo-like kinase 1 (PLK1), ERK and AKT. PLK1 is activated in proliferating cells and promotes proliferation by activating MEK/ERK signal (55–57). Silencing PLK1 ameliorates MTMR14-KO-induced vascular neointimal hyperplasia, indicating MTMR14 inhibits PLK1 activity by interacting with PLK1, suppresses MAPK activity and thus inhibits vascular restenosis (10).
4 Common biological functions of MTMRs
In addition to participating in the occurrence and development of cardiovascular events, MTMRs are also involved in some cellular processes that are critical in cardiovascular regulation. For example, MTMR9/MTMR14 involved in regulating lipid metabolism. MTMR6 negatively regulates ion channels. Additionally, some MTMRs regulate endocytosis, membrane transport during autophagy, and maintaining autophagy flow (Table 2).
4.1 Effect of MTMRs in regulation of metabolism
Increased prevalence and incidence of obesity have garnered considerable attention worldwide (58). Obesity typically manifests as systemic inflammation, metabolic complications, and fatty accumulation (59), leading to increased risk of chronic diseases such as cardiovascular diseases, cancer, and respiratory diseases (60). Previous studies have found that MTMR7 is highly correlated with glucose metabolism and mammalian targets of rapamycin complex 1 (mTORC1). Further experiments confirmed that MTMR7 significantly inhibited glycolysis and mTORC1 activity in PDGF BB-excited VSMCs in vitro, so it was concluded that MTMR7 inhibited glucose metabolism and thus inhibited VSMC proliferation and migration and vascular intimal proliferation (61). PPARγ is a member of the nuclear receptor superfamily that plays a key role in the differentiation, maintenance, and function of adipocytes (62). In addition, PPARγ also plays an important role in pulmonary hypertension, atherosclerotic and right heart failure cardiovascular disease (45, 63). MTMR7 can interact with PPAR, which indirectly indicates that MTMR7 regulates cardiovascular disease by regulating metabolism. Additionaly, Johnson et al. demonstrate that MTMR9 is located at 8p23-p22 segment and is associated with the obesity phenotype (64). Hotta et al. confirm a close relationship between body mass index (BMI) and single nucleotide polymorphism (SNP) RS2293855. Further analysis shows that the transcription level of MTMR9 in the mouse hypothalamic region is increased after fasting while decreased after high-fat diet, suggesting that genetic variants in MTMR9 may cause obesity and hypertension by regulating hypothalamic neuropeptides (65). SNP RS2293855 on the MTMR9 gene intron is associated with increased HbA1c level, insulin sensitivity, and insulin secretion. However, this association disappears after the recovery of blood glucose, indicating this association is mediated by glycaemic pathways (66). Moreover, MTMR9 overexpression can reduce the surface expression of Wnt/β-catenin signaling gene WNT3A (20). And the rs752107 polymorphism of WNT3A gene is significantly associated with susceptibility to Essential hypertension (EH), and is also associated with the risk of heart failure (HF) and ischemic stroke (IS), suggesting that MTMR9 may be a target between Wnt signaling and cardiovascular diseases (67).
In addition to MTMR9, catalytically active MTMRs also play a role in regulating lipid metabolism. The weight of adult MTMR14-KO mice increases more quickly than that of wild type mice (68), indicating that MTMR14 is involved in obesity. Further analysis shows that MTMR14 deletion results in fatty accumulation, inflammation, and metabolic disorder by releasing serum cytokines, abnormal regulation of several modulatory genes and the PI3K/AKT and ERK signaling pathways. There are also studies demonstrating that elder MTMR14-KO mice display more severe fatty accumulation and metabolic disorder, suggesting that MTMR14-mediated inflammation and metabolic disorder are age-dependent (51) (68).
4.2 Regulates potassium channels
There are four genes encoding Ca2+-activated K+ channels with small or intermediate conductance, including KCa2.1, KCa2.2, KCa2.3, and KCa3.1. KCa3.1 (also known as KCa4, IKCa1, hIK1, or SK4) can be activated by intracellular Ca2+ and the gating of KCa3.1 is voltage-independent (69). It has been reported that KCa3.1 mRNA expression was significantly increased in the coronary arteries of pigs with early atherosclerosis (70) or in rats with myocardial infarction (71) and hypertension (72). Therefore, inhibition of KCa3.1 activity is essential for the development of cardiovascular diseases. CHO-KCa3.1 is a cell line stably expressing KCa3.1. The KCa3.1 current is significantly decreased after co-transfection of CHO-KCa3.1 with GFP-labeled MTMR6, suggesting that MTMR6 inhibits the activity of KCa3.1 (73). MTM1 does not display inhibitory effect on KCa3.1, however, the chimeric MTM1 in which the CC domain is exchanged with the MTMR6 CC domain also inhibits KCa3.1, demonstrating that this inhibition is mediated by the CC domain (73). MTMR6 is known to form heterodimer with MTMR9 to exert its function (5). However, silencing MTMR6 instead of MTMR9 attenuates the lethality of Vps34 mutation in C. elegans, indicating that MTMR6 can also function in a MTMR9-independent manner (73). Whether MTMR9 is essential for the inhibitory role of MTMR6 in KCa3.1 also needs further investigation. MTMR6 selectively dephosphorylates PI(3)P and leads to declined PI(3)P in lipid microdomains adjacent to K(Ca)3.1. Further analysis shows that KCa3.1 activity is also suppressed by PI3K inhibitors, and this suppression can be reversed by the supplement of PI(3)P instead of other phosphoinositides. Additionally, MTMR6-mediated inhibition of K(Ca)3.1 is also rescued by the supplement of PI(3)P (73). Taken together, these data suggest that MTMR6-mediated inhibition of K(Ca)3.1 by dephosphorylating and decreasing PI(3)P may participant in the development of some cardiovascular diseases.
4.3 Role of MTMRs in autophagy/apoptosis
Autophagy and apoptosis are closely regulated processes in cellular and tissue homeostasis, development, and disease (74). Autophagy is a evolutionarily conserved cellular process that depends on lysosomal degradation of cytoplasmic components (75). Serving as an important cellular survival mechanism under stress, autophagy plays a critical role in maintaining cellular homeostasis and function (76). Autophagy or necessary proteins involved in the autophagy process may promote cell death, either by decomposing cells to promote apoptosis or by activating the necrosis program to promote cell death (77). So autophagy is inseparable from programmed cell death. There are many lines of evidences that autophagy/apoptosis is involved in the regulation of CVDs, such as atherosclerosis, hypertension, myocardial infarction, and cardiomyopathy, etc. (78, 79). Depending on PI(3)P- and PI(3,5)P2-mediated degradation of membranes during autophagosome-lysosome fusion, macromolecules are digested by lysosomal enzymes and transported to the cytoplasm for anabolic activities (80). Therefore, enzymes that facilitate the conversion or production of PIs are important for autophagy. MTMRs are known to modulate membrane trafficking (81) and maintain autophagic flux (82) during autophagy and endocytosis. For instance, suppression of MTM1, MTMR1, MTMR2, and MTMR3 in mammal cells, zebrafish, mice, and fruit flies both increase the amount of autophagosome (83–85). And Overexpression of MTM1 can inhibit granulosa cell proliferation and promote cell apoptosis in polycystic ovary syndrome (86). MTMR3 is one of the main genes involved in the regulation of autophagy pathway in mammalian cells, and MTMR3 induces autophagy by inducing or down-regulating mTORC1 (87), while up-regulation leads to the reduction of autophagosomes, thereby inhibiting autophagy (36, 88, 89). MTMR2/MTMR5 is a heterodimer that suppresses autophagy and is crucial for autophagy initiation and autophagosome maturation (90). MTM1, MTMR6 and MTMR9 in C. elegans promote fluid-phase endocytosis. Moreover, Allen et al. demonstrate that CG3530 (dMTMR6), which affects autophagy in fruit flies, is homologous to the human MTMR6, exerts as a regulator of autophagy flux in Drosophila cells, and shares similar function to MTMR8 in mammal cells (82). Further analysis shows that dMTMR6 and MTMR8 function as positive regulators of autophagosome-lysosome homeostasis and positively regulate late autophagy. Downregulation of dMTMR6 and MTMR8 leads to accumulation of autophagic vesicles and disorder of phagocytosis, which finally impairs lysosomal homeostasis (82). In spite of the effect on promoting late autophagy, MTMR6 also exerts as an antagonism under stress via interference with PI3K signal pathway and inhibiting formation of autophagosome (4). There have also been reports that deletion of either the MTMR6 or the MTMR8/MTMR9 complex leads to an increase in autophagy (43, 91). Wang et al. found that negative regulation of MTMR6 can inhibit the proliferation of ovarian cancer cells and promote apoptosis. MTMR7 and MTMR8 are homologous to MTMR6 and can also interact with MTMR9 (19, 46). MTMR7 inhibits insulin signaling and negatively regulates autophagy in colorectal cell line (8), while MTMR8/MTMR9 regulates the PI(3)P pool and positively modulates the level of p62, whose degradation within autophagosome serves as a hallmark of autophagy. However, silencing MTMR8 or MTMR9 alone does not affect autophagy (19). MTMR14 is able to suppress basal autophagy instead of stress-induced autophagy (4). Knocking down MTMR14 leads to accumulation of autophagosome and increased level of LC3Ⅱ, which prevents the subsequent degradation of autophagy macromolecules and provides evidence that MTMR14 is a positive regulator of autophagy (92). MTMR14 can also regulate cardiomyocyte enlargement and programmed cell death through the PI3K \/AKT pathway, as well as inhibit nuclear transcription factor (NF)- κ B) signal transduction reduces cell death and inflammatory response, serving as a protective factor against hepatic ischemia-reperfusion injury. Moreover, knocking out MTMR14 can promote tumor cell apoptosis and inhibit cell migration.
5 Conclusion
The current review summarized the roles and mechanisms of MTMRs in CVDs. MTMRs are a kind of phosphatases that are involved in many biological processes. Mutations in MTMRs are associated with several lines of diseases, including neural disorders, skeletal muscle defects, cancers and CVDs. The canonical biological roles of MTMRs include forming homodimer or heterodimer and regulating PI3K/AKT pathway, by which MTMRs exert their regulatory roles in cardiovascular system. In addition to directly participating in the development of cardiovascular events, MTMRs are also involved in a series of cellular processes that are critical in cardiovascular regulation, including autophagy, metabolism and ion channel (Figure 2). However, the current opinions on MTMRs-dependent cardiovascular regulation are still insufficient. For example, since several MTMRs regulate proliferation in cancer cells and neurocytes, what about the role of these MTMRs in the proliferation of VSMCs, vascular adventitial fibroblasts and vascular intimal hyperplasia? Do MTMRs modulate the development of CVDs via regulating mechanism, autophagy or potassium channels? Taken collectively, a better understanding of the functions of MTMs/MTMRs might significantly contribute to develop novel targets for preventing CVDs.
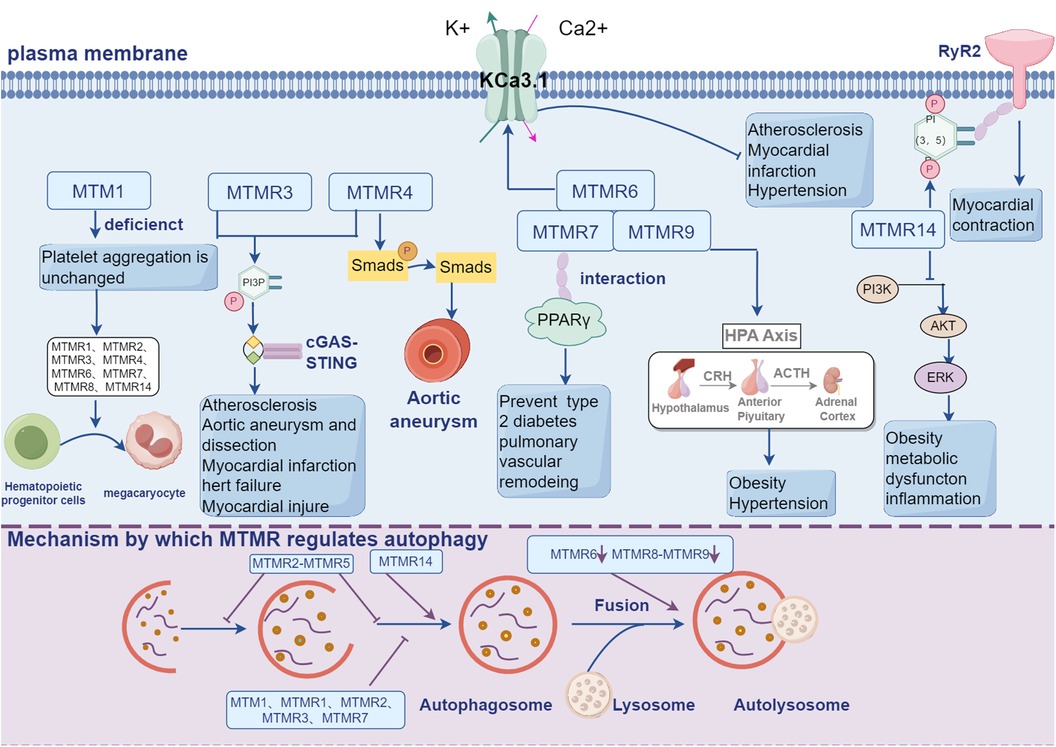
Figure 2. MTMRs not only exert their regulatory effects in the cardiovascular system through the PI3K-AKT signaling pathway, but also participate in a series of cellular processes crucial for cardiovascular regulation, including autophagy, metabolism, and ion channels.
Author contributions
JW: Writing – original draft, Writing – review & editing. WG: Writing – review & editing. QW: Writing – review & editing. XS: Funding acquisition, Writing – review & editing. YY: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
This work was supported by International Natural Science Foundation (82100419 to XS) and (82070289 to YY).
Acknowledgments
Thanks to the Figdraw provided by the Home for Researchers, the production of “Figure 2” comes from Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wishart MJ, Dixon JE. Pten and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. (2002) 12(12):579–85. doi: 10.1016/s0962-8924(02)02412-1
2. Hsu F, Mao Y. The structure of phosphoinositide phosphatases: insights into substrate specificity and catalysis. Biochim Biophys Acta. (2015) 1851(6):698–710. doi: 10.1016/j.bbalip.2014.09.015
3. Lecompte O, Poch O, Laporte J. Ptdins5p regulation through evolution: roles in membrane trafficking? Trends Biochem Sci. (2008) 33(10):453–60. doi: 10.1016/j.tibs.2008.07.002
4. Manzeger A, Tagscherer K, Lorincz P, Szaker H, Lukacsovich T, Pilz P, et al. Condition-dependent functional shift of two Drosophila mtmr lipid phosphatases in autophagy control. Autophagy. (2021) 17(12):4010–28. doi: 10.1080/15548627.2021.1899681
5. Kim SA, Vacratsis PO, Firestein R, Cleary ML, Dixon JE. Regulation of myotubularin-related (mtmr)2 phosphatidylinositol phosphatase by Mtmr5, a catalytically inactive phosphatase. Proc Natl Acad Sci U S A. (2003) 100(8):4492–7. doi: 10.1073/pnas.0431052100
6. Li S, Zhang M, Zhang B. Mtmr14 protects against hepatic ischemia-reperfusion injury through interacting with akt signaling in vivo and in vitro. Biomed Pharmacother. (2020) 129:110455. doi: 10.1016/j.biopha.2020.110455
7. Bolis A, Zordan P, Coviello S, Bolino A. Myotubularin-related (mtmr) phospholipid phosphatase proteins in the peripheral nervous system. Mol Neurobiol. (2007) 35(3):308–16. doi: 10.1007/s12035-007-0031-0
8. Weidner P, Söhn M, Gutting T, Friedrich T, Gaiser T, Magdeburg J, et al. Myotubularin-related protein 7 inhibits insulin signaling in colorectal cancer. Oncotarget. (2016) 7(31):50490–506. doi: 10.18632/oncotarget.10466
9. Guerrero-Valero M, Grandi F, Cipriani S, Alberizzi V, Di Guardo R, Chicanne G, et al. Dysregulation of myelin synthesis and actomyosin function underlies aberrant myelin in Cmt4b1 neuropathy. Proc Natl Acad Sci U S A. (2021) 118(10):e2009469118. doi: 10.1073/pnas.2009469118
10. Kong LY, Liang C, Li PC, Zhang YW, Feng SD, Zhang DH, et al. Myotubularin-related Protein14 prevents neointima formation and vascular smooth muscle cell proliferation by inhibiting polo-like kinase1. J Am Heart Assoc. (2022) 11(21):e026174. doi: 10.1161/jaha.122.026174
11. Zhang JL, Zhang DH, Li YP, Wu LM, Liang C, Yao R, et al. Myotubularin-related protein 14 suppresses cardiac hypertrophy by inhibiting akt. Cell Death Dis. (2020) 11(2):140. doi: 10.1038/s41419-020-2330-6
12. Previtali SC, Zerega B, Sherman DL, Brophy PJ, Dina G, King RH, et al. Myotubularin-related 2 protein phosphatase and neurofilament light chain protein, both mutated in cmt neuropathies, interact in peripheral nerve. Hum Mol Genet. (2003) 12(14):1713–23. doi: 10.1093/hmg/ddg179
13. Berger P, Berger I, Schaffitzel C, Tersar K, Volkmer B, Suter U. Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2. Hum Mol Genet. (2006) 15(4):569–79. doi: 10.1093/hmg/ddi473
14. Nandurkar HH, Layton M, Laporte J, Selan C, Corcoran L, Caldwell KK, et al. Identification of myotubularin as the lipid phosphatase catalytic subunit associated with the 3-phosphatase adapter protein, 3-pap. Proc Natl Acad Sci U S A. (2003) 100(15):8660–5. doi: 10.1073/pnas.1033097100
15. Gupta VA, Hnia K, Smith LL, Gundry SR, McIntire JE, Shimazu J, et al. Loss of catalytically inactive lipid phosphatase myotubularin-related protein 12 impairs myotubularin stability and promotes centronuclear myopathy in zebrafish. PLoS Genet. (2013) 9(6):e1003583. doi: 10.1371/journal.pgen.1003583
16. Berger P, Schaffitzel C, Berger I, Ban N, Suter U. Membrane association of myotubularin-related protein 2 is mediated by a pleckstrin homology-gram domain and a coiled-coil dimerization module. Proc Natl Acad Sci U S A. (2003) 100(21):12177–82. doi: 10.1073/pnas.2132732100
17. Berger P, Tersar K, Ballmer-Hofer K, Suter U. The Cmt4b disease-causing proteins Mtmr2 and Mtmr13/Sbf2 regulate akt signalling. J Cell Mol Med. (2011) 15(2):307–15. doi: 10.1111/j.1582-4934.2009.00967.x
18. Lorenzo O, Urbe S, Clague MJ. Systematic analysis of myotubularins: heteromeric interactions, subcellular localisation and endosome related functions. J Cell Sci. (2006) 119(Pt 14):2953–9. doi: 10.1242/jcs.03040
19. Zou J, Zhang C, Marjanovic J, Kisseleva MV, Majerus PW, Wilson MP. Myotubularin-related protein (mtmr) 9 determines the enzymatic activity, substrate specificity, and role in autophagy of Mtmr8. Proc Natl Acad Sci U S A. (2012) 109(24):9539–44. doi: 10.1073/pnas.1207021109
20. Doubravská L, Dostál V, Knop F, Libusová L, Macůrková M. Human myotubularin-related protein 9 regulates er-to-Golgi trafficking and modulates Wnt3a secretion. Exp Cell Res. (2020) 386(1):111709. doi: 10.1016/j.yexcr.2019.111709
21. Abeyrathna P, Su Y. The critical role of akt in cardiovascular function. Vascul Pharmacol. (2015) 74:38–48. doi: 10.1016/j.vph.2015.05.008
22. Taylor GS, Maehama T, Dixon JE. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. (2000) 97(16):8910–5. doi: 10.1073/pnas.160255697
23. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. Foxo3 controls autophagy in skeletal muscle in vivo. Cell Metab. (2007) 6(6):458–71. doi: 10.1016/j.cmet.2007.11.001
24. Razidlo GL, Katafiasz D, Taylor GS. Myotubularin regulates akt-dependent survival signaling via phosphatidylinositol 3-phosphate. J Biol Chem. (2011) 286(22):20005–19. doi: 10.1074/jbc.M110.197749
25. Cao C, Backer JM, Laporte J, Bedrick EJ, Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal pi(3)P and growth factor receptor trafficking. Mol Biol Cell. (2008) 19(8):3334–46. doi: 10.1091/mbc.e08-04-0367
26. Domin J, Harper L, Aubyn D, Wheeler M, Florey O, Haskard D, et al. The class ii phosphoinositide 3-kinase Pi3k-C2beta regulates cell migration by a Ptdins3p dependent mechanism. J Cell Physiol. (2005) 205(3):452–62. doi: 10.1002/jcp.20478
27. Biancalana V, Caron O, Gallati S, Baas F, Kress W, Novelli G, et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet. (2003) 112(2):135–42. doi: 10.1007/s00439-002-0869-1
28. Mansour R, Severin S, Xuereb JM, Gratacap MP, Laporte J, Buj-Bello A, et al. Expression of myotubularins in blood platelets: characterization and potential diagnostic of X-linked myotubular myopathy. Biochem Biophys Res Commun. (2016) 476(3):167–73. doi: 10.1016/j.bbrc.2016.04.127
29. Schweinfurth N, Hohmann S, Deuschle M, Lederbogen F, Schloss P. Valproic acid and all trans retinoic acid differentially induce megakaryopoiesis and platelet-like particle formation from the megakaryoblastic cell line meg-01. Platelets. (2010) 21(8):648–57. doi: 10.3109/09537104.2010.513748
30. Lornage X, Sabouraud P, Lannes B, Gaillard D, Schneider R, Deleuze JF, et al. Novel speg mutations in congenital myopathy without centralized nuclei. J Neuromuscul Dis. (2018) 5(2):257–60. doi: 10.3233/jnd-170265
31. Amoasii L, Hnia K, Chicanne G, Brech A, Cowling BS, Müller MM, et al. Myotubularin and Ptdins3p remodel the sarcoplasmic reticulum in muscle in vivo. J Cell Sci. (2013) 126(Pt 8):1806–19. doi: 10.1242/jcs.118505
32. Hnia K, Tronchère H, Tomczak KK, Amoasii L, Schultz P, Beggs AH, et al. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J Clin Invest. (2011) 121(1):70–85. doi: 10.1172/jci44021
33. Dowling JJ, Joubert R, Low SE, Durban AN, Messaddeq N, Li X, et al. Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis Model Mech. (2012) 5(6):852–9. doi: 10.1242/dmm.009746
34. Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, et al. Speg interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet. (2014) 95(2):218–26. doi: 10.1016/j.ajhg.2014.07.004
35. Karolczak S, Deshwar AR, Aristegui E, Kamath BM, Lawlor MW, Andreoletti G, et al. Loss of Mtm1 causes cholestatic liver disease in a model of X-linked myotubular myopathy. J Clin Invest. (2023) 133(18):e166275. doi: 10.1172/jci166275
36. Ayoub SE, Shaker OG, Aboshama RA, Etman MK, Khalefa AA, Khamiss Abd Elguaad MM, et al. Expression profile of lncrna anril, mir-186, mir-181a, and mtmr-3 in patients with preeclampsia. Non-coding RNA Research. (2023) 8(4):481–6. doi: 10.1016/j.ncrna.2023.06.001
37. Dimitriadis E, Rolnik DL, Zhou W, Estrada-Gutierrez G, Koga K, Francisco RPV, et al. Pre-eclampsia. Nature Reviews Disease Primers. (2023) 9(1):8. doi: 10.1038/s41572-023-00417-6
38. Lee Y-K, Sala L, Mura M, Rocchetti M, Pedrazzini M, Ran X, et al. Mtmr4 snvs modulate Ion channel degradation and clinical severity in congenital long qt syndrome: insights in the mechanism of action of protective modifier genes. Cardiovasc Res. (2021) 117(3):767–79. doi: 10.1093/cvr/cvaa019
39. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-qt syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. (2001) 103(1):89–95. doi: 10.1161/01.cir.103.1.89
40. Yu J, Pan L, Qin X, Chen H, Xu Y, Chen Y, et al. Mtmr4 attenuates transforming growth factor beta (tgfbeta) signaling by dephosphorylating R-smads in endosomes. J Biol Chem. (2010) 285(11):8454–62. doi: 10.1074/jbc.M109.075036
41. Jones JA, Spinale FG, Ikonomidis JS. Transforming growth factor-beta signaling in thoracic aortic aneurysm development: a paradox in pathogenesis. J Vasc Res. (2009) 46(2):119–37. doi: 10.1159/000151766
42. Moushi A, Pillar N, Keravnou A, Soteriou M, Shomron N, Cariolou MA, et al. Micrornas in ascending thoracic aortic aneurysms. Biosci Rep. (2020) 40(7):BSR20200218. doi: 10.1042/bsr20200218
43. Zhou Z, Ou-yang C, Chen Q, Ren Z, Guo X, Lei M, et al. Trafficking and effect of released DNA on cgas-sting signaling pathway and cardiovascular disease. Front Immunol. (2023) 14:1287130. doi: 10.3389/fimmu.2023.1287130
44. Weidner P, Söhn M, Schroeder T, Helm L, Hauber V, Gutting T, et al. Myotubularin-related protein 7 activates peroxisome proliferator-activated receptor-gamma. Oncogenesis. (2020) 9(6):59. doi: 10.1038/s41389-020-0238-8
45. Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, et al. An erk/Cdk5 axis controls the diabetogenic actions of ppargamma. Nature. (2015) 517(7534):391–5. doi: 10.1038/nature13887
46. Mochizuki Y, Majerus PW. Characterization of myotubularin-related protein 7 and its binding partner, myotubularin-related protein 9. Proc Natl Acad Sci U S A. (2003) 100(17):9768–73. doi: 10.1073/pnas.1333958100
47. Hnia K, Vaccari I, Bolino A, Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol Med. (2012) 18(6):317–27. doi: 10.1016/j.molmed.2012.04.004
48. Calvier L, Chouvarine P, Legchenko E, Hoffmann N, Geldner J, Borchert P, et al. Pparγ links Bmp2 and Tgfβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. (2017) 25(5):1118–34.e7. doi: 10.1016/j.cmet.2017.03.011
49. Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M, et al. Ppargamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. (2018) 10(438):eaao0303. doi: 10.1126/scitranslmed.aao0303
50. Sanchez-Juan P, Bishop MT, Aulchenko YS, Brandel JP, Rivadeneira F, Struchalin M, et al. Genome-wide study links Mtmr7 gene to variant creutzfeldt-jakob risk. Neurobiol Aging. (2012) 33(7):1487.e21–8. doi: 10.1016/j.neurobiolaging.2011.10.011
51. Lv Y, Xue L, Cai C, Liu QH, Shen J. Deficiency of myotubularin-related protein 14 influences body weight, metabolism, and inflammation in an age-dependent manner. Cell Biosci. (2015) 5:69. doi: 10.1186/s13578-015-0062-6
52. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. (2005) 115(8):2108–18. doi: 10.1172/JCI24682
53. Touchberry CD, Bales IK, Stone JK, Rohrberg TJ, Parelkar NK, Nguyen T, et al. Phosphatidylinositol 3,5-bisphosphate (pi(3,5)P2) potentiates cardiac contractility via activation of the ryanodine receptor. J Biol Chem. (2010) 285(51):40312–21. doi: 10.1074/jbc.M110.179689
54. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114(4):590–600. doi: 10.1093/cvr/cvy010
55. Wilson JL, Wang L, Zhang Z, Hill NS, Polgar P. Participation of Plk1 and Foxm1 in the hyperplastic proliferation of pulmonary artery smooth muscle cells in pulmonary arterial hypertension. PLoS One. (2019) 14(8):e0221728. doi: 10.1371/journal.pone.0221728
56. Rezey AC, Gerlach BD, Wang R, Liao G, Tang DD. Plk1 mediates paxillin phosphorylation (ser-272), centrosome maturation, and airway smooth muscle layer thickening in allergic asthma. Sci Rep. (2019) 9(1):7555. doi: 10.1038/s41598-019-43927-8
57. de Cárcer G, Wachowicz P, Martínez-Martínez S, Oller J, Méndez-Barbero N, Escobar B, et al. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat Med. (2017) 23(8):964–74. doi: 10.1038/nm.4364
58. Karaman S, Hollmén M, Robciuc MR, Alitalo A, Nurmi H, Morf B, et al. Blockade of vegf-C and vegf-D modulates adipose tissue inflammation and improves metabolic parameters under high-fat diet. Mol Metab. (2015) 4(2):93–105. doi: 10.1016/j.molmet.2014.11.006
59. Pang J, Rhodes DH, Pini M, Akasheh RT, Castellanos KJ, Cabay RJ, et al. Increased adiposity, dysregulated glucose metabolism and systemic inflammation in galectin-3 ko mice. PLoS One. (2013) 8(2):e57915. doi: 10.1371/journal.pone.0057915
60. Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. (2010) 314(1):1–16. doi: 10.1016/j.mce.2009.07.031
61. Sun X, Yang Y, Zhao W, Wang M, Chen Y, Wang J, et al. Mtmr7 suppresses the phenotypic switching of vascular smooth muscle cell and vascular intimal hyperplasia after injury via regulating P62/Mtorc1-mediated glucose metabolism. Atherosclerosis. (2024) 390:117470. doi: 10.1016/j.atherosclerosis.2024.117470
62. Hernandez-Quiles M, Broekema MF, Kalkhoven E. Ppargamma in metabolism, immunity, and cancer: unified and diverse mechanisms of action. Front Endocrinol (Lausanne). (2021) 12:624112. doi: 10.3389/fendo.2021.624112
63. Sorice M. Crosstalk of autophagy and apoptosis. Cells. (2022) 11(9):1479. doi: 10.3390/cells11091479
64. Johnson L, Luke A, Adeyemo A, Deng HW, Mitchell BD, Comuzzie AG, et al. Meta-analysis of five genome-wide linkage studies for body mass Index reveals significant evidence for linkage to chromosome 8p. Int J Obes (Lond. (2005) 29(4):413–9. doi: 10.1038/sj.ijo.0802817
65. Hotta K, Kitamoto T, Kitamoto A, Mizusawa S, Matsuo T, Nakata Y, et al. Association of variations in the fto, Scg3 and Mtmr9 genes with metabolic syndrome in a Japanese population. J Hum Genet. (2011) 56(9):647–51. doi: 10.1038/jhg.2011.74
66. Tang L, Tong Y, Cao H, Xie S, Yang Q, Zhang F, et al. The Mtmr9 Rs2293855 polymorphism is associated with glucose tolerance, insulin secretion, insulin sensitivity and increased risk of prediabetes. Gene. (2014) 546(2):150–5. doi: 10.1016/j.gene.2014.06.028
67. Ren H, Luo J-Q, Ouyang F, Cheng L, Chen X-P, Zhou H-H, et al. Wnt3a Rs752107(C > T) polymorphism is associated with an increased risk of essential hypertension and related cardiovascular diseases. Front Cardiovasc Med. (2021) 8:413–9. doi: 10.3389/fcvm.2021.675222
68. Yin L, Yong-bo P, Meng-Fei Y, Weiwei C, Ping Z, Lu X, et al. Mice lacking myotubularin-related protein 14 show accelerated high-fat diet-induced lipid accumulation and inflammation. J Physiol Biochem. (2016) 73(1):17–28. doi: 10.1007/s13105-016-0520-6
69. Zhang XD, Thai PN, Lieu DK, Chiamvimonvat N. Cardiac small-conductance calcium-activated potassium channels in health and disease. Pflugers Arch. (2021) 473(3):477–89. doi: 10.1007/s00424-021-02535-0
70. Tharp DL, Wamhoff BR, Turk JR, Bowles DK. Upregulation of intermediate-conductance Ca2+-activated K+ channel (Ikca1) mediates phenotypic modulation of coronary smooth muscle. American Journal of Physiology Heart and Circulatory Physiology. (2006) 291(5):H2493–503. doi: 10.1152/ajpheart.01254.2005
71. Saito T, Fujiwara Y, Fujiwara R, Hasegawa H, Kibira S, Miura H, et al. Role of augmented expression of intermediate-conductance Ca2+-activated K+ channels in postischaemic heart. Clin Exp Pharmacol Physiol. (2002) 29(4):324–9. doi: 10.1046/j.1440-1681.2002.03652.x
72. Terata Y, Saito T, Fujiwara Y, Hasegawa H, Miura H, Watanabe H, et al. Pitavastatin inhibits upregulation of intermediate conductance calcium-activated potassium channels and coronary arteriolar remodeling induced by long-term blockade of nitric oxide synthesis. Pharmacology. (2003) 68(4):169–76. doi: 10.1159/000070455
73. Srivastava S, Li Z, Lin L, Liu G, Ko K, Coetzee WA, et al. The phosphatidylinositol 3-phosphate phosphatase myotubularin- related protein 6 (Mtmr6) is a negative regulator of the Ca2+-activated K+ channel Kca3.1. Mol Cell Biol. (2005) 25(9):3630–8. doi: 10.1128/MCB.25.9.3630-3638.2005
74. Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. (2018) 20(10):1110–7. doi: 10.1038/s41556-018-0201-5
75. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. (2004) 6(4):463–77. doi: 10.1016/s1534-5807(04)00099-1
76. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. (2007) 13(5):619–24. doi: 10.1038/nm1574
77. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. (2014) 15(2):81–94. doi: 10.1038/nrm3735
78. Salabei JK, Hill BG. Autophagic regulation of smooth muscle cell biology. Redox Biol. (2015) 4:97–103. doi: 10.1016/j.redox.2014.12.007
79. Pedro JM B-S, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. (2017) 120(11):1812–24. doi: 10.1161/CIRCRESAHA.117.311082
80. Schink KO, Tan KW, Stenmark H. Phosphoinositides in control of membrane dynamics. Annu Rev Cell Dev Biol. (2016) 32:143–71. doi: 10.1146/annurev-cellbio-111315-125349
81. Nicot AS, Laporte J. Endosomal phosphoinositides and human diseases. Traffic. (2008) 9(8):1240–9. doi: 10.1111/j.1600-0854.2008.00754.x
82. Allen EA, Amato C, Fortier TM, Velentzas P, Wood W, Baehrecke EH. A conserved myotubularin-related phosphatase regulates autophagy by maintaining autophagic flux. J Cell Biol. (2020) 219(11):e201909073. doi: 10.1083/jcb.201909073
83. Al-Qusairi L, Prokic I, Amoasii L, Kretz C, Messaddeq N, Mandel JL, et al. Lack of myotubularin (Mtm1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. Faseb J. (2013) 27(8):3384–94. doi: 10.1096/fj.12-220947
84. Dowling JJ, Low SE, Busta AS, Feldman EL. Zebrafish Mtmr14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet. (2010) 19(13):2668–81. doi: 10.1093/hmg/ddq153
85. Hao F, Itoh T, Morita E, Shirahama-Noda K, Yoshimori T, Noda T. The Ptdins3-phosphatase Mtmr3 interacts with Mtorc1 and suppresses its activity. FEBS Lett. (2016) 590(1):161–73. doi: 10.1002/1873-3468.12048
86. Xu X, Guan R, Gong K, Xie H, Shi L. Circ_furin knockdown assuages testosterone-induced human ovarian Granulosa-like tumor cell disorders by sponging mir-423-5p to reduce Mtm1 expression in polycystic ovary syndrome. Reprod Biol Endocrinol. (2022) 20(1):32. doi: 10.1186/s12958-022-00891-9
87. Dossou AS, Basu A. The emerging roles of Mtorc1 in macromanaging autophagy. Cancers (Basel). (2019) 11(10):1422. doi: 10.3390/cancers11101422
88. Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, et al. Modulation of local Ptdins3p levels by the pi phosphatase Mtmr3 regulates constitutive autophagy. Traffic. (2010) 11(4):468–78. doi: 10.1111/j.1600-0854.2010.01034.x
89. Sun G, Li Z, He Z, Wang W, Wang S, Zhang X, et al. Circular rna Mctp2 inhibits cisplatin resistance in gastric cancer by mir-99a-5p-mediated induction of Mtmr3 expression. J Exp Clin Cancer Res. (2020) 39(1):246. doi: 10.1186/s13046-020-01758-w
90. Chua JP, Bedi K, Paulsen MT, Ljungman M, Tank EMH, Kim ES, et al. Myotubularin-related phosphatase 5 is a critical determinant of autophagy in neurons. Curr Biol. (2022) 32(12):2581–95.e6. doi: 10.1016/j.cub.2022.04.053
91. Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase jumpy. EMBO J. (2009) 28(15):2244–58. doi: 10.1038/emboj.2009.159
Keywords: cardiovascular diseases, MTMR, phosphoinositide, PI3K/AKT, autophagy
Citation: Wang J, Guo W, Wang Q, Yang Y and Sun X (2024) Recent advances of myotubularin-related (MTMR) protein family in cardiovascular diseases. Front. Cardiovasc. Med. 11:1364604. doi: 10.3389/fcvm.2024.1364604
Received: 4 January 2024; Accepted: 27 February 2024;
Published: 11 March 2024.
Edited by:
Venkatesh Sundararajan, West Virginia University, United StatesReviewed by:
Sithara Thomas, University of Texas Health Science Center at Houston, United StatesHarikrishnan Venugopal, Albert Einstein College of Medicine, United States
Soumya Krishnan, Downstate Health Sciences University, United States
© 2024 Wang, Guo, Wang, Yang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiongshan Sun shan19910927@sina.com Yongjian Yang yyj10001@126.com
†These authors have contributed equally to this work