Research progress on the improvement of cardiovascular diseases through the autonomic nervous system regulation of the NLRP3 inflammasome pathway
 Yuchi Hu
Yuchi Hu Songyuan Dai
Songyuan Dai - Department of Cardiology, First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, China
Cardiovascular disease stands as a leading global cause of mortality. Nucleotide-binding Oligomerization Domain-like Receptor Protein 3 (NLRP3) inflammasome is widely acknowledged as pivotal factor in specific cardiovascular disease progression, such as myocardial infarction, heart failure. Recent investigations underscore a close interconnection between autonomic nervous system (ANS) dysfunction and cardiac inflammation. It has been substantiated that sympathetic nervous system activation and vagus nerve stimulation (VNS) assumes critical roles withinNLRP3 inflammasome pathway regulation, thereby contributing to the amelioration of cardiac injury and enhancement of prognosis in heart diseases. This article reviews the nexus between NLRP3 inflammasome and cardiovascular disorders, elucidating the modulatory functions of the sympathetic and vagus nerves within the ANS with regard to NLRP3 inflammasome. Furthermore, it delves into the potential therapeutic utility of NLRP3 inflammasome to be targeted by VNS. This review serves as a valuable reference for further exploration into the potential mechanisms underlying VNS in the modulation of NLRP3 inflammasome.
1 Introduction
Cardiovascular diseases exhibit high global prevalence (57.2%) (1), with escalating incidence and mortality rates (2). Extensive research has clarified the pathological mechanisms at the core of these diseases, encompassing myocardial cell apoptosis, ventricular remodeling, myocardial inflammatory infiltration, fibrosis, and metabolic disorders (3–6). Notably, Inflammation can induce damage to cardiomyocytes, leading to cardiac dysfunction and disease progression (7). It is crucial to recognize that inflammatory infiltration in myocardial tissue can also contribute to myocardial fibrosis, myocardial cell apoptosis, and disruptions in energy metabolism (8). This inflammatory process stands as a pivotal factor contributing to an unfavorable prognosis and elevated mortality rates in cardiovascular diseases. Mounting evidence emphasizes the significance of the inflammatory cascade reaction initiated by NLRP3 inflammasome activation, serving as a major driver of inflammatory infiltration within myocardial tissue, and a key contributor to specific cardiovascular disorder progression, such as myocardial infarction, heart failure (9–11). Consequently, intervening in the pathological progression in cardiovascular disorders by modulating NLRP3 inflammasome pathway emerges as a crucial protective measure.
Currently, ANS served as pivotal avenue for cardiovascular diseases treatment (12). Numerous studies, particularly in areas including myocardial infarction (MI), as well as heart failure, have illuminated the crucial role of interfering with the NLRP3 pathway in mediating therapeutic effects through the ANS (13). This article aims to comprehensively review the intricate correlation between cardiovascular disorders and NLRP3 inflammasome, delving into the regulatory functions exerted by sympathetic and vagus nerves within the ANS on NLRP3 inflammasome. Furthermore, it explores potential therapeutic targets for VNS in modulating NLRP3 inflammasome, offering valuable insights for subsequent interventions in cardiovascular diseases through ANS-mediated NLRP3 inflammasome pathways.
2 NLRP3 inflammasome: new-established therapeutic target within cardiovascular diseases
2.1 NLRP3 inflammasome
Inflammasome is a multiprotein oligomer present in immune cells comprising Nucleotide-binding oligomerization domain (NOD)-like receptors (NLR), Pyrin domain (PYD), and caspase activation and recruitment domain (CARD). NLRP3 inflammasome constitutes a focal point of research in cardiovascular diseases (14). Notably, extensive investigation have been conducted on NLRP3 inflammasome. NLRP3, identified as a nod-like receptor protein, functions as an intracellular pattern recognition receptor primarily linked to the inflammasome pathway. It possesses the ability to detect both pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) (15, 16). The primary defensive response during infections or tissue damage entails the discharge of cellular contents into the myocardial interstitium and the initiation of intracellular stress pathways. These pathways encompass Reactive oxygen species (ROS) production, oxidative stress, autophagy, and innate immunity triggering. Collectively, these processes contribute to the triggering of NLRP3 inflammasome (17–19). Mitochondrial fission plays a crucial role in enhancing ROS generation and releasing mitochondrial DNA into the cytoplasm, thereby facilitating the activation of NLRP3 inflammasome (17–19). NLRP3 inflammasome acts as a nexus, fostering inflammatory responses and functioning as an effector to initiate the cascade of inflammatory reactions (Figure 1). A critical step in this biological cascade is Caspase-1 activation, an enzyme precursor cleaved into an active form during NLRP3 inflammasome oligomerization (20). Caspase-1 is subsequently responsible for the maturation of pro-Interleukin-1β (pro-IL-1β) and pro-IL-18 into their active form. Gasdermin D (GSDMD)-NT pores formation triggers the processing and secretion of IL-1β and −18. Caspase-1 facilitates pore formation by cleaving various substrates associated with crucial metabolic pathways, actively facilitating the release of IL-1β and IL-18 (21).This culminates in a cascade of inflammatory reactions and the inflammatory infiltration of the myocardium, recognized as pyroptosis (22).
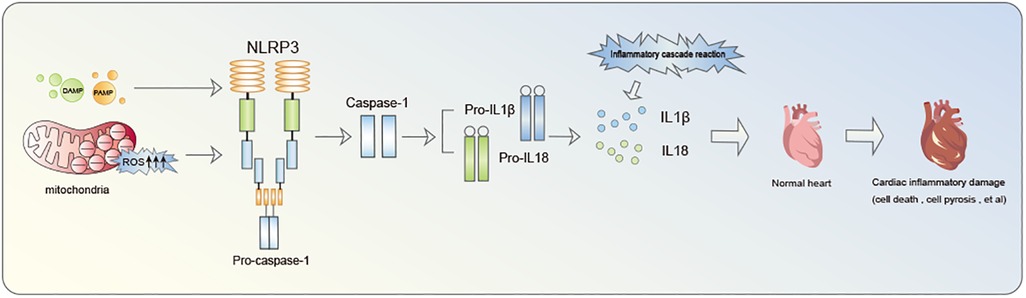
Figure 1. The inflammatory cascade initiated by NLRP3 inflammasome notably contributes to cardiac damage.
2.2 NLRP3 inflammasome as a significant risk factor for myocardial infarction
MI constitutes as a cardiovascular emergency precipitated by arterial thrombosis, resulting in a drastic reduction in myocardial blood flow (23, 24). The pathological features of MI delineate that, signaling pathways involving endothelial cell proliferation, apoptosis, and autophagy, as well as those of fibroblasts, orchestrate myocardial cell injury and the progression of MI. Notably, the NLRP3/Caspase-1-mediated pyroptosis pathway has emerged as a pivotal contributor (25). Research findings (26, 27) highlight that activation of the NLRP3 inflammasome fosters the release of IL-1β and IL-18 from myocardial and arterial endothelial cells, ultimately causing coronary artery impairment and exacerbation of MI. Furthermore, NLRP3 inflammasome activation prompts the release of IL-1β from fibroblasts, thereby instigating fibrotic alterations and promoting the enlargement of infarcted myocardial scars (28). It has been reported that oxidized LDL (oxLDL) within infarcted cardiomyocytes catalyzes cholesterol crystallization and induces the transcription of NLRP3 inflammasome and pro-IL-1β. Subsequent NLRP3 inflammasome activation triggers inflammatory cascade via the Nuclear factor kappa-B (NF-κB) pathway, leading to endothelial cell dysfunction and fostering atherosclerosis (29). Additionally, in accordance with Sandanger et al. (30), upregulation of NLRP3 inflammasome expression alongside pro-inflammatory cytokines IL-1 and IL-18 was documented in MI mice. This affirms, that inflammasome activation in fibroblasts exacerbates myocardial fibrosis and apoptosis, thereby aggravating MI. In summary, NLRP3 emerges as a critical risk factor for MI.
2.3 Correlation between NLRP3 inflammasome and congestive heart failure
The initiation of NLRP3 inflammasome resulted in purinergic receptor P2X7 (P2X7R) activation through Adenosine triphosphate (ATP) and diverse toxins. This activation sets off processes like efflux of potassium, the generation of ROS, and mitochondrial DNA release. This cascade results in myocardial damage, myocardial cell necrosis, and the exacerbation of congestive heart failure (CHF) (31). Research indicates that NLRP3 inflammasome activation facilitates IL-1β and −18 extensive release, both of which have the potential to impair myocardial contractility (32). Concurrently, they interfere with the regulation of cytoplasmic calcium by altering the synthesis of proteins, inducing diastolic dysfunction, and subsequently impairing cardiac pumping function, thereby worsening heart failure or potentially inducing heart failure (33, 34). Xin-Li formula (XLF) is a traditional Chinese medicine formulation showing promise in treating heart failure. A study conducted on rats with heart failure induced by combined hyperlipidemia and MI revealed that XLF possesses anti-inflammatory properties. These effects are attributed to its ability to inhibit mammalian target of rapamycin (mTOR) phosphorylation, increasing the population of Foxp3+ T-regulatory cells (Tregs), and inhibiting NLRP3 activation in THP-1 macrophages. This leads to the inhibition of the inflammatory response mediated by NLRP3 inflammasomes, offering potential benefits for patients with heart failure (35). Moreover, Canakinumab is a therapeutic monoclonal antibody targeting IL-1β (36), holds promise in reducing tissue or cell inflammation by blocking IL-1β activity, and potentially inhibiting the inflammatory pathway triggered by the NLRP3 inflammasome. A sub-analysis conducted within the CANTOS trial demonstrated a reduction in the rates of heart failure hospitalization with the administration of canakinumab administration. The MRC-ILA-Heart study (37), which included 182 patients experiencing small acute myocardial infarction (AMI), revealed that NLRP3 inflammasome activation and the heightened IL-1 activity during the acute phase of myocardial infarction correlated with escalating heart failure risk. Canakinumab, conversely, demonstrated the ability to block NLRP3 inflammasome and IL-1β activations, highlighting IL-1-targeted therapy potential in preventing the recurrence of atherosclerotic thrombotic events and heart failure happening (38). In summary, NLRP3 inflammasome activation is implicated in the generation and exacerbation of chronic heart failure (CHF). Intervening within NLRP3 inflammasome pathway may hold promise for improving the prognosis of CHF.
3 Correlation between excessive sympathetic nervous system activation and NLRP3 activation
3.1 Cardiovascular diseases pathogenesis: dysregulation of the autonomic nervous system
ANS regulation governing cardiac function can be categorized into two systems. Firstly, the central nervous system comprises sympathetic and parasympathetic nerves extending from the brain and spinal cord to the heart. Secondly, the intrinsic cardiac nervous system, constituted by multiple ganglion plexuses (GP) distributed in the epicardial fat pad and within the Marshall ligament, encompassing both sympathetic and parasympathetic nerves. A crucial factor within cardiovascular disease progression is the imbalance in the functions of cardiac sympathetic and parasympathetic nerves. Pathophysiologically, ANS activation, including Sympathetic nervous system (SNS) and renin-angiotensin-aldosterone system (RAAS), serves as determinant within progression and recurrence of cardiovascular diseases (39). Both animal and clinical studies provide substantial evidence indicating that autonomic imbalance in cardiovascular diseases advances from vagal dominance in the early stages to sympathetic dominance in the later stages (40–42). The initial surge in sympathetic activity aims to maintain cardiac output. however, prolonged SNS can lead to increased adrenaline secretion, precipitating changes such as cardiac structural remodeling, deterioration of cardiac function, myocardial cell apoptosis, myocardial tissue fibrosis, and cardiac electrical remodeling. These alterations may manifest as symptoms such as chest tightness, shortness of breath, restricted exercise capacity, or difficulty lying down. Often, these factors can overlap or be causally linked, resulting in a substantial decrease in ejection fraction and severe clinical symptoms and consequences. The unfavorable clinical prognosis observed in patients with cardiovascular diseases is associated with the activation of the SNS, diminished parasympathetic nervous system activity, and reduced heart rate variability (43).
3.2 Induction of NLRP3 inflammasome activation by excessive sympathetic nervous system stimulation
SNS excessive activation is a crucial factor in initiating inflammation and damage to heart. It's stated that the non-selective β-adrenergic receptor agonist isoproterenol (ISO) triggers various pro-inflammatory cytokine expressions within myocardial cells, including tumor necrosis factor (TNF)-α, IL-6, −1β, and −18, as a result of chronic β-adrenergic stimulation (44, 45). These pro-inflammatory cytokines are recognized contributors to myocardial injury and contribute to long-term pathological remodeling, such as myocardial fibrosis. A study on IL-18 in β-adrenergic injury-induced cardiac inflammation and fibrosis validated the findings of the previously mentioned research (46).. This study found that when the heart is excessively stimulated by stress hormones (acute β-adrenergic overstimulation), it triggers certain inflammatory response in heart cell, resulting in harmful changes in the heart structure. This process involves reactive oxygen and receptors called β1-adrenergic receptors. It sets off a chain reaction of cytokines and the infiltration of immune cells (macrophages), ultimately causing negative changes in the heart (Figure 2). SNS has receptors called α1-adrenergic receptors (α1-AR). In a significant experiment, using a substance called phenylephrine (PE) that activates these α1-adrenergic receptors in mice resulted in heart dysfunction and inflammation (47). Notably, PE injection markedly increased the expression levels of caspase-1, NLRP3, and IL-18 in the heart. In summary, The overstimulation of SNS can trigger the activation of NLRP3, setting off a series of inflammatory responses and ultimately leading to pyroptosis in myocardial cells.

Figure 2. Regulation of cardiac function by ANS: influence mechanism based on NLRP3 inflammasome pathway.
Conversely, additional studies indicated that interrupting β-adrenergic receptor (β-AR) signaling can effectively inhibit IL-1β, NLRP3, and IL-18 activations, subsequently preventing their mediation of cardiac fibrosis (13). Consequently, within cardiovascular disease progression, NLRP3 inflammasome activation induced by excessive sympathetic activation is intricately linked to the progression of cardiovascular diseases. Strategies for inhibiting the overactivation of SNS and promoting VNS to maintain ANS balance present new avenues for therapeutic intervention in cardiac disease treating.
4 Mechanisms of VNS within cardiovascular diseases treatment via modulating NLRP3 inflammasome
VNS can effectively inhibit SNS activation by enhancing vagal nerve stimulation, thereby restoring the balance of ANS function. This therapeutic methodology is employed in heart diseases treatment. As previously discussed, excessive SNS activity is closely linked to cardiac inflammation, myocardial injury, NLRP3 inflammasome activation. Recent years have witnessed multiple studies indicating that VNS serves as a determinant in ameliorating cardiac inflammation infiltration, and enhancing ventricular remodeling and improving cardiac function (Figure 2).
4.1 Anti-drug induced cardiotoxic effects
Medications, especially cancer drugs, have been known to potentially cause harmful effects on the heart (48). Prolonged administration of drugs may result in myocardial injury and compromised cardiac function, potentially resulting in cardiovascular diseases progression and, in severe cases, progressing to end-stage heart failure. Recent research (49) suggests that VNS therapy may possess cardioprotective properties. It achieves this by alleviating cardiac dysfunction mediated by the NLRP3 inflammasome, enhancing myocardial energy metabolism, reducing inflammatory infiltration, and safeguarding cardiac function in the aftermath of drug-induced injury.
4.1.1 Mitigating doxorubicin-induced cardiotoxicity
Doxorubicin (DOX) serves as one of the common chemotherapy meditation applied for treating various cancers like soft tissue sarcoma, osteosarcoma, breast cancer, bladder cancer, gastric cancer, and thyroid cancer (50). However, a major downside of using DOX is its potential harm to the heart.
In a recent study by Nanthip Prathumsap and colleagues (51), they explored the impact and mechanisms of VNS on DOX-induced cardiotoxicity. The findings confirmed that VNS could confer cardiac protection by enhancing the balance of cardiac sympathetic/parasympathetic nerves, reducing myocardial cell apoptosis, thereby improving left ventricular function, and mitigating cardiac damage. Mechanistically, DOX disrupts cardiac mitochondrial function and dynamics, subsequently activating autophagy in myocardial cells and mitochondria, leading to cardiac inflammation (52). In the course of this process, NLRP3 inflammasome expression and activation increase, promoting pyroptosis within Wistar rats myocardium, which emerges as cell death primary form within Wistar rats myocardial cells processed by DOX (53). Study outcomes indicated that VNS treatment significantly ameliorated DOX-induced mitochondrial and myocardial cell dysfunction, almost entirely abrogating the increase in levels of pyroptosis-related proteins and inflammatory factors induced by NLRP3 inflammasome activation. Additionally, the study revealed that the beneficial effects of VNS could be completely nullified by employing muscarinic acetylcholine receptor (mAChR) or nicotinic acetylcholine receptor (nAChR) inhibitors. In summary, VNS alleviates DOX-induced cardiotoxicity by obstructing NLRP3 inflammasome pathway, playing crucial protective role, potentially mediated through acetylcholine receptors. Although the study suggests that DOX primarily induces myocardial cell death through pyroptosis, additional research is necessary. Techniques like electron microscopy and immunohistochemistry should be employed to clarify the different types of cell death mediated by DOX.
4.1.2 Alleviating trastuzumab-induced cardiotoxicity
Trastuzumab (Trz) is a targeted anticancer medication specifically formulated for the treatment of tumors characterized by human epidermal growth factor receptor 2 (HER2) positivity. While Trz has proven effective in enhancing cancer patient survival, research has substantiated that prolonged use of Trz may result in cardiotoxicity and subsequent cardiac dysfunction (54). Furthermore, extended administration of Trz has been associated with impaired cardiac function, with potential mechanisms of Trastuzumab-induced cardiotoxicity (TIC) encompassing mitochondrial dysfunction, oxidative stress, and inflammation within the heart (55).
In the recent study by Thawatchai Khuanjing et al., they conducted the implantation and stimulation of VNS devices in rats treated with Trz. Additionally, they introduced a treatment group that combined Trz with donepezil (TDPZ), an acetylcholinesterase inhibitor used to mimic VNS functionality (56). The research findings suggest that the hearts of rats treated with Trz exhibit an imbalance between sympathetic and vagal activity, leading to left ventricular dysfunction. Moreover, rats with TIC experience significant damage to mitochondrial function and dynamics, while Trz increases cardiomyocyte death through inducing ferroptosis, apoptosis, and pyroptosis. However, within Trz-treated rats subjected to VNS and TDPZ, mitochondrial ROS levels decrease. The phosphorylation of dynamin-related protein 1 (Drp1), associated with mitochondrial dynamics, and the expression of mitochondrial Drp1 significantly decrease, while fusion protein optic atrophy 1 (OPA1) expression notably increases. These findings affirm that both VNS and donepezil can significantly enhance mitochondrial function and alleviate oxidative stress in cardiomyocytes, offering protection to the heart. Moreover, a crucial factor contributing to cardiomyocyte death is NLRP3 inflammasome-mediated pyroptosis, closely associated with levels of ROS and mitochondrial function (57). Research results indicate that VNS and donepezil similarly alleviate ferroptosis, apoptosis, and pyroptosis in cardiac myocytes.
Collectively, research suggests that VNS or acetylcholinesterase inhibitor application in the context of doxorubicin-induced TIC provides protective effects for the heart. This protection is attributed to the reduction of inflammatory infiltration, improvement in mitochondrial function, suppression of oxidative stress, and inhibition of pyroptosis mediated by NLRP3 inflammasome. Such interventions could potentially be valuable in future treatments for TIC. However, it's important to note that this study simulated VNS effects through acetylcholinesterase inhibitors and did not directly target acetylcholine receptors with specific drugs. VNS therapy is still awaited to be researched.
4.2 Cardiac protection against myocardial injury induced by thermal damage
Thermal injury pathogenesis is intricate, involving the confluence of factors such as systemic alterations in cytokine expression, free radical damage, and inadequate tissue perfusion. Thermal injury has the potential to initiate a systemic inflammatory response (SIRS), which may be instigated by the cascade reaction mediated by NLRP3 activation, leading to substantial release of IL-1β (58). Moreover, severe thermal injury has the potential to induce cardiac stress by causing a surge in plasma catecholamines. This leads to stress reactions and heightened sympathetic nerve tension due to increased metabolism, ultimately resulting in myocardial cell damage through the mitochondrial pathway (59). The mitochondrial pathway is a prominent mechanism for myocardial cell death (60). It promotes inflammatory cells infiltration into myocardial tissue by inducing oxidative stress, loss of growth factors, hypoxia, and triggering NLRP3 activation and DNA damage.
In a murine model of thermal injury, Xiaojiong Lu and colleagues delved into the deleterious effects of thermal injury on myocardial tissue and examined VNS impact on myocardial damage. The study underscored the pivotal role of VNS in safeguarding mitochondrial function and suppressing inflammation in myocardial tissue (61). Research findings unveiled notable pathological alterations in mouse myocardial tissue at 6 h and 24 h post-burn injury. At the molecular level, there were observable signs of mitochondrial swelling and dysfunction, coupled by an escalation in ROS linked to oxidative stress and an elevation in NLRP3 inflammasome expression. These observations highlight that thermal injury can lead to significant inflammatory infiltration and damage to the heart, suggesting the potential for post-burn cardiac dysfunction and heart failure (62). Importantly, mice subjected to VNS showed noticeable improvement in myocardial pathology. Critically, VNS effectively reduced thermal injury-induced swelling of myocardial mitochondria and NLRP3 inflammasome expression. Collectively, VNS demonstrates the ability to alleviate myocardial damage caused by thermal injury, potentially by reducing inflammatory infiltration in myocardial tissue induced by NLRP3 inflammasomes. Additionally, the study proposed that VNS might offer protection against cardiac injury through M3-AchR, though further investigation is required.
4.3 Application of VNS in myocardial ischemia-reperfusion injury
VNS improves cardiac function by connecting electrodes to the vagus nerve trunk in the neck and a pulse generator (63). In recent years, some studies have advanced VNS devices to enhance therapeutic effectiveness. Sun Yu and colleagues developed a closed-loop self-powered LL-VNS system using triboelectric nanogenerator technology (64). Their results indicated that, even with stimulation intensity much lower than traditional VNS, the system still produced significant therapeutic effects. Moreover, this system significantly reduced inflammatory infiltration of myocardial cells, thus preventing additional damage to the heart cells.
In recent research, Yao Lu and colleagues introduced a magnetic vagus nerve stimulation (mVNS) device. They replaced the traditional vagus nerve electrode with an injectable magnetic hydrogel containing superparamagnetic iron oxide (SPIO) nanoparticles, aiming for more precise and effective vagus nerve stimulation (65). Furthermore, they applied mVNS in an SD rat model and investigated its mechanism of action in myocardial ischemia-reperfusion injury (I/R). The research results revealed that mVNS treatment significantly alleviated myocardial I/R injury and the release of inflammatory cytokines, while inhibiting the expression of OGDHL (a protein associated with increased mitochondrial ROS levels) (66). This reduction in OGDHL expression contributed to less myocardial necrosis caused by myocardial I/R injury. Hence, mVNS could serve as a novel therapeutic approach to inhibit myocardial necrosis, significantly influencing the I/R injury process and improving the prognosis of ischemia-reperfusion myocardium. Additionally, the researchers used the M2AChR antagonist Otenzepad to explore the anti-necrotic mechanism of mVNS. The results showed that Otenzepad attenuated mVNS's function within limiting myocardial infarct size and inhibiting NLRP3 inflammasome activation. Consequently, mVNS may effectively inhibit myocardial necrosis by modulating the M2AChR/OGDHL/ROS axis, protecting the heart, and improving the prognosis of myocardial I/R injury. However, more investigation seems to be necessary to fully delve into the precise mechanism by which acetylcholine receptor activation reduces OGDHL expression.
5 Potential therapeutic targets of VNS
Our article has underscored the pivotal role of NLRP3 inflammasome within heart injury and cardiac diseases pathological progression. Moreover, This article elucidates the mechanism by which blocking NLRP3 inflammasome activation via ANS can be utilized to regulate the treatment of heart disease. For instance, studies have unveiled that the excessive activation of β-AR triggers NLRP3 inflammasome in myocardial cells through the β1-AR/ROS signaling pathway, leading to cardiac inflammation and fibrosis. Notably, the blockade of β-AR in C57BL/6 mice significantly alleviates cardiac inflammation, accompanied by a substantial reduction in NLRP3 inflammasome activity (46). Similarly, the inhibition of α1-AR in mice treated with PE markedly diminishes heart inflammation induced by NLRP3 inflammasome pathway, resulting in a noticeable improvement in heart function (47).Additionally, VNS exhibits the capacity to curb the overactivation of the SNS and enhance VNS, thereby restoring the balance of ANS function and consequently ameliorating cardiac inflammation and function. Hence, VNS may realize therapeutic objectives in treating heart diseases by diminishing the expression of α1-AR and β-AR in the SNS, thereby inhibiting NLRP3 inflammasome pathway.
The fusion and dysfunction of mitochondria play pivotal roles in the pathological changes associated with heart diseases, resulting in ROS accumulation, where ROS serves as an upstream regulatory factor within NLRP3 inflammasome activation (67). Pertinent researches substantiated that VNS can ameliorate mitochondrial fusion and dysfunction while diminishing ROS production (56, 57, 61). Consequently, mitochondria emerge as a focal point for VNS in cardiac inflammation treatment. Acetylcholine, primary neurotransmitter of the vagus nerve, acts on mAchR or nAchR (68). VNS modulates relevant inflammatory pathways by augmenting acetylcholine release and the expression of mAchR and nAchR. Research indicated that the M2AChR/OGDHL/ROS axis effectively inhibited myocardial cell necrosis (65), affirming the efficacy of this pathway and participation of mAchR. 5 subtypes of muscarinic acetylcholine receptors (M1-5 receptors) have been identified and extensively studied in cardiovascular research. M3AchR is also recognized as a contributor to improve mitochondrial function (69), rendering it a novel and effective potential treating target.
6 Conclusion
NLRP3 inflammasome emerges as a pivotal factor triggering cardiac inflammation, exerting unignorable influences on initiation and developing of heart diseases. Dysregulation of the ANS stands out as crucial mediator within NLRP3 inflammasome pathway. This article reviews the roles of SNS activation and VNS in NLRP3 inflammasome and underscores that, despite extensive research on SNS activation and VNS in NLRP3 inflammasome, the precise mechanisms through which VNS regulates NLRP3 inflammasome to ameliorate cardiovascular diseases remain uncertain. In conclusion, the potential of VNS targeting NLRP3 inflammasome shows promise in the treatment of the cardiovascular system. Subsequent research endeavors into the intricate mechanisms governing how VNS regulates NLRP3 inflammasome are imperative for a more comprehensive understanding.
Author contributions
YH: Conceptualization, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft. SD: Resources, Writing – review & editing. LuZ: Writing – review & editing. LiZ: Writing – review & editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article.
This research was funded by National Natural Science Foundation (grant no. 82160080); Yunnan Province Revitalizing Yunnan Talents Support Plan Project (grant no. YNWR-MY-2020-011); Yunnan Province Revitalizing Yunnan Talents Support Plan Project (grant no. XDYC-QNRC-2022-0315).
Acknowledgments
The authors thank all the medical and technology team.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Global Cardiovascular Risk Consortium, Magnussen C, Ojeda FM, Leong DP, Alegre-Diaz J, Amouyel P, et al. Global effect of modifiable risk factorson cardiovascular disease and mortality. N Engl J Med. (2023) 389:1273–85. doi: 10.1056/NEJMoa2206916
2. Read SH, Wild SH. Prevention of premature cardiovascular death worldwide. Lancet. (2020) 395:758–60. doi: 10.1016/s0140-6736(19)32034-3
3. Ajoolabady A, Chiong M, Lavandero S, Klionsky DJ, Ren J. Mitophagy incardiovascular diseases: molecular mechanisms, pathogenesis, and treatment. Trends Mol Med. (2022) 28:836–49. doi: 10.1016/j.molmed.2022.06.007
4. Corbalan JJ, Vatner DE, Vatner SF. Myocardial apoptosis in heart disease: does the emperor have clothes? Basic Res Cardiol. (2016) 111:31. doi: 10.1007/s00395-016-0549-2
5. Wang L, Yuan D, Zheng J, Wu X, Wang J, Liu X, et al. Chikusetsu saponin IVa attenuates isoprenaline-induced myocardial fibrosis in mice through activation autophagy mediated by AMPK/mTOR/ULK1 signaling. Phytomedicine. (2019) 58:152764. doi: 10.1016/j.phymed.2018.11.024
6. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. CardiovascRes. (2017) 113:389–98. doi: 10.1093/cvr/cvx012
7. Suetomi T, Miyamoto S, Brown JH. Inflammation in nonischemic heart disease: initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am J Physiol Heart Circ Physiol. (2019) 317:H877–90. doi: 10.1152/ajpheart.00223.2019
8. Wang H, Jing ZC. Inflammation and cardiovascular diseases. Chronic Dis Transl Med. (2020) 6:215–8. doi: 10.1016/j.cdtm.2020.09.003
9. Wen ZG, Jie S, Feng G. Roles of pyroptosis in myocardial ischemia/reperfusion injury diseases. J Adv Health. (2019) 01:184–9. doi: 10.3724/sp.j.2640-8686.2019.0102
10. Balanescu S, Barbu E, Georgescu C, Popescu AC. NLRP3 inflammasome in cardiovascular disease: David’s stone against Goliath? Rom J Cardiol. (2021) 31:517–27. doi: 10.47803/rjc.2021.31.3.517
11. Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu Z, et al. NLRP3: a novel mediator in cardiovascular disease. J Immunol Res. (2018) 2018:1–8. doi: 10.1155/2018/5702103
12. Lu M, Yu Z, Li Q, Gong M, An L, Xu T, et al. Electroacupuncture stimulation regulates adipose lipolysis via catecholamine signaling mediated by NLRP3 suppression in obese rats. Front Endocrinol. (2021) 12:773127. doi: 10.3389/fendo.2021.773127
13. Yang HJ, Kong B, Shuai W, Zhang JJ, Huang H. Knockout of MD1 contributes to sympathetic hyperactivity and exacerbates ventricular arrhythmias following heart failure with preserved ejection fraction via NLRP3 inflammasome activation. Exp Physiol. (2020) 105:966–78. doi: 10.1113/EP088390
14. Mauro AG, Bonaventura A, Mezzaroma E, Quader M, Toldo S. NLRP3 inflammasome in acute myocardial infarction. J Cardiovasc Pharmacol. (2019) 74:175–87. doi: 10.1097/FJC.0000000000000717
15. Kim YK, Shin JS, Nahm MH. NOD-like receptors in infection, immunity, and diseases. Yonsei Med J. (2016) 57:5–14. doi: 10.3349/ymj.2016.57.1.5
16. Amin J, Boche D, Rakic S. What do we know about the inflammasome in humans? Brain Pathol. (2017) 27:192–204. doi: 10.1111/bpa.12479
17. Mu Y, Yin TL, Zhang Y, Yang J, Wu YT. Diet-induced obesity impairs spermatogenesis: the critical role of NLRP3 in sertoli cells. Inflamm Regen. (2022) 42:24. doi: 10.1186/s41232-022-00203-z
18. Darwesh AM, Keshavarz-Bahaghighat H, Jamieson KL, Seubert JM. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase ameliorates cardiac ischemia/reperfusion injury by attenuating NLRP3 inflammasome activation. Int J Mol Sci. (2019) 20:null. doi: 10.3390/ijms20143502
19. Shrivastava G, Visoso-Carvajal G, Garcia-Cordero J, Leon-Juarez M, Chavez-Munguia B, Lopez T, et al. Dengue virus serotype 2 and its non-structural proteins 2A and 2B activate NLRP3 inflammasome. Front Immunol. (2020) 11:352. doi: 10.3389/fimmu.2020.00352
20. Diaz-Del-Olmo I, Worboys J, Martin-Sanchez F, Gritsenko A, Ambrose AR, Tannahill GM, et al. Internalization of the membrane attack complex triggers NLRP3 inflammasome activation and IL-1β secretion in human macrophages. Front Immunol. (2021) 12:720655. doi: 10.3389/fimmu.2021.720655
21. Wang L, Jiao XF, Wu C, Li XQ, Sun HX, Shen XY, et al. Trimetazidine attenuates dexamethasone-induced muscle atrophy via inhibiting NLRP3/GSDMD pathway-mediated pyroptosis. Cell Death Discov. (2021) 7:251. doi: 10.1038/s41420-021-00648-0
22. Sun W, Lu H, Dong S, Li R, Chu Y, Wang N, et al. Beclin1 controls caspase-4 inflammsome activation and pyroptosis in mouse myocardial reperfusion-induced microvascular injury. Cell Commun Signal. (2021) 19:107. doi: 10.1186/s12964-021-00786-z
23. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
24. Golforoush P, Yellon DM, Davidson SM. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res Cardiol. (2020) 115:73. doi: 10.1007/s00395-020-00829-5
25. Zhang Q, Wang L, Wang S, Cheng H, Xu L, Pei G, et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct Target Ther. (2022) 7:78. doi: 10.1038/s41392-022-00925-z
26. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. (2011) 108:19725–30. doi: 10.1073/pnas.1108586108
27. Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem. (2007) 282:36321–9. doi: 10.1074/jbc.M708182200
28. Karen VS, Meng D, Ting JPY. The NLRP3 inflammasome: molecularactivation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
29. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
30. Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2013) 99:164–74. doi: 10.1093/cvr/cvt091
31. Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, et al. Α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. (2014) 20:350–8. doi: 10.2119/molmed.2013.00117
32. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
33. Buckley LF, Abbate A. Interleukin-1 blockade in cardiovascular diseases: a clinical update. Eur Heart J. (2018) 39:2063–9. doi: 10.1093/eurheartj/ehy128
34. Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1β induces a reversible cardiomyopathy in the mouse. Inflamm Res. (2013) 62:637–40. doi: 10.1007/s00011-013-0625-0
35. Lan T, Zeng Q, Zhu Y, Zheng G, Chen K, Jiang W, et al. Xin-Li formula attenuates heart failure induced by a combination of hyperlipidemia and myocardial infarction in rats via treg immunomodulation and NLRP3 inflammasome inhibition. J Tradit Complement Med. (2023) 13:441–53. doi: 10.1016/j.jtcme.2023.03.009
36. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. CANTOS trial group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
37. Abbate A, Trankle CR, Buckley LF, Lipinski MJ, Appleton D, Kadariya D, et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J Am Heart Assoc. (2020) 9:e014941. doi: 10.1161/JAHA.119.014941
38. Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-STelevation acute coronary syndromes: the MRC-ILA heart study. Eur Heart J. (2015) 36:377–84. doi: 10.1093/eurheartj/ehu272
39. Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, et al. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression ina canine high-rate pacing model. Circ Heart Fail. (2009) 2:692–9. doi: 10.1161/CIRCHEARTFAILURE.109.873968
40. Karim S, Chahal A, Khanji MY, Petersen SE, Somers VK. Autonomic cardiovascular control in health and disease. Compr Physiol. (2023) 13:4493–511. doi: 10.1002/cphy.c210037
41. DePace NL, Colombo J, Mandal K, Eisen HJ. Autonomic testing optimizes therapy for heart failure and related cardiovascular disorders. Curr Cardiol Rep. (2022) 24:1699–709. doi: 10.1007/s11886-022-01781-7
42. Gold MR, Van Veldhuisen DJ, Hauptman PJ, Borggrefe M, Kubo SH, Lieberman RA, et al. Vagus nerve stimulation for the treatment of heart failure: the INOVATE-HF trial. J Am Coll Cardiol. (2016) 68:149–58. doi: 10.1016/j.jacc.2016.03.525
43. Sabbah HN, Ilsar I, Zaretsky A, Rastogi S, Wang M, Gupta RC. Vagus nerve stimulation in experimental heart failure. Heart Fail Rev. (2011) 16:171–8. doi: 10.1007/s10741-010-9209-z
44. Chandrasekar B, Marelli-Berg FM, Tone M, Bysani S, Prabhu SD, Murray DR. Beta-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Commun. (2004) 319:304–11. doi: 10.1016/j.bbrc.2004.04.185
45. Frangogiannis NG. Inflammation in cardiac injury, repair and regeneration. CurrOpin Cardiol. (2015) 30:240–5. doi: 10.1097/HCO.0000000000000158
46. Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur Heart J. (2018) 39:60–9. doi: 10.1093/eurheartj/ehx261
47. Xin JZ, Wu JM, Hu GM, Gu HJ, Feng YN, Wang SX, et al. α1-AR overactivation induces cardiac inflammation through NLRP3 inflammasome activation. Acta Pharmacol Sin. (2020) 41:311–8. doi: 10.1038/s41401-019-0305-x
48. Lyon AR. Heart failure resulting from cancer treatment: still serious but an opportunity for prevention. Heart. (2019) 105:6–8. doi: 10.1136/heartjnl-2018-313493
49. Ahmed M, Nudy M, Bussa R, Weigel F, Naccarelli G, Maheshwari A. Non-pharmacologic autonomic neuromodulation for treatment of heart failure: a systematic review and meta-analysis of randomized controlled trials. Trends Cardiovasc Med. (2024) 34:101–7. doi: 10.1016/j.tcm.2022.09.007
50. Sohail M, Sun Z, Li Y, Gu X, Xu H. Research progress in strategies to improve theefficacy and safety of doxorubicin for cancer chemotherapy. Expert Rev Anticancer Ther. (2021) 21:1385–98. doi: 10.1080/14737140.2021.1991316
51. Prathumsap N, Ongnok B, Khuanjing T, Arinno A, Maneechote C, Apaijai N, et al. Vagus nerve stimulation exerts cardioprotection against doxorubicin-induced cardiotoxicity through inhibition of programmed cell death pathways. Cell Mol Life Sci. (2022) 80(1):21. doi: 10.1007/s00018-022-04678-4
52. Maneechote C, Khuanjing T, Ongnok B, Arinno A, Prathumsap N, Chunchai T, et al. Promoting mitochondrial fusion in doxorubicin-induced cardiotoxicity: a novel therapeutic target for cardioprotection. Clin Sci. (2022) 136:841–60. doi: 10.1042/CS20220074
53. Shi S, Chen Y, Luo Z, Nie G, Dai Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun Signal. (2023) 21:61. doi: 10.1186/s12964-023-01077-5
54. Banke A, Fosbøl EL, Ewertz M, Videbæk L, Dahl JS, Poulsen MK, et al. Long-term risk of heart failure in breast cancer patients after adjuvant chemotherapy with or without trastuzumab. JACC Heart Fail. (2019) 7:217–24. doi: 10.1016/j.jchf.2018.09.001
55. Choksey A, Timm KN. Cancer therapy-induced cardiotoxicity-A metabolic perspective on pathogenesis, diagnosis and therapy. Int J Mol Sci. (2021) 23:441. doi: 10.3390/ijms23010441
56. Khuanjing T, Maneechote C, Ongnok B, Prathumsap N, Arinno A, Chunchai T, et al. Vagus nerve stimulation and acetylcholinesterase inhibitor donepezil provide cardioprotection against trastuzumab-induced cardiotoxicity in rats by attenuating mitochondrial dysfunction. Biochem Pharmacol. (2023) 217:115836. doi: 10.1016/j.bcp.2023.115836
57. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. (2020) 34:101523. doi: 10.1016/j.redox.2020.101523
58. Hofmann E, Holzer-Geissler JCJ, Tiffner KI. 365 A human ex vivo burn model—its characterization and use for preclinical testing of innovative wound dressings. J Invest Dermatol. (2021) 141:S212. doi: 10.1016/j.jid.2021.08.374
59. Hatef DA, Hollier LH. Pathophysiologic response to severe burn injury. J Craniofac Surg. (2009) 20:1300–1. doi: 10.1097/scs.0b013e3181ae2210
60. Yu JD, Miyamoto S. Molecular signaling to preserve mitochondrial integrity against ischemic stress in the heart: rescue or remove mitochondria in danger. Cells. (2021) 10:3330. doi: 10.3390/cells10123330
61. Lu X, Costantini T, Lopez NE, Wolf PL, Hageny AM, Putnam J, et al. Vagal nerve stimulation protects cardiac injury by attenuating mitochondrial dysfunction in a murine burn injury model. J Cell Mol Med. (2013) 17:664–71. doi: 10.1111/jcmm.12049
62. Niederbichler AD, Hoesel LM, Ipaktchi K, Olivarez L, Erdmann M, Vogt PM, et al. Burn-induced heart failure: lipopolysaccharide binding protein improves burn and endotoxin-induced cardiac contractility deficits. J Surg Res. (2011) 165:128–35. doi: 10.1016/j.jss.2009.06.012
63. Hamann JJ, Ruble SB, Stolen C, Wang M, Gupta RC, Rastogi S, et al. Vagusnerve stimulation improves left ventricular function in a canine model of chronic heart failure. Eur J Heart Fail. (2013) 15:1319–26. doi: 10.1093/eurjhf/hft118
64. Sun Y, Chao S, Ouyang H, Zhang W, Luo W, Nie Q, et al. Hybrid nanogenerator based closed-loop self-powered low-level vagus nerve stimulation system for atrial fibrillation treatment. Sci Bull. (2022) 67:1284–94. doi: 10.1016/j.scib.2022.04.002
65. Lu Y, Chen K, Zhao W, Hua Y, Bao S, Zhang J, et al. Magnetic vagus nerve stimulation alleviates myocardial ischemia-reperfusion injury by the inhibition of pyroptosis through the M2AChR/OGDHL/ROS axis in rats. J Nanobiotechnology. (2023) 21:421. doi: 10.1186/s12951-023-02189-3
66. Sherrill JD, Kc K, Wang X, Wen T, Chamberlin A, Stucke EM, et al. Whole-exome sequencing uncovers oxidoreductases DHTKD1 and OGDHL as linkers between mitochondrial dysfunction and eosinophilic esophagitis. JCI Insight. (2018) 3:e99922. doi: 10.1172/jci.insight.99922
67. Zhou Z, Wang Z, Guan Q, Qiu F, Li Y, Liu Z, et al. PEDF inhibits the activation of NLRP3 inflammasome in hypoxia cardiomyocytes through PEDF receptor/phospholipase A2. Int J Mol Sci. (2016) 17:2064. doi: 10.3390/ijms17122064
68. Li DL, Liu JJ, Liu BH, Hu H, Sun L, Miao Y, et al. Acetylcholine inhibits hypoxia-induced tumor necrosis factor-α production via regulation of MAPKs phosphorylation in cardiomyocytes. J Cell Physiol. (2011) 226:1052–9. doi: 10.1002/jcp.22424
Keywords: NLRP3, ANS, VNS, SNS, cardiovascular disease
Citation: Hu Y, Dai S, Zhao L and Zhao L (2024) Research progress on the improvement of cardiovascular diseases through the autonomic nervous system regulation of the NLRP3 inflammasome pathway. Front. Cardiovasc. Med. 11:1369343. doi: 10.3389/fcvm.2024.1369343
Received: 16 January 2024; Accepted: 28 March 2024;
Published: 8 April 2024.
Edited by:
Suet Nee Chen, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Junping Zhang, First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, ChinaRaffaella Lombardi, University of Naples Federico II, Italy
© 2024 Hu, Dai, Zhao and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lulu Zhao 757432706@qq.com Ling Zhao zhaoling580@126.com