 Fadila Benhamed1,2,3
Fadila Benhamed1,2,3 Gaelle Filhoulaud1,2,3Sandrine Caron4,5,6,7
Gaelle Filhoulaud1,2,3Sandrine Caron4,5,6,7 Philippe Lefebvre4,5,6,7
Philippe Lefebvre4,5,6,7 Bart Staels4,5,6,7
Bart Staels4,5,6,7 Catherine Postic1,2,3*
Catherine Postic1,2,3*- 1U1016, Institut Cochin, INSERM, Paris, France
- 2UMR 8104, CNRS, Paris, France
- 3Sorbonne Paris Cité, Université Paris Descartes, Paris, France
- 4European Genomic Institute for Diabetes (EGID), Lille, France
- 5UMR 1011, INSERM, Lille, France
- 6Univ Lille 2, Lille, France
- 7Institut Pasteur de Lille, Lille, France
Accumulating evidence suggests that O-GlcNAc transferase, an enzyme responsible for O-GlcNAc post-translational modification acts as a nutrient sensor that links glucose and the hexosamine biosynthetic pathway to the regulation of transcriptional factors involved in energy homeostasis. In liver, glucose signaling is mediated by carbohydrate response element-binding protein (ChREBP), which stimulates glycolytic and lipogenic gene expression through its binding on a specific ChoRE DNA sequence. Modulation of ChREBP by O-GlcNAcylation increases its DNA binding affinity and its activity. ChREBP transcriptional activity also depends on the presence of several other co-factors and transcriptional factors. Among them, the nuclear Farnesoid X Receptor (FXR), a key transcription factor of bile acid metabolism involved in the gut–liver axis homeostasis was recently shown to directly interact with ChREBP, acting as a repressor on the ChoRE of glycolytic genes. Interestingly, similarly to ChREBP, FXR is O-GlcNAcylated in response to glucose. This review discusses the importance of ChREBP and FXR modifications through O-GlcNAcylation in liver and how glucose can modify their mutual affinity and transcriptional activity.
Introduction
The liver plays a central role in the control of energy homeostasis. In the liver, glucose does not only serve as an energy source but also acts as a signaling molecule to control the expression of key genes of glucose, fatty acid, and bile acid metabolism. Once in the hepatocyte, glucose is converted into glucose 6-phosphate (G6P) by the glucokinase enzyme (GK) leading, in turn, to the activation of glycolytic and lipogenic enzymes including l-pyruvate kinase (l-PK), acetyl-CoA carboxylase (ACC), and fatty-acid synthase (FAS). The positive effects of glucose on gene expression are mediated by the transcription factor carbohydrate-responsive element-binding protein (ChREBP). ChREBP, which belongs to the Mondo family of bHLH/Zip transcription factors, is a large protein (864 a.a) that contains several regulatory domains including a nuclear localization signal (NLS, amino acids 158–173) near the N-terminus, polyproline domains, a bHLH/LZ domain (amino acids 660–737), and a leucine zipper-like (Zip-like) domain (amino acids 807–847) (1). A conserved consensus sequence, named carbohydrate response element (ChoRE), the ChREBP-binding site, is required for glucose responsiveness. Modulation of ChREBP expression and/or activity by glucose occurs at multiple levels. In the presence of high glucose concentrations, ChREBP mRNA levels are increased (2, 3). ChREBP is also regulated at the post-translational level: in response to high glucose concentrations, ChREBP translocates into the nucleus (4) where the protein undergoes several post-translational modifications (PTMs), including acetylation and O-GlcNAcylation, which stimulates ChREBP activity and affinity for ChoRE sequences (5–7). The O-GlcNAc modification requires the hexosamine biosynthetic pathway (HBP): in response to high glucose concentrations, HBP synthesizes N-acetyl-glucosamine (UDP-GlcNAc), an obligatory substrate for β-N-acetylglucosaminyltransferase (OGT), a key enzyme allowing O-GlcNAcylation of proteins (8). This modification is reversible and is able to alter several protein properties such as stability, degradation, and/or modulation of transcriptional activity. Recently, key transcription factors involved in energy homeostasis, including ChREBP, have been reported to be modified by O-GlcNAc in liver. Among them, the nuclear receptor Farnesoid X receptor (FXR) (9) is a regulator of gene expression involved in bile acid synthesis and transport in the liver and the intestine (10). Interestingly, FXR was also recently reported to be involved in the control of glucose homeostasis via its direct interaction with ChREBP (9, 11). This review will discuss how ChREBP and FXR, both regulated by O-GlcNAcylation, modulate the signaling pathway that controls glucose homeostasis.
ChREBP: A Key Regulator of Glucose Homeostasis
The discovery of ChREBP as key regulator of glycolysis and lipogenesis has shed light on the mechanism by which glucose transcriptionally regulates gene expression. ChREBP stimulates the expression of several genes involved in glucose and lipid metabolism such as l-PK, FAS, and ACC not only in the liver (3, 12) and also in adipose tissue (13) and in pancreatic β cells (14). ChREBP directly binds a conserved consensus sequence, ChoRE, present on the promoter region of its target genes (15, 16). The ChoRE sequence is composed of a tandem E box element separated by 5 nucleotides (5’-CACGTGnnnnnCACGTG-3’). ChREBP interacts with Max-like protein X (Mlx), its functional partner to form a heterodimer. The association of two heterodimers is necessary to bind the ChoRE motif and to provide a transcriptional complex regulated by glucose (17).
Several Key Glucose Metabolites Activate ChREBP in Response to Glucose
The regulation of ChREBP activity by glucose is complex and brings in different steps [see Ref. (18) for review]. The laboratory of K. Uyeda was the first to describe a mechanism of activation dependent on a glucose metabolite. Kabashima et al. (19) demonstrated that xylulose-5-phosphate (X5P), a metabolite of the pentose phosphate pathway (PPP), is central for ChREBP translocation and DNA binding activity in response to glucose. Under high glucose concentrations, X5P activates the protein phosphatase PP2A, which dephosphorylates ChREBP on the serine residue 196 (Ser196), allowing its translocation to the nucleus. In a second step also occurring in a X5P and PP2A-dependent manner, ChREBP is dephosphorylated on the threonine residue 666 (Thr666) leading to its binding to DNA and to transactivation (19). However, this mechanism is controversial as several distinct hypotheses were proposed to explain the glucose-mediated activation of ChREBP [see Ref. (18) for review]. For instance, a structure–function analysis of the ChREBP protein identified an N-terminal domain, named the glucose-sensing module (GSM), a highly conserved sequence through evolution (20). The GSM contains two domains, the low glucose inhibitory domain (LID, residues 1–197) and the glucose-response activated conserved element, residues 197–298 (GRACE), both implicated in the regulation of ChREBP in response to glucose (21). Under low glucose concentrations, the GRACE domain is inhibited by the LID domain, leading to a lack of induction of ChREBP activity. Under high glucose concentrations, the inhibitory effect of the LID is relieved, thereby allowing the GRACE domain to stimulate ChREBP activity. In agreement with this hypothesis, deletion of the 196 first amino acids encompassing most of the LID yields a constitutive active form of ChREBP, independent of glucose concentrations (21). Interestingly, McFerrin and Atshley (22) identified a G6P binding pocket using structure prediction of the ChREBP protein. G6P, produced by the GK enzyme, after binding onto the GSM could induce a conformation change, dissociating the LID from the GRACE domain and therefore supporting ChREBP transactivation. More importantly, G6P could “open/derepress” the ChREBP protein structure allowing interaction with co-activators such as CBP and p300 (22). Arguments in favor of a role for G6P in activating ChREBP in hepatocytes and other cell types were reported. Overexpression of glucose-6-phosphate dehydrogenase (G6PDH), a rate limiting enzyme of the PPP in the pancreatic β cell line INS1 deprives cells from G6P and inhibits ChREBP transcriptional activity. In contrast, G6P accumulation driven by the specific inhibition of G6PDH activity increases ChREBP transcriptional activity in these cells (23). Using G6PDH overexpression and silencing approaches in hepatocytes, our laboratory showed that G6P, but not X5P, is required for ChREBP translocation to the nucleus and transactivation, suggesting that G6P is necessary and sufficient to induce ChREBP activity (24). The glucose-mediated activation of ChREBP remains complex and additional studies will be required to elucidate the exact contribution of the proposed metabolites. A step forward concerning ChREBP regulation was recently made when Herman and colleagues identified a novel variant of ChREBP named ChREBP-β (13). This variant arises from an alternative promoter located in exon1b of the ChREBP gene. This new transcript, which results from the splicing of exon 1b to exon 2, is translated at the next start-site located in exon 4 and produces a shorter protein of 687 amino acids (ChREBP-β) compared to the full-length protein ChREBP, re-named ChREBP-α (13). According to the hypothesis raised by Herman and co-workers, glucose metabolism (potentially G6P) would first induce the transcription of ChREBP-α. ChREBP-α would in turn bind the ChoRE identified in exon1b to enhance ChREBP-β transcription. In adipose tissue, ChREBP-β was described as a much more potent transcriptional regulator than ChREBP-α. In the liver, ChREBP-β expression seems to be less sensitive than that of ChREBP-α to nutritional regulations (fasting versus refeeding). However, the physiological contribution of ChREBP-β to the glucose-induced transcriptional response in the liver remains to be determined.
A Central Role for ChREBP in Regulating Hepatic Glycolysis and Lipogenesis
Convincing in vitro and in vivo evidences revealed that ChREBP is required for the induction of glycolytic and lipogenic genes in response to glucose (3, 12). Stimulation of primary cultured hepatocytes with high glucose concentrations (25 mM) leads to the induction of ChREBP expression and activity allowing stimulation of its target genes (25). In contrast, inhibition of ChREBP expression by a siRNA approach prevents this induction and blunts the accumulation of lipids in response to glucose (3). Importantly, global inactivation or liver-specific inhibition of ChREBP leads to a decrease in glycolytic and lipogenic gene expression associated with a significant decrease in triglyceride synthesis under both physiological and pathophysiological conditions (12, 26). The mirror experiment in which the ChREBP protein was overexpressed through an adenoviral approach in liver of mice led to an exacerbation of the glycolysis and lipogenesis pathways associated with the development of hepatic steatosis (27).
FXR: A New Modulator of Glucose Homeostasis
Once activated by its ligands such as natural bile acids, FXR binds, alone or with its partner Retinoid X receptor (RXR), onto its response elements (FXRE) to regulate its target genes. While largely implicated in the transcriptional control of genes controlling bile acid metabolism, FXR also recently emerged as a novel modulator of glucose homeostasis (28, 29). FXR is necessary for the control of blood glucose concentrations in response to starvation in mice. FXR knockout mice (FXR−/−, whole body inactivation) were reported to be hypoglycemic in response to a short time (6 h) fasting. This phenotype can be, in part, explained by an alteration of the expression of phosphoenolpyruvate carboxykinase (PEPCK), a key enzyme of gluconeogenesis. Interestingly, the response to longer fasting (24–48 h) was not affected in the absence of FXR, suggesting a defective adaptative response in FXR−/− mice. Surprisingly, activation of FXR by GW4064, a specific synthetic ligand, did not increase PEPCK expression in primary mouse hepatocytes (30). However, primary human and rat hepatocytes stimulated with GW4064 displayed an increase in PEPCK expression that was correlated with enhanced glucose output (31) suggesting either species differences or dependence on changes such as nutritional or environmental stimuli.
Interestingly, FXR−/− mice respond more rapidly to high carbohydrate feeding with an accelerated induction of glycolytic and lipogenic genes without, however, any difference in ChREBP mRNA levels (29). In addition, nuclear translocation of ChREBP protein was not affected by FXR activation. FXR was shown to directly interact with the ChREBP protein in different cell lines. In vitro GST pulldown experiments showed that FXR interacts with ChREBP, irrespective of its ligation to GW4064. Analysis of FXR deletion mutants revealed that FXR interacts with ChREBP via its N-terminal activation function-1 (AF-1) domain (amino acids 1–127) and via the N-terminal part of its ligand-binding domain (amino acids 215–300) (11).
At the functional level, treatment of primary hepatocytes with the FXR agonist GW4064 decreased the glucose-induced expression of l-PK, ACC, and FAS. This inhibition was prevented in FXR−/− hepatocytes. Importantly, using a ChoRE luciferase promoter construct, the authors reported that FXR transfection and/or activation prevented the stimulation of ChoRE-driven target genes. Gel shift analysis revealed that FXR was indeed able to bind to the L3 site (contained within the ChoRE) but not to the L4 site of l-PK promoter (29). These results were confirmed in the immortalized human hepatocyte (IHH) cell line: when activated by either its most potent natural ligand (CDCA: chenodeoxycholic acid) or synthetic agonists (GW4064; INT747; and WAY362450), FXR was able to bind the l-PK promoter. Finally, using chromatin immunoprecipitation (ChIP) assays, Caron et al. (11) were able to demonstrate the concomitant recruitment of ChREBP, HNF4α, p300, CBP, and FXR on the genomic L4/L3 region of the l-PK promoter in the presence of high glucose concentrations. According to this model, agonist-mediated activation of FXR leads to the release of CBP and p300, while allowing the recruitment of the co-repressor SMRT (Figure 1). This study reveals that FXR acts as a transrepressor and provides a novel mechanism by which FXR directly controls ChREBP-dependent genes, such as the l-PK gene.
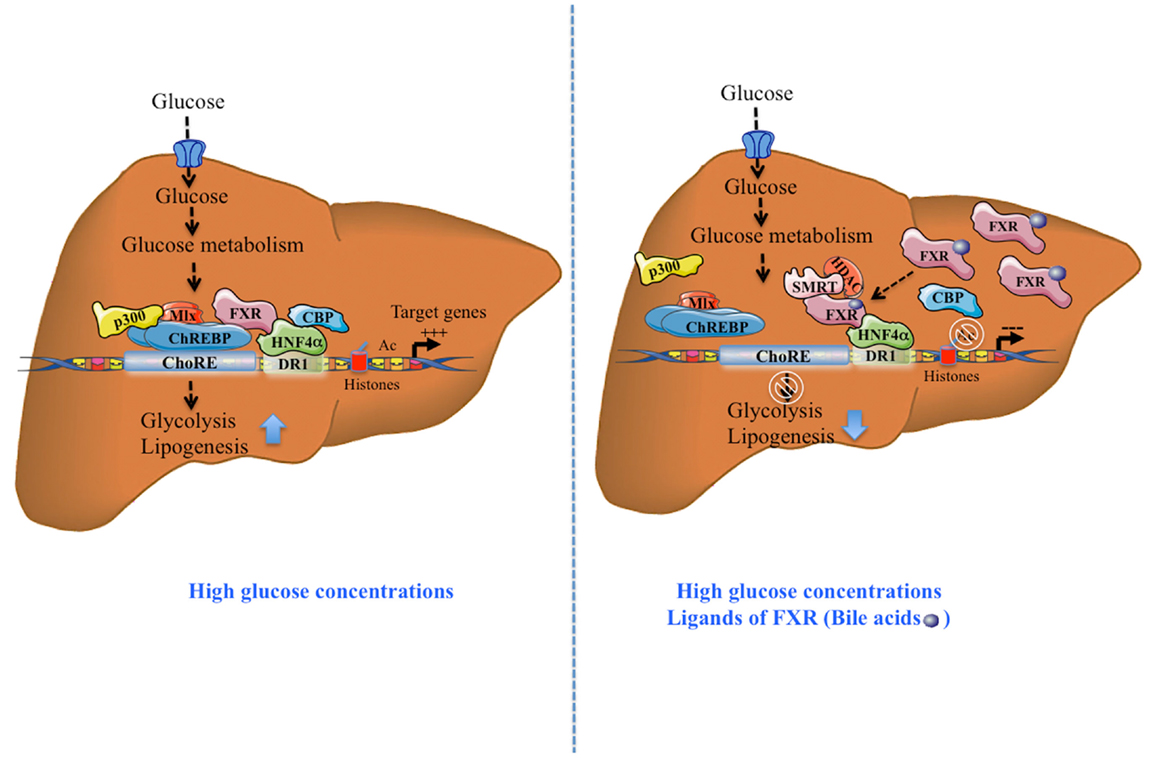
Figure 1. Activation and transrepression of ChREBP-target genes by ChREBP and FXR. After a meal, in the presence of high glucose concentrations, without FXR activation, ChREBP binds together with HNF4α, to ChoRE region of the l-PK promoter and transactivates gene expression, in part due to the recruitment of co-activators p300 and CBP. Due to its direct interaction with ChREBP and HNF4α, FXR interacts with this complex. The complex formation leads to the stimulation of the glycolytic and lipogenic pathways. The synergistic presence of high glucose concentrations and FXR ligands (bile acids, CDCA), activated FXR recruits the co-repressor SMRT. This recruitment leads to the release of ChREBP, CBP, and p300 leading to the inhibition of ChREBP-target gene expression. Tethered to the promoter through its interaction with HNF4α, FXR recruits the transcriptional co-inhibitor SMRT and represses transcription through the recruitment of HDACs and deacetylation of H3 histones. This effect leads to inhibition of the glycolytic and lipogenic pathways.
The structural base of the repressive activity of FXR on ChREBP activity may rely on the existence of the LxQLLT motif, called the nuclear receptor box (NRB) within the ChREBP protein (22). This NRB matches the consensus LXXLL motif primarily found in co-activators of nuclear receptors that confers agonist-induced binding to nuclear receptors suggesting a potential ligand-dependent interaction between ChREBP and nuclear receptors such as FXR (22) and recruitment of FXR on ChoRE-bound ChREBP. SMRT tethering to the FXR–ChREBP complex could then occur through the second co-activator binding motif specifically found in FXR (32), although this awaits formal investigation. It would be of interest to mutate this NRB motif and study the modification of interaction between ChREBP and FXR as well as the consequences on the transcriptional regulation of the l-PK gene. One can also speculate that under appropriate conditions, ChREBP might serve as a FXR co-regulator, hence conferring glucose responsiveness to FXR. Such a possibility could be investigated by high resolutive genomic binding studies such as ChIP-Exo assays. Another hypothesis is that glucose metabolism could act as a signal that activates FXR independently of ChREBP via PTMs such as O-GlcNAcylation, as discussed below.
ChREBP and FXR are O-GlcNAcylated
O-GlcNAcylation Stabilizes the ChREBP Protein and Stimulates Its Transcriptional Activity
Approximately 2–5% of total glucose in the cell is used through the HBP. O-GlcNAcylation is a dynamic reaction catalyzed by two enzymes: (i) O-GlcNAc transferase (OGT), which adds a monosaccharide to serine/threonine residues of target proteins; (ii) the O-GlcNAc hydrolase (OGA), which hydrolyzes the monosaccharide. Sakiyama and co-workers (33) first showed that when Hepa1–6 hepatoma cells are treated with PUGNAC, a drug that increases O-GlcNAc, the transcriptional activity of ChREBP is exacerbated under high glucose concentrations, without any change in protein levels. In contrast, in cells treated with DON (6-diazo-5-oxo-l-norleucine), a drug that decreases O-GlcNAc, the stimulatory effect of high glucose on ChREBP activity is prevented (33). Our laboratory showed that ChREBP directly interacts with OGT in HEK293 cells and hepatocytes (6). ChREBP is O-GlcNAcylated in hepatocytes treated with high concentrations of glucose or glucosamine and in the liver of refed mice, demonstrating a nutritional regulation of ChREBP O-GlcNAcylation (ChREBPOG). In mouse hepatocytes, overexpression of OGA led to an inhibition of ChREBP-target genes associated with a decrease of lipid droplets under high glucose concentrations. In vivo, OGT overexpression in mouse liver was associated with an increase of ChREBPOG, correlated with the induction of l-PK expression and with ChREBP recruitment to the l-PK promoter. OGT overexpression also increased ChREBP protein content without modifying ChREBP mRNA levels suggesting that the protein may be stabilized by O-GlcNAcylation (6). Interestingly, in a follow-up study, Ido-Kitamura and co-workers suggested that ChREBP poly-ubiquitination (ChREBPub) was reduced when ChREBP is O-GlcNAcylated (7) suggesting that these two PTMs may interfere to regulate ChREBP stability.
FXR is Regulated by O-GlcNAcylation
While the impact of nutrients and glucose on bile acid homeostasis is not fully understood (10), it was recently shown that FXR can be modified through O-GlcNAcylation in response to high glucose concentration level (9). Berrabah et al. revealed that FXR is modified by O-GlcNAcylation through its interaction with OGT, which catalyzed this reaction in response to high glucose level. O-GlcNAcylation leads to an increase of FXR protein stability, transcriptional activity, and chromatin binding through SMRT inactivation. O-GlcNAcylation of FXR occurs on serine 62 (Ser62) within the AF-1 domain. In agreement, mutation of Ser62 decreased FXR O-GlcNAcylation that correlated with an inhibition of its transcriptional activity. In vivo, nutritional experiments reveals that FXR is O-GlcNAcylated under fed conditions, which correlates with an induction of its target genes (Shp, Cyp7A1) and a decrease in hepatic bile acid content. Interestingly, a recent study reported that FXR can also be modified by SUMOylation (34). Ligand-dependent SUMOylation of FXR leads to a decrease of FXR transcriptional activity and consequently to a down regulation of its target genes. Interestingly, O-GlcNAcylation (Ser62) and sumoylation (Lys122) of FXR occur within the same domain, the A/B-domain known to play gene-specific role in transactivation and cofactor recruitment (11). It would be of interest to determine whether O-GlcNAcylation of FXR prevents and/or interferes with its SUMOylation and vice versa.
Relevance of ChREBP and/or FXR O-GlcNAcylation to Physiopathology
Hyperglycemia and diabetes result in an increased flux through the HBP, which, in turn, increases PTM of Ser/Thr residues of proteins by O-GlcNAcylation. Altered O-GlcNAc signaling has been implicated in the pathogenesis of diabetes and may play an important role in its complications including non-alcoholic fatty liver disease (NAFLD), diabetic nephropathy, and/or retinopathy (35). Indeed, we have reported that the hepatic content of ChREBPOG is increased in liver of diabetic db/db mice, and correlated to the pathophysiology of hepatic steatosis in this mouse model. OGA overexpression in the liver of db/db mice reduced ChREBPOG concentrations leading to an inhibition of its target genes involved in de novo lipogenesis. Consequently, hepatic steatosis was prevented and correlated to an improvement of several physiological parameters (improved glucose tolerance and insulin sensitivity). The improved phenotype in OGA-treated db/db mice was also associated with a significant decrease in O-GlcNAcylation of the transcriptional co-activator CRTC2 (6) involved in the control of gluconeogenic genes (36). Recently, the role of ChREBPOG in diabetic nephropathy was also investigated (37). Treatment with high glucose concentrations increased cellular O-GlcNAc and ChREBPOG levels in mesangial cells compared with low glucose concentrations. PUGNAc treatment increased ChREBP-target expression in mesangial cells, whereas DON blunted the stimulatory effect of high glucose. Mechanistically, O-GlcNAc augmented protein stability, transcriptional activity, and nuclear translocation of ChREBP in these cells, leading to an exacerbated lipid accumulation. Importantly, in a pathophysiological context, ChREBPOG was elevated in mesangial cells from streptozotocin-induced diabetic rats. Altogether, this study suggests that the hyperglycemia-mediated induction of ChREBP O-GlcNAcylation in mesangial cells may drive excess lipid accumulation and fibrosis, characteristic features of diabetic nephropathy (37). The potential contribution of FXR O-GlcNAcylation to the pathophysiology of liver and/or of other organs has not yet been addressed. Interestingly, FXR deficiency was previously reported to improve several of the metabolic abnormalities observed in ob/ob mice. Indeed, FXR−/− mice crossed on ob/ob background are less obese, more tolerant to glucose and more sensitive to insulin than controls (38). The finding that FXR is modified by O-GlcNAcylation (9) further links bile acid metabolism to nutrient availability as observed in human in physiology (39), but also in a context of metabolic dysfunctions such as type 2 diabetes (40). In fasting–refeeding experiments, an FXR-dependent correlation between hepatic bile acid content, FXR transcriptional activity, and plasma glucose concentration has been established, suggesting that O-GlcNAcylation of FXR might regulate bile acid production (9). However, this awaits a formal demonstration using O-GlcNAcylation-deficient FXR in vivo. Importantly, the concomitant regulation of ChREBP and FXR by O-GlcNAcylation in liver cells in response to hyperglycemia may trigger and/or enhance their physical interaction, modulating in turn the transcriptional regulation of their common target genes involved in glycolysis, lipogenesis, and/or bile acid metabolism (Figure 2). Interestingly, FXR was reported to interact with the ChREBP protein through its AF-1 domain (11), a domain also shown to be the site of FXR O-GlcNAcylation (9). Further analysis of this interaction in response to high glucose concentrations, as well as the identification of O-GlcNAc residues within the ChREBP protein should provide a better understanding of the relevance of the coordinated O-GlcNAcylation of ChREBP and FXR under physiological and pathophysiological conditions (Figure 2).
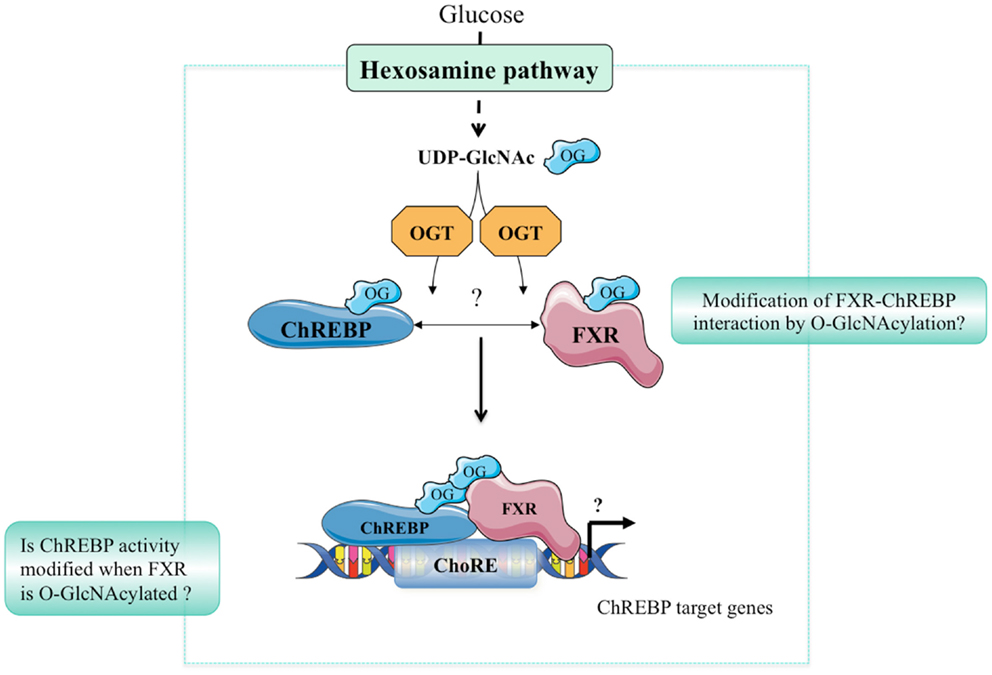
Figure 2. Hypothetical model of ChREBP and FXR interaction. In response to high glucose concentrations, the hexosamine biosynthesis (HBP) pathway is activated leading to UDP-GlcNAc production. ChREBP and FXR are in turn O-GlcNAcylated through direct binding with the OGT enzyme. O-GlcNAcylation of ChREBP and FXR may represent an important feature of their interaction. The physiological or pathophysiological consequences of such a modification remains, however, unknown.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Associate Editor Tarik Issad declares that, despite being affiliated to the same institution as the authors Fadila Benhamed, Gaelle Filhoulaud and Catherine Postic, the review process was handled objectively and no conflict of interest exists.
Acknowledgments
The work from the Institut Cochin INSERM U1016 was performed within the Département Hospitalo-Universitaire (DHU) AUToimmune and HORmonal diseaseS and supported by grants from the Agence Nationale de la Recherche (Crisalis, Genopath), Fondation Française de la Recherche Médicale (FRM, Labélisation Equipe) and the EU Grant FLORINASH (FP7). The work performed at Institut Pasteur INSERM UMR1011 was supported by grants from the EU Grant HEPADIP (no. 018734), the Region Nord-Pas-de-Calais/FEDER, the Agence Nationale de la Recherche (no. 11 BSV1 032 01), and European Genomic Institute for Diabetes (no. ANR-10-LABX-46). Bart Staels is a member of the Institut Universitaire de France.
Abbreviations
ACC, acetyl-CoA carboxylase; AF-1, activation function-1; ChIP, chromatin immunoprecipitation; CBP, CREB-binding protein; ChoRE, carbohydrate response element; ChREBP, carbohydrate-responsive element-binding protein; FAS, fatty-acid synthase; FXR, farnesoid X receptor; FXRE, FXR response element; G6P, glucose-6-phosphate; GSM, glucose-sensing module; GK, glucokinase; G6PDH, glucose 6-phosphate dehydrogenase; GRACE, glucose-response activated conserved element; HBP, hexosamine biosynthetic pathway; LID, low glucose inhibitory domain; l-PK, l-pyruvate kinase; Mlx, max-like protein X; NAFLD, non-alcoholic fatty liver disease; NRB, nuclear receptor box; OGT, β-N-acetylglucosaminyltransferase; p300, histone acetyl transferase p300; PPP, pentose phosphate pathway; PEPCK, phosphoenolpyruvate carboxykinase; PTM, post-translational modification; UDP-GlcNAc, N-acetyl-glucosamine; NLS, nuclear localization signal; RXR, retinoid X receptor; X5P; xylulose-5-phosphate.
References
1. Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A (2001) 98(16):9116–21. doi:10.1073/pnas.161284298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem (2002) 277(6):3829–35. doi:10.1074/jbc.M107895200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem (2004) 279(19):20314–26. doi:10.1074/jbc.M312475200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. Glucose and cAMP regulate the l-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci U S A (2001) 98(24):13710–5. doi:10.1073/pnas.231370798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest (2010) 120(12):4316–31. doi:10.1172/JCI41624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes (2011) 60(5):1399–413. doi:10.2337/db10-0452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ido-Kitamura Y, Sasaki T, Kobayashi M, Kim HJ, Lee YS, Kikuchi O, et al. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of ChREBP O-glycosylation. PLoS One (2012) 7(10):e47231. doi:10.1371/journal.pone.0047231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem (2011) 80:825–58. doi:10.1146/annurev-biochem-060608-102511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Berrabah W, Aumercier P, Gheeraert C, Dehondt H, Bouchaert E, Alexandre J, et al. Glucose sensing O-GlcNAcylation pathway regulates the nuclear bile acid receptor farnesoid X receptor (FXR). Hepatology (2014) 59(5):2022–33. doi:10.1002/hep.26710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev (2009) 89(1):147–91. doi:10.1152/physrev.00010.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Caron S, Huaman Samanez C, Dehondt H, Ploton M, Briand O, Lien F, et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol Cell Biol (2013) 33(11):2202–11. doi:10.1128/MCB.01004-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A (2004) 101(19):7281–6. doi:10.1073/pnas.0401516101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature (2012) 484(7394):333–8. doi:10.1038/nature10986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Noordeen NA, Khera TK, Sun G, Longbottom ER, Pullen TJ, da Silva Xavier G, et al. Carbohydrate-responsive element-binding protein (ChREBP) is a negative regulator of ARNT/HIF-1beta gene expression in pancreatic islet beta-cells. Diabetes (2010) 59(1):153–60. doi:10.2337/db08-0868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Jeong YS, Kim D, Lee YS, Kim HJ, Han JY, Im SS, et al. Integrated expression profiling and genome-wide analysis of ChREBP targets reveals the dual role for ChREBP in glucose-regulated gene expression. PLoS One (2011) 6(7):e22544. doi:10.1371/journal.pone.0022544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Rufo C, Teran-Garcia M, Nakamura MT, Koo SH, Towle HC, Clarke SD. Involvement of a unique carbohydrate-responsive factor in the glucose regulation of rat liver fatty-acid synthase gene transcription. J Biol Chem (2001) 276(24):21969–75. doi:10.1074/jbc.M100461200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Ma L, Robinson LN, Towle HC. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem (2006) 281(39):28721–30. doi:10.1074/jbc.M601576200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab (2013) 24(5):257–68. doi:10.1016/j.tem.2013.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A (2003) 100(9):5107–12. doi:10.1073/pnas.0730817100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Li MV, Chang B, Imamura M, Poungvarin N, Chan L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes (2006) 55(5):1179–89. doi:10.2337/db05-0822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Li MV, Chen W, Poungvarin N, Imamura M, Chan L. Glucose-mediated transactivation of carbohydrate response element-binding protein requires cooperative actions from Mondo conserved regions and essential trans-acting factor 14-3-3. Mol Endocrinol (2008) 22(7):1658–72. doi:10.1210/me.2007-0560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. McFerrin LG, Atchley WR. A novel N-terminal domain may dictate the glucose response of Mondo proteins. PLoS One (2012) 7(4):e34803. doi:10.1371/journal.pone.0034803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Li MV, Chen W, Harmancey RN, Nuotio-Antar AM, Imamura M, Saha P, et al. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem Biophys Res Commun (2010) 395(3):395–400. doi:10.1016/j.bbrc.2010.04.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Dentin R, Tomas-Cobos L, Foufelle F, Leopold J, Girard J, Postic C, et al. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in liver. J Hepatol (2011) 56(1):199–209. doi:10.1016/j.jhep.2011.07.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A (2004) 101(44):15597–602. doi:10.1073/pnas.0405238101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Iizuka K, Miller B, Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am J Physiol Endocrinol Metab (2006) 291(2):E358–64. doi:10.1152/ajpendo.00027.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest (2012) 122(6):2176–94. doi:10.1172/JCI41636
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, et al. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes (2004) 53(4):890–8. doi:10.2337/diabetes.53.4.890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, et al. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem (2005) 280(33):29971–9. doi:10.1074/jbc.M501931200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk T, Grefhorst A, Bouchaert E, et al. Transient impairment of the adaptive response to fasting in FXR-deficient mice. FEBS Lett (2005) 579(19):4076–80. doi:10.1016/j.febslet.2005.06.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology (2005) 146(3):984–91. doi:10.1210/en.2004-0965
32. Mi LZ, Devarakonda S, Harp JM, Han Q, Pellicciari R, Willson TM, et al. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell (2003) 11(4):1093–100. doi:10.1016/S1097-2765(03)00112-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Sakiyama H, Fujiwara N, Noguchi T, Eguchi H, Yoshihara D, Uyeda K, et al. The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem Biophys Res Commun (2010) 402(4):784–9. doi:10.1016/j.bbrc.2010.10.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Balasubramaniyan N, Luo Y, Sun AQ, Suchy FJ. SUMOylation of the farnesoid X receptor (FXR) regulates the expression of FXR target genes. J Biol Chem (2013) 288(19):13850–62. doi:10.1074/jbc.M112.443937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Ma J, Hart GW. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev Proteomics (2013) 10(4):365–80. doi:10.1586/14789450.2013.820536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Dentin R, Hedrick S, Xie J, Yates J III, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science (2008) 319(5868):1402–5. doi:10.1126/science.1151363
37. Park MJ, Kim DI, Lim SK, Choi JH, Han HJ, Yoon KC, et al. High glucose-induced O-GlcNAcylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis: the possible role in the development of diabetic nephropathy. J Biol Chem (2014) 289(19):13519–30. doi:10.1074/jbc.M113.530139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Zhang Y, Ge X, Heemstra LA, Chen WD, Xu J, Smith JL, et al. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol Endocrinol (2012) 26(2):272–80. doi:10.1210/me.2011-1157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Prawitt J, Caron S, Staels B. Glucose-lowering effects of intestinal bile acid sequestration through enhancement of splanchnic glucose utilization. Trends Endocrinol Metab (2014) 25(5):235–44. doi:10.1016/j.tem.2014.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Matysik S, Martin J, Bala M, Scherer M, Schaffler A, Schmitz G. Bile acid signaling after an oral glucose tolerance test. Chem Phys Lipids (2011) 164(6):525–9. doi:10.1016/j.chemphyslip.2011.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ChREBP, FXR, glucose-sensing, O-GlcNAcylation, liver metabolism
Citation: Benhamed F, Filhoulaud G, Caron S, Lefebvre P, Staels B and Postic C (2015) O-GlcNAcylation links ChREBP and FXR to glucose-sensing. Front. Endocrinol. 5:230. doi: 10.3389/fendo.2014.00230
Received: 09 October 2014; Accepted: 12 December 2014;
Published online: 13 January 2015.
Edited by:
Tarik Issad, University Paris Descartes, FranceReviewed by:
Heike M. Hermanns, Rudolf-Virchow-Center, GermanyChad Slawson, University of Kansas Medical Center, USA
Sabire Ozcan, University of Kentucky, USA
Copyright: © 2015 Benhamed, Filhoulaud, Caron, Lefebvre, Staels and Postic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Postic, 24 rue du Faubourg Saint Jacques, Paris, France e-mail: catherine.postic@inserm.fr