 Crystal D. C. Kamilaris
Crystal D. C. Kamilaris Constantine A. Stratakis
Constantine A. Stratakis- Section on Endocrinology and Genetics and Endocrinology Inter-institute Training Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
Multiple endocrine neoplasia type 1 (MEN1) is a rare hereditary tumor syndrome inherited in an autosomal dominant manner and characterized by a predisposition to a multitude of endocrine neoplasms primarily of parathyroid, enteropancreatic, and anterior pituitary origin, as well as nonendocrine neoplasms. Other endocrine tumors in MEN1 include foregut carcinoid tumors, adrenocortical tumors, and rarely pheochromocytoma. Nonendocrine manifestations include meningiomas and ependymomas, lipomas, angiofibromas, collagenomas, and leiomyomas. MEN1 is caused by inactivating mutations of the tumor suppressor gene MEN1 which encodes the protein menin. This syndrome can affect all age groups, with 17% of patients developing MEN1-associated tumors before 21 years of age. Despite advances in the diagnosis and treatment of MEN1-associated tumors, patients with MEN1 continue to have decreased life expectancy primarily due to malignant neuroendocrine tumors. The most recent clinical practice guidelines for MEN1, published in 2012, highlight the need for early genetic and clinical diagnosis of MEN1 and recommend an intensive surveillance approach for both patients with this syndrome and asymptomatic carriers starting at the age of 5 years with the goal of timely detection and management of MEN1-associated neoplasms and ultimately decreased disease-specific morbidity and mortality. Unfortunately, there is no clear genotype-phenotype correlation and individual mutation-dependent surveillance is not possible currently.
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal dominant hereditary tumor syndrome with a high degree of penetrance, that is caused by inactivating mutations of the tumor suppressor gene MEN1, and is characterized by a predisposition to a multitude of endocrine and nonendocrine tumors (1). MEN1 was first described as early as 1903 by Erdheim and was defined by Underdahl and Werner ~5 decades later (2–4). The syndrome is classically comprised of hyperplasia and/or tumors of parathyroid, enteropancreatic, and/or anterior pituitary origin, that develop in 90, 30–70, and 30–40% of patients, respectively, by age 40 (5–8). Other endocrine tumors noted with increased frequency in MEN1 include foregut carcinoid tumors, such as thymic and bronchial carcinoid, and gastric enterochromaffin-like tumors, which each have a penetrance of 2%, 2%, and 10%, respectively by 40 years of age. Adrenocortical tumors develop in 40% of affected individuals by this age, whereas pheochromocytomas are rare (<1%). Nonendocrine manifestations are predominantly comprised of neoplasms of the central nervous system including, meningiomas, ependymomas, and schwannomas, cutaneous lesions such as lipomas, angiofibromas, and collagenomas, and smooth muscle tumors such as leiomyomas (9–15). Women with MEN1 appear to have an increased risk of breast cancer (16). MEN1 can affect all age groups with an estimated prevalence of 2 per 100,000 and no apparent gender predilection (17). Though extensive data is lacking regarding MEN1 in young individuals, in a cohort of 924 patients with MEN1, 160 patients, or 17%, developed MEN1-associated tumors before 21 years of age. In these young patients, primary hyperparathyroidism (PHPT) was diagnosed in 75% by age 21, followed by pituitary adenomas in 34%, pancreatic neuroendocrine tumors (pNETs) in 34% (14% non-functional pNETs [NF-pNETs], 12% insulinomas, and 2% gastrinomas), adrenal tumors in 1% (2 patients), and thymic neuroendocrine tumor (th-NET) in 1% (1 patient). MEN1-associated neoplasms were diagnosed as early as within the first 5 years of life, though the majority were diagnosed after the age of 10 with increasing disease penetrance with age (18).
Despite advances in the diagnosis and treatment of MEN1-associated tumors, patients with this syndrome continue to have a decreased life expectancy compared to the rest of the population, with a mean age of death of 55–60 years (19, 20). Furthermore, delayed MEN1 diagnosis has been associated with potential harm to patients, as demonstrated in a recent study where family members of MEN1 index cases developed metastatic NETs before or during the lag time (median lag time 3.5 years) between the diagnosis of MEN1 in the index case and genetic testing of family members for the disease (21, 22). The cause of death in 50–70% of patients with MEN1 is related to the disease itself (23–26). Through the advent of new treatments, the most common cause of death has shifted from the complications of hormone-excess states, primarily due to gastrinomas, to malignant NETs, most notably pNETs and th-NETs. The most recent Endocrine Society clinical practice guidelines for MEN1 published in 2012, recommend an intensive surveillance approach for patients with MEN1 and asymptomatic carriers starting at the age of 5 years, based on the assumption that early detection and management of MEN1-associated neoplasms may lead to decreased morbidity and mortality (8).
Genetics and Molecular Pathogenesis
MEN1 may be inherited in an autosomal dominant manner, with 90% of individuals diagnosed with this disease having an affected parent, and only 10% having a de novo MEN1 germline mutation (27). The MEN1 gene, located on chromosome 11 (11q13), was first identified in 1997, and spans ~9,000 base pairs of genomic DNA containing 10 exons. This gene encodes the protein menin (1, 28). Germline heterozygous mutations in MEN1, are identified in 70–90% of familial MEN1 cases with the frequency of finding a de novo mutation being significantly lower in sporadic MEN1 cases (27). More than 1,200 germline mutations in the MEN1 gene have been identified, which are scattered over the entire coding region of the gene without any significant hot spots or genotype-phenotype correlations (27, 29). The majority of MEN1 germline mutations (69%) are predicted to be pathogenic due to either premature truncation of menin due to frame-shift mutations (42%) and nonsense mutations (14%), or exon region deletions which are attributed to splicing defects (10.5%) and large deletions (2.5%) (27, 29). Other MEN1 germline mutations include missense mutations (25.5%) and single or few amino acid in-frame deletions or insertions (5.5%), which require further investigation to determine their pathogenicity.
Approximately 5–25% of patients with MEN1 may not have mutations in the MEN1 coding region. These individuals may have whole or partial gene deletions, and it has been postulated that mutations may also occur in the promoter or untranslated regions (27, 30, 31). In addition, the occurrence of phenocopies, or patients that develop disease manifestations typically associated with mutations in the MEN1 gene but instead are due to another etiology, has been described in 5–10% of MEN1 kindreds (32–34). These phenocopies may occur in individuals with a family history of MEN1 and one MEN1-associated tumor or in patients with two MEN1-associated tumors with other gene involvement.
MEN1 phenocopies can be attributed to multiple endocrine neoplasia type 4 (MEN4) in 1–2% of cases. This syndrome results from inactivating mutations of the tumor suppressor gene CDKNIB, that encodes the p27kip1 inhibitor of cyclin dependent kinase 2, with manifestations such as parathyroid and pituitary adenomas, neuroendocrine tumors and various benign, and malignant tumors. Less common germline mutations that may be identified in another 1–2% of phenocopies include those attributed to mutations in genes encoding additional members of the cyclin-dependent kinase inhibitor (CDKN) family, such as CDKN1A (P21cip1), CDKN2B (p15Ink4b), or CDKN2C (p18Ink4c) (34–37). These CDKN genetic defects should be evaluated in patients that present as MEN1-like phenocopies.
Additional genes to be considered for screening in phenocopies include CDC73 (also known as HRPT2) which is a tumor suppressor gene that encodes the protein parafibromin (mutations implicated in hyperparathyroid-jaw tumor syndrome), CaSR which encodes the calcium sensing receptor (mutations associated with familial benign hypocalciuric hypercalcemias), GNA11 that encodes the G-protein alpha 11, and AP2S1 which encodes the adaptor-related protein complex 2, sigma 1 subunit, particularly in patients with familial hyperparathyroidism. Defects in AIP, which encodes the aryl hydrocarbon receptor interacting protein and is associated with familial isolated pituitary adenomas, should also be evaluated in children and adolescents with prolactinoma or somatotropinoma (8). Germline MEN1 mutations have also been noted in families with a parathyroid only disorder, familial isolated primary hyperparathyroidism, where there is a higher frequency of missense mutations compared to patients with the MEN1 syndrome (27, 29, 38, 39). Similarly, germline MEN1 mutations have been reported in 5 cases of “sporadic” pNETs (40).
MEN1 acts as a tumor suppressor gene. Patients with germline inactivating mutations in MEN1 demonstrate loss of heterozygosity (LOH) in more than 90% of their tumors, though LOH involving chromosome 11q13 has also been observed in 5–50% of sporadic endocrine tumors (27). Neoplasms develop (as described in Knudson's two-hit hypothesis), when a second somatic inactivating mutation occurs in one MEN1 allele in the setting of the preexisting germline inactivating mutation in the alternate allele (41). The protein product of MEN1, menin, is implicated in the regulation of transcription, genome stability, cell division, and cell proliferation, though the exact role of menin in tumorigenesis is yet to be elucidated (1, 27, 28, 42). Menin is composed of 610 amino acids, and is a highly conserved, ubiquitously expressed, predominantly nuclear protein, that does not show homology to any other protein. Two main nuclear localization signals (NLS), NLS1 and NLS2, as well as a third accessory NLS, NLSa, are harbored within the amino acid sequence of menin (43, 44). Mutations in MEN1 that lead to premature protein truncation may lead to functional inactivation of menin through loss of one or both main NLSs. Menin has not been demonstrated to have intrinsic enzymatic activity, but studies of protein-protein interaction by multiple groups have identified more than 50 proteins that could partner with menin. Furthermore, the crystal structure of menin demonstrates a deep pocket that can serve as a binding site for interacting proteins (45–47). Menin is predicted to be a multi-functional protein that plays a role in epigenetic regulation and gene transcription through interaction with proteins in chromatin-associated protein complexes and transcription factors, with regulation of gene expression of target genes such as those that control cell proliferation. Similarly, through its protein partners, menin has also been implicated in playing a possible role in DNA-repair associated with response to DNA damage, cell signaling, cytoskeletal structure, cell division, cell adhesion, and/or cell motility (42, 48–50). In genetically engineered mouse models, germline targeted deletion of both copies of the Men1 gene leads to death in utero, whereas germline targeted deletion of one copy of the Men1 gene results in live mice that develop endocrine tumors similar to those in humans (47, 51).
Screening for MEN1 mutations in the appropriate setting has several benefits including confirmation of the clinical diagnosis of MEN1, identification of family members that are carriers of MEN1 mutations, so that appropriate screening and/or treatment can be implemented, and identification of the 50% of family members that do not harbor a pathogenic mutation in MEN1 and thus do not require screening. Genetic testing for defects in the MEN1 gene currently includes PCR-based screening for mutations in the coding region and splice junctions. If a mutation is not identified by the aforementioned method, then multiplex ligation probe amplification (MLPA)-based screening is performed in order to detect large deletions of the MEN1 gene. This testing should be performed in a clinical genetics laboratory that is accredited to perform MEN1 mutational analysis. Twenty-four different polymorphisms of the MEN1 gene have been described and should be distinguished from mutations (27). According to the most recent clinical practice guidelines published in 2012 by Thakker et al. (8) MEN1 mutational analysis should be performed in index cases with two or more MEN1-associated endocrine tumors (parathyroid, pancreatic, or pituitary tumors), asymptomatic first-degree relatives of a known MEN1 mutation carrier, first-degree relatives of a MEN1 mutation carrier expressing familial MEN1 (in order to exclude phenocopies), and in patients with suspicious or atypical MEN1 (individuals with parathyroid adenomas occurring prior to 30 years of age or multi-gland parathyroid disease, gastrinoma, or multiple pNET at any age, or those who have at least two MEN1-associated tumors not part of the classical triad of parathyroid, entero-pancreatic, and anterior pituitary tumors) (Table 1). When possible, this testing should be undertaken in the first decade of life, and as early as before the age of 5, with the goal of detecting and preventing significant morbidity and even mortality (8, 18, 52). All individuals should be provided genetic counseling prior to and following testing.

Table 1. Indications for MEN1 mutational analysis* (8).
Clinical Manifestations and Diagnosis
The diagnosis of MEN1 can be made on the basis of clinical, familial, and/or genetic criteria (Table 2). Per the most recent clinical practice guidelines, a patient can be diagnosed with MEN1 by meeting any one of the following three conditions: the occurrence of at least two primary MEN1-associated endocrine tumors (i.e., parathyroid adenoma, enteropancreatic tumor, and pituitary adenoma); the development of one MEN1-associated tumor in a first degree relative of a patient with a clinical diagnosis of MEN1; and the identification of a germline MEN1 mutation in an individual, who may be asymptomatic without biochemical or radiological evidence of MEN1 (8). Genetic evaluation in family members of patients with MEN1 was recently shown to result in the diagnosis of MEN 1, 10 years earlier than clinical or biochemical diagnosis (53).

Table 2. Diagnostic criteria for MEN1* (8).
Parathyroid Tumors
Primary hyperparathyroidism (PHPT), due to parathyroid hyperplasia and/or adenoma, is the most common and often earliest endocrine manifestation in MEN1 and occurs primarily in the third decade of life with 100% penetrance by the age of 50 (5, 8). Though PHPT in children is predominantly diagnosed after the age of 10, asymptomatic PHPT has been described in patients with MEN1 as young as 4 years-old, with symptomatic disease diagnosed as early as 8 years of age (18). Furthermore, it is estimated that 1–18% of patients diagnosed with PHPT will have MEN1. The degree of hypercalcemia in these individuals is usually mild and patients may be asymptomatic or present with polydipsia, polyuria, constipation, malaise, altered mentation, hypertension, shortened QT interval, peptic ulcer disease, urolithiasis, and/or decreased bone mineral density with increased fracture risk. There is a high prevalence of urolithiasis and early bone mineral loss in young individuals with MEN1-associated PHPT, and bone or renal complications are progressively more frequent, extensive, and severe in long-standing PHPT cases and in those associated with gastrinoma (54–58). Severe hypercalcemia or parathyroid cancer are rare (59). Compared to sporadic PHPT, parathyroid disease in MEN1 occurs at an earlier age (20–25 years of age vs. 55 years) with an equal male to female ratio (1:1 vs. 1:3), and typically has multiple gland involvement, with ultimate involvement of all four glands (as compared to 80–85% of patients with sporadic PHPT that have single gland disease). MEN1-associated PHPT has a high recurrence rate after apparently successful subtotal parathyroidectomy that reaches up to 50% 12 years postoperatively (compared to 4–16% recurrence in sporadic PHPT) (60). In addition, a greater decline in bone mineral density is noted in patients with PHPT due to MEN1 when compared to patients with sporadic PHPT, with decreased bone mineral density recovery in the lumbar spine, femur, and particularly in the distal radius when compared to sporadic cases 1 year postoperatively (8, 60–63).
Parathyroid tumors in patients with multi-gland disease are asynchronous and vary in size, with each tumor likely representing a different clonal adenoma (64). In addition, a “negative feedback loop” between miR-24-1 and menin, has recently been suggested in MEN1-associated parathyroid tumorigenesis. This “negative feedback loop” mimics the second hit in Knudson's hypothesis by silencing the expression of the second MEN1 wild type allele through a post-transcriptional, reversible, epigenetic effect, which may precede the permanent genetic deletion or inactivation of the second wild type allele (65).
PHPT is often diagnosed incidentally by biochemical testing that demonstrates hypercalcemia in association with inappropriately increased parathyroid hormone (PTH) levels. Given the multi-glandular nature of parathyroid involvement in MEN1, preoperative imaging to localize the parathyroid tumors, such as ultrasound of the neck, Tc99m-sestamibi parathyroid scintigraphy, or chest computed tomography (CT), is of limited value, with the exception of possible use in cases of recurrent or persistent PHPT. However, some groups perform pre-operative imaging in order to evaluate for ectopic parathyroid glands, or more recently, in order to plan for less extensive surgeries, but these protocols need to be validated by large series and post-operative long-term follow-up (66–69). Bilateral neck exploration during surgery should be planned in an attempt to identify all pathological parathyroid glands (70).
Surgery is the treatment of choice for PHPT though the optimal timing, type, and extent of parathyroid surgery are unclear and should be decided on a case by case basis. Whether early parathyroid surgery reduces morbidity and mortality in patients with MEN1 is yet to be elucidated. Some clinicians favor early intervention to reduce the years a patient is at risk of developing bone disease, while others prefer delaying surgery as much as possible with the goal of avoiding the risk of postsurgical hypoparathyroidism and limiting the number of surgeries a patient may need to undergo for management of recurrent PHPT. Indications for parathyroidectomy in patients with MEN1 are similar to those for individuals with sporadic PHPT and include symptomatic or marked hypercalcemia/hypercalciuria, nephrolithiasis, and/or evidence of bone disease (8, 71–74). Furthermore, parathyroidectomy may be required in the treatment of patients with severe peptic ulcer disease or other symptoms caused by gastrinoma (the Zollinger-Ellison syndrome [ZES]) that are poorly controlled with medical management, as hypercalcemia typically worsens hypergastrinemia (57, 75). The recommended surgical approach for the treatment of MEN1-associated PHPT is subtotal parathyroidectomy with removal of 3.5 parathyroid glands. Total parathyroidectomy with heterotopic auto-transplantation of fresh or cryopreserved normal parathyroid tissue into the forearm or neck may also be an option for certain patients, particularly those that have extensive disease either at initial or repeat surgery (8). Previous observational studies have noted that the total parathyroidectomy approach has been associated with lower rates of persistent hyperparathyroidism but higher rates of hypoparathyroidism. This was demonstrated in a review of 18 reports that included 2–73 patients with MEN1 followed for 4–12 years after subtotal parathyroidectomy with or without cervical thymectomy, where persistent hyperparathyroidism was noted in 0–33%, recurrent hyperparathyroidism in 0–36%, and persistent hypoparathyroidism in 0–35% of patients. The same review analyzed 10 reports that included 4–36 patients with MEN1 who underwent total parathyroidectomy with cervical thymectomy and heterotopic auto-transplantation of a small parathyroid graft, where after a mean follow up of 6–10 years, patients were noted to have persistent hyperparathyroidism in 0–3%, recurrent hyperparathyroidism in 0–55%, and hypoparathyroidism in 0–46% (76). Furthermore, in prior observational studies, patients who underwent less than subtotal parathyroidectomy more commonly had persistent disease, with 42% of patients who underwent <3 gland parathyroidectomy having persistent disease compared to 0–12% of those who had three or more parathyroid glands resected (57, 76–78). However, a recent retrospective study from a single institution that compared 8 patients with MEN1 that underwent “unilateral clearance” (preoperative localization of a single enlarged parathyroid gland with resection of that parathyroid gland, the remaining ipsilateral parathyroid gland, as well as ipsilateral transcervical thymectomy) to 16 patients with MEN1 who underwent subtotal parathyroidectomy, showed that one patient from each group had persistent disease (12.5 and 6.25%, respectively), 13% of patients with unilateral clearance and 31% of patients with subtotal parathyroidectomy had recurrent PHPT after a mean follow up of 47 (2–112) and 68 (1–223) months, whereas no unilateral clearance patients and 2 (12.5%) patients who underwent subtotal parathyroidectomy had permanent hypoparathyroidism (69). During parathyroidectomy, intraoperative monitory of PTH is suggested in order to monitor the correct ablation of all adenomatous and/or hyperplastic parathyroid glands (79). Furthermore, transcervical thymectomy is recommended in all patients with MEN1 during initial parathyroid surgery, as supernumerary glands are identified in 6–20% of MEN1 patients, and intrathymic parathyroid tissue may be a source of PTH excess (80, 81). Calcimimetics, such as cinacalcet, have been used to treat patients with hyperparathyroidism in whom surgery has failed or who were poor surgical candidates (82). Though data is limited in patients with MEN1, cinacalcet has been shown to have calcium lowering effects, but current evidence does not show an improvement in bone mineral density in the spine or femur or in hypercalciuria (83).
Enteropancreatic Neuroendocrine Tumors
Pancreatic neuroendocrine tumors (pNETs) are estimated to occur in 30–80% of patients with MEN1, and in up to 80–100% of patients in postmortem studies. MEN1 is the most common hereditary syndrome associated with pNETs with approximately 10% of all pNETs being associated with MEN1 (84). In addition, somatic MEN1 mutations can be observed in >40% of sporadic pNETs (40, 85). These tumors may secrete excessive quantities of hormone such as gastrin, insulin, vasoactive intestinal polypeptide (VIP), glucagon, or somatostatin and may be associated with distinct clinical syndromes, with approximately one third of these neoplasms becoming clinically apparent. Also, these tumors may be nonfunctional (NF-pNET), either non-secretory, or releasing hormonally inactive peptides such as pancreatic polypeptide, chromogranin A, neurotensin, neuron-specific enolase, or ghrelin (8). By the age of 40, the prevalence of gastrinoma is 30–40%, that of insulinoma is 10%, and that of other functioning pNETs such as glucagonoma, VIPoma, somatostatinoma, etc. is 2%, whereas the prevalence of non-functioning enteropancreatic tumors including those that secrete pancreatic polypeptide is 20–55% (5). Pancreatic neuroendocrine tumors have an earlier age of onset in patients with MEN1, when compared to sporadic cases (10–50 years of age vs. 50–80 years), and tend to be multiple (as opposed to single in sporadic pNETs), most commonly leading to diffuse microadenomatosis (tumors <0.5 cm), with <13% of patients developing larger tumors (>2 cm) which are often nonfunctional (5, 6, 86–88). As previously mentioned, given the effective treatment of PHPT, pituitary disease, and Zollinger-Ellison syndrome (ZES), the malignant potential of enteropancreatic NETs, particularly NF-pNETs, is now the primary cause of death in patients with MEN1. Several MEN1 genotype-phenotype correlations regarding pNET growth and/or malignant potential have been identified (mutations in JunD, CHES1, truncating mutations in the N- or C- terminal regions of the MEN1 gene [exons 2,8,9] and the CDKN1B V109G polymorphism), and particular pathological characteristics such as high mitotic count in large NF-pNETs, may characterize more aggressive tumors, however these have not been studied prospectively and are not broadly used (7, 89–95).
Gastrinoma is the most common functional enteropancreatic tumor in MEN1 and may lead to ZES (hypergastrinemia with recurrent peptic ulcerations), with ZES being the initial clinical manifestation in up to 40% of patients with MEN1. Up to 60% of patients with MEN1 have either ZES or asymptomatic elevation in serum gastrin concentration (96). Furthermore, 20–60% of patients with ZES have underlying MEN1 (97). ZES may present with gastroesophageal reflux disease (GERD) and peptic ulcer disease (PUD), which are often refractory, with or without diarrhea. In a study of 160 patients with MEN1 below the age of 21, 6 patients (or 2%), developed ZES before the age of 21, with the youngest patient to develop the disease being 6 years of age (18). Gastrinomas in MEN1 are typically diagnosed by the age of 40 (on average 10 years earlier than sporadic gastrinomas) and are most commonly microadenomas that occur primarily in the duodenum (>80%), and less frequently in the pancreas. They may be complicated by lymph node metastases in 34–85% of cases, and hepatic metastases in 6–16% of cases at the time of diagnosis (98–100). The diagnosis of gastrinoma is indicated by elevated fasting serum gastrin concentration (gastrin levels 10 times greater than the upper limit of normal) in the presence of hyperchloridria or gastric pH <2. If fasting gastrin levels are below this threshold (1,000 pg/ml), then gastrin stimulation (either by secretin or calcium infusion) may be necessary for diagnosis. Localization, which should include evaluation for metastases, may be comprised of gastroscopy and endoscopic ultrasound (EUS), particularly for duodenal gastrinomas, as well as other imaging procedures (ultrasound [US], CT, magnetic resonance imaging [MRI], selective abdominal angiography, somatostatin receptor scintigraphy [with In-DPTA-octreotide or octreoscan], 68Ga-DOTATATE-PET [positron emission tomography]/CT), with combined EUS and MRI being the most sensitive for pancreatic disease. The combined use of intraarterial calcium injections with hepatic venous gastrin sampling may also be used for diagnosis (101–103). Though they typically grow slowly, gastrinomas can frequently metastasize to the peripancreatic lymph nodes and less commonly to the liver, with a higher mortality rate than insulinomas but not than other pNETs (26). Lymph node metastases do not inevitably result in a poor prognosis or a high probability that clinically significant metastases will occur, and the risk of death from lymph node metastases due to MEN1 associated gastrinoma is less than that for sporadic cases (104). The prognosis of patients with MEN1-associated gastrinomas is associated with tumor size and the presence of hepatic metastases, with increased risk of hepatic metastases with increasing tumor size (103, 105). Treatment of MEN1-associated gastrinomas is primarily comprised of medical therapy which aims to decrease gastric acid secretion through the use of histamine 2-receptor antagonists, proton pump inhibitors, and/or somatostatin analogs (SSAs). These medications have shown long-term effectiveness and safety in controlling hypergastrinemia and the complications of ZES. Furthermore, SSAs have been demonstrated to have an anti-neoplastic effect, though data is lacking regarding their effectiveness in malignant and/or metastatic gastrinoma (106, 107). Surgical treatment of gastrinomas/ZES in patients with MEN1 is controversial given the multiplicity and small size of these tumors, that leads to rare surgical cure without aggressive resections. Surgery is suggested when concomitant NF-NETs double in size over 6 months or exceed 2 centimeters in diameter, with possible surgical approaches including Thompson's procedure (excision of the duodenal gastrinoma through a longitudinal excision), duodeno-pancreatectomy, or pancreas-preserving surgery (8, 108). The use of surgical resection is also limited in advanced metastatic disease, with treatment involving several approaches that may include medical therapy (with everolimus, tyrosine kinase inhibitors such as sunitinib, or chemotherapy), peptide radioreceptor therapy with lutetium-177-labeled somatostatin analog, chemotherapy, or liver directed therapies (such as embolization, chemoembolization, or radioembolization) (109). As aforementioned, hypercalcemia from concomitant PHPT may worsen the symptoms of ZES, with parathyroidectomy leading to decreased fasting and secretin simulated gastrin levels and basal acid secretion (57). Another special consideration in patients with ZES is the increased incidence of Cushing syndrome that has been described in these patients, which is due to a corticotroph pituitary adenoma causing Cushing disease, as opposed to ectopic ACTH secretion from an islet cell tumor which is more commonly seen in conjunction with sporadic gastrinomas (110).
Insulinomas are the second most common type of functional enteropancreatic neoplasm in patients with MEN1, representing 10–30% of pNETs in this group (8). They are the most frequent cause of endogenous hyperinsulinemic hypoglycemia in adult patients, with approximately 4% of insulinomas occurring in patients with MEN1. These tumors comprise the first MEN1-associated manifestation in 10% of patients with the syndrome. In contrast to sporadic insulinomas, which tend to occur after the age of 40, MEN1-associated insulinomas occur primarily in patients younger than 40 years-old, with many of these tumors arising in patients younger than 20 years of age (5, 17, 18, 86, 111). In the aforementioned cohort of 160 patients with MEN1 below the age of 21, insulinoma occurred in 10% of patients and as early as 5 years of age (18). These tumors are solitary in 85% and multiple in 6–13% of cases, and are associated with other pNETs at the time of diagnosis in 10% of patients (112). They are scattered across the pancreas and tend to be small, with 82% of these neoplasms measuring less than 2 centimeters and 47% measuring less than 1 centimeter, which may make them difficult to localize (113). Diagnosis is established with a 72-hour fast through documentation of hypoglycemia with characteristic symptoms that are rapidly reversed by glucose administration and with demonstration of inappropriately increased plasma insulin, c-peptide, and/or proinsulin, in the setting of hypoglycemia (8). Localization is pursued with ultrasound, CT, and/or MRI, with EUS demonstrating a sensitivity of 94% (114). If these studies fail to localize the insulinoma(s), more invasive techniques such as selective angiography with intra-arterial calcium stimulation and hepatic venous sampling for insulin levels may localize more than 80% of these neoplasms (115, 116). In addition, more novel somatostatin receptor-targeting PET modalities such as 68Ga-DOTATATE-PET/CT, may aide in localization, with PET imaging with labeled glucagon-like peptide 1 (GLP1) analogs being a promising possible future modality (117–120). Medical therapy is often unsuccessful in patients with MEN1-associated insulinomas, with surgery being the treatment of choice. Unlike patients with MEN1-associated gastrinoma/ZES that are rarely cured with pNET enucleation or local gastrinoma excision, and often require more extensive resections, those with other functional pNETs including insulinomas are often cured without extensive surgical resections, but may have recurrent disease (108). There is no consensus on the optimal extent of surgical resection in patients with insulinoma, which can range from enucleation of a single tumor, to distal pancreatectomy or partial pancreatectomy, to excision of all the macroscopic pancreatic neoplasms and enucleation of nodules in the remaining pancreas (8). Monitoring of the insulin/glucose ratio during surgery may be helpful in assessing successful insulinoma excision (121). Many experts suggest distal pancreatic resection associated with enucleation of any additional pancreatic head tumors as the treatment of choice (122, 123). Metastatic insulinoma is rare, occurring in 4–14% of cases, with the goal of treatment, as in other malignant functioning pNETs, being symptom control and tumor volume reduction with possible resection of lesions that are amenable to surgery (124, 125). In cases with unresectable disease or in patients that need further symptom control, adjuvant therapy may vary from diazoxide and SSAs to everolimus and chemotherapy, as well as local therapies (radiofrequency ablation [RFA], chemoembolization, or radiotherapy) or peptide receptor radionuclide therapy (PRRT) (112).
Glucagonomas occur in <3% of MEN1 patients, though NF-pNETs may have positive immunostaining for glucagon (5, 111, 121, 126). These tumors most frequently occur in the tail of the pancreas, and 50–80% of patients have metastases at the time of diagnosis (127). Patients may present with characteristic symptoms such as skin rash (necrolytic migratory erythema), weight loss, anemia, and stomatitis (8). Asymptomatic patients may be detected incidentally through findings on imaging and/or biochemical evidence of glucose intolerance or hyperglucagonemia. VIP-secreting tumors or VIPomas, are rare in MEN1 and present with watery diarrhea, hypokalemia, and achlorhydria (WDHA) (127). Like glucagonomas, these neoplasms are primarily located in the pancreatic tail. The diagnosis of a VIPoma is made through evidence of a fasting stool volume >1–2 liters per day and a significantly increased plasma VIP concentration, in the absence of laxative or diuretic use (8). The treatment approach for both glucagonomas and VIPomas is similar to that for insulinomas as described above (112).
Non-functioning pNETs are among the most common enteropancreatic tumors in MEN1 (6, 7, 128, 129). Detection of NF-pNETs has significantly increased in the setting of standardized biochemical and imaging protocols in patients with MEN1 (8, 128, 130). Furthermore, pathology from pancreatic resections in patients with MEN1 has revealed numerous microtumors that were not shown preoperatively on imaging (131). NF-pNETs have been detected in patients with MEN1 as early as 12–14 years of age, and patients between the ages of 10 and 20 have been reported as developing large tumors (18, 87, 132, 133). EUS is the most sensitive modality for detecting NF-pNETS, with a combined strategy of EUS and MRI being the most useful. 68Ga-DOTATATE-PET/CT has been reported to have a high sensitivity for detecting NETs in MEN1, at times leading to a change in management. This study may be added to identify metastases in patients with NF-pNETs (119, 134). In addition, 18F-fluorodeoxyglucose (FDG) PET/CT imaging can be valuable in predicting the malignant potential of pNETs (135). There is lack of agreement regarding the optimal indications for surgery in patients with NF-pNETs, with major considerations including the size of the neoplasm and other considerations being the growth rate, presence of metastases, and tumor histology. Previous studies have shown an increased rate of metastases in patients with larger tumors (6, 7, 87, 136). However, there is controversy among experts regarding the tumor size cutoff for offering surgery, with the suggested cutoff ranging between 1 and 3 centimeters (8, 112, 137). There is a consensus that withholding surgery for tumors that reach or exceed 3 centimeters in size leads to a negative prognosis, however the indication for surgical intervention in smaller-sized tumors is unclear (138). The Endocrine Society clinical practice guidelines for MEN1 suggest surgical resection for NF-pNETs >1 cm in size, or tumors <1 cm in size with significant growth, such as doubling of tumor size over 3–6 months and exceeding 1 cm in size (8). The 1-centimeter cutoff for surgical NF-pNET resection has also been suggested by several other groups of experts including the Uppsala group, the Marburg group, and the MEN consortium in Japan (139–141). However, the European Neuroendocrine Tumor Society and the Groupe d'Etude des Tumeurs Endocrines (GTE) suggest conservative management for tumors <2 cm in size that do not demonstrate characteristics of aggressive behavior such as rapid growth (108, 136). Furthermore, two recent studies advocating for conservative management for NF-pNETs that are 2 cm or less in size, demonstrated low disease-specific mortality without loss of oncologic safety at this threshold (138, 142). In terms of surgical approach, the least invasive procedure is typically considered best, with lymph node dissection in addition to pancreatic resection according to the type and location of the tumors (108). Treatment options for unresectable pNETs or advanced metastatic disease may include biotherapies such as SSAs, interferon alpha, mechanistic target of rapamycin (mTOR) inhibitors, receptor tyrosine kinase (RTK) inhibitors, including platelet-derived growth factor receptor (PDGFR) and vascular endothelial growth factor receptor (VEGFR) inhibitors, and vascular endothelial growth factor A (VEGFA) antibodies. Chemotherapy may be appropriate for patients with metastatic pNETs with a high tumor burden, high proliferative index, rapid tumor progression, and/or symptoms not controlled with biotherapy. Radiological therapies such as PRRT, or localized interventional radiological treatment using RFA, transarterial embolization (TAE), transarterial chemoembolization (TACE) and selective internal radiation therapy (SIRT) may also be considered in select cases (143).
Anterior Pituitary Tumors
The prevalence of anterior pituitary tumors in patients with MEN1 varies widely among series between 10 and 60%, and was noted to be 42% and 38.1% in two large cohorts (144–146). Pituitary tumors (PITs) have been reported as early as the age of 5 years and can occur as late as the ninth decade of life, with onset primarily in the fourth decade (30, 52). Furthermore, these tumors were noted in 35% of patients in a large cohort of pediatric patients, comprising the second most common MEN1-associated neoplasm in this group, and the first lesion identified in 21% of cases (18). Overall, PITs may be the first MEN1-associated lesion to develop in at least 10–15% of cases, and up to 3% of patients with the diagnosis of a PIT will have MEN1 (111, 147–149). Approximately 60% of these tumors secrete prolactin (PRL), 25% release growth hormone (GH), 5% secrete ACTH, and the remaining are primarily nonfunctional (NFTs) (147, 150, 151). This profile is similar to that of sporadic PITs but differs from that of other hereditary pituitary tumors (due to GNAS, AIP, or PRKAR1A defects) that secrete primarily GH. In a study that included 324 patients, MEN1-associated PITs were described as being larger (macroadenomas in 85 vs. 42% in sporadic cases), with more aggressive behavior (one third of MEN1-associated cases were observed to have invasive features), and reduced response to medical therapy (42 vs. 90% in sporadic cases), but without increased prevalence of pituitary carcinoma (146, 147, 151). However, in a subsequent study that included 323 patients with MEN1, two thirds of the PITs were microadenomas. Tumors detected by screening were predominantly nonfunctional microadenomas, that did not require treatment and were stable in size over time. Symptom development that required treatment was rare during long-term follow-up (median 6 years). Furthermore, the response rate to medical therapy of prolactinomas was more than 90% (144). MEN1-associated PITs have been shown to demonstrate plurihormonal expression more frequently than sporadic tumors (146, 151). However, in contrast with other MEN1-associated neoplasms, multiple pituitary tumors are rare (152). Clinical manifestations of PITs in MEN1 are similar to those with sporadic PITs, and include those that are due to hormone excess, and those that are related to the size of the tumor. Patients with prolactinomas may present with amenorrhea, galactorrhea and infertility in women, or erectile dysfunction and infertility in men. Those with excess GH or ACTH secretion may present with signs or symptoms of gigantism or acromegaly, or Cushing syndrome, respectively. Individuals with enlarging PITs may experience headaches and signs of compression of adjacent structures such visual field disturbances in the setting of optic chiasm compression, and/or hypopituitarism. Biochemical evaluation can lead to the diagnosis of hormone excess syndromes, whereas MRI of the pituitary gland is the imaging study of choice to diagnose PITs (8). Treatment of PITs in patients with MEN1 is similar to treatment in sporadic disease. Medical therapy with dopamine agonists is first-line for prolactinomas, whereas transsphenoidal surgery (TSS) and radiotherapy are reserved for drug-resistant tumors. For GH- and ACTH-secreting tumors the initial treatment of choice is often TSS. When surgery is indicated, TSS is also the primary treatment for non-functioning PITs, particularly in macroadenomas associated with visual signs and symptoms or that are in close proximity to the optic chiasm and/or those that demonstrate rapid growth.
Other Tumors
Several other neoplasms occur with increased frequency in MEN1 including foregut carcinoid tumors, adrenocortical tumors, cutaneous tumors, meningiomas and ependymomas, tumors of smooth muscle, and very rarely pheochromocytomas.
MEN1-associated carcinoid tumors include thymic, bronchial, and gastric enterochromaffin-like cell NETs (8). Thymic NETs (th-NETs), occur in 2–8% of patients with MEN1, and are associated with strong heritability and a poor prognosis with frequent recurrence and increased risk of death (153–159). They occur mostly in men, whereas carcinoids in women are primarily bronchial (157, 160, 161). Heavy smoking may be a risk factor (161). Th-NETs have been identified as early as 16 years of age and may be fatal even in young patients (18). These tumors are primarily nonfunctional and are the most common cause of anterior mediastinal masses in MEN1. Most individuals may be asymptomatic at diagnosis, however liver, bone, and lung metastases may be identified. Biochemical evaluation is not useful diagnostically in these patients with the diagnostic studies of choice being CT or MRI of the chest (112, 162). MEN1-associated th-NETs comprise the second most common MEN1-related cause of death and have a high mortality rate, with the 10-year survival rate in several larger studies ranging between 25 and 45% (153, 157, 163). Treatment is primarily surgical and complete resection of thymic tumors is recommended. Adjuvant radiotherapy may be used for incomplete resection and/or positive surgical margins. Medical treatments that can be considered for unresectable/metastatic disease, include those that are implemented in sporadic th-NETs such as SSAs, chemotherapy, mTOR inhibitors, and PRRT (112). Prophylactic transcervical thymectomy is typically performed at the time of parathyroidectomy for PHPT, however this does not eliminate the risk of development of th-NETs, due to residual thymic tissue (81, 164). The frequency of bronchial NETs (br-NETs) in MEN1 has been reported to range from 3 to 13%, and may even reach 31%, depending on the mode of diagnosis (imaging and/or pathology), with the frequency of histologically proven br-NETs ranging from 4.6 to 6.6% (153, 158, 165–168). Br-NETs have not been described in patients younger than 20 years old, with increasing penetrance with age which reaches 8.1% at 60 years (165). Presenting symptoms may include dyspnea, hemoptysis, cough, and/or flushing, however most patients are asymptomatic (158, 165, 169). Several cases of br-NETs have been described in the same families (165). Most br-NETs are typical or atypical carcinoids, though small- and large-cell neuroendocrine carcinomas were identified in 10% of cases in a cohort of 51 patients (165). Alhough, br-NETs do not decrease overall survival in patients with MEN1, poorly differentiated and aggressive br-NETs can lead to death. Chest CT or MRI may be used to detect these tumors. Management is similar to that for sporadic br-NETs, with surgical resection being the recommended treatment modality, though it is possible that small non-central lesions can be followed without affecting mortality (112). Gastric carcinoids (type II gastric enterochromaffin-like cell [ECL] carcinoids) and ECL proliferation (a precursor to gastric carcinoid) have been described in patients with MEN1 and ZES. In a cohort of 57 patients with MEN1 and ZES, ECL proliferative changes were universally present, whereas advanced changes were noted in 53% and carcinoids were diagnosed in 23% (170). Recent studies have demonstrated that these Type II gastric carcinoids may be aggressive and metastasize to the liver. However, disease is localized in at least 70 to 90% of cases and these tumors can often be excised endoscopically after assessment of the extent of invasion by EUS. In certain patients, tumors are present in excessive numbers or are large and may be invasive. In these patients, more aggressive resection and additional therapy with SSAs or CCK-B receptor antagonists may be considered (171). Annual surveillance with endoscopy is recommended.
Asymptomatic adrenocortical tumors (ACTs) may be detected in 20–73% of patients with MEN1 depending on the imaging modality used for screening (172–175). These lesions are primarily nonfunctioning and may include cortical adenomas that may be multiple, hyperplasia, cysts, or carcinomas. Less than 10% of MEN1-associated ACTs demonstrate hormone hypersecretion, which can lead most commonly to primary hyperaldosteronism and/or ACTH-independent Cushing syndrome (174). Hyperandrogenemia may occasionally be detected, particularly in the setting of an adrenocortical carcinoma (ACC). ACC has been described in 1% of patients, manifesting as early as 3 years of age. In the previously described group of 160 young patients under 21 years of age, 2 patients were diagnosed with ACC (18). Pheochromocytoma is rare with a prevalence of <1% by age 40 (5). Patients with signs or symptoms concerning for a functional ACT or pheochromocytoma, and those with tumors >1 cm on imaging should be evaluated biochemically for hormone excess. The incidence of ACC in tumors >1 cm in size rises to 13% (174). Patients with MEN1 and adrenal tumors should receive annual imaging, with consideration for surgical tumor resection if tumors are larger than 4 centimeters, display atypical radiological features and are 1–4 cm in size, or show significant growth over 6 months. Treatment of functional adrenal tumors in MEN1 is similar to that of sporadic neoplasms (172–174).
Cutaneous manifestations are common in MEN1 and may include lipomas, angiofibromas, and collagenomas, with the penetrance of these lesions by age 40 being 30, 85, and 70%, respectively (5). Angiofibromas and collagenomas tend to be multiple and are more common in patients with MEN1 than in the general population (64% vs. 8% and 62% vs. 5%, respectively). The presence of angiofibromas and/or collagenomas may be a strong indicator of the diagnosis of MEN1. In one study that included 110 patients with gastrinoma, including 48 individuals with MEN1 and 62 without MEN1, the presence angiofibromas or collagenomas (single or multiple), had a sensitivity of 50–65% for MEN1 and a specificity of 92–100%. The combined finding of at least 3 angiofibromas and any collagneoma had a sensitivity of 75% and a specificity of 95% in diagnosing MEN1 (176).
Surveillance
The most recent Endocrine Society clinical practice guidelines for MEN1 recommend a comprehensive surveillance scheme commencing at the age of 5, with the goal of early detection and management of MEN1-associated manifestations and tumors (Table 3) (8). Unfortunately, there is no clear genotype-phenotype correlation and individual mutation-dependent surveillance is not possible at this time.
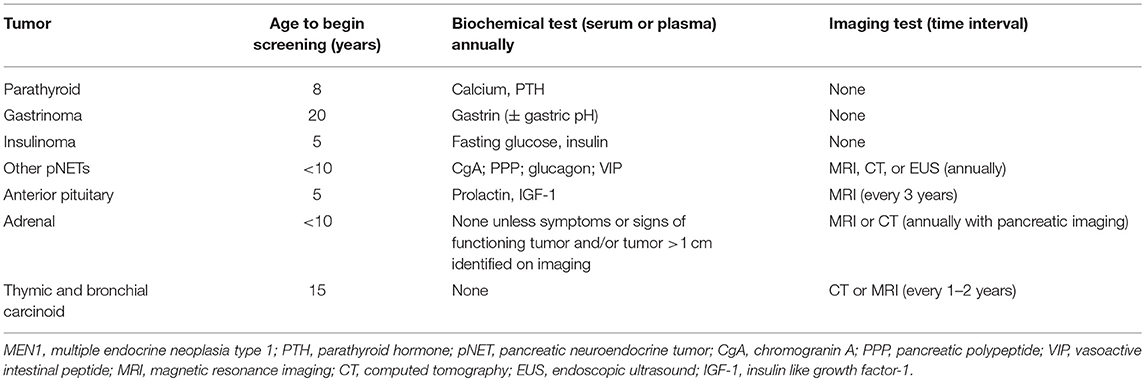
Table 3. Recommended biochemical and radiological surveillance for MEN1-associated tumors in patients with MEN1 and MEN1 mutation carriers (8).
Screening for hyperparathyroidism with annual biochemical evaluation of calcium and PTH is recommended starting at the age of 8, which is the age at which the earliest symptomatic patient with PHPT has been described (asymptomatic hyperparathyroidism has been observed as early as age 4) (18). Biochemical evaluation for gastrinoma is recommended beginning at the age of 20, though 2% of cases have been detected in patients younger than 21 years-old. Screening for gastrinoma is performed through annual evaluation of gastrin (± gastric pH). Evaluation for insulinoma should commence at age 5, which is the age at which the earliest symptomatic case has been reported, and should include annual insulin and glucose measurement. For other pNETs the annual measurement of chromogranin-A, pancreatic polypeptide, glucagon, and VIP is recommended beginning before the age of 10. However, the sensitivity of these measurements to detect foregut NETs is unclear, as two retrospective analyses of the use of chromogranin A, pancreatic polypeptide, and glucagon to screen for the emergence of MEN1-associated neoplasms found that these tests were not effective in early tumor diagnosis, either singly or in combination (177, 178). Imaging with MRI, CT, or EUS, is also recommended annually to evaluate for pNETs beginning before the age of 10 years, as large pancreatic tumors may develop between 10 and 20 years of age. Surveillance for anterior PITs is recommended starting at age 5, which is consistent with the age of the earliest case of MEN1-associated PIT that has been reported and includes annual measurement of serum PRL and insulin-like growth factor 1 (IGF-1) and MRI of the pituitary every 3 years. Evaluation for MEN1-associated adrenal tumors is primarily based on imaging either with CT or MRI that can be performed in conjunction with pancreatic imaging, with biochemical testing indicated only in the setting of symptoms or signs of a functional tumor or the presence of a tumor >1 cm. Screening for adrenal neoplasms is recommended beginning before the age of 10, as ACC has been identified in a child as young as 3 years of age. Lastly, screening for thymic and bronchial NETs is recommended starting at the age of 15, with CT or MRI recommended every 1–2 years. This is consistent with the diagnosis of fatal cases of th-NETs in patients as young as 16 years-old, though br-NETs have not been reported in patients younger than 20 years-old.
Author Contributions
CS and CK wrote and contributed to the manuscript equally.
Conflict of Interest Statement
CS holds patents on PRKAR1A and other genes of the cyclic AMP pathway and their genetics and applications. CS lab has received research funding from Pfizer Inc. for the study of gigantism and/or acromegaly.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the research project Z01-HD008920 (Principal Investigator: CS) of the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, USA.
References
1. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. (1997) 276:404–7. doi: 10.1126/science.276.5311.404
2. Erdheim J. Normal and pathological histology of the glandula thyroidea, parathyroidea and hypophysis. Beitr Zur Pathol Anatomie Zur Allgemeinen Pathol. (1903) 33:158–236.
3. Underdahl LO, Woolner LB, Black BM. Multiple endocrine adenomas; report of 8 cases in which the parathyroids, pituitary and pancreatic islets were involved. J Clin Endocrinol Metab. (1953) 13:20–47. doi: 10.1210/jcem-13-1-20
4. Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. (1954) 16:363–71. doi: 10.1016/0002-9343(54)90353-8
5. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. (2001) 86:5658–71. doi: 10.1210/jcem.86.12.8070
6. Triponez F, Dosseh D, Goudet P, Cougard P, Bauters C, Murat A, et al. Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg. (2006) 243:265–72. doi: 10.1097/01.sla.0000197715.96762.68
7. Kouvaraki MA, Shapiro SE, Cote GJ, Lee JE, Yao JC, Waguespack SG, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg. (2006) 30:643–53. doi: 10.1007/s00268-006-0360-y
8. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230
9. Chigot JP, Bendib S, Turpin G, Benlian P. Characteristic pathological associations in multiple endocrine neoplasia type 1'. Presse Med. (1996) 25:1229–33.
10. Kato H, Uchimura I, Morohoshi M, Fujisawa K, Kobayashi Y, Numano F, et al. Multiple endocrine neoplasia type 1 associated with spinal ependymoma' Intern Med. (1996) 35:285–9. doi: 10.2169/internalmedicine.35.285
11. Giraud S, Choplin H, Teh BT, Lespinasse J, Jouvet A, Labat-Moleur F, et al. A large multiple endocrine neoplasia type 1 family with clinical expression suggestive of anticipation. J Clin Endocrinol Metab. (1997) 82:3487–92. doi: 10.1210/jc.82.10.3487
12. Doumith R, de Gennes JL, Cabane JP, Zygelman N. Pituitary prolactinoma, adrenal aldosterone-producing adenomas, gastric schwannoma and colonic polyadenomas: a possible variant of multiple endocrine neoplasia (MEN) type I. Acta Endocrinol. (1982) 100:189–95. doi: 10.1530/acta.0.1000189
13. Pack S, Turner ML, Zhuang Z, Vortmeyer AO, Boni R, Skarulis M, et al. Cutaneous tumors in patients with multiple endocrine neoplasia type 1 show allelic deletion of the MEN1 gene. J Investig Dermatol. (1998) 110:438–40. doi: 10.1046/j.1523-1747.1998.00140.x
14. McKeeby JL, Li X, Zhuang Z, Vortmeyer AO, Huang S, Pirner M, et al. Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol. (2001) 159:1121–7. doi: 10.1016/S0002-9440(10)61788-9
15. Asgharian B, Chen YJ, Patronas NJ, Peghini PL, Reynolds JC, Vortemyeyer A, et al. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res. (2004) 10:869–80. doi: 10.1158/1078-0432.CCR-0938-3
16. van Leeuwaarde RS, Dreijerink KM, Ausems MG, Beijers HJ, Dekkers OM, de Herder WW, et al. MEN1-dependent breast cancer: indication for early screening? results from the Dutch MEN1 study group. J Clin Endocrinol Metab. (2017) 102:2083–90. doi: 10.1210/jc.2016-3690
17. Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1) (vol 89, pg 653, 1996). Q Mon J Assoc Phys. (1996) 89:957–58. doi: 10.1093/qjmed/89.9.653
18. Goudet P, Dalac A, Le Bras M, Cardot-Bauters C, Niccoli P, Levy-Bohbot N, et al. MEN1 disease occurring before 21 years old: a 160-patient cohort study from the groupe d'etude des tumeurs endocrines. J Clin Endocrinol Metab. (2015) 100:1568–77. doi: 10.1210/jc.2014-3659
19. Ito T, Igarashi H, Uehara H, Berna MJ, Jensen RT. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine. (2013) 92:135–81. doi: 10.1097/MD.0b013e3182954af1
20. Norton JA, Krampitz G, Zemek A, Longacre T, Jensen RT. Better survival but changing causes of death in patients with multiple endocrine neoplasia type 1. Ann Surg. (2015) 261:e147–8. doi: 10.1097/SLA.0000000000001211
21. van Leeuwaarde RS, van Nesselrooij BP M, Hermus AR, Dekkers OM, de Herder WW, et al. Impact of delay in diagnosis in outcomes in MEN1: results from the Dutch MEN1 study group. J Clin Endocrinol Metab. (2016) 101:1159–65. doi: 10.1210/jc.2015-3766
22. de Laat JM, van Leeuwaarde RS, Valk GD. The importance of an early and accurate MEN1 diagnosis. Front Endocrinol. (2018) 9:533. doi: 10.3389/fendo.2018.00533
23. Dean PG, van Heerden JA, Farley DR, Thompson GB, Grant CS, Harmsen WS, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg. (2000) 24:1437–41. doi: 10.1007/s002680010237
24. Doherty GM, Olson JA, Frisella MM, Lairmore TC, Wells SA, Norton JA. Lethality of multiple endocrine neoplasia type I. World J Surg. (1998) 22:581–87. doi: 10.1007/s002689900438
25. Geerdink EA, Van der Luijt RB, Lips CJM. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol. (2003) 149:577–82. doi: 10.1530/eje.0.1490577
26. Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A, Ruszniewski P, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg. (2010) 34:249–55. doi: 10.1007/s00268-009-0290-1
27. Lemos MC, Thakker RV. Multiple endocrine neoplaslia type 1 (MEN 1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutation. (2008) 29:22–32. doi: 10.1002/humu.20605
28. Lemmens I, VandeVen WJM, Kas K, Zhang CX, Giraud S, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Hum Mol Genetics. (1997) 6:1177–83. doi: 10.1093/hmg/6.7.1177
29. Concolino P, Costella A, Capoluongo E. Multiple endocrine neoplasia type 1 (MEN1): an update of 208 new germline variants reported in the last nine years. Cancer Genetics. (2016) 209:36–41. doi: 10.1016/j.cancergen.2015.12.002
30. Sakurai A, Suzuki S, Kosugi S, Okamoto T, Uchino S, Miya A, et al. Multiple endocrine neoplasia type 1 in Japan: establishment and analysis of a multicentre database. Clin Endocrinol. (2012) 76:533–39. doi: 10.1111/j.1365-2265.2011.04227.x
31. Tham E, Grandell U, Lindgren E, Toss G, Skogseid B, Nordenskjold M. Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases. J Clin Endocrinol Metab. (2007) 92:3389–95. doi: 10.1210/jc.2007-0476
32. Burgess JR, Nord B, David R, Greenaway TM, Parameswaran V, Larsson C, et al. Phenotype and phenocopy: the relationship between genotype and clinical phenotype in a single large family with multiple endocrine neoplasia type 1 (MEN 1). Clin Endocrinol. (2000) 53:205–11. doi: 10.1046/j.1365-2265.2000.01032.x
33. Newey PJ, Thakker RV. Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract. (2011) 17:8–17. doi: 10.4158/EP10379.RA
34. Turner JJO, Christie PT, Pearce SH S, Turnpenny PD, Thakker RV. Diagnostic challenges due to phenocopies: lessons from multiple endocrine neoplasia type1 (MEN1). Hum Mutation. (2010) 31:E1089–101. doi: 10.1002/humu.21170
35. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. (2009) 94:1826–34. doi: 10.1210/jc.2008-2083
36. Pellegata NS. MENX and MEN4. Clinics. (2012) 67(Suppl 1):13–8. doi: 10.6061/clinics/2012(Sup01)04
37. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA. (2006) 103:15558–63. doi: 10.1073/pnas.0603877103
38. Hannan FM, Nesbit MA, Christie PT, Fratter C, Dudley NE, Sadler GP, et al. Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab. (2008) 4:53–8. doi: 10.1038/ncpendmet0718
39. Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine. (2002) 81:1–26. doi: 10.1097/00005792-200201000-00001
40. Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. (2017) 543:65–71. doi: 10.1038/nature21063
41. Knudson AG. Mutation and cancer - statistical study of retinoblastoma. Proc Natl Acad Sci USA. (1971) 68:820–3. doi: 10.1073/pnas.68.4.820
42. Matkar S, Thiel A, Hua XX. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci. (2013) 38:394–402. doi: 10.1016/j.tibs.2013.05.005
43. Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA. (1998) 95:1630–34. doi: 10.1073/pnas.95.4.1630
44. La P, Desmond A, Hou Z, Silva AC, Schepp RW, Hua X. Tumor suppressor menin: the essential role of nuclear localization signal domains in coordinating gene expression. Oncogene. (2006) 25:3537–46. doi: 10.1038/sj.onc.1209400
45. Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. (2012) 482:542–6. doi: 10.1038/nature10806
46. Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. (2011) 286:31742–8. doi: 10.1074/jbc.M111.258186
47. Agarwal SK. The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer. (2017) 24:T119–34. doi: 10.1530/ERC-17-0199
48. Balogh K, Patocs A, Hunyady L, Racz K. Menin dynamics and functional insight: take your partners. Mol Cell Endocrinol. (2010) 326:80–4. doi: 10.1016/j.mce.2010.04.011
49. Hendy GN, Kaji H, Canaff L. Cellular functions of menin. Adv Exp Med Biol. (2009) 668:37–50. doi: 10.1007/978-1-4419-1664-8_4
50. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. (2014) 386:2–15. doi: 10.1016/j.mce.2013.08.002
51. Agarwal SK. Exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. Int J Endocr Oncol. (2014) 1:153–61. doi: 10.2217/ije.14.16
52. Stratakis CA, Schussheim DH, Freedman SM, Keil MF, Pack SD, Agarwal SK, et al. Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. (2000) 85:4776–80. doi: 10.1210/jc.85.12.4776
53. Giusti F, Cianferotti L, Boaretto F, Cetani F, Cioppi F, Colao A, et al. Multiple endocrine neoplasia syndrome type 1: institution, management, and data analysis of a nationwide multicenter patient database. Endocrine. (2017) 58:349–59. doi: 10.1007/s12020-017-1234-4
54. Burgess JR, David R, Greenway TM, Parameswaran V, Shephard JJ. Osteoporosis in Multiple Endocrine Neoplasia Type 1: severity, clinical significance, relationship to primary hyperparathyroidism, and response to parathyroidectomy. Arch Surg. (1999) 134:1119–23. doi: 10.1001/archsurg.134.10.1119
55. Christopoulos C, Antoniou N, Thempeyioti A, Calender A, Economopoulos P. Familial multiple endocrine neoplasia type 1:the urologist is first on the scene. BJU Int. (2005) 96:884–7. doi: 10.1111/j.1464-410X.2005.05731.x
56. Lourenco DM Jr., Toledo RA, Mackowiak II, Coutinho FL, Cavalcanti MG, Correia-Deur JE, et al. Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. Eur J Endocrinol. (2008) 159:259–74. doi: 10.1530/EJE-08-0153
57. Norton JA, Venzon DJ, Berna MJ, Alexander HR, Fraker DL, Libutti SK, et al. Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg. (2008) 247:501–10. doi: 10.1097/SLA.0b013e31815efda5
58. Lourenco DM Jr., Coutinho FL, Toledo RA, Montenegro FL, Correia-Deur JE, Toledo SP. Early-onset, progressive, frequent, extensive, and severe bone mineral and renal complications in multiple endocrine neoplasia type-1 associated primary hyperparathyroidism. J Bone Miner Res. (2010) 25:2382–91. doi: 10.1002/jbmr.125
59. Di Meo G, Sgaramella LI, Ferraro V, Prete FP, Gurrado A, Testini M. Parathyroid carcinoma in multiple endocrine neoplasm type 1 syndrome: case report and systematic literature review. Clin Exp Med. (2018) 18:585–93. doi: 10.1007/s10238-018-0512-7
60. Rizzoli R, Green J III, Marx SJ. Primary hyperparathyroidism in familial multiple endocrine neoplasia type I. Long-term follow-up of serum calcium levels after parathyroidectomy. Am J Med. (1985) 78:467–74. doi: 10.1016/0002-9343(85)90340-7
61. Eller-Vainicher C, Chiodini I, Battista C, Viti R, Mascia ML, Massironi S, et al. Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity. J Bone Miner Res. (2009) 24:1404–10. doi: 10.1359/jbmr.090304
62. Coutinho FL, Lourenco DM Jr., Toledo RA, Montenegro FL, Correia-Deur JE, Toledo SP. Bone mineral density analysis in patients with primary hyperparathyroidism associated with multiple endocrine neoplasia type 1after total parathyroidectomy. Clin Endocrinol. (2009) 72:462–8. doi: 10.1111/j.1365-2265.2009.03672.x
63. Silva AM, Vodopivec D, Christakis I, Lyons G, Wei Q, Waguespack SG, et al. Operative intervention for primary hyperparathyroidism offers greater bone recovery in patients with sporadic disease than in those with multiple endocrine neoplasia type 1-related hyperparathyroidism. Surgery. (2017) 161:107–15. doi: 10.1016/j.surg.2016.06.065
64. Dwight T, Nelson AE, Theodosopoulos G, Richardson AL, Learoyd DL, Philips J, et al. Independent genetic events associated with the development of multiple parathyroid tumors in patients with primary hyperparathyroidism. Am J Pathol. (2002) 161:1299–306. doi: 10.1016/S0002-9440(10)64406-9
65. Luzi E, Marini F, Giusti F, Galli G, Cavalli L, Brandi ML. The negative feedback-loop between the Oncomir Mir-24-1 and Menin modulates the men1 tumorigenesis by mimicking the “Knudson's Second Hit”. PLoS ONE. (2012) 7:e39767. doi: 10.1371/journal.pone.0039767
66. Tonelli F, Marini F, Giusti F, Brandi ML. Total and subtotal parathyroidectomy in young patients with multiple endocrine neoplasia type 1-related primary hyperparathyroidism: potential post-surgical benefits and complications. Front Endocrinol. (2018) 9:558. doi: 10.3389/fendo.2018.00558
67. Montenegro FLM, Brescia MDG, Lourenco DM Jr., Arap SS, d'Alessandro AF, de Britto E, Silva Filho G, et al. Coud the less-than subtotal parathyroidectomy be an option for treating young patients with multiple endocrine neoplasia type 1-related hyperparathyroidism? Front Endocrinol. (2019) 10:123. doi: 10.3389/fendo.2019.00123
68. Versnick M, Popadich A, Sidhu S, Sywak M, Robinson B, Delbridge L. Minimally invasive parathyroidectomy provides a conservative surgical option for multiple endocrine neoplasia type1-primary hyperparathyroidism. Surgery. (2013) 154:101–5. doi: 10.1016/j.surg.2013.03.004
69. Kluijfhout WP, Beninato T, Drake FT, Vriens MR, Gosnell J, Shen WT, et al. Unilateral clearance for primary hyperparathyroidism in selected patients with multiple endocrine neoplasia type 1. World J Surg. (2016) 40:2964–69. doi: 10.1007/s00268-016-3624-9
70. Nobecourt PF, Zagzag J, Asare EA, Perrier ND. Intraoperative decision-making and technical aspects of parathyroidectomy in young patinets with MEN1 related hyperparathyroidism. Front Endocrinol. (2018) 9:618. doi: 10.3389/fendo.2018.00618
71. Lourenco DM Jr., Coutinho FL, Toledo RA, Goncalves TD, Montenegro FLM, Toledo SPA. Biochemical, bone and renal patters in hyperparathyroidism associated with multiple endocrine neoplasia type 1. Clinics. (2012) 67:99–108. doi: 10.6061/clinics/2012(Sup01)17
72. Giusti F, Tonelli F, Brandi ML. Primary hyperparathyroidism in multiple endocrine neoplasia type 1: when to perform surgery? Clinics. (2012) 67(Suppl. 1):141–4. doi: 10.6061/clinics/2012(Sup01)23
73. Marini F, Giusti F, Tonelli F, Brandi ML. When parathyroidectomy should be indicated or postponed in adolescents with MEN1-related primary hyperparathyroidism. Front Endocrinol. (2018) 9:597. doi: 10.3389/fendo.2018.00597
74. Marx SJ, Lourenco DM Jr. Questions and controversies about parathyroid pathophysiology in children with multiple endocrine neoplasia type 1. Front Endocrinol. (2018) 9:359. doi: 10.3389/fendo.2018.00359
75. Mai HD, Sanowski RA. Regression of duodenal gastrinomas in a patient with multiple endocrine neoplasia type I after parathyroidectomy. Gastrointest Endosc. (1992) 38:706–8. doi: 10.1016/S0016-5107(92)70571-9
76. Tonelli F, Marcucci T, Giudici F, Falchetti A, Brandi ML. Surgical approach in hereditary hyperparathyroidism. Endocr J. (2009) 56:827–41. doi: 10.1507/endocrj.K09E-204
77. Schreinemakers JM, Pieterman CR, Scholten A, Vriens MR, Valk GD, Rinkes IH. The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg. (2011) 35:1993–2005. doi: 10.1007/s00268-011-1068-9
78. Stalberg P, Carling T. Familial parathyroid tumors: diagnosis and management. World J Surg. (2009) 33:2234–43. doi: 10.1007/s00268-009-9924-6
79. Nilubol N, Weisbrod AB, Weinstein LS, Simonds WF, Jensen RT, Phan GQ, et al. Utility of intraoperative parathyroid hormone monitoring in patients with multiple endocrine neoplasia type 1-associated primary hyperparathyroidism undergoing initial parathyroidectomy. World J Surg. (2013) 37:1966–72. doi: 10.1007/s00268-013-2054-1
80. Langer P, Kann PH, Fendrich V, Richter G, Diehl S, Rothmund M, et al. Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg. (2004) 28:1317–22. doi: 10.1007/s00268-004-7642-7
81. Powell AC, Alexander HR, Pingpank JF, Steinberg SM, Skarulis M, Bartlett DL, et al. The utility of routine transcervical thymectomy for multiple endocrine neoplasia 1-related hyperparathyroidism. Surgery. (2008) 144:878–83; discussion 83–4. doi: 10.1016/j.surg.2008.08.031
82. Moyes VJ, Monson JP, Chew SL, Akker SA. Clinical use of cinacalcet in MEN1 hyperparathyroidism. Int J Endocrinol. (2010) 2010:906163. doi: 10.1155/2010/906163
83. Giusti F, Cianferotti L, Gronchi G, Cioppi F, Masi L, Faggiano A, et al. Cinacalcet therapy in patients affected by primary hyperparathyroidism associated to multiple endocrine neoplasia syndrome type 1 (MEN1). Endocrine. (2016) 52:495–506. doi: 10.1007/s12020-015-0696-5
84. Marx SJ, Simonds WF. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev. (2005) 26:615–61. doi: 10.1210/er.2003-0037
85. Jiao YC, Shi CJ, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. (2011) 331:1199–203. doi: 10.1126/science.1200609
86. Machens A, Schaaf L, Karges W, Frank-Raue K, Bartsch DK, Rothmund M, et al. Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol. (2007) 67:613–22. doi: 10.1111/j.1365-2265.2007.02934.x
87. Newey PJ, Jeyabalan J, Walls GV, Christie PT, Gleeson FV, Gould S, et al. Asymptomatic children with multiple endocrine neoplasia type 1 mutations may harbor nonfunctioning pancreatic neuroendocrine tumors. J Clin Endocrinol Metab. (2009) 94:3640–46. doi: 10.1210/jc.2009-0564
88. Marini F, Giusti F, Tonelli F, Brandi ML. Management impact: effects on quality of life and prognosis in MEN1. Endocr Relat Cancer. (2017) 24:T227–42. doi: 10.1530/ERC-17-0203
89. Bartsch DK, Langer P, Wild A, Schilling T, Celik I, Rothmund M, et al. Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: surgery or surveillance? Surgery. (2000) 128:958–66. doi: 10.1067/msy.2000.109727
90. Bartsch DK, Slater EP, Albers M, Knoop R, Chaloupka B, Lopez CL, et al. Higher risk of aggressive pancreatic neuroendocrine tumors in MEN1 patients with MEN1 mutations affecting the CHES1 interacting MENIN domain. J Clin Endocrinol Metab. (2014) 99:E2387–91. doi: 10.1210/jc.2013-4432
91. Christakis I, Qiu W, Hyde SM, Cote GJ, Grubbs EG, Perrier ND, et al. Genotype-phenotype pancreatic neuroendocrine tumor relationship in multiple endocrine neoplasia type 1 patients: a 23-year experience at a single institution. Surgery. (2018) 163:212–17. doi: 10.1016/j.surg.2017.04.044
92. Circelli L, Ramundo V, Marotta V, Sciammarella C, Marciello F, Del Prete M, et al. Prognostic role of the CDNK1B V109G polymorphism in multiple endocrine neoplasia type 1. J Cell Mol Med. (2015) 19:1735–41. doi: 10.1111/jcmm.12552
93. Conemans EB, Brosens LAA, Raicu-Ionita GM, Pieterman CRC, de Herder WW, et al. Prognostic value of WHO grade in pancreatic neuro-endocrine tumors in multiple endocrine neoplasia type 1: results from the DutchMEN1 study group. Pancreatology. (2017) 17:766–72. doi: 10.1016/j.pan.2017.07.196
94. Thevenon J, Bourredjem A, Faivre L, Cardot-Bauters C, Calender A, Murat A, et al. Higher risk of death among MEN1 patients with mutations in the JunD interacting domain: a Groupe d'etude des Tumeurs Endocrines (GTE) cohort study. Hum Mol Genetics. (2013) 22:1940–8. doi: 10.1093/hmg/ddt039
95. Longuini VC, Lourenço DM Jr., Sekiya T, Meirelles O, Goncalves TD, Coutinho FL, et al. Association between the p27 rs2066827 variant and tumor multiplicity in patients harboring MEN1 germline mutations. Eur J Endocrinol. (2014) 171:335–42. doi: 10.1530/EJE-14-0130
96. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine. (2004) 83:43–83. doi: 10.1097/01.md.0000112297.72510.32
97. Pipeleers-Marichal M, Somers G, Willems G, Foulis A, Imrie C, Bishop AE, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. (1990) 322:723–7. doi: 10.1056/NEJM199003153221103
98. Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. (2008) 113:1807–43. doi: 10.1002/cncr.23648
99. Singh MH, Fraker DL, Metz DC. Importance of surveillance for multiple endocrine neoplasia-1 and surgery in patients with sporadic Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. (2012) 10:1262–9. doi: 10.1016/j.cgh.2012.08.014
100. Yates CJ, Newey PJ, Thakker RV. Challenges and controversies in management of pancreatic neuroendocrine tumours in patients with MEN1. Lancet Diabetes Endocrinol. (2015) 3:895–905. doi: 10.1016/S2213-8587(15)00043-1
101. Barbe C, Murat A, Dupas B, Ruszniewski P, Tabarin A, Vullierme MP, et al. Magnetic resonance imaging versus endoscopic ultrasonography for the detection of pancreatic tumours in multiple endocrine neoplasia type 1. Dig Liver Dis. (2012) 44:228–34. doi: 10.1016/j.dld.2011.09.014
102. Imamura M, Komoto I, Ota S, Hiratsuka T, Kosugi S, Doi R, et al. Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol. (2011) 17:1343–53. doi: 10.3748/wjg.v17.i10.1343
103. Turner JJ, Wren AM, Jackson JE, Thakker RV, Meeran K. Localization of gastrinomas by selective intra-arterial calcium injection. Clin Endocrinol. (2002) 57:821–5. doi: 10.1046/j.1365-2265.2002.01655.x
104. Gibril F, Jensen RT. Advances in evaluation and management of gastrinoma in patients with Zollinger-Ellison syndrome. Curr Gastroenterol Rep. (2005) 7:114–21. doi: 10.1007/s11894-005-0049-2
105. Norton JA, Fraker DL, Alexander HR, Venzon DJ, Doppman JL, Serrano J, et al. Surgery to cure the Zollinger-Ellison syndrome. N Engl J Med. (1999) 341:635–44. doi: 10.1056/NEJM199908263410902
106. Plockinger U. Diagnosis and treatment of gastrinomas in multiple endocrine neoplasia type 1 (MEN-1). Cancers. (2012) 4:39–54. doi: 10.3390/cancers4010039
107. Wolin EM. The expanding role of somatostatin analogs in the management of neuroendocrine tumors. Gastrointest Cancer Res. (2012) 5:161–8.
108. Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, Caplin M, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology. (2016) 103:153–71. doi: 10.1159/000443171
109. Norton JA, Foster DS, Ito T, Jensen RT. Gastrinomas: medical or surgical treatment. Endocrinol Metab Clin North Am. (2018) 47:577–601. doi: 10.1016/j.ecl.2018.04.009
110. Maton PN, Gardner JD, Jensen RT. Cushing's syndrome in patients with the Zollinger-Ellison syndrome. N Engl J Med. (1986) 315:1–5. doi: 10.1056/NEJM198607033150101
111. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab. (2010) 24:355–70. doi: 10.1016/j.beem.2010.07.003
112. Sadowski SM, Cadiot G, Dansin E, Goudet P, Triponez F. The future: surgical advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer. (2017) 24:T243–60. doi: 10.1530/ERC-17-0285
113. Service FJ, McMahon MM, O'Brien PC, Ballard DJ. Functioning insulinoma–incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. (1991) 66:711–9. doi: 10.1016/S0025-6196(12)62083-7
114. McLean AM, Fairclough PD. Endoscopic ultrasound in the localisation of pancreatic islet cell tumours. Best Pract Res Clin Endocrinol Metab. (2005) 19:177–93. doi: 10.1016/j.beem.2004.11.012
115. Brown CK, Bartlett DL, Doppman JL, Gorden P, Libutti SK, Fraker DL, et al. Intraarterial calcium stimulation and intraoperative ultrasonography in the localization and resection of insulinomas. Surgery. (1997) 122:1189–93; discussion 93–4. doi: 10.1016/S0039-6060(97)90226-9
116. Guettier JM, Kam A, Chang R, Skarulis MC, Cochran C, Alexander HR, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab. (2009) 94:1074–80. doi: 10.1210/jc.2008-1986
117. Antwi K, Fani M, Nicolas G, Rottenburger C, Heye T, Reubi JC, et al. Localization of hidden insulinomas with (6)(8)Ga-DOTA-exendin-4 PET/CT: a pilot study. J Nucl Med. (2015) 56:1075–8. doi: 10.2967/jnumed.115.157768
118. Nockel P, Babic B, Millo C, Herscovitch P, Patel D, Nilubol N, et al. Localization of insulinoma using 68Ga-DOTATATE PET/CT scan. J Clin Endocrinol Metab. (2017) 102:195–99. doi: 10.1210/jc.2016-3445
119. Sadowski SM, Millo C, Cottle-Delisle C, Merkel R, Yang LA, Herscovitch P, et al. Results of (68)gallium-DOTATATE PET/CT scanning in patients with multiple endocrine neoplasia type 1. J Am Coll Surg. (2015) 221:509–17. doi: 10.1016/j.jamcollsurg.2015.04.005
120. Sadowski SM, Neychev V, Millo C, Shih J, Nilubol N, Herscovitch P, et al. Prospective study of 68Ga-DOTATATE positron emission tomography/computed tomography for detecting gastro-entero-pancreatic neuroendocrine tumors and unknown primary sites. J Clin Oncol. (2016) 34:588–96. doi: 10.1200/JCO.2015.64.0987
121. Giudici F, Nesi G, Brandi ML, Tonelli F. Surgical management of insulinomas in multiple endocrine neoplasia type 1. Pancreas. (2012) 41:547–53. doi: 10.1097/MPA.0b013e3182374e08
122. Cougard P, Goudet P, Peix JL, Henry JF, Sarfati E, Proye C, et al. [Insulinomas in multiple endocrine neoplasia type 1. Report of a series of 44 cases by the multiple endocrine neoplasia study group]. Ann Chir. (2000) 125:118–23. doi: 10.1016/S0001-4001(00)00112-4
123. Hellman P, Goretzki P, Simon D, Dotzenrath C, Roher HD. Therapeutic experience of 65 cases with organic hyperinsulinism. Langenbecks Arch Surg. (2000) 385:329–36. doi: 10.1007/s004230000148
124. Baudin E, Caron P, Lombard-Bohas C, Tabarin A, Mitry E, Reznick Y, et al. [Malignant insulinoma: recommendations for workup and treatment]. Presse Med. (2014) 43:645–59. doi: 10.1016/j.lpm.2013.08.007
125. Pavel M, Baudin E, Couvelard A, Krenning E, Oberg K, Steinmuller T, et al. ENETS consensus guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. (2012) 95:157–76. doi: 10.1159/000335597
126. Dralle H, Krohn SL, Karges W, Boehm BO, Brauckhoff M, Gimm O. Surgery of resectable nonfunctioning neuroendocrine pancreatic tumors. World J Surg. (2004) 28:1248–60. doi: 10.1007/s00268-004-7609-8
127. Akerstrom G, Hellman P. Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. (2007) 21:87–109. doi: 10.1016/j.beem.2006.12.004
128. Thomas-Marques L, Murat A, Delemer B, Penfornis A, Cardot-Bauters C, Baudin E, et al. Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol. (2006) 101:266–73. doi: 10.1111/j.1572-0241.2006.00367.x
129. Wamsteker EJ, Gauger PG, Thompson NW, Scheiman JM. EUS detection of pancreatic endocrine tumors in asymptomatic patients with type 1 multiple endocrine neoplasia. Gastrointest Endosc. (2003) 58:531–5. doi: 10.1067/S0016-5107(03)01965-5
130. Pieterman CR, Conemans EB, Dreijerink KM, de Laat JM, Timmers HT, Vriens MR, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. (2014) 21:R121–42. doi: 10.1530/ERC-13-0482
131. Anlauf M, Schlenger R, Perren A, Bauersfeld J, Koch CA, Dralle H, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. (2006) 30:560–74. doi: 10.1097/01.pas.0000194044.01104.25
132. Goncalves TD, Toledo RA, Sekiya T, Matuguma SE, Maluf Filho F, Rocha MS, et al. Penetrance of functioning and nonfunctioning pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1 in the second decade of life. J Clin Endocrinol Metab. (2014) 99:E89–96. doi: 10.1210/jc.2013-1768
133. Lairmore TC, Piersall LD, DeBenedetti MK, Dilley WG, Mutch MG, Whelan AJ, et al. Clinical genetic testing and early surgical intervention in patients with multiple endocrine neoplasia type 1 (MEN 1). Ann Surg. (2004) 239:637–45; discussion 45–7. doi: 10.1097/01.sla.0000124383.98416.8d
134. van Treijen MJC, van Beek DJ, van Leeuwaarde RS, Vriens MR, Valk GD. Diagnosing nonfunctional pancreatic NETs in MEN1: the evidence base. J Endocr Soc. (2018) 2:1067–88. doi: 10.1210/js.2018-00087
135. Kornaczewski Jackson ER, Pointon OP, Bohmer R, Burgess JR. Utility of FDG-PET imaging for risk stratification of pancreatic neuroendocrine tumors in MEN1. J Clin Endocrinol Metab. (2017) 102:1926–33. doi: 10.1210/jc.2016-3865
136. Triponez F, Goudet P, Dosseh D, Cougard P, Bauters C, Murat A, et al. Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg. (2006) 30:654–62; discussion 63–4. doi: 10.1007/s00268-005-0354-9
137. Nell S, Borel Rinkes IHM, Verkooijen HM, Bonsing BA, van Eijck CH, et al. Early and late complications after surgery for MEN1-related nonfunctioning pancreatic neuroendocrine tumors. Ann Surg. (2018) 267:352–56. doi: 10.1097/SLA.0000000000002050
138. Nell S, Verkooijen HM, Pieterman CRC, de Herder WW, Hermus AR, et al. Management of MEN1 related nonfunctioning pancreatic NETs: a shifting paradigm: results from the DutchMEN1 study group. Ann Surg. (2018) 267:1155–60. doi: 10.1097/SLA.0000000000002183
139. Hanazaki K, Sakurai A, Munekage M, Ichikawa K, Namikawa T, Okabayashi T, et al. Surgery for a gastroenteropancreatic neuroendocrine tumor (GEPNET) in multiple endocrine neoplasia type 1. Surg Today. (2013) 43:229–36. doi: 10.1007/s00595-012-0376-5
140. Akerstrom G, Stalberg P, Hellman P. Surgical management of pancreatico-duodenal tumors in multiple endocrine neoplasia syndrome type 1. Clinics. (2012) 67(Suppl. 1):173–8. doi: 10.6061/clinics/2012(Sup01)29
141. Bartsch DK, Fendrich V, Langer P, Celik I, Kann PH, Rothmund M. Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg. (2005) 242:757–64, discussion 64–6. doi: 10.1097/01.sla.0000189549.51913.d8
142. Triponez F, Sadowski SM, Pattou F, Cardot-Bauters C, Mirallie E, Le Bras M, et al. Long-term follow-up of MEN1 patients who do not have initial surgery for small </ = 2 cm nonfunctioning pancreatic neuroendocrine tumors, an AFCE and GTE Study: Association Francophone de Chirurgie Endocrinienne and Groupe d'Etude des Tumeurs Endocrines. Ann Surg. (2018) 268:158–64. doi: 10.1097/SLA.0000000000002191
143. Frost M, Lines KE, Thakker RV. Current and emerging therapies for PNETs in patients with or without MEN1. Nat Rev Endocrinol. (2018) 14:216–27. doi: 10.1038/nrendo.2018.3
144. de Laat JM, Dekkers OM, Pieterman CR, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-term natural course of pituitary tumors in patients with MEN1: results from the DutchMEN1 study group (DMSG). J Clin Endocrinol Metab. (2015) 100:3288–96. doi: 10.1210/JC.2015-2015
145. Syro LV, Scheithauer BW, Kovacs K, Toledo RA, Londono FJ, Ortiz LD, et al. Pituitary tumors in patients with MEN1 syndrome. Clinics.(2012) 67(Suppl. 1):43–8. doi: 10.6061/clinics/2012(Sup01)09
146. Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab. (2002) 87:457–65. doi: 10.1210/jcem.87.2.8145
147. Beckers A, Betea D, Valdes Socin H, Stevenaert A. The treatment of sporadic versus MEN1-related pituitary adenomas. J Intern Med. (2003) 253:599–605. doi: 10.1046/j.1365-2796.2003.01164.x
148. Elston MS, McDonald KL, Clifton-Bligh RJ, Robinson BG. Familial pituitary tumor syndromes. Nat Rev Endocrinol. (2009) 5:453–61. doi: 10.1038/nrendo.2009.126
149. Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, et al. Multiple endocrine neoplasia type 1 (MEN1): not only inherited endocrine tumors. Genet Med. (2009) 11:825–35. doi: 10.1097/GIM.0b013e3181be5c97
150. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. (2011) 7:257–66. doi: 10.1038/nrendo.2011.40
151. Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol. (2008) 32:534–43. doi: 10.1097/PAS.0b013e31815ade45
152. Agarwal SK, Ozawa A, Mateo CM, Marx SJ. The MEN1 gene and pituitary tumours. Horm Res. (2009) 71(Suppl. 2):131–8. doi: 10.1159/000192450
153. de Laat JM, Pieterman CR, van den Broek MF, Twisk JW, Hermus AR, Dekkers OM, et al. Natural course and survival of neuroendocrine tumors of thymus and lung in MEN1 patients. J Clin Endocrinol Metab. (2014) 99:3325–33. doi: 10.1210/jc.2014-1560
154. de Laat JM, van der Luijt RB, Pieterman CR, Oostveen MP, Hermus AR, Dekkers OM, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med. (2016) 14:182. doi: 10.1186/s12916-016-0708-1
155. Ferolla P, Falchetti A, Filosso P, Tomassetti P, Tamburrano G, Avenia N, et al. Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series. J Clin Endocrinol Metab. (2005) 90:2603–9. doi: 10.1210/jc.2004-1155
156. Gibril F, Chen YJ, Schrump DS, Vortmeyer A, Zhuang Z, Lubensky IA, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. (2003) 88:1066–81. doi: 10.1210/jc.2002-021314
157. Goudet P, Murat A, Cardot-Bauters C, Emy P, Baudin E, du Boullay Choplin H, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1: a comparative study on 21 cases among a series of 761 MEN1 from the GTE (Groupe des Tumeurs Endocrines). World J Surg. (2009) 33:1197–207. doi: 10.1007/s00268-009-9980-y
158. Singh Ospina N, Thompson GB, Nichols CF III, Cassivi SD, Young WF. Thymic and bronchial carcinoid tumors in multiple endocrine neoplasia type 1: the Mayo Clinic experience from 1977 to 2013. Horm Cancer. (2015) 6:247–53. doi: 10.1007/s12672-015-0228-z
159. Thevenon J, Bourredjem A, Faivre L, Cardot-Bauters C, Calender A, Le Bras M, et al. Unraveling the intrafamilial correlations and heritability of tumor types in MEN1: a Groupe d'etude des Tumeurs Endocrines study. Eur J Endocrinol. (2015) 173:819–26. doi: 10.1530/EJE-15-0691
160. Doherty GM. Multiple endocrine neoplasia type 1. J Surg Oncol. (2005) 89:143–50. doi: 10.1002/jso.20181
161. Teh BT, McArdle J, Chan SP, Menon J, Hartley L, Pullan P, et al. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Medicine. (1997) 76:21–9. doi: 10.1097/00005792-199701000-00002
162. Manoharan J, Albers MB, Bartsch DK. The future: diagnostic and imaging advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer. (2017) 24:T209–25. doi: 10.1530/ERC-17-0231
163. Christakis I, Qiu W, Silva Figueroa AM, Hyde S, Cote GJ, Busaidy NL, et al. Clinical features, treatments, and outcomes of patients with thymic carcinoids and multiple endocrine neoplasia type 1 syndrome at MD Anderson cancer center. Horm Cancer. (2016) 7:279–87. doi: 10.1007/s12672-016-0269-y
164. Welch K, McHenry CR. The role of transcervical thymectomy in patients with hyperparathyroidism. Am J Surg. (2012) 203:292–5; discussion 95–6. doi: 10.1016/j.amjsurg.2011.09.013
165. Lecomte P, Binquet C, Le Bras M, Tabarin A, Cardot-Bauters C, Borson-Chazot F, et al. Histologically proven bronchial neuroendocrine tumors in MEN1: a GTE 51-case cohort study. World J Surg. (2018) 42:143–52. doi: 10.1007/s00268-017-4135-z
166. Bartsch DK, Albers MB, Lopez CL, Apitzsch JC, Walthers EM, Fink L, et al. Bronchopulmonary neuroendocrine neoplasms and their precursor lesions in multiple endocrine neoplasia type 1. Neuroendocrinology. (2016) 103:240–7. doi: 10.1159/000435921
167. Goudet P, Bonithon-Kopp C, Murat A, Ruszniewski P, Niccoli P, Menegaux F, et al. Gender-related differences in MEN1 lesion occurrence and diagnosis: a cohort study of 734 cases from the Groupe d'etude des Tumeurs Endocrines. Eur J Endocrinol. (2011) 165:97–105. doi: 10.1530/EJE-10-0950
168. Sachithanandan N, Harle RA, Burgess JR. Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer. (2005) 103:509–15. doi: 10.1002/cncr.20825
169. Fink G, Krelbaum T, Yellin A, Bendayan D, Saute M, Glazer M, et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. (2001) 119:1647–51. doi: 10.1378/chest.119.6.1647
170. Berna MJ, Annibale B, Marignani M, Luong TV, Corleto V, Pace A, et al. A prospective study of gastric carcinoids and enterochromaffin-like cell changes in multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: identification of risk factors. J Clin Endocrinol Metab. (2008) 93:1582–91. doi: 10.1210/jc.2007-2279
171. Norton JA, Krampitz G, Jensen RT. Multiple endocrine neoplasia: genetics and clinical management. Surg Oncol Clin N Am. (2015) 24:795–832. doi: 10.1016/j.soc.2015.06.008
172. Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg. (2002) 26:891–6. doi: 10.1007/s00268-002-6492-4
173. Schaefer S, Shipotko M, Meyer S, Ivan D, Klose KJ, Waldmann J, et al. Natural course of small adrenal lesions in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Eur J Endocrinol. (2008) 158:699–704. doi: 10.1530/EJE-07-0635
174. Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol. (2012) 166:269–79. doi: 10.1530/EJE-11-0679
175. Scarsbrook AF, Thakker RV, Wass JA, Gleeson FV, Phillips RR. Multiple endocrine neoplasia: spectrum of radiologic appearances and discussion of a multitechnique imaging approach. Radiographics. (2006) 26:433–51. doi: 10.1148/rg.262055073
176. Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab. (2004) 89:5328–36. doi: 10.1210/jc.2004-0218
177. Qiu W, Christakis I, Silva A, Bassett RLJr, Cao L, Meng QH, et al. Utility of chromogranin A, pancreatic polypeptide, glucagon and gastrin in the diagnosis and follow-up of pancreatic neuroendocrine tumours in multiple endocrine neoplasia type 1 patients. Clin Endocrinol. (2016) 85:400–7. doi: 10.1111/cen.13119
Keywords: MEN1, hyperparathyroidism, pituitary adenoma, enteropancreatic tumor, carcinoid, adrenocortical tumor
Citation: Kamilaris CDC and Stratakis CA (2019) Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front. Endocrinol. 10:339. doi: 10.3389/fendo.2019.00339
Received: 24 October 2018; Accepted: 10 May 2019;
Published: 11 June 2019.
Edited by:
Delmar Muniz Lourenco Jr., University of São Paulo, BrazilReviewed by:
Dario Giuffrida, Mediterranean Institute of Oncology (IOM), ItalySabrina Corbetta, University of Milan, Italy
Copyright © 2019 Kamilaris and Stratakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Constantine A. Stratakis, stratakc@mail.nih.gov