Consensus docking aid to model the activity of an inhibitor of DNA methyltransferase 1 inspired by de novo design
 Diana L. Prado-Romero1
Diana L. Prado-Romero1  Alejandro Gómez-García1
Alejandro Gómez-García1  Raziel Cedillo-González1
Raziel Cedillo-González1  Hassan Villegas-Quintero1
Hassan Villegas-Quintero1  Juan F. Avellaneda-Tamayo1
Juan F. Avellaneda-Tamayo1  Edgar López-López1,2
Edgar López-López1,2  Fernanda I. Saldívar-González1
Fernanda I. Saldívar-González1  Ana L. Chávez-Hernández1
Ana L. Chávez-Hernández1  José L. Medina-Franco1*
José L. Medina-Franco1*- 1DIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 2Department of Chemistry and Graduate Program in Pharmacology, Center for Research and Advanced Studies of the National Polytechnic Institute, Mexico City, Mexico
The structure-activity relationships data available in public databases of inhibitors of DNA methyltransferases (DNMTs), families of epigenetic targets, plus the structural information of DNMT1, enables the development of a robust structure-based drug design strategy to study, at the molecular level, the activity of DNMTs inhibitors. In this study, we discuss a consensus molecular docking strategy to aid in explaining the activity of small molecules tested as inhibitors of DNMT1. The consensus docking approach, which was based on three validated docking algorithms of different designs, had an overall good agreement with the experimental enzymatic inhibition assays reported in the literature. The docking protocol was used to explain, at the molecular level, the activity profile of a novel DNMT1 inhibitor with a distinct chemical scaffold whose identification was inspired by de novo design and complemented with similarity searching.
1 Introduction
Epigenetic drug discovery is a promising strategy for treating cancer and other complex diseases. Over the past 20 years, several small molecules with novel chemical scaffolds have been investigated with high affinity and selectivity against specific epigenetic targets (Dueñas-González et al., 2016). In several cases, the epi drugs administered alone are not very potent but are co-administered with other epigenetic drugs in combined therapies (Dueñas-González et al., 2016). Amongst the major clinically validated epigenetic targets are the DNA methyltransferases (DNMTs) including the two de novo methyltransferases: DNMT3A and DNMT3B, and the maintenance methyltransferase DNMT1. The latter, which is the most abundant of the three, duplicates the pattern of DNA methylation during replication, and it is essential for proper mammalian development. Since DNA methylation represents a crucial epigenetic mechanism for gene regulation, the development of inhibitors of DNMTs (DNMTis) represents promising perspectives for new therapies. Of the three, DNMT1 has been proposed as the most interesting target for experimental cancer treatments (Dueñas-González et al., 2016; Yu et al., 2019; Zhang et al., 2022).
Azacitidine and 5-aza-decitabine (Figure 1) are two DNMT1 FDA-approved inhibitors for the treatment of myelodysplastic syndrome. However, both drugs are non-specific and have several pharmacokinetic issues (Stresemann and Lyko, 2008). Many other small molecules have been investigated by our and other research groups (Medina-Franco et al., 2015; Giri and Aittokallio, 2019; Hu et al., 2021; Ala et al., 2023) whose structure-activity data is freely accessible in large public databases such as ChEMBL (Davies et al., 2015; Mendez et al., 2019). In the current release of ChEMBL (33), the most active DNMT1 inhibitor has a reported IC50 value of 0.3 nM, although the value is inconclusive. Computational approaches including molecular docking, molecular dynamics, and a broad range of chemoinformatics methods, collectively called “epi-informatics” (Medina-Franco, 2016), have contributed to identifying or developing novel DNMT and other epigenetic targets’ modulators (Sessions et al., 2020). Of note, de novo design is being employed extensively to identify novel epigenetic drug candidates (Prado-Romero and Medina-Franco, 2021) although it has not been pursued (or at least published) to guide the design of DNMT inhibitors.
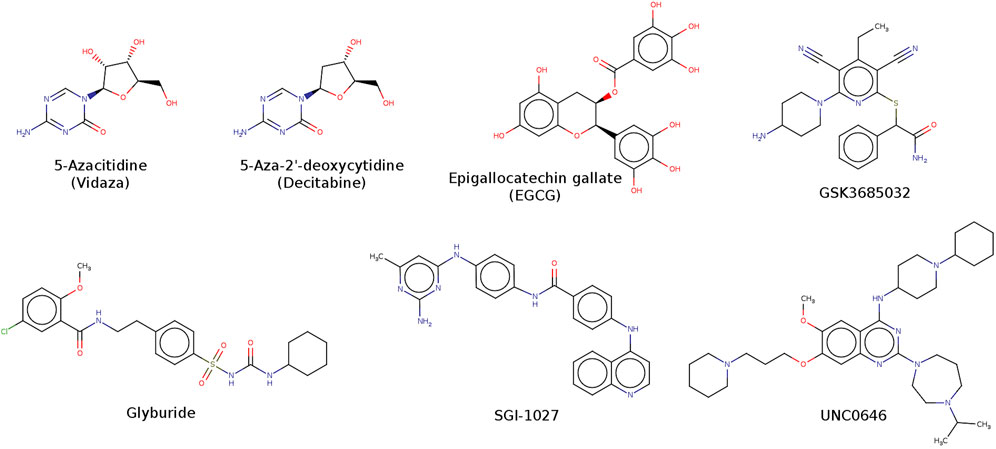
FIGURE 1. Chemical structures of representative inhibitors of DNMT1.
In addition to a large amount of enzymatic inhibition assays’ data of small molecules, since the first crystallographic structure of the catalytic domain of DNMT1 was published (Song et al., 2011) other three-dimensional (3D) coordinates of DNMTs (Syeda et al., 2011; Cheng et al., 2015; Li et al., 2018; Horton et al., 2022; Kikuchi et al., 2022) are available at the Protein Data Bank (Berman et al., 2000). This information has boosted the application of structure-based design data to understand the activity of small molecules at the structural level and to select small molecules for testing among large chemical libraries.
Structure-based virtual screening (SBVS) is a useful technique for drug discovery (Lionta et al., 2014). SBVS aims to predict the best interaction mode between two molecules to form a stable complex, and it uses scoring functions to estimate the force of non-covalent interactions between ligands against a molecular target. As a result, the ligands are ranked according to their predicted affinity to the target. The next goal is to develop hit compounds into leads that then can enter into preclinical studies as drug candidates (Lionta et al., 2014). SBVS relies on the availability of a 3D structure of the target protein. Remarkably, the pose prediction and scoring functions are major factors for the success or failure of the SBVS, not to mention that it is possible to obtain different results from different software using the same input. To reduce the number of false positives (Maia et al., 2020), consensus virtual screening (CVS) has been used (Houston and Walkinshaw, 2013).
SBVS has guided the identification of hit compounds with epigenetic targets. For example, Chen et al. uncover the first selective inhibitor against the disruptor of telomeric silencing 1-like (DOT1L) (Chen et al., 2016), the most studied non-SET-containing methyltransferase that is responsible for the mono-, di- and trimethylation of lysine 79 of histone H3 - H3K79 (Feoli et al., 2022). Zheng et al. reported the combination of high-throughput screening, SBVS, and molecular dynamics to identify computational hits against a histone methyltransferase (Zheng et al., 2021). Kong et al. used SBVS to uncover astemizole as an inhibitor of EZH2/EED (Kong, et al., 2014). Yu et al. reported the SBVS of a commercial screening library, followed by in vitro assays to identify a low micromolar DNMT3A inhibitor with a distinct chemical scaffold (Yu, Chai, et al., 2022). The experimentally validated hit compound was later used in a ligand-based virtual screening (LBVS) based on structural similarity to uncover a submicromolar DNMT3A inhibitor with selectivity against DNMT1, DNMT3B, and G9a. The hit compounds also showed activity in a cancer cell proliferation assay (Yu et al., 2022). In a recent study, Ala et al. reported an SBVS based on molecular docking and dynamics of three databases to identify four compounds with potential inhibitory activity of DNMT1 (Ala et al., 2023).
The goal of this study was to develop a consensus docking protocol to analyze DNMT1 inhibitors. The protocol was based on a combination of well-validated search algorithms, molecular docking scores, and data fusion. We also report a novel DNMT1 inhibitor with a distinct chemical scaffold whose design was based on de novo design and similarity searching. In this study, we did not test directly the compounds designed de novo because of the additional time and economic resources that require the chemical synthesis. Instead, as the first approach to reduce costs and speed up time (as explained in the Methods Section), we combined the results of de novo design with similarity searching of a commercial chemical library. The docking protocol helped to suggest a binding mode with DNMT1. Unexpectedly, new activators of the enzymatic activity of DNMT1 were also found.
2 Methods
The general approach to developing the consensus docking protocol is outlined in Figure 2A, followed by a docking-based analysis of a novel DNMT1 inhibitor whose identification was inspired by de novo design (Figure 2B). In general, the docking protocol comprised six key steps: 1) Target selection; 2) Target preparation; 3) Dataset preparation; 4) Molecular docking; 5) Ranking and re-scoring; and 6) Data fusion (consensus scoring). Details of the protocol are explained in the following sections.
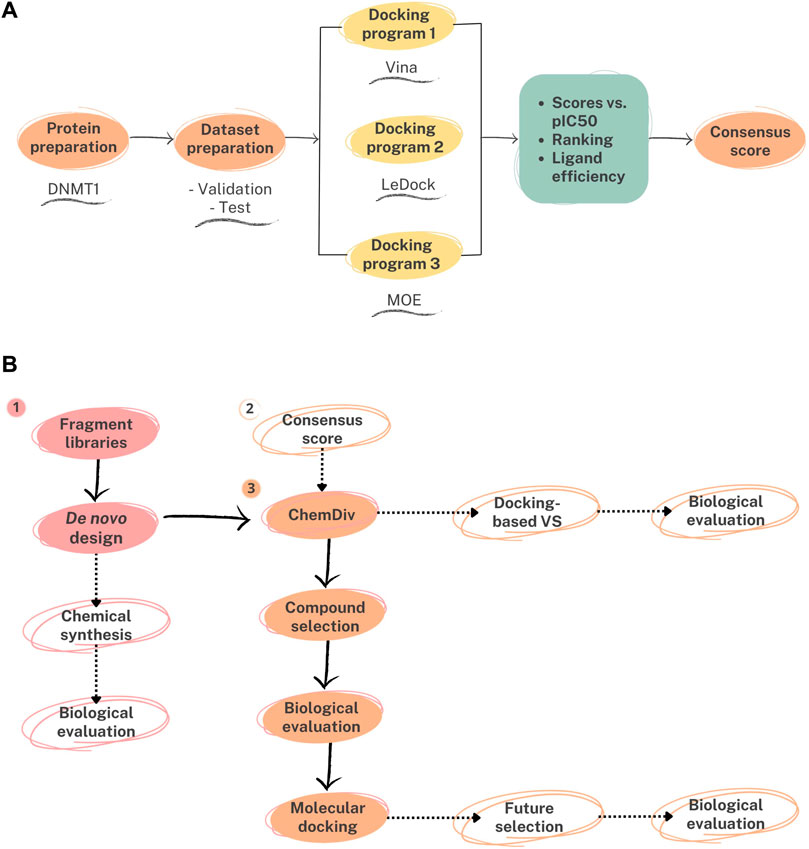
FIGURE 2. General workflow of the strategies implemented in this work. (A) Consensus molecular docking based on Autodock Vina (Vina), LeDock, and Molecular Operating Environment (MOE). (B) Structure-based analysis of a de novo inspired compound. The de novo compound was obtained from fragment libraries retrieved from active compounds. As a first approach, similar compounds from a commercial and ready available library (ChemDiv) were selected for purchase and testing. Continuous black arrows represent the steps followed in this study. Dashed arrows denote perspectives of this work and alternative strategies to identify active molecules: (1) chemical synthesis and testing of compounds designed de novo; (2) virtual screening of the commercial library (including ChemDiv) using the consensus docking protocol; (3) structure-based design and selection of additional candidate compounds based on the docking results of the newly identified compound.
The consensus molecular docking (Figure 2A) was used to generate a binding model of a DNMT1 inhibitor with a novel chemical scaffold identified from an independent de novo design approach combined with similarity searching (method schematically presented in Figure 2B). In Figure 2B, we mark with dashed arrows alternative strategies that will be pursued in forthcoming studies to identify DNMT1 inhibitors based on the outcomes of this study, namely, chemical synthesis and testing of compounds designed de novo; virtual screening of a commercial library using the consensus docking protocol; and structure-based design and selection of additional candidate compounds based on the docking results of the newly identified compound.
Hereunder, we describe first the specific methods used to develop the docking protocol (Sections 2.1–2.8) and this is followed by the description of the selection of the newly tested compounds and the enzymatic inhibition assay (Sections 2.9–2.10).
In all steps, MarvinSketch 22.18 was used for drawing and displaying chemical structures (“MarvinSketch 22.18, Chemaxon, 2023”). Datasets and code for the analysis are available on GitHub at https://github.com/DIFACQUIM/DNMT1-Protocol.
2.1 Targets selection and preparation
The crystallographic structure of human DNMT1 (PDB ID: 4WXX) was retrieved from the RCSB Protein Data Bank (PDB) available online: https://www.rcsb.org/ (accessed on 30 June 2023) (Berman et al., 2000). Among the different crystallographic structures of DNMT1 available on PDB we selected PDB ID: 4WXX because it contains a co-crystallized molecule of S-adenosyl-L-homocysteine (SAH), and was diffracted with a resolution of 2.62 Å. SAH is reported to be a potent inhibitor of both DNA and histone transmethylation (Halsted and Medici, 2016), therefore this 3D structure could be of interest as a model for inhibitory interactions (Alkaff et al., 2021). The protein preparation was made with default settings of the QuickPrep module of Molecular Operating Environment (MOE) v. 2022.02 (“Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2023”): addition of all the lacking hydrogen atoms, protonation state at pH 7, elimination of water molecules 4.5 Å farther from the protein and inside the SAH cavity, addition of missing amino acids residues (breaks of up to ten residues and terminal out gaps of up to five residues) and for larger gaps, neutralization of the endpoints adjoining empty residues and energy minimization. The parameters employed for the energy minimization stage were from the AMBER14:EHT forcefield [ff14SB (Maier et al., 2015) for the protein; MAB forcefield (Gerber and Müller, 1995), and AM1-BCC charges for SAH (Jakalian et al., 2002)]. The energy minimization of the protein in MOE is carried out with three successive nonlinear methods: steepest descent, conjugate gradient, and truncated Newton.
2.2 Dataset selection and preparation
The 153 ligands with reported enzymatic activity against DNMT1 in a biochemical assay were obtained from ChEMBL API v. 32 (Davies et al., 2015; Mendez et al., 2019). Only molecules with binding assay type and unequivocally assigned IC50 were selected. Compounds with nucleoside scaffolds (Supplementary Figure S1) were removed using RDKit library (Landrum et al., 2023) substructure search with SMARTS. Before docking (vide infra), the 153 ligands were built and their geometry was energy minimized using MFF94x forcefield implemented on MOE software. For every ligand, the dominant protonation state at physiological pH (7.4) was chosen (“Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2023”).
2.3 Docking with Vina
The file with the prepared ligands was split with the LeFrag module (Lephar Research, 2023), and Open Babel v.3.1.1 (O’Boyle et al., 2011) was used to convert to .pdb format. Protein and ligands were converted to.pdbqt with MGLTools v.1.5.6. The molecular docking was carried out with Vina v.1.2.3 (Trott and Olson, 2010; Eberhardt et al., 2021) with an exhaustiveness of 8 and 5 binding modes to output. The best score for each ligand was selected for further analysis, with the code freely available at https://github.com/DIFACQUIM/Docking. The grid box was centered in the coordinates: -47.673, 61.885, 6.256 (x, y, z) with a search space of 17 × 25 × 14 Å.
2.4 Docking with LeDock
Docking with Ledock (Kirkpatrick et al., 1983) was carried out in the SAH cavity with the default settings of the software: the grid centered 4 Å around the co-crystallized SAH, twenty docking runs for every ligand and 1 Å for the root mean square deviation (RMSD) clustering. For further data analysis, the best score for every ligand was selected with the code available at https://github.com/DIFACQUIM/Docking.
2.5 Docking with MOE
Docking with MOE v. 2022.02 was centered on the SAH cavity and molecular docking was carried out with the default settings: placement (method: triangle matcher, score function: London dG) and refinement (method: rigid receptor, score function: GBVI/WSA dG) (Vilar et al., 2008). Using the “Triangle Matcher” method, the compounds were subjected to 30 search steps and the default values for the other parameters. The clusters with an RMSD <2 Å were visually explored. During the docking, the receptor was considered rigid and the ligands flexible. The conformations with the lowest binding energy were selected for additional analysis.
2.6 Validation of docking protocol
For this analysis, ligands with IC50 equal to, or lower than 10 μM (pIC50 ≥ 5) were labeled as ‘active’, otherwise they were considered ‘inactive’. Notably, a 10 μM value has been used as a general threshold to define active/inactive molecules in other large-scale studies (Sun et al., 2017; López-López et al., 2022). To develop the current consensus docking protocol, we made the approximation that the enzymatic inhibition assays and the activity values reported in ChEMBL are comparable. The RMSD between the docked and co-crystallized binding conformation of SAH was calculated with Open Babel v. 3.1.1 (O’Boyle et al., 2011). From molecular docking scores and the positive class probabilities, receiver operating characteristic (ROC) curves were generated using KNIME software version 4.6.0 (Gómez-García and Medina-Franco, 2022; Berthold et al., 2009). The results were recorded in a comma-delimited CSV file, which included the scores of each docking program and their ligand efficiency (LE) (vide infra).
2.7 Re-scoring
Docked ligands were ranked according to their predicted scores in ascending order, compounds with higher rank have more negative values, thus better predicted affinity against DNMT1. pIC50 values were also ranked in descending order since a higher value represents a more potent compound (pIC50 ranking). Additionally, LE was calculated individually for each molecular docking score (obtained by Vina, LeDock, or MOE software) with the equation:
In Equation 1, the Heavy Atom Count for each ligand was calculated using RDKit library (Landrum et al., 2023). Correlations and graphs were obtained with SciPy (Virtanen et al., 2020), Matplotlib (Hunter, 2007), and seaborn (Waskom, 2021) libraries using Python programming language version 3.10.12.
2.8 Consensus scoring
Since there is not a single “best” scoring function and docking program, it has been established that combining results from different docking programs increases the likelihood of identifying correct docking poses and improve the performance of docking-based virtual screening (Charifson et al., 1999; Houston and Walkinshaw, 2013; Perez-Castillo et al., 2019; Blanes-Mira et al., 2022). In this study, docking scores and LE values from Vina, LeDock, and MOE were used to calculate seven data fusion metrics: maximum, minimum, arithmetic mean, geometric mean, harmonic mean, median, and Euclidean norm (Bajusz et al., 2019). The data fusion metrics were calculated employing SciPy (Virtanen et al., 2020).
2.9 De novo inspired selection of compounds
Automated de novo design was carried out with alvaBuilder v.1.0.6 (Mauri and Bertola, 2023). Briefly, alvaBuilder combines structural fragments which are obtained from the training sets chosen by the user. The new sets of molecules constructed from the fragments are scored with a scoring function, also chosen by the user (vide infra). Two different training sets were selected as the source of fragments used as construction blocks. The first dataset was retrieved from ChEMBL 31 (Davies et al., 2015; Mendez et al., 2019) selecting compounds with IC50 against DNMT1 equal to, or lower than 10 μM. The second is the diversity subset (PS6) of 5,000 compounds from Life Chemicals (“Diversity Screening Libraries, 2021”) (accessed in August 2021). Both datasets were curated with the same protocol. Briefly, compounds were standardized, the largest component was retained, and compounds were neutralized and reionized to generate canonical SMILES and remove duplicates, as previously published by our group (Sánchez-Cruz et al., 2019; DIFACQUIM, 2020). A random subset of 285 compounds from Life Chemicals was used to match the number of ‘active’ molecules from ChEMBL after curation. We set the scoring function with ranges of descriptors calculated from the molecules with reported biological activity, using alvaDesc 2.0.10 (Mauri, 2020) (the values used for the scoring function are in the Supplementary Table S1): molecular weight (MW), hydrogen bond donors and acceptors, consensus partition coefficient (logP), aqueous solubility (ESOL), synthetic accessibility (SAscore), topological polar surface area (TPSA). The aggregation method was an arithmetic mean with a population size of 70 and 100 iterations. For each training set, 700 molecules were computed. Finally, 1,398 compounds remained after curation.
De novo compounds were used for similarity searching with the commercial library from the Epigenetics Focused Set of ChemDiv (“ChemDiv, 2023”), with 25,883 compounds. Morgan fingerprints of radius 2 (Morgan2) and 3 (Morgan3) (Rogers and Hahn, 2010), along with MACCS keys (166-bit) fingerprint (Durant et al., 2002) were calculated for all compounds with RDKit (Landrum et al., 2023), and similarity was computed with the Tanimoto coefficient. Molecules from ChemDiv that exhibit one of the following similarity values to at least one compound de novo designed were selected for additional analysis: equal to, or higher than 0.30 for Morgan fingerprints radius 2 or 3; or equal to, or higher than 0.80 for MACCS keys. The selection of these thresholds was based on typical values of intuitive high structure similarity for each fingerprint (Medina-Franco, 2012). Similarity values, along with commercial availability criteria, were used to purchase compounds for further evaluation (vide infra).
2.10 Enzymatic DNMT1 inhibition assay
Compounds obtained from ChemDiv were experimentally tested at the company Reaction Biology in an enzymatic inhibition methyltransferase assay (“Reaction Biology Corporation, 2023”) using the HotSpotSM platform. Our research group has reported the methodology and results of this biochemical assay, including the identification of 7-amino alkoxy-quinazolines (Figure 1) (Medina-Franco et al., 2022). Briefly, HotSpotSM is a low-volume radioisotope-based assay that employs tritium-labeled AdoMet (3H-SAM) as a methyl donor. The test compounds diluted in dimethyl sulfoxide were added using acoustic technology (Echo550, Labcyte, San Jose, CA, United States) into an enzyme/substrate mixture in the nano-liter range. The reactions were started by adding 3H-SAM and incubated at 30 °C. Total final methylations on the substrate (Poly dI-dC) were identified by a filter binding method implemented in Reaction Biology. Data analysis was conducted with GraphPad Prism software available at Reaction Biology (La Jolla, CA, United States) for curve fits. The enzymatic inhibition assays were carried out at 1 μM of SAM. The standard positive control was SAH. The compounds were tested in 10-concentration IC50 (effective concentration to inhibit enzymatic activity by 50%) with a threefold serial dilution starting at 100 μM and 200 μM only for F447-0397. Activity percentage values and dose-response curve are reported as provided by the testing laboratory in Figure 8, Supplementary Table S4, respectively.
3 Results and discussion
First, we present the results of the docking protocol with DNMT1 (validation and consensus approach), followed by the results of the newly identified DNMT1 inhibitor with a distinct chemical scaffold. Since the chemical synthesis of de novo compounds requires more time investment, a similarity searching was performed as a first approach to identify novel scaffolds de novo inspired. To have an insight about the possible mechanism of action of the new inhibitor, the docking protocol was used to identify possible key interactions.
Three different algorithms to generate conformers were employed: MOE (Triangle Matcher), Ledock (simulated annealing) (Kirkpatrick et al., 1983), and Vina (Iterated Local Search global optimizer) (Baxter, 1981; Blum et al., 2008). In the Triangle Matcher method, the conformers generated for every ligand are placed inside a space of approximately 5 Å around SAH, this space is permeated with alpha spheres, and, the poses are generated by aligning ligand triplets of atoms on triplets of receptor site points in a systematic way. The receptor site points are alpha sphere centers representing tight packing locations (“Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2023”). The docking run begins with a random conformation, and the move consists of random perturbations of rotatable bonds and the search of the conformational space is carried out using molecular mechanics force fields, with a final rejection test for each molecular move to find an optimal solution (Vilar et al., 2008).
The simulated annealing method of Ledock initially generates an aleatory conformer from which the neighborhood of conformers is generated in search of the one with the most favorable binding energy. Nonetheless, during the first iterations, the generation of conformers will not always move in search of the most favorable binding energy but can move towards the generation of conformers with less favorable binding energy. This is intending to expand the region of search in conformational space (Kirkpatrick et al., 1983). The Iterated Local Search global optimizer of Vina consists of a succession of steps of a mutation and a local optimization, with each step being accepted according to the Metropolis criterion (Trott and Olson, 2010). It uses the Broyden-Fletcher-Goldfarb-Shanno method (Nocedal and Wright, 2006) for local optimization, which is an efficient quasi-Newton method (Trott and Olson, 2010).
In MOE, two different scoring functions were employed: London dG for the initial conformer generation and GBVI/WSA dG for the conformer refinement. London dG takes into account the average gain/loss of rotational and translational entropy, the energy due to the loss of flexibility of the ligand (calculated from ligand topology only), the hydrogen bond energy, and the desolvation energy. GBVI/WSA dG also considers the gain/loss of rotational and translational entropy, the Coulombic electrostatic energy, van der Waals interactions, and the solvation electrostatic energy. The exposed surface area of the ligand is penalized (“Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2023”). The scoring function of Ledock takes into account the Coulombic electrostatic energy, van der Waals interactions, the hydrogen bond energy, the intra-molecular clashes, and torsion strain (Kirkpatrick et al., 1983). The scoring function of Vina (Trott and Olson, 2010) is inspired by the scoring function X-CSCORE which takes into account the van der Waals interactions, hydrogen bonding, deformation penalty, and the hydrophobic effect (Wang et al., 2002).
3.1 Validation of docking protocol and re-scoring
The validation of the molecular docking protocol was done with two approaches: RMSD values between the docked and co-crystallized binding conformation of SAH, and ROC curves (as detailed in the Methods Section).
The RMSD values for SAH were lower than 2 Å for all docking programs (Vina: 1.586 Å (second pose); LeDock: 1.291 Å; and MOE: 1.214 Å), Supplementary Figure S2 shows the 3D predicted pose of SAH with each software. The calculated values suggest that the docking protocols are able to identify the experimental 3D conformation of SAH found in the crystallographic structure.
Figure 3 shows the ROC curves for all three docking software using the docking scores and the LE. The ROC curves indicated that MOE’s binding scores led to better identification of true positives, in contrast with Ledock and Vina. However, the calculation of LE (Equation 1) is detrimental to the area under the curve (AUC) (Vina: 0.295; LeDock: 0.250; and MOE: 0.084), this suggests that LE does not contribute to discarding inactive molecules (Figure 3B). These results highlight the relevance of considering the ligand size, herein with the heavy atom count, to evaluate the performance of the docking programs.
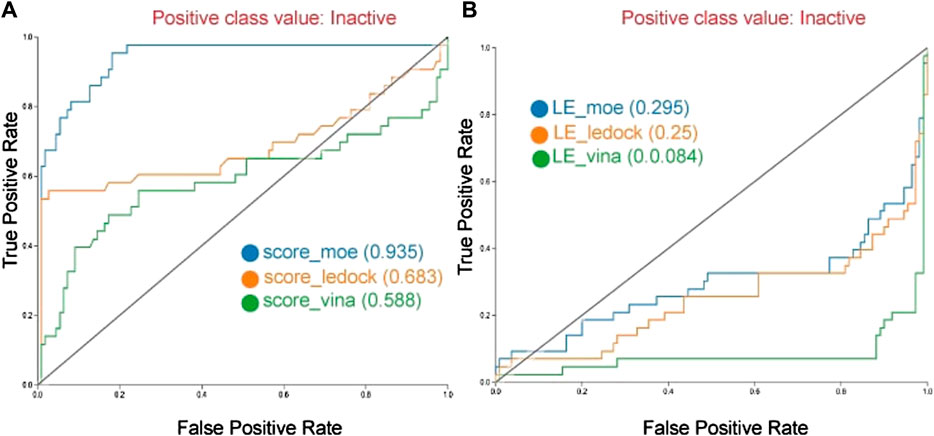
FIGURE 3. Receiver operating characteristic (ROC) curves of the docking with DNMT1 with three different docking programs. Curves are generated with scoring (A), and with ligand efficiency (B).
To have an insight into the data distribution, correlation plots are shown in Figure 4: Vina (4A), LeDock (4B), and MOE (4C). In each plot, the horizontal axis represents the pIC50 ranking. Docking scores, scores’ ranking, and LE are shown in the vertical axis for each docking software. Spearman correlation (ρ) was computed for each plot. ‘Active’ and ‘inactive’ compounds against DNMT1 are represented in different colors. Supplementary Figure S3 shows the correlation plots for all three docking programs plotting the pIC50 values on the horizontal axis.
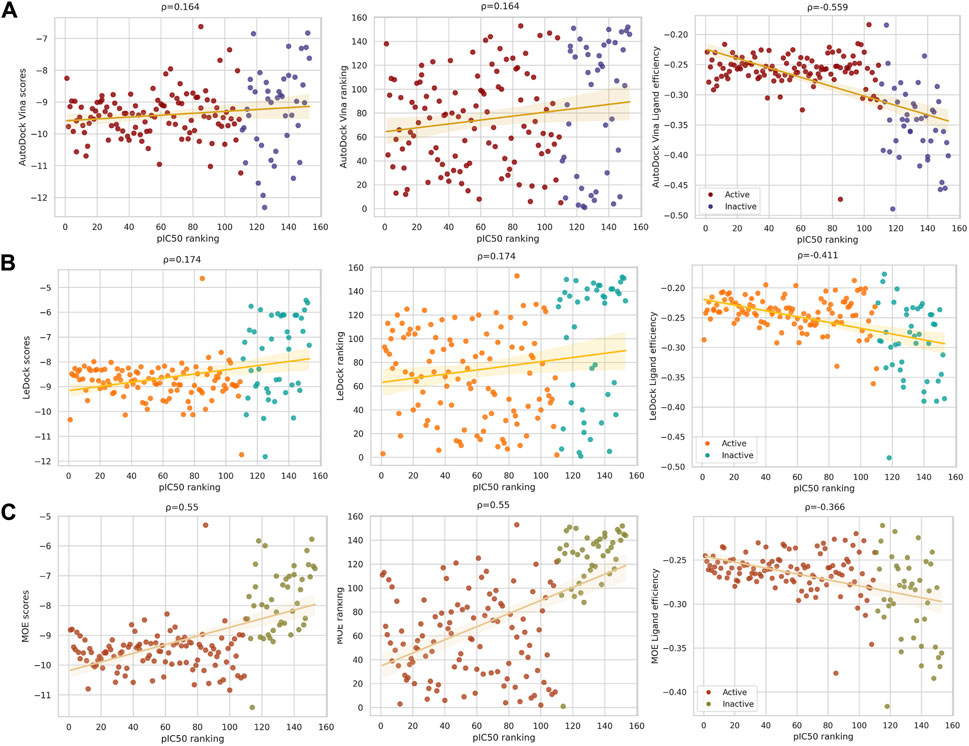
FIGURE 4. Docking scores and ligand efficiency (LE) correlations with ranked pIC50 of compounds with activity against DNMT1. Compounds labeled as active are in red, orange, or firebrick, and inactive compounds are in blue, cyan, or olive green. Spearman’s correlation is shown above each graph. From left to right: binding scores, scores’ ranking, and LE calculated with (A) Vina, (B) LeDock, and (C) MOE. Despite the low correlation (maximum 0.55), taking into account the ligand’s size improves the performance of the docking program.
Although ρ values for docking scores and scores’ ranking are equal for each program, as expected due to the transformation to rank variables, the distribution of the data is more scattered when plotting the scores’ ranking. Despite the fact the highest correlation observed is low (0.55), the correlation plots in Figure 4 indicate that, overall, considering the ligand size improves the performance of the docking program. This is particularly noticeable in the results obtained with Vina and LeDock where the ρ values improved when considering the LE (Figure 4). The observations obtained with the correlation plots agreed with the conclusions obtained from the ROC curves (Figure 3).
3.2 Consensus docking
As discussed in the Introduction, consensus SBVS could be more accurate at identifying active compounds as compared to individual methods (Wang and Wang, 2001; Houston and Walkinshaw, 2013). Data fusion also helps to rationalize the relationships between the chemical, physicochemical, and biological features that explain in more detail the possible binding mechanism of different kinds of inhibitors (López-López and Medina-Franco, 2023). Data generated with consensus docking has been useful in developing new drug candidates (Maia et al., 2020; Morris et al., 2022). The advantage of using different docking programs is that the variety of generated conformers is enriched because every program has its own conformer generation algorithm and scoring functions.
Analysis of consensus docking results with data fusion metrics has been shown to improve the results of individual docking (Bajusz et al., 2019; Triches et al., 2022; López-López and Medina-Franco, 2023). Table 1 summarizes the resulting correlations (ρ) of the pIC50 ranking and different data fusion metrics implemented in this work (see Methods Section for details). The resulting correlations (ρ) with pIC50 can be found in the Supplementary Table S2. The best performances were achieved with the median and the minimum rules for the docking scores and LE, respectively. There is a higher correlation with LE, in concordance with the results before the consensus.
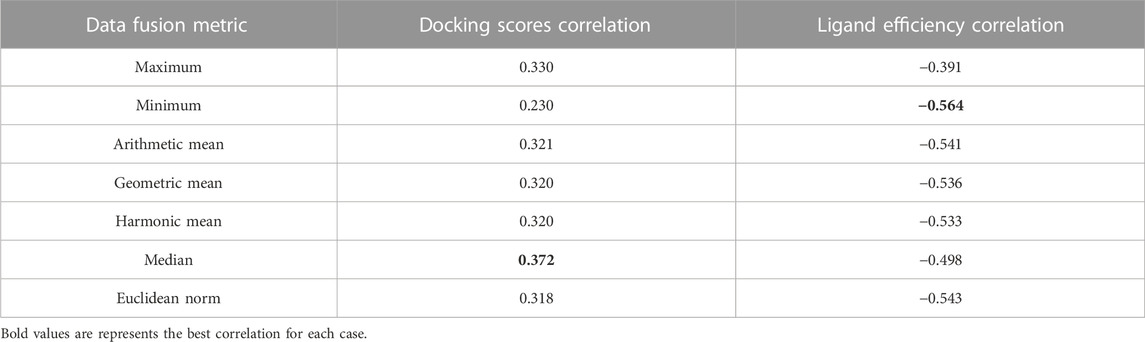
TABLE 1. Results of data fusion metrics and their correlations with pIC50 ranking.
Figure 5 shows the correlation between different consensus docking approaches obtained with different data fusion rules (described in the Methods Section) and the bioactivity of DNMT1 inhibitors reported in the literature. The two best correlations are shown as calculated with Spearman’s correlation: the pIC50 values with the median docking score (ρ = 0.372) and with the minimum LE (ρ = −0.564). In agreement with the results discussed in Section 3.1, LE had the best correlations, which further emphasizes the convenience of accounting for the size of the ligand while doing docking analysis with DNMTis.
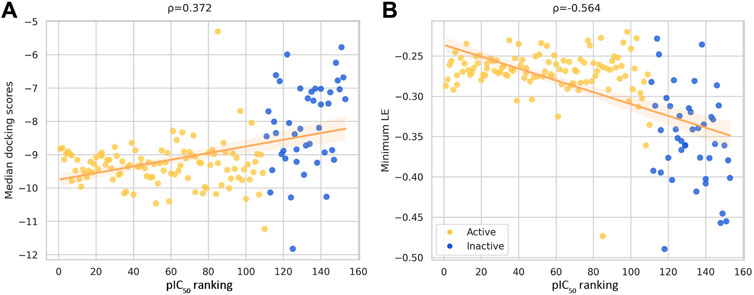
FIGURE 5. Correlation plots between ranked pIC50 values reported in ChEMBL for DNMT1 inhibitors and (A) docking scores and (B) minimum ligand efficiency (LE). The Spearman’s correlation coefficient is indicated in the plots. Compounds with IC50 values lower/greater than 10 μM are represented with a different color.
As observed from Figure 5, correlation values increased from individual docking scores in the case of Vina and LeDock. Minimum LE also shows a better correlation than LeDock and MOE alone. Nevertheless, the calculated correlation for Vina LE has a close value (ρ = 0.559).
The corresponding ROC curves are shown in Figure 6 emphasizing the improved performance of the consensus median and consensus minimum.
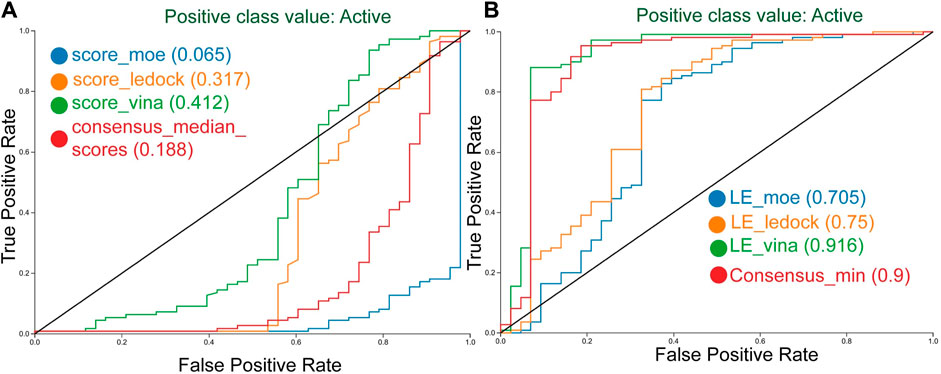
FIGURE 6. Receiver operating characteristic (ROC) curves of the docking with DNMT1 with three different docking programs. Curves generated with scoring (A), and with ligand efficiency (B), including the best consensus metric.
3.3 Distinct DNMT1 inhibitor inspired by de novo design
As a result of the similarity searching using the 1,398 molecules proposed with de novo design, six compounds from ChemDiv were purchased. The results of the similarity calculations are provided as supplementary .csv files. Figure 7 shows the chemical structures of the newly tested compounds with DNMT1, as well as the most similar de novo compound according to Morgan fingerprint of radius 2 (Morgan2) (Rogers and Hahn, 2010). Most similar de novo compounds according to Morgan fingerprint of radius 3 and MACCS keys are in Supplementary Table S5. The database of the de novo-designed molecules is available at https://github.com/DIFACQUIM/DNMT1-Protocol/tree/main/De-Novo_inspired. Compound F447-0397 had inhibition against DNMT1 in the enzymatic assays, with an IC50 of 41.3 ± 2.1 μM. Dose-response curves used for the calculation of IC50 are in Figure 8. The data used by the testing laboratory (Reaction Biology) to obtain the IC50 is in Supplementary Table S4. It should be noted that, although one point was excluded from the curve fit, F447-0397 inhibited in more than 99% the enzymatic activity of DNMT1 at the highest concentration tested. Nevertheless, the Hill slope of the IC50 curve is not close to 1.0 as it occurs for the positive and internal control, SAH for which the assay conditions to measure DNMT1 were developed by the testing laboratory. Based on these results it is important to conduct additional biochemical and orthogonal assays (e.g., in a cellular context) to further confirm the activity of the compound F447-0397. This compound exhibits a novel scaffold, not previously published among DNMT1 inhibitors to our knowledge. This was shown as no matching molecule was found after the substructure search with the Murcko scaffold of F447-0397 as implemented in RDKit (Landrum et al., 2023), using the curated dataset of 743 molecules with biological activities against DNMT1 found in ChEMBL 33 (Davies et al., 2015; Mendez et al., 2019). Supplementary Table S3 summarizes the results of the enzymatic inhibition assays of the six compounds.
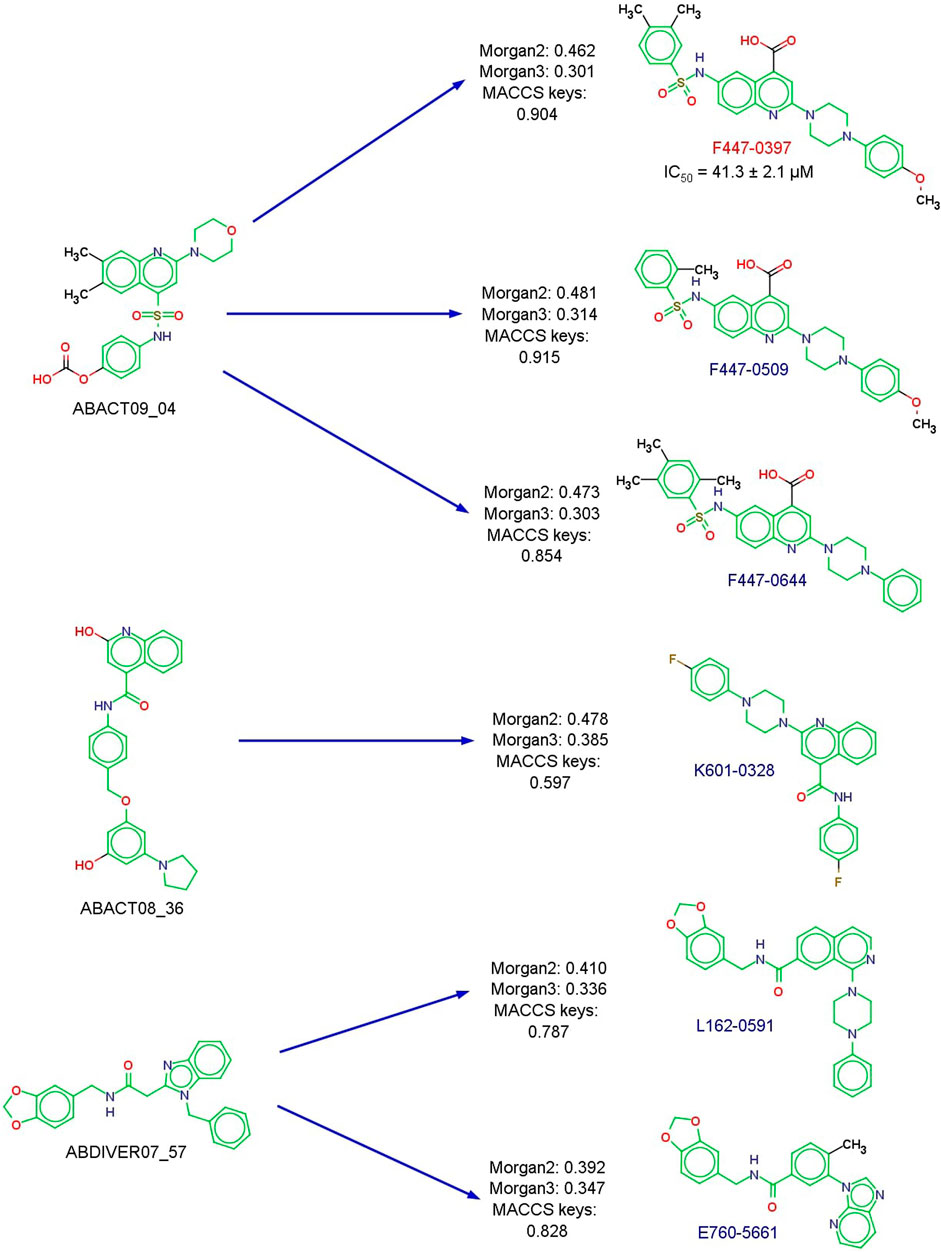
FIGURE 7. Chemical structures of newly tested compounds with DNMT1 according to commercial availability. The Murcko scaffold is marked in green. The most similar de novo compound (Morgan2 representation) to the commercially available molecule (from ChemDiv) is shown on the left, alongside the similarity values calculated with the Tanimoto coefficient. The IC50 value of the active compound is indicated.
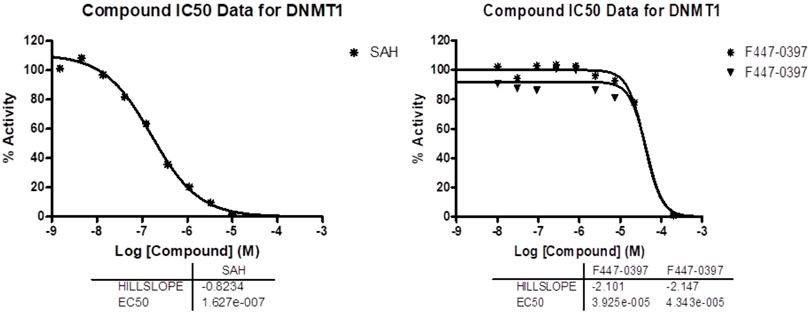
FIGURE 8. Dose-response curves used to calculate the IC50 values. Data and curves provided by the testing laboratory, Reaction Biology. Data for the positive control SAH (left) and ChemDiv compound F447-0397 (right). F447-0397 was tested in a 10-dose IC50 mode with 3-fold serial dilution, starting at 200 µM.
The compounds F447-0397, F447-0509, and F447-0644 are quite similar, in particular, F447-0397 and F447-0509 (an additional methyl group and substitution pattern in the sulfonamide phenyl ring). The docking scores and LE of the three molecules are also similar (Supplementary Table S3), as could be anticipated from their structural similarity. However, the percentage of enzymatic activity at 100 μM is quite different, with F447-0397 being the only inhibitor (12.76%). These are good examples of activity cliffs: compounds with similar chemical structures but very unexpected activity differences (Maggiora, 2006). Although the IC50 of F447-0397 indicated that it is not a very potent compound (41.3 μM - and could be considered “inactive,” the scaffold is novel and could be an interesting starting point for optimization). The novel DNMT1 inhibitor has a “long scaffold” e.g., four-ring systems connected with one-to-three bond linkers. This is in line with other DNMT1 inhibitors with “long or extended scaffolds,” such as the 4-aminoquinoline SGI-1027 and its analogs (Datta et al., 2009; Gros et al., 2015) and glyburide (Juárez-Mercado et al., 2020) (Figure 1). However, unlike SGI-1027 and glyburide, F447-0397 was identified by a combination of de novo design and similarity searching.
Figure 9 shows the predicted binding mode of F447-0397 with DNMT1 generated with Vina, the docking program that had, overall, the best performance of all three docking programs (as shown in Figure 3). The predicted pose shows a hydrogen bond between Glu1168 and the piperazine ring of F447-0397. This could be a key interaction since the co-crystallized SAH also makes a hydrogen bond interaction with Glu1168. Re-docking of SAH, with the three software, predicted the same interaction (Supplementary Figure S4). The predicted binding mode with Vina also exhibits a hydrogen bond between Arg1310 and the oxygens of the sulfonamide from F447-0397. MOE predicted pose also showed the hydrogen bond with the oxygens of the inhibitor’s carboxylic acid (Supplementary Figures S5, S6). Interactions with Arg1310 and computational hits were previously observed in separate docking studies with DNMT1 (Bashir et al., 2023), and also between Arg1310 and EGCG (Figure 1) (Assumpção et al., 2020). Of note, LeDock and MOE predicted interactions between Asn1578 and F447-0397, the interaction with this particular residue could provide selectivity towards DNMT1 versus DNMT3A (Yu et al., 2019). This suggests that F447-0397 could be the starting point of an optimization project toward selective DNMT1 inhibitors.
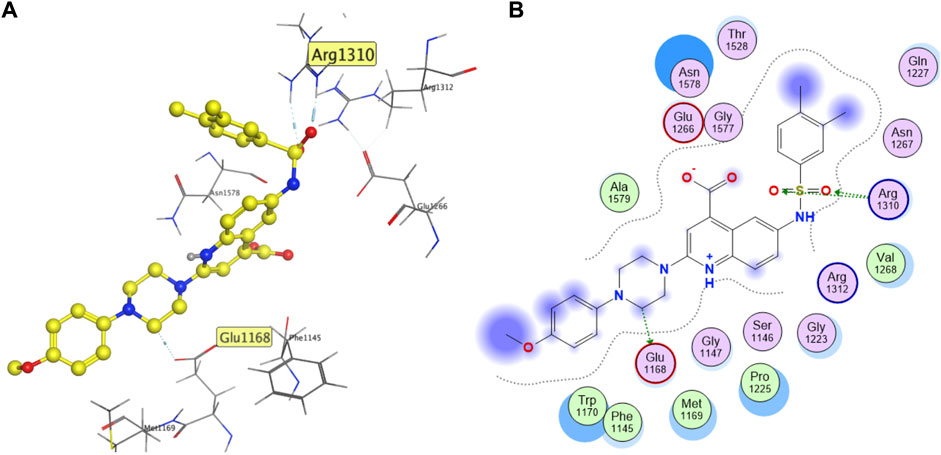
FIGURE 9. The predicted binding mode (Vina) of F447-0397 with DNMT1 (PDB ID: 4WXX), showing (A) 3D and (B) 2D binding models.
Compound E760-5661 was an activator (142% enzymatic activity under the assay conditions), followed by the structurally related molecule L162-0591 (132% activity, Supplementary Table S3). Although this is an unexpected result (as we were looking for inhibitors), at least there is agreement that compounds structurally similar have similar (activation) profiles. The activation of DNMT1 also has clinical implications, as DNA hypomethylation has been related to various human diseases, like cancer, and cardiovascular diseases (Wilson et al., 2007; Pogribny and Beland, 2009). Other activators, also identified serendipitously, have been recently published (Rodríguez-Mejía et al., 2022). It would remain to confirm the capabilities of compounds E760-5661 and L162-0591 in a cellular context. To this end, a global human DNA methylation assay could be performed, as recently reported by Rodríguez-Mejía et al. that recently identified two DNMT1 activators (Rodríguez-Mejía et al., 2022). It also remains to explore, at the structural level, the activity cliffs identified in this work. Preliminary structural comparisons of the three compounds (F447-0397, F447-0509, and F447-0644) suggest a quite precise protein-ligand interaction of the active compound - F447-0397 - with DNMT1. The structural analogs could be binding in a different binding region that activates the enzymatic activity of DNMT1 at a certain level, possibly by relaxing allosteric autoinhibition of human DNMT1, as recently proposed for two activators of DNMT1. The mechanism of activation of DNMT1 is out of the scope of this study.
4 Conclusion and perspectives
This study contributes to the further development of inhibitors of DNMT1 through a comprehensive analysis of docking protocols and analysis of the individual vs. consensus results. Herein, we also used different structure-based analyses to suggest the binding mode of a new DNMT1 inhibitor whose design was inspired by a de novo ligand-based design. Noteworthy, there are no previous reports of inhibitors of DNMTs proposed with de novo design. We concluded that, overall, out of the three docking programs, Vina had the best performance concerning the docking poses, as measured by the LE. Calculation or consideration of LE significantly enhanced the performance of Vina, Ledock, and MOE to prioritize compounds in SBVS of the 153 DNMT1is in ChEMBL. Regarding the consensus protocol, the best data fusion rules were the median and, more significantly, the minimum fusion, particularly considering the LE. The results emphasize the significance of considering the size of the ligand as part of the results of the docking analysis.
We also report a small molecule (F447-0397) with a chemical scaffold that had not been previously published as a DNMT1 inhibitor. Docking simulations suggested a binding mode of the new inhibitor making interactions with Glu1168 (like co-crystallized SAH) and Arg1310 (like previous hits). As part of the study, we uncovered two activity cliffs: compounds with a chemical structure similar to F447-0397 but a very different activity profile.
One of the main perspectives of this work is performing additional biochemical assays at different testing concentrations of F447-0397 and conducting orthogonal assays to confirm its DNMT1 inhibitory activity. To this end, the whole genome methylation profiling could be assessed with techniques such as High-Performance Liquid Chromatography Ultraviolet (HPLC-UV), Liquid Chromatography coupled with tandem Mass Spectrometry (LC-MS/MS), ELISA-Based Methods, LINE-1+Pyrosequencing, PCR-based amplification fragment length polymorphism (AFLP), restriction fragment length polymorphism (RFLP) or a combination of both, or luminometric methylation assay (LUMA) (Kurdyukov and Bullock, 2016; Pechalrieu et al., 2017). The activators of DNMT1 encourage investigating these compounds as potential biochemical probes to explore the role of DNMT1. Another perspective is to perform virtual screenings of chemical libraries with the newly developed consensus docking protocol (Figure 2B), including the screening of ChemDiv. Also, it can be pursued the chemical synthesis and testing of compounds designed de novo and the structure-based optimization (including chemical synthesis and testing) of the active compound identified in this work, F447-0397 (the latter two perspectives also outlined in Figure 2B). Of note, since several successful SBVS to identify DNMT1 inhibitors have been reported, a key point in future screenings is filtering chemical libraries that had not previously been screened, including newly developed focused libraries.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
DP-R: Data curation, Formal Analysis, Methodology, Validation, Visualization, Conceptualization, Investigation, Writing–original draft, Writing–review and editing. AG-G: Investigation, Writing–original draft, Writing–review and editing, Data curation, Formal Analysis, Methodology, Validation, Visualization. RC-G: Investigation, Writing–original draft, Writing–review and editing, Data curation, Formal Analysis, Methodology, Validation, Visualization. HV-Q: Investigation, Writing–review and editing, Data curation, Formal Analysis, Methodology, Validation, Visualization. JA-T: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–review and editing. EL-L: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–review and editing, Writing–original draft. FS-G: Investigation, Methodology, Writing–review and editing. AC-H: Investigation, Methodology, Writing–review and editing, Data curation, Visualization. JM-F: Investigation, Writing–review and editing, Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing–original draft.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. We thank DGAPA, UNAM, Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT), grants No. IN201321 (to test the compounds) and IV200121 (to purchase the MOE’s academic license). We also thank the innovation space UNAM-HUAWEI the computational resources to use their supercomputer under project-7 “Desarrollo y aplicación de algoritmos de inteligencia artificial para el diseño de fármacos aplicables al tratamiento de diabetes mellitus y cáncer.”
Acknowledgments
DP-R, AG-G, RC-G, JA-T, EL-L, FS-G, and AC-H thank Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCyT), Mexico, for the postgraduate scholarships 888207, 912137, 1099206, 1270553, 894234, 848061, 847870. HV-Q is grateful to UNAM-HUAWEI for the scholarship under the project no. 7, “Desarrollo y aplicación de algoritmos de inteligencia artificial para el diseño de fármacos aplicables al tratamiento de diabetes mellitus y cáncer”. We acknowledge K. Eurídice Juárez-Mercado for providing the code for the similarity searching. We also thank Marvin for the Research License. MarvinSketch was used for drawing and displaying chemical structures, MarvinSketch 22.18, Chemaxon (https://www.chemaxon.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2023.1261094/full#supplementary-material
Abbreviations
3D, three-dimensional; AUC, area under the curve; CVS, consensus virtual screening; DNMT, DNA methyltransferase; DNMTis, inhibitors of DNA methyltransferases; DS, docking score; LBVS, ligand-based virtual screening; LE, ligand efficiency; MOE, Molecular Operating Environment; MW, molecular weight; PDB, Protein Data Bank; RMSD, root mean square deviation; ROC, Receiver Operating Characteristic; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosyl-L-methionine; SBVS, structure-based virtual screening; TPSA, topological polar surface area; Vina, AutoDock Vina; VS, virtual screening.
References
Ala, C., Joshi, R. P., Gupta, P., Ramalingam, S., and Sankaranarayanan, M. (2023). Discovery of potent DNMT1 inhibitors against sickle cell disease using structural-based virtual screening, MM-GBSA and molecular dynamics simulation-based approaches. J. Biomol. Struct. Dyn., 1–13. doi:10.1080/07391102.2023.2199081
Alkaff, A. H., Saragih, M., Imana, S. N., Nasution, M. A. F., and Tambunan, U. S. F. (2021). Identification of DNA methyltransferase-1 inhibitor for breast cancer therapy through computational fragment-based drug design. Molecules 26, 375. doi:10.3390/molecules26020375
Assumpção, J. H. M., Takeda, A. A. S., Sforcin, J. M., and Rainho, C. A. (2020). Effects of propolis and phenolic acids on triple-negative breast cancer cell lines: potential involvement of epigenetic mechanisms. Molecules 25, 1289. doi:10.3390/molecules25061289
Bajusz, D., Rácz, A., and Héberger, K. (2019). Comparison of data fusion methods as consensus scores for ensemble docking. Molecules 24, 2690. doi:10.3390/molecules24152690
Bashir, Y., Noor, F., Ahmad, S., Tariq, M. H., Qasim, M., Tahir Ul Qamar, M., et al. (2023). Integrated virtual screening and molecular dynamics simulation approaches revealed potential natural inhibitors for DNMT1 as therapeutic solution for triple negative breast cancer. J. Biomol. Struct. Dyn., 1–11. doi:10.1080/07391102.2023.2198017
Baxter, J. (1981). Local optima avoidance in depot location. J. Oper. Res. Soc. 32 (9), 815–819. doi:10.2307/2581397
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein Data Bank. Nucleic Acids Res. 28 (1), 235–242. doi:10.1093/nar/28.1.235
Berthold, M. R., Cebron, N., Dill, F., Gabriel, T. R., Kötter, T., Meinl, T., et al. (2009). KNIME - the Konstanz information miner: version 2.0 and beyond. SIGKDD Explor 11 (1), 26–31. doi:10.1145/1656274.1656280
Blanes-Mira, C., Fernández-Aguado, P., de Andrés-López, J., Fernández-Carvajal, A., Ferrer-Montiel, A., and Fernández-Ballester, G. (2022). Comprehensive survey of consensus docking for high-throughput virtual screening. Molecules 28, 1. doi:10.3390/molecules28010175
Blum, C., Roli, A., and Sampels, M. (2008). Hybrid metaheuristics: an emerging approach to optimization. Berlin: Springer Science and Business Media.
Charifson, P. S., Corkery, J. J., Murcko, M. A., and Walters, W. P. (1999). Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 42 (25), 5100–5109. doi:10.1021/jm990352k
ChemDiv (2023). ChemDiv. Available at: https://www.chemdiv.com/catalog/focused-and-targeted-libraries/epigenetics-focused-set/ (Accessed March 13, 2023).
Chen, S., Li, L., Chen, Y., Hu, J., Liu, J., Liu, Y.-C., et al. (2016). Identification of novel disruptor of telomeric silencing 1-like (DOT1L) inhibitors through structure-based virtual screening and biological assays. J. Chem. Inf. Model. 56 (3), 527–534. doi:10.1021/acs.jcim.5b00738
Cheng, J., Yang, H., Fang, J., Ma, L., Gong, R., Wang, P., et al. (2015). Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat. Commun. 6, 7023. doi:10.1038/ncomms8023
Datta, J., Ghoshal, K., Denny, W. A., Gamage, S. A., Brooke, D. G., Phiasivongsa, P., et al. (2009). A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 69 (10), 4277–4285. doi:10.1158/0008-5472.CAN-08-3669
Davies, M., Nowotka, M., Papadatos, G., Dedman, N., Gaulton, A., Atkinson, F., et al. (2015). ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 43 (W1), W612–W620. doi:10.1093/nar/gkv352
DIFACQUIM (2020). IFG_General: repository for the work functional group and diversity analysis of biofacquim: a Mexican natural product database. Available at: https://github.com/DIFACQUIM/IFG_General (Accessed July 12, 2023).
Diversity Screening Libraries (2021). Diversity screening libraries. Available at: https://lifechemicals.com/screening-libraries/pre-plated-diversity-sets (Accessed August 26, 2021).
Dueñas-González, A., Jesús Naveja, J., and Medina-Franco, J. L. (2016). “Chapter 1 - introduction of epigenetic targets in drug discovery and current status of epi-drugs and epi-probes,” in Epi-informatics. Editor J. L. Medina-Franco (Boston: Academic Press), 1–20. doi:10.1016/B978-0-12-802808-7.00001-0
Durant, J. L., Leland, B. A., Henry, D. R., and Nourse, J. G. (2002). Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42 (6), 1273–1280. doi:10.1021/ci010132r
Eberhardt, J., Santos-Martins, D., Tillack, A. F., and Forli, S. (2021). AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61 (8), 3891–3898. doi:10.1021/acs.jcim.1c00203
Feoli, A., Viviano, M., Cipriano, A., Milite, C., Castellano, S., and Sbardella, G. (2022). Lysine methyltransferase inhibitors: where we are now. RSC Chem. Biol. 3 (4), 359–406. doi:10.1039/d1cb00196e
Gerber, P. R., and Müller, K. (1995). MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput.-Aided Mol. Des. 9 (3), 251–268. doi:10.1007/BF00124456
Giri, A. K., and Aittokallio, T. (2019). DNMT inhibitors increase methylation in the cancer genome. Front. Pharmacol. 10, 385. doi:10.3389/fphar.2019.00385
Gómez-García, A., and Medina-Franco, J. L. (2022). Progress and impact of Latin American natural product databases. Biomolecules 12, 1202. doi:10.3390/biom12091202
Gros, C., Fleury, L., Nahoum, V., Faux, C., Valente, S., Labella, D., et al. (2015). New insights on the mechanism of quinoline-based DNA Methyltransferase inhibitors. J. Biol. Chem. 290 (10), 6293–6302. doi:10.1074/jbc.M114.594671
Halsted, C. H., and Medici, V. (2016). “Chapter 8 - vitamin B regulation of alcoholic liver disease,” in Molecular aspects of alcohol and nutrition. Editor V. B. Patel (San Diego: Academic Press), 95–106. doi:10.1016/B978-0-12-800773-0.00008-2
Horton, J. R., Pathuri, S., Wong, K., Ren, R., Rueda, L., Fosbenner, D. T., et al. (2022). Structural characterization of dicyanopyridine containing DNMT1-selective, non-nucleoside inhibitors. Structure 30, 793–802.e5. doi:10.1016/j.str.2022.03.009
Houston, D. R., and Walkinshaw, M. D. (2013). Consensus docking: improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 53 (2), 384–390. doi:10.1021/ci300399w
Hu, C., Liu, X., Zeng, Y., Liu, J., and Wu, F. (2021). DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin. Epigenet. 13, 166. doi:10.1186/s13148-021-01154-x
Hunter, J. D. (2007). Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9 (3), 90–95. doi:10.1109/MCSE.2007.55
Jakalian, A., Jack, D. B., and Bayly, C. I. (2002). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23 (16), 1623–1641. doi:10.1002/jcc.10128
Juárez-Mercado, K. E., Prieto-Martínez, F. D., Sánchez-Cruz, N., Peña-Castillo, A., Prada-Gracia, D., and Medina-Franco, J. L. (2020). Expanding the structural diversity of DNA methyltransferase inhibitors. Pharmaceuticals 14, 17. doi:10.3390/ph14010017
Kikuchi, A., Onoda, H., Yamaguchi, K., Kori, S., Matsuzawa, S., Chiba, Y., et al. (2022). Structural basis for activation of DNMT1. Nat. Commun. 13 (1), 7130. doi:10.1038/s41467-022-34779-4
Kirkpatrick, S., Gelatt, C. D., and Vecchi, M. P. (1983). Optimization by simulated annealing. Science 220, 671–680. doi:10.1126/science.220.4598.671
Kong, X., Chen, L., Jiao, L., Jiang, X., Lian, F., Lu, J., et al. (2014). Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of Polycomb Repressive Complex 2. J. Med. Chem. 57 (22), 9512–9521. doi:10.1021/jm501230c
Kurdyukov, S., and Bullock, M. (2016). DNA methylation analysis: choosing the right method. Biology 5, 3. doi:10.3390/biology5010003
Landrum, G., Tosco, P., Kelley, B., Cosgrove, D., Kawashima, E., Dalke, A., et al. (2023). Rdkit/rdkit: 2023_03_2 (Q1 2023) release. doi:10.5281/zenodo.8053810
Lephar Research (2023). Software. Available at: http://www.lephar.com/software.htm (Accessed July 12, 2023).
Li, T., Wang, L., Du, Y., Xie, S., Yang, X., Lian, F., et al. (2018). Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res. 46 (6), 3218–3231. doi:10.1093/nar/gky104
Lionta, E., Spyrou, G., Vassilatis, D. K., and Cournia, Z. (2014). Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 14 (16), 1923–1938. doi:10.2174/1568026614666140929124445
López-López, E., Fernández-de Gortari, E., and Medina-Franco, J. L. (2022). Yes SIR! On the structure-inactivity relationships in drug discovery. Drug Discov. Today 27 (8), 2353–2362. doi:10.1016/j.drudis.2022.05.005
López-López, E., and Medina-Franco, J. L. (2023). Towards decoding hepatotoxicity of approved drugs through navigation of multiverse and consensus chemical spaces. Biomolecules 13, 176. doi:10.3390/biom13010176
Maggiora, G. M. (2006). On outliers and activity cliffs--why QSAR often disappoints. J. Chem. Inf. Model. 46 (4), 1535. doi:10.1021/ci060117s
Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A. M., and Taranto, A. G. (2020). Structure-based virtual screening: from classical to artificial intelligence. Front. Chem. 8, 343. doi:10.3389/fchem.2020.00343
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11 (8), 3696–3713. doi:10.1021/acs.jctc.5b00255
MarvinSketch 22.18, Chemaxon (2023). MarvinSketch 22.18, Chemaxon. Available at: https://www.chemaxon.com (Accessed July, 2023).
Mauri, A. (2020). “alvaDesc: a tool to calculate and analyze molecular descriptors and fingerprints,” in Ecotoxicological QSARs. Editor K. Roy (New York: Springer), 801–820. doi:10.1007/978-1-0716-0150-1_32
Mauri, A., and Bertola, M. (2023). AlvaBuilder: a software for de novo molecular design. J. Chem. Inf. Model. doi:10.1021/acs.jcim.3c00610
Medina-Franco, J. (2016). Epi-informatics: discovery and development of small molecule epigenetic drugs and probes. Elsevier Science.
Medina-Franco, J. L. (2012). Scanning structure-activity relationships with structure-activity similarity and related maps: from consensus activity cliffs to selectivity switches. J. Chem. Inf. Model. 52 (10), 2485–2493. doi:10.1021/ci300362x
Medina-Franco, J. L., López-López, E., and Martínez-Fernández, L. P. (2022). 7-Aminoalkoxy-Quinazolines from epigenetic focused libraries are potent and selective inhibitors of DNA methyltransferase 1. Molecules 27, 2892. doi:10.3390/molecules27092892
Medina-Franco, J. L., Méndez-Lucio, O., Dueñas-González, A., and Yoo, J. (2015). Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today 20 (5), 569–577. doi:10.1016/j.drudis.2014.12.007
Mendez, D., Gaulton, A., Bento, A. P., Chambers, J., De Veij, M., Félix, E., et al. (2019). ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 47 (D1), D930–D940. doi:10.1093/nar/gky1075
Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada (2023). Molecular operating environment (MOE). Chemical computing group Inc.: Montreal, QC, Canada. Available at: http://www.chemcomp.com (Accessed March, 2023).
Morris, C. J., Stern, J. A., Stark, B., Christopherson, M., and Della Corte, D. (2022). MILCDock: machine learning enhanced consensus docking for virtual screening in drug discovery. J. Chem. Inf. Model. 62 (22), 5342–5350. doi:10.1021/acs.jcim.2c00705
Nocedal, J., and Wright, S. (2006). Numerical optimization. New York: Springer Science and Business Media.
O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: an open chemical toolbox. J. Cheminf. 3, 33. doi:10.1186/1758-2946-3-33
Pechalrieu, D., Etievant, C., and Arimondo, P. B. (2017). DNA methyltransferase inhibitors in cancer: from pharmacology to translational studies. Biochem. Pharmacol. 129 (1), 1–13. doi:10.1016/j.bcp.2016.12.004
Perez-Castillo, Y., Sotomayor-Burneo, S., Jimenes-Vargas, K., Gonzalez-Rodriguez, M., Cruz-Monteagudo, M., Armijos-Jaramillo, V., et al. (2019). CompScore: boosting structure-based virtual screening performance by incorporating docking scoring function components into consensus scoring. J. Chem. Inf. Model. 59 (9), 3655–3666. doi:10.1021/acs.jcim.9b00343
Pogribny, I. P., and Beland, F. A. (2009). DNA hypomethylation in the origin and pathogenesis of human diseases. Cell. Mol. Life Sci. 66 (14), 2249–2261. doi:10.1007/s00018-009-0015-5
Prado-Romero, D. L., and Medina-Franco, J. L. (2021). Advances in the exploration of the epigenetic relevant chemical space. ACS Omega 6 (35), 22478–22486. doi:10.1021/acsomega.1c03389
Reaction Biology Corporation (2023). Reaction Biology corporation. Available at: http://www.reactionbiology.com (Accessed July 08, 2023).
Rodríguez-Mejía, L. C., Romero-Estudillo, I., Rivillas-Acevedo, L. A., French-Pacheco, L., Silva-Martínez, G. A., Alvarado-Caudillo, Y., et al. (2022). The DNA methyltransferase inhibitor RG108 is converted to activator following conjugation with short peptides. Int. J. Pept. Res. Ther. 28 (3), 79. doi:10.1007/s10989-022-10390-5
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inf. Model. 50 (5), 742–754. doi:10.1021/ci100050t
Sánchez-Cruz, N., Pilón-Jiménez, B. A., and Medina-Franco, J. L. (2019). Functional group and diversity analysis of BIOFACQUIM: a Mexican natural product database. F1000Res. 8, Chem Inf Sci-2071. doi:10.12688/f1000research.21540.2
Sessions, Z., Sánchez-Cruz, N., Prieto-Martínez, F. D., Alves, V. M., Santos, H. P., Muratov, E., et al. (2020). Recent progress on cheminformatics approaches to epigenetic drug discovery. Drug Discov. Today 25 (12), 2268–2276. doi:10.1016/j.drudis.2020.09.021
Song, J., Rechkoblit, O., Bestor, T. H., and Patel, D. J. (2011). Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–1040. doi:10.1126/science.1195380
Stresemann, C., and Lyko, F. (2008). Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer. 123 (1), 8–13. doi:10.1002/ijc.23607
Sun, J., Jeliazkova, N., Chupakin, V., Golib-Dzib, J.-F., Engkvist, O., Carlsson, L., et al. (2017). ExCAPE-DB: an integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminf. 9, 17. doi:10.1186/s13321-017-0203-5
Syeda, F., Fagan, R. L., Wean, M., Avvakumov, G. V., Walker, J. R., Xue, S., et al. (2011). The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J. Biol. Chem. 286 (17), 15344–15351. doi:10.1074/jbc.M110.209882
Triches, F., Triches, F., and Lino de Oliveira, C. (2022). Consensus combining outcomes of multiple ensemble dockings: examples using dDAT crystalized complexes. MethodsX 9, 101788. doi:10.1016/j.mex.2022.101788
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Vilar, S., Cozza, G., and Moro, S. (2008). Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 8 (18), 1555–1572. doi:10.2174/156802608786786624
Virtanen, P., Gommers, R., Oliphant, T. E., Haberland, M., Reddy, T., Cournapeau, D., et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 17 (3), 261–272. doi:10.1038/s41592-019-0686-2
Wang, R., Lai, L., and Wang, S. (2002). Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput.-Aided Mol. Des. 16 (1), 11–26. doi:10.1023/a:1016357811882
Wang, R., and Wang, S. (2001). How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 41 (5), 1422–1426. doi:10.1021/ci010025x
Waskom, M. (2021). Seaborn: statistical data visualization. J. Open Source Softw. 6, 3021. doi:10.21105/joss.03021
Wilson, A. S., Power, B. E., and Molloy, P. L. (2007). DNA hypomethylation and human diseases. Biochim. Biophys. Acta. 1775 (1), 138–162. doi:10.1016/j.bbcan.2006.08.007
Yu, J., Chai, X., Pang, J., Wang, Z., Zhao, H., Xie, T., et al. (2022). Discovery of novel non-nucleoside inhibitors with high potency and selectivity for DNA methyltransferase 3A. Eur. J. Med. Chem. 242, 114646. doi:10.1016/j.ejmech.2022.114646
Yu, J., Xie, T., Wang, Z., Wang, X., Zeng, S., Kang, Y., et al. (2019). DNA methyltransferases: emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug Discov. Today 24 (12), 2323–2331. doi:10.1016/j.drudis.2019.08.006
Zhang, Z., Wang, G., Li, Y., Lei, D., Xiang, J., Ouyang, L., et al. (2022). Recent progress in DNA methyltransferase inhibitors as anticancer agents. Front. Pharmacol. 13, 1072651. doi:10.3389/fphar.2022.1072651
Keywords: consensus scoring, data fusion, De novo design, DNMT, molecular docking, drug discovery, epigenetics
Citation: Prado-Romero DL, Gómez-García A, Cedillo-González R, Villegas-Quintero H, Avellaneda-Tamayo JF, López-López E, Saldívar-González FI, Chávez-Hernández AL and Medina-Franco JL (2023) Consensus docking aid to model the activity of an inhibitor of DNA methyltransferase 1 inspired by de novo design. Front. Drug Discov. 3:1261094. doi: 10.3389/fddsv.2023.1261094
Received: 18 July 2023; Accepted: 30 November 2023;
Published: 11 December 2023.
Edited by:
Yudibeth Sixto-López, University of Granada, SpainReviewed by:
Tigran Abramyan, Atomwise Inc., United StatesConrad Veranso Simoben, University of Buea, Cameroon
Copyright © 2023 Prado-Romero, Gómez-García, Cedillo-González, Villegas-Quintero, Avellaneda-Tamayo, López-López, Saldívar-González, Chávez-Hernández and Medina-Franco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José L. Medina-Franco, medinajl@unam.mx