Rapid screening of chemicals for their potential to cause specific toxidromes
 Ruifeng Liu1,2
Ruifeng Liu1,2  Mohamed Diwan M. AbdulHameed1,2*
Mohamed Diwan M. AbdulHameed1,2*  Zhen Xu1,2 Benjamin Clancy1,2 Valmik Desai1,2
Zhen Xu1,2 Benjamin Clancy1,2 Valmik Desai1,2  Anders Wallqvist1*
Anders Wallqvist1*- 1Department of Defense Biotechnology High Performance Computing Software Applications Institute, Telemedicine and Advanced Technology Research Center, U.S. Army Medical Research and Development Command, Frederick, MD, United States
- 2The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., Bethesda, MD, United States
Toxidromes constitute patterns of symptoms and signs caused by specific toxic effects that guide emergency treatments. Computational identification of chemicals that cause different toxidromes allows us to rapidly screen novel compounds and compound classes as to their potential toxicity. The aim of the current study was to create a computational toolset that can map chemicals to their potential toxidromes. Hence, we evaluated the performance of a state-of-the-art deep learning method—the recently developed communicative message passing neural network (CMPNN)—for its ability to overcome the use of small datasets for training deep learning models. Our results indicated that multi-task training—a technique known for its ability to use multiple small datasets to train conventional deep neural networks—works equally well with CMPNN. We also showed that CMPNN-based ensemble learning results in more reliable predictions than those obtained using a single CMPNN model. In addition, we showed that the standard deviations of individual model predictions from an ensemble of CMPNN models correlated with the errors of ensemble predictions and could be used to estimate the reliability of ensemble predictions. For toxidromes that do not have well-defined molecular mechanisms or sufficient data to train a deep learning model, we used the similarity ensemble approach to develop molecular structural similarity-based toxidrome models. We made the toolset developed in this study publicly accessible via a web user interface at https://toxidrome.bhsai.org/.
Introduction
Rapid identification of adverse health effects of chemicals is important for identifying and classifying their potential toxicity. Experimentally testing and classifying thousands of commercial product components, drugs and drug candidates, as well as newly developed synthetic chemicals for their potential to cause health hazards is not feasible, triggering the development of multiple alternative approaches based on high-throughput-assay data, chemoinformatics, and computational methods (Vinken, 2013; Allen et al., 2014; Oki et al., 2016; Liu et al., 2017; Schyman et al., 2017; Liu et al., 2018b; Strickland et al., 2018; Wang et al., 2019; AbdulHameed et al., 2021; Liu et al., 2022).
The concept of toxidromes has been proposed to focus on acute intoxication signs and symptoms as a means to quickly identify the underlying compound class and guide treatments. Toxidromes are a set of adverse health signs and symptoms that are caused by specific classes of chemicals eliciting similar identifiable sets of signs and symptoms (Holstege and Borek, 2012). Figure 1 shows nine major toxidromes defined in the U.S. Department of Homeland Security’s “Report on the Toxic Chemical Syndrome. Definitions and Nomenclature Workshop” (2012). Table 1 summarizes the known molecular mechanisms for five toxidromes that are traceable to chemical interactions with specific receptor-chemical interactions, e.g., the opioid toxidrome is due to chemical activation of the mu, delta, and/or kappa opioid receptors (Strickler et al., 2018). Similarly, the cholinergic and anticholinergic toxidromes are due to increased and decreased acetylcholine activity because of activation or inhibition of muscarinic and nicotinic receptors and acetylcholinesterase, respectively (Lott and Jones, 2023). All the receptors involved in these toxidromes are drug targets, and the activities of many chemicals at these targets have been experimentally evaluated. The last four toxidromes listed in Table 1 have molecular mechanisms that are less well defined and/or pose an additional challenge as the number of chemicals investigated for their potential to cause these toxidromes is too small for training robust machine learning models.
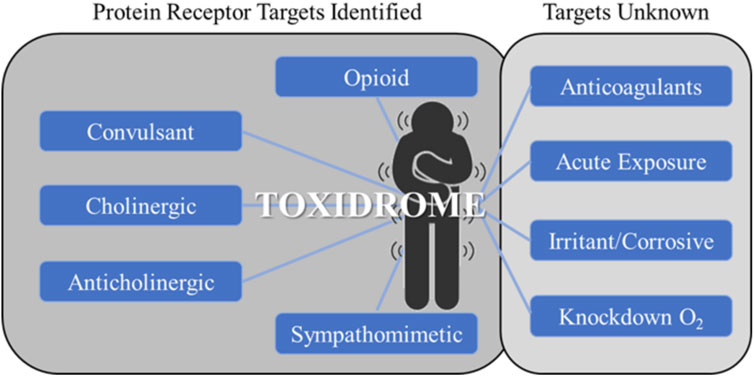
FIGURE 1. Toxidromes listed in the 2012 U.S. Department of Homeland Security workshop on toxic chemical syndromes (Oki et al., 2016). The toxidromes on the left have well-known molecular mechanisms through protein-receptor interactions. These are typically known drug targets with a relatively large amount of available experimentally determined data. The toxidromes on the right either do not have well-defined molecular mechanisms or lack sufficient experimental data for deep learning training.
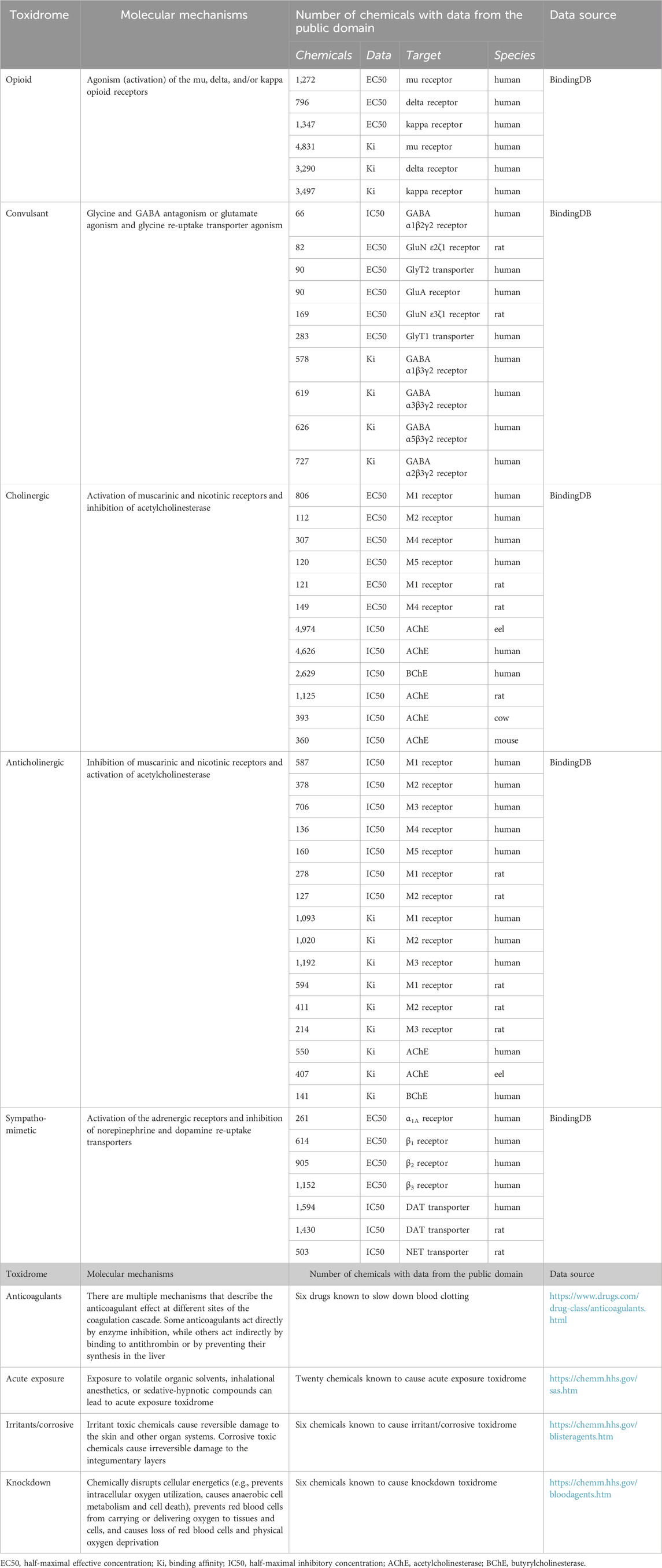
TABLE 1. Summary of molecular mechanisms, protein targets, and number of chemicals with relevant experimental data collected from the public domain.
Here, we developed computational tools for rapid screening of chemicals with potential to cause these toxidromes. We created the tools based on recently developed communicative message passing neural networks (CMPNNs) (Song et al., 2020) for the toxidromes with well-known molecular mechanisms and using the similarity ensemble approach (SEA) (Keiser et al., 2007) for the other toxidromes. To deploy the toolset developed in this study for public access, we created a web user interface at https://toxidrome.bhsai.org.
Materials and methods
Molecular activity data
We downloaded relevant molecular binding and functional activity data as described in Table 1 from BindingDB—a public, web-accessible database of measured binding affinities and functional activities (Gilson et al., 2016). The data in BindingDB were extracted by the BindingDB project from the literature and from patents, selected PubChem confirmatory BioAssays, and ChEMBL entries for which a well-defined protein target is provided. For each of the protein targets listed in Table 1, we retrieved compounds with relevant functional activities in all organisms and removed duplicate entries and entries with uncertain numerical activity values, such as those larger or smaller than a threshold value. The relevant functional activities include the half-maximal effective concentrations (EC50) or half-maximal inhibitory concentrations (IC50), expressed as moles/L (M). We faced a major challenge in that the number of chemicals with relevant functional activity data for a specific protein receptor of a specific organism may be too low to train a robust deep learning model. Thus, we also retrieved additional large chemical binding-affinity datasets and used them as auxiliary datasets for multi-task training to help develop more robust CMPNN models with the limited functional activity data. Table 1 summarizes the number of chemicals with functional and binding activity data for each protein target and organism. We converted EC50, IC50, and Ki (binding affinity) data into pEC50, pIC50, and pKi before using them to train deep learning models. All the raw data used for model development are provided in Supplementary Table S1 (Toxidrome Data. xlsx) and at https://github.com/BHSAI/Toxidrome.
Methods
For predicting molecular bioactivities, the directed message passing neural network (DMPNN) has been shown to outperform most other DNNs (Yang et al., 2019). Recently, the communicative message passing enhancement to the DMPNN method was shown to improve prediction results, establishing it as a powerful deep learning method for mining molecular bioactivity data (Song et al., 2020). In this study, we chose to use this state-of-the-art CMPNN method to develop quantitative structure-activity relationship models for rapid screening of chemicals for their potential to cause toxidromes.
Table 1 shows that for many toxidrome-relevant protein targets, the number of chemicals with experimentally determined functional activity data is small. To develop robust models with a small number of training data and large number of DNN parameters, we evaluated and applied multi-task training and ensemble learning techniques. Figure 2A shows conventional single-task training where all model parameters are optimized by a single dataset, and Figure 2B shows multi-task training where a larger number of model parameters are shared by all the tasks. In the latter case, the models are trained by all the datasets, equivalent to being trained on a combined larger training set. Studies using conventional feed-forward DNNs have shown that this multi-task training technique leads to more robust models, especially for tasks with limited data (Ramsundar et al., 2017). In this study, we first evaluated the effectiveness of this approach using the CMPNN method. We then applied this technique using large chemical binding-affinity datasets as auxiliary datasets to develop functional activity prediction models.
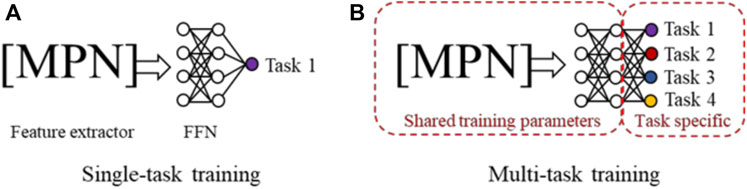
FIGURE 2. (A) In single-task training, all model parameters in both the message passing network (MPN) and the feed-forward network (FFN) are trained by the data from a single task. (B) In multi-task training, a portion of the network and parameters are shared and trained using the datasets from all tasks.
DNNs employ a large number of model parameters and a stochastic training algorithm. As a result, we end up with a different set of model parameters each time the networks are trained. The ensemble learning technique trains an ensemble of models and combines individual model predictions to generate an ensemble prediction. The simplest way to do this is to take the average of all the individual model predictions. In principle, ensemble predictions are better in the sense that they reduce the variance in individual model predictions. Additionally, as observed in previous DNN studies with other deep learning methods, ensemble predictions may be better than any individual model prediction (Hansen and Salamon, 1990).
In this study, we evaluated the performance of CMPNN-based ensemble learning and applied it to develop robust toxidrome models. First, we evaluated the performance of single-task CMPNN and ensemble models. For this, we used the mu receptor binding-affinity data with nearly 4,831 chemicals—the largest dataset in this study—and the kappa receptor activation data with 1,347 chemicals. We randomly split each dataset into a 60% training set, a 20% validation set, and a 20% test set. We used the training and validation sets to train an ensemble of single-task CMPNN models, with the number of models ranging from 1 to 15. Each training ran 30 epochs, and the model parameters of the epoch that gave the lowest root mean squared error (RMSE) of the validation set were restored as the final trained model. We then calculated the RMSE of the test sets using the ensemble models. Next, we compared the single-task and multi-task CMPNN models. For this, we first randomly split the datasets of multiple tasks of the same toxidrome into 60% for training, 20% for validation, and 20% for testing. We used the training and validation sets to train a multi-task model and calculated the RMSE of the test sets using the multi-task models. We then trained the corresponding single-task models using the same training and validation sets and calculated the RMSE of the test sets of the specific tasks. For these computations, we used ensemble learning as described in the previous section.
Multiple ways exist to estimate prediction accuracies (Liu et al., 2018a; Liu et al., 2019; Liu and Wallqvist, 2019), but previous studies have shown that the standard deviation of individual model predictions is correlated with the reliability of an ensemble model prediction (Tetko et al., 2008). In this study, we evaluated whether in the CMPNN method, the standard deviation of the individual model prediction also correlated with the prediction error. For this, we ran a 5-fold cross validation for a multi-task model trained with the opioid datasets (three opioid receptor subtype activation datasets and three opioid receptor subtype binding-affinity datasets). In each step of the 5-fold cross validation, we reserved 20% of the molecules in the datasets as test molecules and made predictions for these test molecules using models trained with data from the rest of the molecules. In the end, we made ∼15,000 predictions of the receptor subtype activation and binding affinities. We also calculated the standard deviations of the ensemble-model predictions and the mean absolute errors of the ensemble predictions. We binned the prediction errors using a 0.05 standard deviation interval and calculated an average of the prediction errors in each bin. If we assume the midpoint of each bin as the mean standard deviation of the bin, we used linear regression to derive an equation relating the standard deviation (σ) and the mean absolute error of prediction:
We used Eq. 1 to estimate the mean prediction errors of our ensemble predictions in this study.
We used the default hyperparameters in the CMPNN code as they have been shown to be reasonable hyperparameters for most molecular datasets (Song et al., 2020). We did not perform a hyperparameter search for two reasons: 1) many of the individual molecular activity datasets are too small for a reliable hyperparameter search using a single dataset, and 2) our strategy to train networks using small datasets is to use multi-task training, in which a large part of the network architecture and most of the model parameters are shared across all the tasks. An optimal set of hyperparameters for one dataset may be inferior for another dataset, which renders an individual dataset-based hyperparameter search futile for multi-task learning.
SEA for toxidromes without well-defined molecular mechanisms or with an insufficient number of chemicals for deep learning
For the toxidromes that do not have well-defined molecular mechanisms or have too few chemicals with experimental data, it is challenging to develop machine-learning models based on assay data alone. Therefore, we used the SEA to develop a computational tool for rapid screening of chemicals for these toxidromes. The SEA was developed to assess the relatedness between two drug receptors A and B based on an overall molecular structural similarity score between all the ligands of the two receptors (Keiser et al., 2007; Schyman et al., 2016). It is based on the Tanimoto similarity approach (TSA), but unlike TSA that assesses the similarity between two molecules only, SEA assesses the overall similarity between two groups of molecules. In the application of SEA in this study, we based our predictions on the overall similarity between a molecule and a group of molecules known to cause a toxidrome (see Eqs 2–4). Briefly, the raw similarity score between the query molecule and the set of compounds (m) known to cause a toxidrome is calculated using a raw similarity score RS using Tanimoto similarity (TS) as
where TS0 is a minimum threshold similarity set to 0.57. The z-score for a query molecule is calculated as
where μ(m) = 4.24·10−4 m and σ(m) = 4.49·10−3 m0.665 and converted into a p-value as
where Г’(1) is 0.577215665.
To assess the feasibility and compare the performance of SEA versus TSA for our purpose, we used the St. Jude malaria high-throughput screening (HTS) dataset (Guiguemde et al., 2010), which contains a library of 305 K screened compounds with 1,524 active compounds identified. In our evaluation of the TSA and SEA methods, we first randomly selected 153 (10%) of the active compounds as “known” actives and embedded the remaining actives (1,371) in the rest of the 305 K compounds. Next, we ranked the 305 K compounds based on their highest Tanimoto similarity value to the “known” actives (TSA method) and on their lowest p-values (SEA method). We then checked the percentages of the remaining actives in increasing fractions of the highest ranked screening library with both methods and calculated the active retrieval efficiency as defined by Eq. 5:
Finally, to test the resilience of the methods to false actives, we randomly picked 45 inactive compounds from the screening library and combined them with the 153 “known” actives as false positives and repeated the TSA and SEA calculations.
Toxidrome web application
The toxidrome web application runs on an Apache Tomcat server and utilizes a three-tiered architecture consisting of a front-end, a database, and a controller. For the front-end, we used Flutter (https://docs.flutter.dev/) to provide user input, information, and graphics. The database is a standard PostgreSQL relational database that stores compounds provided by the user as well as all calculated models results. This information is stored for 2 weeks, at which point it is deleted from the database. None of the stored information is used to train new models or collect user data. The Java-based controller works to pass the information between the front-end and the database. The controller is also responsible for executing a Python-based command-line tool that runs both the CMPNN model and the SEA for the toxidromes.
Software availability
We trained and validated the CMPNN models using the Python code that is freely available for download on the GitHub website (https://github.com/SY575/CMPNN). To calculate the standard deviations for an ensemble of CMPNN model predictions, which are needed for estimating the mean absolute prediction errors, we modified a function of the Python code named make_predictions.py in the/chemprop/folder in the installation directory. Our modified make_predictions.py code is given in the Supplementary Material for anyone interested in reproducing our studies. All the models developed in this work as well as the raw data and codes used for the command-line tool are available at https://github.com/BHSAI/Toxidrome. We developed an easy-to-use, publicly accessible, web user interface that allows users to submit their query compounds and make toxidrome predictions using our models (available at https://toxidrome.bhsai.org/).
Results
The primary goal of this work was to develop a tool to predict the potential of a chemical to cause specific toxidromes. For this purpose, we chose to use the recently reported CMPNN approach for molecular property prediction. Song et al. (2020), the developers of this approach, performed a comparative analysis with other methods and reported that CMPNN performed better than the other methods.
Performance of a CMPNN single model versus an ensemble of models
Previous studies using DNN methods have shown that ensemble-model predictions perform better than single-model predictions (Hansen and Salamon, 1990). We assessed if this was also true with CMPNNs using the mu receptor binding-affinity data—the largest dataset in this study—and the kappa receptor activation data. Figure 3 shows the plots of RMSE versus the number of models in the ensemble models, indicating that the RMSE of a single model was the highest. With an increasing number of models, the RMSE decreased and fluctuated in a narrow range when the number of models approached 10 or more. Based on this observation, we used ensemble models incorporating 10 models in the rest of our study.
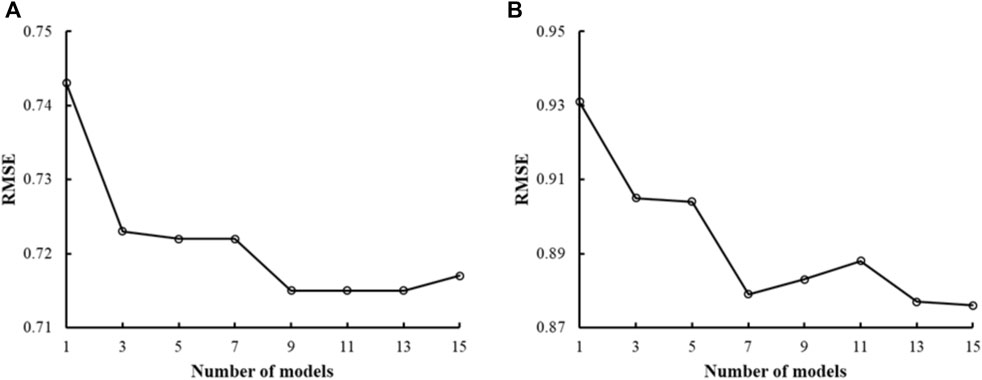
FIGURE 3. Plots of the root mean squared error (RMSE) of ensemble predictions vs. the number of models in the ensemble. (A) Results from human mu receptor binding affinity models. (B) Results from human kappa receptor activation models. Both plots show that ensemble learning (number of models >1) performs better than single-model learning (number of models = 1). We chose to use 10 models in our ensemble learning as the plots show that the RMSE decreased and fluctuated in a narrow range when the number of models approached 10 or more.
Single-task versus multi-task training
Using the classic multi-layer perceptron (fully connected feed-forward DNNs), Sadawi et al. (2019) have shown that multi-task training results in better models than single-task models in most cases, especially for tasks with limited training data. Thus, we explored whether multi-task training would be a viable approach for us to mitigate against small datasets for training CMPNN models. Before adopting this approach, we assessed the performance of CMPNN-based single- and multi-task training. Table 2 summarizes the results of our evaluation using the opioid toxidrome datasets, and Table 3 summarizes the results using the convulsant toxidrome datasets. The results using each of the datasets showed that the RMSEs of the multi-task models were smaller than or similar to those of the single-task models (22% and 4% RMSE reduction for the convulsant and opioid datasets, respectively). In addition, for tasks with a small number of training compounds (<100), the RMSEs of the multi-task models were significantly smaller than those of the single-task models (34% and 13% RMSE reduction for the convulsant datasets). However, the multi-task RMSEs of the binding-affinity datasets, which contain significantly more compounds, were relatively closer to those of the corresponding single-task models (10% versus 20% closer). As we based the toxidrome prediction on functional potencies of the chemicals, we included the binding-affinity data as auxiliary datasets in the multi-task training to create robust functional activity models.
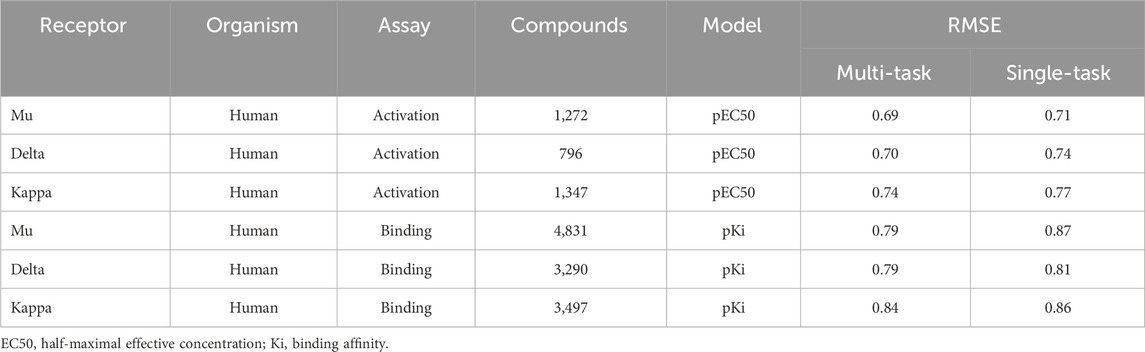
TABLE 2. Root mean squared errors (RMSE) of single- and multi-task ensemble communicative message passing neural network (CMPNN) predictions for the opioid datasets, showing improved (lower RMSE) performance of multi-task training.
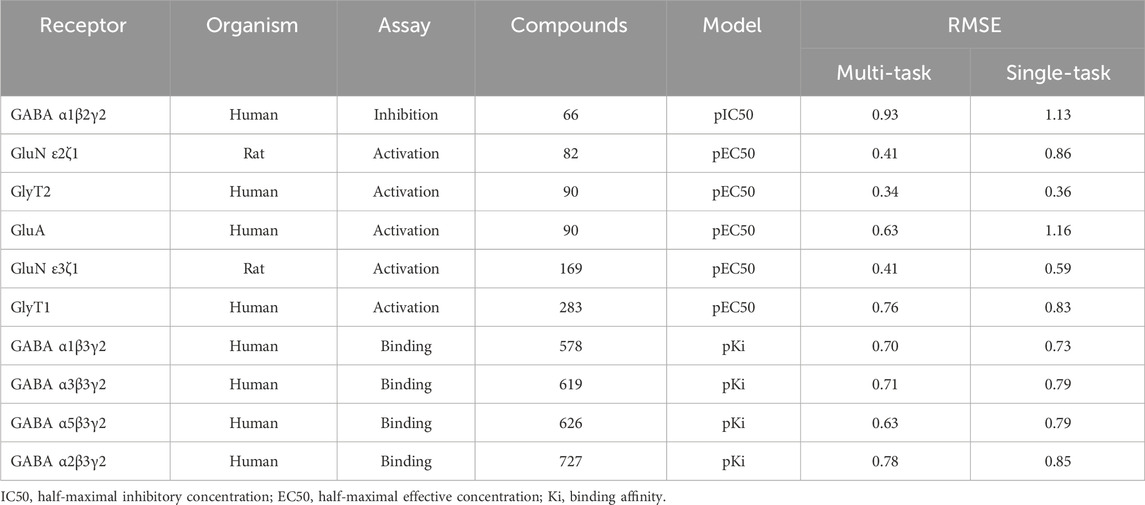
TABLE 3. Root mean squared errors (RMSE) of single-task and multi-task ensemble communicative message passing neural network (CMPNN) predictions for the convulsant toxidrome datasets, showing improved (lower RMSE) performance of multi-task training.
Estimating prediction reliability
Previous studies using associated artificial neural networks have reported that the standard deviation of all individual model predictions from an ensemble of models correlates with the prediction error, therefore, the magnitude of the standard deviation can serve as a domain applicability measure (Tetko et al., 2008). In this study, we observed that with the CMPNN method, the standard deviation of the individual model prediction also correlated with the prediction error. Table 4 and Figure 4 show a correlation between the standard deviation and the mean absolute error of predictions: the larger the standard deviation, the larger the mean prediction error.
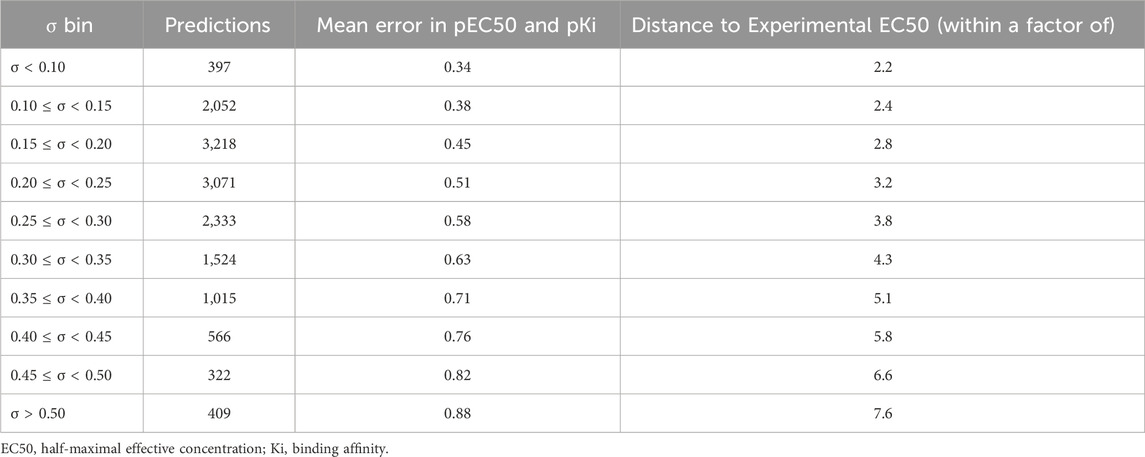
TABLE 4. Number of predictions in each standard deviation (σ) bin and the mean absolute errors of multi-task ensemble communicative message passing neural network (CMPNN) models for the opioid toxidrome.
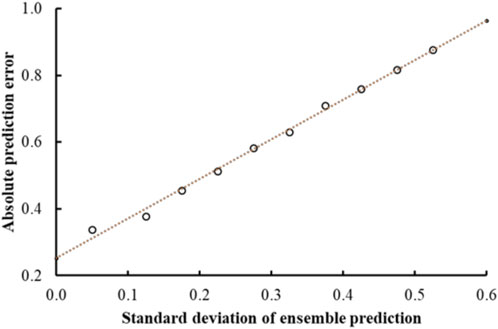
FIGURE 4. Mean absolute error of predictions as a function of the standard deviation of the ensemble prediction. The graph shows a clear linear correlation between the error and the standard deviations of the ensemble predictions that is used to make estimates of the prediction accuracies.
Distance to model approaches that use the standard deviation of an ensemble of classifiers are widely used to define applicability domain (Tetko et al., 2008; Mathea et al., 2016). Here, we used the standard deviation of predictions given by an ensemble of models as an applicability domain measure. In the toxidrome web tool, any prediction with a standard deviation less than one is considered to be within the applicability domain of the model. In the similarity ensemble models, any prediction with a p-value less than 0.05 is considered to be within the applicability domain.
Use of the similarity ensemble approach
The TSA is commonly used in selecting compounds from a large compound library for bioactivity testing. Starting with compounds with a desirable activity at a drug target, the TSA selects other compounds with a high molecular structural similarity and considers them active based on their similar structure using the similar activity principle. However, the TSA has a potential caveat, i.e., it is based on individual Tanimoto similarity between a library compound and an active compound, not the overall similarity between a library compound and all active compounds. The SEA is based on the overall similarity between two sets of compounds and, therefore, should perform better than the TSA as there could be false positives in any active compound set. We compared SEA and TSA approaches using the St. Jude malaria HTS dataset.
Figure 5 shows the results of this computational experiment, illustrating that if a large fraction (>5%) of the compound library was screened, the performances of the TSA and SEA were essentially the same. However, when a very small fraction of the huge library (>300 K compounds) was screened, SEA performed significantly better than TSA, especially when there were false actives in the “known” active set of compounds. Contrary to TSA, which is very sensitive to false actives, SEA is surprisingly insensitive to random false actives because structurally dissimilar false actives do not change SEA p-values. Our results indicate that SEA performed better than TSA for molecular structural similarity-based active retrieval.

FIGURE 5. Active-compound retrieval efficiencies determined using the Tanimoto similarity approach (TSA) and the similarity ensemble approach (SEA), with and without false positives (noise) among the “known” actives. The active retrieval efficiency is defined as the fraction of actives retrieved (number of actives retrieved/total actives) divided by the fraction of the library screened (number of chemicals screened/total number of chemicals in the library). Because the fraction of the library screened is small, and both TSA and SEA enrich actives among the top-ranked samples, the active retrieval efficiency can exceed 100%. To generate the data, we used 153 (10%) compounds randomly chosen from the actives identified in the St. Jude malaria high-throughput screening campaign as “known” actives, with and without 45 randomly selected inactive compounds as false positives (30% noise). The remainder of the actives (90%) were embedded into the screening library of ∼305 K compounds. We used the TSA and SEA methods to rank the compounds in the screening library and evaluated the active retrieval efficiencies in increasing fractions of the ranked screening library. The data show that when a small fraction of the library is screened, the performance of SEA is better than for TSA, with or without randomly selected false positives (noise).
Web interface to the toxidrome screening tools
The aim of our study was to create a publicly accessible computational toolset for rapid screening of chemicals with potential to cause toxidromes. Hence, we created a web-based user interface that allows access to the deep learning models and SEA-based screening tools as a means to rapidly identify chemicals with toxidrome-causing potential.
Figure 6 shows the login page where the user is given the option of creating a new account, logging in to an existing account, or logging in to a guest account. Once logged in, the user will be brought to a home page (Figure 7), where they can create a new job or review the results of a previously submitted job. If submitting a new job, the user will be able to give the job a name and description as well as input a list of compounds in the simplified molecular-input line-entry system (SMILES) format. The SMILES can be pasted directly into the browser or uploaded via a csv file. The user can also draw molecular structures as input to the application using the Marvin chemical drawing tool (Chemaxon, Boston, MA). After a job is submitted, it will be queued for processing, and once the results are ready, the user can view them by clicking the “View Results” button beside the job. The results can be viewed and downloaded for 2 weeks, after which they will be removed from the database.
FIGURE 6. Login page for the web-based user interface (https://toxidrome.bhsai.org/). The system supports registration of users as well as limited guest accounts to explore the system.
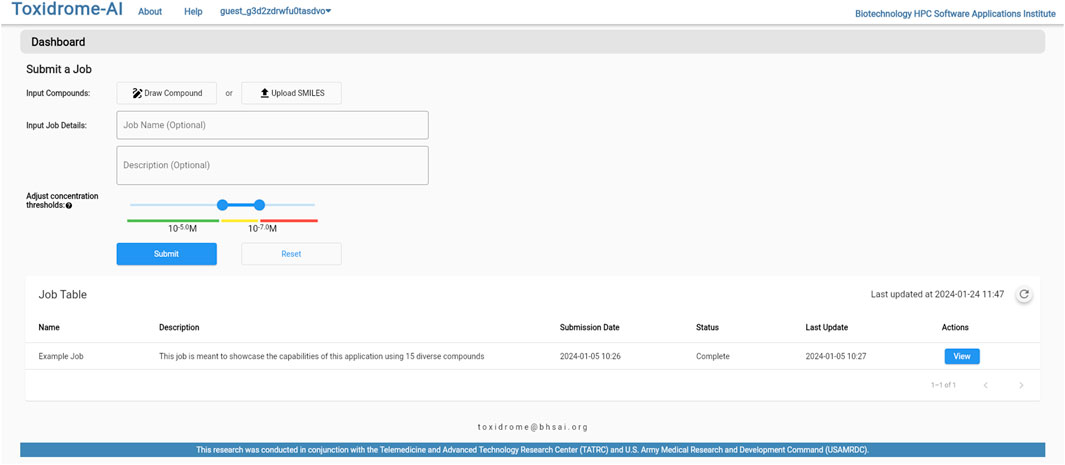
FIGURE 7. Job submission control page. Compounds can be uploaded from cvs-separated lists of SMILES or drawn using a chemical drawing tool to generate any desired compound structure.
Clicking the “View Results” button will bring the user to the results page (Figure 8), which gives an overview of each compound and its toxidrome-causing potential. To visualize the potential of a chemical to cause a toxidrome, we first determined a 60% and 80% potency threshold from the distribution of experimental potency values. Then, when a chemical is predicted to be more potent than the 80% threshold, it is considered highly likely (color red) to cause the toxidrome; between the 60% and the 80% thresholds, it is predicted to be likely (color yellow); and for values below that it is predicted to be unlikely (color green). For the SEA prediction, we call a toxidrome if the p-value is less than 0.05 and color the corresponding cell red, otherwise, we color it green.
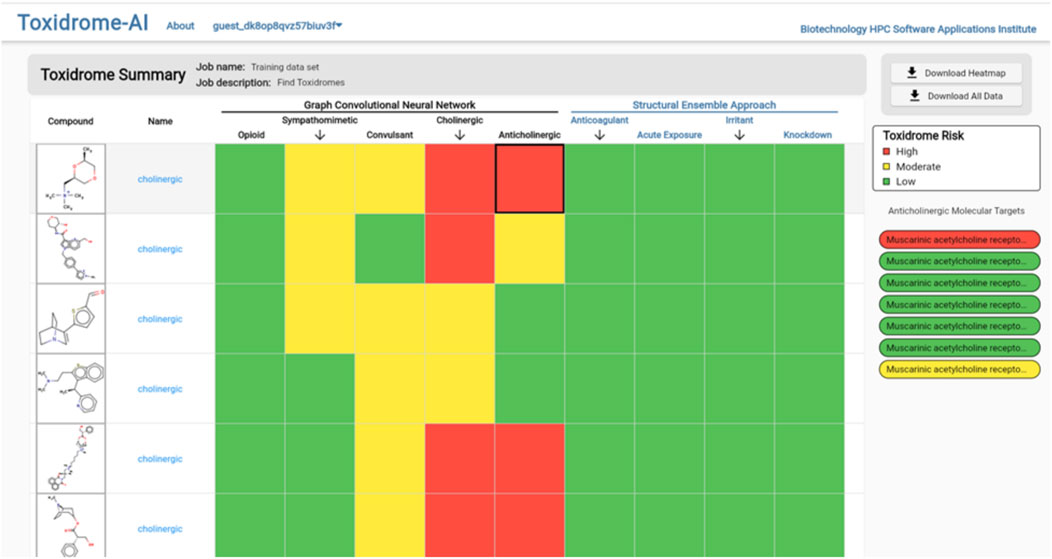
FIGURE 8. Toxidrome overall results page for a series of compounds displaying input structures and names and color-coded assessments of the likelihood of each chemical to cause a particular toxidrome. The results can be downloaded and stored by the user.
To view the individual values calculated by the models, the user can either hover over a cell in the table (which will display values in a column on the right), download the complete set of data in csv format via the button at the top of the page, or click on the compound they wish to view, which will bring them to the compound page (Figure 9). The compound page displays an image of the structure, a rose plot summarizing toxidrome predictions, and a table listing the predicted values and estimated prediction errors associated with each receptor.
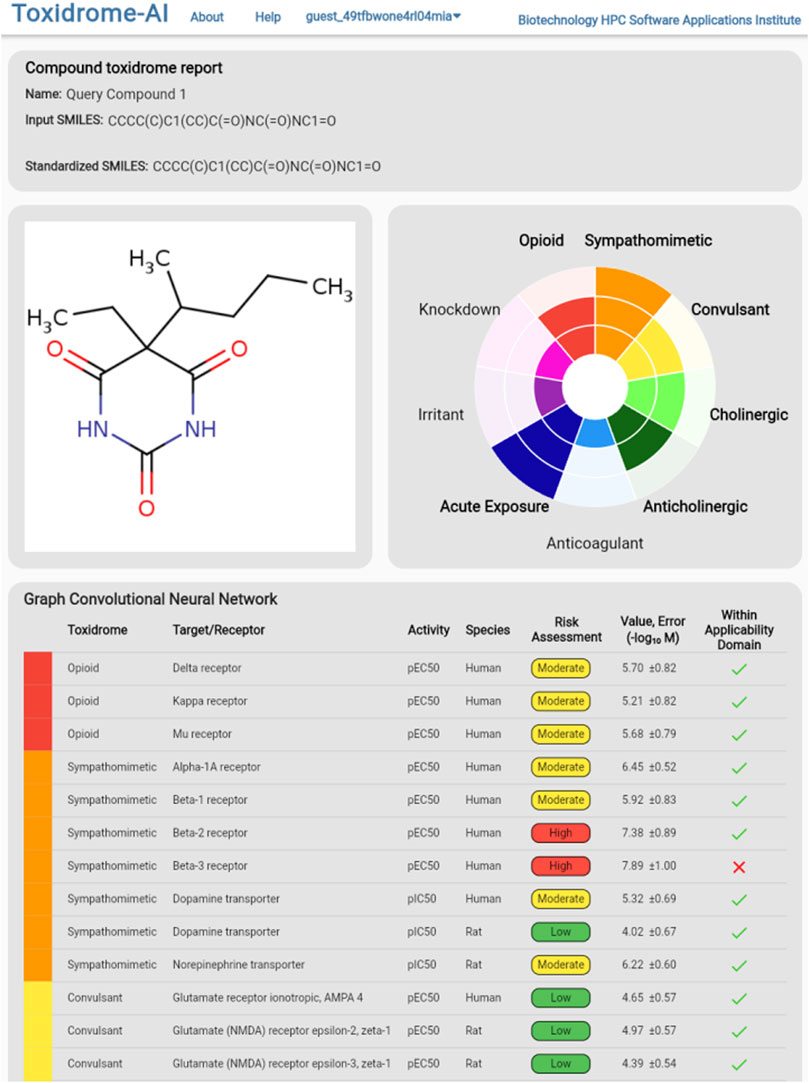
FIGURE 9. Detailed model break-down results for an individual chemical displaying each underlying target/receptor, activity types, species, and estimated risks based on the predicted values. An estimated 95% error is also shown.
As the CMPNN code was written to run on graphics processing units (GPUs) and at times GPU resource may be limited, we also created corresponding DMPNN models. Thus, depending on the GPU loads, some of the outputs may be based on DMPNN model predictions.
Summary
Our objective of this study was to create a publicly accessible computational toolset for rapid in silico identification of chemicals with potential to cause toxidromes. Toward this aim, we evaluated the performance of the current state-of-the-art deep learning method—the CMPNN—for its ability to help mitigate the challenge of small datasets for deep learning. Our results indicated that multi-task training, a technique with potential to mitigate the small dataset challenge using conventional DNNs, worked equally well with the recently developed CMPNN. We also showed that CMPNN-based ensemble learning resulted in more reliable predictions than those given by any single CMPNN model. In addition, we showed that the standard deviation of individual model predictions from an ensemble of CMPNN models correlated with the error of ensemble model predictions and can be used to estimate the reliability of ensemble predictions. For toxidromes that do not have well-defined molecular mechanisms or sufficient data to train deep learning models, we used the similarity ensemble approach instead of the common Tanimoto similarity approach to develop computational tools for rapid identification of chemicals with potential to cause these toxidromes. We made these resources publicly available via a web-user interface to facilitate future studies on chemical toxicity.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
RL: Conceptualization, Writing–review and editing, Formal Analysis, Writing–original draft, Investigation, Methodology. MA: Conceptualization, Software, Writing–review and editing, Investigation, Project administration, Visualization. ZX: Data curation, Software, Writing–review and editing, Visualization. BC: Data curation, Software, Writing–review and editing, Visualization. VD: Data curation, Software, Writing–review and editing, Visualization. AW: Conceptualization, Supervision, Writing–review and editing, Funding acquisition, Project administration, Visualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the U.S. Army Medical Research and Development Command under Contract No. W81XWH20C0031 and by Defense Threat Reduction Agency Grant CBCall14-CBS-05-2-0007.
Conflict of interest
Authors RL, MA, ZX, BC, and VD were employed by The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the United States (U.S.) Army, the U.S. Department of Defense, or The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. Distribution Statement A. Approved for public release: Distribution is unlimited.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2024.1324564/full#supplementary-material
References
AbdulHameed, M. D. M., Liu, R., Schyman, P., Sachs, D., Xu, Z., Desai, V., et al. (2021). ToxProfiler: toxicity-target profiler based on chemical similarity. Comput. Toxicol. 18, 100162. doi:10.1016/j.comtox.2021.100162
Allen, T. E., Goodman, J. M., Gutsell, S., and Russell, P. J. (2014). Defining molecular initiating events in the adverse outcome pathway framework for risk assessment. Chem. Res. Toxicol. 27, 2100–2112. doi:10.1021/tx500345j
Gilson, M. K., Liu, T., Baitaluk, M., Nicola, G., Hwang, L., and Chong, J. (2016). BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44, D1045–D1053. doi:10.1093/nar/gkv1072
Guiguemde, W. A., Shelat, A. A., Bouck, D., Duffy, S., Crowther, G. J., Davis, P. H., et al. (2010). Chemical genetics of Plasmodium falciparum. Nature 465, 311–315. doi:10.1038/nature09099
Hansen, L. K., and Salamon, P. (1990). Neural network ensembles. IEEE Trans. Pattern Anal. Mach. Intell. 12, 993–1001. doi:10.1109/34.58871
Holstege, C. P., and Borek, H. A. (2012). Toxidromes. Crit. Care Clin. 28, 479–498. doi:10.1016/j.ccc.2012.07.008
Keiser, M. J., Roth, B. L., Armbruster, B. N., Ernsberger, P., Irwin, J. J., and Shoichet, B. K. (2007). Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206. doi:10.1038/nbt1284
Liu, R., AbdulHameed, M. D. M., Kumar, K., Yu, X., Wallqvist, A., and Reifman, J. (2017). Data-driven prediction of adverse drug reactions induced by drug-drug interactions. BMC Pharmacol. Toxicol. 18, 44. doi:10.1186/s40360-017-0153-6
Liu, R., Glover, K. P., Feasel, M. G., and Wallqvist, A. (2018a). General approach to estimate error bars for quantitative structure-activity relationship predictions of molecular activity. J. Chem. Inf. Model 58, 1561–1575. doi:10.1021/acs.jcim.8b00114
Liu, R., Laxminarayan, S., Reifman, J., and Wallqvist, A. (2022). Enabling data-limited chemical bioactivity predictions through deep neural network transfer learning. J. Comput. Aided Mol. Des. 36, 867–878. doi:10.1007/s10822-022-00486-x
Liu, R., Madore, M., Glover, K. P., Feasel, M. G., and Wallqvist, A. (2018b). Assessing deep and shallow learning methods for quantitative prediction of acute chemical toxicity. Toxicol. Sci. 164, 512–526. doi:10.1093/toxsci/kfy111
Liu, R., and Wallqvist, A. (2019). Molecular similarity-based domain applicability metric efficiently identifies out-of-domain compounds. J. Chem. Inf. Model 59, 181–189. doi:10.1021/acs.jcim.8b00597
Liu, R., Wang, H., Glover, K. P., Feasel, M. G., and Wallqvist, A. (2019). Dissecting machine-learning prediction of molecular activity: is an applicability domain needed for quantitative structure-activity relationship models based on deep neural networks? J. Chem. Inf. Model 59, 117–126. doi:10.1021/acs.jcim.8b00348
Lott, E. L., and Jones, E. B. (2023). “Cholinergic toxicity,” in StatPearls (Treasure Island, FL, USA: StatPearls Publishing).
Mathea, M., Klingspohn, W., and Baumann, K. (2016). Chemoinformatic classification methods and their applicability domain. Mol. Inf. 35, 160–180. doi:10.1002/minf.201501019
Oki, N. O., Nelms, M. D., Bell, S. M., Mortensen, H. M., and Edwards, S. W. (2016). Accelerating adverse outcome pathway development using publicly available data sources. Curr. Environ. Health Rep. 3, 53–63. doi:10.1007/s40572-016-0079-y
Ramsundar, B., Liu, B., Wu, Z., Verras, A., Tudor, M., Sheridan, R. P., et al. (2017). Is multitask deep learning practical for pharma? J. Chem. Inf. Model 57, 2068–2076. doi:10.1021/acs.jcim.7b00146
Sadawi, N., Olier, I., VanSchoren, J., Van Rijn, J. N., Besnard, J., Bickerton, R., et al. (2019). Multi-task learning with a natural metric for quantitative structure activity relationship learning. J. Cheminform 11, 68. doi:10.1186/s13321-019-0392-1
Schyman, P., Liu, R., Desai, V., and Wallqvist, A. (2017). vNN web server for ADMET predictions. Front. Pharmacol. 8, 889. doi:10.3389/fphar.2017.00889
Schyman, P., Liu, R., and Wallqvist, A. (2016). General purpose 2D and 3D similarity approach to identify hERG blockers. J. Chem. Inf. Model 56, 213–222. doi:10.1021/acs.jcim.5b00616
Song, Y., Zheng, S., Niu, Z., Fu, Z.-H., Lu, Y., and Yang, Y. (2020). “Communicative representation learning on attributed molecular graphs,” in Proceedings of the Twenty-Ninth International Joint Conference on Artificial Intelligence (IJCAI-20), Yokohama, Japan, July, 2020, 2831–2838.
Strickland, J., Clippinger, A. J., Brown, J., Allen, D., Jacobs, A., Matheson, J., et al. (2018). Status of acute systemic toxicity testing requirements and data uses by U.S. regulatory agencies. Regul. Toxicol. Pharmacol. 94, 183–196. doi:10.1016/j.yrtph.2018.01.022
Strickler, J., James, A., O'Leary, S., and Dube-Clark, G. (2018). Portrait of an epidemic: acute opioid intoxication in adults. Nursing 48, 40–43. doi:10.1097/01.NURSE.0000541389.52104.65
Tetko, I. V., Sushko, I., Pandey, A. K., Zhu, H., Tropsha, A., Papa, E., et al. (2008). Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model 48, 1733–1746. doi:10.1021/ci800151m
U.S. Department of Homeland Security (2012). Report on the toxic chemical syndrome. Definitions and nomenclature workshop. Available at: https://chemm.hhs.gov/Report_from_Toxic_Syndrome_Workshop_final_with_ACMT_edits_cover.pdf (accessed on October 17, 2023).
Vinken, M. (2013). The adverse outcome pathway concept: a pragmatic tool in toxicology. Toxicology 312, 158–165. doi:10.1016/j.tox.2013.08.011
Wang, H., Liu, R., Schyman, P., and Wallqvist, A. (2019). Deep neural network models for predicting chemically induced liver toxicity endpoints from transcriptomic responses. Front. Pharmacol. 10, 42. doi:10.3389/fphar.2019.00042
Keywords: toxidrome, deep learning, ensemble learning, graph neural networks, multi-task training, similarity ensemble approach
Citation: Liu R, AbdulHameed MDM, Xu Z, Clancy B, Desai V and Wallqvist A (2024) Rapid screening of chemicals for their potential to cause specific toxidromes. Front. Drug Discov. 4:1324564. doi: 10.3389/fddsv.2024.1324564
Received: 19 October 2023; Accepted: 12 January 2024;
Published: 05 February 2024.
Edited by:
Patric Schyman, Eikon Therapeutics, Inc., United StatesReviewed by:
Jianjun Tan, Beijing University of Technology, ChinaRodolpho C. Braga, InsilicAll, Brazil
Copyright © 2024 Liu, AbdulHameed, Xu, Clancy, Desai and Wallqvist. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Diwan M. AbdulHameed, mabdulhameed@bhsai.org; Anders Wallqvist, sven.a.wallqvist.civ@health.mil