 Milad Rouhimoghadam
Milad Rouhimoghadam Anh S. Lu
Anh S. Lu Aliasger K. Salem
Aliasger K. Salem Edward J. Filardo
Edward J. Filardo- 1Department of Surgery, University of Iowa, Carver College of Medicine, Iowa City, IA, United States
- 2Holden Comprehensive Cancer Center, University of Iowa, Iowa City, IA, United States
- 3College of Pharmacy, University of Iowa, Iowa City, IA, United States
Estrogens exert their physiological and pathophysiological effects via cellular receptors, named ERα, ERβ, and G-protein coupled estrogen receptor (GPER). Estrogen-regulated physiology is tightly controlled by factors that regulate estrogen bioavailability and receptor sensitivity, while disruption of these control mechanisms can result in loss of reproductive function, cancer, cardiovascular and neurodegenerative disease, obesity, insulin resistance, endometriosis, and systemic lupus erythematosus. Restoration of estrogen physiology by modulating estrogen bioavailability or receptor activity is an effective approach for treating these pathological conditions. Therapeutic interventions that block estrogen action are employed effectively for the treatment of breast and prostate cancer as well as for precocious puberty and anovulatory infertility. Theoretically, treatments that block estrogen biosynthesis should prevent estrogen action at ERs and GPER, although drug resistance and ligand-independent receptor activation may still occur. In addition, blockade of estrogen biosynthesis does not prevent activation of estrogen receptors by naturally occurring or man-made exogenous estrogens. A more complicated scenario is provided by anti-estrogen drugs that antagonize ERs since these drugs function as GPER agonists. Based upon its association with metabolic dysregulation and advanced cancer, GPER represents a therapeutic target with promise for the treatment of several critical health concerns facing Western society. Selective ligands that specifically target GPER have been developed and may soon serve as pharmacological agents for treating human disease. Here, we review current forms of estrogen therapy and the implications that GPER holds for these therapies. We also discuss existing GPER targeted drugs, additional approaches towards developing GPER-targeted therapies and how these therapies may complement existing modalities of estrogen-targeted therapy.
Introduction
This review is organized in three general sections. First, we review basic information regarding estrogen bioavailability and its receptors. Second, we discuss the impact that GPER has upon our understanding of the influence of estrogen on human disease, and its implications for anti-estrogen therapy. Finally, we review existing pharmacological compounds that selectively target GPER and outline future potential approaches for targeting GPER.
Estrogen and Its Receptors
Estrogens are gonadocorticoids and the primary female sex hormones. Their actions promote the development of female reproductive tissue and secondary sexual characteristics, and they influence all phases of reproduction including conception, fetal development, parturition, and nursing. Hence, estrogens exert their effects not only on reproductive tissue but on a wide range of physiological systems, including integumentary, central nervous, cardiovascular, skeletal, immune, metabolic, and excretory systems (1, 2). In humans, three forms of estrogen are synthesized. They are defined by their common 18 carbon (C-18) estrane ring structure and are numbered E1- E3 to reflect the number of hydroxyl groups linked to the estrane ring (Figure 1). Accordingly, they are named estrone (E1), estradiol (E2), and estriol (E3). Each of these endogenous estrogens is lipophilic and is presumed to exit and enter cells through their ability to freely diffuse across the plasma membrane. All endogenous estrogens are synthesized in the smooth endoplasmic reticulum in a shared pathway of steroidogenesis from cholesterol (C-27) (Figure 2). In this pathway, cholesterol is metabolized through a variety of enzymatic steps into (C-21) progestogens and (C-19) androgens that serve as the immediate steroid intermediate for estrogens. E1 and E2 are primarily secreted by ovarian granulosa cells in response to stimulation by neuroendocrine glycoprotein hormones, including luteinizing releasing hormone (LHRH), luteinizing hormone (LH), and follicle stimulating hormone (FSH), which are released from the hypothalamus and pituitary (3). During reproductive years, E1 and E2 are the two most common circulating estrogens found in plasma, with scant amounts of E3 measured. Estrogens can also be synthesized in a variety of non-ovarian tissues, including, adrenal gland, fat, brain, bone, skin, vascular smooth muscle and intestine (2). However, in these tissues, estrogens must be directly synthesized from androgens, as these tissues lack the necessary enzymatic machinery to synthesize C-19 androgens. E3 is synthesized at low levels in the liver and intestine by 16α-hydroxylation of E1 or E2 by cytochrome P450 enzymes, such as CYP3A4 (4). During pregnancy, E3 becomes the primary estrogen as it is synthesized at high levels by the placenta, far exceeding that of E1 or E2 in plasma. While its role in fetal development is not clear, low levels of E3 in maternal serum or urine is prognostic of poor perinatal health and congenital anomalies (5, 6).
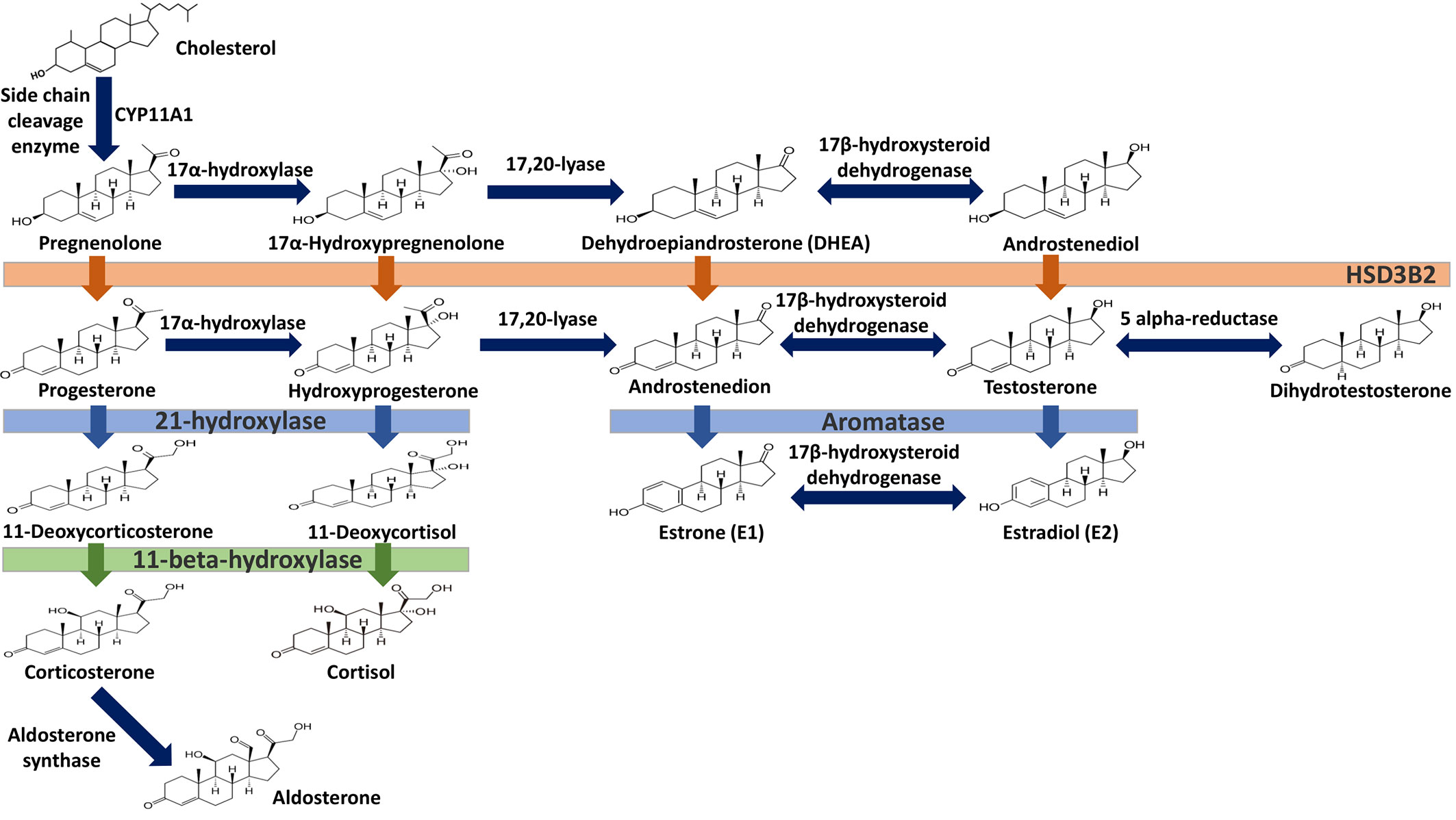
Figure 1 Steroid hormone synthesis and metabolism. The diagram designates key enzymatic steps in steroidogenesis.

Figure 2 Estrogen metabolism. This schematic identifies key intermediates in the metabolism of estrone and estradiol.
The process by which estrogens are transported throughout the body and exert their biologic effects in target tissues is not completely understood. The vast majority of synthesized estrogen circulates in the plasma bound to either serum albumin or sex hormone binding globulin (SHBG) (7, 8). Only a small fraction (~ 1 to 2%) is unbound or “free” and available to bind to its receptors (9). E1 and E3 each bind SHBG with much lower affinity than E2 and likewise each of these estrogens also shows a much lower affinity and potency for its receptors than E2 (10). SHBG also binds dihydrotestosterone (DHT) and testosterone (T) but with relative binding affinities that are 20- and 5-fold higher than for E2 (11, 12). In premenopausal women, SHBG levels are twice as high as in men and this has been suggested to limit their androgen and estrogen exposure (9, 13). SHBG concentrations decrease following menopause but increase during the sixth decade of life (14), and low serum levels of SHBG have been associated with hyperandrogenism and endometrial cancer (13). Ultimately, estrogens are eliminated from the body following their metabolic conversion to inactive metabolites, which poorly bind SHBG, and are excreted in urine and feces. Metabolic conversion occurs primarily in the liver but also in other tissues, and involves their biotransformation via enzyme-mediated conjugation to glucuronide, glutathione, methyl, and/or sulfate moieties, modifications which enhance their solubility in plasma and enhance its absorbability by tissues (15) (Figure 2). Among these estrogen conjugates, estrone sulfate (E1-S) is the most predominant in plasma, and its reclamation by steroid sulfatase is yet another route by which estrogen biosynthesis may occur in extragonadal tissue (16).
The physiological effects of estrogen are manifested through the integrated action of cellular receptors that belong to the nuclear steroid hormone receptor (SHR) and G-protein coupled receptor (GPCR) superfamilies. This paradigm of coordinated signaling by estrogen through SHRs and GPCRs is evolutionarily conserved (17) and is also employed by progestogens (18, 19) and androgens (20). ER and GPER transmit intracellular signals via fundamentally distinct mechanisms that occur with distinct kinetics and involve unique signaling effectors (21) (Figure 3). In general, ERs are localized intracellularly and function as estrogen-inducible transcription factors, while GPER exhibits all the hallmarks of a plasma membrane receptor that manifests its actions through heterotrimeric G-proteins, which in turn transactivate plasma membrane receptors and enzymes (22). Evidence also exists that ERs may function similarly to GPER, and this has been reviewed elsewhere (23). Despite their differences in cellular location and mechanism of action, SHRs and GPCRs each undergo allosteric modulation in response to binding their cognate ligands, with signaling activity of SHRs and GPCRs enhanced by the physical interaction of their cognate ligands at specific receptor contact sites. The estrogen binding characteristics of GPER and ER are distinct, and they demonstrate a different dissociation constant, Kd, in radiotracer assays using 3H-estradiol (Table 1). As discussed in detail (30), it is important to recognize that the relative binding affinities (RBAs) of ERα, ERβ and GPER cannot be readily compared due to the fact that ERs and GPER are expressed at different levels and they exist in different physicochemical environments; ER isolated in detergent-free cytosolic homogenates versus GPER enriched in lipid-rich plasma membrane preparations. Thus, the lower Kd that is measured for E2 in ER binding assays relative to GPER binding assays does not suggest that E2 has a higher affinity for ER relative to GPER. Because SHRs are readily isolated from the soluble fraction of cellular homogenates, crystallization and identification of physical ligand contact sites encoded with the structure of SHRs has been achieved (31–33). Crystal structures at resolutions of 2.6 angstroms for ER liganded to E2 or the ER antagonist, raloxifene (RAL), have been determined (34). These results show that E2 and RAL share contact sites with different binding modes and that each induces distinct conformations within the ER transactivation domain. The findings from these studies illustrate that the principal ligand contact sites of ER are defined within a hydrophobic cavity consisting of twelve helices (H1-12). Recognition of E2 within the ligand binding domain is achieved through a combination of hydrogen bond formation by the phenolic hydroxyls with polar residues contained within H3. H6 and H11, as well as alignment of the nonpolar character of estrane ring with hydrophobic residues that comprise these helices. As GPCRs are integral membrane proteins, purification is more challenging, and crystallization of GPER has yet to be achieved. In some regards, the hydrophobic environment provided within the closely aligned seven transmembrane helices of GPER is somewhat similar to the structure of the ER ligand binding domain. Several studies relying upon in silico molecular docking simulations have calculated principal binding interactions within the exoplasmic and/or transmembrane of GPER (35, 36). However, the role of these predicted ligand contact sites still needs to be evaluated by genetic studies which examine the influence of amino acid substitutions on GPER binding and signaling activity.
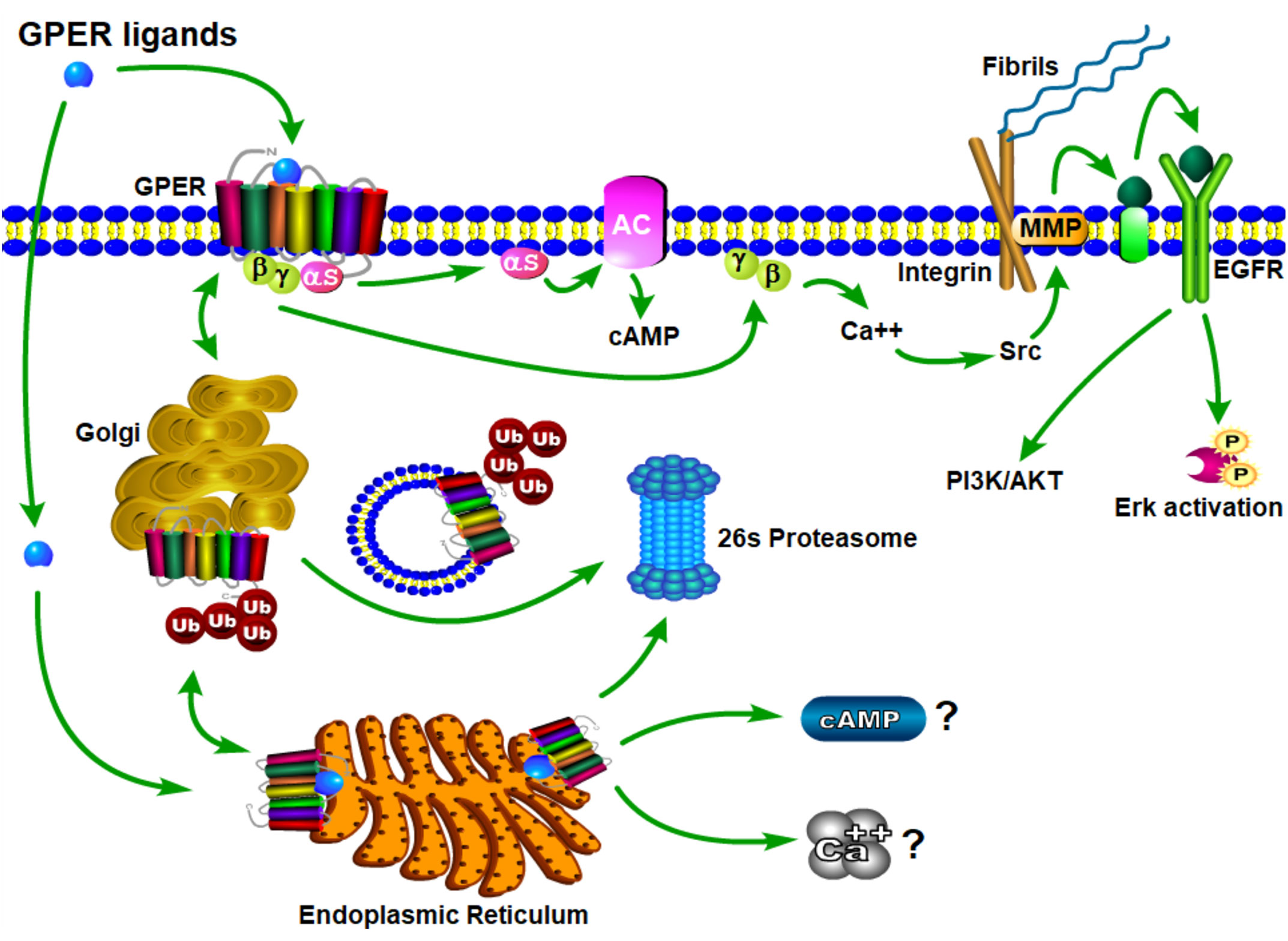
Figure 3 Schematic model of GPER trafficking and signaling. Nascent GPER is biosynthesized in the endoplasmic reticulum (ER) where it undergoes carbohydrate addition, editing and dimerization prior to forward trafficking through the Golgi apparatus during its transport to the plasma membrane. Misfolded GPER is polyubiquitinated and degraded at the 26S-proteasome. At the plasma membrane GPER exists as a high affinity GDP-coupled Gαβγ heterotrimer. Upon engagement of estrogenic ligands, GPER assumes an activated confirmation resulting in the dissociation of Gαs and Gβγ subunit proteins, which in turn, stimulate adenylyl cyclase and integrin-dependent release of membrane-tethered EGF-ligands, respectively. Independent studies evaluating retrograde trafficking of GPER suggest that it undergoes constitutive endocytosis and degradation via a ubiquitin-transGolgi-proteasome pathway. It is not yet clear whether sustained GPER signaling is observed from intracellular receptor (question marks).
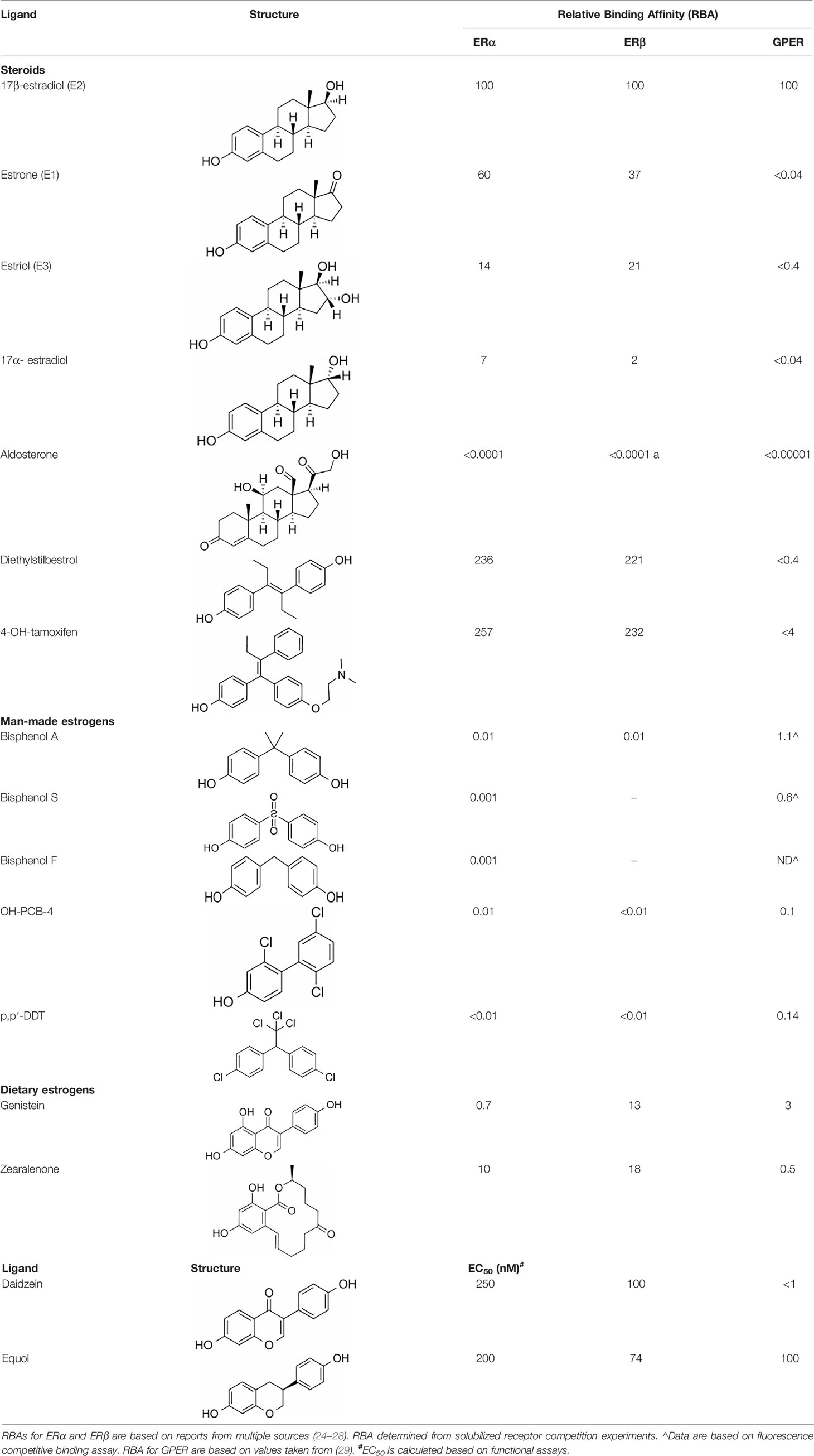
Table 1 Relative binding affinities of estrogenic ligands to estrogen receptors.
GPER in Metabolic Disease and Cancer
Studies using knockout mice indicate that ER and GPER play different roles in estrogen physiology, with ER or GPER null mice primarily exhibiting reproductive (37, 38) and metabolic (39) deficits, respectively. This simple dichotomous description clearly oversimplifies the influence of each receptor type on estrogen physiology. However, collectively, the phenotypes of ER-null (40) and GPER-null (41) mice reflect the loss of reproductive function and metabolic homeostasis that is attributed to decreased ovarian estrogen biosynthesis accompanying menopause. While it is well appreciated that the metabolic effects of estrogen are manifested through ERs (42) and GPER (43), preclinical results published earlier this year in a study led by Sharma and Prossnitz, showed that chronic administration of the synthetic GPER selective agonist, G-1/Tespria, could restore fat, glucose and lipid homeostasis (44). This result indicates that targeting GPER may be an effective means for treating diabetes and obesity, and extends prior work that showed G-1 can ameliorate atherosclerosis in mice (45). The observation that chronic GPER signaling may alter metabolic activity has potential significance regarding a role for GPER in cancer as prolonged, uninterrupted estrogen exposure (46) and metabolic syndrome (47) are independent risk factors for cancer. Thus, GPER may serve as a centrally positioned factor that drives estrogen-induced carcinogenesis through chronic signaling that promotes metabolic disorder. In support of this concept, studies have linked GPER expression to clinical indices that predict advanced disease in breast cancer including increased tumor size, the presence of distant metastases, and tamoxifen-resistance (48–51). Similar results have been obtained in ovarian (52), endometrial (53), and testicular cancers (54) with GPER directly linked to poor survival. However, other reports suggest an inverse relationship between GPER and cancer progression (similar to that demonstrated by ER) (55, 56). The most likely explanation for the differences observed in the analysis of human cancer and GPER resides in the lack of a standardized procedure for its immunohistochemical detection and quantification in tumor biopsy specimens. For instance, some studies have set an absolute threshold for GPER expression among tumors, while others have focused on the relative difference between GPER in tumors and adjacent normal tissue in individual patients (55, 57, 58). Neither have laboratory studies resolved whether GPER is pro-oncogenic. Several observations strongly support that it is. First, GPER is required for the survival of xenograft-derived cancer stem cells and metastatic disease (59). Second, in breast cancer cells, GPER integrates assembly of the fibronectin matrix (60) with the release of EGF (61); thus satisfying two basic requirements or cellular survival: attachment to the extracellular matrix and responsiveness to growth factors. Third, in a preclinical model, breast cancer is less aggressive when GPER is genetically inactivated (62). Finally, the GPER selective antagonist, G36, delays the growth of type II endometrial cancer in mice (63). Nevertheless, other studies have suggested that GPER is tumor suppressive (64, 65). Specifically, stimulation with GPER-selective agonist, G-1 leads to pro-apoptotic signaling, as well as decreased proliferation and migration by cancer cells. Limitations of the latter studies are that G-1 was used at a 100-fold higher concentration than its reported Ki or EC50 (65) and receptor knockdown strategies were not used to test for off-target effects. In addition, these studies did not determine whether the G-1 responses also occur when the endogenous estrogen, E2 is applied, or for effects of selective GPER antagonists (G15 or G36). The latter point is particularly relevant because studies reporting GPER as tumor suppressive measured inhibitory biological responses. Other studies have reported that the GPER promoter is methylated in a small percentage of cancer biopsies (66). Then again, genetic silencing is observed for many genes in cancer specimens, and this could be explained by genomic instability. Indeed, promoter methylation of ESR-1 (ERα) is common in breast cancer (67, 68). Notably, epigenetic silencing of GPER as an anti-cancer mechanism is at odds with data in public repositories, showing that GPER is widely expressed, and rarely mutated, in solid or hematopoietic cancers and in cancer cell lines. Thus, the conclusion that GPER is “tumor suppressive” is inconsistent with the widely accepted concept that a tumor suppressor gene requires genetic inactivation or epigenetic silencing. Furthermore, the idea that GPER is anti-oncogenic does not fit well with findings which suggest an active role for GPER in cancer progression in the tumor microenvironment (21). Specifically, the hypoxic environment created by proliferating cancer cells favors increased expression of GPER and local estrogen production. Breast cancer cells and cancer-associated fibroblasts (CAFs) upregulate GPER expression via hypoxia-inducing factor-1α (HIF-1α)-regulated transcriptional control (69). Increased AP-1 mediated aromatase transcription and activity is measured in breast cancer cells following estradiol or tamoxifen-mediated stimulation of GPER (70). Nor does an anti-oncogenic role for GPER reconcile with bioinformatic analyses that show that its expression correlates directly with pro-metastatic signaling pathways in estrogen receptor negative breast cancer (71). Nevertheless, the discrepancy between the pro-oncogenic and tumor suppressive activities of GPER has been discussed (72) and underscores the need to define the mechanisms that drive GPER activity and their relationship to oncogenesis.
Implications of GPER for Anti-Estrogen Therapy
ERs and GPER act independently but coordinately to maintain homeostasis of estrogen-responsive tissue. Thus, it is likely that neoplasms that arise from these tissues may either continue to direct estrogen action through both receptor types or lose control of one or both receptor mechanisms during their evolution. In fact, this is the pattern that is observed in breast cancer with treatment-naive tumors containing both receptors, one or the other receptor, or neither receptor (73). From a clinical perspective, GPER disrupts the ER-centric, binary rubric which categorizes breast cancer as either estrogen responsive or nonresponsive, with nearly, 20% of all breast cancers expressing GPER in the absence of ER. Interestingly, a preponderance of these ER-GPER+ tumors are triple negative breast cancers that lack ER, progesterone receptor (PR) and her2/neu (74).
Therapeutic interventions that reduce bioavailable estrogen should be an effective means to prevent the biological action of ERα, ERβ, and GPER. At present, three common methods are employed for reducing bioavailable estrogen: i) ovarian ablation by ovariectomy or radiation, ii) ovarian suppression by bolus administration of a gonadotrophin releasing hormone (GnRH) superagonist, such as goserelin or leuprolide, or iii) chemical inhibition by administration of aromatase inhibitors (AIs), such as exemestane, letrozole or anastrazole. Each of these three treatment interventions are used for the treatment of breast cancer. However, no single method for reducing estrogen is failproof and each of these approaches induces premature menopause, which is associated with long-term mortality risks, including increased risk of cardiovascular disease (75) and loss of bone density (76), as well as menopausal symptoms that can impact on quality of life (77). Elimination of ovarian function, either permanently by ablation or temporarily by interrupting the neuroendocrine circuit of estrogen biosynthesis, does not interfere with nonovarian biosynthesis. AIs are effective in this manner in that their effects prevent estrogen biosynthesis independent of tissue origin. While AIs effectively delay breast cancer progression in approximately 50% of breast cancer patients, their beneficial value in the remaining patients is offset by their high rate of acquired and de novo resistance (78). In evaluating the efficacy of blockade of estrogen biosynthesis in the context of either GPER (or ER), it is important to point out that nuclear steroid hormone receptors (SHRs) and G-protein coupled receptors (GPCRs) are allosterically regulated receptors that are capable of ligand-independent action (79, 80). Thus, inhibition of estrogen biosynthesis may not be effective for patients whose tumors contain mutant receptors that lose ligand binding activity but retain constitutive signaling. Although ligand binding mutants have not yet been defined for GPER, they have been identified for other GPCRs (81) and for ER (82).
An important concern regarding therapies that block estrogen biosynthesis is that theoretically they should effectively increase the ability of exogenous estrogens to interact with their cellular receptors. Albeit, it is not known whether or not AIs alter the interaction of exogenous estrogens with either GPER or ER, as this has not yet been tested experimentally. This idea is particularly interesting in light of the fact that although xenoestrogens show low binding affinities relative to 17β-estradiol for ER. The same is not true for GPER, as xenoestrogens show much higher relative binding affinities for GPER (Table 1). In order to illustrate their potential effect on anti-estrogen therapy in the context of GPER, a few of the more abundant exogenous estrogens that are relevant for this discussion are mentioned here. For example, in independent assays, the dietary soy isoflavone, daidzein (DZN) exhibits a high relative potency for GPER relative to ER, with an EC50 in the subnanomolar range compared with an EC50 that is more than 100- to 200-fold higher for nuclear ERs. Dietary exposure to soy is not trivial, in fact, measurements of postprandial serum concentrations of DZN can exceed preovulatory levels of E2 by 10-fold (83). Adding further complexity to the influence of phytoestrogens on breast cancer is the popular belief that a soy-rich diet is breast cancer protective (84). Epidemiological studies have placed emphasis on whether metabolism of DZN to S-equol, which is exclusively mediated by the gut microbiome is a critical factor in influencing estrogen physiology and ER-targeted therapy (85). This concept is interesting in light of the finding that Eastern women, whom show a two-fold reduced risk for developing breast cancer relative to Western women are twice as likely to harbor gut bacteria that metabolize DZN to S-equol (86). However, the oncogenic activity of DZN and S-equol is unclear as DZN exerts pro- and anti-oncogenic activity in mice, while other studies suggest that S-equol is anti-oncogenic (84). The influence of dietary estrogens on estrogen-targeted therapies is controversial (87). A recent guidance statement from the American Association of Clinical Endocrinologists (AACE) suggests that a soy-rich diet may be used as an alternative approach for estrogen replacement therapy (88) indicating that endogenous estrogens and phytoestrogens are biologically equivalent. Yet, an oft quoted study of 524 postmenopausal Chinese women with breast cancer showed improved survival and less recurrence in patients with the highest quartile of soy intake relative to counterparts in the lowest quartile of soy consumption (89). Significantly, this study showed a significant risk increase for patients receiving tamoxifen compared to those that received anastrozole. These data have been interpreted to indicate that soy may act competitively to block binding of tamoxifen to ER. Alternatively, these findings may suggest that the poorer survival observed in the tamoxifen arm of the study may be due to the fact that tamoxifen and soy isoflavones function as GPER agonists. Moreover, the Kang study did not control for obesity nor bacterial metabolism of DZN. Nonetheless, in humans avoidance of dietary soy or ingestion of DZN supplements by breast cancer patients receiving estrogen targeted therapy is encouraged (90) despite the fact that the RBA of DZN is 0.003% for ERα and 0.05% for Erβ (91). The question of whether soy isoflavones show enhanced carcinogenicity in the absence of endogenous estrogen has not yet been carefully addressed. Human and mouse studies which control for phytoestrogen intake, gut metabolome, and obesity in the presence or absence of AIs are necessary to evaluate the carcinogenicity of soy isoflavones in the face of AI therapy.
GPER also provides similar concerns regarding the carcinogenicity of the plasticizer, bisphenol A (BPA), the highest volume chemical produced world-wide (92). Human exposure to BPA is significant as >90% of the US population contains measurable amounts of BPA, with highest levels in children (93). BPA exhibits an RBA for GPER that is 100-fold greater than that measured for nuclear ERs (Table 1). In vitro studies indicate that BPA potency for GPER is high, with biological effects measured in the low nanomolar range in breast cancer cells and breast cancer-associated fibroblasts (60, 94) and in human seminoma and testicular cancer cells (95). Exposure to BPA is associated with many human diseases, including obesity, diabetes and cancer, and is able to induce toxicological effects in tissues and cultured cells (96). The Environmental Protection Agency and the Food and Drug Administration agree upon a safe reference dose (RfD) for BPA in humans at 50 μg/kg/day that was scaled from toxicology studies in rodents (97). Carcinogenicity testing at doses below and above the RfD in mice has yielded mixed results. While BPA is not considered a robust carcinogen, early life exposures in rodents at the RfD is associated with prostate and breast cancer (98). These authors duly underscore that the most vexing variable in the analysis of BPA carcinogenicity is the acknowledged error of scaling RfD between man and rodent due to the fact that BPA exhibits nonmonotonic dose responses in many biochemical and biological assays (99). Even more significant with regards to GPER, urinary concentrations of BPA in participants in the National Health and Nutrition Examination Survey (NHANES) demonstrated a positive association with metabolic syndrome (100). Moreover, exposure to BPA correlates with an increase in serum SHBG, even though BPA shows poor binding affinity for SHBG (12). Thus, theoretically, for a patient receiving AIs, BPA is a particularly potent GPER agonist. However, this has yet-to-be addressed in studies in which dietary estrogen intake, obesity, and gut metabolome are carefully controlled. Nonetheless, BPA is a particularly troubling environmental estrogen due to the fact that it is a malleable chemical structure that has been manipulated by chemists to produced more than 40 analogues. Many of these BPA similar are detected in humans at even higher concentrations than BPA (93, 101), and at least seven BPA analogues exhibit similar RBAs and relative potencies for GPER in breast cancer cells (35).
ER antagonism, using a selective estrogen receptor modulator (SERM), such as tamoxifen or a selective estrogen receptor degrader (SERD), such as fulvestrant, is yet another form of anti-estrogen therapy that is widely effective in the treatment of breast cancer, providing greater than 10 year survival in postmenopausal women with early stage, ER-positive cancer (102). Still, not all of these patients respond to ER antagonists, as de novo resistance occurs, and this may be due to many reasons, including: i) the presence of constitutively active ER mutants, ii) hyperactive growth factor signaling, or iii) the presence of an alternative estrogen receptor, i.e. GPER (103). GPER adds further complexity to anti-estrogen therapy in that ER antagonists, including tamoxifen, faslodex and raloxifene function as GPER agonists (21, 29). Furthermore, ER antagonism or AIs are not effective for postmenopausal women with late stage disease or for premenopausal women (104). Consistent with this idea, results from the SOFT (Suppression of Ovarian Function Trial) suggest that even further supplementation of estrogen-targeted therapy (Tamoxifen or AI) by adding ovarian suppression for premenopausal ER-positive breast cancer, while effective in reducing serum estrogen and disease relapse had no effect on overall survival (78). In this study, patients were not further stratified by whether their tumors expressed GPER. However, an argument could be made that patients whose tumors lacked GPER [approximately one-third of ER+ tumors (73)] may be more likely to respond to ER antagonism plus ovarian suppression. Further confusion regarding the role of estrogen and its receptors in female reproductive cancer comes from the disconnect between menopausal status and proliferative index, as measured by Ki-67 in tumor biopsy tissue. Breast tumors from patients with intact ovaries, show high mitotic indices, while postmenopausal women with ER-positive breast cancer are assigned either anti-estrogen therapy regardless of Ki-67 index (105). Chemotherapeutic agents, which are toxic but target rapidly proliferating cells are layered on top of anti-estrogen therapy for patients with aggressive estrogen-dependent cancers (106), without consideration of their GPER status, which has been tied to chemotherapeutic resistance via its capacity to trigger EGFR transactivation (107). Recent results from the PALOMA-III trials, further showed that addition of palbociclib, which targets cyclin-dependent kinases, CDK4 and CDK6, to ER-targeted therapy (fulvestrant) provides increased overall survival for patients with advanced ER-positive breast cancer (108). Early results achieved with palbociclib in metastatic breast cancer are encouraging. Yet they do not resolve whether palbociclib selectively targets proliferation in fulvestrant- resistant, ER-positive breast cancer cells, or whether its actions directly influence GPER-dependent cellular responses associated with tumor cell metastasis and disease progression. Collectively, these examples indicate that definition of GPER status for patients with breast cancer may help to select patient populations which are best able to respond to existing anti-estrogen therapies, either ovarian suppression, ER antagonism or aromatase inhibitor.
Existing and Future Pharmacological Compounds That Target GPER
For all of the above reasons, therapeutic approaches that block GPER action hold great promise for the treatment of cancer. After all, nearly one-third of all FDA-approved drugs target GPCRs (109). While GPCR targeted drugs have been predominately used for the treatment of cardiovascular disease and diabetes, the concept of developing GPCR targeted cancer therapeutics has gained traction over the past decade (110). This is largely due to preclinical studies which link GPCRs to cancer growth and metastasis, often in a scenario where the GPCR involved is chronically exposed to local or circulating agonist. Examples of this include, the bioactive lipid, lysophosphatidic acid, and its receptor, LPAR-1 in breast cancer (111), chemokines, CXCL8/IL8 and CXCR1 and CXCR2 in melanoma, pancreatic cancer and gastric tumors (112) and CXCL12 and CXCR4 in multiple cancers (113). Consistent with the notion that chronic estrogen exposure may drive GPER oncogenesis, breast tumors with increased GPER plasma membrane density show poor prognosis (51). This may be consistent with the concept that GPCRs often demonstrate a hyperbolic relationship between ligand occupancy and receptor response (114). This is widely described as “fractional occupancy” and suggests that a small change in GPER plasma receptor density could result in a more than linear increase in GPER activity. It is also important to consider that GPER shows specific binding activity to estrogenic ligands, natural or synthetic, which are hydrophobic and/or lipophilic and easily diffuse through or insert themselves into a lipid bilayer. In fact, it has previously published that crude membrane fractions exhibit specific GPER binding activity (Thomas et al, 2005). Whether intracellular interaction between GPER and its ligands allows for sustained intracellular signaling or plays a role in the proper folding and transport of GPER to the plasma membrane has not yet been determined. In this regard, it is important to recognize that an intracellular staining pattern is observed in most, but not all, cell types (115). However, a plasma membrane staining pattern by immunohistochemical (IHC) analysis of microtome-sectioned, archival paraffin-embedded tissue is not easily detected unless the majority of the receptor is at the plasma membrane, and little is detected intracellularly. With this in mind, slight differences in GPER ligand sensitivity would be difficult to detect by IHC, however, measurement of GPER plasma membrane density by flow cytometric analysis of intact breast cancer cells (116) may provide a better handle as whether to apply anti-estrogen therapy in the context of GPER-targeted therapies described below.
Small Molecule GPER Antagonists
Several GPER antagonists have been developed (Table 2). While many of these first-generation drugs hold promise, we review below two GPER antagonists with half-maximal inhibitory concentration (IC50) within the nanomolar range. The first GPER antagonist in this class, named G15, was developed by Prossnitz and colleagues using a combination of virtual and biomolecular screening steps (117). First these authors used a software-assisted virtual screen of the NIH Molecular Libraries Small Molecule Repository (MLSMR) of 144, 457 molecules. From this primary screen 57 compounds were isolated that were similar in structure to the GPER selective agonist, G-1, a substituted dihydroquinolone (24). These compounds were tested subsequently for their capacity to inhibit E2-mediated calcium mobilization in human SKBR3 breast cancer cells that express endogenous GPER but lack ERα and ERβ. G15 emerged from this screen based on its: i) structure and presumed ability to interact competitively with E2, ii) ability to block E2-dependent calcium signaling, and iii) measured binding affinity (Kd = 20 nM) for GPER, which was assessed using I125-labelled G-1 as radiotracer. G-15 displays relatively low binding affinity for ERα and ERβ as measured in a competition assay employing an Alexa 633-estradiol conjugate as fluorotracer (Ki > 10 nM). In vivo testing has shown that G15 blocks a proliferative response in uterine epithelial cells (117). A G15 derivative, named G36 was subsequently synthesized by Dennis and Prossnitz, with even lower affinity interactions with ERα (118). G36 inhibits E2 and G-1-dependent calcium mobilization as well as erk-1/2 activation in SKBR3 cells (IC50 = 200 nM) and blocks the growth of transplanted estrogen-dependent type II endometrial cancer cells (63). Recently, Chris Arnatt and David Wang have collaborated to report a new GPER antagonist that protects ovariectomized ERα null mice from estrogen-induced cholesterol gallstones (119). Using a receptor-ligand interaction computational screen, a novel series of GPER-selective antagonists were generated, including one new compound, 2-cyclohexyl-4-isopropyl-N-(4-methoxybenzyl) aniline (CIMBA). that shows strong antagonism with selectivity for GPER. Specifically, CIMBA inhibits G-1 dependent calcium mobilization in HL60 cells (IC50 = 75 nM), with a binding activity for ERα or ERβ <10 μM in fluorescence polarization assays. Some differences were noted by Arnatt and colleagues with regards to the efficacy of G15, G36 and CIMBA to inhibit calcium mobilization, although all three GPER antagonists each showed inhibitory capacity for G-1 induced cAMP accumulation by homogenous time resolved fluorescence (HTRF). Thus, while the computational algorithms that yielded the G-series based and methoxybenzyl aniline based GPER antagonists were inherently distinct, both show similar capacity to inhibit G-1 induced GPER signaling, with each showing efficacy for reducing estrogen-induced pathology in mice.
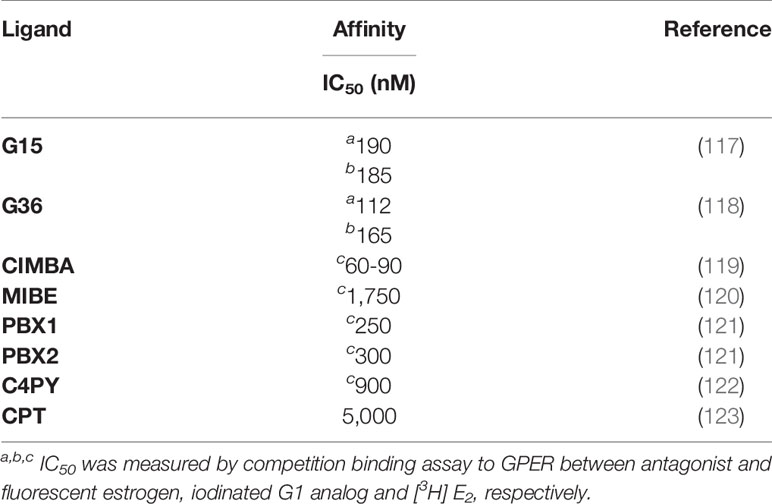
Table 2 IC50 for GPER antagonists.
Targeting G Proteins
An alternative approach to developing selective agents that block GPER action is to employ pharmaceutical compounds that directly target G proteins (124, 125). This strategy has the added benefit that although GPER is a driving force in the genesis of metabolic disorder and cancer, these are complex diseases in which multiple GPCRs are involved. Primary examples include chemokine receptors (CXCR1, CXCR2, CXCR4, CCR5, CCR7) that drive chronic inflammatory responses common to both obesity and cancer. The premise by which G protein blockade is effective as a therapeutic is the ability of these agents to preferentially inhibit signaling pathways shared by more than one GPCR. Towards this end, cell permeant pharmacological agents have been developed that interfere with conformational activation of the GPCR-Gαβγ complex following ligand binding. To date, pharmacological compounds that specifically inhibit Gα-GTPase have been limited to the Gαq proteins and include YM-254890 (126) and FR900359 (127). Gαq inhibitors show good preclinical success in thrombosis (128), asthma (129) and melanoma (130). In contrast, Gβγ inhibitors, which were initially based upon the carboxyl terminal domain structure of G-protein receptor kinase 2 but now also include M119 and gallein, show efficacy in preclinical models of opioid analgesia, chronic inflammatory disease, heart failure (124). Blockade of GPER-dependent EGFR transactivation in breast cancer cells is effective using a Gβγ-sequestrant peptide (131), and further study is needed to evaluate whether Gβγ-inhibitors are effective in mouse models of metabolic disorder and cancer.
“Biased” agonists that stabilize a GPCR conformation that preferentially activates one signaling pathway over another (132) represents a related approach towards selective inhibition of G protein dependent signaling. Oliceridine, a biased agonist for μ-opioid receptor was developed to favor Gαi-inhibition of adenylyl cyclase over Gβγ-dependent activation of β-arrestin (133) and has been evaluated in clinical trials for chronic pain. Although recent reports indicate that low agonist efficacy, rather than receptor bias, may explain the low side effect profile of oliceridine (134). Similarly, biased agonists have been developed and characterized for angiotensin I receptors that preferentially recruit β-arrestin for their potential use in reducing hypertension (135). Biased agonists have yet to make their way into the clinic. However, it is unclear at the moment whether the biased agonist conformation is unique to certain GPCRs or whether it has broad application. Still, our environment is replete with compounds that function as estrogen mimetics, and it may be possible by high throughput analysis of synthetic and nutraceutical compounds to identify biased GPER agonists that may have therapeutic value.
Targeting Downstream Signaling Effectors of GPER
Via GPER, estrogens trigger an epidermal growth factor (EGF)-autocrine loop (22) that holds significance for breast carcinoma, and potentially other malignancies that arise from epithelial tissue. In breast cancer this holds particular significance due to the reciprocal relationship that is often observed between ER and epidermal growth factor receptors (EGFRs) in primary tumors. This relationship has fostered the dichotomous categorization of breast cancers as either estrogen responsive or growth factor responsive. While GPER disrupts this simple binary scheme, GPER holds potential diagnostic value in selecting patients that may best benefit from either erbB1 or erbB2/her2/neu targeted therapy, particularly among premenopausal women. Assessment of GPER expression also may suggest the appropriate combinatorial assignment of AI or GPER antagonist with EGFR targeted antibody treatment. As discussed in section 3, GPER is expressed in a majority of TNBC, an aggressive subtype of breast cancer with no known molecular targets. erbB1/EGFR is also commonly overexpressed in TNBC, although results from numerous clinical trials reveal low response rates to anti-EGFR therapy for patients with TNBC (136). However, some patients do respond well, which may suggest a need to stratify patients for EGFR responsiveness and to develop combinatorial therapies. In both regards, GPER may have value. First, as a theranostic index. Second, GPER targeted therapeutics may fit well as part of a combinatorial anti-EGFR therapy for patients with erbB1 overexpressing TNBC.
Phosphoinositide 3 (PI3) kinase/AKT signaling lies downstream of erbB1/erbB2, and is activated following GPER stimulation (137). Activation of PI3K/AKT signaling occurs commonly in breast cancer and is associated with endocrine resistance and worse prognosis (138). Pan-PI3K inhibitors have fared poorly in clinical trials due to their toxicity, while the isoform-specific PI3K inhibitor, alpelisib, has been approved by the FDA as co-therapy with fulvestrant for patients with ER-positive, PI3Kalpha mutated advanced breast cancer (139). FDA approval of alpelisib with fulveestrant followed the results of the SOLAR-1 trial that showed that patients receiving alpelisib with fulvestrant showed a median increase of 6 months of progression free survival. Future studies that include a more comprehensive view of patients which are estrogen responsive by including analysis of GPER, may lead to similarly designed clinical trials that combine either AIs or GPER targeted therapy with alpelisib.
Antibodies
Traditionally, small molecules have dominated as the preferred means to target GPCRs but recent pharmaceutical trends that favor immunotherapeutic approaches have led to the development of GPCR-targeted antibodies for clinical use (Table 3). The most significant progress has been made in the development of antibodies that block the binding of chemokines to their cognate GPCRs in cancer and inflammatory disease (144, 145). Notably, mogamulizumab/Poteligeo, an anti-CCR4 targeted therapy for refractory adult T cell leukemia and mycosis fungoides has received FDA approval (146). Likewise, the FDA has also approved erenumab/Aimovig, targeting Calcitonin Gene-Related Peptide Receptor (CGRPR) as a prophylactic treatment for migraine headaches (141). In addition, the angiogenesis/tumor metastasis-associated receptor, CXCR4, targeted by ulocuplumab (Bristol Myers Squibb), a fully humanized antibody that blocks binding of stromal-derived factor 1 (SDF-1) in adult myeloid leukemia has entered phase II trials (143). The CCR5-targeted antibody, leronlimab is currently under phase III investigation as an HIV therapy and has entered phase II testing to relieve chronic lung inflammation that accompanies COVID 19 infection (142). CCR2 targeted mAB, MLN1202/plozalizumab (Millenium/Takeda Oncology) has been evaluated in multiple clinical trials for cancer and other indications (147).
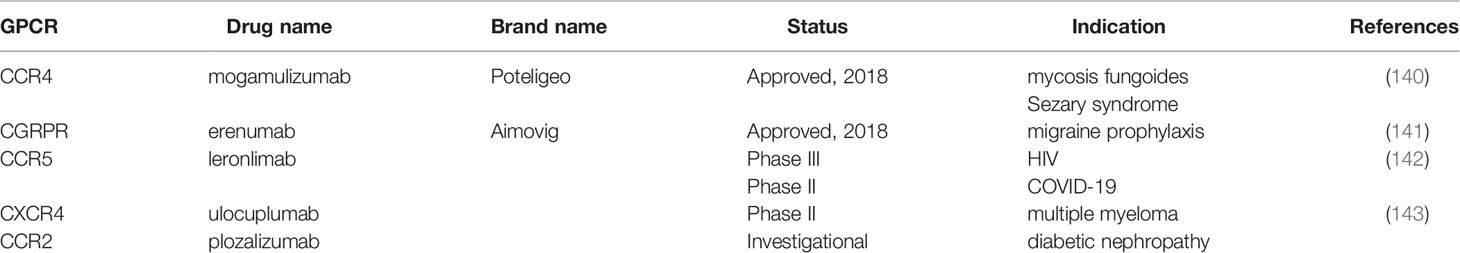
Table 3 Status of GPCR therapeutic antibodies.
Once considered difficult to target via antibody-based approaches, the combined use of lipid-enriched GPCR preparations and the development of recombinant phage display technology has allowed for the rapid growth and development of antibodies that target GPCRs. The fact that GPCR heterodimerization is a widely accepted paradigm that adds diversity and complexity to GPCR functionality is an additional reason why antibody-based therapeutic approaches have gained traction relative to small molecule antagonists.
Antibodies that target GPCRs could also be used to deliver anti-cancer agents by conjugating the antibodies to nanoparticles (148). Such nanoparticles can be formed from biodegradable polymers and can physically entrap the anti-cancer agent throughout the nanoparticle (149). Through diffusion and degradation of the polymer, the drug can be released in a controlled manner to the target cancer (149). Polymers used to prepare these particles include but are not limited to poly lactic-co-glycolic acid (149), polysulfenamides (150), and polyanhydrides (151). Agents that can be loaded into the particles include proteins such as cancer antigens (152), nucleic acid based molecules like plasmid DNA (153) and CpG (154) and small molecule drugs like paclitaxel and doxorubicin (149, 155).
Conclusions
Anti-estrogen therapies are successfully employed for the treatment of breast cancer and anovulatory infertility. Still, at present, decisions regarding the appropriate assignment of anti-estrogen therapy in breast cancer are limited strictly upon the detection of ER in tumor biopsy specimens. This ER-centric perspective ignores the fact that 20% of breast cancers express GPER and in the absence of ER (73), and that GPER is expressed in a majority of TNBCs (74). Despite the relative success of ER antagonists, aromatase inhibitors and ovarian ablation/suppression strategies for postmenopausal women with early stage ER- positive cancer, resistance occurs. A further confounding variable for the assignment of anti-estrogen therapy is the fact that ER antagonists (both SERMS and SERDs) function as GPER agonists, which aligns with the finding that GPER is associated with tamoxifen resistance in breast cancer patients (103). The realization that daidzein (156) and environmental bisphenols (35) potently activate GPER further alters our perspective regarding the appropriate assignment of anti-estrogen therapy. In addition, recent clinical trials evaluating AI or TAM with ovarian suppression have shown a median increase in progression free survival suggesting that some patients may respond favorably to tandem anti-estrogen blockade. However, these studies did not include patients whose tumors are GPER- positive and ER-negative. Our current perspective for determining which patients may respond to anti-estrogen therapy is evolving, and is bolstered by findings that show that GPER associates with cancer progression variables (48, 52, 53), activates cellular receptors that facilitate cancer cell survival (54), promotes the survival of patient-derived breast cancer stem cells (59), and acts in the tumor microenvironment to drive cancer metastasis (62).
The development of GPER targeted therapies holds the promise of expanding our existing arsenal of estrogen-targeted therapies. GPER is a therapeutic target that holds particular promise for the treatment of several critical health concerns facing Western society, including obesity, diabetes, vascular pathology and advanced cancer. In the preclinical setting, chronic administration of G1/Tespria restores fat, lipid, and glucose homeostasis in obese and diabetic mice without uterotropic effects (44). Analysis of human cancer and in mice suggest that GPER is linked to advanced cancer, and chronic estrogen exposure and metabolic syndrome are independent risk factors for many cancers. Thus, GPER provides a likely mechanism by which metabolic disorder may be part of the landscape for estrogen-driven malignancies. The selective GPER antagonists, G15 delays the growth of endometrial cancer (63) and exciting new data indicates that a new GPER antagonist, CIMBA, can prevent estrogen-induced gallstones (119). Additional methodologies for targeting GPER may also include direct blockade of G-proteins, the development of biased agonists and therapeutic antibodies. Collectively, these approaches may complement existing anti-estrogen therapies and improve our approach towards treating patients suffering from estrogen-driven malignancies and disease.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
EJF and AKS acknowledge the NIH NCI P30 CA086862 Cancer Center support grant as well as I-award funds from the Holden Comprehensive Cancer Center, including an Oberley award and monies from the Breast Cancer Research Interest Group. EJF acknowledges funds from the Department of Surgery at the University of Iowa. AKS is the Lyle and Sharon Bighley Chair in Pharmaceutical Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors appreciate senior guidance and constructive comments provided by Drs. Ronald Weigel and George Weiner.
References
1. Carreau S, Silandre D, Bourguiba S, Hamden K, Said L, Lambard S, et al. Estrogens and male reproduction: a new concept. Braz J Med Biol Res (2007) 40(6):761–8. doi: 10.1590/S0100-879X2007000600003
2. Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med (2013) 19(3):197–209. doi: 10.1016/j.molmed.2012.12.007
3. Farzaneh S, Zarghi A. Estrogen Receptor Ligands: A Review (2013-2015). Sci Pharm (2016) 84(3):409–27. doi: 10.3390/scipharm84030409
4. Lee AJ, Cai MX, Thomas PE, Conney AH, Zhu BT. Characterization of the oxidative metabolites of 17β-estradiol and estrone formed by 15 selectively expressed human cytochrome P450 isoforms. Endocrinology (2003) 144(8):3382–98. doi: 10.1210/en.2003-0192
5. Chatuphonprasert W, Jarukamjorn K, Ellinger I. Physiology and Pathophysiology of Steroid Biosynthesis, Transport and Metabolism in the Human Placenta. Front Pharmacol (2018) 9:1027–55. doi: 10.3389/fphar.2018.01027
6. Berkane N, Liere P, Oudinet J-P, Hertig A, Lefèvre G, Pluchino N, et al. From pregnancy to preeclampsia: a key role for estrogens. Endocr Rev (2017) 38(2):123–44. doi: 10.1210/er.2016-1065
7. Knochenhauer ES, Boots LR, Potter HD, Azziz R. Differential binding of estradiol and testosterone to SHBG. Relation to circulating estradiol levels. J Reprod Med (1998) 43(8):665–70.
8. Balogh A, Karpati E, Schneider AE, Hetey S, Szilagyi A, Juhasz K, et al. Sex hormone-binding globulin provides a novel entry pathway for estradiol and influences subsequent signaling in lymphocytes via membrane receptor. Sci Rep (2019) 9(1):4. doi: 10.1038/s41598-018-36882-3
9. Hammond GL. Diverse roles for sex hormone-binding globulin in reproduction. Biol Reprod (2011) 85(3):431–41. doi: 10.1095/biolreprod.111.092593
10. Smiley DA, Khalil RA. Estrogenic compounds, estrogen receptors and vascular cell signaling in the aging blood vessels. Curr Med Chem (2009) 16(15):1863–87. doi: 10.2174/092986709788186093
11. Round P, Das S, Wu T-S, Wähälä K, Van Petegem F, Hammond GL. Molecular interactions between sex hormone–binding globulin and nonsteroidal ligands that enhance androgen activity. J Biol Chem (2020) 295(5):1202–11. doi: 10.1074/jbc.RA119.011051
12. Xiong Q, Liu X, Shen Y, Yu P, Chen S, Hu J, et al. Elevated serum Bisphenol A level in patients with dilated cardiomyopathy. Int J Environ Res Public Health (2015) 12(5):5329–37. doi: 10.3390/ijerph120505329
13. Saez-Lopez C, Brianso-Llort L, Torres-Torronteras J, Simo R, Hammond GL, Selva DM. Resveratrol Increases Hepatic SHBG Expression through Human Constitutive Androstane Receptor: a new Contribution to the French Paradox. Sci Rep (2017) 7(1):12284. doi: 10.1038/s41598-017-12509-x
14. Maggio M, Lauretani F, Basaria S, Ceda GP, Bandinelli S, Metter EJ, et al. Sex hormone binding globulin levels across the adult lifespan in women–the role of body mass index and fasting insulin. J Endocrinol Invest (2008) 31(7):597–601. doi: 10.1007/BF03345608
15. Samavat H, Kurzer MS. Estrogen metabolism and breast cancer. Cancer Lett (2015) 356(2 Pt A):231–43. doi: 10.1016/j.canlet.2014.04.018
16. Reed M, Purohit A, Woo LL, Newman SP, Potter BV. Steroid sulfatase: molecular biology, regulation, and inhibition. Endocr Rev (2005) 26(2):171–202. doi: 10.1210/er.2004-0003
17. Norman AW, Mizwicki MT, Norman DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov (2004) 3(1):27–41. doi: 10.1038/nrd1283
18. Zhu Y, Rice CD, Pang Y, Pace M, Thomas P. Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci U S A (2003) 100(5):2231–6. doi: 10.1073/pnas.0336132100
19. Zhu Y, Bond J, Thomas P. Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc Natl Acad Sci U S A (2003) 100(5):2237–42. doi: 10.1073/pnas.0436133100
20. Heinlein CA, Chang C. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol Endocrinol (2002) 16(10):2181–7. doi: 10.1210/me.2002-0070
21. Filardo EJ. A role for G-protein coupled estrogen receptor (GPER) in estrogen-induced carcinogenesis: Dysregulated glandular homeostasis, survival and metastasis. J Steroid Biochem Mol Biol (2018) 176:38–48. doi: 10.1016/j.jsbmb.2017.05.005
22. Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab (2005) 16(8):362–7. doi: 10.1016/j.tem.2005.08.005
23. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol (2016) 17(12):783–97. doi: 10.1038/nrm.2016.122
24. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol (2006) 2(4):207–12. doi: 10.1038/nchembio775
25. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology (1997) 138(3):863–70. doi: 10.1210/endo.138.3.4979
26. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology (1998) 139(10):4252–63. doi: 10.1210/endo.139.10.6216
27. Muthyala RS, Ju YH, Sheng S, Williams LD, Doerge DR, Katzenellenbogen BS, et al. Equol, a natural estrogenic metabolite from soy isoflavones: convenient preparation and resolution of R- and S-equols and their differing binding and biological activity through estrogen receptors alpha and beta. Bioorg Med Chem (2004) 12(6):1559–67. doi: 10.1016/j.bmc.2003.11.035
28. Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, et al. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands. Toxicol Sci (2000) 54(1):138–53. doi: 10.1093/toxsci/54.1.138
29. Prossnitz ER, Arterburn JB. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev (2015) 67(3):505–40. doi: 10.1124/pr.114.009712
30. Filardo EJ, Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology (2012) 153(7):2953–62. doi: 10.1210/en.2012-1061
31. Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem (2000) 275(34):26164–71. doi: 10.1074/jbc.M004571200
32. Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci U S A (1998) 95(11):5998–6003. doi: 10.1073/pnas.95.11.5998
33. Hard T, Kellenbach E, Boelens R, Maler BA, Dahlman K, Freedman LP, et al. Solution structure of the glucocorticoid receptor DNA-binding domain. Science (1990) 249(4965):157–60. doi: 10.1126/science.2115209
34. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature (1997) 389(6652):753–8. doi: 10.1038/39645
35. Cao LY, Ren XM, Li CH, Zhang J, Qin WP, Yang Y, et al. and Bisphenol B Exert Higher Estrogenic Effects than Bisphenol A via G Protein-Coupled Estrogen Receptor Pathway. Environ Sci Technol (2017) 51(19):11423–30. doi: 10.1021/acs.est.7b03336
36. Mendez-Luna D, Martinez-Archundia M, Maroun RC, Ceballos-Reyes G, Fragoso-Vazquez MJ, Gonzalez-Juarez DE, et al. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J Biomol Struct Dyn (2015) 33(10):2161–72. doi: 10.1080/07391102.2014.994102
37. Hewitt SC, Harrell JC, Korach KS. Lessons in estrogen biology from knockout and transgenic animals. Annu Rev Physiol (2005) 67:285–308. doi: 10.1146/annurev.physiol.67.040403.115914
38. Hewitt SC, Korach KS. Oestrogen receptor knockout mice: roles for oestrogen receptors alpha and beta in reproductive tissues. Reproduction (2003) 125(2):143–9. doi: 10.1530/rep.0.1250143
39. Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology (2013) 154(11):4136–45. doi: 10.1210/en.2013-1357
40. Walker VR, Korach KS. Estrogen receptor knockout mice as a model for endocrine research. ILAR J (2004) 45(4):455–61. doi: 10.1093/ilar.45.4.455
41. Prossnitz ER, Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol (2015) 153:114–26. doi: 10.1016/j.jsbmb.2015.06.014
42. Hevener AL, Clegg DJ, Mauvais-Jarvis F. Impaired estrogen receptor action in the pathogenesis of the metabolic syndrome. Mol Cell Endocrinol (2015) 418 Pt 3:306–21. doi: 10.1016/j.mce.2015.05.020
43. Sharma G, Mauvais-Jarvis F, Prossnitz ER. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J Steroid Biochem Mol Biol (2018) 176:31–7. doi: 10.1016/j.jsbmb.2017.02.012
44. Sharma G, Hu C, Staquicini DI, Brigman JL, Liu M, Mauvais-Jarvis F, et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci Transl Med (2020) 12:528–41. doi: 10.1126/scitranslmed.aau5956
45. Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci Rep (2014) 4:7564. doi: 10.1038/srep07564
46. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol (2011) 7(12):715–26. doi: 10.1038/nrendo.2011.122
47. Sharma G, Prossnitz ER. G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis. Adv Exp Med Biol (2017) 1043:427–53. doi: 10.1007/978-3-319-70178-3_20
48. Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, et al. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res (2006) 12(21):6359–66. doi: 10.1158/1078-0432.CCR-06-0860
49. Arias-Pulido H, Royce M, Gong Y, Joste N, Lomo L, Lee SJ, et al. GPR30 and estrogen receptor expression: new insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res Treat (2010) 123(1):51–8. doi: 10.1007/s10549-009-0631-7
50. Ignatov T, Claus M, Nass N, Haybaeck J, Seifert B, Kalinski T, et al. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res Treat (2019) 174(1):121–7. doi: 10.1007/s10549-018-5064-8
51. Sjostrom M, Hartman L, Grabau D, Fornander T, Malmstrom P, Nordenskjold B, et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res Treat (2014) 145(1):61–71. doi: 10.1007/s10549-014-2936-4
52. Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ, et al. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol (2009) 114(3):465–71. doi: 10.1016/j.ygyno.2009.05.015
53. Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, et al. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol (2007) 196(4):386 e1–9; discussion e9-11. doi: 10.1016/j.ajog.2007.01.004
54. Rago V, Romeo F, Giordano F, Maggiolini M, Carpino A. Identification of the estrogen receptor GPER in neoplastic and non-neoplastic human testes. Reprod Biol Endocrinol (2011) 9:135. doi: 10.1186/1477-7827-9-135
55. Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD, Yang X, et al. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-kappaB signals. Mol Oncol (2016) 10(6):775–88. doi: 10.1016/j.molonc.2016.01.002
56. Rochefort H, Platet N, Hayashido Y, Derocq D, Lucas A, Cunat S, et al. Estrogen receptor mediated inhibition of cancer cell invasion and motility: an overview. J Steroid Biochem Mol Biol (1998) 65(1–6):163–8. doi: 10.1016/s0960-0760(98)00010-7
57. Liang S, Chen Z, Jiang G, Zhou Y, Liu Q, Su Q, et al. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-kappaB/IL-6 signals. Cancer Lett (2017) 386:12–23. doi: 10.1016/j.canlet.2016.11.003
58. Luo H, Yang G, Yu T, Luo S, Wu C, Sun Y, et al. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr Relat Cancer (2014) 21(2):355–69. doi: 10.1530/ERC-13-0237
59. Chan YT, Lai AC, Lin RJ, Wang YH, Wang YT, Chang WW, et al. GPER-induced signaling is essential for the survival of breast cancer stem cells. Int J Cancer (2020) 146(6):1674–85. doi: 10.1002/ijc.32588
60. Magruder HT, Quinn JA, Schwartzbauer JE, Reichner J, Huang A, Filardo EJ. The G protein-coupled estrogen receptor-1, GPER-1, promotes fibrillogenesis via a Shc-dependent pathway resulting in anchorage-independent growth. Horm Cancer (2014) 5(6):390–404. doi: 10.1007/s12672-014-0195-9
61. Quinn JA, Graeber CT, Frackelton AR Jr., Kim M, Schwarzbauer JE, Filardo EJ. Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. Mol Endocrinol (2009) 23(7):1052–64. doi: 10.1210/me.2008-0262
62. Marjon NA, Hu C, Hathaway HJ, Prossnitz ER. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol Cancer Res (2014) 12(11):1644–54. doi: 10.1158/1541-7786.MCR-14-0128-T
63. Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, et al. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int (2013) 2013:472720. doi: 10.1155/2013/472720
64. Weissenborn C, Ignatov T, Ochel HJ, Costa SD, Zenclussen AC, Ignatova Z, et al. GPER functions as a tumor suppressor in triple-negative breast cancer cells. J Cancer Res Clin Oncol (2014) 140(5):713–23. doi: 10.1007/s00432-014-1620-8
65. Ignatov T, Modl S, Thulig M, Weissenborn C, Treeck O, Ortmann O, et al. GPER-1 acts as a tumor suppressor in ovarian cancer. J Ovarian Res (2013) 6(1):51. doi: 10.1186/1757-2215-6-51
66. Weissenborn C, Ignatov T, Nass N, Kalinski T, Dan Costa S, Zenclussen AC, et al. GPER Promoter Methylation Controls GPER Expression in Breast Cancer Patients. Cancer Invest (2017) 35(2):100–7. doi: 10.1080/07357907.2016.1271886
67. Gaudet MM, Campan M, Figueroa JD, Yang XR, Lissowska J, Peplonska B, et al. DNA hypermethylation of ESR1 and PGR in breast cancer: pathologic and epidemiologic associations. Cancer Epidemiol Biomarkers Prev (2009) 18(11):3036–43. doi: 10.1158/1055-9965.EPI-09-0678
68. Stone A, Zotenko E, Locke WJ, Korbie D, Millar EK, Pidsley R, et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat Commun (2015) 6:7758. doi: 10.1038/ncomms8758
69. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S, et al. GPER mediates activation of HIF1alpha/VEGF signaling by estrogens. Cancer Res (2014) 74(15):4053–64. doi: 10.1158/0008-5472.CAN-13-3590
70. Catalano S, Giordano C, Panza S, Chemi F, Bonofiglio D, Lanzino M, et al. Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res Treat (2014) 146(2):273–85. doi: 10.1007/s10549-014-3017-4
71. Talia M, De Francesco EM, Rigiracciolo DC, Muoio MG, Muglia L, Belfiore A, et al. The G Protein-Coupled Estrogen Receptor (GPER) Expression Correlates with Pro-Metastatic Pathways in ER-Negative Breast Cancer: A Bioinformatics Analysis. Cells (2020) 9(3):622–34. doi: 10.3390/cells9030622
72. Hsu LH, Chu NM, Lin YF, Kao SH. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int J Mol Sci (2019) 20(2):306–321. doi: 10.3390/ijms20020306
73. Filardo EJ, Quinn JA, Sabo E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids (2008) 73(9-10):870–3. doi: 10.1016/j.steroids.2007.12.025
74. Phadke S, Mott S, Bashir A, Bellizzi A, Resnick M, Sturtevant A, et al. Distribution of G-protein coupled estrogen receptor in treatment-naive triple negative breast cancer and association with clinicopathologic characteristics. Cancer Res (2020) 80(4):P2-11-16. Proceedings of the 2019 San Antonio Breast Cancer Symposium; 2019 Dec 10-14; San Antonio, TX. Philadelphia (PA). doi: 10.1158/1538-7445.SABCS19-P2-11-16
75. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ (2017) 8(1):33. doi: 10.1186/s13293-017-0152-8
76. Weitzmann MN, Pacifici R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest (2006) 116(5):1186–94. doi: 10.1172/JCI28550
77. Whiteley J, DiBonaventura M, Wagner JS, Alvir J, Shah S. The impact of menopausal symptoms on quality of life, productivity, and economic outcomes. J Womens Health (Larchmt) (2013) 22(11):983–90. doi: 10.1089/jwh.2012.3719
78. Nourmoussavi M, Pansegrau G, Popesku J, Hammond GL, Kwon JS, Carey MS. Ovarian ablation for premenopausal breast cancer: A review of treatment considerations and the impact of premature menopause. Cancer Treat Rev (2017) 55:26–35. doi: 10.1016/j.ctrv.2017.02.005
79. Weigel NL, Zhang Y. Ligand-independent activation of steroid hormone receptors. J Mol Med (Berl) (1998) 76(7):469–79. doi: 10.1007/s001090050241
80. Levoye A, Dam J, Ayoub MA, Guillaume JL, Jockers R. Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep (2006) 7(11):1094–8. doi: 10.1038/sj.embor.7400838
81. Stoy H, Gurevich VV. How genetic errors in GPCRs affect their function: Possible therapeutic strategies. Genes Dis (2015) 2(2):108–32. doi: 10.1016/j.gendis.2015.02.005
82. Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov (2017) 7(3):277–87. doi: 10.1158/2159-8290.CD-15-1523
83. King RA, Bursill DB. Plasma and urinary kinetics of the isoflavones daidzein and genistein after a single soy meal in humans. Am J Clin Nutr (1998) 67(5):867–72. doi: 10.1093/ajcn/67.5.867
84. Rice S, Whitehead SA. Phytoestrogens and breast cancer–promoters or protectors? Endocr Relat Cancer (2006) 13(4):995–1015. doi: 10.1677/erc.1.01159
85. Lampe JW. Emerging research on equol and cancer. J Nutr (2010) 140(7):1369S–72S. doi: 10.3945/jn.109.118323
86. He F-J, Chen J-Q. Consumption of soybean, soy foods, soy isoflavones and breast cancer incidence: differences between Chinese women and women in Western countries and possible mechanisms. Food Sci Hum Wellness (2013) 2(3-4):146–61. doi: 10.1016/j.fshw.2013.08.002
87. Messina MJ, Wood CE. Soy isoflavones, estrogen therapy, and breast cancer risk: analysis and commentary. Nutr J (2008) 7:17. doi: 10.1186/1475-2891-7-17
88. Randel A. AACE releases guidelines for menopausal hormone therapy. Am Family Physician (2012) 86(9):864–8.
89. Kang X, Zhang Q, Wang S, Huang X, Jin S. Effect of soy isoflavones on breast cancer recurrence and death for patients receiving adjuvant endocrine therapy. CMAJ Can Med Assoc J (2010) 182(17):1857–62. doi: 10.1503/cmaj.091298
90. Hilakivi-Clarke L, Andrade JE, Helferich W. Is soy consumption good or bad for the breast? J Nutr (2010) 140(12):2326S–34S. doi: 10.3945/jn.110.124230
91. Jiang Y, Gong P, Madak-Erdogan Z, Martin T, Jeyakumar M, Carlson K, et al. Mechanisms enforcing the estrogen receptor beta selectivity of botanical estrogens. FASEB J (2013) 27(11):4406–18. doi: 10.1096/fj.13-234617
92. Björnsdotter MK, de Boer J, Ballesteros-Gómez A, Bisphenol A. and replacements in thermal paper: A review. Chemosphere (2017) 182:691–706. doi: 10.1016/j.chemosphere.2017.05.070
93. Lehmler HJ, Liu B, Gadogbe M, Bao W. Exposure to Bisphenol A, Bisphenol F, and Bisphenol S in U.S. Adults and Children: The National Health and Nutrition Examination Survey 2013-2014. ACS Omega (2018) 3(6):6523–32. doi: 10.1021/acsomega.8b00824
94. Pupo M, Pisano A, Lappano R, Santolla MF, De Francesco EM, Abonante S, et al. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ Health Perspect (2012) 120(8):1177–82. doi: 10.1289/ehp.1104526
95. Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect (2009) 117(7):1053–8. doi: 10.1289/ehp.0800367
96. Rezg R, El-Fazaa S, Gharbi N, Mornagui B. Bisphenol A and human chronic diseases: current evidences, possible mechanisms, and future perspectives. Environ Int (2014) 64:83–90. doi: 10.1016/j.envint.2013.12.007
97. NationalToxicologyProgram. Carcinogenesis Bioassay of Bisphenol A (CAS No. 80-05-7) in F344 Rats and B6C3F1 Mice (Feed Study). Natl Toxicol Program Tech Rep Ser (1982) 215:1.
98. Seachrist DD, Bonk KW, Ho SM, Prins GS, Soto AM, Keri RA. A review of the carcinogenic potential of bisphenol A. Reprod Toxicol (2016) 59:167–82. doi: 10.1016/j.reprotox.2015.09.006
99. Hill CE, Myers JP, Vandenberg LN. Nonmonotonic Dose-Response Curves Occur in Dose Ranges That Are Relevant to Regulatory Decision-Making. Dose Response (2018) 16(3). doi: 10.1177/1559325818798282 1559325818798282.
100. Teppala S, Madhavan S, Shankar A. Bisphenol A and Metabolic Syndrome: Results from NHANES. Int J Endocrinol (2012) 2012:598180. doi: 10.1155/2012/598180
101. Chen D, Kannan K, Tan H, Zheng Z, Feng YL, Wu Y, et al. Bisphenol Analogues Other Than BPA: Environmental Occurrence, Human Exposure, and Toxicity-A Review. Environ Sci Technol (2016) 50(11):5438–53. doi: 10.1021/acs.est.5b05387
102. Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther (2018) 186:1–24. doi: 10.1016/j.pharmthera.2017.12.012
103. Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat (2011) 128(2):457–66. doi: 10.1007/s10549-011-1584-1
104. Group EBCTC. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet (2005) 365(9472):1687–717. doi: 10.1016/S0140-6736(05)66544-0
105. Nishimura R, Osako T, Okumura Y, Hayashi M, Toyozumi Y, Arima N. Ki-67 as a prognostic marker according to breast cancer subtype and a predictor of recurrence time in primary breast cancer. Exp Ther Med (2010) 1(5):747–54. doi: 10.3892/etm.2010.133
106. Lumachi F, Santeufemia DA, Basso SM. Current medical treatment of estrogen receptor-positive breast cancer. World J Biol Chem (2015) 6(3):231–9. doi: 10.4331/wjbc.v6.i3.231
107. Yin H, Zhu Q, Liu M, Tu G, Li Q, Yuan J, et al. GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int J Oncol (2017) 51(4):1191–8. doi: 10.3892/ijo.2017.4117
108. Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med (2018) 379(20):1926–36. doi: 10.1056/NEJMoa1810527
109. Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov (2011) 10(8):579–90. doi: 10.1038/nrd3478
110. Nieto Gutierrez A, McDonald PH. GPCRs: Emerging anti-cancer drug targets. Cell Signal (2018) 41:65–74. doi: 10.1016/j.cellsig.2017.09.005
111. Panupinthu N, Lee HY, Mills GB. Lysophosphatidic acid production and action: critical new players in breast cancer initiation and progression. Br J Cancer (2010) 102(6):941–6. doi: 10.1038/sj.bjc.6605588
112. Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics (2017) 7(6):1543–88. doi: 10.7150/thno.15625
113. Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev (2010) 29(4):709–22. doi: 10.1007/s10555-010-9256-x
114. Salahudeen MS, Nishtala PS. An overview of pharmacodynamic modelling, ligand-binding approach and its application in clinical practice. Saudi Pharm J (2017) 25(2):165–75. doi: 10.1016/j.jsps.2016.07.002
115. Gaudet H, Cheng S, Christensen E, Filardo E. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol Cell Endocrinol (2015) 418:207–19. doi: 10.1016/j.mce.2015.07.016
116. Pillai V, Dorfman DM. Flow Cytometry of Nonhematopoietic Neoplasms. Acta Cytol (2016) 60(4):336–43. doi: 10.1159/000448371
117. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol (2009) 5(6):421–7. doi: 10.1038/nchembio.168
118. Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol (2011) 127(3-5):358–66. doi: 10.1016/j.jsbmb.2011.07.002
119. DeLeon C, Wang HH, Gunn J, Wilhelm M, Cole A, Arnett S, et al. A novel GPER antagonist protects against the formation of estrogen-induced cholesterol gallstones in female mice. J Lipid Res (2020) 61(5):767–77. doi: 10.1194/jlr.RA119000592
120. Lappano R, Santolla MF, Pupo M, Sinicropi MS, Caruso A, Rosano C, et al. MIBE acts as antagonist ligand of both estrogen receptor alpha and GPER in breast cancer cells. Breast Cancer Res (2012) 14(1):R12. doi: 10.1186/bcr3096
121. Maggiolini M, Santolla MF, Avino S, Aiello F, Rosano C, Garofalo A, et al. Identification of two benzopyrroloxazines acting as selective GPER antagonists in breast cancer cells and cancer-associated fibroblasts. Future Med Chem (2015) 7(4):437–48. doi: 10.4155/fmc.15.3
122. Lappano R, Rosano C, Pisano A, Santolla MF, De Francesco EM, De Marco P, et al. A calixpyrrole derivative acts as an antagonist to GPER, a G-protein coupled receptor: mechanisms and models. Dis Model Mech (2015) 8(10):1237–46. doi: 10.1242/dmm.021071
123. Shi D, Zhao P, Cui L, Li H, Sun L, Niu J, et al. Inhibition of PI3K/AKT molecular pathway mediated by membrane estrogen receptor GPER accounts for cryptotanshinone induced antiproliferative effect on breast cancer SKBR-3 cells. BMC Pharmacol Toxicol (2020) 21(1):32. doi: 10.1186/s40360-020-00410-9
124. Campbell AP, Smrcka AV. Targeting G protein-coupled receptor signalling by blocking G proteins. Nat Rev Drug Discov (2018) 17(11):789–803. doi: 10.1038/nrd.2018.135
125. Ghanemi A. Targeting G protein coupled receptor-related pathways as emerging molecular therapies. Saudi Pharm J (2015) 23(2):115–29. doi: 10.1016/j.jsps.2013.07.007
126. Zhang H, Xiong XF, Boesgaard MW, Underwood CR, Brauner-Osborne H, Stromgaard K. Structure-Activity Relationship Studies of the Cyclic Depsipeptide Natural Product YM-254890, Targeting the Gq Protein. ChemMedChem (2017) 12(11):830–4. doi: 10.1002/cmdc.201700155
127. Schrage R, Schmitz AL, Gaffal E, Annala S, Kehraus S, Wenzel D, et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat Commun (2015) 6:10156. doi: 10.1038/ncomms10156
128. Kawasaki T, Taniguchi M, Moritani Y, Hayashi K, Saito T, Takasaki J, et al. Antithrombotic and thrombolytic efficacy of YM-254890, a G q/11 inhibitor, in a rat model of arterial thrombosis. Thromb Haemost (2003) 90(3):406–13. doi: 10.1160/TH03-02-0115
129. Matthey M, Roberts R, Seidinger A, Simon A, Schroder R, Kuschak M, et al. Targeted inhibition of Gq signaling induces airway relaxation in mouse models of asthma. Sci Transl Med (2017) 9(407):2288–98. doi: 10.1126/scitranslmed.aag2288
130. Onken MD, Makepeace CM, Wang S, Kaltenbronn KM, Kanai SM, Broekelmann TJ, et al. Pharmacologic targeting of Gq reveals new pathways in uveal melanoma. Cancer Res (2018) 78(13 Suppl): Abstract nr 4363. Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14-18; Chicago, IL. Philadelphia (PA). doi: 10.1158/1538-7445.AM2018-4363
131. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol (2000) 14(10):1649–60. doi: 10.1210/mend.14.10.0532
132. Michel MC, Charlton SJ. Biased Agonism in Drug Discovery-Is It Too Soon to Choose a Path? Mol Pharmacol (2018) 93(4):259–65. doi: 10.1124/mol.117.110890
133. DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther (2013) 344(3):708–17. doi: 10.1124/jpet.112.201616
134. Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal (2020) 13(625):3140–57. doi: 10.1126/scisignal.aaz3140
135. Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther (2010) 335(3):572–9. doi: 10.1124/jpet.110.173005
136. Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res (2016) 6(8):1609–23.
137. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (5715) 2005) 307:1625–30. doi: 10.1126/science.1106943
138. Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol (2019) 30 Suppl 10:x12–20. doi: 10.1093/annonc/mdz381
139. Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med (2019) 380(20):1929–40. doi: 10.1056/NEJMoa1813904
140. Ollila TA, Sahin I, Olszewski AJ. Mogamulizumab: a new tool for management of cutaneous T-cell lymphoma. Onco Targets Ther (2019) 12:1085–94. doi: 10.2147/OTT.S165615
141. Zhu C, Guan J, Xiao H, Luo W, Tong R. Erenumab safety and efficacy in migraine: A systematic review and meta-analysis of randomized clinical trials. Med (Baltimore) (2019) 98(52):e18483. doi: 10.1097/MD.0000000000018483
142. Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs (2019) 11(2):219–38. doi: 10.1080/19420862.2018.1556465
143. Ghobrial IM, Liu CJ, Redd RA, Perez RP, Baz R, Zavidij O, et al. A Phase Ib/II Trial of the First-in-Class Anti-CXCR4 Antibody Ulocuplumab in Combination with Lenalidomide or Bortezomib Plus Dexamethasone in Relapsed Multiple Myeloma. Clin Cancer Res (2020) 26(2):344–53. doi: 10.1158/1078-0432.CCR-19-0647
144. Hutchings CJ, Koglin M, Olson WC, Marshall FH. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat Rev Drug Discov (2017) 16(9):787–810. doi: 10.1038/nrd.2017.91
145. Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol (2015) 6:12. doi: 10.3389/fimmu.2015.00012
146. Subramaniam JM, Whiteside G, McKeage K, Croxtall JC. Mogamulizumab: first global approval. Drugs (2012) 72(9):1293–8. doi: 10.2165/11631090-000000000-00000
147. Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum (2008) 58(7):1931–9. doi: 10.1002/art.23591
148. Jin H, Pi J, Zhao Y, Jiang J, Li T, Zeng X, et al. EGFR-targeting PLGA-PEG nanoparticles as a curcumin delivery system for breast cancer therapy. Nanoscale (2017) 9(42):16365–74. doi: 10.1039/c7nr06898k
149. Ebeid K, Meng X, Thiel KW, Do AV, Geary SM, Morris AS, et al. Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat Nanotechnol (2018) 13(1):72–81. doi: 10.1038/s41565-017-0009-7
150. Geary SM, Hu Q, Joshi VB, Bowden NB, Salem AK. Diaminosulfide based polymer microparticles as cancer vaccine delivery systems. J Control Release (2015) 220(Pt B):682–90. doi: 10.1016/j.jconrel.2015.09.002
151. Wafa EI, Geary SM, Ross KA, Goodman JT, Narasimhan B, Salem AK. Single Dose of a Polyanhydride Particle-Based Vaccine Generates Potent Antigen-Specific Antitumor Immune Responses. J Pharmacol Exp Ther (2019) 370(3):855–63. doi: 10.1124/jpet.118.252809
152. Joshi VB, Geary SM, Salem AK. Biodegradable particles as vaccine delivery systems: size matters. AAPS J (2013) 15(1):85–94. doi: 10.1208/s12248-012-9418-6
153. Abbas AO, Donovan MD, Salem AK. Formulating poly(lactide-co-glycolide) particles for plasmid DNA delivery. J Pharm Sci (2008) 97(7):2448–61. doi: 10.1002/jps.21215
154. Makkouk A, Joshi VB, Wongrakpanich A, Lemke CD, Gross BP, Salem AK, et al. Biodegradable microparticles loaded with doxorubicin and CpG ODN for in situ immunization against cancer. AAPS J (2015) 17(1):184–93. doi: 10.1208/s12248-014-9676-6
155. Chitphet K, Geary SM, Chan CH, Simons AL, Weiner GJ, Salem AK. Combining Doxorubicin-Loaded PEGylated Poly (Lactide-co-glycolide) Nanoparticles with Checkpoint Inhibition Safely Enhances Therapeutic Efficacy in a Melanoma Model. ACS Biomater Sci Eng (2019) 6(5):2659–67. doi: 10.1021/acsbiomaterials.9b01108
Keywords: GPER, estrogen receptors, therapeutics, anti-estrogens, cancer
Citation: Rouhimoghadam M, Lu AS, Salem AK and Filardo EJ (2020) Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front. Endocrinol. 11:591217. doi: 10.3389/fendo.2020.591217
Received: 04 August 2020; Accepted: 19 October 2020;
Published: 23 November 2020.
Edited by:
Sarah H. Lindsey, Tulane University, United StatesReviewed by:
Marcello Maggiolini, University of Calabria, ItalyRajeshwar Rao Tekmal, The University of Texas Health Science Center at San Antonio, United States
Helen Hathaway, University of New Mexico, United States
Copyright © 2020 Rouhimoghadam, Lu, Salem and Filardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward J. Filardo, edward-filardo@uiowa.edu