Ligand-based discovery of a novel GATA2 inhibitor targeting acute myeloid leukemia cells
 Juan Bautista Menendez-Gonzalez1,2,3
Juan Bautista Menendez-Gonzalez1,2,3  Kathryn E. Strange1,4,5 Marcella Bassetto6
Kathryn E. Strange1,4,5 Marcella Bassetto6  Andrea Brancale7,8†
Andrea Brancale7,8†  Neil P. Rodrigues1*†
Neil P. Rodrigues1*†  Salvatore Ferla9*†
Salvatore Ferla9*†- 1European Cancer Stem Cell Research Institute, School of Biosciences, Cardiff University, Cardiff, United Kingdom
- 2Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, Cambridge, MA, United States
- 3Center for Regenerative Medicine, Massachusetts General Hospital, Boston, MA, United States
- 4Anthony Nolan Research Institute, Royal Free Hospital, London, United Kingdom
- 5UCL Cancer Institute, University College London, London, United Kingdom
- 6Department of Chemistry, Faculty of Science and Engineering, Swansea University, Swansea, United Kingdom
- 7School of Pharmacy and Pharmaceutical Sciences, Cardiff University, Cardiff, United Kingdom
- 8Vysoká Skola Chemicko-Technologická v Praze, Prague, Czechia
- 9Medical School, Faculty of Medicine, Health and Life Science, Swansea University, Swansea, United Kingdom
Despite major therapeutic advances leading to improved patient outcomes for other haematological malignancies, development of new therapeutics to improve prognosis for acute myeloid leukemia (AML) patients remains an area of unmet clinical need. Overexpression of GATA2, a member of the GATA family of zinc finger transcription factors, has been implicated in AML. In settings where GATA2 is overexpressed in human AML cells, K7174, a proteasome inhibitor that inhibits GATA2, induces apoptosis and enhances the killing activity of AML chemotherapeutics in vitro yet targeting the proteasome has been associated with high toxicity in the clinic. Using an in silico approach, we embarked on a screen to identify specific GATA2 inhibitors that will target AML cells independently of the proteasome. A shape-based virtual screening of an in-house library of small molecules was performed using a low-energy conformation of K7174. The virtual hit compounds were subsequently filtered according to their potential selectivity for GATA2 over the proteasome. From 15 selected compounds evaluated for their ability to kill AML cells in vitro, one compound, an asymmetrical substituted piperazine with Hepatitis C antiviral activity, exhibited superior ability to induce apoptosis and reduce cell cycling in AML cells without proteasome inhibition. This compound was also able to promote cell death of the relapse propagating leukemic stem cell (LSC) compartment while sparing Gata2 knockout LSCs, crucially demonstrating specificity to inhibit GATA2. We have identified a GATA2 specific inhibitor with promising capability to target AML cells in vitro, including LSCs that underpin poor prognosis in AML.
Introduction
Acute myeloid leukemia (AML) is an aggressive blood cancer where myeloid progenitors are blocked in their ability to differentiate into specialized myeloid blood cells, instead generating immature abnormal blast cells that competitively suppress the ability of the bone marrow to make these and other blood cell types (Arber et al., 2016).
AML is treated with a combination of high-dose induction chemotherapy to kill proliferating leukemia blasts and consolidation chemotherapy to keep the leukemia at bay for an extended period, with the further option of maintenance therapy (Döhner et al., 2017). For younger or fitter AML patients (generally <65 years old), high-dose chemotherapy followed by rescue of the hematopoietic system with allogeneic bone marrow transplantation (BMT) is the only curative option available (Döhner et al., 2017). Yet, most patients diagnosed with AML are older (>65 years) or less healthy and ineligible for BMT due to the high toxicity and long-term side-effects associated with treatment, which includes Graft versus Host Disease (GvHD) and the emergence of secondary malignancies caused by the impact of immunosuppression on physiologic immunosurveillance mechanisms (Horowitz et al., 1990). Thus, for most patients afflicted with AML, treatments have remained largely non-specific and toxic, targeting leukemia and normal cells alike, and ineffective at eliciting cure.
The quest to develop more specific, targeted therapies for AML has been encumbered by 1) the genetic and epigenetic mutational complexity and heterogeneity observed in AML patients (Papaemmanuil et al., 2016), and 2) a subset of leukemia cells termed leukemia stem cells/initiating cells (LSCs), which are the root cause of AML disease, enabling AML cells to prosper, and which notably resist treatment by standard chemotherapy, ultimately causing relapse in AML (Bonnet and Dick 1997). We have addressed these issues by focusing on understanding the oncogenic function of haematopoietic specific transcription factor GATA2, which is overexpressed across several subtypes of AML and approximately 25% of all AML cases (Lahortiga et al., 2005; Menendez-Gonzalez et al., 2019b). In mouse models of AML driven by the co-expression of Meis1a and Hoxa9, we found that genetic deletion of Gata2 causes apoptosis of LSCs via reduction in the expression of anti-apoptotic factor BCL2 while also facilitating myeloid differentiation, overcoming the myeloid differentiation block normally observed in AML (Menendez-Gonzalez et al., 2019a). To further explore the potential for clinical targeting of GATA2 in AML, we reduced GATA2 expression using shRNAi in human AML cell lines overexpressing GATA2, which enhanced apoptosis and forestalled leukemic cell proliferation (Menendez-Gonzalez et al., 2019b). Of therapeutic relevance, a proteasome inhibitor (K7174) that inhibits GATA2 was able to successfully induce apoptosis in AML cell lines overexpressing GATA2 when combined with standard of care AML chemotherapy (Majik et al., 2012; Menendez-Gonzalez et al., 2019b). These proof of principle data collectively suggest the potential of targeting GATA2 in AML and, more specifically, in AML LSCs. While proteasome targeting approaches are clearly effective for treating other blood cancers, as evidenced by the clinical use of bortezomib for treating multiple myeloma and mantle cell lymphoma (Richardson et al., 2003), they also lead to substantial side-effects (Richardson et al., 2003; Csizmar et al., 2016; Aplenc et al., 2020) and we showed K7174 lacks direct specificity to inhibit GATA2 (Menendez-Gonzalez et al., 2019b). Together, this highlights the necessity to identify specific inhibitors of GATA2 to target AML.
The crystal structure of GATA2 has been only partially solved (Nurmohamed et al., 2017), minimal structural information is currently available regarding a potential druggable site, and the GATA2 binding area of K7174 has yet to be elucidated, making the rational discovery of novel and selective small molecule inhibitors of GATA2 a challenging proposition. Ligand-based virtual screening is an in silico technique widely used as an alternative for the discovery of novel potential active molecules against a biological target when its crystal structure has not been resolved or its active site is not known (Hawkins et al., 2007; Bassetto et al., 2017a; Ferla et al., 2018). This technique compares the shape and properties of a known ligand of a biological target (e.g. natural substrates or known inhibitors) with the shape of a series of small molecules, evaluating two-dimensional (i.e. same functional groups and chemical properties) and/or three-dimensional similarities (i.e. molecule occupational volume). For a molecule to have a biological effect, it needs to have shape/properties complementarity with the target binding site; therefore, the shape-based approach relies on the premise that two molecules with similar 3D shape are highly likely to occupy the same binding site and cause a similar biological effect (Kumar and Zhang 2018). In this study, to identify novel, specific GATA2 inhibitors that will specifically target AML cells independently of the proteasome, we applied a ligand-based virtual screening using the structure of K7174. This approach unveiled a novel small molecule that specifically inhibits GATA2 with capability to target AML cells in vitro. This new compound represents a starting point for the future development of AML therapeutics directed against GATA2.
Methods
Molecular Modelling. All molecular modelling studies were performed on a Viglen Genie Intel∗i7-3770 vPro CPU@ 3.40 GHz × 8 running Ubuntu 18.04. Molecular Operating Environment (MOE) 2022.02 Molecular Operating Environment (2022), Maestro (Schrödinger Release 2022) (Schrödinger Release 2022), and OpenEye Scientific Software OpenEye Scientific Software (1997) were used as molecular modelling software. Shape-based virtual screening was performed with ROCS version 3.4.3.0 (OpenEye Scientific Software, 1997). Both shape and colour screen criteria were applied to the query. Output conformations were ranked according to the Tanimoto Combo score, which combines shape-density overlapping volumes score (ShapeTanimoto) with pharmacophore features match score (ColorTanimoto). The internal library of small molecules was built with MOE using the MOE Builder tool, saved as sdf format, and prepared using the conformations generator Omega version 4.1.2.0 (OpenEye Scientific Software, 1997). The following parameters have been used in Omega: maxconfs 600 -flipper true -strictstereo false-strictatomtyping false -strictfrags true. The conformation of K7174 has been selected among multiple low-energy three-dimensional states generated using the MOE Conformational Search tool, using the default parameters and a LowModeMD search method (forcefield: MMFF94x in vacuum).
The proteasome structure was retrieved from the PDB data bank (http://www.rcsb.org/; PDB code 4EU2) (Kikuchi et al., 2013). The protein was pre-processed using the Schrödinger Protein Preparation Wizard by assigning bond orders, adding hydrogens, and performing a restrained energy minimization of the added hydrogens using the OPLS_2005 force field. The structure of compound 11 was built with MOE and then prepared using the Maestro LigPrep tool by energy minimizing the structure (OPLS_2005 force filed), generating possible ionization states at pH 7 ± 2, generating tautomers and low-energy ring conformers. After isolating the β4 and β5 subunits, a 20 Å docking grid (inner-box 10 Å and outer-box 30 Å) was prepared using as centroid the co-crystallized K7174. Molecular docking studies were performed using Glide SP precision keeping the default parameters and setting 10 as number of output poses per input ligand to include in the solution. The output database was saved as mol2 file. The docking results were visually inspected evaluating the potential ability of compound 11 to bind the active site of the proteasome. The ligand-protein interactions were calculated using the MOE contact’s tool.
Cell lines and compounds. K562 and THP1 (Menendez-Gonzalez et al., 2019b) were cultured in RMPI 1640 supplemented with 10% FBS and 1% l-glutamine. Murine WT and Gata2 KO Meis1a/Hoxa9 acute myeloid leukemia stem cells (Menendez-Gonzalez et al., 2019a) were cultured in IMDM 10% FBS, 1% l-Glutamine, 20 ng/ml SCF, 10 ng/ml IL3, and 10 ng/ml IL6.
Compounds were reconstituted in DMSO at stock concentration of 10 mM.
Primary screening assay. K562 cells were cultured in 96-well plates and treated with compounds for 48 h. Apoptosis was measured using Annexin V and measured with a BD Fortessa.
Cell-cycle. K562 and THP1 cells were resuspended in PBS with 0.1% NP40 and 5 μg/ml DAPI, incubated for 5 min and analysed in a BD Fortessa.
Proteasome assay. K562 cells were cultured with Bortezomib, K7174 and AB-042 for 4-h. Proteasome activity was measured using the Proteasome-Glo cell-based assay from Promega. Proteasome substrate was added to the cells and incubated for 1 h before adding the luciferase substrate and analysing the Glo-luminescence 10 minutes later.
In vivo mouse model of leukemia stem cells. All mice were housed at the Heath Park Unit, Cardiff University. All animal experiments were executed under PPL 30/3380 in accordance with the 1986 Animals (Scientific Procedures) Act. Two thousand Meis1/Hoxa9 AML LSCs were transplanted to irradiated secondary recipients (Menendez-Gonzalez et al., 2019a). When peripheral blood engraftment reached 1%, mice were treated every-day for 5 days with compound 11.
Results
Shape-based virtual screening
A shape-based virtual screening of an internal library of approximately 3,000 compounds, previously prepared with the Omega conformations generator, was performed using ROCS 3D (OpenEye Scientific Software, 1997). The conformation of the query molecule plays an essential role in shape-based screening since all analysed molecules will be evaluated for their overlap with the query. Multiple low-energy conformations were generated for K7174, and, among them, the most representative and most likely to exist at physiological conditions and to interact with a biological target was selected (Figure 1 and Supplementary Figure S1).
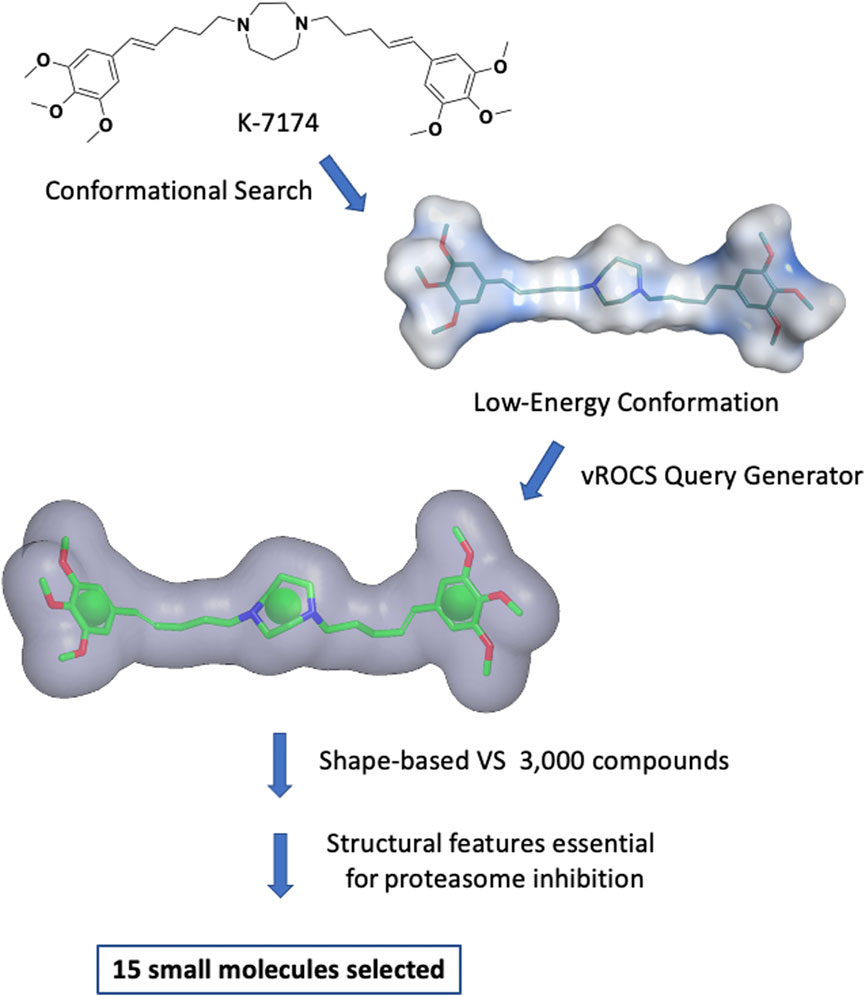
FIGURE 1. Shape-based virtual screening flow. Multiple low-energy conformations of K7174 were generated. The most representative and most likely conformation to exist at physiological conditions and to interact with a biological target was selected. A query was generated using vROCS. The overall molecular shape of K7174 is represented as a grey surface, while the chemical features considered are one ring in correspondence of the homopiperazine (green sphere), and two hydrophobic/aromatic centers representing (green spheres). After the shape-based virtual screening and further filtering to avoid any structural features likely to cause proteasome inhibition, 15 small molecules were selected for biological evaluation.
The query generated using this conformation included the occupational volume of K7174, a mono-cyclic, aliphatic pharmacophore feature corresponding to the homopiperazine, and two hydrophobic/aromatic features representing the two trimethoxy phenyl groups (Figure 1). No other chemical features were considered due to the lack of information on the binding mode of K7174 to GATA2. The query was used to filter the above-mentioned library, and the best 60 molecules matching the shape and the pharmacophore features according to Tanimoto Combo score (cut-off 0.60) were selected (OpenEye Scientific Software, 1997). These molecules were further filtered by removing any compound containing structural features previously reported to be essential for proteasome inhibition by K7174 derivatives (Kikuchi et al., 2013): the combined presence of a methoxy group in position three of a phenyl ring, which forms an important hydrogen bond with the OH group of proteasome Thr1, separated by a five-carbon atom chain from the central homopiperazine ring, which imparts to the compounds the correct size to fit the hydrophobic grooves of proteasome. Thus, any molecule presenting a hydrogen bond acceptor substituent in position three of a phenyl ring separated by five atoms from a central core was discarded from the screen (e.g. compound 16 and 17; Supplementary Table S1). This left 15 molecules (Supplementary Table S1) with hypothetical selectivity for GATA2 over the proteasome. Notably, all these molecules, apart from one, belong to a family of non-toxic piperazine-based Hepatitis C antiviral agents, reported previously by our group (Bassetto et al., 2017b).
Identification of a hit compound targeting GATA2 in AML cells in vitro by enhancing apoptosis and reducing cell cycling
To assess the ability of the selected compounds to elicit apoptosis of AML cells, varying doses (10, 15 or 20 μM) of compound or vehicle (DMSO) were added in vitro to K562, a myeloid leukemia cell line that overexpresses GATA2 (Menendez-Gonzalez et al., 2019b), and after 48 h of exposure to each compound cells were harvested, and apoptosis was evaluated by the flow cytometry based Annexin V assay. Compounds 1 and 10 displayed solubility issues and were excluded from the assay (data not shown). Among the 13 remaining compounds tested, compound 11 induced apoptosis in K562 cells to the greatest extent across all doses in comparison to DMSO and, notably, outperformed K7I74 at a concentration of 20 μM (Figures 2A, 3A). We also independently assessed the capacity of 11 to induce apoptosis in an AML line that overexpresses GATA2, THP1 (Menendez-Gonzalez et al., 2019b). Compound 11 induced apoptosis to THP1 to a similar extent as K562 (Figure 3B). In combination with apoptosis, reduced cell cycling occurs in response to chemotherapy as leukemic cells are killed. After 48 h of exposure to 11, the proportion of cycling K562 and THP1 cells was reduced at a concentration of 20 μM (Figures 3C,D). In striking contrast to K7174 which has potential off-target effects (Menendez-Gonzalez et al., 2019b), in vitro exposure to compound 11 alone induces both apoptosis and a reduction in cell cycling in AML cells consistent with potent anti-leukemia capability.
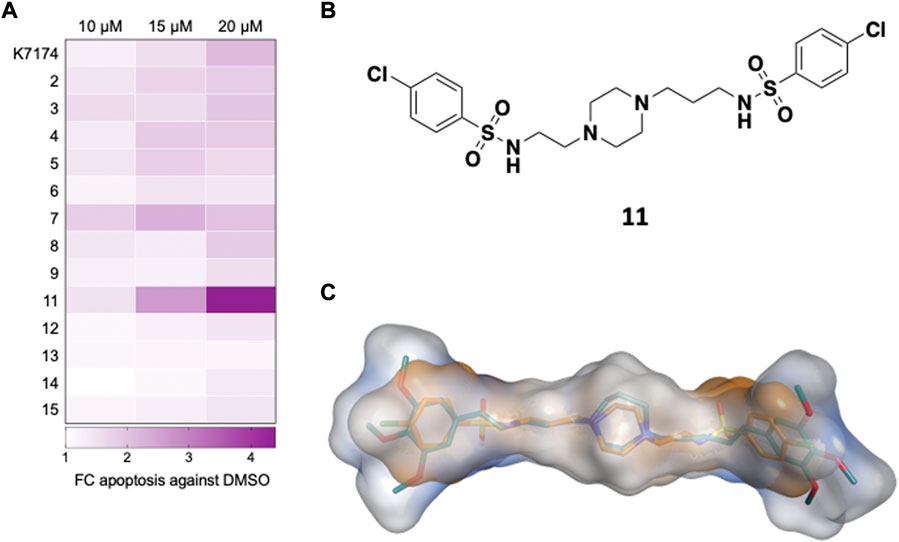
FIGURE 2. Identification of a compound targeting GATA2 in AML cells in vitro (A) Primary screen in K562. K562 were treated with compounds and apoptosis (Annexin+) was measured 48 h later (n = 3) (B) Compound 11 is an asymmetrically disubstituted piperazine. (C) Structural overlap between K7174 and 11. K7174 carbon atoms are shown in teal, while compound 11 carbon atoms are represented in orange. The overall molecular shape of K7174 is represented as a light blue surface, while molecular shape of 11 is shown as orange surface.
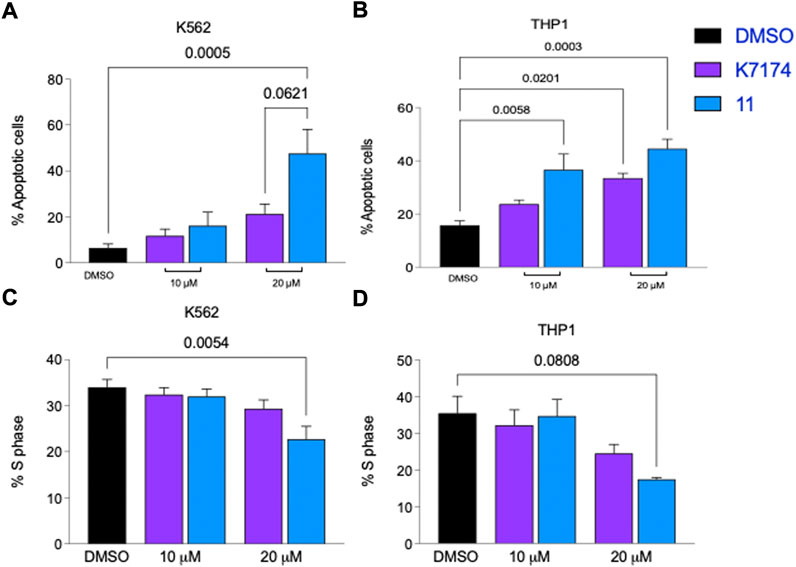
FIGURE 3. Compound 11 has greater anti-leukemic activity than K7174 in AML cells. Frequency of apoptosis cells in (A) K562 and (B) THP1 cells. Frequency of cells in S-phase cells in (C) K562 and (D) THP1. n = 4.
Compound 11 is characterised by an asymmetrical structure, with a central piperazine nucleus presenting an aliphatic linker formed by two carbon atoms at one side, and by three carbon atoms on the other side, connected by a sulphonamide group to two 4-chloro phenyl groups (Figure 2B). Although these structural features are also present on some of the other tested compounds (e.g. 4-chloro phenyl groups in compound 7, asymmetry in 14, a two-carbon side chain in 5) (Supplementary Table S1), it is possible that the combination of all these chemical features together found in 11 is responsible for the anti-leukemic behaviour observed here. Figure 2C shows the structural overlap obtained from ROCS 3D between K7174 and 11. The central piperazine ring is overlapping the homopiperazine, while the three-carbon aliphatic chain together with the sulphonamide group form a five-atom spacer that allow a perfect overlapping between the 4-chloro phenyl ring and the trimethoxy phenyl group. On the contrary, the other lateral chain of 11 is shorter, impeding a correct overlap of the two phenyl rings, which reflects the lower Tanimoto Combo score obtained for 11 in comparison with the other derivatives (Supplementary Table S1).
Effectiveness of compound 11 to target LSCs in vitro and in vivo
Poor prognosis for most AML patients is, in part, ascribed to the inability of current therapeutic regimens to eradicate LSCs, which in turn, drive disease relapse, therapy resistance and mortality (Bonnet and Dick 1997). In a mouse model of AML where transformation of haematopoietic progenitors is mediated by retroviral co-expression of Meis1a and Hoxa9 and subsequent transplantation of these cells into irradiated recipients (Vukovic et al., 2015), we prospectively isolated LSCs from these recipients and cultured them for 48 h with compound 11 or K7174, to assess the ability of 11 to target LSCs. Both 11 and K7174 induced apoptosis in LSCs compared to treatment with vehicle (DMSO), demonstrating that 11 was effective in targeting LSCs (Supplementary Figure S2A). To test the ability of 11 to target Gata2 specifically, we exposed LSCs which lack Gata2 (Gata2 KO LSCs) (Menendez-Gonzalez et al., 2019a) to 11 and assessed apoptosis after 48 h of culture. The proportion of Gata2 KO LSCs undergoing apoptosis with 11 was markedly reduced in comparison to WT LSCs (control) (Supplementary Figure S2A), highlighting the specificity of the compound to kill only LSCs expressing GATA2. In marked contrast, K7174 exposure was equally effective in inducing apoptosis in WT LSCs and Gata2 KO LSCs, confirming that K7174 can target LSCs with or without GATA2 expression (Supplementary Figure S2A). These data together show that compound 11 can specifically target GATA2 in LSCs in vitro.
To test whether 11 was effective at targeting LSCs in vivo, Meis1a/Hoxa9 LSCs were prospectively isolated from primary recipients and transplanted into secondary recipients to re-initiate AML (Supplementary Figure S2B). When engraftment of LSCs reached 1% in the peripheral blood (Supplementary Figures S2B, 2C), these recipients were treated every-day for 5 days with 1, 10 or 100 mg/kg of compound 11 or vehicle control. Kaplan-Meier survival analysis revealed that mice treated at these doses of the compound succumbed to AML within a comparable timeframe to those mice treated with vehicle (Supplementary Figure S2D). Within the context of this specific treatment regimen, compound 11 was not effective at targeting LSCs in vivo, and further structural optimization of this compound and/or the treatment regimen will be required.
Compound 11 does not target the proteasome in vitro
As alluded to above, a significant drawback of using a proteasome inhibitor in the clinic is their high-grade toxicity (Menashe 2007; Csizmar et al., 2016; Pancheri et al., 2020). We therefore tested whether compound 11 targets the proteasome in K562 cells using the Cell-Based Proteasome-Glo™ assay, which is based on the generation of luminescence from the breakdown of luminogenic substrates linked to proteasome activity. As expected, exposure of K562 cells to bortezomib showed clear inhibition of the proteasome, evidenced by a precipitous decrease in luminescence as the concentration of bortezomib increased (Figure 4A). K7174 showed a less striking dose-dependent reduction in luminescence in K562 cells (Figure 4A). In marked contrast, K562 cells exposed to increasing doses of 11 sustained similar levels of luminescence to DMSO treated K562 cells (Figure 4A), indicating that 11 does not inhibit the proteasome.
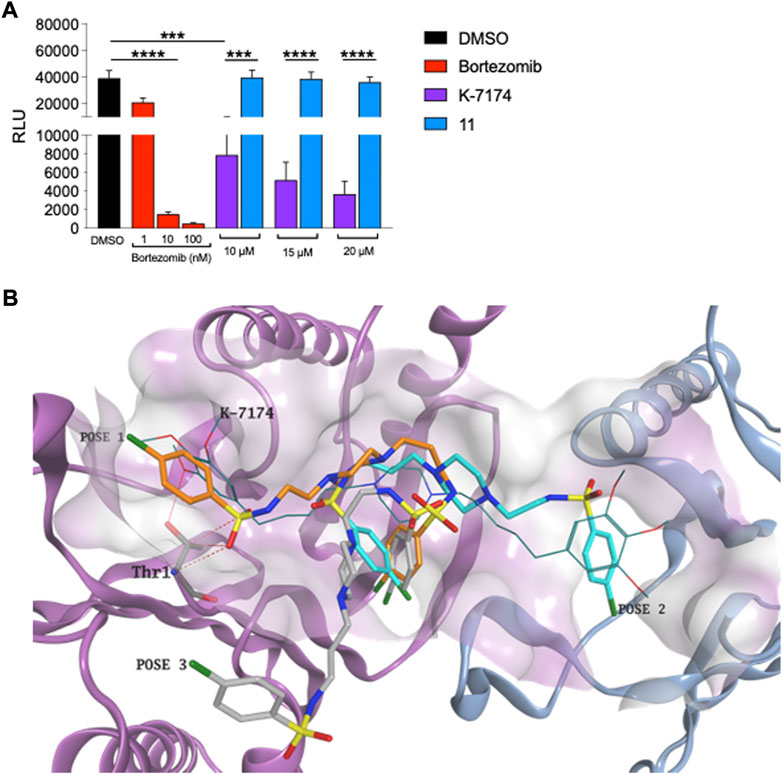
FIGURE 4. Compound 11 does not inhibit proteasome in leukemic cells (A) Proteasome activity assay. Bortezomib was employed as positive proteasome inhibitor (n = 3). (B) Three different binding conformations obtained for 11. Due to the shorter length, the compound does not interact with Thr1 and does not fit the hydrophobic grooves of proteasome at the same time. Compound 11 carbon atoms are represented in orange, torques and grey in the docking poses obtained. K7174 is shown as thin bonds. The proteasome β4 subunit is represented as pigeon blue ribbon, while the β5 subunit is shown as light purple ribbon. K7174 binding site is shown as molecular surface.
To independently gauge whether compound 11 targets the proteasome, we also conducted a molecular docking simulation on the K7174 proteasome binding site. Docking results demonstrated that, due to the shorter length, 11 is not able to entirely occupy the K7174 binding site. Furthermore, due to the high flexibility of the two asymmetrical lateral chains, different possible binding poses were obtained for 11 but, importantly, none of them interacts with Thr1 and fits the hydrophobic grooves of the proteasome at the same time, which are the two essential features for proteasome binding and inhibition (Figure 4B). Overall, the inability of 11 to correctly occupy the K7174 proteasome binding site may reflect the lack of proteasome inhibition found for compound 11.
Discussion
Most AML patients must withstand the rigours of current chemotherapy regimens, where remission largely fails to endure due to the lack of therapeutic targeting of specific driver genetic/epigenetic abnormalities that occur and the therapy resistance of AML LSCs (Craddock et al., 2013; Paczulla et al., 2019). In this report, we have attempted to resolve these challenges by focusing on the potential targeting of GATA2, which is broadly overexpressed across multiple subtypes of AML, and functionally regulates AML LSCs (Menendez-Gonzalez et al., 2019a). Whereas our previous work focused on targeting GATA2 in AML cells using the proteasome inhibitor K7174 (Menendez-Gonzalez et al., 2019b), and given the acknowledged toxicity associated with targeting the proteasome clinically (Menashe 2007; Pancheri et al., 2020), our current work sought to identify a GATA2 inhibitor that operates independently of the proteasome. Utilizing a shape-based screening approach, we identified a compound developed in house, 11, an asymmetrical substituted piperazine with Hepatitis C antiviral activity (Bassetto et al., 2017b), which induces apoptosis and reduces proliferation of leukemic cell blasts in vitro leaving the proteasome intact, consistent with the idea that targeting Gata2 specifically leads to the repression of anti-apoptotic pathways in leukemia cells (Menendez-Gonzalez et al., 2019a). Interestingly, 11 specifically targets Gata2 in LSCs in vitro but lacks efficacy to target LSCs in vivo. Further work will be needed to investigate the reasons for the discordance between results from the in vitro and in vivo LSC systems, which may be explained by 1) sub-optimal dosing with compound 11, 2) timing/duration of intervention with 11, 3) the impact of the in vivo leukemic niche on drug effectiveness, and 4) the metabolic stability of 11 in vivo. Combinatorial in vivo targeting of GATA2 using 11 together with standard AML chemotherapy may also prove more effective in targeting AML LSCs.
There are further caveats to acknowledge in targeting GATA2 in AML. GATA2 expression is needed for normal blood production from hematopoietic stem/progenitor cells and the consequence of sustained lowering of GATA2 expression is borne out in germline GATA2 haploinsufficiency syndromes that promote immunodeficiency which evolves into myelodysplastic syndrome and AML (Hahn et al., 2011; Hsu et al., 2011). Yet, with GATA2 targeting leading to cell death in both leukemic blast cells (as demonstrated by the data in K562 and THP1 cells) and LSCs in vitro (as demonstrated by the data in Meis1/Hoxa9 transformed LSCs), the data presented here continue to support the idea of a regimen of transient targeting of GATA2 in AML during different phases of treatment. The effectiveness of compound 11 to induce apoptosis in AML cells when used alone suggests that lower doses of currently used AML chemotherapeutics could be used in combination with 11 during induction chemotherapy to kill leukemia blasts. During consolidation/maintenance treatment, this compound could be evaluated together with agents that have shown incomplete effectiveness in targeting LSCs (e.g. azacitidine) (Craddock et al., 2013), with the aspiration that this combination of drugs could potentiate eradication of LSCs.
The identification of compound 11 represents a promising starting point for further structure-activity relationship studies, to enhance potency, selectivity, and specificity for targeting GATA2 in AML that could improve the woefully inadequate prognosis observed in these patients. Our efforts will focus on investigating the metabolic stability of compound 11, and addressing the lack of in vivo effect. Finally, we will apply our successful shape-based approach to different commercial drug-like compound libraries, to explore a wider chemical diversity and novel chemical scaffolds to be used for the future development of novel selective GATA2 inhibitors.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by The United Kingdom Home Office under project licence (PPL) 30/3380.
Author contributions
JM-G designed and performed the leukaemia cell-based experiments, analyzed/ interpreted the data, prepared figures, and contributed to manuscript preparation. KS performed the leukaemia cell-based experiments. MB and SF performed the shape based virtual screen, and associated analysis, contributed significantly to overall experimental design, data analysis/interpretation and figure preparation. MB synthesised all the small molecules evaluated. NR conceived the project with AB and SF. NR designed the experiments and analyzed/interpreted the data. NR and SF co-wrote the manuscript. All authors reviewed the manuscript.
Funding
This project was supported by Life Sciences Research Network, Wales (to JM-G and NR) and the Leukemia Cancer Society (to NR). SF was supported by the Ser Cymru II program which part-funded by Swansea University and the European Regional Development Fund through the Welsh Government.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.1013229/full#supplementary-material
References
Aplenc, R., Meshinchi, S., Sung, L., Alonzo, T., Choi, J., Fisher, B., et al. (2020). Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: A report from the children’s oncology group. Haematologica 105 (7), 1879–1886. doi:10.3324/haematol.2019.220962
Arber, D. A., Orazi, A., Hasserjian, R., Thiele, J., Borowitz, M. J., Le Beau, M. M., et al. (2016). The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20), 2391–2405. doi:10.1182/blood-2016-03-643544
Bassetto, M., Leyssen, P., Neyts, J., Yerukhimovich, M. M., Frick, D. N., and Brancale, A. (2017a). Shape-based virtual screening, synthesis and evaluation of novel pyrrolone derivatives as antiviral agents against HCV. Bioorg. Med. Chem. Lett. 27 (4), 936–940. doi:10.1016/j.bmcl.2016.12.087
Bassetto, M., Leyssen, P., Neyts, J., Yerukhimovich, M. M., Frick, D. N., Courtney-Smith, M., et al. (2017b). In silico identification, design and synthesis of novel piperazine-based antiviral agents targeting the hepatitis C virus helicase. Eur. J. Med. Chem. 125, 1115–1131. doi:10.1016/j.ejmech.2016.10.043
Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3 (7), 730–737. doi:10.1038/nm0797-730
Craddock, C., Quek, L., Goardo, N. N., , , Freeman, S., Siddique, S., Raghavan, M., , et al. (2013). Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia 27 (5), 1028–1036. doi:10.1038/leu.2012.312
Csizmar, C. M., Kim, D. H., and Sachs, Z. (2016). The role of the proteasome in AML. Blood Cancer J. 6 (12), e503. doi:10.1038/bcj.2016.112
Döhner, H., Estey, E., Grimwade, D., Amadori, S., Appelbaum, F. R., Buchner, T., et al. (2017). Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129 (4), 424–447. doi:10.1182/blood-2016-08-733196
Ferla, S., Netzler, N. E., Veronese, S., Tuipulotu, D. E., Guccione, S., et al. (2018). In silico screening for human norovirus antivirals reveals a novel non-nucleoside inhibitor of the viral polymerase. Sci. Rep. 8 (1), 4129. doi:10.1038/s41598-018-22303-y
Hahn, C. N., Chong, C. E., Carmichael, C. L., Wilkins, E. J., Brautigan, P. J., Li, X. C., et al. (2011). Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 43 (10), 1012–1017. doi:10.1038/ng.913
Hawkins, P. C. D., Skillman, A. G., and Nicholls, A. (2007). Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 50 (1), 74–82. doi:10.1021/jm0603365
Horowitz, M. M., Gale, R. P., Sondel, P. M., Goldman, J. M., Kersey, J., Kolb, H. J., et al. (1990). Graft-versus-leukemia reactions after bone marrow transplantation. Blood 75 (3), 555–562. doi:10.1182/blood.v75.3.555.555
Hsu, A. P., Sampaio, E. P., Khan, J., Calvo, K. R., Lemieux, J. E., Patel, S. Y., et al. (2011). Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 118 (10), 2653–2655. doi:10.1182/blood-2011-05-356352
Kikuchi, J., Shibayama, N., Yamada, S., Wada, T., Nobuyoshi, M., Izumi, T., et al. (2013). Homopiperazine derivatives as a novel class of proteasome inhibitors with a unique mode of proteasome binding. PLoS ONE 8 (4), e60649. doi:10.1371/journal.pone.0060649
Kumar, A., and Zhang, K. Y. J. (2018). Advances in the development of shape similarity methods and their application in drug discovery. Front. Chem. 6, 315. doi:10.3389/fchem.2018.00315
Lahortiga, I., Vazquez, I., Marcotegui, N., Bandres, E., Malumbres, R., Larrayoz, M. J., et al. (2005). GATA2 is overexpressed in 46% of patients with AML and normal karyotype. The mutational pattern FLT3-ITD/GATA2/WT1 could define a group of patients with normal karyotype and AML-M1 subtype. Blood 106 (11), 2378. doi:10.1182/blood.v106.11.2378.2378
Majik, M. S., Yu, J. H., and Jeong, L. S. (2012). A straightforward synthesis of K7174, a GATA-specific inhibitor. Bull. Korean Chem. Soc. 33 (9), 2903–2906. doi:10.5012/bkcs.2012.33.9.2903
Menashe, J. (2007). Managing and avoiding bortezomib toxicity. Community Oncol. 4 (8), 480–484. doi:10.1016/S1548-5315(11)70481-8
Menendez-Gonzalez, J. B., Vukovic, M., Abdelfattah, A., Saleh, L., Almotiri, A., Thomas, L. A., et al. (2019a). Gata2 as a crucial regulator of stem cells in adult hematopoiesis and acute myeloid leukemia. Stem Cell Rep. 13 (2), 291–306. doi:10.1016/j.stemcr.2019.07.005
Menendez-Gonzalez, J. B., Sinnadurai, S., Gibbs, A., Thomas, L. A., Konstantinou, M., Garcia-Valverde, A., et al. (2019b). Inhibition of GATA2 restrains cell proliferation and enhances apoptosis and chemotherapy mediated apoptosis in human GATA2 overexpressing AML cells. Sci. Rep. 9 (1), 12212. doi:10.1038/s41598-019-48589-0
Molecular Operating Environment (MOE) (2022). Chemical computing group ULC. Montreal, Quebec, Canada: Molecular Operating Environment. https://www.chemcomp.com.
Nurmohamed, S. S., Broadhurst, R. W., May, G., and Enver, T. (2017). Solution NMR structure of human GATA2 C-terminal zinc finger domain. Leuka.
OpenEye Scientific Software (1997). OpenEye Scientific Software, Santa Fe, NM. Available at: https://www.eyesopen.com.
Paczulla, A. M., Rothfelder, K., Raffel, S., Konantz, M., Steinbacher, J., Wang, H., et al. (2019). Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 572 (7768), 254–259. doi:10.1038/s41586-019-1410-1
Pancheri, E., Guglielmi, V., Wilczynski, G. M., Malatesta, M., Tonin, P., Tomelleri, G., et al. (2020). Non-hematologic toxicity of bortezomib in multiple myeloma: The neuromuscular and cardiovascular adverse effects. Cancers 12 (9), E2540. doi:10.3390/cancers12092540
Papaemmanuil, E., Gerstung, M., Bullinger, L., Gaidzik, V. I., Paschka, P., Roberts, N. D., et al. (2016). Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374 (23). doi:10.1056/nejmoa1516192
Richardson, P. G., Barlogie, B., Berenson, J., Singhal, S., Jagannath, S., Irwin, D., et al. (2003). A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 348 (26), 2609–2617. doi:10.1056/nejmoa030288
Schrödinger Release (2022). Schrödinger Release. New York, NY: Maestro, Schrödinger, LLC. Available at: https://www.schrodinger.com.
Keywords: GATA2, acute myeloid leukaemia, virtual shape based screen, in silico, proteasome inhibitor
Citation: Menendez-Gonzalez JB, Strange KE, Bassetto M, Brancale A, Rodrigues NP and Ferla S (2022) Ligand-based discovery of a novel GATA2 inhibitor targeting acute myeloid leukemia cells. Front. Drug. Discov. 2:1013229. doi: 10.3389/fddsv.2022.1013229
Received: 06 August 2022; Accepted: 14 September 2022;
Published: 05 October 2022.
Edited by:
Carmen Cerchia, University of Naples Federico II, ItalyReviewed by:
Sayed K. Goda, University of Derby, United KingdomMatteo Pavan, University of Padua, Italy
Giovanni Ribaudo, University of Brescia, Italy
Copyright © 2022 Menendez-Gonzalez, Strange, Bassetto, Brancale, Rodrigues and Ferla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neil P. Rodrigues, RodriguesN@cardiff.ac.uk; Salvatore Ferla, salvatore.ferla@swansea.ac.uk
†These authors have contributed equally to this work