 Janak L. Pathak
Janak L. Pathak Nathalie Bravenboer
Nathalie Bravenboer Jenneke Klein-Nulend
Jenneke Klein-Nulend- 1Key Laboratory of Oral Medicine, Guangzhou Institute of Oral Disease, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou, China
- 2Department of Clinical Chemistry, Amsterdam University Medical Centers, Amsterdam Movement Sciences, Vrije Universiteit Amsterdam, Amsterdam, Netherlands
- 3Department of Oral Cell Biology, Academic Centre for Dentistry Amsterdam, Amsterdam Movement Sciences, University of Amsterdam and Vrije Universiteit Amsterdam, Amsterdam, Netherlands
Osteocytes are the most abundant (~95%) cells in bone with the longest half-life (~25 years) in humans. In the past osteocytes have been regarded as vestigial cells in bone, since they are buried inside the tough bone matrix. However, during the last 30 years it has become clear that osteocytes are as important as bone forming osteoblasts and bone resorbing osteoclasts in maintaining bone homeostasis. The osteocyte cell body and dendritic processes reside in bone in a complex lacuno-canalicular system, which allows the direct networking of osteocytes to their neighboring osteocytes, osteoblasts, osteoclasts, bone marrow, blood vessels, and nerves. Mechanosensing of osteocytes translates the applied mechanical force on bone to cellular signaling and regulation of bone adaptation. The osteocyte lacuno-canalicular system is highly efficient in transferring external mechanical force on bone to the osteocyte cell body and dendritic processes via displacement of fluid in the lacuno-canalicular space. Osteocyte mechanotransduction regulates the formation and function of the osteoblasts and osteoclasts to maintain bone homeostasis. Osteocytes produce a variety of proteins and signaling molecules such as sclerostin, cathepsin K, Wnts, DKK1, DMP1, IGF1, and RANKL/OPG to regulate osteoblast and osteoclast activity. Various genetic abnormality-associated rare bone diseases are related to disrupted osteocyte functions, including sclerosteosis, van Buchem disease, hypophosphatemic rickets, and WNT1 and plastin3 mutation-related disorders. Meticulous studies during the last 15 years on disrupted osteocyte function in rare bone diseases guided for the development of various novel therapeutic agents to treat bone diseases. Studies on genetic, molecular, and cellular mechanisms of sclerosteosis and van Buchem disease revealed a role for sclerostin in bone homeostasis, which led to the development of the sclerostin antibody to treat osteoporosis and other bone degenerative diseases. The mechanism of many other rare bone diseases and the role of the osteocyte in the development of such conditions still needs to be investigated. In this review, we mainly discuss the knowledge obtained during the last 30 years on the role of the osteocyte in rare bone diseases. We speculate about future research directions to develop novel therapeutic drugs targeting osteocyte functions to treat both common and rare bone diseases.
Introduction
Bone mainly contains three types of cells, i.e., osteocytes, osteoblasts, and osteoclasts. The osteocytes are the most abundant cells comprising 95% of the total cell population in bone with an average half-life of 25 years (1, 2). The bone-forming osteoblasts and bone-resorbing osteoclasts account for only ~5% of the total bone cell population, and live for only a few days to weeks. The characteristics and function of osteoblasts and osteoclasts during physiological bone remodeling and bone diseases have been extensively studied (3–6). However, the cellular and molecular mechanisms of osteocyte-mediated effects on skeletal health have not been fully elucidated. Five decades ago the osteocytes were still regarded as inert cells buried alive inside the bone matrix, despite the fact that the healthy human skeleton contains ~42 billion osteocytes (7). The mechanosensing property of osteocytes has been reported for the first time about three decades ago (8). With the advancement of new technologies in molecular and cellular mechanisms, imaging, transgenic approaches, and RNA sequencing, important functions of osteocytes and their role in bone homeostasis and vital systemic functions have become clear in the last two decades (1). Osteocytes are descendants of osteoblasts. During the bone mineralization process, some osteoblasts bury themselves in the bone matrix. They regulate mineralization, develop connective dendritic processes, and become osteocytes. Although osteocytes are buried deep inside the bone matrix, their dendritic processes are well-connected with neighboring osteocytes, osteoblasts, blood vessels, nerve cells, and bone marrow. The osteocyte cell body resides in a lacunar space inside the bone matrix. From the cell body 50–60 dendritic processes radiate in canaliculi space, forming a complex osteocyte lacuno-canalicular system (9). Mechanical loading of bone triggers interstitial fluid flow in this lacuno-canalicular system. Osteocyte dendritic processes sense the fluid flow, resulting in cellular signaling (10–12). In response to mechanical stimuli, osteocytes release nitric oxide (NO), prostaglandins (PGs), and ATP (within milliseconds), which affects many other cellular signaling pathways including interleukin-6 (IL-6), receptor activator of nuclear factor κB ligand/osteoprotegerin (RANKL/OPG), Wnt/β-catenin, and calcium signaling pathways (10, 11, 13–15). During the last 30 years various mechanisms of osteocyte mechanotransduction have been reported. Calcium oscillation in osteocytes has been shown to be a critical regulator of osteocyte mechanotransduction (16–18). Recently, mechanical loading-induced Ca2+ oscillation has been shown to cause the release of extracellular vesicles from osteocytes and to promote bone regeneration (19). Loading-induced Ca2+ oscillation in osteocytes triggers the release of downstream signaling molecules, e.g., NO (14, 20–22), prostaglandin E2 (PGE2) (23), matrix extracellular phosphoglycoprotein (MEPE), insulin-like growth factor-1 (IGF-1) (24), and β-catenin (25). Similarly, primary cilia on the osteocyte cell body as well as the dendritic processes play a regulatory role in the mechanotransduction process in osteocytes (26). Focal adhesions are macromolecular complexes consisting of multiple actin-associated proteins, such as paxillin, vinculin, connexin-43, integrins, and talin, that serve as physical linkages between a cell's cytoskeleton and the ECM. The mechanism of focal adhesion-mediated osteocyte mechanotransduction has been partly unraveled (27–30).
Osteocytes produce various signaling proteins such as sclerostin, WNT1, WNT3a, Dickkopf-related protein 1 (DKK1), phosphate regulating endopeptidase homolog X-linked (PHEX), RANKL, MEPE, fibroblast growth factor-23 (FGF23), sclerostin, and vascular endothelial growth factor (VEGF) (31–34). These proteins and growth factors not only play a crucial role in bone biology, but also in other organs such as kidney, and in fat metabolism (34, 35). Disruption of the production of these proteins by impaired osteocyte function causes bone diseases, including rare bone diseases (36–40). Osteocyte-specific release of growth factors and signaling molecules is disturbed during long-term unloading, such as occurs in astronauts during space traveling and long-term bed rest (11). Similarly, inflammatory conditions caused by various inflammatory diseases also affect osteocyte function and signaling (41, 42). Various genetic abnormality-associated rare bone diseases are related to disrupted osteocyte functions.
Wnt signaling plays a vital role in skeletal health, mainly via osteogenic differentiation of precursor cells, osteocyte viability, and osteocyte signaling to other bone cells (43, 44). Wnt/β-catenin activation in osteocytes mainly contributes to the anabolic effect in bone (45). Mechanical loading-induced early release of PGE2 causes rapid activation of Wnt/β-catenin signaling in osteocytes (46, 47). Wnt ligand co-receptor LRP5 is essential for osteocyte mechanotransduction and mechanical loading-induced bone formation (43, 48–50). This suggests a crucial role of osteocytic Wnt signaling in the process of mechanotransduction. The consequence of disturbed Wnt signaling in osteocytes is demonstrated by a mutation in the WNT1 gene, which causes autosomal-recessive osteogenesis imperfecta, a childhood rare bone disease (51). The osteocyte is the main source of sclerostin, a negative regulator of Wnt/β-catenin signaling. Mechanical loading reduces, while proinflammatory cytokines enhance sclerostin production in osteocytes (31, 41). Sclerostin deficiency in various rare genetic bone diseases, such as sclerosteosis and van Buchem disease, causes osteopetrosis, a high bone mass phenotype (36).
Parathyroid hormone (PTH) signaling contributes via PTH-related protein (PTHrP)-derived peptides, to the mechanical loading-induced osteocyte-mediated adaptation of bone tissue composition (52, 53). Inherited hypoparathyroidism is a rare disease that reduces bone turnover causing higher bone mineral density (BMD) and brittle bone (54). However, the osteocyte mechanotransduction-mediated bone adaptation in inherited hypoparathyroidism is still unknown. Similarly, mechanical loading upregulates insulin growth factor-1 (IGF1) expression in osteocytes, and IGF1 signaling plays an important role in the osteogenic response to mechanical loading (24, 55, 56). Moreover, IGF1 regulates PTH/PTHrP signaling in osteocytes, and bone regeneration (57–61). Osteocytic IGF1 signaling in rare bone diseases still needs to be investigated. Osteocytes produce RANKL and OPG to regulate osteoclastogenesis and osteoclast activity (6, 62). The RANKL/OPG ratio in osteocytes is upregulated by proinflammatory cytokines (31, 41, 63), but reduced by mechanical loading (64). Mechanical loading of osteocytes downregulates the expression of most proinflammatory cytokines, except IL-6. Interestingly, mechanical loading upregulates IL-6 expression in parallel with PGE2 production in bone cells (63, 65). However, the exact role of mechanical loading-induced osteocytic IL-6 signaling in bone biology and rare bone diseases is poorly understood. Osteocytes not only regulate osteoblast and osteoclast formation and activity, but also phosphate homeostasis and the function of vital organs in an endocrine fashion (62, 66). Osteocytes respond to PTH by inducing osteolysis that releases calcium in the bloodstream to maintain the systemic mineral homeostasis (67). During lactation, osteocytic sclerostin modulates the production of the osteoclast markers tartrate-resistant acid phosphatase (TRAP), cathepsin K, and carbonic anhydrase-2 in osteocytes to regulate the release of calcium from bone (68). Mutation of the cathepsin K encoding gene causes a rare autosomal recessive osteochondrodysplasia (69). Although cathepsin K is mainly required for osteoclastic bone resorption, osteocytes also release cathepsin K and regulate mechanotransduction (70). Osteocytes release FGF23, dentin matrix acidic phosphoprotein 1 (DMP1), PHEX, and MEPE, and act as endocrine cells to regulate phosphate metabolism (1, 71–73). Osteocytes release sclerostin to control bone mineralization via the modulation of DMP1, PHEX, MEPE, and FGF-23 expression (74, 75). The osteocyte is a critical player in chronic kidney disease-associated adverse effects on bone and heart (76). Osteocyte-derived DMP1 reduces FGF23 expression and enhances bone mineralization (35). Chronic kidney disease reduces DMP1 expression in osteocytes, while DMP1 supplementation prevents osteocyte apoptosis, lowers FGF23 expression, increases serum phosphate, and prevents the development of left ventricular hypertrophy in a chronic kidney disease mice model (35, 76). PHEX indirectly regulates FGF23, and PHEX gene mutation causes hypophosphatemic rickets, a rare hereditary bone disease (39). The MEPE-PHEX interaction regulates bone turnover, mineralization, and bone-renal vascularization (77). MEPE is highly expressed in human osteocytes embedded within mineralized bone (78). MEPE−/− mice develop increased bone mass, hyperphosphatemia and creatinine-clearence, and transgenic overexpression of MEPE C-terminal acidic serine aspartate-rich MEPE-associated (ASARM)-motif corrects these abnormalities (79). C-terminal ASARM-motif plays a major role in regulation of bone mass and renal function in aging mice showing the association of MEPE in age-dependent osteoporosis. This unveils the endocrine function of osteocytes affecting the function of distant organs such a kidney and heart. Thus, osteocytes play a vital role in bone homeostasis, and several osteocyte-specific proteins are involved in the pathogenesis of rare bone diseases. In this review, we mainly focus on the role of disturbed development and activity of osteocytes in rare bone diseases. We will discuss the existing insights on the role of osteocytes in the pathophysiology of rare metabolic bone disorders as well as the consequences of these rare metabolic bone disorders for the development and function of osteocytes.
Disturbed Osteocyte Function Can Cause Metabolic Bone Diseases
Many factors, including aging, osteoporosis, inflammatory diseases, and systemic diseases, disrupt osteocyte functions (2, 41, 76, 80). Aging causes 15–30% reduction in lacunar density or osteocyte numbers (81). Smaller and more round osteocyte lacunae are common in aged mice compared to young mice (82). The age-related decrease in lacunar density is accompanied by osteocyte death, hypermineralization, and micropetrosis (83). Aging also reduces the number of osteocyte dendrites and canaliculi by ~30% (80, 84). The remarkable decrease in osteocyte lacunar density, canaliculi, and dendrites number will reduce the entire osteocyte network connectivity that affects osteocyte function and bone homeostasis. Since the osteocyte lacuno-canalicular system plays a crucial role in mechanotransduction, abnormalities in this system might directly affect osteocyte mechanotransduction-mediated bone adaptation and remodeling (85). Estrogen, PTH, bisphosphonates, and muscle-derived irisin increase osteocyte survival (86–88). Postmenopausal estrogen deficiency, imbalance in PTH signaling, long-term glucocorticoid treatment, and oxidative stress caused by disuse may cause osteocyte death resulting in imbalanced bone remodeling and decreased bone mass (89). Systemic inflammatory conditions, such as periodontitis, rheumatoid arthritis, chronic kidney disease, and cancer, affect osteocyte function mainly via elevated levels of proinflammatory cytokines.
Advanced glycation end products (AGEs) are inflammatory mediators in diabetes. AGEs induce osteocyte apoptosis and upregulate osteocytic expression of IL-6 and VEGF (90, 91). Periodontitis-mediated inflammation causes sclerostin production and NF-κβ activation in alveolar osteocytes (92). Diabetic rats with periodontitis show a higher expression of sclerostin, RANKL, tumor necrosis factor-α (TNFα), and IL-1β in osteocytes, which affects osteoblast and osteoclast function (93–95). Brucella abortus infection is a common cause of osteomyelitis, which not only inhibits connexin-43 expression in osteocytes, but also induces osteocyte apoptosis and upregulates expression of inflammatory mediators RANKL, TNFα, and IL-6 in osteocytes (96). Multiple myeloma, a cancer that directly affects bone, induces osteocyte apoptosis and osteocyte-derived sclerostin and RANKL expression (97). Osteocytic sclerostin and FGF23 expression are highly upregulated in chronic kidney disease (98). In rheumatoid arthritis, a systemic inflammatory disease, elevated levels of inflammatory cytokines enhance IL-1β, TNFα, sclerostin (SOST), and DKK1 gene expression in osteocytes (31).
Rare Bone Diseases and Osteocyte Function
Genetic defects cause various rare bone diseases such as Paget disease, fibrous dysplasia, pycnodysostosis, sclerosteosis, osteogenesis imperfecta, X-linked hypophosphatemia, and hypophosphatasia. Osteocyte functions are disturbed in many genetic defect-mediated rare bone diseases (99, 100). Possible mechanisms of disrupted osteocyte functions in rare bone diseases are depicted in Figure 1. An impaired activity/function of osteoblasts, osteoclasts, and/or osteocytes could lead to alterations in the mechanical environment of osteocytes, variations in ECM structure, and de-regulation of mechanotransduction-related pathways, resulting in disturbed mechanotransduction possibly via primary cilium, calcium channels, physical deformation of bone matrix, canalicular fluid flow, shear stress, adhesion molecules, and/or cytoskeleton.
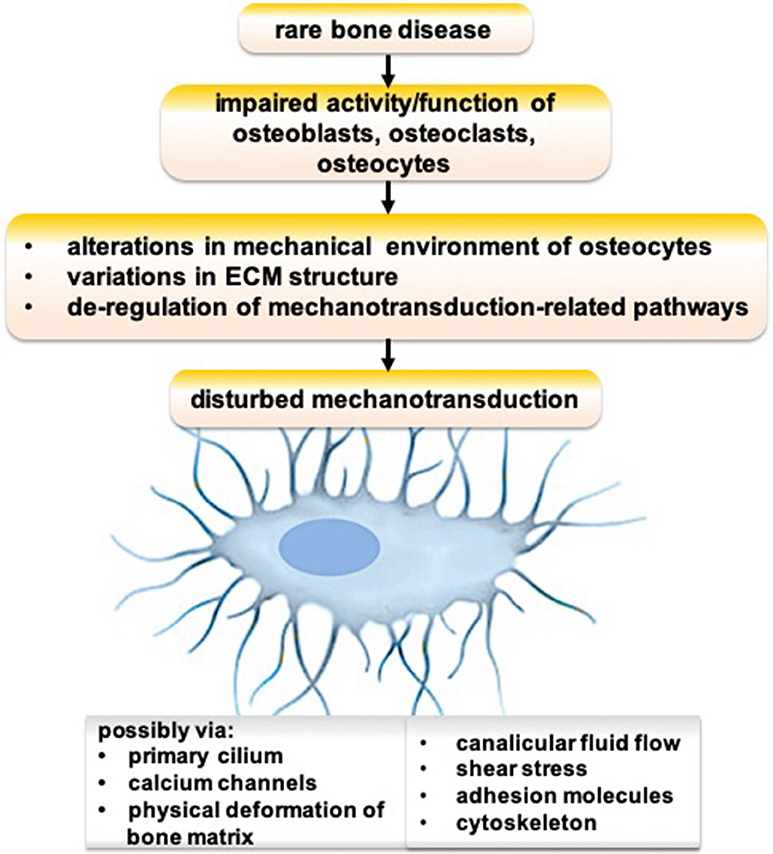
Figure 1. Schematic showing the possible mechanism of disrupted osteocyte functions in rare bone diseases.
Sclerosteosis and van Buchem Disease
Sclerosteosis and van Buchem disease are autosomal recessive skeletal dysplasia causing deficiency of sclerostin protein and progressive skeletal growth (36). Sclerosteosis is primarily reported in the descendants of Dutch settlers from the seventeenth century in South Africa (101). Van Buchem disease is mainly found in a Dutch population in The Netherlands (102, 103). Skeletal manifestations of sclerosteosis and van Buchem disease are similar, including increased thickening of skull, jaw bones, long bones, and ribs. Gigantism, and hand abnormalities in sclerosteosis are distinguishing features between sclerosteosis and gigantism (104). SOST, the gene responsible for sclerosteosis and van Buchem disease, is localized on chromosome 17q12-q21, and encodes sclerostin protein. A point mutation in the SOST gene causes sclerosteosis, and a 52 kb deletion of the downstream gene of SOST causes van Buchem disease (36, 37). A study on the genetics and pathophysiology of sclerosteosis and van Buchem disease led to the discovery of sclerostin and its function that contributed to the development of an anti-sclerostin drug to treat osteoporosis (105). Mature osteoblasts produce sclerostin to some extent, but osteocytes are the primary source of sclerostin (106). Activation of Wnt/β-catenin signaling enhances osteogenic differentiation and bone formation (107). Sclerostin, a potent Wnt inhibitor, controls osteogenic differentiation of precursor cells and bone formation (108). On the other hand, Wnt inhibition causes overexpression of RANKL and deregulation of OPG resulting in osteoclastogenesis (38). Studies on rare bone diseases, sclerosteosis, and van Buchem disease, have unraveled the role of sclerostin in bone homeostasis (99). In the case of sclerostin deficiency, osteocytes become like a “snake without fang” and are unable to control new bone deposition by osteoblasts (36, 37). Sclerostin deficiency results in excessive bone formation (109), as observed in sclerosteosis and van Buchem disease. Since both sclerosteosis and van Buchem disease are genetic diseases caused by osteocytic sclerostin deficiency, the osteocyte could be the possible target cell to treat these diseases.
Hypophosphatemic Rickets
Hypophosphatemic rickets is a hereditary disease with a prevalence of 1/20,000. PHEX gene mutations have been reported to cause hypophosphatemia and a hypomineralized bone phenotype (39, 40). Hypophosphatemic rickets is characterized by a generalized bone mineralization defect resulting in a decreased total volumetric bone mineral density (vBMD) at the radius and tibia, and lower cortical vBMD and cortical thickness at the radius compared to healthy adults (110). However, the exact mechanism of PHEX gene mutation-mediated FGF23 upregulation, hypophosphatemia, and development of rickets is still unclear. Both PHEX and FGF23 are mainly produced by osteocytes (111). One autosomal recessive hypophosphatemic rickets family carried a mutation affecting the dentin matrix protein (DMP1) start codon (112). DMP1 is essential for osteocyte maturation, while DMP1 mutation leads to altered skeletal mineralization and disturbed phosphate homeostasis associated with increased FGF23 production via an effect on the function of osteocytes (112). A combination of oral phosphorous supplementation and active vitamin D analogs is the conventional therapy to counteract the consequences of excessive FGF23 in hypophosphatemic rickets (113). Anti-FGF23 antibody or gene therapy targeting DMP1, FGF23, or PHEX, could be a future direction to treat hypophosphatemic rickets. This has been demonstrated already in children with X-linked hypophosphatemia, where treatment with anti-FGF23 antibody Burosumab improved linear growth and physical function, and reduced the pain and the severity of rickets (114).
WNT1 and PLS3 Mutation
WNT1 is a key ligand of the canonical WNT signaling pathway, which is the most important signaling pathway in bone (115). The WNT family contains a total of 19 WNT proteins, including WNT1, which are essential for fetal bone development and maintenance of postnatal bone health (38). The plastin protein family belongs to the actin bundling proteins and is ubiquitously expressed in solid tissue, including neurons in the brain, osteoblasts and osteocytes in bone, hematopoietic cells, and many cancer cell types (116). Plastin3 (PLS3) expression in mesenchymal stem cells and osteoblasts increases during osteogenic differentiation (117, 118). Missense mutation c.652T>G (p.C218G) in WNT1, and an X-linked form resulting from a splice mutation c.73-24T>A in PLS3 are associated with osteoporosis in children (115, 119). The role of WNT1 and PLS3 in the function of osteocytes is not yet fully understood. WNT1 mutation affects WNT/β-catenin signaling that might affect osteocyte function, and causes an imbalance in bone homeostasis resulting in osteoporosis (51). PLS3 has been suggested to play a role in osteocyte dendrite function and mechanotransduction (120). High FGF23 expression has been reported in osteocytes of a patient with a WNT1 mutation compared to a PLS3 mutation (121). The expression pattern of DMP1, sclerostin, and phospo-β-catenin is similar in patients with a WNT1 and PLS3 mutation (121). This suggests that WNT1 and PLS3-mediated osteoporosis might have a similar mechanism of disease progression. Osteocyte-derived WNT1 is a key regulator of osteoblast function and bone homeostasis (122). Deletion of Wnt1 in osteocytes results in low bone mass and increased fracture risk, similar as WNT1 mutation-related osteoporosis (122). Interestingly, Wnt1 overexpression in osteocytes stimulates bone formation by increasing the osteoblast number and activity partly via activation of mTORC1 signaling (122). Anti-sclerostin antibody robustly increases bone mass and reduces the fracture rate in Wnt1 global knockout mice (122). These findings suggest that WNT1 mutation-related osteoporosis is caused in part by a loss of WNT1 signaling in osteocytes, which decreases mTORC1-dependent osteoblast formation and bone regeneration. The sclerostin antibody has been suggested to be an effective treatment option for WNT1 mutation-related osteoporosis (122). However, osteocytic mechanotransduction in patients with a WNT1 mutation is not yet fully understood. Microgravity, or unloading, decreases WNT3a, WNT5a, DKK1, cyclinD1, LEF-1, and CX43, but increases WNT1 and SOST expression in osteocytes (11, 123). Microgravity dramatically reduces the number of F-actin filaments in osteocytes (123). This suggests a role for WNT1 in the formation of the osteocyte cytoskeleton and in osteocyte mechanosensitivity. PLS3 mutation or deficiency causes low bone mass, possibly via hyperactivity of osteoclasts. PLS3-deficient mice show no effect in trabecular bone, but cortical bone mass is highly reduced (124). Normal osteocyte morphology is observed in PLS3-deficient mice (125). Bone marrow stem cells from PLS3-deficient mice show compromised osteogenic differentiation with reduced expression of osteocalcin, Wnt16, and Sfrp4 mRNA (125). This indicates a role of PLS3 in bone regeneration via osteoblast differentiation and function (125). A lack of PLS3 has been shown to decrease the expression of NFkB repressing factor, thereby augmenting Nfatc1 transcription and osteoclastogenesis, indicating osteoclast-mediated bone loss in PLS3-deficient mice (124). The actin cytoskeleton and focal adhesions play an important role in osteocyte mechanotransduction. Since the plastin protein family belongs to the actin bundling protein, plastin might have a direct or focal adhesion-mediated indirect effect on osteocyte mechanotransduction. However, the role of PLS3 in osteocyte functions, such as mechanotransduction, osteocyte-to-osteoblast communication, and osteocyte-to-osteoclast signaling, and its cellular and molecular influence on bone remodeling has not been investigated yet.
Osteogenesis Imperfecta
Osteogenesis imperfecta is mainly an autosomal dominant disease of connective tissue that lowers bone mass and causes fracture. Very few cases of recessive and X-chromosome-linked forms of osteogenesis imperfecta have been reported so far. Osteogenesis imperfecta is one of the most common bone fragility disorders with an incidence of about 1/15–20,000 (126). It is a brittle bone disease directly related to abnormalities of type I collagen primary posttranslational modification, folding, structure, strength, and quantity (127). Mutations in the COL1A1 or COL1A2 gene, encoding the α1(I) or α2(I) chain of type I collagen, are associated with ~85% of osteogenesis imperfecta cases (128). Mutation-mediated alteration in processing, structure, and secretion of type I collagen, as well as ER stress causes a subclinical to lethal skeletal phenotype. Loss-of-function mutations in WNT1 lead to moderately severe and progressive forms of osteogenesis imperfecta (119, 129). Since osteocytes are embedded in the bone ECM, ECM-to-osteocyte interaction plays a vital role in bone homeostasis. The effect of deregulated collagen matrix-to-osteocyte interaction in osteogenesis imperfecta could influence the severity of bone fragility. However, the role of osteocytes in osteogenesis imperfecta disease progression has rarely been investigated yet. Future studies focusing on the role of the collagen matrix-to-osteocyte interaction in osteocytes function, including mechanotransduction, and osteoblast-to-osteoclast communication could guide in the development of new therapeutic targets to treat osteogenesis imperfecta.
Pycnodysostosis
Pycnodysostosis (OMIM 265800) is a rare autosomal recessive osteochondrodysplasia with a prevalence rate of 1–1.7/million and without gender specificity (130). Pycnodysostosis is characterized by a short stature with increased bone mineral density and an increased bone fragility phenotype (105, 131). Cortical and trabecular osteosclerosis with increased cortical width and high bone mineral density is observed in patients with pycnodysostosis (11, 12). Gelb et al. reported mutation of the gene encoding cathepsin K in chromosome 1q21 in patients with pycnodysostosis (69). Cathepsin K degrades bone matrix proteins, including collagen type I, and is therefore essential for osteoclastic bone resorption (132). A study on the genetics and pathophysiology of pycnodysostosis revealed the role of cathepsin K in osteoclast activity that led to the development of cathepsin K inhibitors to treat osteoporosis by inhibiting osteoclastic bone resorption (105). Unfortunately cathepsin K inhibitors did not lead to new osteoporosis medication because of serious side effects (stroke). In pycnodysostosis the number of osteoclasts is not affected, but bone resorption is highly reduced (133). Osteoclastic bone resorption is essential for bone homeostasis, as old and cracked bone is removed as well as the fibrous extracellular matrix that provides the signal to osteoblasts to deposit new bone and increase bone strength. Cathepsin K is also produced by osteoblasts and osteocytes (70, 134). Osteocytic cathepsin K is responsible for lactation-induced bone loss (135). Mechanical loading increases cathepsin K expression in cortical bone of wild type mice (70). Globally knocking out of cathepsin K enhances mechanotransduction signals resulting in cortical bone formation (70). Cathepsin K regulates bone remodeling not only by enhancing osteoclast activity, but also by inhibiting osteogenic differentiation via modulation of Wnt signaling (70). Cathepsin K deficiency in osteoclasts increases sphingosine kinase 1 (Sphk1) that catalyzes the phosphorylation of sphingosine to sphingosine-1-phosphate (S1P) (136, 137). S1P promotes osteoblast differentiation, bone regeneration (136), and osteocytic mechanotransduction (138). New research approaches reducing the mechanosensitivity of osteocytes by inhibiting S1P could be important to develop therapeutics for the treatment of cathepsin K deficiency-mediated high bone mass phenotype.
Cathepsin K regulates bone remodeling and cortical bone formation by degrading periostin (139). Periostin is mainly expressed in the periosteum and in osteocytes, and enhances bone formation via activation of Wnt signaling (70). Bonnet et al. nicely depicted the role of osteoblastic and osteocytic periostin in cathepsin K-mediated bone modeling and remodeling (70). Osteocyte-mediated periostin could be a possible target in pycnodystostosis.
Analysis of Osteocyte Function
Multiple approaches have been developed to analyze osteocyte morphology (80). Confocal laser scanning electron microscopy (CLSM) (140), scanning electron microscopy (SEM) (141), ultra-high voltage electron microscopy, tomography on silver stained bone sections (117, 142), and SEM of acid-etching technique of non-decalcified bone samples (143) have been developed to visualize osteocyte density, morphology, and osteocyte lacuno-canalicular network in bone biopsies from patients. Van Hove and colleagues nicely show differences in osteocyte morphology in patients with osteoarthritis, osteopenia, and osteopetrosis using CLSM (144). Schneider and colleagues developed serial focused ion beam/SEM imaging for quantitative 3D-assessment of the osteocyte lacuno-canalicular network (145). Micropetrotic lacunae, as seen in old age, in cortical and trabecular bone can be visualized by transmission electron microscopy (TEM) and SEM (81). High power backscattered SEM images of a bone tissue section visualizes the mineralized micropetrotic lacunae (146). Osteocyte-specific expression of proteins such as sclerostin, IL-1β, TNFα, DKK1, DMP1, and FGF23 is altered in different disease conditions. Immunohistochemistry using specific antibodies easily visualizes the expression pattern in bone sections (33, 121, 147). Serum sclerostin is a key marker of osteocyte function in different disease conditions (148, 149). Serum sclerostin levels are upregulated in osteoporosis and downregulated in high bone mass conditions (150). Enzyme-linked immune assays and automated chemiluminescent assays have been developed and validated for high precision analysis of serum sclerostin (151, 152). Spinal cord injury causes patient immobilization and bed rest that mimics unloading conditions. Serum of patients with spinal cord injury contains increased periostin and decreased sclerostin levels (153). Since sclerostin and periostin are mainly secreted by osteocytes, these proteins could possibly be used as serum markers to analyze osteocyte function in different diseases.
Osteocyte mechanotransduction alters in different disease conditions, such as aging, osteoporosis, and inflammatory diseases (82, 154–157). Various in vitro and ex vivo methods have been developed to analyze osteocyte functions (158). However, most of these methods are invasive and difficult to perform routinely in clinical setting. Non-invasive bone loading methods are available to analyze osteocyte functions in murine models (59, 159, 160). Future research is recommended to develop non-invasive approaches to analyze osteocyte mechanotransduction in vivo.
Recently, extracellular vesicles and exosomes are regarded as the key cargo-carrying organelles affecting the local and systemic cellular activities. Exosomes are released from living cells and carry miRNAs, circular RNAs, mRNAs, and various proteins from one cell to other cells. Osteocyte-derived exosomes detected in the circulation are enriched with osteocyte-specific miRNAs (161). A possible role of extracellular vesicles and exosomes in bone biology has been presented nicely in a recent review from Tao and Guo (162). Mechanically loaded osteocytes release exosomes with bone regenerating potential, via Ca2+ oscillation (19). Proteomic analysis of exosomes from cortical bone osteocytes provide a clear picture of osteocyte function in different disease conditions, including rare bone diseases (32). The osteocyte transcriptome is extensively deregulated in a mouse model of osteogenesis imperfecta (163). Transcriptome and proteomic analysis in osteocytic exosomes could unravel the role of exosomes in the pathophysiology of rare bone diseases. Recent advancements in RNA sequencing, functional analysis tools, and bioinformatic tools reveal a role of non-coding RNAs such as miRNAs, circular RNAs, piRNAs, and lncRNAs in various cellular signaling and biological activities including development and diseases (164–168). Various mRNAs and their translated proteins play a role in osteocyte function (36, 56). Only few studies address the role of non-coding RNAs in osteocyte function (161, 169, 170). Disruption of the Cx43/miR21 pathway results in osteocyte apoptosis and increases osteocyte-mediated osteoclastogenesis in old-age subjects (170). miR-29b-3p regulates osteogenic differentiation of precursor cells via modulating IGF1 secretion in mechanically loaded osteocytes (169). The role of circular RNAs, piRNAs, lncRNAs, and other miRNAs on osteocyte functions in physiological and disease conditions is poorly understood. The differential expression pattern of non-coding RNAs in osteocytes during rare bone diseases has not been investigated yet. Altered expression pattern of non-coding RNAs in osteocytes during rare bone diseases could play role in disease development and pathophysiology. We believe that this research direction could guide the development of new targets and techniques to analyze the function of osteocytes in patients.
Therapies to Improve Osteocyte Function
Intermittent PTH therapy enhances bone regeneration and bone mineral density (171). PTH signaling affects the function of osteoblasts, osteoclasts, and osteocytes. Intermittent PTH treatment enhances the commitment of precursor cells to an osteogenic fate (172). PTH signaling in osteocytes regulates sclerostin expression and controls osteocyte-mediated osteoblastogenesis (58, 87, 173, 174). PTH treatment (teriparatide, PTH1-34) in osteogenesis imperfecta increases bone mineral density and vertebral strength (175, 176). PTH inhibits Notch signaling in osteoblasts and osteocytes, which might exert the anabolic effect on bone (177).
Studies on sclerostin deficiency-related high bone mass phenotype illustrate the role of sclerostin in bone biology guiding the development of anti-sclerostin bone anabolic agents. Anti-sclerostin monoclonal antibody has the potency to treat diseases with low bone mass phenotype, including osteoporosis (178, 179). There is increasing evidence suggesting a role of sclerostin in myeloma bone diseases and breast cancer bone metastasis-mediated complications (149, 180). In the bone niche, sclerostin is mainly produced by mature osteoblasts and osteocytes (181). Interestingly, multiple myeloma cells and breast cancer cells also produce sclerostin that might have a catabolic effect on bone (180, 181). Furthermore, cancer metastasis-induced inflammation upregulates osteocytic sclerostin that inhibits osteoblast function (181). Therefore, sclerostin monoclonal antibody could be beneficial to reduce myeloma and breast cancer-mediated complications in bone (182–184). Sclerostin antibody romosozumab clears a phase III trial with satisfactory outcomes and already got approval for osteoporosis treatment (185). This sclerostin antibody has shown promising potential to treat osteogenesis imperfecta (127, 186, 187). Therefore, romosozumab might be beneficial to treat rare bone disease patients with low bone mass phenotypes, such as osteogenesis imperfecta, Wnt1 mutation, and PLS3 mutation.
DKK1 is another potent Wnt inhibitor, that is also mainly produced by osteocytes in bone. Similar to sclerostin, DKK1 is also produced by breast, prostate, and multiple myeloma cancer cells (188–190). Increased levels of DKK1 in various cancers cause osteolytic bone disease and inhibit osteoblast function (188, 189). DKK1 is an osteocyte-specific target to treat osteoporosis and other low bone mass diseases (191). In systemic inflammation, the neutralization of DKK1 reduces sclerostin expression and protects systemic bone loss (192). Monoclonal antibodies against DKK1 showed DKK1 inhibitory potential in vitro and increased bone mass in vivo (192). Moreover, a bispecific antibody targeting both sclerostin and DKK1 shows higher efficiency on bone formation and fracture repair (193). Phase I and phase II clinical trials have been performed to test the efficacy of anti-DKK1 antibodies on myeloma and myeloma-induced skeletal events (194, 195).
Studies on the role of osteocytic RANKL in bone homeostasis have led to the development of an anti-RANKL monoclonal antibody to treat common metabolic bone diseases, including osteoporosis (196, 197). During the last 10 years, the use of denosumab proved to be satisfactory with rare adverse effects (198). An imbalance in RANK-RANKL-OPG signaling is also observed in many rare bone diseases such as Juvenile Paget disease, fibrous dysplasia, Hajdu Cheney syndrome, and Langerhans cell histiocytosis (199). Therefore, denosumab has also been used off-label in rare metabolic bone diseases, including Paget's disease, osteogenesis imperfecta, and aneurysmal bone cysts (200). Bisphosphonate treatment prevents bone loss and fractures caused by rare bone disease-mediated osteogenesis imperfecta (201–203). Physical therapy/rehabilitation regimes in children with osteogenesis imperfecta improved mobility and bone mineral density, and thereby prevented fractures (175). Most treatment approaches for rare bone diseases directly act on osteoblast or osteoclast activity, and are symptomatic treatments.
The meticulous research on the molecular mechanism of osteocytic sclerostin on bone remodeling led to the development of anti-sclerostin antibodies to treat osteoporosis and other skeletal disorders demanding an increase in bone mass. Anti-sclerostin antibody primarily targets bone-lining cells, rather than the osteocytes imbedded in bone matrix (204). Anti-sclerostin antibody activates selected canonical Wnt target genes in a mature osteoblast subpopulation and increases bone formation (204). Sclerostin monoclonal antibody romosozumab treatment significantly increases bone mineral density in postmenopausal women with low bone mineral density and reduces fracture risk in postmenopausal women with osteoporosis (205). However, adverse side effects of a loss of sclerostin are osteoarthritis (206), TNF-dependent inflammatory joint destruction (207), negative effect on B cells (208), and risk of cardiac failure (205), which should be carefully evaluated before romosozumab treatment is considered. Although research on the cellular and molecular mechanisms of sclerosteosis and van Buchem disease guided the development of anti-sclerostin antibody to treat osteoporosis, an osteocyte function-targeted therapy for sclerosteosis and van Buchem disease has not yet been developed. Genetic disorders disrupt the expression of osteocytic proteins that play a role in the pathophysiology of various rare bone diseases (Figure 2). Since osteocyte functions play a crucial role in bone homeostasis, and since these functions are disrupted in many rare bone diseases, a better understanding of the molecular mechanisms of disrupted osteocyte functions in rare bone diseases may guide to discover novel targets to treat these rare bone diseases.
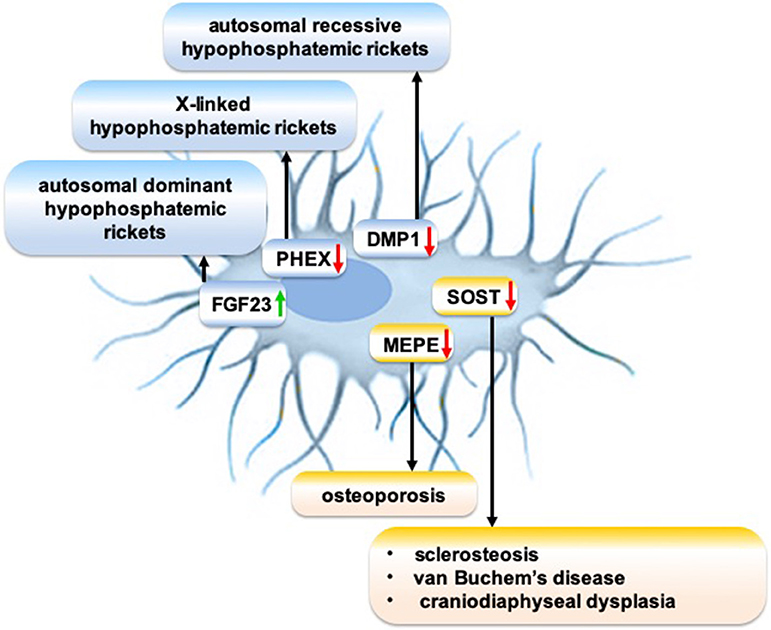
Figure 2. Schematic showing the role of disrupted expression of osteocytic proteins on the pathophysiology of rare bone diseases. Green arrow: Gain-of-function mutation; red arrow: Loss-of-function mutation.
Conclusions
Genetic and pathophysiological research on three rare bone diseases, i.e., sclerosteosis, pycnodysostosis, and van Buchem disease, provided new effective interventions to treat osteoporosis. The current available therapeutic approaches for rare bone diseases are symptomatic and mainly target osteoblast and osteoclast formation and activity. Since osteocytes play a vital role in bone homeostasis, and because their function is disrupted in many rare bone diseases, it would be wise to focus on unraveling the osteocyte-specific targets to treat rare bone diseases. The role of coding RNAs (mRNAs) in osteocyte function during pathophysiological conditions has been widely investigated. Non-coding RNAs (piRNAs, circRNAs, lncRNAs, shRNAs, etc.) represent 97% of the total RNA in the cell, and recent technological advances have unveiled a crucial role of non-coding RNAs in various biological processes including bone homeostasis. Therefore, meticulous research focusing on the role of non-coding RNAs in osteocyte functions under physiological conditions and in various bone diseases including rare bone diseases could be the future research direction. The results of this research could provide clues for the discovery of novel osteocyte-specific targets to treat rare bone diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a Project of Education of Guangdong Province, China (2017KQNCX162) and High-Level University Construction Talents of Guangzhou Medical University (B185006003014 and B19502003017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Wei Cao for help with the illustrations.
References
2. Bonewald LF. The role of the osteocyte in bone and nonbone disease. Endocrinol Metab Clin North Am. (2017) 46:1–18. doi: 10.1016/j.ecl.2016.09.003
3. Chen H, Senda T, Kubo KY. The osteocyte plays multiple roles in bone remodeling and mineral homeostasis. Med Mol Morphol. (2015) 48:61–8. doi: 10.1007/s00795-015-0099-y
4. Xiong J, O'Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res. (2012) 27:499–505. doi: 10.1002/jbmr.1547
5. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. (2011) 17:1235–41. doi: 10.1038/nm.2448
6. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE. (2015) 10:e0138189. doi: 10.1371/journal.pone.0138189
7. Buenzli PR, Sims NA. Quantifying the osteocyte network in the human skeleton. Bone. (2015) 75:144–50. doi: 10.1016/j.bone.2015.02.016
8. Pead MJ, Suswillo R, Skerry TM, Vedi S, Lanyon LE. Increased 3H-uridine levels in osteocytes following a single short period of dynamic bone loading in vivo. Calcif Tissue Int. (1988) 43:92–6. doi: 10.1007/BF02555153
9. Thi MM, Suadicani SO, Schaffler MB, Weinbaum S, Spray DC. Mechanosensory responses of osteocytes to physiological forces occur along processes and not cell body and require alphaVbeta3 integrin. Proc Natl Acad Sci USA. (2013) 110:21012–7. doi: 10.1073/pnas.1321210110
10. Bacabac RG, Smit TH, Van Loon JJWA, Zandieh Doulabi B, Helder MN, Klein-Nulend J. Bone cell responses to high-frequency vibration stress: does the nucleus oscillate within the cytoplasm? FASEB J. (2006) 20:858–64. doi: 10.1096/fj.05-4966.com
11. Burger EH, Klein-Nulend J. Microgravity and bone cell mechanosensitivity. Bone. (1998) 22(Suppl. 5):127–30S. doi: 10.1016/S8756-3282(98)00010-6
12. Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA, Nijweide PJ, et al. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. (1995) 9:441–5. doi: 10.1096/fasebj.9.5.7896017
13. Klein-Nulend J, Bacabac RG, Bakker AD. Mechanical loading and how it affects bone cells: the role of the osteocyte cytoskeleton in maintaining our skeleton. Eur Cells Mater. (2012) 24:278–91. doi: 10.22203/eCM.v024a20
14. Vatsa A, Smit TH, Klein-Nulend J. Extracellular NO signalling from a mechanically stimulated osteocyte. J Biomech. (2007) 40:S89–95. doi: 10.1016/j.jbiomech.2007.02.015
15. Klein-Nulend J, Bakker AD, Bacabac RG, Vatsa A, Weinbaum S. Mechanosensation and transduction in osteocytes. Bone. (2013) 54:182–90. doi: 10.1016/j.bone.2012.10.013
16. Hung CT, Allen FD, Pollack SR, Brighton CT. Intracellular Ca2+ stores and extracellular Ca2+ are required in the real-time Ca2+ response of bone cells experiencing fluid flow. J Biomech. (1996) 29:1411–7. doi: 10.1016/0021-9290(96)84536-2
17. Lewis KJ, Frikha-Benayed D, Louie J, Stephen S, Spray DC, Thi MM, et al. Osteocyte calcium signals encode strain magnitude and loading frequency in vivo. Proc Natl Acad Sci USA. (2017) 114:11775–80. doi: 10.1073/pnas.1707863114
18. Santos A, Bakker AD, Klein-Nulend J. The role of osteocytes in bone mechanotransduction. Osteoporos Int. (2009) 20:1027–31. doi: 10.1007/s00198-009-0858-5
19. Morrell AE, Brown GN, Robinson ST, Sattler RL, Baik AD, Zhen G, et al. Mechanically induced Ca(2+) oscillations in osteocytes release extracellular vesicles and enhance bone formation. Bone Res. (2018) 6:6. doi: 10.1038/s41413-018-0007-x
20. Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, et al. Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res. (1999) 14:1123–31. doi: 10.1359/jbmr.1999.14.7.1123
21. Vatsa A, Mizuno D, Smit TH, Schmidt CF, MacKintosh FC, Klein-Nulend J. Bio imaging of intracellular NO production in single bone cells after mechanical stimulation. J Bone Miner Res. (2006) 21:1722–8. doi: 10.1359/jbmr.060720
22. Klein-Nulend J, van Oers RF, Bakker AD, Bacabac RG. Nitric oxide signaling in mechanical adaptation of bone. Osteoporos Int. (2014) 25:1427–37. doi: 10.1007/s00198-013-2590-4
23. Ajubi NE, Klein-Nulend J, Alblas MJ, Burger EH, Nijweide PJ. Signal transduction pathways involved in fluid flow-induced PGE2 production by cultured osteocytes. Am J Physiol. (1999) 276:E171–8. doi: 10.1152/ajpendo.1999.276.1.E171
24. Lean JM, Jagger CJ, Chambers TJ, Chow JW. Increased insulin-like growth factor I mRNA expression in rat osteocytes in response to mechanical stimulation. Am J Physiol. (1995) 268(Pt 1):E318–27. doi: 10.1152/ajpendo.1995.268.2.E318
25. Kamel MA, Picconi JL, Lara-Castillo N, Johnson ML. Activation of beta-catenin signaling in MLO-Y4 osteocytic cells versus 2T3 osteoblastic cells by fluid flow shear stress and PGE2: implications for the study of mechanosensation in bone. Bone. (2010) 47:872–81. doi: 10.1016/j.bone.2010.08.007
26. Xiao Z, Dallas M, Qiu N, Nicolella D, Cao L, Johnson M, et al. Conditional deletion of Pkd1 in osteocytes disrupts skeletal mechanosensing in mice. FASEB J. (2011) 25:2418–32. doi: 10.1096/fj.10-180299
27. Santos A, Bakker AD, Zandieh-Doulabi B, de Blieck-Hogervorst JMA, Klein-Nulend J. Early activation of the beta-catenin pathway in osteocytes is mediated by nitric oxide, phosphatidyl inositol-3 kinase/Akt, and focal adhesion kinase. Biochem Bioph Res Commun. (2010) 391:364–9. doi: 10.1016/j.bbrc.2009.11.064
28. Geoghegan IP, Hoey DA, McNamara LM. Integrins in osteocyte biology and mechanotransduction. Curr Osteoporos Rep. (2019) 17:195–206. doi: 10.1007/s11914-019-00520-2
29. Geoghegan IP, Hoey DA, McNamara LM. Estrogen deficiency impairs integrin alphavbeta3-mediated mechanosensation by osteocytes and alters osteoclastogenic paracrine signalling. Sci Rep. (2019) 9:4654. doi: 10.1038/s41598-019-41095-3
30. Vatsa A, Semeins CM, Smit TH, Klein-Nulend J. Paxillin localisation in osteocytes–is it determined by the direction of loading? Biochem Biophys Res Commun. (2008) 377:1019–24. doi: 10.1016/j.bbrc.2007.12.174
31. Pathak JL, Bakker AD, Luyten FP, Verschueren P, Lems WF, Klein-Nulend J, et al. Systemic inflammation affects human osteocyte-specific protein and cytokine expression. Calcif Tissue Int. (2016) 98:596–608. doi: 10.1007/s00223-016-0116-8
32. Zhang C, Xu S, Zhang S, Liu M, Du H, Sun R, et al. Ageing characteristics of bone indicated by transcriptomic and exosomal proteomic analysis of cortical bone cells. J Orthop Surg Res. (2019) 14:129. doi: 10.1186/s13018-019-1163-4
33. Shah KM, Stern MM, Stern AR, Pathak JL, Bravenboer N, Bakker AD. Osteocyte isolation and culture methods. Bonekey Rep. (2016) 5:838. doi: 10.1038/bonekey.2016.65
34. Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell and more. Endocr Rev. (2013) 34:658–90. doi: 10.1210/er.2012-1026
35. Dussold C, Gerber C, White S, Wang X, Qi L, Francis C, et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res. (2019) 7:12. doi: 10.1038/s41413-019-0051-1
36. Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. (2001) 10:537–43. doi: 10.1093/hmg/10.5.537
37. Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. (2002) 39:91–7. doi: 10.1136/jmg.39.2.91
38. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. (2013) 19:179–92. doi: 10.1038/nm.3074
39. Goldsweig BK, Carpenter TO. Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep. (2015) 13:88–97. doi: 10.1007/s11914-015-0259-y
40. Pavone V, Testa G, Gioitta Iachino S, Evola FR, Avondo S, Sessa G. Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol. (2015) 25:221–6. doi: 10.1007/s00590-014-1496-y
41. Zhou M, Li S, Pathak JL. Pro-inflammatory cytokines and osteocytes. Curr Osteoporos Rep. (2019) 17:97–104. doi: 10.1007/s11914-019-00507-z
42. Bakker AD, Klein-Nulend J, Tanck E, Heyligers IC, Albers GH, Lips P, et al. Different responsiveness to mechanical stress of bone cells from osteoporotic versus osteoarthritic donors. Osteoporos Int. (2006) 17:827–33. doi: 10.1007/s00198-006-0072-7
43. Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. (2011) 17:684–91. doi: 10.1038/nm.2388
44. Bullock WA, Pavalko FM, Robling AG. Osteocytes and mechanical loading: the Wnt connection. Orthod Craniofac Res. (2019) 22(Suppl. 1):175–9. doi: 10.1111/ocr.12282
45. Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, et al. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc Natl Acad Sci USA. (2015) 112:E478–86. doi: 10.1073/pnas.1409857112
46. Lara-Castillo N, Kim-Weroha NA, Kamel MA, Javaheri B, Ellies DL, Krumlauf RE, et al. In vivo mechanical loading rapidly activates beta-catenin signaling in osteocytes through a prostaglandin mediated mechanism. Bone. (2015) 76:58–66. doi: 10.1016/j.bone.2015.03.019
47. Klein-Nulend J, Burger EH, Semeins CM, Raisz LG, Pilbeam CC. Pulsating fluid flow stimulates prostaglandin release and inducible prostaglandin G/H synthase mRNA expression in primary mouse bone cells. J Bone Miner Res. (1997) 12:45–51. doi: 10.1359/jbmr.1997.12.1.45
48. Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. (2006) 281:23698–711. doi: 10.1074/jbc.M601000200
49. Saxon LK, Jackson BF, Sugiyama T, Lanyon LE, Price JS. Analysis of multiple bone responses to graded strains above functional levels, and to disuse, in mice in vivo show that the human Lrp5 G171V High Bone Mass mutation increases the osteogenic response to loading but that lack of Lrp5 activity reduces it. Bone. (2011) 49:184–93. doi: 10.1016/j.bone.2011.03.683
50. Zhao L, Shim JW, Dodge TR, Robling AG, Yokota H. Inactivation of Lrp5 in osteocytes reduces young's modulus and responsiveness to the mechanical loading. Bone. (2013) 54:35–43. doi: 10.1016/j.bone.2013.01.033
51. Kampe AJ, Makitie RE, Makitie O. New genetic forms of childhood-onset primary osteoporosis. Horm Res Paediatr. (2015) 84:361–9. doi: 10.1159/000439566
52. Gardinier JD, Al-Omaishi S, Morris MD, Kohn DH. PTH signaling mediates perilacunar remodeling during exercise. Matrix Biol. (2016) 52–54:162–75. doi: 10.1016/j.matbio.2016.02.010
53. Camirand A, Goltzman D, Gupta A, Kaouass M, Panda D, Karaplis A. The role of Parathyroid Hormone-Related Protein (PTHrP) in osteoblast response to microgravity: mechanistic implications for osteoporosis development. PLoS ONE. (2016) 11:e0160034. doi: 10.1371/journal.pone.0160034
54. Clarke BL. Bone disease in hypoparathyroidism. Arq Bras Endocrinol Metabol. (2014) 58:545–52. doi: 10.1590/0004-2730000003399
55. Reijnders CM, Bravenboer N, Tromp AM, Blankenstein MA, Lips P. Effect of mechanical loading on insulin-like growth factor-I gene expression in rat tibia. J Endocrinol. (2007) 192:131–40. doi: 10.1677/joe.1.06880
56. Lau KHW, Baylink DJ, Zhou XD, Rodriguez D, Bonewald LF, Li ZH, et al. Osteocyte-derived insulin-like growth factor I is essential for determining bone mechanosensitivity. Am J Physiol Endocr Metab. (2013) 305:E271–81. doi: 10.1152/ajpendo.00092.2013
57. Saini V, Marengi DA, Barry KJ, Fulzele KS, Heiden E, Liu X, et al. Parathyroid hormone (PTH)/PTH-related peptide type 1 receptor (PPR) signaling in osteocytes regulates anabolic and catabolic skeletal responses to PTH. J Biol Chem. (2013) 288:20122–34. doi: 10.1074/jbc.M112.441360
58. Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res. (2011) 26:1035–46. doi: 10.1002/jbmr.304
59. Gross TS, Srinivasan S, Liu CC, Clemens TL, Bain SD. Noninvasive loading of the murine tibia: an in vivo model for the study of mechanotransduction. J Bone Miner Res. (2002) 17:493–501. doi: 10.1359/jbmr.2002.17.3.493
60. Kesavan C, Wergedal JE, Lau KH, Mohan S. Conditional disruption of IGF-I gene in type 1alpha collagen-expressing cells shows an essential role of IGF-I in skeletal anabolic response to loading. Am J Physiol Endocrinol Metab. (2011) 301:E1191–7. doi: 10.1152/ajpendo.00440.2011
61. Sakata T, Wang Y, Halloran BP, Elalieh HZ, Cao J, Bikle DD. Skeletal unloading induces resistance to insulin-like growth factor-I (IGF-I) by inhibiting activation of the IGF-I signaling pathways. J Bone Miner Res. (2004) 19:436–46. doi: 10.1359/JBMR.0301241
62. Tan SD, de Vries TJ, Kuijpers-Jagtman AM, Semeins CM, Everts V, Klein-Nulend J. Osteocytes subjected to fluid flow inhibit osteoclast formation and bone resorption. Bone. (2007) 41:745–51. doi: 10.1016/j.bone.2007.07.019
63. Pathak JL, Bravenboer N, Luyten FP, Verschueren P, Lems WF, Klein-Nulend J, et al. Mechanical loading reduces inflammation-induced human osteocyte-to-osteoclast communication. Calcif Tissue Int. (2015) 97:169–78. doi: 10.1007/s00223-015-9999-z
64. You L, Temiyasathit S, Lee P, Kim CH, Tummala P, Yao W, et al. Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone. (2008) 42:172–9. doi: 10.1016/j.bone.2007.09.047
65. Sanchez C, Gabay O, Salvat C, Henrotin YE, Berenbaum F. Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarthritis Cartilage. (2009) 17:473–81. doi: 10.1016/j.joca.2008.09.007
66. Vezeridis PS, Semeins CM, Chen Q, Klein-Nulend J. Osteocytes subjected to pulsating fluid flow regulate osteoblast proliferation and differentiation. Biochem Biophys Res Commun. (2006) 348:1082–8. doi: 10.1016/j.bbrc.2006.07.146
67. Tsourdi E, Jahn K, Rauner M, Busse B, Bonewald LF. Physiological and pathological osteocytic osteolysis. J Musculoskelet Neuronal Interact. (2018) 18:292–303.
68. Kogawa M, Wijenayaka AR, Ormsby RT, Thomas GP, Anderson PH, Bonewald LF, et al. Sclerostin regulates release of bone mineral by osteocytes by induction of carbonic anhydrase 2. J Bone Miner Res. (2013) 28:2436–48. doi: 10.1002/jbmr.2003
69. Gelb BD, Edelson JG, Desnick RJ. Linkage of pycnodysostosis to chromosome 1q21 by homozygosity mapping. Nat Genet. (1995) 10:235–7. doi: 10.1038/ng0695-235
70. Bonnet N, Brun J, Rousseau JC, Duong LT, Ferrari SL. Cathepsin K controls cortical bone formation by degrading periostin. J Bone Miner Res. (2017) 32:1432–41. doi: 10.1002/jbmr.3136
71. Fujii O, Tatsumi S, Ogata M, Arakaki T, Sakaguchi H, Nomura K, et al. Effect of osteocyte-ablation on inorganic phosphate metabolism: analysis of bone-kidney-gut axis. Front Endocrinol. (2017) 8:359. doi: 10.3389/fendo.2017.00359
72. Tresguerres FGF, Torres J, Lopez-Quiles J, Hernandez G, Vega JA, Tresguerres IF. The osteocyte: a multifunctional cell within the bone. Ann Anat. (2020) 227:151422. doi: 10.1016/j.aanat.2019.151422
73. Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. (2003) 285:E1–9. doi: 10.1152/ajpendo.00016.2003
74. Sapir-Koren R, Livshits G. Osteocyte control of bone remodeling: is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos Int. (2014) 25:2685–700. doi: 10.1007/s00198-014-2808-0
75. Ryan ZC, Ketha H, McNulty MS, McGee-Lawrence M, Craig TA, Grande JP, et al. Sclerostin alters serum vitamin D metabolite and fibroblast growth factor 23 concentrations and the urinary excretion of calcium. Proc Natl Acad Sci USA. (2013) 110:6199–204. doi: 10.1073/pnas.1221255110
76. Martin A. Bone and heart health in chronic kidney disease: role of dentin matrix protein 1. Curr Opin Nephrol Hypertens. (2019) 28:297–303. doi: 10.1097/MNH.0000000000000512
77. David V, Martin A, Hedge AM, Rowe PSN. Matrix Extracellular Phosphoglycoprotein (MEPE) is a new bone renal hormone and vascularization modulator. Endocrinology. (2009) 150:4012–23. doi: 10.1210/en.2009-0216
78. Nampei A, Hashimoto J, Hayashida K, Tsuboi H, Shi K, Tsuji I, et al. Matrix extracellular phosphoglycoprotein (MEPE) is highly expressed in osteocytes in human bone. J Bone Miner Metab. (2004) 22:176–84. doi: 10.1007/s00774-003-0468-9
79. Zelenchuk LV, Hedge AM, Rowe PS. Age dependent regulation of bone-mass and renal function by the MEPE ASARM-motif. Bone. (2015) 79:131–42. doi: 10.1016/j.bone.2015.05.030
80. Tiede-Lewis LM, Dallas SL. Changes in the osteocyte lacunocanalicular network with aging. Bone. (2019) 122:101–13. doi: 10.1016/j.bone.2019.01.025
81. Busse B, Djonic D, Milovanovic P, Hahn M, Puschel K, Ritchie RO, et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell. (2010) 9:1065–75. doi: 10.1111/j.1474-9726.2010.00633.x
82. Hemmatian H, Bakker AD, Klein-Nulend J, van Lenthe GH. Aging, osteocytes, and mechanotransduction. Curr Osteoporos Rep. (2017) 15:401–11. doi: 10.1007/s11914-017-0402-z
83. Rolvien T, Schmidt FN, Milovanovic P, Jahn K, Riedel C, Butscheidt S, et al. Early bone tissue aging in human auditory ossicles is accompanied by excessive hypermineralization, osteocyte death and micropetrosis. Sci Rep. (2018) 8:1920. doi: 10.1038/s41598-018-19803-2
84. Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, Dusevich V, et al. Degeneration of the osteocyte network in the C57BL/6 mouse model of aging. Aging. (2017) 9:2190–208. doi: 10.18632/aging.101308
85. Burger EH, Klein-Nulend J. Mechanotransduction in bone–role of the lacuno-canalicular network. FASEB J. (1999) 13:S101–12. doi: 10.1096/fasebj.13.9001.s101
86. Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, et al. Irisin mediates effects on bone and fat via alphav integrin receptors. Cell. (2018) 175:1756–68 e17. doi: 10.1016/j.cell.2018.10.025
87. Bellido T, Plotkin LI. Novel actions of bisphosphonates in bone: preservation of osteoblast and osteocyte viability. Bone. (2011) 49:50–5. doi: 10.1016/j.bone.2010.08.008
88. Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. (2013) 54:264–71. doi: 10.1016/j.bone.2012.11.038
89. Jilka RL, O'Brien CA. The role of osteocytes in age-related bone loss. Curr Osteoporos Rep. (2016) 14:16–25. doi: 10.1007/s11914-016-0297-0
90. Chen H, Liu W, Wu X, Gou M, Shen J, Wang H. Advanced glycation end products induced IL-6 and VEGF-A production and apoptosis in osteocyte-like MLO-Y4 cells by activating RAGE and ERK1/2, P38 and STAT3 signalling pathways. Int Immunopharmacol. (2017) 52:143–9. doi: 10.1016/j.intimp.2017.09.004
91. Tanaka K, Yamaguchi T, Kanazawa I, Sugimoto T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem Biophys Res Commun. (2015) 461:193–9. doi: 10.1016/j.bbrc.2015.02.091
92. Pacios S, Xiao W, Mattos M, Lim J, Tarapore RS, Alsadun S, et al. Osteoblast lineage cells play an essential role in periodontal bone loss through activation of nuclear factor-kappa B. Sci Rep. (2015) 5:16694. doi: 10.1038/srep16694
93. Graves DT, Alshabab A, Albiero ML, Mattos M, Correa JD, Chen SS, et al. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J Clin Periodontol. (2018) 45:285–92. doi: 10.1111/jcpe.12851
94. Kim JH, Kim AR, Choi YH, Jang S, Woo GH, Cha JH, et al. Tumor necrosis factor-alpha antagonist diminishes osteocytic RANKL and sclerostin expression in diabetes rats with periodontitis. PLoS ONE. (2017) 12:e0189702. doi: 10.1371/journal.pone.0189702
95. Kim JH, Lee DE, Woo GH, Cha JH, Bak EJ, Yoo YJ. Osteocytic sclerostin expression in alveolar bone in rats with diabetes mellitus and ligature-induced periodontitis. J Periodontol. (2015) 86:1005–11. doi: 10.1902/jop.2015.150083
96. Pesce Viglietti AI, Arriola Benitez PC, Gentilini MV, Velasquez LN, Fossati CA, Giambartolomei GH, et al. Brucella abortus invasion of osteocytes modulates connexin 43 and integrin expression and induces osteoclastogenesis via receptor activator of NF-kappaB ligand and tumor necrosis factor alpha secretion. Infect Immun. (2016) 84:11–20. doi: 10.1128/IAI.01049-15
97. Delgado-Calle J, Bellido T, Roodman GD. Role of osteocytes in multiple myeloma bone disease. Curr Opin Support Palliat Care. (2014) 8:407–13. doi: 10.1097/SPC.0000000000000090
98. Morena M, Jaussent I, Dupuy AM, Bargnoux AS, Kuster N, Chenine L, et al. Osteoprotegerin and sclerostin in chronic kidney disease prior to dialysis: potential partners in vascular calcifications. Nephrol Dial Transplant. (2015) 30:1345–56. doi: 10.1093/ndt/gfv081
99. van Bezooijen RL, Bronckers AL, Gortzak RA, Hogendoorn PC, van der Wee-Pals L, Balemans W, et al. Sclerostin in mineralized matrices and van Buchem disease. J Dent Res. (2009) 88:569–74. doi: 10.1177/0022034509338340
100. Sebastian A, Loots GG. Genetics of Sost/SOST in sclerosteosis and van Buchem disease animal models. Metabolism. (2018) 80:38–47. doi: 10.1016/j.metabol.2017.10.005
101. Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet. (2003) 63:192–7. doi: 10.1034/j.1399-0004.2003.00036.x
102. Van Buchem FS, Hadders HN, Ubbens R. An uncommon familial systemic disease of the skeleton: hyperostosis corticalis generalisata familiaris. Acta radiol. (1955) 44:109–20. doi: 10.3109/00016925509170789
103. Van Hul W, Balemans W, Van Hul E, Dikkers FG, Obee H, Stokroos RJ, et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet. (1998) 62:391–9. doi: 10.1086/301721
104. Beighton P, Barnard A, Hamersma H, van der Wouden A. The syndromic status of sclerosteosis and van Buchem disease. Clin Genet. (1984) 25:175–81. doi: 10.1111/j.1399-0004.1984.tb00481.x
105. Appelman-Dijkstra NM, Papapoulos SE. From disease to treatment: from rare skeletal disorders to treatments for osteoporosis. Endocrine. (2016) 52:414–26. doi: 10.1007/s12020-016-0888-7
106. Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. (2005) 19:1842–4. doi: 10.1096/fj.05-4221fje
107. Case N, Rubin J. beta-catenin-a supporting role in the skeleton. J Cell Biochem. (2010) 110:545–53. doi: 10.1002/jcb.22574
108. van Bezooijen RL, ten Dijke P, Papapoulos SE, Lowik CW. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytok Growth Factor Rev. (2005) 16:319–27. doi: 10.1016/j.cytogfr.2005.02.005
109. Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone. (2017) 96:29–37. doi: 10.1016/j.bone.2016.10.007
110. Shanbhogue VV, Hansen S, Folkestad L, Brixen K, Beck-Nielsen SS. Bone geometry, volumetric density, microarchitecture, and estimated bone strength assessed by HR-pQCT in adult patients with hypophosphatemic rickets. J Bone Miner Res. (2015) 30:176–83. doi: 10.1002/jbmr.2310
111. Lambert AS, Linglart A. Hypocalcaemic and hypophosphatemic rickets. Best Pract Res Clin Endocrinol Metab. (2018) 32:455–76. doi: 10.1016/j.beem.2018.05.009
112. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. (2006) 38:1310–5. doi: 10.1038/ng1905
113. Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. (2014) 3:R13–30. doi: 10.1530/EC-13-0103
114. Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab therapy in children with x-linked hypophosphatemia. N Engl J Med. (2018) 378:1987–98. doi: 10.1056/NEJMoa1714641
115. Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. (2013) 368:1809–16. doi: 10.1056/NEJMoa1215458
116. Shinomiya H. Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. Int J Cell Biol. (2012) 2012:213492. doi: 10.1155/2012/213492
117. Kamioka H, Sugawara Y, Honjo T, Yamashiro T, Takano-Yamamoto T. Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. J Bone Miner Res. (2004) 19:471–8. doi: 10.1359/JBMR.040128
118. Kim JM, Kim J, Kim YH, Kim KT, Ryu SH, Lee TG, et al. Comparative secretome analysis of human bone marrow-derived mesenchymal stem cells during osteogenesis. J Cell Physiol. (2013) 228:216–24. doi: 10.1002/jcp.24123
119. Laine CM, Wessman M, Toiviainen-Salo S, Kaunisto MA, Mayranpaa MK, Laine T, et al. A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J Bone Miner Res. (2015) 30:510–8. doi: 10.1002/jbmr.2355
120. Boudin E, Fijalkowski I, Hendrickx G, Van Hul W. Genetic control of bone mass. Mol Cell Endocrinol. (2016) 432:3–13. doi: 10.1016/j.mce.2015.12.021
121. Wesseling-Perry K, Makitie RE, Valimaki VV, Laine T, Laine CM, Valimaki MJ, et al. Osteocyte protein expression is altered in low-turnover osteoporosis caused by mutations in WNT1 and PLS3. J Clin Endocr Metab. (2017) 102:2340–8. doi: 10.1210/jc.2017-00099
122. Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, Munivez E, et al. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. (2017) 127:2678–88. doi: 10.1172/JCI92617
123. Yang X, Sun LW, Liang M, Wang X, Fan Y-B. The response of wnt/ß-catenin signaling pathway in osteocytes under simulated microgravity. Micrograv Sci Technol. (2015) 27:473–83. doi: 10.1007/s12217-015-9439-8
124. Neugebauer J, Heilig J, Hosseinibarkooie S, Ross BC, Mendoza-Ferreira N, Nolte F, et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Human Mol Genet. (2018) 27:4249–62. doi: 10.1093/hmg/ddy318
125. Yorgan TA, Sari H, Rolvien T, Windhorst S, Failla AV, Kornak U, et al. Mice lacking plastin-3 display a specific defect of cortical bone acquisition. Bone. (2020) 130:115062. doi: 10.1016/j.bone.2019.115062
126. Martin E, Shapiro JR. Osteogenesis imperfecta:epidemiology and pathophysiology. Curr Osteoporos Rep. (2007) 5:91–7. doi: 10.1007/s11914-007-0023-z
127. Tauer JT, Robinson ME, Rauch F. Osteogenesis imperfecta: new perspectives from clinical and translational research. JBMR Plus. (2019) 3:e10174. doi: 10.1002/jbm4.10174
128. Morello R. Osteogenesis imperfecta and therapeutics. Matrix Biol. (2018) 71–72:294–312. doi: 10.1016/j.matbio.2018.03.010
129. Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. (2013) 92:590–7. doi: 10.1016/j.ajhg.2013.02.009
130. Arman A, Bereket A, Coker A, Kiper PO, Guran T, Ozkan B, et al. Cathepsin K analysis in a pycnodysostosis cohort: demographic, genotypic and phenotypic features. Orphanet J Rare Dis. (2014) 9:60. doi: 10.1186/1750-1172-9-60
131. Maroteaux P, Lamy M. [2 cases of a condensing osseous disease: pynodysostosis]. Arch Fr Pediatr. (1962) 19:267–74.
132. Vaaraniemi J, Halleen JM, Kaarlonen K, Ylipahkala H, Alatalo SL, Andersson G, et al. Intracellular machinery for matrix degradation in bone-resorbing osteoclasts. J Bone Miner Res. (2004) 19:1432–40. doi: 10.1359/JBMR.040603
133. Motyckova G, Fisher DE. Pycnodysostosis: role and regulation of cathepsin K in osteoclast function and human disease. Curr Mol Med. (2002) 2:407–21. doi: 10.2174/1566524023362401
134. Mandelin J, Hukkanen M, Li TF, Korhonen M, Liljestrom M, Sillat T, et al. Human osteoblasts produce cathepsin K. Bone. (2006) 38:769–77. doi: 10.1016/j.bone.2005.10.017
135. Lotinun S, Ishihara Y, Nagano K, Kiviranta R, Carpentier VT, Neff L, et al. Cathepsin K-deficient osteocytes prevent lactation-induced bone loss and parathyroid hormone suppression. J Clin Invest. (2019) 129:3058–71. doi: 10.1172/JCI122936
136. Lotinun S, Kiviranta R, Matsubara T, Alzate JA, Neff L, Luth A, et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest. (2013) 123:666–81. doi: 10.1172/JCI64840
137. Ryu J, Kim HJ, Chang EJ, Huang H, Banno Y, Kim HH. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. (2006) 25:5840–51. doi: 10.1038/sj.emboj.7601430
138. Zhang JN, Zhao Y, Liu C, Han ES, Yu X, Lidington D, et al. The role of the sphingosine-1-phosphate signaling pathway in osteocyte mechanotransduction. Bone. (2015) 79:71–8. doi: 10.1016/j.bone.2015.05.017
139. Bonnet N, Garnero P, Ferrari S. Periostin action in bone. Mol Cell Endocrinol. (2016) 432:75–82. doi: 10.1016/j.mce.2015.12.014
140. Blouin S, Roschger A, Varga F, Misof B, Spitzer S, Roschger P, et al. Confocal laser scanning microscopy-a powerful tool in bone research. Wien Med Wochenschr. (2018) 168:314–21. doi: 10.1007/s10354-018-0639-x
141. Boyde A. Scanning electron microscopy of bone. Methods Mol Biol. (2019) 1914:571–616. doi: 10.1007/978-1-4939-8997-3_31
142. Kamioka H, Murshid SA, Ishihara Y, Kajimura N, Hasegawa T, Ando R, et al. A method for observing silver-stained osteocytes in situ in 3-microm sections using ultra-high voltage electron microscopy tomography. Microsc Microanal. (2009) 15:377–83. doi: 10.1017/S1431927609990420
143. Lampi T, Dekker H, Ten Bruggenkate CM, Schulten E, Mikkonen JJW, Koistinen A, et al. Acid-etching technique of non-decalcified bone samples for visualizing osteocyte-lacuno-canalicular network using scanning electron microscope. Ultrastruct Pathol. (2018) 42:74–9. doi: 10.1080/01913123.2017.1384418
144. van Hove RP, Nolte PA, Vatsa A, Semeins CM, Salmon PL, Smit TH, et al. Osteocyte morphology in human tibiae of different bone pathologies with different bone mineral density - is there a role for mechanosensing? Bone. (2009) 45:321–9. doi: 10.1016/j.bone.2009.04.238
145. Schneider P, Meier M, Wepf R, Muller R. Serial FIB/SEM imaging for quantitative 3D assessment of the osteocyte lacuno-canalicular network. Bone. (2011) 49:304–11. doi: 10.1016/j.bone.2011.04.005
146. Milovanovic P, Zimmermann EA, Vom Scheidt A, Hoffmann B, Sarau G, Yorgan T, et al. The formation of calcified nanospherites during micropetrosis represents a unique mineralization mechanism in aged human bone. Small. (2017) 13. doi: 10.1002/smll.201602215
147. Santos FR, Moyses RM, Montenegro FL, Jorgetti V, Noronha IL. IL-1beta, TNF-alpha, TGF-beta, and bFGF expression in bone biopsies before and after parathyroidectomy. Kidney Int. (2003) 63:899–907. doi: 10.1046/j.1523-1755.2003.00835.x
148. Chatzopoulos GS, Mansky KC, Lunos S, Costalonga M, Wolff LF. Sclerostin and WNT-5a gingival protein levels in chronic periodontitis and health. J Periodontal Res. (2019) 54:555-65. doi: 10.1111/jre.12659
149. Costa AG, Cremers S, Bilezikian JP. Sclerostin measurement in human disease: validity and current limitations. Bone. (2017) 96:24–8. doi: 10.1016/j.bone.2016.10.012
150. Durosier C, van Lierop A, Ferrari S, Chevalley T, Papapoulos S, Rizzoli R. Association of circulating sclerostin with bone mineral mass, microstructure, and turnover biochemical markers in healthy elderly men and women. J Clin Endocrinol Metab. (2013) 98:3873–83. doi: 10.1210/jc.2013-2113
151. McNulty M, Singh RJ, Li X, Bergstralh EJ, Kumar R. Determination of serum and plasma sclerostin concentrations by enzyme-linked immunoassays. J Clin Endocrinol Metab. (2011) 96:E1159–62. doi: 10.1210/jc.2011-0254
152. Drake MT, Fenske JS, Blocki FA, Zierold C, Appelman-Dijkstra N, Papapoulos S, et al. Validation of a novel, rapid, high precision sclerostin assay not confounded by sclerostin fragments. Bone. (2018) 111:36–43. doi: 10.1016/j.bone.2018.03.013
153. Maimoun L, Ben Bouallegue F, Gelis A, Aouinti S, Mura T, Philibert P, et al. Periostin and sclerostin levels in individuals with spinal cord injury and their relationship with bone mass, bone turnover, fracture and osteoporosis status. Bone. (2019) 127:612–9. doi: 10.1016/j.bone.2019.07.019
154. Hemmatian H, Jalali R, Semeins CM, Hogervorst JMA, van Lenthe GH, Klein-Nulend J, et al. Mechanical loading differentially affects osteocytes in fibulae from lactating mice compared to osteocytes in virgin mice: possible role for lacuna size. Calcif Tissue Int. (2018) 103:675–85. doi: 10.1007/s00223-018-0463-8
155. Bakker AD, Kulkarni RN, Klein-Nulend J, Lems WF. IL-6 alters osteocyte signaling toward osteoblasts but not osteoclasts. J Dent Res. (2014) 93:394–9. doi: 10.1177/0022034514522485
156. Bakker AD, Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, Lin YC, et al. Tumor necrosis factor alpha and interleukin-1beta modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum. (2009) 60:3336–45. doi: 10.1002/art.24920
157. Yan Y, Wang L, Ge L, Pathak JL. Osteocyte-mediated translation of mechanical stimuli to cellular signaling and its role in bone and non-bone-related clinical complications. Curr Osteoporos Rep. (2020) 18:67–80. doi: 10.1007/s11914-020-00564-9
158. Hinton PV, Rackard SM, Kennedy OD. In vivo osteocyte mechanotransduction: recent developments and future directions. Curr Osteoporos Rep. (2018) 16:746–53. doi: 10.1007/s11914-018-0485-1
159. Seref-Ferlengez Z, Basta-Pljakic J, Kennedy OD, Philemon CJ, Schaffler MB. Structural and mechanical repair of diffuse damage in cortical bone in vivo. J Bone Miner Res. (2014) 29:2537–44. doi: 10.1002/jbmr.2309
160. Seref-Ferlengez Z, Maung S, Schaffler MB, Spray DC, Suadicani SO, Thi MM. P2X7R-Panx1 complex impairs bone mechanosignaling under high glucose levels associated with type-1 diabetes. Plos ONE. (2016) 11:e0155107. doi: 10.1371/journal.pone.0155107
161. Sato M, Suzuki T, Kawano M, Tamura M. Circulating osteocyte-derived exosomes contain miRNAs which are enriched in exosomes from MLO-Y4 cells. Biomed Rep. (2017) 6:223–31. doi: 10.3892/br.2016.824
162. Tao SC, Guo SC. Extracellular vesicles in bone: “dogrobbers” in the “eternal battle field”. Cell Commun Signal. (2019) 17:6. doi: 10.1186/s12964-019-0319-5
163. Zimmerman SM, Dimori M, Heard-Lipsmeyer ME, Morello R. The osteocyte transcriptome is extensively dysregulated in mouse models of osteogenesis imperfecta. JBMR Plus. (2019) 3:e10171. doi: 10.1002/jbm4.10171
164. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. (2019) 20:675–91. doi: 10.1038/s41576-019-0158-7
165. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
166. Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. (2015) 16:4. doi: 10.1186/s13059-014-0571-3
167. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15:550. doi: 10.1186/s13059-014-0550-8
168. Qian DY, Yan GB, Bai B, Chen Y, Zhang SJ, Yao YC, et al. Differential circRNA expression profiles during the BMP2-induced osteogenic differentiation of MC3T3-E1 cells. Biomed Pharmacother. (2017) 90:492–9. doi: 10.1016/j.biopha.2017.03.051
169. Zeng Q, Wang Y, Gao J, Yan Z, Li Z, Zou X, et al. miR-29b-3p regulated osteoblast differentiation via regulating IGF-1 secretion of mechanically stimulated osteocytes. Cell Mol Biol Lett. (2019) 24:11. doi: 10.1186/s11658-019-0136-2
170. Davis HM, Pacheco-Costa R, Atkinson EG, Brun LR, Gortazar AR, Harris J, et al. Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell. (2017) 16:551–63. doi: 10.1111/acel.12586
171. Thomas T. Intermittent parathyroid hormone therapy to increase bone formation. Joint Bone Spine. (2006) 73:262–9. doi: 10.1016/j.jbspin.2005.11.007
172. Wang YH, Liu Y, Rowe DW. Effects of transient PTH on early proliferation, apoptosis, and subsequent differentiation of osteoblast in primary osteoblast cultures. Am J Physiol Endocrinol Metab. (2007) 292:E594–603. doi: 10.1152/ajpendo.00216.2006
173. Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O'Brien CA, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. (2005) 146:4577–83. doi: 10.1210/en.2005-0239
174. Wein MN. Parathyroid hormone signaling in osteocytes. JBMR Plus. (2018) 2:22–30. doi: 10.1002/jbm4.10021
175. Hoyer-Kuhn H, Semler O, Stark C, Struebing N, Goebel O, Schoenau E. A specialized rehabilitation approach improves mobility in children with osteogenesis imperfecta. J Musculoskelet Neuronal Interact. (2014) 14:445–53.
176. Gatti D, Rossini M, Viapiana O, Povino MR, Liuzza S, Fracassi E, et al. Teriparatide treatment in adult patients with osteogenesis imperfecta type I. Calcif Tissue Int. (2013) 93:448–52. doi: 10.1007/s00223-013-9770-2
177. Zanotti S, Canalis E. Parathyroid hormone inhibits Notch signaling in osteoblasts and osteocytes. Bone. (2017) 103:159–67. doi: 10.1016/j.bone.2017.06.027
178. McClung MR. Sclerostin antibodies in osteoporosis: latest evidence and therapeutic potential. Ther Adv Musculoskelet Dis. (2017) 9:263–70. doi: 10.1177/1759720X17726744
179. Ke HZ, Richards WG, Li XD, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. (2012) 33:747–83. doi: 10.1210/er.2011-1060
180. Colucci S, Brunetti G, Oranger A, Mori G, Sardone F, Specchia G, et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. (2011) 1:e27. doi: 10.1038/bcj.2011.22
181. Mendoza-Villanueva D, Zeef L, Shore P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFbeta-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res. (2011) 13:R106. doi: 10.1186/bcr3048
182. Kleber M, Ntanasis-Stathopoulos I, Dimopoulos MA, Terpos E. Monoclonal antibodies against RANKL and sclerostin for myeloma-related bone disease: can they change the standard of care? Expert Rev Hematol. (2019) 12:651–63. doi: 10.1080/17474086.2019.1640115
183. Toscani D, Bolzoni M, Ferretti M, Palumbo C, Giuliani N. Role of osteocytes in myeloma bone disease: anti-sclerostin antibody as new therapeutic strategy. Front Immunol. (2018) 9:2467. doi: 10.3389/fimmu.2018.02467
184. Hesse E, Schroder S, Brandt D, Pamperin J, Saito H, Taipaleenmaki H. Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness. JCI Insight. (2019) 5:e125543. doi: 10.1172/jci.insight.125543
185. Markham A. Romosozumab: first global approval. Drugs. (2019) 79:471–6. doi: 10.1007/s40265-019-01072-6
186. Surowiec RK, Battle LF, Ward FS, Schlecht SH, Khoury BM, Robbins C, et al. A xenograft model to evaluate the bone forming effects of sclerostin antibody in human bone derived from pediatric osteogenesis imperfecta patients. Bone. (2020) 130:115118. doi: 10.1016/j.bone.2019.115118
187. Scheiber AL, Barton DK, Khoury BM, Marini JC, Swiderski DL, Caird MS, et al. Sclerostin antibody-induced changes in bone mass are site specific in developing crania. J Bone Miner Res. (2019) 34:2301–10. doi: 10.1002/jbmr.3858
188. Voorzanger-Rousselot N, Goehrig D, Journe F, Doriath V, Body JJ, Clezardin P, et al. Increased Dickkopf-1 expression in breast cancer bone metastases. Br J Cancer. (2007) 97:964–70. doi: 10.1038/sj.bjc.6603959
189. Rachner TD, Gobel A, Benad-Mehner P, Hofbauer LC, Rauner M. Dickkopf-1 as a mediator and novel target in malignant bone disease. Cancer Lett. (2014) 346:172–7. doi: 10.1016/j.canlet.2014.01.010
190. Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. (2003) 349:2483–94. doi: 10.1056/NEJMoa030847
191. Glantschnig H, Hampton RA, Lu P, Zhao JZ, Vitelli S, Huang L, et al. Generation and selection of novel fully human monoclonal antibodies that neutralize Dickkopf-1 (DKK1) inhibitory function in vitro and increase bone mass in vivo. J Biol Chem. (2010) 285:40135–47. doi: 10.1074/jbc.M110.166892
192. Heiland GR, Zwerina K, Baum W, Kireva T, Distler JH, Grisanti M, et al. Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis. (2010) 69:2152–9. doi: 10.1136/ard.2010.132852
193. Florio M, Gunasekaran K, Stolina M, Li X, Liu L, Tipton B, et al. A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat Commun. (2016) 7:11505. doi: 10.1038/ncomms11505
194. Iyer SP, Beck JT, Stewart AK, Shah J, Kelly KR, Isaacs R, et al. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br J Haematol. (2014) 167:366–75. doi: 10.1111/bjh.13056
195. Sousa S, Clezardin P. Bone-targeted therapies in cancer-induced bone disease. Calcif Tissue Int. (2018) 102:227–50. doi: 10.1007/s00223-017-0353-5
196. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. (2011) 17:1231–4. doi: 10.1038/nm.2452
197. Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE. (2011) 6:e25900. doi: 10.1371/journal.pone.0025900
198. Zaheer S, LeBoff M, Lewiecki EM. Denosumab for the treatment of osteoporosis. Expert Opin Drug Metab Toxicol. (2015) 11:461–70. doi: 10.1517/17425255.2015.1000860
199. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. (2014) 5:511. doi: 10.3389/fimmu.2014.00511
200. Polyzos SA, Makras P, Tournis S, Anastasilakis AD. Off-label uses of denosumab in metabolic bone diseases. Bone. (2019) 129:115048. doi: 10.1016/j.bone.2019.115048
201. Rauch F, Travers R, Plotkin H, Glorieux FH. The effects of intravenous pamidronate on the bone tissue of children and adolescents with osteogenesis imperfecta. J Clin Invest. (2002) 110:1293–9. doi: 10.1172/JCI0215952
202. Alcausin MB, Briody J, Pacey V, Ault J, McQuade M, Bridge C, et al. Intravenous pamidronate treatment in children with moderate-to-severe osteogenesis imperfecta started under three years of age. Horm Res Paediatr. (2013) 79:333–40. doi: 10.1159/000351374
203. Bishop N, Adami S, Ahmed SF, Anton J, Arundel P, Burren CP, et al. Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial. Lancet. (2013) 382:1424–32. doi: 10.1016/S0140-6736(13)61091-0
204. Nioi P, Taylor S, Hu R, Pacheco E, He YDD, Hamadeh H, et al. Transcriptional profiling of laser capture microdissected subpopulations of the osteoblast lineage provides insight into the early response to sclerostin antibody in rats. J Bone Miner Res. (2015) 30:1457–67. doi: 10.1002/jbmr.2482
205. Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. (2017) 377:1417–27. doi: 10.1056/NEJMoa1708322
206. Bouaziz W, Funck-Brentano T, Lin H, Marty C, Ea HK, Hay E, et al. Loss of sclerostin promotes osteoarthritis in mice via beta-catenin-dependent and -independent Wnt pathways. Arthritis Res Ther. (2015) 17:24. doi: 10.1186/s13075-015-0540-6
207. Wehmeyer C, Frank S, Beckmann D, Bottcher M, Cromme C, Konig U, et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci Transl Med. (2016) 8:330ra35. doi: 10.1126/scitranslmed.aac4351
Keywords: osteocyte, rare bone disease, mechanotransduction, bone remodeling, niche, sost/sclerostin, phosphate-homeostasis, RANKL
Citation: Pathak JL, Bravenboer N and Klein-Nulend J (2020) The Osteocyte as the New Discovery of Therapeutic Options in Rare Bone Diseases. Front. Endocrinol. 11:405. doi: 10.3389/fendo.2020.00405
Received: 10 February 2020; Accepted: 20 May 2020;
Published: 08 July 2020.
Edited by:
Wim Van Hul, University of Antwerp, BelgiumReviewed by:
Niklas Rye Jørgensen, Copenhagen University Hospital, DenmarkReinhold Gottfried Erben, University of Veterinary Medicine Vienna, Austria
Copyright © 2020 Pathak, Bravenboer and Klein-Nulend. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenneke Klein-Nulend, j.kleinnulend@acta.nl