 Alicja M. Lacinska
Alicja M. Lacinska Michael T. Styles
Michael T. Styles Keith Bateman
Keith Bateman Matthew Hall2
Matthew Hall2- 1Minerals and Waste Team, British Geological Survey, Nottingham, United Kingdom
- 2Faculty of Engineering, The University of Nottingham, Nottingham, United Kingdom
In situ sequestration of CO2 in mantle peridotites has been proposed as a method to alleviate the amount of anthropogenic CO2 in the atmosphere. This study presents the results of 8-month long laboratory fluid-rock experiments on representative mantle rocks from the Oman-United Arab Emirates ophiolite to investigate this process. Small core samples (3 cm long) were reacted in wet supercritical CO2 and CO2-saturated brine at 100 bar and 70°C. The extent of carbonate formation, and hence the degree of carbon sequestration, varied greatly depending on rock type, with serpentinite (lizardite-dominated) exhibiting the highest capacity, manifested by the precipitation of magnesite MgCO3 and ferroan magnesite (Mg,Fe)CO3. The carbonate precipitation occurred predominantly on the surface of the core and subordinately within cross-cutting fractures. The extent of the CO2 reactions appeared to be principally controlled by the chemical and mineralogical composition of the rock, as well as the rock texture, with all these factors influencing the extent and rate of mineral dissolution and release of Mg and Fe for subsequent reaction with the CO2. It was calculated that ≈0.7 g of CO2 was captured by reacting ≈23 g of serpentinite, determined by the mass of magnesite formed. This equates to ≈30 kg CO2 per ton of host rock, equivalent to ≈3% carbonation in half a year. However, recycling of carbonate present in veins within the original rock sample could mean that the overall amount is around 2%. The increased reactivity of serpentinite was associated with preferential dissolution of more reactive types of serpentine minerals and brucite that were mainly present in the cross-cutting veins. The bulk of the serpentinite rock was little affected. This study, using relatively short term experiments, suggests that serpentinite might be a good host rock for CO2 sequestration, although long term experiments might prove that dunite and harzburgite could be as effective in an engineered system of CCSM. Wet scCO2 proved to be chemically more aggressive than CO2-saturated brine and its ingress along fractures and grain boundaries resulted in greater host rock dissolution and subsequent carbonate precipitation.
Introduction
In situ sequestration of CO2 in mafic and ultramafic rocks (otherwise known as carbon capture and storage by mineralization, CCSM, or mineral carbonation) is a method proposed to alleviate the amount of anthropogenic CO2 in the atmosphere (IPCC, 2011). Although in principle, in situ CCSM provides an opportunity for long-term, safe sequestration, it has yet to be implemented on an industrial scale. The barriers to large-scale deployment include concerns about the process technologies and operational costs.
The fundamental process of in situ CCSM involves the injection of CO2 into a suitable geological formation, defined as a carbon repository (Kaszuba et al., 2005), where it reacts with the host rock to form carbonate minerals. Inevitably, a natural rock repository constitutes a complex structural package with diverse geochemical environments, incorporating variable groundwater chemistries, host rock compositions, etc. Mafic and ultramafic rocks have been proposed as the main host rocks for CCSM (Kelemen and Matter, 2008; Gislason et al., 2010; Kelemen et al., 2011 and references therein). These rocks are composed largely of Mg (plus Ca and Fe, in mafic rocks) silicate minerals that exhibit relatively high reactivity, and hence are prone to dissolution in acidic and neutral environments (van Noort et al., 2013), thereby providing divalent cations (predominantly Mg2+) that are utilized in the precipitation of carbonate minerals for the permanent sequestration of CO2.
Over the past couple of decades, fundamental studies on CCSM have focused largely on isolated mineral grains, such as olivine, and their interaction with CO2 under conditions relevant to in situ injection (Giammar et al., 2005; Olsson et al., 2012; Johnson et al., 2014). Whilst such studies are essential, enabling the determination of specific reaction kinetics and the factors that control mineral dissolution under hydrothermal conditions; it is also important to perform experiments within a system that approximates more closely to the subsurface environment, i.e., that of the host rock at an injection site (Hovelmann et al., 2012; van Noort et al., 2013). The reactivity of minerals within complex rocks will not be the same as that of isolated mineral grains, being influenced by rock texture, including grain size, microfractures, and compositional heterogeneity resulting from different types of adjacent minerals and mineral veins. Hence, an understanding of the behavior of minerals in complex rock is critical to the choice of a host rock (Oelkers et al., 2008).
There is a wide range of Mg-rich rocks that could be used as a host for CCSM and Styles et al. (2014) have described the variety. To appraise the reactivity of different Mg-dominated host rocks, we devised a series of batch experiments using dunite, harzburgite, and serpentinite under conditions relevant for injection and conducted at 70°C and 100 bar CO2 pressure. The pressure was appropriate for CO2 injection depths, i.e., ≈1 km. However, the temperature selected was somewhat greater than that expected at this depth (≈40–50°C), but chosen to enhance the probability of significant reaction within the constrained time frame available for the experiments. The experiments were run continuously for 246 days (≈8-months) with supercritical (sc) CO2 or in CO2-saturated brine (synthetic sea water), as well as under an N2 atmosphere as a control.
Materials and Methods
Materials
Three types of candidate host-rocks were tested, i.e., dunite (Dun), harzburgite (Hzb), and serpentinite (Srp). All samples originated from the Oman—United Arab Emirates (UAE) ophiolite and are typical of the rocks encountered in an ophiolite mantle injection site as suggested by Kelemen and Matter (2008). The rock samples were collected from small quarries and natural outcrops where there is negligible weathering in the desert environment of the UAE and the rocks are likely to be similar to those at considerable depth. The cores used in the experiments were drilled from a single piece of each rock type but these types of rock, as do many others, show variation on a cm to mm scale. This means that the two pieces of core will be slightly different in detail and the sub-sample for analysis might be very slightly different from the pieces used in the experiment. Several thin sections were examined from each rock type and the range of variation observed is described in the following section.
Dunite (Figures 1A1–A3) was partially serpentinised and predominantly composed of olivine (forsterite) with minor interstitial clinopyroxene, trace amphibole, and scattered crystals of chromite (Table 1). The degree of serpentinisation of olivine varied slightly but is generally around 40%. A few veins traverse the samples and these are mostly serpentine with a very few thin (10–20 μm) veins of secondary granular carbonate.

Figure 1. Starting materials: (A1–A3) dunite core, thin section scan with scattered black crystals of chromite and crossed-polar optical image with fractured, partially serpentinised dunite; (B1–B3) harzburgite core, thin section scan with <5 mm large orthopyroxenes in partially serpentinited matrix and crossed-polar optical image showing partially serpentinised pyroxene+ olivine dominated rock, locally cross-cut by carbonate veins; (C1–C3) serpentinite core, thin section scan with scattered magnetite and localized Fe-oxide staining and crossed-polar optical image showing blocky, pseudomorphic serpentinite, cross-cut by a perpendicular array of serpentine-dominated veins. Core dimensions: 27 mm long and 17 mm diameter. Note that the fields of view of images 2 and 3 are not from the same part of the thin section.
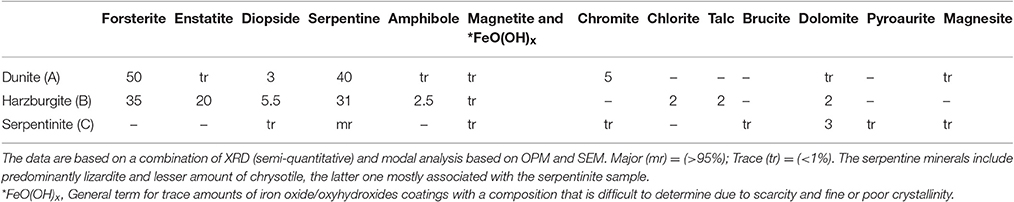
Table 1. Mineralogical composition of the starting materials.
Harzburgite (Figures 1B1–B3) samples show the most minor variation, both within and between subsamples. They consist dominantly of olivine (forsterite), with orthopyroxene (enstatite) around 20% and a small amount of clinopyroxene that ranges from around 1–4%, minor amphibole, chlorite, talc, and trace interstitial chromite (Table 1). The olivine was partially serpentinised (≈30–40%). The amphibole and talc are related to microshears and are only a trace component in some sections. There are several serpentine veins and a very small number of thin carbonate veins in all samples.
Serpentinite (Figures 1C1–C3) is almost entirely composed of fine-grained massive serpentine minerals, including lizardite and chrysotile (Table 1). The ghost texture suggests the original rock type was a dunite. There are scattered chromites throughout and a few relics of clinopyroxene in one sample. The ghost textures show that the original olivine cores were replaced by relatively coarse serpentine, probably lizardite. The outer parts of the original olivine were replaced by a very fine fibrous serpentine that has thin veins (20 μm) that mark the original grain boundaries. The rock is cut by several thicker (100–200 μm) veins of secondary fibrous serpentine locally intermixed with brucite. There are probably at least three different varieties of serpentine minerals. A few thin veins that appear to be largely carbonate cut across the serpentine veins. The type and accurate amounts of serpentine minerals could not be discerned by XRD due to severe peak overlaps and poor pattern fit. However, it was inferred from optical and scanning electron microscopy (SEM) that the bulk rock serpentine is dominantly lizardite, whilst chrysotile, and possibly other serpentine polymorphs and polytypes were present in the cross-cutting veins. Dolomite and trace amounts of pyroaurite and magnesite, magnetite, and chromite were also detected.
It was difficult to estimate the proportion of the sample that comprises veins rather than the main rock type, but it is probably only a few percent. Carbonate veins appear to be less than 2% in all samples.
The whole rock chemistry of the samples selected for the experiments is summarized in Table 2. Prior to experimentation, cores ≈27 mm long by 17 mm diameter were cut into each rock type and subsequently fractured to enhance the reactive surface area and enable a comparison of fresh and old mineral sealed fractures (Figures 1A1–C1, Table 3). The fracturing was carried out using a compression load frame applying an unconfined maximum load of 30,000 N for dunite and harzburgite, and 10,000 N for serpentinite, to form a few fractures within each core. The maximum load varied according to the strength of the rock and was controlled so that the number of fractures produced in each core was similar. Geometric surface areas, based on reconstructions of the X-ray Computed Tomography (XRCT) data, were calculated for the core samples that were used to provide data for reaction rate calculations. Accordingly, values of 0.0027 m2 for serpentinite, 0.0022 m2 for dunite, and 0.0018 m2 for harzburgite were determined. The dimensions and surface area of the cores used for all experiments are shown in Table 3.
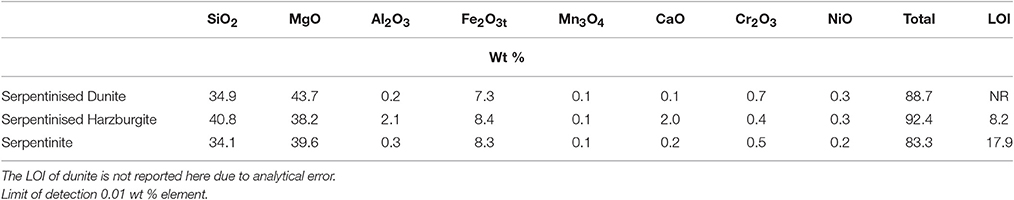
Table 2. Whole rock chemistry, as determined by XRF (Ti, Na, K, P, Sr, Zr, Ba, Cu, Zn, Pb, V, Hf, F, and Cl were below detection).
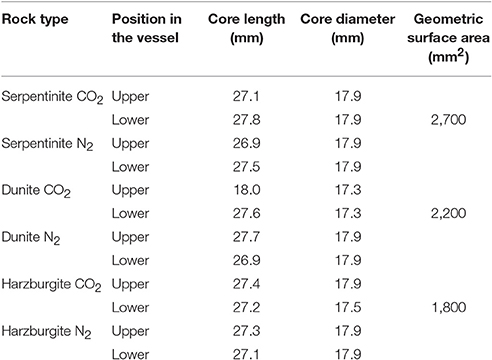
Table 3. Dimensions of all the experimental cores and geometric surface areas for cores used to calculate reaction rates.
Experimental Methodology
Carbonation experiments were carried out in pressure vessels at 70°C (±5°C), connected to CO2 or N2 (control) gas lines at 100 bar (±5 bar), for 8-months (Figure 2). The experimental conditions and fluid composition are summarized in Table 4. The vessels were made from 316 stainless steel, with wetted parts lined with polytetrafluoroethylene (PTFE); gas inlet and sampling tubes were made from 316 stainless steel or polyetheretherketone (PEEKTM) and the O-ring seals were made from Viton®. Each vessel contained a magnetic stirrer used for 2 min every 4 h to agitate the solution.
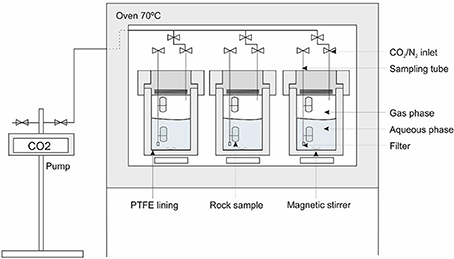
Figure 2. Schematic representation of the experimental set-up.
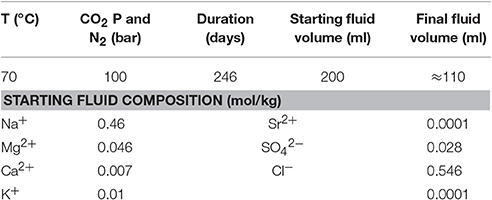
Table 4. Summary of the conditions for the CO2 and N2 control experiments.
The fractured cores were wrapped in gold wire to hold the pieces together so that they could be mounted on the sampling tubes for insertion into the pressure vessels. Two vessels per rock type were used with one pressurized with CO2 and the other with N2 as a control. Each vessel contained two cores of each sample, one immersed in the aqueous phase (CO2-saturated or N2-equilibrated brine) and one located above the solution, in the headspace filled with wet supercritical CO2 or wet N2. Two hundred milliliters of synthetic seawater solution was used in each vessel. The seawater composition was modified from that given by Millero (1996) to contain only major cations and anions (Table 4). Each experiment was sampled 11 times (including the final solution). Approximately 10 ml of fluid was extracted for the first seven samples and ≈5 ml was extracted for the remaining samples. On termination of the experiments, the vessels were cooled, depressurized, and opened to collect the final solution and solid samples. The solids were photographed in their post-test state. Any precipitates were collected for analysis, whilst the cores were subsampled for microanalysis. Reference material was kept in a desiccator.
Analytical Methodology
SEM was performed using an FEI Quanta 600, with quantitative chemical analyses of mineral phases using an Oxford Instruments energy dispersive X-ray spectroscopy (EDS) system, following ASTM recommendations (ASTM E1508-12a, 2012). Samples were analyzed in the form of carbon sputter coated (≈25 nm thick) polished thin sections. Post-experimental, polished thin sections were cut perpendicular to the length of each core, to include the outer surface, induced and natural fractures, and the partially reacted host rock. The SEM was operated under conditions of high vacuum (< 1 × 10−5 torr) at 20 kV accelerating voltage, with optimal analytical 10 mm working distance and an X-ray detector take-off angle of 45°. For EDS analyses, conditions of 1.5–7 nA beam current and 30 s acquisition time were used, giving ≈1 × 106 total X-ray counts. These optimal conditions of beam current, acquisition time, and spot mode (rather than raster mode) provided for the analysis of major elements, free of any significant mineral damage under the electron beam.
X-ray diffractometry (XRD) was performed using a PANalytical X'Pert Pro in Bragg-Brentano geometry, equipped with a cobalt-target tube and generator operated at 45 kV and 40 mA, and an X'Celerator detector. Complementary X-ray fluorescence (XRF) spectroscopy investigations were performed by PANalytical Ltd., Environmental Science Centre, Nottingham. Nine grams of PANalytical pre-fused 66/34 Li2B4O7/LiBO2 flux was weighed into a 95/5 Pt/Au crucible and fused with 0.9000 g of sample powder at 1,200°C. Loss of ignition (LOI) was determined after 1 h at 1,050°C. The composition of the experimental leachates was determined using a Perkin Elmer Optima 7,300 DV (Dual View) Inductively Coupled Plasma—Atomic Emission Spectrometer (ICP-AES), with WinLab 32 for ICP (Version 5.5) software, in the inorganic geochemistry laboratories of the British Geological Survey (BGS). PHREEQC 3.4 geochemical code (Parkhurst and Appelo, 2013) was used to model the ionic speciation of experimental solutions, based on ionic concentrations measured in the leachates. This, in combination with data from the Bureau de Recherches Géologiques et Minières (BRGM) database for phases of interest, was used to calculate the saturation indices (SI) of potential phases.
Results
Chemistry of the Experimental Leachates
The evolution of Mg, Si, and Ca in the experimental leachates is shown in Figures 3–5 as a function of time (corrected for decreasing volume), for both the CO2 and N2-pressurized (control) vessels. The “volume corrected time” is a term used to account for the progressive decrease of fluid in the experimental vessel, affecting the solid to volume ratio during the experiment (Bevan and Savage, 1989). In all cases, except for Ca in the Srp experiment, the changes in elemental concentration for the N2 control experiments were insignificant, as expected, with little visible evidence for any alteration.
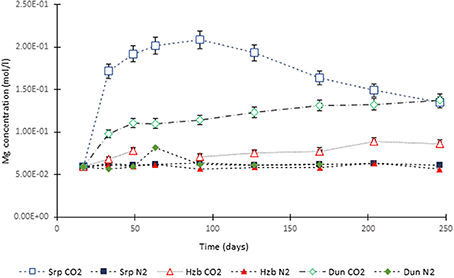
Figure 3. Mg concentration in the experimental leachates as a function of time for dunite, harzburgite, and serpentinite.
For the CO2-pressurized experiments, the most distinct concentration changes were recorded in the leachates extracted from serpentinite, as illustrated by the sudden increase in Mg concentration during the initial 2 weeks of the experiment, i.e., the 1st sampling event. The initial relatively high Mg concentration peaked at the 4th sampling event with just over 0.2 (±5%) mol/l (Figure 3). Thereafter, a steady decrease in Mg concentration continued until the end of the experiment. The Si concentration also showed distinct variations with time, marked by two distinct levels. On the 1st sampling event (≈17th day) it reached ≈0.0018 (±5%) mol/l and stayed steady. Between the 7 and 8th sampling events it increased to ≈0.005 (±5%) mol/l (Figure 4). In contrast to the Mg and Si levels, the concentration of Ca decreased with time, in both the CO2 and N2-pressurized vessels of serpentinite (Figure 5).
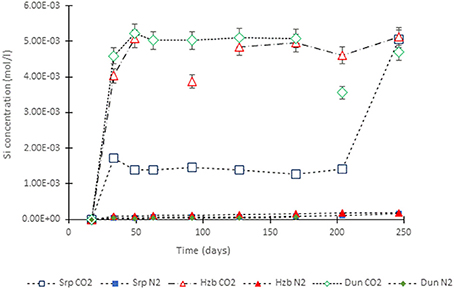
Figure 4. Si concentration in the experimental leachates as a function of time for dunite, harzburgite, and serpentinite.
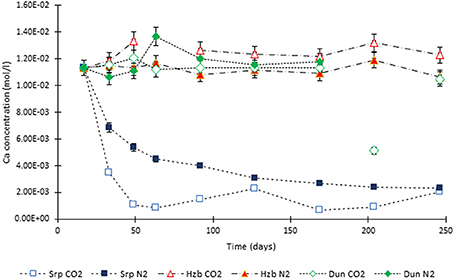
Figure 5. Ca concentration in the experimental leachates as a function of time for dunite, harzburgite, and serpentinite.
The temporal variation of Mg concentration in the leachate of dunite and harzburgite in the CO2-pressurized vessels was less pronounced than serpentinite. A general steady increase was observed for the dunite, reaching 0.14 (±5%) mol/l at the last sampling point. The harzburgite leachate showed the lowest Mg concentration of ≈0.08 (±5%) mol/l at the end of the experiment, and although this curve displayed several small fluctuations, it maintained a general flat trend (Figure 3). The temporal evolution of Si for both dunite and harzburgite exhibited general similarities, with a rapid increase by the first sampling event up to ≈0.004–0.005 (±5%) mol/l, after which several minor fluctuations were observed, although the concentration rarely fell below 0.004 mol/l (Figure 4).
The concentration of Ca changed significantly for the serpentinite samples; the decrease of Ca in the N2-pressurized was caused by the precipitation of aragonite on the core surface and formation of calcite in the CO2-pressurized experiment. This was confirmed by the microanalysis.
The concentration of Ca in the dunite and harzburgite experiments remained largely invariant.
The complete data set of fluid chemistry is given in the Supplementary Tables S1, S2.
Rate of Mg Release
The rate (R) of net release of metal (i) from serpentinite, dunite, and harzburgite was calculated using the following equation (Brantley et al., 2008):
Where V is the volume of the solution, corrected for its reduction with time; GSA is the geometric surface area (m2/g); and dC/dt is the change of elemental concentration (mol) with time (s).
Table 5 provides a summary of rates of net Mg release for the CO2-pressurized experiments. In addition, Table 5 shows pH values for all experimental solutions, corresponding to in situ conditions at 70°C and 100 bar, as calculated using PHREEQC, along with values obtained from direct measurement at ambient P and T. For the CO2-pressurized experiments, the calculated pH values were 3.1–3.6 compared to the measured pH values of 6–7, with the latter being higher due to CO2 degassing during sampling under ambient conditions. For comparison, the modeling of pH in situ in the N2-pressurized vessels returned values of pH 6–8, in close agreement with the values measured in solution post-sampling.
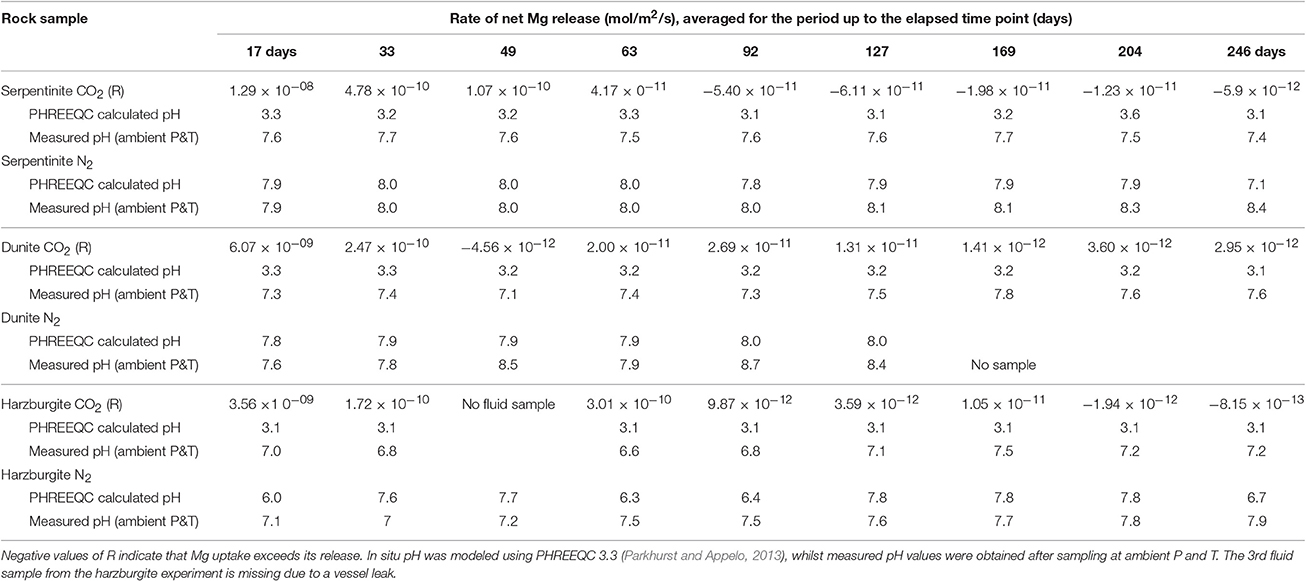
Table 5. The net rates (R) of Mg release for the CO2-pressurized samples.
The values for the rate of net1 Mg release (Figure 6) gives an indication of two competing processes, with positive values indicating that Mg release dominates, whilst negative values suggest that Mg uptake prevails, i.e., mineral precipitation.
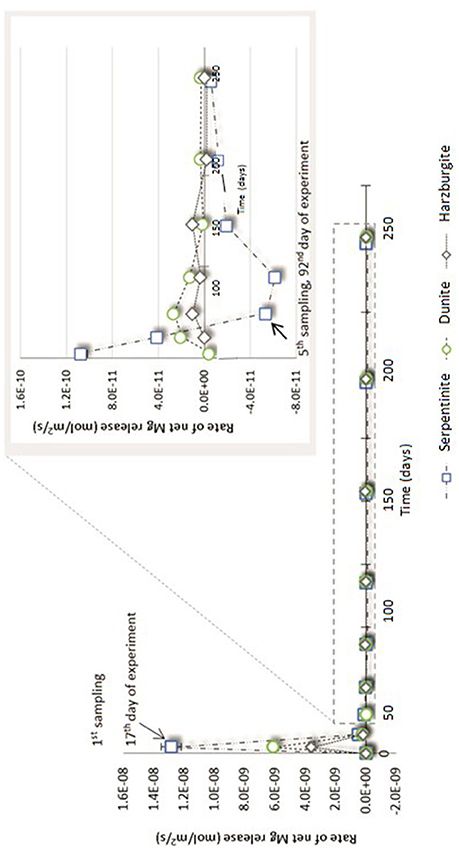
Figure 6. Net Mg release rate (R) for serpentinite, dunite, and harzburgite processed in the CO2-pressurized vessels.
Figure 6 shows that the initial net Mg release rate was high in the serpentinite experiment, with respect to the overall rate measured throughout the experiment, reaching ≈1.3 × 10−8 mol/m2/s for serpentinite, ≈6 × 10−9 mol/m2/s for dunite, and ≈2 × 10−9 mol/m2/s for harzburgite. The initial release rate was approximately an order of magnitude higher for serpentinite than for the other two samples. A sudden drop in rate was observed after the 1st sampling event, with the dunite and harzburgite remaining roughly at the same level until the end of the experiments, whilst serpentinite exhibited significant variation.
The inset of Figure 6 shows that the Mg release rate rarely dropped below zero for dunite and harzburgite, i.e., the release of Mg exceeded uptake at most time points. For serpentinite, however, the net Mg release rate fell below zero between the 4 and 5th sampling points (≈70th day of the experiment) and remained in the negative field until the end of the experiment. This shows that Mg uptake was greater than release, largely due to the precipitation of magnesite, the presence of which was confirmed by microanalysis of the final solids (Figure 8C1).
The high initial release rates may be associated with: (a) fast dissolution of the surface layer of the cores and inner open fractures upon exposure to reactive fluid, but before any newly formed minerals could inhibit fluid interaction; and/or (b) fast dissolution of carbonate minerals present in veins within serpentinite and harzburgite (as observed for serpentinite from the N2-pressurized experiment, Reactions in the control N2-pressurized vessel).
Predicted Phases
The ionic speciation of the experimental solutions was modeled using PHREEQC 3.2 (Parkhurst and Appelo, 2013), based on the concentrations measured in the liquids extracted at different time points and the pH of the solution measured at 70°C and 100 bar, with the redox conditions controlled by a HS−/ redox couple. This data, together with data for phases of interest from the BRGM database (Base de Donnee Thermoddem_V1.10), was used to calculate the saturation indices (SI) of potentially precipitating phases.
Figure 7 shows the SI for magnesite and amorphous silica, as a function of time, in the CO2-pressurized experiments.
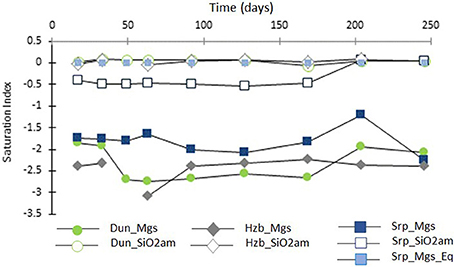
Figure 7. Saturation indices of magnesite (filled symbols) and amorphous silica (transparent symbols) as a function of time (days) for the CO2-pressurized experiments.
The SI of magnesite ranged from SI = −3.1 to −1.2, with the lowest values for harzburgite and the highest for serpentinite. This suggested that the solutions were close to saturation with respect to magnesite, without actually reaching it where SI is expected to be ≥0. However, post-processing analyses of the cores revealed the presence of magnesite for both the dunite and the serpentinite experiments. It has been shown previously that magnesite requires a critical SI of between 0.25 to 1.14 to precipitate at 95°C and 100 bar pCO2 (i.e., temperature conditions slightly higher than that used in this study; Giammar et al., 2005). The discrepancy between the presence of magnesite in the cores and negative SI could have several causes: the small variation in composition between the various sub-samples, analytical errors including those associated with sample preparation, e.g., dilution factor and the ICP AES analysis of the fluid itself. The possibility of imprecise data in the thermodynamic database when used with the combination of components encountered here must also be considered.
Figure 7 also shows that the solutions were saturated or close to saturation (in the case of serpentinite) with respect to amorphous silica; this is consistent with the microanalytical observations of the post-processing samples, as discussed in Section Post-experimental Materials.
Post-experimental Materials
Macro-Scale
The rock cores before and after 8-month reaction with CO2-saturated brine (lower cores) or scCO2 (upper cores) are shown in Figure 8. For the case of dunite (Figures 8A1,A2), the transformations at a macro-scale were manifested by the presence of a thin (< 1 mm), white coating of magnesite (confirmed by XRD in all cases) on the upper core and a brown ferruginous veneer on the lower core. For the case of harzburgite, the upper core had newly formed magnesite, but it was less prominent than on the dunite; only occurring locally as scattered nodules on the surface and where some fractures intersected the core surface. The lower core was again covered by a brown ferruginous veneer.

Figure 8. Pre- and post-experimental cores from the CO2-pressurized vessels: (A) dunite, (B) harzburgite, and (C) serpentinite. The upper cores are those reacted with wet supercritical CO2 whilst the lower cores were immersed in CO2-saturated brine. The lower cores were covered by a thin ferruginous veneer and magnesite for the case of serpentinite. Magnesite was the dominant phase in the white crust for all the cores.
Serpentinite (Figure 8C) showed the most prominent macro-scale transformations; a magnesite crust covered both upper and lower cores. The lower core displayed a sequence of mineral precipitation with a ferruginous veneer forming the first newly-precipitated coating, similar to those observed in dunite and harzburgite, but in this case overgrown by a carbonate crust. The carbonate crust thickness was uneven, generally <3 mm, highly porous and friable due to loose crystal packing. Interestingly, the binocular microscope observations revealed that the very first layer of carbonate mimicked the dendritic network of the original serpentine veinlets. The upper serpentinite core had undergone most textural transformation during the experiment, resulting in the loss of ≈1/3 of the core that had fallen to the base of the vessel. When the pressure vessel was opened at the end of the experiment, small fragments of serpentinite (maximum size 1–2 mm) were found amongst the carbonate debris. It is not known when or how this part of the core became detached. However, the outer surface of the upper core was completely covered by the carbonate crust and hence, it is assumed that disintegration took place sometime prior to the end of the experiment, allowing time for further carbonate growth. It is postulated that the core fragments did not come off as one large piece, but probably as several small pieces that became detached as new carbonate formed along the fractures. The sudden increase in Si concentration (Figure 4) toward the end of the experiment might indicate the timing of the major disintegration event.
The newly precipitated crust on the serpentinite core was dominated by morphologically variable magnesite and ferroan magnesite, with a lesser amount of calcite, mainly observed in the serpentinite core. Magnesite exhibited <1 μm equant crystals and <500 μm, but mostly <50 μm, nodular crystal aggregates (Figure 9A). Calcite displayed typical dog tooth (scalenohedral) morphology (Figure 9B).

Figure 9. Morphology of crystals exemplified by the serpentinite crust. Magnesite exhibited <1 μm equant crystals and <500 μm (here <50 μm shown) nodular crystal aggregates (A); calcite displayed typical dog tooth (scalenohedral) morphology (B).
Micro-Scale
A petrographic examination was made using polished thin sections of all the cores recovered after completion of the experiments. The bulk rock visually appeared to be largely unaltered. Localized detailed changes were analyzed and a selection of BSE and SE images illustrating representative micro-scale features of the reacted cores are presented in Figures 10–12. Table 6 summarizes the compositional and textural transformations in all cores after completion of the experiments. The following paragraphs focus on the samples that exhibited the most prominent transformations (bold in Table 6).

Figure 10. BSE (A–C) and SE (D–F) images showing micro-scale features within reacted dunite (CO2-pressurized vessel). (A) Preferentially dissolved olivine and associated secondary porosity; (B) finely crystalline magnesite within an induced fracture; (C) details of dissolution pits in olivine and magnesite infill; (D) partially dissolved olivine on the surface of an induced fracture; (E,F) details of newly formed phases, magnesite, and amorphous silica, displaying alignment with olivine cleavage planes.

Figure 11. BSE images presenting details of the micro-scale transformations in serpentinite: (A,B) from the core reacted with wet sc-CO2; (C–E) from the core reacted with CO2-saturated brine. (A) General view of an induced fracture in serpentinite. The BSE bright phases are chromite (e.g., large crystal in top left corner) and magnetite. The fracture is infilled by several generations of Mg carbonate, including hemispherical ferroan magnesite and magnesite, and finely crystalline, generally corroded Mg carbonate (as detailed in B); (B) details of the induced fracture infill: cores of hemispherical magnesite are typically Fe-enriched. Second generation Mg carbonate, postdating magnesite, is microcrystalline and shows signs of corrosion. The double arrow shows two matching sides of host rock that has been pushed-apart; (C,D), details of partially corroded dolomite (probably primary), locally overgrown by <2 μm crystals of authigenic Mg carbonate. The Si-rich fibrous phase is probably a relic of peripheral dissolution of the serpentinite host-rock; (E) and associated X-ray maps show preferential Mg dissolution from the veins rather than bulk-rock serpentinite.

Figure 12. Newly formed carbonate minerals in serpentinite following reaction with saline solution in an N2 pressurized vessel: (A) crossed polarized light OM image of acicular aragonite on the outer surface of serpentinite; (B) SE image of acicular aragonite crystals with typical chisel-like terminations; (C) SE image showing details of platy Mg-carbonate precipitated on the surface of the serpentinite core.
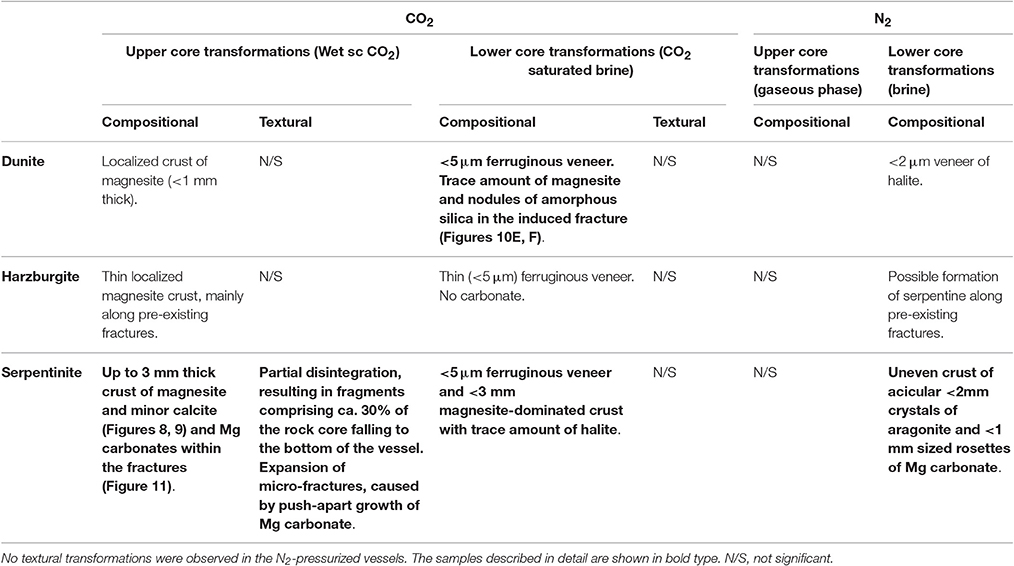
Table 6. Summary of the compositional and textural transformation observed in the reacted rock cores.
Dunite processed under CO2-saturated brine
It was evident that olivine underwent preferential dissolution, resulting in the development of secondary porosity. This was observed close to the core surface (Figure 10A) and on the induced fracture surfaces where fluid access was extensive (Figure 10B). Figures 10B,C show that olivine developed <20 μm deep etch pits. These etch pits, the larger scale secondary porosity and the fractures were partially filled with very fine-grained magnesite (Figures 10B,C). Figures 10D–F show in detail the results of olivine transformation by dissolution and precipitation of <10 μm sized Mg carbonate nodules and <3 μm sized amorphous silica nodules. These newly formed phases displayed preferential alignment along the olivine cleavage planes. This suggests that their precipitation position was related to the relict minerals' original structure rather than the transport of components to and from the site.
Serpentinite
Details of the micro-scale transformations in serpentinite cores following reaction with wet scCO2 are presented in Figures 11A,B and following reaction with CO2-saturated brine in Figures 11C–E. The first two images show an induced fracture, partially filled with reaction products, i.e., magnesite and an unidentified Mg-carbonate. The magnesite, typically exhibits a hemispherical morphology and was widely distributed, attached to the surface of the core or margins of the induced fractures. It grew in open spaces not replacing other minerals. The other Mg-carbonate occurred as partially corroded, branching aggregates of fine <1 μm crystals. The scarcity and fine-grained nature of this mineral prevented its precise identification. However, it is evident from appraisal of the texture that this post-dated the magnesite and probably had a high crystallization force; pushing apart serpentinite slivers from the sides of the induced fracture zone (Figure 11B).
The mineral formation in an open induced fracture in the serpentinite core (following reaction with CO2-saturated brine) is shown in Figures 11C–E along with associated X-ray elemental distribution maps. The dolomite in this fracture is probably remnant from the thin carbonate veins observed in the starting material. Here, it appears to be partially corroded, with the crystal cores leached. The newly formed phase is an assemblage of <3 μm rhombic crystals of Mg-carbonate that grew preferentially on the dolomite crystals (Figure 11D). Again, the very small size of the Mg-carbonate prevented its precise identification. The originally serpentine and brucite-filled veins became strongly Mg-depleted, and to a lesser extent Si-depleted, as evidenced by the associated X-ray elemental distribution maps (Figure 11E). The veins developed micropores and the components, once removed, served most probably as the main source of Mg for the precipitation of newly formed Mg-carbonates. The solid rock serpentinite, as distinct from the later veins, was not greatly affected by the dissolution. The only exception being a <5 μm thick layer of a fibrous Si-rich phase (Figure 11D) that has probably crystallized from the fluid on the wall of the fracture.
Reactions in the Control N2-Pressurized Vessel
Micro-scale petrographical observations agree with the results of the solution chemistry, in that only negligible dissolution-carbonation reactions took place within the N2-pressurized vessels. Amongst the cores investigated experimentally, the only changes observed were on serpentinite that reacted with saline solution, where; a crust of acicular aragonite and lesser amounts of platy, almost rosette-like aggregates of Mg-carbonate crystals were formed. It is considered that these carbonates precipitated from components derived from the dissolution of dolomite, pyroaurite, and other minerals present in the veins of the starting materials.
Discussion
The primary aim of this study was to ascertain the extent of mineral carbonation reactions, i.e., the degree of transformation of Mg silicate to Mg carbonate, under conditions approximating to the injection of CO2 at a depth of ≈1 km into three different types of mantle rock. It has been shown that several mineralogical and textural changes occurred during the 8-month duration of these experiments and that their extent depended greatly on the starting rock composition and the experimental conditions applied. They are summarized in Table 6.
Mineralogical Transformations
Evaluation of the temporal evolution of fluid chemistry, rate of net Mg release, and petrographic data for the rock samples demonstrated that most of the reactions observed were as expected, promoted by the presence of CO2 compared to the N2-pressurized control experiments. The extent and rate of reaction varied between the three rock types investigated and the extent of changes decreased from:
The generic dissolution and carbonation reactions reflecting the processes observed for olivine can be written as:
and for serpentine:
These reactions resulted in the formation of two types of solid phases, i.e., Mg carbonate (the majority identified as magnesite) and amorphous silica, both were observed in situ on the surface of partially dissolved olivine in dunite (Figure 13A-pre-experiment and Figure 13B-post-experiment).
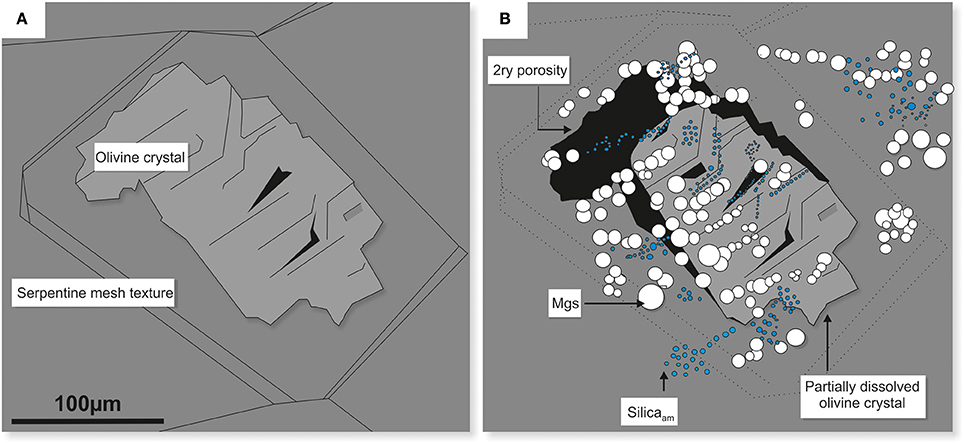
Figure 13. Schematic diagram illustrating the effects of congruent dissolution of olivine in dunite. The original olivine crystal (A) dissolved stoichiometrically, providing components of Mg2+ and H4SiO4 for the subsequent in situ precipitation of magnesite (white) and amorphous silica nodules (blue) (B). The dissolution of olivine also resulted in the development of secondary porosity (black zones). Plate B corresponds to plate E in Figure 10.
The reactivity of the rock depended primarily on the composition and spatial relationships of the minerals. The study of the dual system of olivine-serpentine in dunite shows that at 70°C and 100 bar CO2 pressure, olivine (Fo87) dissolved partially but congruently (providing a substrate for in situ precipitation of carbonate and silica, Figure 13) whilst serpentine dissolution was negligible. This is consistent with the observations made by van Noort et al. (2013), who used Oman-UAE peridotite (partially serpentinised and containing 51% of olivine and 2–3% orthopyroxene), under experimental conditions of 150°C and 10 MPa CO2, and also reported the dissolution of olivine but not serpentine. This preferential dissolution may be related to the different crystal structures of the two minerals, with the isolated Si-tetrahedra in olivine promoting faster dissolution than the polymerised Si-tetrahedra in serpentine. In addition, olivine is a higher temperature mineral and is thermodynamically less stable than serpentine under the conditions applied here.
The dissolution of olivine resulted in the development of secondary porosity that provided additional pathways for fluid ingress. These pathways may have become interconnected, aiding the progressive dissolution of rock components and the release of Mg into solution.
The petrographic evidence for congruent dissolution of olivine, however, was not supported by the leachate chemistry that showed Mg concentrations up to two orders of magnitude higher than that of Si, rather than the expected similar amounts. This suggests that other components of the rock (e.g., serpentine minerals) dissolved incongruently or significant amounts of silica had dropped out of the solution and precipitated, not only as well-defined nodules (Figure 13), but also as a fine coating layer. Such layers are considered to be passivating, limiting the number of reactive sites and having overall negative influence on the dissolution of both mineral and rock (Daval et al., 2011; Johnson et al., 2014 and references therein). Although it is possible that a passivating layer formed during the initial stages of these experiments, as proposed also by Johnson et al. (2014), no evidence for such a layer was found at this scale of observation, i.e., thickness >1 μm, and techniques with much better resolution would be required. Fibers of a Si-dominated phase (<5 μm long) were observed on the surface of the induced fracture in serpentinite (Figure 11D), but this is a new precipitate not a passivating layer.
It is noted that although the experimental study of Johnson et al. (2014) was conducted under similar conditions to those used here, the source materials were different, i.e., isolated olivine grains vs. olivine crystals embedded in a matrix of serpentine (dunite). As suggested by van Noort et al. (2013), the microtexture of polymineralic peridotites (such as dunite used here) may have a strong influence on the apparent dissolution rates, as compared to the dissolution of a (powdered) single mineral phase (such as the olivine, used by Johnson et al., idem.). In mineralogically heterogeneous and texturally complex rocks, the reactive fluids are never in direct and continuous contact with all the phases present, unlike isolated homogenous mineral grains. This may result in the generation of reaction fronts with varied geochemical micro-regimes, with localized pH differences and solution compositions, in turn leading to variable degrees of dissolution and precipitation, as observed in this study.
In the serpentinite that consisted of various types of serpentine, the extent of dissolution and early reaction rates were greater than in the partially serpentinised dunite. The major cation source in serpentinite was an array of serpentine and brucite-filled veins, rather than the bulk rock serpentinite (Figure 11, X-ray maps). Our related investigation (Lacinska et al., 2016) demonstrated that many serpentine vein minerals are either chrysotile and/or those exhibiting wide inter-crystalline spacing or poor crystallinity. These types, due to their crystal structure, are generally less stable in the presence of acid (weak acid present here) than those formed earlier during the replacement of primary Mg-silicates that are the most abundant type in serpentinite and dunite. The preferential release of Mg from the original serpentine veins is also consistent with the distribution of magnesite on the surface of the reacted core that follows the distribution of the veins. Although no clear evidence was found in these experiments, it is likely that after initial vein dissolution and with a continuous supply of fresh solution (unlike in our experiment that was run as a close system), bulk rock serpentine minerals may start to dissolve. Nevertheless, the preferential leaching of Mg from serpentine veins resulted in a porous siliceous residuum that provided pathways for fluid ingress into deeper parts of the sample and increased the overall dissolution of the sample. This positive feedback is of high importance to the real CCSM scenario because it has the capacity of creating higher surface area for dissolution-carbonation processes.
Harzburgite, containing olivine and serpentine, in addition to the major component of pyroxene, showed the lowest extent and slowest rate of rock dissolution. The chemistry of the experimental leachates agreed with the observations of the reacted rock in hand-specimen and thin-section, with both suggesting minimal transformation. It was observed that neither serpentine nor olivine from the harzburgite reacted during the experiment. The starting harzburgite rock contained lesser amounts of original, relatively-reactive serpentine veins and a lower percentage of olivine and serpentine minerals, as well as a higher proportion of pyroxene (ortho- and clinopyroxene collectively at ≈23%), that are generally less reactive (Styles et al., 2014). All these factors contributed to the lower degree of dissolution.
Comparison between CO2-Saturated Brine (Lower Cores) and Wet scCO2 (Upper Cores)
One of the scenarios for industrial CCSM is to inject CO2 as a compressed supercritical fluid (McGrail et al., 2006), at conditions exceeding the CO2 critical point of 31.1°C and 7.38 MPa. The scCO2 exhibits very low viscosity that makes it highly diffusive (Kwak et al., 2011) and consequently (once wet) highly reactive. Gaining improved understanding of the chemical interactions between wet scCO2 and rocks is essential for evaluating the effectiveness and safety of CO2 sequestration (Kerisit et al., 2012). Hence, conditions allowing for the persistence of supercritical CO2 were used in this study, enabling the evaluation of its effect on host rock transformations.
Observations of the reacted cores in hand-specimens on the micro-scale demonstrated that the reactivity in the two CO2-regimes, i.e., CO2-saturated brine (lower cores) and wet scCO2 (upper cores), was different and manifested by the presence of Fe oxide/oxyhydroxides on the lower cores and partial disintegration of the upper serpentinite core.
CO2-Saturated Brine (Lower Core) and Goethite
Goethite was only present on the surface of cores that were immersed in CO2-saturated brine. It formed the very first layer, <1 μm thick, of secondary precipitate. It was subsequently overgrown by carbonate minerals, both best displayed on the serpentinite core. The temporal evolution of Fetotal concentration in the leachate solutions showed a rapid initial release and subsequent decrease within the first 33 days of the experiment. The absence of goethite on the surface of the upper cores, as well as the cores from the N2 control vessels, suggests its formation resulted from a combination of dissolved oxygen and CO2 in the brine. The CO2 when dissolved forms carbonic acid, causing a drop of pH that may enhance the dissolution of minerals (Gaus, 2010). The ferrous iron released into saline solution reacted with the dissolved oxygen and underwent rapid oxidation to ferric iron that then precipitated as goethite; a naturally insoluble mineral under these conditions. Several mineral phases might have provided the ferrous iron, including the original Mg, Fe silicates (serpentine and olivine), ferroan carbonate (pyroaurite), clay minerals, or Fe-oxyhydroxide formed from the earlier alteration of the primary silicates.
The dunite sample that reacted with CO2-saturated brine showed that the goethite and Mg-carbonate precipitates were mutually exclusive. In the goethite-zone, apart from minor etch pits on the very edge of the sample, no other evidence for olivine dissolution was observed. This is consistent with observations reported elsewhere (Wogelius and Walther, 1991; Olsson et al., 2012) and as suggested by Wogelius and Walther (idem), the precipitation of secondary Fe3+-bearing phases decreases the number of rapidly reacting surface sites, thereby inhibiting further dissolution. It appears that initial minor dissolution of olivine at the very surface of the dunite core provided ferrous iron into solution. This oxidized immediately to ferric iron and precipitated as goethite, forming a barrier, and hindering further dissolution.
The initial precipitation of goethite requires further investigation because inhibiting the formation of carbonate may have a negative influence on the reactivity of rock used for CCSM and hence its capacity to sequester CO2.
Textural Disintegration
Textural evidence showed that reactions in wet scCO2 had an impact on sample integrity. The two serpentinite cores that reacted in the CO2 vessel varied greatly in their resultant textures. While the lower core in CO2-saturated brine retained its integrity, the upper core had partially but significantly disintegrated, with approximately a third of the original sample falling off during the experiment, leaving detached fragments in the lower part of the vessel. The mechanisms for chemical transformation of host rock minerals exposed to wet scCO2 are still poorly understood (Schaef et al., 2011; Thompson et al., 2013).
It has been suggested that mineral reactions in wet supercritical CO2 are fundamentally different to those of the same minerals in aqueous solution, being manifested by localized mineral replacements or transformations, rather than dissolution/re-precipitation involving solution phase ion transport (McGrail et al., 2009). Further, wet scCO2-dominated fluids can drive some mineral reactions via dehydration or hydration processes, which could lead to a mineral volume change and thereby affecting the rock's porosity and permeability and hence long term integrity (Loring et al., 2013). Wet scCO2 is chemically aggressive and due to its low viscosity and wetting capability has a higher relative capacity to permeate through the rock than brine. This possibly allowed a greater ingress of fluid along veins/fractures and grain boundaries and subsequent dissolution of the serpentinite sample, reducing the overall integrity of the rock and resulting in loss core from the upper sample by the end of the experiment. In addition, since the amount of oxygen dissolved in wet scCO2 fluid was low, no goethite, a potential reaction-inhibitor was formed.
It is noted that varying degrees of textural disintegration might also be related to core sample inhomogeneity and the distribution of fractures and veins, as well as the arrangement of the gold wire cage used for sample mounting, despite attempts to keep these factors to a minimum.
Implications for Engineered In situ CCSM
Carbonate Recycling
Several lines of petrographic evidence indicated that the stability of carbonate minerals was disturbed under the conditions investigated in this study (70°C, 100 bar CO2). This was typically manifested by the presence of corrosion patterns on the surface of both original and secondary carbonates (Figures 11B,D), as well as the presence of newly formed carbonate minerals in the N2-pressurized system. For example, the precipitation of aragonite and unidentified Mg-carbonates (Figure 12) on the surface of the serpentinite that had reacted with N2-pressurized saline solution was probably facilitated by the initial dissolution of original carbonate minerals, e.g., dolomite, calcite, and/or pyroaurite that had been present in thin veins in the starting rock sample. Furthermore, microanalysis of the newly formed carbonates within the induced fractures of serpentinite (CO2-saturated brine) showed evidence for significant crystal corrosion and dissolution during the experiment (Figures 11, 14). The extent of carbonate recycling needs to be understood more fully, to be able to estimate the efficiency of mineral carbonation in an engineered system.
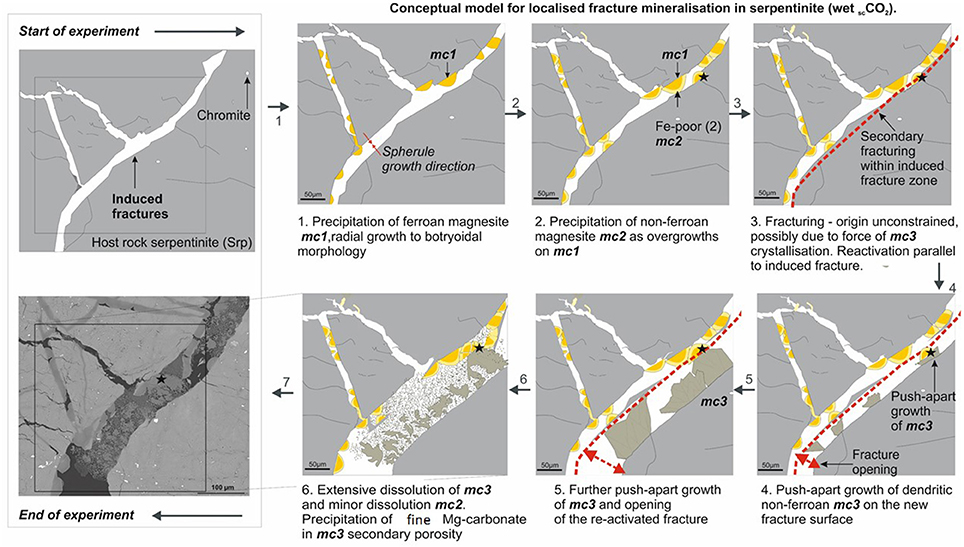
Figure 14. Conceptual model of induced fracture mineralization in serpentinite reacted with wet scCO2. Detailed explanation of the paragenetic stages is given in the accompanying plate. In summary, several stages of precipitation and dissolution of carbonate minerals were observed; the force of crystallization of carbonate “mc3” was sufficient to re-activate and “push-apart” a previously generated fracture (mechanical fracturing prior to experiment), but not to initiate any new fracturing.
Reaction Driven Cracking
Several authors have reported on mechanisms for self-propagating fractures, induced by the force of crystallization, commonly termed reaction driven cracking (Evans, 2004; Røyne et al., 2011; Plumper et al., 2012). Recently, Kelemen and Hirth (2012) suggested that, where initial permeability is sufficient to assure the continuous supply of aqueous fluids, positive feedback between reaction-driven cracking, fluid supply and reactive surface area will result most likely in nearly 100% carbonation at 185°C (accordingly, this temperature was suggested for in situ accelerated CCSM). Analysis of the reacted cores in this study suggested that the force of crystallization of Mg-carbonate was sufficient to push apart slivers of weakened wall rock in the pre-existing fracture zones, but not to create new fractures (Figure 14).
Amount of Carbonate Formed
The serpentinite core that reacted with the CO2-saturated solution showed the maximum extent of carbonation. It is desirable to estimate the extent of carbonation, but due to the nature of the friable carbonate crust it was not possible to measure it directly. Alternatively, we chose to use the changes in composition of the leachate solution to calculate the amount of carbonate formed.
The plot of Mg concentration in the CO2-pressurized serpentinite (Figure 15) is a simplification of Figure 3 and shows that, from ≈70th day of experiment, the Mg uptake exceeds that of release i.e., precipitation of a Mg-bearing phase was taking place. The calculation of the chemical affinities of potential phases and the analysis of post-experimental solids confirmed that the only Mg-bearing secondary phase formed in the serpentinite was magnesite. The amount formed can be calculated from the change in Mg concentration between the peak of Mg concentration (C1) at 0.21 mol Mg/l and the end of the experiment (C2).
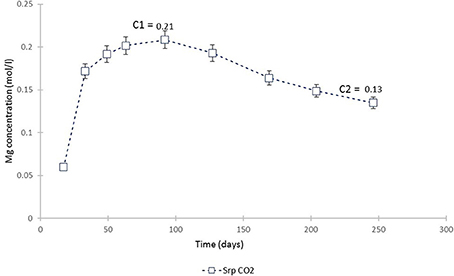
Figure 15. Mg concentration in the fluid from the serpentinite CO2 experiment. C1 marks the point from which the uptake of Mg by precipitating phase (magnesite) exceeded its release (dissolution of serpentine) and C2 is the end of experiment. The change in Mg concentration between the two points was used to calculate the amount of magnesite formed during the experiment.
Thus, 0.084 M Mg was utilized in the precipitation of magnesite. The weight of magnesite, as calculated from stoichiometry and adjusted to the volume of solution used in this study (i.e., 200 ml), was 1.4 g. By mass, this constitutes ≈8% of the ≈17.3 g starting mass of the serpentinite core. However, as revealed at the cessation of the experiment, ≈one third of the upper serpentinite core disintegrated and fell down the vessel. Although we are not able to estimate how much of the upper core contributed to lower core carbonation, we attempt to take this into account. Thus, 5.4 g from the upper core is added to the 17.3 g starting weight of the lower serpentinite.
As a result, direct measurement of the change in fluid composition showed that 0.7 g CO2 was captured (calculated from the stoichiometry of 1.4 g magnesite), utilizing a portion of the serpentine minerals from 22.7 g (17.3 + 5.4 g) of serpentinite, in sea water at 70°C pressurized with 100 bar CO2. If the entire rock weight of 22.7 g is used to calculate the rate of carbonation, then after conversion to the amount of CO2 sequestered, a value of ≈30.8 kg of CO2 per ton serpentinite is realized after ≈6.5-months, corresponding to ≈3% carbonation. In the experiments carried out here it appears that around 1% carbonate present in the starting rocks is recycled to contribute to the 3% final carbonate formed, giving an overall amount of new carbonate that captures CO2 reduced to 2%. Due to the complexity of this fluid-rock system, there are several caveats that could affect the accuracy of this calculation:
• Chlorine was present in the original fluid, and chlorine could act as another sink for magnesium, by forming MgCl2. This could potentially lead to an overestimate of the amount of CO2 captured, however no evidence for any MgCl2 precipitation was found. Hence, the assumption that magnesite was the only authigenic magnesium sink is considered reasonable.
• There is an issue with contamination of the fluid with precipitates and fragments of host rock core from the headspace that fell into the solution. It is not known at what stage of the experiment this occurred, nor the effect it might have had on solution chemistry. The estimated amount of core that fell into the solution has been taken in to consideration and included as part of the total mass of serpentinite undergoing dissolution and carbonation.
The extent of laboratory carbonation was compared with two estimates of natural mineral carbonation, i.e., carbonation of chrysotile mine tailings at Clinton Creek, Canada (Wilson et al., 2006), and carbonation of peridotite in Oman (Kelemen et al., 2011), as presented in Table 7.

Table 7. Extent of laboratory mineral carbonation compared to natural processes represented by Clinton Creek chrysotile mine tailings and peridotite in Oman, data expressed as x kg CO2 captured per ton of rock, over 6 months, assuming a linear rate.
Although the values in Table 7 represent a rough estimate of the extent of the reaction, the comparison shows that the laboratory carbonation of serpentinite, as pursued in this study, greatly exceeded the extent of natural carbonation of peridotite. Studies of the rate of weathering of silicate minerals in nature report that natural reactions are 1–3 orders of magnitude slower than those in laboratory experiments. This may result from differences in reactivity between freshly-crushed and weathered minerals, errors in estimating the exposed surface areas of minerals in weathered rock, and differences in solution composition (Swoboda-Colberg and Drever, 1993; Drever et al., 1994). Some parts of the weathering process are similar to carbonation and such factors could be applicable to this study.
The experiments reported here were at 70°C and 100 bar compared to ambient conditions for the natural examples and this will increase the rate of reaction. Kelemen and Matter (2008), Kelemen et al. (2011) noted that accelerated carbonation, using conditions of 185°C and 150 bar CO2 might result in a rate ≈106 times higher than that of natural carbonation. Our data fits in between the two extreme rate predictions suggested by these authors.
It should be noted that the estimates above do not compare the same rock types. In this study, serpentinite might initially be much more reactive due to the preferential dissolution of serpentine minerals and brucite from the cross-cutting veins that may have contributed to the high extent of reaction. This would make it more analogous to the reaction of chrysotile mine tailings. The minimal carbonation of harzburgite would give reaction extent many orders of magnitude lower, more in line with that estimated by Kelemen et al. (2011).
What is not known from these 8-month long experiments is how much the main body of the serpentinite, or indeed the other rock types, will react given a longer period of time. This data is needed to predict behavior in a more realistic engineered CCSM scenario. It is known from natural analog studies of serpentinite and peridotite pebbles in a serpentinite sand matrix that total carbonation of serpentine and peridotite can take place in brine under near ambient conditions (Lacinska et al., 2014), but there is no control on the time taken; it could be thousands of years. Experiments over a much longer time are required to get an understanding of the likelihood of extensive proportions of the rocks being converted to carbonate and whether the carbonation rates achieved are suitable for industrial scale CO2 injection.
Conclusions
This study has focused on the changes observed and the reactivity of dunite, harzburgite, and serpentinite during 8-month, closed-system experiments, using simulated seawater at 70°C and 100 bar CO2. These conditions approximate the in situ injection of CO2 into partially serpentinised mantle peridotite for reaction with silicate minerals, to release Mg for the formation of carbonate and the long term capture of CO2. We have shown that rock reactivity depends on a complex interplay between composition and texture, and the experimental conditions. Control experiments using N2 showed essentially no changes.
We conclude that:
• Serpentinite was the most reactive—resulting in the fastest initial rates of net Mg release and the greatest amount of new carbonate formed. Dunite showed low reactivity and harzburgite was virtually unchanged.
• The increased reactivity of serpentinite was associated with the preferential dissolution of veins filled with more reactive types of serpentine minerals and brucite. The dissolution of minerals from these previously sealed fractures provided pathways for further fluid ingress, thereby enhancing overall CCSM efficiency.
• The extent of carbonation based on the analysis of serpentinite that reacted with CO2-saturated brine was estimated to be ≈3% per half year. This is 100 times greater than the extent of natural carbonation of Clinton Creek mine tailings in Canada (Wilson et al., 2006) and about 200,000 times greater than the natural carbonation of peridotite in Oman (Kelemen and Matter, 2008). The estimate suggested from this study is associated with several types of uncertainty. Around 1% of the carbonate formed might be from recycling of pre-existing carbonate, giving a net figure of 2%. Further, it is stressed that the rate of serpentinite carbonation may be reduced significantly once the most reactive, i.e., vein material, is exhausted. The rate or extent of carbonation over a period of many years cannot be predicted from these experiments.
• Ferrous iron, mainly originating from the dissolution of ferromagnesian minerals present in the host-rock, undergoes rapid transformation to ferric iron upon contact with oxidative solutions. In this study, it precipitated as goethite; forming a very thin surface layer that hindered further rock dissolution by covering rapidly reacting surface sites.
• Wet scCO2 is highly reactive and rock dissolution and carbonate precipitation, affected the integrity of the rock core, increasing the reactive surface area, and promoting further geochemical transformations.
• Although the results of PHREEQC geochemical modeling were in general agreement with the microanalysis of post-experimental solids and leachate liquids, some discrepancies were found in the values of saturation indices and the phases present. These discrepancies, albeit small, were assigned to a combination of analytical errors and possible imprecisions in the database.
• Reaction driven cracking only occurred in the zone around induced fractures, but not within previously undisturbed areas.
• The choice of the most suitable reservoir rock for in situ CCSM is essential and will significantly impact the efficiency of any proposed technology, as there appear to be very large differences in the likelihood of extensive carbonation. Assumptions on rock composition should not be made and detailed petrographical and geochemical studies are essential as a part of the site selection of any proposed CCSM repository. This study of relatively short term experiments suggests that serpentinite might be a very good host rock. However, although the reactivity of dunite and harzburgite was poor compared to that of serpentinite, long term experiments might prove that these rocks might be an effective host for reaction with CO2 in an engineered system of CCSM. Longer term laboratory investigation is thus required to test this possibility.
• This study demonstrates the difficulties, complexities and uncertainties that arise when carrying out experiments on natural rocks compared to simpler systems of mineral separates. Such studies are essential to gain an understanding of the likely behavior at an industrial scale CO2 injection site.
Author Contributions
AL: carried out the experiments and interpreted the results. MS: assisted with mineralogical and petrographical study. KB: assisted with geochemical study. PB and MH: assisted with microanalysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was funded by the British Geological Survey, the University of Nottingham (jointly the GeoEnergy Research Centre) and Caterpillar Inc. C. Rochelle, G. Purser, and A. Kilpatrick are acknowledged for their support in the hydrothermal laboratories at BGS; A. Kilpatrick is also thanked for reviewing the manuscript. J. Fletcher and P. Neep are thanked for the preparation of polished thin sections; D. Wagner for assistance with XRD analyses; and M Watts and I Mountaney for facilitating the chemical analyses. The authors publish with the permission of the Executive Director of the British Geological Survey (NERC).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/feart.2017.00037/full#supplementary-material
Footnotes
1. ^Net Mg release corresponds to the amount of Mg in solution, considering all reactions taking place at a specific time point in the experiment, arising from the simultaneous dissolution and precipitation of Mg-bearing minerals.
References
ASTM E1508-12a (2012). Standard Guide for Quantitative Analysis by Energy-Dispersive Spectroscopy, ASTM International, West Conshohocken, PA. Available online at: www.astm.org
Bevan, J., and Savage, D. (1989). The effect of organic acids on the dissolutiotion of K-feldspar under conditions relevant to burial diagenesis. Mineral. Mag. 53, 415–425. doi: 10.1180/minmag.1989.053.372.02
Brantley, S. L., Kubicki, J. D., and White, A. F. (2008). Kinetics of Water-Rock Interaction. New York, NY: Springer Science+Business Media.
Daval, D., Sissmann, O., Menguy, N., Saldi, G. D., Guyot, F., Martinez, I., et al. (2011). Influence of amorphous silica layer formation on the dissolution rate of olivine at 90 degrees C and elevated pCO2. Chem. Geol. 284, 193–209. doi: 10.1016/j.chemgeo.2011.02.021
Drever, J. I., Murphy, K. M., and Clow, D. W. (1994). Field weathering rates versus laboratory dissolution rates: an update. Mineral. Mag. 58A, 239–240. doi: 10.1180/minmag.1994.58a.1.126
Evans, B. W. (2004). The serpentinite multisystem revisited: chrysotile is metastable. Int. Geol. Rev. 46, 479–506. doi: 10.2747/0020-6814.46.6.479
Gaus, I. (2010). Role and impact of CO2-rock interactions during CO2 storage in sedimentary rocks. Int. J. Greenhouse Gas Control 4, 73–89. doi: 10.1016/j.ijggc.2009.09.015
Giammar, D. E., Bruant, R. G., and Peters, C. A. (2005). Forsterite dissolution and magnesite precipitation at conditions relevant for deep saline aquifer storage and sequestration of carbon dioxide. Chem. Geol. 217, 257–276. doi: 10.1016/j.chemgeo.2004.12.013
Gislason, S. R., Wolff-Boenisch, D., Stefansson, A., Oelkers, E. H., Gunnlaugsson, E., Sigurdardottir, H., et al. (2010). Mineral sequestration of carbon dioxide in basalt: a pre-injection overview of the CarbFix project. Int. J. Greenhouse Gas Control 4, 537–545. doi: 10.1016/j.ijggc.2009.11.013
Hovelmann, J., Austrheim, H., and Jamtveit, B. (2012). Microstructure and porosity evolution during experimental carbonation of a natural peridotite. Chem. Geol. 334, 254–265. doi: 10.1016/j.chemgeo.2012.10.025
IPCC (2011). Summary for Policymakers. IPCC Special Report on Renewable Energy Sources and Climate Change Mitigation, eds by O. Edenhofer, R. Pichs-Madruga, Y. Sokona, K. Seyboth, P. Matschoss, S. Kadner et al. (Cambridge; New York, NY: Cambridge University Press).
Johnson, N. C., Thomas, B., Maher, K., Rosenbauer, R. J., Bird, D., and Brown, G. E. Jr. (2014). Olivine dissolution and carbonation under conditions relevant for in situ carbon storage. Chem. Geol. 373, 93–105. doi: 10.1016/j.chemgeo.2014.02.026
Kaszuba, J. P., Janecky, D. R., and Snow, M. G. (2005). Experimental evaluation of mixed fluid reactions between supercritical carbon dioxide and NaCl brine: relevance to the integrity of a geologic carbon repository Chem. Geol. 217, 277–293. doi: 10.1016/j.chemgeo.2004.12.014
Kelemen, P. B., and Hirth, G. (2012). Reaction-driven cracking during retrograde metamorphism: olivine hydration and carbonation. Earth Planet. Sci. Lett. 345–348, 81–89. doi: 10.1016/j.epsl.2012.06.018
Kelemen, P. B., and Matter, J. (2008). in situ carbonation of peridotite for CO2 storage. Proc. Natl. Acad. Sci. U.S.A. 105, 17295–17300. doi: 10.1073/pnas.0805794105
Kelemen, P. B., Matter, J., Streit, E. E., Rudge, J. F., Curry, W. B., and Blusztajn, J. (2011). Rates and mechanisms of mineral carbonation in peridotite: natural processes and recipes for enhanced, in situ CO2 capture and storage. Annu. Rev. Earth Planet. Sci. 39, 545–576. doi: 10.1146/annurev-earth-092010-152509
Kerisit, S., Weare, J. H., and Felmy, A. R. (2012). Structure and dynamics of forsterite-scCO2/H2O interfaces as a function of water content. Geochim. Cosmochim. Acta 84, 137–151. doi: 10.1016/j.gca.2012.01.038
Kwak, J. H., Hu, J. Z., Turcu, R. V. F., Rosso, K. M., Ilton, E. S., Wang, C., et al. (2011). The role of H2O in the carbonation of forsterite in supercritical CO2. Int. J. Greenhouse Gas Control 5, 1081–1092. doi: 10.1016/j.ijggc.2011.05.013
Lacinska, A. M., Styles, M. T., and Farrant, A. (2014). Near-surface diagenesis of ophiolite-derived conglomerates of the Barzaman Formation, United Arab Emirates: a natural analogue for permanent CO2 sequestration via mineral carbonation of ultramafic rocks. Geol. Soc. Special Edition-Tectonics of Oman Mountains, 392, 343–360. doi: 10.1144/SP392.18
Lacinska, A. M., Styles, M. T., Bateman, K., Wagner, D., Hall, M. R., Gowing, C., et al. (2016). Acid-dissolution of antigorite, chrysotile and lizardite for ex situ carbon capture and storage by mineralisaiton. Chem. Geol. 437, 153–169. doi: 10.1016/j.chemgeo.2016.05.015
Loring, J. S., Schaef, H. T., Thomson, C. J., Turcu, R. V., Miller, Q. R., Chen, J., et al. (2013). Clayhydration/dehydration in dry to water–saturated supercritical CO2: implications for caprock integrity. Energy Proced. 37, 5443–5448. doi: 10.1016/j.egypro.2013.06.463
McGrail, B. P., Schaef, H. T., Glezakou, V. A., Dang, L. X., and Owen, A. T. (2009). Water reactivity in the liquid and supercritical CO2 phase: has half the story been neglected? Energy Proced. 1, 3415–3419. doi: 10.1016/j.egypro.2009.02.131
McGrail, B. P., Schaef, H. T., Ho, A. M., Chien, Y.-J., Dooley, J. J., and Davidson, C. L. (2006). Potential for carbon dioxide sequestration in flood basalts. J. Geophys. Res. Solid Earth 111, B12201. doi: 10.1029/2005JB004169
Oelkers, E. H., Gislason, S. R., and Matter, J. (2008). Mineral carbonation of CO2. Elements 4, 333–337. doi: 10.2113/gselements.4.5.333
Olsson, J., Bovet, N., Makovicky, E., Bechgaard, K., Balogh, Z., and Stipp, S. L. S. (2012). Olivine reactivity with CO2 and H2O on a microscale: implications for carbon sequestration. Geochim. Cosmochim. Acta 77, 86–97. doi: 10.1016/j.gca.2011.11.001
Parkhurst, D. L., and Appelo, C. A. J. (2013). Description of Input and Examples for Phreeqc Version 3 – a Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport and Inverse Geochemical Calculations. U.S. Geological Survey, Techniques and Methods 6–A43, 497, Denver, CO.
Plumper, O., Royne, A., Magraso, A., and Jamtveit, B. (2012). The interface-scale mechanism of reaction-induced fracturing during serpentinization. Geology 40, 1103–1106. doi: 10.1130/G33390.1
Røyne, A., Meakin, P., Malthe-Sorenssen, A., Jamtveit, B., and Dysthe, D. K. (2011). Crack propagation driven by crystal growth. Europhys. Lett. 96:24003. doi: 10.1209/0295-5075/96/24003
Schaef, H., Windisch, T. C. Jr., McGrail, F. B., Martin, P. P. F., and Rosso, K. M. (2011). Brucite [Mg9OH02] carbonation in wet supercritical CO2: an in situ high pressure x-ray diffraction study. Geochim. Cosmochim. Acta 75, 7458–7471. doi: 10.1016/j.gca.2011.09.029
Styles, M. T., Sanna, A., Lacinska, A. M., Naden, J., and Maroto-Valer, M. (2014). The variation in composition of ultramafic rocks and the effect on their suitability for carbon dioxide sequestration by mineralization following acid leaching. Greenhouse Gases Sci. Technol. 4, 440–451. doi: 10.1002/ghg.1405
Swoboda-Colberg, N. G., and Drever, J. I. (1993). Mineral dissolution rates in plot-scale field and laboratory experiments. Chem. Geol. 105, 51–69. doi: 10.1016/0009-2541(93)90118-3
Thompson, C. J., Loring, J. S., Rosso, K. M., and Wang, Z. (2013). Comparative reactivity study of forsterite and antigorite in wet supercritical CO2 by in situ infrared spectroscopy. Int. J. Greenhouse Gas Control 18, 246–255. doi: 10.1016/j.ijggc.2013.07.007
van Noort, R., Spiers, C. J., Drury, M. R., and Kandianis, M. T. (2013). Peridotite dissolution and carbonation rates at fracture surfaces under conditions relevant for in situ mineralization of CO2. Geochim. Cosmochim. Acta 106, 1–24. doi: 10.1016/j.gca.2012.12.001
Wilson, S. A., Raudsepp, M., and Dipple, G. M. (2006). Verifying and quantifying carbon fixation in minerals from serpentine-rich mine tailings using the Rietveld method with X-ray powder diffraction data. Am. Mineral. 91, 1331–1341. doi: 10.2138/am.2006.2058
Keywords: CO2 sequestration, ultramafic rocks, serpentinites, carbonation, fluid-rock interaction
Citation: Lacinska AM, Styles MT, Bateman K, Hall M and Brown PD (2017) An Experimental Study of the Carbonation of Serpentinite and Partially Serpentinised Peridotites. Front. Earth Sci. 5:37. doi: 10.3389/feart.2017.00037
Received: 14 November 2016; Accepted: 28 April 2017;
Published: 02 June 2017.
Edited by:
Benjamin Alexander Black, City College of New York, United StatesReviewed by:
Chiara Boschi, Institute of Geosciences and Earth Resources (CNR), ItalyReinier Van Noort, Institute for Energy Technology, Norway
Copyright © 2017 Lacinska, Styles, Bateman, Hall and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alicja M. Lacinska, YWxjaUBiZ3MuYWMudWs=