Non-neuronal cell-derived acetylcholine, a key modulator of the vascular endothelial function in health and disease
 Takashi Sonobe
Takashi Sonobe Yoshihiko Kakinuma
Yoshihiko Kakinuma- Department of Bioregulatory Science, Graduate School of Medicine, Nippon Medical School, Tokyo, Japan
Vascular endothelial cells play an important role in regulating peripheral circulation by modulating arterial tone in the microvasculature. Elevated intracellular Ca2+ levels are required in endothelial cells to induce smooth muscle relaxation via endothelium-dependent mechanisms such as nitric oxide production, prostacyclin, and endothelial cell hyperpolarization. It is well established that exogenous administration of acetylcholine can increase intracellular Ca2+ concentrations, followed by endothelium-dependent vasodilation. Although endogenous acetylcholine's regulation of vascular tone remains debatable, recent studies have reported that endogenously derived acetylcholine, but not neuronal cell-derived acetylcholine, is a key modulator of endothelial cell function. In this minireview, we summarize the current knowledge of the non-neuronal cholinergic system (NNCS) in vascular function, particularly vascular endothelial cell function, which contributes to blood pressure regulation. We also discuss the possible pathophysiological impact of endothelial NNCS, which may induce the development of vascular diseases due to endothelial dysfunction, and the potential of endothelial NNCS as a novel therapeutic target for endothelial dysfunction in the early stages of metabolic syndrome, diabetes, and hypertension.
1 Introduction
Vascular endothelial cells, the key players in maintaining cardiovascular homeostasis, actively modulate the arterial tone in the microvasculature. Our modern lifestyles, often characterized by sedentary behavior, are strongly linked to the development of endothelial dysfunction, a significant contributing factor (1–3). This dysfunction has emerged as a fundamental factor in the pathogenesis of cardiovascular diseases and metabolic complications, including hypertension, obesity, and diabetes (4–6). Various factors, such as hypertension, hyperlipidemia, obesity, diabetes, and aging, induce excess shear stress, oxidative stress, and local inflammation in the vascular endothelial cells, leading to endothelial cell damage. In experimental and clinical studies, endothelial dysfunction is typically characterized by impaired endothelium-dependent vascular relaxation. The physiological response of endothelial cells to exogenous acetylcholine (ACh) is vasodilation (7), which is markedly attenuated in individuals with hypertension (4) and diabetes (8). Reduced responsiveness to ACh is frequently used as a marker of endothelial dysfunction (9, 10). Recent studies have highlighted the importance of “endogenous” ACh, whose origin differs from the classical neurotransmitter ACh in the nervous system (11, 12). The non-neuronal cholinergic system (NNCS) is comprised of various cell types, including immune (13), myocardial (14, 15), and endothelial cells (16), all of which can synthesize and respond to ACh. Endothelial cells respond to exogenously administered ACh to initiate vasodilator signaling. Endothelial function can be modulated by endothelial cells by synthesizing and releasing ACh in an autocrine manner. Considering this, a notable correlation may exist between endothelium-derived ACh and the impaired modulation of endothelial function in the development of cardiovascular disease.
This minireview provides an overview of the roles of non-neuronal cell-derived ACh in regulating vascular endothelial function under physiological and pathophysiological conditions. In particular, we focused on NNCS in endothelial cells, which affect arterial tone. In addition, we discuss the potential therapeutic implications of NNCS function, emphasizing hypertension. One of the main objectives of the current review is to provide additional perspectives on strategies focusing on endothelium-derived ACh to promote vascular health and reduce the risk of developing cardiovascular diseases.
2 Effects of ACh on vascular function
The effect of ACh on intact endothelial blood vessels has been well-established since Furchgott’s seminal experiments indicated endothelium-dependent vasodilation (7). Exogenously applied ACh is frequently used to explore endothelial function. ACh administration triggers intracellular Ca2+-dependent signaling pathways in vascular endothelial cells (17). This process involves activating the M3 muscarinic ACh receptor on endothelial cell membranes (18). Activation of the M3 receptor triggers the inositol triphosphate (IP3) signaling pathway, resulting in intracellular Ca2+ transients from intracellular Ca2+ stores and the subsequent production of nitric oxide (NO) (17, 19). ACh-induced endothelium-dependent Ca2+ signaling not only activates NO production via endothelial NO synthase (eNOS) but also activates cyclooxygenase, which mediates prostacyclin (PGI2) (20) and opens Ca2+-activated K+ channels (in particular small-conductance, SK3, and intermediate-conductance, IK1), which induces endothelial-derived hyperpolarization (EDH) (21, 22). These three components (i.e., NO, PGI2, and EDH) involved in ACh-dependent vasodilation are used to estimate how the endothelial cell capacity is balanced between healthy and impaired modifications of peripheral artery beds (23, 24) given that the magnitude of the contribution of NO, PGI2, and EDH differs depending on the artery size (25) as well as the condition of health or disease. For example, exercise training enhances ACh-dependent endothelium-dependent vasodilation under healthy conditions in rats (26, 27). A NO-dependent mechanism mediates training-induced endothelial cell adaptation (26, 28). Furthermore, exercise training has been shown to upregulate NO-dependent and prostacyclin (29)- and EDH (30, 31)-dependent mechanisms. Meanwhile, it is well-known that ACh-dependent vasodilation is substantially altered following endothelial dysfunction induced by hypertension (32), obesity (33), and diabetes (9, 10, 31). Altered ACh-dependent vasodilation may be improved by exercise training, potentially maintaining endothelial cell homeostasis (28). We recently demonstrated that low-intensity exercise training could improve endothelial function in rats with obesity and type 2 diabetes by increasing the contribution of EDH to hind limb arterioles (10). These findings indicate an association between homeostasis of endothelial function and the ACh-dependent signaling pathway in endothelial cells.
3 Endogenous source of non-neuronal ACh
Neurons, particularly cholinergic neurons, are the primary source of ACh in the central and peripheral nervous systems. ACh is synthesized, stored in vesicles, and released into the synapses upon neuronal stimulation. As a substrate for ACh synthesis, choline is taken up from the extracellular environment by the high-affinity choline transporter (CHT) (34). Choline acetyltransferase (ChAT), a key enzyme in ACh synthesis, catalyzes the transfer of an acetyl group from coenzyme acetyl-CoA to choline. Vesicular ACh transporter (VAChT) is a member of the proteins constituting the cholinergic system. The VAChT is responsible for loading ACh into secretory vesicles. Acetylcholinesterase (AChE) is responsible for ACh degradation. Experimental studies conducted over the last two decades have revealed that ACh is synthesized and released by various cell types that possess the enzymes and proteins necessary for establishing the local cholinergic system (12). For instance, cardiomyocytes have been found to express these enzymes and proteins, suggesting a role for endogenous ACh in modulating cardiac function independent of neuronal input (14, 15). Notably, ChAT overexpression in murine cardiomyocytes enhances the integrity of brain endothelial function (35). This upregulation of the cardiac cholinergic system, that is, increased ACh synthesis in the heart, does not directly interact with endothelial cells via M3 receptors but may activate ascending vagus nerve signaling (35, 36).
Two major sources of non-neuronal ACh directly affect endothelial function: lymphocytes and endothelial cells. Endothelial cells are discussed in a separate section.
3.1 Lymphocytes
Given that ACh can be measured in the human blood (13), Kawashima et al. explored the origin of ACh and identified T lymphocytes as the source of circulating ACh (13, 37). Lymphocytes that express ChAT have been characterized as a subset of T-helper cells, such as CD4+, CD44hi, and CD62 Llo (38). Lymphocytes regulate blood pressure via endothelium-dependent mechanisms by increasing the bioavailability of ACh in the blood. In a small animal experiment, infusion of ChAT-overexpressing Jurkat T cells reduced the mean arterial blood pressure in mice (38). Moreover, human ChAT-positive T cells have been shown to release ACh and promote arterial relaxation through cholinergic mechanisms (39, 40). Although T-cell-derived ACh can induce endothelium- and NO-dependent vasodilation, it is unlikely that T-cells are constantly activated to release ACh to modulate endothelial function and blood pressure under normal physiological conditions. Additional investigations are needed to establish the effects of T cell-derived ACh on cardiovascular diseases, particularly inflammation-related vascular dysfunction (40, 41).
4 ACh synthesized in endothelial cells
The ability of ACh to stimulate endothelium-dependent vasodilation has been demonstrated in several vascular beds (9, 10, 42). The origin of ACh present in vivo, particularly in the bloodstream, acting on vascular endothelial cells, has long been debated (43–45). First, the parasympathetic nerve ending is a typical source of ACh; however, few blood vessels are innervated by parasympathetic nerves (46). Second, autonomic nerves are usually located on the adventitial side of the blood vessel wall, and any ACh released from parasympathetic nerve endings faces the basal lamina barrier before reaching the endothelial cell membrane, where the M3 ACh receptors are localized. Third, in the 1930s, researchers identified the presence of ACh in ox blood (47, 48), and subsequent studies also reported the presence of ACh in the blood, as discussed in the previous section (13). Even if physiologically sufficient concentrations of ACh are present in the blood, it is unlikely that endothelial cells will be persistently activated by circulating ACh because of its inactivation by cholinesterase, which rapidly deactivates ACh into acetate and choline (49). ACh spillover is an intriguing idea in which ACh is released via activation of the motor nerve and spills over onto muscarinic receptors on the arteries of skeletal muscles, resulting in vasodilatation (50). This is reasonable because active skeletal muscles require adequate blood supply, which the peripheral vascular tone regulates.
Milner et al. reported that endothelial cells can produce and release ACh (51, 52). Using a modified chemiluminescence assay, the authors detected the ACh content in the perfusate of rat Langendorff heart preparations during hypoxia (51). In a subsequent study, they also demonstrated a substantial increase in ACh content in the perfusate from cultured umbilical vein endothelial cells during a high-flow perfusion period under shear stress conditions (52). The presence of ACh derived from endothelial cells has also been documented by Kawashima et al., who demonstrated its presence in the culture supernatant of bovine arterial endothelial cells using a radioimmunoassay (53). The presence of endothelial ACh was confirmed by electrochemical detection combined with high-performance liquid chromatography, which identified an ACh-derived peak in human umbilical vein endothelial cells (54).
Endothelial cells express ChAT, similar to other tissues, such as immune cells, in which NNCS exists. In the 1980s, Parnavelas et al. demonstrated that ChAT immunoreactivity is localized in the endothelial cells of capillaries and small vessels in the rat brain (43). Notably, the authors also suggested the synthesis and release of ACh from endothelial cells. Endothelial cells also express other essential components of the cholinergic system, including VAChT (55), CHT (56), and AChE (57, 58), which can develop self-containing NNCS in endothelial cells. Increasing evidence has indicated that vascular endothelial cells are an important source of ACh in the NNCS, thereby establishing an autocrine regulatory loop for endothelial function via the M3 receptor in the vascular microenvironment (16). The local production of ACh by endothelial cells contributes to fine-tuning vascular tone under physiological conditions. Wilson et al. suggested that autocrine-like regulation of endothelial function mediated by ACh is required to facilitate flow-mediated vasodilation via shear stress-dependent activation of the intracellular Ca2+ signaling pathway in endothelial cells (16). The autocrine-like modulation of endothelial function via local endothelial ACh may support the role of small peripheral arteries, which necessitate local EDH to induce ascending vasodilation (59). This mechanism may share the signaling pathways of mechanosensitive modulation of endothelial function, which also activates intracellular Ca2+ signaling pathways in response to shear stress (60–62). Although the mechanisms by which ACh release is activated by changes in flow (shear stress) remain unclear, local ACh signaling potentially influences exercise-induced hyperemia triggered by local vasodilation and vasodilation during skeletal muscle contraction (59).
5 Endothelial ACh in cardiovascular disease
Arterial vasodilation caused by exogenous ACh administration is impaired in several cardiovascular diseases (63). An impaired vasodilator response is often defined as endothelial dysfunction because an endothelium-dependent mechanism mediates the vasodilator effect of ACh (7). ACh is a pharmacological tool for examining whether endothelial cells respond to external stimuli mediated by ACh receptors. However, recent evidence suggests impaired endothelial function is associated with NNCS dysfunction.
Considering lymphocytes, in which the NNCS is known to exist, levels of ChAT expression and ACh production decreased in spontaneously hypertensive rats (SHR) (64). Similarly, the genetic ablation of ChAT in ChAT-positive T cells results in hypertension in mice, which is attributed to an endothelium-eNOS-dependent mechanism (38). These findings suggest that NNCS lymphocyte dysfunction triggers the development of hypertension in an ACh endothelial axis-dependent manner. Interesting experimental approaches have been employed to overcome hypertension in rodents (65). In mice with angiotensin II-induced hypertension, systemic administration of recombinant ChAT substantially reduced the mean arterial blood pressure. Notably, this ChAT-induced decrease in blood pressure in hypertensive mouse models was reversed by inhibiting NO production, indicating that the effect of exogenous ChAT administration on systemic blood pressure is NO-dependent (65). Different experimental studies have demonstrated that exogenous choline administration delays the progression of hypertension in SHR, presumably by enhancing vagus nerve activity (66). Moreover, serum ACh levels were substantially lower in SHR than in control Wistar Kyoto rats (WKY); choline administration restored these levels. This concept of increasing the bioavailability of circulating ACh, which directly acts on ACh receptors in endothelial cells, may offer a potential strategy to prevent the development of hypertension caused by depletion or dysfunction of the circulating ACh source. The bioavailability of circulating ACh may be clinically relevant because low plasma ACh levels and endothelial dysfunction are associated with increased mortality in critically ill patients with poor cardiovascular outcomes (67–69).
As discussed in the previous section, vascular endothelial cells are an important source of ACh. Therefore, a new hypothesis was proposed: endothelial NNCS dysfunction is responsible for endothelial dysfunction. Although there is limited experimental evidence supporting this hypothesis, a study by Zou et al. revealed that the expression of VAChT protein, an NNCS component, was markedly lower in the aortic ring of SHR than in the aortic ring of control WKY (70). Additionally, the concentration of ACh in the aortic ring was lower in SHR than in WKY after choline incubation. These findings suggest that the decreased synthesis and release of ACh from endothelial cells may impair endothelial function, impairing arterial tone regulation, thereby contributing to hypertension (Figure 1). Additionally, evidence not directly related to the endothelium revealed that NNCS in the heart is downregulated in mice and humans with obese type 2 diabetes (71). Considering these findings, NNCS may be down-regulated in the endothelial cells under diabetic conditions. Further studies are required to clarify the relationship between endothelial NNCS and diabetes-induced microvascular dysfunction.
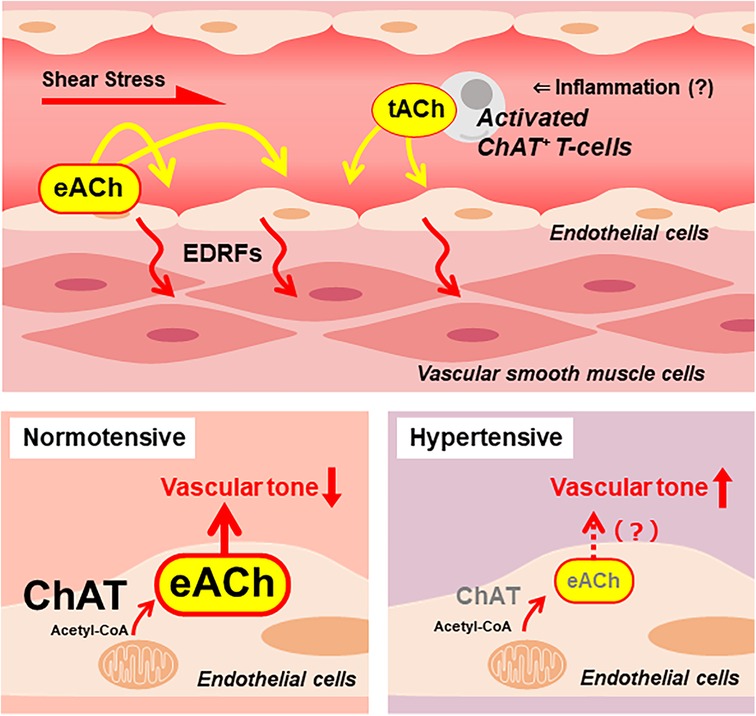
Figure 1. A putative endothelial non-neuronal cholinergic system that regulates endothelium-derived relaxing factors (EDRFs) modulating vascular tone. Increased shear stress stimulates vascular endothelial cells to release ACh. The endothelial cell-derived ACh (eACh) affects the endothelial function in an autocrine manner. Local inflammation recruits activated ChAT-positive T cells, which release ACh. The T cell-derived ACh (tACh) is also suggested to affect endothelial function. In a healthy, normotensive condition, ChAT in endothelial cells contributes to the synthesis of eACh. It is hypothesized that adequate storage and release of eACh maintains basal vascular tone. Meanwhile, endothelial ChAT decreases in the condition of hypertension. Thus, it causes decreased eACh synthesis that results in increased vascular tone.
The clearly defined clinical manifestations of endothelial NNCS dysfunction in humans remain poorly understood. Instead, the implications of endothelial dysfunction have been frequently discussed in the context of its role in various clinically relevant cardiovascular diseases, including hypertension and diabetic microangiopathy. Future studies are needed to shed light on endothelial NNCS as potential therapeutic targets for these diseases.
6 Conclusion
In conclusion, understanding NNCS in the vascular system is increasingly important as it may contribute to the development of cardiovascular disease by modulating arterial tone. Recent findings indicate that endogenous ACh is pivotal in regulating endothelial signaling mechanisms. Investigating the pathophysiological significance of endothelial NNCS dysfunction may improve our understanding of the early stages of vascular diseases caused by endothelial dysfunction. These findings suggest that endothelial NNCS may be a potential therapeutic target for conditions such as metabolic syndrome, diabetes, and hypertension and may lead to the development of novel therapeutic strategies.
Author contributions
TS: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. YK: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (grant numbers: 21K11358 and 24K14410 for TS).
Acknowledgments
This manuscript has been checked and edited for language by Editage, Cactus Communications Co. Ltd, Tokyo, Japan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. La Favor JD, Dubis GS, Yan H, White JD, Nelson MA, Anderson EJ, et al. Microvascular endothelial dysfunction in sedentary, obese humans is mediated by NADPH oxidase: influence of exercise training. Arterioscler Thromb Vasc Biol. (2016) 36(12):2412–20. doi: 10.1161/ATVBAHA.116.308339
2. Boyle LJ, Credeur DP, Jenkins NT, Padilla J, Leidy HJ, Thyfault JP, et al. Impact of reduced daily physical activity on conduit artery flow-mediated dilation and circulating endothelial microparticles. J Appl Physiol (1985). (2013) 115(10):1519–25. doi: 10.1152/japplphysiol.00837.2013
3. Restaino RM, Holwerda SW, Credeur DP, Fadel PJ, Padilla J. Impact of prolonged sitting on lower and upper limb micro- and macrovascular dilator function. Exp Physiol. (2015) 100(7):829–38. doi: 10.1113/EP085238
4. Panza JA, Quyyumi AA, Brush JE, Epstein SE. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med. (1990) 323(1):22–7. doi: 10.1056/NEJM199007053230105
5. McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, et al. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. (1992) 35(8):771–6. doi: 10.1007/BF00429099
6. Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. (1996) 97(11):2601–10. doi: 10.1172/JCI118709
7. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. (1980) 288(5789):373–6. doi: 10.1038/288373a0
8. Kingwell BA, Formosa M, Muhlmann M, Bradley SJ, McConell GK. Type 2 diabetic individuals have impaired leg blood flow responses to exercise: role of endothelium-dependent vasodilation. Diabetes Care. (2003) 26(3):899–904. doi: 10.2337/diacare.26.3.899
9. Sonobe T, Tsuchimochi H, Schwenke DO, Pearson JT, Shirai M. Treadmill running improves hindlimb arteriolar endothelial function in type 1 diabetic mice as visualized by x-ray microangiography. Cardiovasc Diabetol. (2015) 14:51. doi: 10.1186/s12933-015-0217-0
10. Sonobe T, Tsuchimochi H, Maeda H, Pearson JT. Increased contribution of KCa channels to muscle contraction induced vascular and blood flow responses in sedentary and exercise trained ZFDM rats. J Physiol. (2022) 600(12):2919–38. doi: 10.1113/JP282981
11. Grando SA, Kawashima K, Wessler I. Introduction: the non-neuronal cholinergic system in humans. Life Sci. (2003) 72(18–19):2009–12. doi: 10.1016/S0024-3205(03)00063-8
12. Grando SA, Kawashima K, Wessler I. A historic perspective on the current progress in elucidation of the biologic significance of non-neuronal acetylcholine. Int Immunopharmacol. (2020) 81:106289. doi: 10.1016/j.intimp.2020.106289
13. Fujii T, Yamada S, Misawa H, Tajima S, Fujimoto K, Suzuki T, et al. Expression of choline acetyltransferase mRNA and protein in T-lymphocytes. Proc Jpn Acad Series B. (1995) 71(7):231–5. doi: 10.2183/pjab.71.231
14. Kakinuma Y, Akiyama T, Sato T. Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. (2009) 276(18):5111–25. doi: 10.1111/j.1742-4658.2009.07208.x
15. Kakinuma Y, Tsuda M, Okazaki K, Akiyama T, Arikawa M, Noguchi T, et al. Heart-specific overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia-inducible factor-1α-related defense mechanisms. J Am Heart Assoc. (2013) 2(1):e004887. doi: 10.1161/JAHA.112.004887
16. Wilson C, Lee MD, McCarron JG. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J Physiol. (2016) 594(24):7267–307. doi: 10.1113/JP272927
17. Wang X, Lau F, Li L, Yoshikawa A, van Breemen C. Acetylcholine-sensitive intracellular Ca2+ store in fresh endothelial cells and evidence for ryanodine receptors. Circ Res. (1995) 77(1):37–42. doi: 10.1161/01.RES.77.1.37
18. Lamping KG, Wess J, Cui Y, Nuno DW, Faraci FM. Muscarinic (M) receptors in coronary circulation: gene-targeted mice define the role of M2 and M3 receptors in response to acetylcholine. Arterioscler Thromb Vasc Biol. (2004) 24(7):1253–8. doi: 10.1161/01.ATV.0000130661.82773.ca
19. Doyle MP, Duling BR. Acetylcholine induces conducted vasodilation by nitric oxide-dependent and -independent mechanisms. Am J Physiol. (1997) 272(3 Pt 2):H1364–71. doi: 10.1152/ajpheart.1997.272.3.H1364
20. Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol. (2005) 146(6):834–45. doi: 10.1038/sj.bjp.0706390
21. Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. (2012) 336(6081):597–601. doi: 10.1126/science.1216283
22. Peixoto-Neves D, Yadav S, MacKay CE, Mbiakop UC, Mata-Daboin A, Leo MD, et al. Vasodilators mobilize SK3 channels in endothelial cells to produce arterial relaxation. Proc Natl Acad Sci U S A. (2023) 120(31):e2303238120. doi: 10.1073/pnas.2303238120
23. Kamata K, Sugiura M, Kasuya Y. Decreased Ca2+ influx into the endothelium contributes to the decrease in endothelium-dependent relaxation in the aorta of streptozotocin-induced diabetic mice. Res Commun Mol Pathol Pharmacol. (1995) 90(1):69–74.8581350
24. Matsumoto T, Kobayashi T, Kamata K. Alterations in EDHF-type relaxation and phosphodiesterase activity in mesenteric arteries from diabetic rats. Am J Physiol Heart Circ Physiol. (2003) 285(1):H283–91. doi: 10.1152/ajpheart.00954.2002
25. Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. (1996) 28(5):703–11. doi: 10.1097/00005344-199611000-00014
26. Delp MD, McAllister RM, Laughlin MH. Exercise training alters endothelium-dependent vasoreactivity of rat abdominal aorta. J Appl Physiol (1985). (1993) 75(3):1354–63. doi: 10.1152/jappl.1993.75.3.1354
27. Delp MD. Effects of exercise training on endothelium-dependent peripheral vascular responsiveness. Med Sci Sports Exerc. (1995) 27(8):1152–7.7476059
28. Green DJ, Maiorana A, O'Driscoll G, Taylor R. Effect of exercise training on endothelium-derived nitric oxide function in humans. J Physiol. (2004) 561(Pt 1):1–25. doi: 10.1113/jphysiol.2004.068197
29. Zoladz JA, Majerczak J, Duda K, Chłopicki S. Endurance training increases exercise-induced prostacyclin release in young, healthy men–relationship with VO2max. Pharmacol Rep. (2010) 62(3):494–502. doi: 10.1016/S1734-1140(10)70305-4
30. Woodman CR, Thompson MA, Turk JR, Laughlin MH. Endurance exercise training improves endothelium-dependent relaxation in brachial arteries from hypercholesterolemic male pigs. J Appl Physiol (1985). (2005) 99(4):1412–21. doi: 10.1152/japplphysiol.00293.2005
31. Minami A, Ishimura N, Harada N, Sakamoto S, Niwa Y, Nakaya Y. Exercise training improves acetylcholine-induced endothelium-dependent hyperpolarization in type 2 diabetic rats, otsuka long-evans tokushima fatty rats. Atherosclerosis. (2002) 162(1):85–92. doi: 10.1016/S0021-9150(01)00685-2
32. Lüscher TF, Raij L, Vanhoutte PM. Endothelium-dependent vascular responses in normotensive and hypertensive dahl rats. Hypertension. (1987) 9(2):157–63. doi: 10.1161/01.HYP.9.2.157
33. Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in zucker obese and zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. (2006) 291(4):H1780–7. doi: 10.1152/ajpheart.01297.2005
34. Parikh V, Sarter M. Cortical choline transporter function measured in vivo using choline-sensitive microelectrodes: clearance of endogenous and exogenous choline and effects of removal of cholinergic terminals. J Neurochem. (2006) 97(2):488–503. doi: 10.1111/j.1471-4159.2006.03766.x
35. Oikawa S, Kai Y, Mano A, Sugama S, Mizoguchi N, Tsuda M, et al. Potentiating a non-neuronal cardiac cholinergic system reinforces the functional integrity of the blood brain barrier associated with systemic anti-inflammatory responses. Brain Behav Immun. (2019) 81:122–37. doi: 10.1016/j.bbi.2019.06.005
36. Oikawa S, Kai Y, Tsuda M, Ohata H, Mano A, Mizoguchi N, et al. Non-neuronal cardiac cholinergic system influences CNS via the vagus nerve to acquire a stress-refractory propensity. Clin Sci (Lond). (2016) 130(21):1913–28. doi: 10.1042/CS20160277
37. Fujii T, Tsuchiya T, Yamada S, Fujimoto K, Suzuki T, Kasahara T, et al. Localization and synthesis of acetylcholine in human leukemic T cell lines. J Neurosci Res. (1996) 44(1):66–72. doi: 10.1002/(SICI)1097-4547(19960401)44:1%3C66::AID-JNR9%3E3.0.CO;2-G
38. Olofsson PS, Steinberg BE, Sobbi R, Cox MA, Ahmed MN, Oswald M, et al. Blood pressure regulation by CD4+ lymphocytes expressing choline acetyltransferase. Nat Biotechnol. (2016) 34(10):1066–71. doi: 10.1038/nbt.3663
39. Chester AH, McCormack A, Miller EJ, Ahmed MN, Yacoub MH. Coronary vasodilation mediated by T cells expressing choline acetyltransferase. Am J Physiol Heart Circ Physiol. (2021) 321(5):H933–H9. doi: 10.1152/ajpheart.00694.2020
40. Tarnawski L, Shavva VS, Kort EJ, Zhuge Z, Nilsson I, Gallina AL, et al. Cholinergic regulation of vascular endothelial function by human ChAT+ T cells. Proc Natl Acad Sci U S A. (2023) 120(14):e2212476120. doi: 10.1073/pnas.2212476120
41. Vijayaraghavan S, Karami A, Aeinehband S, Behbahani H, Grandien A, Nilsson B, et al. Regulated extracellular choline acetyltransferase activity- the plausible missing link of the distant action of acetylcholine in the cholinergic anti-inflammatory pathway. PLoS One. (2013) 8(6):e65936. doi: 10.1371/journal.pone.0065936
42. Sonobe T, Schwenke DO, Pearson JT, Yoshimoto M, Fujii Y, Umetani K, et al. Imaging of the closed-chest mouse pulmonary circulation using synchrotron radiation microangiography. J Appl Physiol (1985). (2011) 111(1):75–80. doi: 10.1152/japplphysiol.00205.2011
43. Parnavelas JG, Kelly W, Burnstock G. Ultrastructural localization of choline acetyltransferase in vascular endothelial cells in rat brain. Nature. (1985) 316(6030):724–5. doi: 10.1038/316724a0
44. Dora KA. Conducted dilatation to ATP and K+ in rat skeletal muscle arterioles. Acta Physiol (Oxf). (2017) 219(1):202–18. doi: 10.1111/apha.12656
45. Vanhoutte PM. Endothelial muscarinic M3 -receptors: a Σ-target? Acta Physiol (Oxf). (2019) 226(1):e13273. doi: 10.1111/apha.13273
46. Rhoden A, Speiser J, Geertz B, Uebeler J, Schmidt K, de Wit C, et al. Preserved cardiovascular homeostasis despite blunted acetylcholine-induced dilation in mice with endothelial muscarinic M3 receptor deletion. Acta Physiol (Oxf). (2019) 226(1):e13262. doi: 10.1111/apha.13262
47. Dudley HW. The alleged occurrence of acetylcholine in ox blood. J Physiol. (1933) 79(3):249–54. doi: 10.1113/jphysiol.1933.sp003048
48. Ettinger GH, Hall GE. Acetylcholine in ox and dog blood. J Physiol. (1934) 82(1):38–40. doi: 10.1113/jphysiol.1934.sp003157
49. Altiere RJ, Travis DC, Thompson DC. Characterization of acetylcholinesterase in rabbit intrapulmonary arteries. Am J Physiol. (1994) 267(6 Pt 1):L745–52. doi: 10.1152/ajplung.1994.267.6.L745
50. Welsh DG, Segal SS. Coactivation of resistance vessels and muscle fibers with acetylcholine release from motor nerves. Am J Physiol. (1997) 273(1 Pt 2):H156–63. doi: 10.1152/ajpheart.1997.273.1.H156
51. Milner P, Ralevic V, Hopwood AM, Fehér E, Lincoln J, Kirkpatrick KA, et al. Ultrastructural localisation of substance P and choline acetyltransferase in endothelial cells of rat coronary artery and release of substance P and acetylcholine during hypoxia. Experientia. (1989) 45(2):121–5. doi: 10.1007/BF01954843
52. Milner P, Kirkpatrick KA, Ralevic V, Toothill V, Pearson J, Burnstock G. Endothelial cells cultured from human umbilical vein release ATP, substance P and acetylcholine in response to increased flow. Proc Biol Sci. (1990) 241(1302):245–8. doi: 10.1098/rspb.1990.0092
53. Kawashima K, Watanabe N, Oohata H, Fujimoto K, Suzuki T, Ishizaki Y, et al. Synthesis and release of acetylcholine by cultured bovine arterial endothelial cells. Neurosci Lett. (1990) 119(2):156–8. doi: 10.1016/0304-3940(90)90822-Q
54. Kirkpatrick CJ, Bittinger F, Unger RE, Kriegsmann J, Kilbinger H, Wessler I. The non-neuronal cholinergic system in the endothelium: evidence and possible pathobiological significance. Jpn J Pharmacol. (2001) 85(1):24–8. doi: 10.1254/jjp.85.24
55. Haberberger RV, Bodenbenner M, Kummer W. Expression of the cholinergic gene locus in pulmonary arterial endothelial cells. Histochem Cell Biol. (2000) 113(5):379–87. doi: 10.1007/s004180000153
56. Lips KS, Pfeil U, Reiners K, Rimasch C, Kuchelmeister K, Braun-Dullaeus RC, et al. Expression of the high-affinity choline transporter CHT1 in rat and human arteries. J Histochem Cytochem. (2003) 51(12):1645–54. doi: 10.1177/002215540305101208
57. Carvalho FA, Graça LM, Martins-Silva J, Saldanha C. Biochemical characterization of human umbilical vein endothelial cell membrane bound acetylcholinesterase. FEBS J. (2005) 272(21):5584–94. doi: 10.1111/j.1742-4658.2005.04953.x
58. Santos SC, Vala I, Miguel C, Barata JT, Garção P, Agostinho P, et al. Expression and subcellular localization of a novel nuclear acetylcholinesterase protein. J Biol Chem. (2007) 282(35):25597–603. doi: 10.1074/jbc.M700569200
59. Sinkler SY, Segal SS. Rapid versus slow ascending vasodilatation: intercellular conduction versus flow-mediated signalling with tetanic versus rhythmic muscle contractions. J Physiol. (2017) 595(23):7149–65. doi: 10.1113/JP275186
60. Köhler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. (2006) 26(7):1495–502. doi: 10.1161/01.ATV.0000225698.36212.6a
61. Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, et al. Piezo1 integration of vascular architecture with physiological force. Nature. (2014) 515(7526):279–82. doi: 10.1038/nature13701
62. Yamamoto K, Korenaga R, Kamiya A, Ando J. Fluid shear stress activates Ca(2+) influx into human endothelial cells via P2X4 purinoceptors. Circ Res. (2000) 87(5):385–91. doi: 10.1161/01.RES.87.5.385
63. Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf). (2009) 196(2):193–222. doi: 10.1111/j.1748-1716.2009.01964.x
64. Fujimoto K, Matsui M, Fujii T, Kawashima K. Decreased acetylcholine content and choline acetyltransferase mRNA expression in circulating mononuclear leukocytes and lymphoid organs of the spontaneously hypertensive rat. Life Sci. (2001) 69(14):1629–38. doi: 10.1016/S0024-3205(01)01237-1
65. Stiegler A, Li JH, Shah V, Tsaava T, Tynan A, Yang H, et al. Systemic administration of choline acetyltransferase decreases blood pressure in murine hypertension. Mol Med. (2021) 27(1):133. doi: 10.1186/s10020-021-00380-6
66. Liu L, Lu Y, Bi X, Xu M, Yu X, Xue R, et al. Choline ameliorates cardiovascular damage by improving vagal activity and inhibiting the inflammatory response in spontaneously hypertensive rats. Sci Rep. (2017) 7:42553. doi: 10.1038/srep42553
67. Tao G, Min-Hua C, Feng-Chan X, Yan C, Ting S, Wei-Qin L, et al. Changes of plasma acetylcholine and inflammatory markers in critically ill patients during early enteral nutrition: a prospective observational study. J Crit Care. (2019) 52:219–26. doi: 10.1016/j.jcrc.2019.05.008
68. Duffy MJ, Mullan BA, Craig TR, Shyamsundar M, MacSweeney RE, Thompson G, et al. Impaired endothelium-dependent vasodilatation is a novel predictor of mortality in intensive care. Crit Care Med. (2011) 39(4):629–35. doi: 10.1097/CCM.0b013e318206bc4a
69. Fischer D, Rossa S, Landmesser U, Spiekermann S, Engberding N, Hornig B, et al. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J. (2005) 26(1):65–9. doi: 10.1093/eurheartj/ehi001
70. Zou Q, Leung SW, Vanhoutte PM. Transient receptor potential channel opening releases endogenous acetylcholine, which contributes to endothelium-dependent relaxation induced by mild hypothermia in spontaneously hypertensive rat but not Wistar-Kyoto rat arteries. J Pharmacol Exp Ther. (2015) 354(2):121–30. doi: 10.1124/jpet.115.223693
Keywords: ACh, endothelium-dependent vasodilation, the non-neuronal cholinergic system, cardiovascular disease, exercise training
Citation: Sonobe T and Kakinuma Y (2024) Non-neuronal cell-derived acetylcholine, a key modulator of the vascular endothelial function in health and disease. Front. Cardiovasc. Med. 11:1388528. doi: 10.3389/fcvm.2024.1388528
Received: 20 February 2024; Accepted: 6 May 2024;
Published: 15 May 2024.
Edited by:
Luciana Venturini Rossoni, University of São Paulo, BrazilReviewed by:
Maria Sancho, University of Vermont, United StatesSteyner F. Cortes, Federal University of Minas Gerais, Brazil
© 2024 Sonobe and Kakinuma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Sonobe, t-sonobe@nms.ac.jp