 Nazia Parveen
Nazia Parveen Sangeeta Dhawan*
Sangeeta Dhawan*- Department of Translational Research and Cellular Therapeutics, Arthur Riggs Diabetes and Metabolism Research Institute, City of Hope, Duarte, CA, United States
Pancreatic beta cells play a central role in regulating glucose homeostasis by secreting the hormone insulin. Failure of beta cells due to reduced function and mass and the resulting insulin insufficiency can drive the dysregulation of glycemic control, causing diabetes. Epigenetic regulation by DNA methylation is central to shaping the gene expression patterns that define the fully functional beta cell phenotype and regulate beta cell growth. Establishment of stage-specific DNA methylation guides beta cell differentiation during fetal development, while faithful restoration of these signatures during DNA replication ensures the maintenance of beta cell identity and function in postnatal life. Lineage-specific transcription factor networks interact with methylated DNA at specific genomic regions to enhance the regulatory specificity and ensure the stability of gene expression patterns. Recent genome-wide DNA methylation profiling studies comparing islets from diabetic and non-diabetic human subjects demonstrate the perturbation of beta cell DNA methylation patterns, corresponding to the dysregulation of gene expression associated with mature beta cell state in diabetes. This article will discuss the molecular underpinnings of shaping the islet DNA methylation landscape, its mechanistic role in the specification and maintenance of the functional beta cell phenotype, and its dysregulation in diabetes. We will also review recent advances in utilizing beta cell specific DNA methylation patterns for the development of biomarkers for diabetes, and targeting DNA methylation to develop translational approaches for supplementing the functional beta cell mass deficit in diabetes.
Introduction
All of the distinct cell types in a multicellular organism share the same DNA sequence, and yet the phenotype of each individual cell type is unique, and dictated by the cell-type specific patterns of gene expression. In addition, an individual cell type can often adapt its behavior in response to changing environment by modifying its gene expression. This context specific interpretation of the genome is facilitated through a set of mechanisms that impact the accessibility of genomic regions. These mechanisms include covalent modifications of DNA and DNA binding proteins such as histones, as well as RNA species such as microRNAs and long non coding RNAs, with DNA methylation being the most well-known direct modification of the genome (1). Collectively, these mechanisms are referred to as epigenetic mechanisms. The term ‘epigenetics’ was first used by Waddington in 1942 to describe the influence exerted by the environment on the genome towards shaping the cellular phenotype during development (2). More recently, Arthur Riggs gave a mechanistic dimension to this term, and defined epigenetics as “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” (3). In its present form, this term has come to encompass all transient or stable changes in chromatin structure that mark or perpetuate altered activity, in response to an environmental cue (4). Epigenetic changes permit a cell to alter its gene expression and phenotype in response to any changes in the environment, in contexts such as generating cellular diversity in response to morphogens during development, or adaptation of cellular behavior to changing nutrient availability or exposure to stress stimuli.
DNA methylation is one of the earliest and most well studied epigenetic modifications (5, 6). Methylation of DNA occurs predominantly at the cytosine residue within CpG dinucleotides in mammalian cells and serves to regulate gene expression (7, 8). DNA methylation levels at repetitive elements and CpG dense regions around promoters do not vary much between different cell types. However, the patterns of DNA methylation at regions that regulate gene expression, such as enhancers, are very cell type specific and essential for the maintenance of cellular phenotype [reviewed in (9)]. This suggests that any changes in DNA methylation patterns can potentially impact cell-fate and function. Depending on the stimuli triggering such changes and the genes impacted by them, altered DNA methylation patterns can either facilitate adaptation to the changing environment, or disrupt cellular function and lead to pathological changes. This view is supported by the plastic nature of DNA methylation and the existence of enzymatic mechanisms that can erase DNA methylation marks (10). A large number of studies have demonstrated that stage specific patterning of DNA methylation signatures is essential for cell-fate specification and functional maturation of pancreatic beta cells, the major cell type that regulates glucose homeostasis [reviewed in (9, 11)].
Beta cell failure plays a central role in the pathogenesis of diabetes, a disease that impairs the body’s ability to regulate blood glucose. Accumulating evidence suggests that loss of beta cell identity and functional maturity contribute to beta cell dysfunction in diabetes (12). Comparison of genome-wide DNA methylation profiles in islets from diabetic and non-diabetic subjects reveals that DNA methylation patterns related to beta cell function and identity are altered in diabetic islets (13). The formation and erasure of DNA methylation patterns is tightly linked to cellular metabolism. It is therefore likely that an altered metabolic milieu in diabetes drives epigenetic disruptions underlying beta cell failure. The influence of environmental factors such as diet and exercise on metabolic health and diabetes risk is now well recognized, and further underscores the importance of epigenetic regulation in glucose homeostasis (14, 15). In this review, we focus on the essential role of DNA methylation in the establishment and maintenance of the functional beta cell phenotype, and the relevance of altered DNA methylation patterns to diabetes pathophysiology. We will also discuss emerging approaches that aim to harness beta cell specific DNA methylation signatures as biomarkers for diabetes, and highlight proof-of-principle studies that demonstrate the therapeutic potential of targeting DNA methylation to promote beta cell function and regeneration.
DNA Methylation and Beta Cell Homeostasis
DNA Methylation: A Key Epigenetic Module
DNA methylation is a heritable epigenetic modification that involves the covalent addition of a methyl group to the bases, most predominantly at the 5th carbon of a cytosine within the CpG dinucleotides, leading to the formation of 5-methylcytosine (5mC) (16). 5mC is the most well characterized and abundant direct modification of DNA in the mammalian genome. In contrast, methylation of non-CpG sites occurs at a very low frequency (17), and is beyond the scope of this review. The addition of methyl group on cytosine is carried out by a family of enzymes called DNA methyltransferases (Dnmts), that includes Dnmt1, Dnmt2, Dnmt3a, Dnmt3b and Dnmt3l [reviewed in (18, 19)]. Dnmt1, Dnmt3a, and Dnmt3b are the bona fide DNA methyltransferases that have the catalytic activity, while Dnmt2 and Dnmt3l lack DNA methyltransferase activity of their own, and are likely to play an allosteric regulatory role (19). DNA methylation has two distinct regulatory layers: de novo methylation, or the formation of new methylation patterns (20–22), and maintenance methylation, or the post-replication restoration of existing methylation patterns on the nascent strand (22–24) (Figure 1). De novo methylation is primarily carried by Dnmt3a and Dnmt3b (21), while Dnmt1, the first methyltransferase to be discovered, serves to maintain methylation patterns through replication and prefers hemi-methylated CpG sites (23, 25).
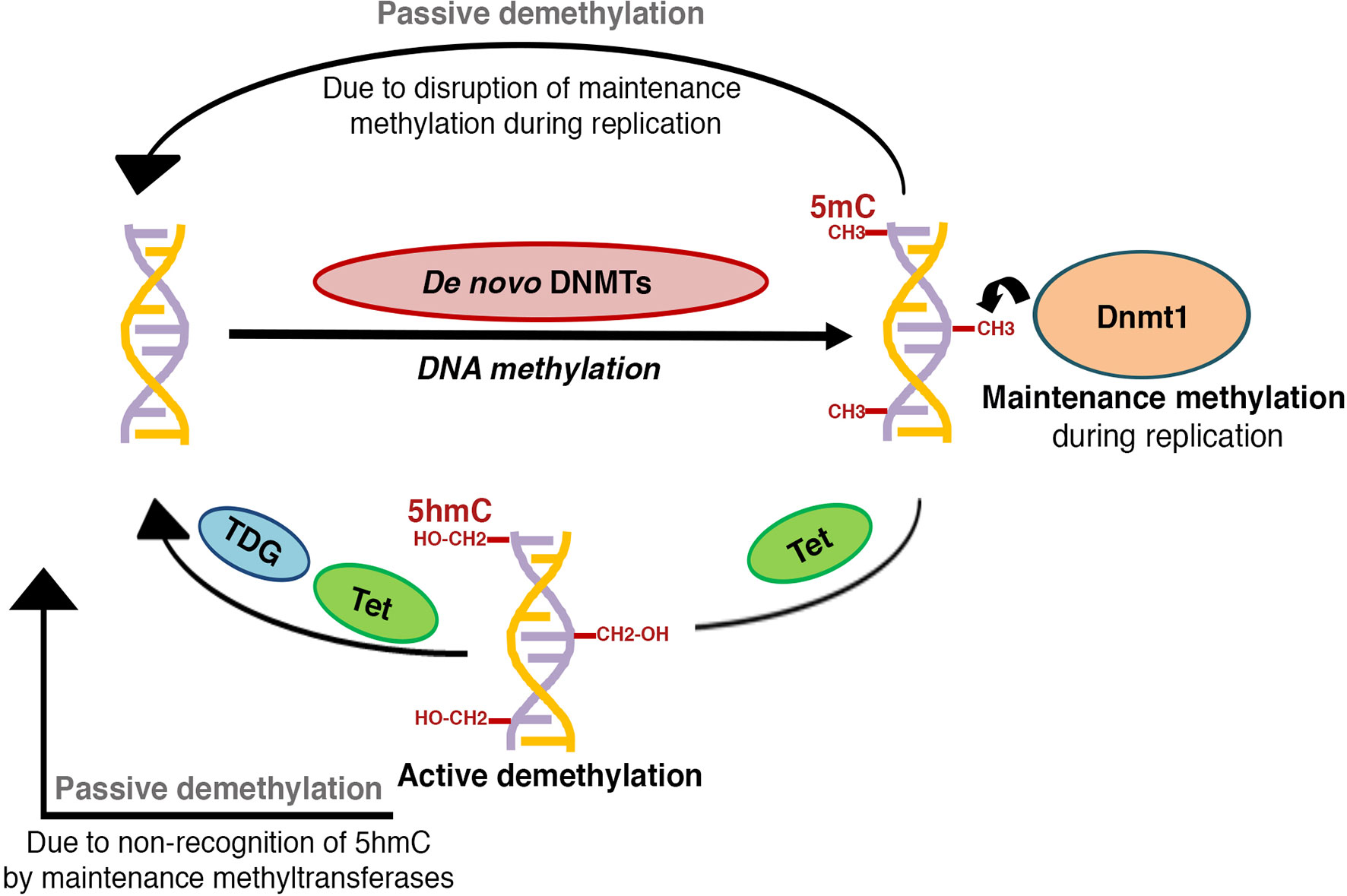
Figure 1 Schematic representation of DNA methylation patterning: The establishment of new DNA methylation patterns during development is regulated by the activity of de novo DNA methyltransferases, while activity of maintenance DNA methyltransferases serves to perpetuate these patterns during successive rounds of cell division. DNA methylation marks can be reversed through active or passive demethylation. Active demethylation involves the successive enzymatic oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC),5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) by TET (Ten-eleven translocation) dioxygenases, followed by thymine DNA glycosylase (TDG) dependent removal of 5fC and 5caC, coupled with base-excision repair to a cytosine (C). A hemi-methylated 5hmC is not recognized by the maintenance DNA methyltransferases and can get diluted and lost during replication, thus contributing to passive demethylation. Disruption of maintenance methyltransferase activity can similarly result in replication dependent dilution of DNA methylation.
DNA methylation plays a central role in the regulation of gene expression and maintenance of genomic integrity, and directs processes such as gene repression, silencing of transposons, genomic imprinting, and X- chromosome inactivation [reviewed in (7, 8, 26, 27)]. It is noteworthy that the target of DNA methylation regulation, the CpG dinucleotides, represent only about 1% of the mammalian genome. A large proportion of CpG residues are clustered in CpG-rich regions called the CpG islands (CGIs), which are typically devoid of methylation [reviewed in (28)]. About half of the CGIs are located proximal to the transcription start sites (TSSs), and need to remain hypomethylated for promoter activity (29). While the hypomethylation of promoter-associated CGIs is common to most cell types, the DNA methylation patterns at enhancer are cell type specific and indicate the status of enhancer activity (9). In contrast to the association of promoter CGI methylation with gene repression, gene body methylation typically indicates active transcription [reviewed in (16)]. This suggests that DNA methylation can repress, permit, or modify gene expression, depending on the genomic context. The overall low frequency of CpG residues and observed paucity of CGI methylation has likely evolved to protect against mutational loss of CpGs due to 5mC deamination, while creating an in-built selectivity to DNA methylation dependent regulation. Transcriptional regulation by DNA methylation involves the recognition of methylated CpGs, residues by DNA methylation “readers” such as the “methyl-CpG binding domain” proteins or MBD proteins, which recruit chromatin regulatory enzymes that establish appropriate epigenetic modifications to create a specific regulatory chromatin milieu [reviewed in (8, 30)]. Additional specificity of DNA methylation is achieved by interaction of MBD complexes and/or methylated regions with stage-specific transcription factors, to regulate DNA methylation at their binding sites (31–33).
While it has long been recognized that DNA methylation is dynamic in nature, the mechanisms mediating the active removal of 5mC remained largely understudied until a decade ago (10, 34). 5mC marks can be reversed through two mechanisms: active demethylation and passive demethylation [reviewed in (10, 16, 34)] (Figure 1). Active demethylation of CpG residues involves the step-wise enzymatic oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) by TET (Ten-eleven translocation) dioxygenases (35, 36). 5fC and 5caC can then undergo base excision mediated by thymine DNA glycosylase (TDG) leading to active erasure of methyl marks [reviewed in (10)]. Alternative pathways, involving Growth Arrest and DNA Damage-inducible (GADD) 45 proteins (GADD45A and GADD45B), AID (activation induced cytidine deaminase) and APOBEC (apolipoprotein B mRNA editing enzyme, catalytic subunit) have been reported [reviewed in (10, 37)], but the extent of their contribution to DNA demethylation remains unclear. Passive demethylation, on the other hand, can result from dilution of 5mC during replication due to the failure of maintenance methylation, followed by dilution through subsequent rounds of replication [reviewed in (10)]. Collectively, both active and passive demethylation contribute to the homeostasis and remodeling of DNA methylation.
The establishment and maintenance of DNA methylation patterns is essential for embryonic development and differentiation. Early embryonic development in mammals is marked by two waves of global demethylation and re-methylation, the first, which occurs immediately after fertilization and constitutes the pre-implantation reprogramming, and the second, which happens in the primordial germ cells (PGCs) during gametogenesis [reviewed in (38–40). Such demethylation creates a clean epigenetic slate that underlies developmental multipotency, and can then be used to establish stage specific DNA methylation programs during differentiation. However, this also creates a particularly sensitive developmental window, during which any adverse environmental exposures can have a significant impact on cell phenotypes in later life. During embryonic development, the de novo methyltransferases establish the new methylation patterns in the inner cell mass (ICM) after implantation, and maintenance of these patterns by Dnmt1 ensures continuity of the epigenetic state through cell division (38–40). Lineage specific refinement of DNA methylation patterning of promoters and enhancers then guides the cell-type-specific gene-expression patterns throughout the differentiation program (reviewed in [9)]. Overall, de novo and maintenance methylation perform an indispensable role in shaping mammalian development through guiding the differentiation and growth of different organs, including pancreas.
DNA Methylation in Pancreas Development and Beta Cell Specification
The pancreas is a multifunctional organ, performing both exocrine and endocrine functions, namely the secretion of digestive enzymes and hormones (41). The exocrine component of the pancreas comprises of acinar cells that synthesize digestive enzymes, and ductal epithelium that transports those enzymes into the gut. On the other hand, the endocrine pancreas consists of alpha- (α), beta- (β), gamma- (γ), delta- (δ), and epsilon- (ϵ) cells, which cluster together in the islets of Langerhans to regulate glucose homeostasis by secreting hormones-glucagon, insulin, pancreatic polypeptide, somatostatin, and ghrelin, respectively into the blood stream [reviewed in (42, 43)]. Pancreas is an endodermal lineage organ, and after gastrulation, a series of developmental steps lead to the formation of the primitive gut tube from endoderm (44). Pancreatic development begins with the emergence of dorsal and ventral anlagen that harbor multipotent progenitor cells (MPCs), on opposite sides of the foregut endoderm at embryonic day 9.0 (E9.0). Following the rotation of the gut tube, these buds fuse around E12.5 to form the full organ, with continued expansion and branching of the pancreatic epithelium, accompanied by the differentiation of pancreatic progenitors into endocrine, acinar, and ductal lineages [reviewed in (42–46)]. The differentiation of pancreatic progenitors into the endocrine lineage involves an intermediate, Neurogenin3 (Neurog3) expressing endocrine progenitor stage. By the end of gestation, the developing pancreas has acquired its typical structure, with acinar rosettes surrounding the ends of the ductal tree and clusters of endocrine cells scattered throughout the organ [reviewed in (41, 42)].
Dynamic regulation of DNA methylation plays an important role in the development of pancreas from definitive endoderm and further specification of endocrine cells. De novo methylation patterning plays a critical role in this stepwise differentiation process; with Dnmt3a being the primary de novo DNA methyltransferase in the endocrine lineage, while Dnmt3b is restricted to the acinar lineage (47). A recent study on the in vitro differentiation of human pluripotent stem cells (hPSCs) into pancreatic islets revealed that pluripotency genes undergo DNA hypermethylation to enable chromatin silencing, and that hypomethylation of lineage specific genes drives stage-specific chromatin activation during endocrine specification (48). The hyper- and hypo-methylation of genomic regions coincides with the repression or activation of the nearest gene, and enables stage-specific gene expression during different stages of endocrine differentiation (48). DNA methylation and demethylation patterns also play a central role in regulating the recruitment and activity of pioneer transcription factors (TFs), such as the forkhead box proteins FOXA1 and 2, which direct lineage specific chromatin remodeling and enhancer priming during endocrine differentiation (49–55). Genome-wide mapping of 5mC and 5hmC patterns during differentiation of human embryonic stem cells (hESCs) to pancreatic lineage shows that global 5hmC levels are reduced, with a corresponding increase in 5mC during the formation of definitive endoderm. However, 5hmC levels gradually increase during subsequent differentiation steps, with a concomitant 5mC decline (54, 56), similar to the differentiation of other progenitors (57). 5hmC enrichment also positively correlates with increased chromatin accessibility and lineage specific enhancer activity (54). In line with this, deletion of all three TET enzymes in hESCs leads to impaired differentiation of pancreatic endoderm, corresponding to the disruption of stage-specific 5hmC patterning (55). Thus, pioneer factors such as FOXA2 guide the establishment of lineage specific 5hmC patterns on specific genes, which in turn govern chromatin accessibility and maintenance of the epigenetic landscape in active genomic regions at a given developmental stage. This suggests that an enrichment of global 5hmC levels is a conserved hallmark of cell differentiation. In alignment with this, the differentiated alpha and beta cells appear to be globally hypomethylated compared to various preceding developmental stages during hPSC differentiation (48).
Genetic manipulation studies have provided valuable insights into the role of DNA methylation in pancreatic endocrine development. Loss of the maintenance DNA methyltransferase, Dnmt1, in both zebrafish and mice, leads to defects in pancreas development (58, 59). Dnmt1 not only preserves epigenetic information in replicating cells, but also maintains genomic integrity via centromere methylation (60, 61), such that loss of Dnmt1 causes genomic instability during cell division and triggers p53 dependent cell-death. In mice, Dnmt1 is required to repress p53 expression and promote progenitor survival during pancreatic progenitor differentiation. Dnmt1 ablation in mouse pancreatic progenitors leads to activation of p53 dependent apoptosis that results in pancreatic atrophy, a defect that can be rescued by Trp53 haploinsufficiency (59). This suggests that maintenance of DNA methylation signatures is important for the regulation of key checkpoints in the rapidly dividing pancreatic progenitors (Figure 2). Pancreatic progenitor give rise to endocrine cells via a Neurog3 expressing endocrine progenitor stage [reviewed in (41, 42)]. The differentiation of endocrine progenitors to specific islet cell-fates depends on differential DNA methylation of lineage restricted enhancers, with no apparent differences in promoter methylation between different endocrine cell types (62, 63). The promoter regions of insulin and glucagon genes are rapidly demethylated during endocrine progenitor differentiation, regardless of the specific destined endocrine lineage. Accordingly, promoters of genes encoding insulin and glucagon lack DNA methylation in alpha-, beta- and delta-cells, independent of their expression in these lineages. Instead, endocrine cells exhibit differential methylation in the enhancer regions of these genes, indicating that endocrine cell identity is regulated by enhancer methylation (62). Genome-wide comparison of human alpha- and beta-cell methylomes shows the conservation of this phenomenon in humans, such that lineage specific methylation differences are concentrated in enhancers (62). The shared promoter hypomethylation of lineage-specific genes across endocrine cell-types likely subserves the changes in endocrine identity that occurs in the context of beta cell regeneration and diabetes pathogenesis.
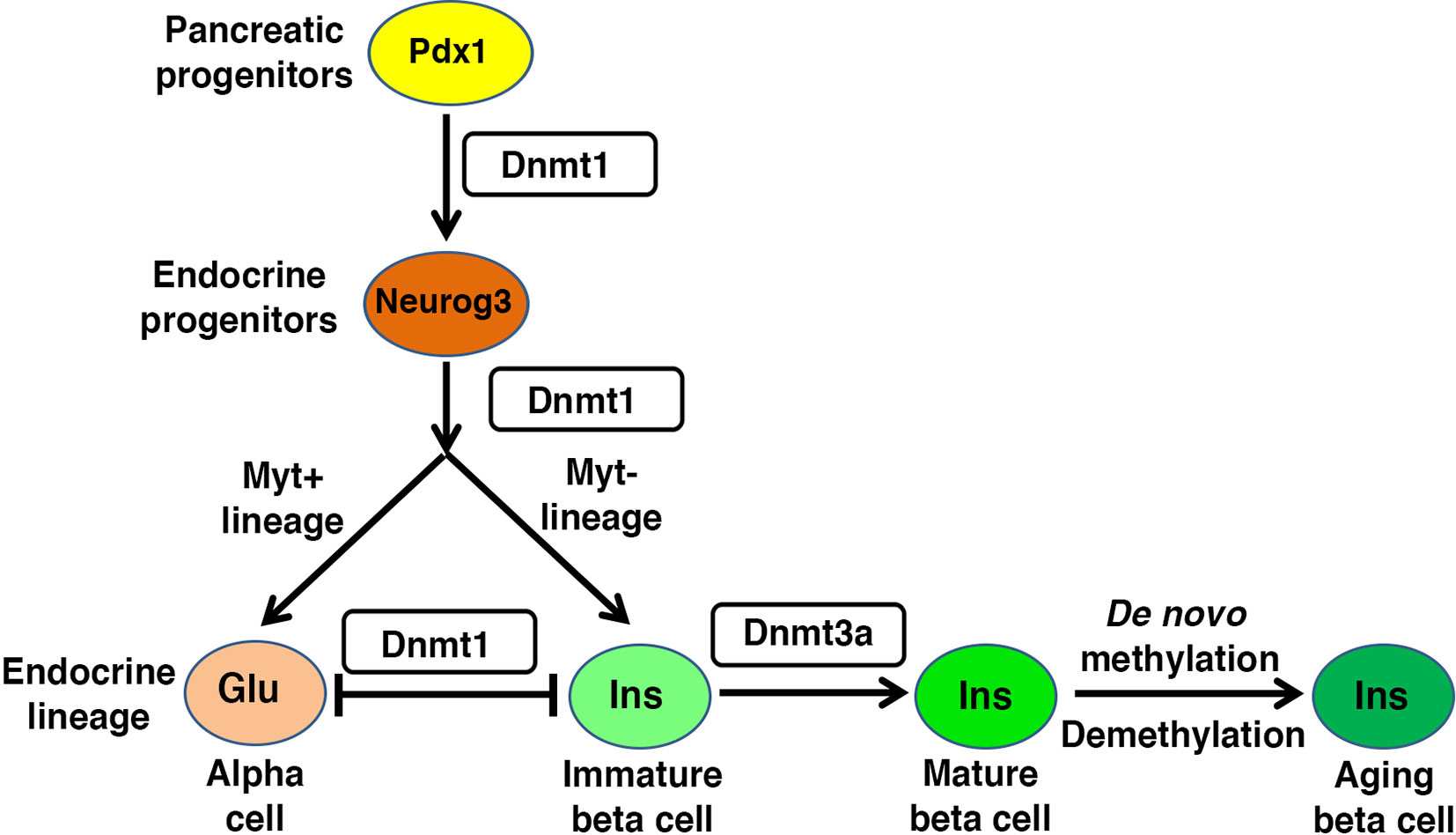
Figure 2 Regulatory role of DNA methylation in beta cell homeostasis: Pancreas morphogenesis requires intact maintenance methylation activity of Dnmt1 in Pdx1+ pancreatic progenitors, and loss of Dnmt1 in pancreatic progenitors leads to pancreatic atrophy. Specification of different endocrine lineages from the Neurog3+ endocrine progenitors involves the DNA methylation dependent regulation of the expression of lineage-specific transcription factors. The endocrine progenitors that co-express Myt1, are marked by high expression of Dnmt1 and hypermethylation of the enhancer region of Arx, a key alpha cell lineage determinant. This leads to repression of Arx and commitment of the Neurog3+ Myt1+ sub-population to beta cell lineage, while the Neurog3+ Myt1- sub-population acquires alpha cell lineage. Functional maturation of beta cells is neonatal life depends on DNA methylation patterning of genes involved in metabolism and replication. The fully differentiated beta cell phenotype is guarded by maintenance methylation through replication, and loss of Dnmt1 in beta cells leads to their trans-differentiation into alpha cells. The proliferative capacity and function of beta cell changes with age, and involves age dependent changes in the beta cell methylome.
DNA methylation directs the specification of different endocrine cell-fates during early pancreatic development by modulating expression of key genes encoding cell-fate regulatory transcription factors (31, 63–65) (Figure 2). For instance, the specification of beta cell identity is regulated by the DNA methylation dependent repression of Aristaless homeobox (Arx) locus, which encodes a master regulator of alpha cell identity (31, 64). The DNA methylation dependent repression of Arx is established by the recruitment of a complex containing Dnmt3a with transcription factors Nkx2.2, Nkx6.1, Grg3 and histone deacetyalse-Hdac1 to the Arx promoter in beta cells (31). Beta cell lineage specification also requires the TET1 dependent hypomethylation at enhancer regions of beta cell specification genes (55), such as Pax4 which represses Arx expression in beta cells (66–68). This suggests that epigenetic regulation directs lineage specification in coordination with transcription factor recruitment. However, lineage specific factors such as Arx and Pax4 are co-expressed in nascent Neurog3+ endocrine progenitors (67, 68), suggesting the existence of mechanisms that restrict factors such as Arx and Pax4 to different lineages. Recent data suggests that the epigenetic roadmap for such restriction is established prior to endocrine progenitor specification. Pancreatic progenitors represent a transcriptionally homogenous pool before differentiating into the Neurog3+ lineage, but differ in DNA methylation pattern at the enhancers of key cell-identity genes, which primes them towards distinct endocrine lineage programs. This epigenetic heterogeneity is regulated by Myt1, such that Neurog3+ cells that co-express Myt1 are marked by higher Dnmt1 levels and hypermethylation of Arx, which biases them to a beta cell-fate (63) (Figure 2). These data point to a surprising role for maintenance methylation in directing the differentiation of endocrine progenitors. Besides Dnmt1, epigenetic regulators such as arginine methyltransferase Prmt5, histone deacetylase Hdac3, and lysine demethylase Kdm4a are differentially partitioned between the two endocrine progenitor sub-populations (63), suggesting that DNA methylation patterning works in conjunction with other epigenetic modules to direct endocrine cell-fate choices. However, the spatio-temporal and signaling events that guide such epigenetic heterogeneity, and the relative roles of de novo versus maintenance methylation in this process remain to be elucidated. Emerging advances in single cell epigenome profiling methods will be useful in clarifying some of these issues. Overall, endocrine differentiation of cells involves the dynamic and highly stage specific patterning of 5mC and 5hmC marks, which is coordinated by an interplay of stage specific activities of pioneer factors, lineage determinant transcription factors, DNA methyltransferases, and DNA demethylases. This not only determines the sequence specificity of DNA methylation, but also guides the epigenetic priming of lineage specific regulatory regions to orchestrate the stage-specific developmental program in response to instructive signals during pancreatic differentiation.
DNA Methylation in Beta Cell Maturation and the Maintenance of Full Functional Beta Cell Phenotype
Beta cell mass expands significantly by replication in the late fetal to early postnatal life corresponding to the high rates of growth, in both mice and humans (69–73). However, beta cells during this growth phase are functionally immature and display a lower threshold of glucose concentration for insulin secretion. Accordingly, they secrete insulin secretion even at low glucose levels and are therefore not glucose responsive (47, 74, 75). Immature beta cells utilize anaerobic glycolysis and higher amino acid metabolism (76–79), which allows a low glucose threshold and supports the high rates of replication in this phase (47, 77, 79). Beta cells acquire glucose sensitivity as they exit cell-cycle, suggesting an inverse correlation between beta cell replication and functional maturity (80). In mice, the process of functional maturation begins around postnatal day 7 and is complete at weaning (75, 81, 82), while in humans, beta cell maturation occurs within the first 2-3 years of life (73, 83). This transition from a replicative, glucose non-responsive to a quiescent, glucose-responsive state is coupled with a metabolic changes that allow beta cells to establish a higher threshold for glucose, and amplify ATP production through oxidative phosphorylation (84). This metabolic switch from anaerobic- to aerobic-glycolysis is directed by the de novo DNA methyltransferase Dnmt3a. Methylation at distal promoter regions of the so-called “disallowed” genes, such as the low Km hexokinases-1 and -2 (Hk1, Hk2) and lactate dehydrogenase A (Ldha), leads to their repression and allows the coupling of insulin secretion to glucose levels (47). Ablation of Dnmt3a in beta cells leads to higher basal insulin secretion even in adult life, similar to neonatal beta cells, and renders them functionally immature. This process is in part controlled by changes in the levels of Dnmt and Tet enzymes; such that Dnmt3a and Tet levels decline after maturation (47, 53). Thus, establishment of the functional beta cell phenotype is dependent upon the DNA methylation mediated repression of glucose-secretion decoupling genes (47).
The changes in beta cell metabolic program during maturation coincide with and reflect a change in nutrient quality and accessibility (85). Studies in mice have shown that weaning induces a distinct, final step of functional maturation that is coupled with a dietary switch from protein rich milk to carbohydrate rich chow, highlighting the involvement of a metabolic shift in maturation (81).The change in nutrient quality due to dietary transition at weaning triggers a switch from the nutrient sensing mTORC1 (Target of Rapamycin) pathway to the energy sensing AMPK (Adenosine Monophosphate-activated Protein Kinase) signaling, which in turn supports the establishment of the functionally mature beta cell phenotype (86). mTORC1 signaling is essential for DNA methylation dependent repression of disallowed genes and the developmental metabolic reprogramming underlying beta cell functional maturation (87). Beta cell specific loss of Raptor, a key subunit of mTORC1, was shown to cause reduced expression of Dnmt3a. The consequent hypomethylation and de-repression of disallowed genes (Hk1, Dlk1, Pdgfra, Oat and Mylk) and beta cell immaturity genes (Dlk1 and MafB) led to impaired glucose stimulated insulin secretion (GSIS) and reduced beta cell mass due to beta cell-death (87). These data suggest that nutrient sensing pathways direct metabolic changes underlying beta cell maturation via DNA methylation patterning. The link between metabolic changes and DNA methylation is underscored by the dependence of DNA methyltransferase and DNA demethylases on metabolites such as S-adenosyl methionine (SAM) and a-ketoglutarate (a-KG), respectively (88), the levels of which depend on nutrient quality such as the extent of carbohydrate, protein, and fat intake, as well as flux through the TCA cycle. Altogether, changes in nutrient sensing and metabolic pathways during the neonatal growth phase orchestrate DNA methylation dependent changes in metabolism that define beta cell functional maturity.
The process of functional maturation also coincides with beta cells acquiring the capacity for compensatory expansion in response to increased insulin demand (81). The functionally mature beta cell phenotype is therefore not only GSIS competent, but also capable of adapting to metabolic challenges. Beta cell replication is a key mechanism that maintains beta cell mass in postnatal life by contributing to growth and adaptive expansion (71). Besides the regulation of genes involved in beta cell metabolism, DNA methylation also controls the expression of genes involved in proliferation (65). The capacity of beta cells to undergo adaptive proliferation declines with age (89, 90), and corresponds to increased GSIS due to age related changes in the methylome of the endocrine pancreas (65). Age-dependent changes in DNA methylation patterning are now well-recognized to reflect functional cellular aging (91, 92). Genome-wide methylome profiling of beta cells with age shows that promoters of genes encoding pro-proliferation proteins, such as Mki67, Cyclin D3, and Cyclin dependent kinases (Cdks), undergo de novo methylation. Age-associated de novo DNA methylation in islets has also been shown to loosen the epigenetic barrier between endocrine and acinar identities (93), and points to an epigenetic drift towards acinar epigenomic fate with age. On the other hand, distal regulatory regions of genes involved in the maintenance of beta cell identity and function such as Pdx1, Nkx6.1, NeuroD1, Foxa2, Mnx1, Kcnj11, Abcc8, and Gck become demethylated with age, corresponding to age-related improvement in GSIS (65). The distal regions that lose methylation with age, are typically marked with signatures of active enhancers in young mice, which likely primes them for future demethylation (65).
While the mechanisms that regulate age-associated DNA methylation changes are far from clear, Dnmt3a mRNA levels appear to be much higher in islets from older mice (6-26 months) compared to early neonatal life (1-2 weeks) (94). This is surprising, given the reduction of Dnmt3a during maturation. It is likely that Dnmt3a mRNA expression is downregulated upon maturation, and increases again in the older adult mice to support the de novo methylation and repression of cell-cycle genes. DNA methylation has been shown to intersect with polycomb protein dependent chromatin organization (95), and polycomb proteins play an integral part in age-dependent regulation of beta cell proliferation and function (96–98). It is, therefore, likely that the age-dependent changes in the proliferative and functional capacity of beta cells are co-regulated by an interplay of polycomb complexes and DNA methylation. In line with this, the distal regulatory regions that undergo age-dependent DNA demethylation in beta cells, are also marked by the loss of polycomb-dependent histone 3 lysine 27 trimethylation (H3K27me3) (65). Collectively, dynamic changes in the methylome lead to repression of the proliferative program and up-regulation of the beta cell functional programs with age in mice. Interestingly, while mouse and human beta cells follows the same trend for age-dependent changes in cell-cycle genes expression, the expression of beta cell function genes declines with age in humans, unlike in mouse beta cells (99). This may likely be a reflection of differences in physiological factors such as the body size and lifespan of the two species.
In addition to age, biological sex is another key variable that dictates DNA methylation patterns; in large part due to the fact that females display a DNA methylation dependent inactivation of the extra X-chromosome to regulate the dosage of X-linked gene in females (100). Several studies suggest that females secrete more insulin compared to males and are more insulin-sensitive (101, 102). A comparison of global DNA methylation patterns in human islets from males and females identified DNA methylation differences corresponding to 61 genes located on X-chromosomes and 18 autosomal loci, which also display sex-related differences in gene-expression (103). Three such genes, NKAP, APLN (both X-linked), and SPESP1 (autosomal), that showed higher methylation and reduced mRNA in female islets, were found to regulate insulin secretion in functional assays. In addition, this study identified two miRNA loci that were differentially methylated and expressed between male and female islets, correlating with the observed sex differences in islet expression of multiple target genes for these miRNAs (103). These data point to the existence of sex-specific differences in human islet methylome that are highly relevant to the islet functional phenotype, and can partly explain the sex-specific differences in metabolic homeostasis.
Replication of a terminally differentiated cell type (such as beta cells) presents a unique challenge for the cell to faithfully transmit the epigenetic patterns that define the differentiated phenotype, from the parent- to daughter-cells. The maintenance methyltransferase Dnmt1 serves to maintain beta cell identity during cell division by reestablishing the DNA methylation patterns required for the continued repression of alpha cell lineage, such as the methylation of Arx (64). Consequently, the inactivation of Dnmt1 in beta cells leads to the de-repression of Arx and trans-differentiation of beta- into alpha-cells, causing a reduction in beta cell mass (64). The DNA methylation dependent repression of Arx in beta cells involves the recruitment of a complex containing the methyl-binding protein MeCP2 and histone methyltransferase Prmt6 to the methylated region of Arx, which establishes a repressive chromatin profile. In the absence of Dnmt1, loss of DNA methylation prevents restoration of the corresponding repressive chromatin state in the daughter cells (64). Notably, beta cell specific loss of Dnmt1 does not result in a large-scale de-repression of silent genes; yet again pointing to the lineage specificity of epigenetic programs. Instead, Dnmt1 appears to maintain the barrier between developmentally related cell lineages, such as alpha- and beta-cells. The conversion of beta- to alpha-cells driven by the loss of Dnmt1 is a slow process, due to the gradual dilution of DNA methylation marks with each round of cell division, and a refractory period before a beta cell can re-enter cell-cycle. Combined with the age-related decline of beta cells undergoing successive rounds of cell division, these factors amount to the observed slow rates of beta cell trans-differentiation. Of note, alpha cell specific loss of Dnmt1 does not result in their trans-differentiation to beta cells, and requires the combined ablation of Dnmt1 and Arx (104), suggesting that alpha cell identity maybe the default epigenetic endocrine state. This is noteworthy, as diabetes is associated with beta-to-alpha trans-differentiation, which occurs at a slow rate (105). A recent study suggests that inhibition of binding of transcription factor Foxo1 to the Arx promoter is essential for Arx hypermethylation to maintain beta cell identity (106), suggesting that transcription factors can regulate the maintenance of endocrine identity in a DNA methylation dependent manner (Figure 3).

Figure 3 Key players involved in DNA methylation dependent regulation of beta cell identity: Hypermethylation of the alpha cell lineage regulator gene, Arx, represses its expression in beta cells, while hypomethylation of Pdx1 and Pax4 in beta cells is essential to maintain their expression. The specificity of DNA methylation patterning is ensured by the interaction of DNA methyltransferase Dnmt3a with transcription factors Nkx2.2 and Nkx6.1, allowing recruitment to specific sites. Once established, the beta cell specific DNA methylation patterns are maintained by the maintenance methyltransferase Dnmt1.
In light of the above discussion, it is conceivable that changes in nutrition and metabolism at any point in postnatal life can alter the DNA methylation patterns underlying beta cell identity and maturity, which can in turn impact the functional response of beta cells. Such plasticity of the beta cell epigenome is essential for adaptation to changing insulin demands. However, altered nutrition during the sensitive growth phase in early life, or sustained high demand for insulin in adult life, can disrupt the epigenetic program and lead to an insufficiency of functional beta cell mass, due to poor growth or poor adaptation, respectively.
Developmental Origins of Diabetes Risk: The Role of DNA Methylation
A large number of studies across multiple species point to a strong link between developmental perturbations such as poor intrauterine nutrition and fetal growth impairment, and diabetes development in later life, and suggest that early epigenetic events can contribute to disease risk and pathogenesis (14, 107–110). For example, studies on the Dutch Hunger Winter famine revealed that intrauterine under-nutrition and low birth weight predispose to an increased risk for developing diabetes in subsequent generations (111). This phenomenon, termed the “thrifty phenotype hypothesis”, proposes that under-nutrition and poor growth during fetal life can drive permanent changes in the glucose-insulin axis (112). On the other hand, maternal over-nutrition and gestational diabetes can also have an adverse impact on the metabolic fitness of the offspring (14). Epidemiological data show that exposure to maternal obesity or diabetes during fetal life predisposes the offspring to insulin resistance, impaired beta cell function, and high risk of diabetes (113, 114). Studies in rat models have shown that severe maternal diabetes leads to acute fetal hyperglycemia and consequent beta cell hyperplasia, as well as increased insulin output and inability to undergo adaptive beta-cell expansion in adult life (115, 116). Similarly, a recent study showed that maternal exposure to a Western style diet in non-human primates results in aberrant islet composition and insulin hypersecretion in the offspring (117). Collectively, these studies demonstrate that optimal nutrition during development is essential for future metabolic health of the offspring.
As discussed earlier, DNA methylation homeostasis directly coupled with cellular metabolism and nutrient quality. The availability of the Dnmt co-factor S-adenosyl methionine (SAM). Cellular levels of SAM are determined by biochemical reactions involving methyl-donor vitamins such as folate and vitamin B12, amino acids such as methionine, choline, taurine, as well as flux through the TCA cycle (118). Thus, altered levels of these micro-and macro-nutrients during the growth phase can impact the epigenome and organ development. Indeed, folate and vitamin B12 excess in pregnant mice leads to islet dysfunction and impaired beta cell mass in in the offspring (119, 120). Of note, S-adenosyl homocysteine (SAH), the by-product of SAM utilization during the methylation reaction, is an inhibitor of DNA methylation (121). Importantly, the levels of SAH are elevated in obesity (122, 123), and can contribute to aberrant DNA methylation in the offspring in response to maternal obesity. Therefore, nutritional imbalances during fetal or neonatal life can alter the epigenetic program responsible for the development, growth, and function of metabolic organs, thus predisposing the offspring to diabetes (14, 124). This is especially concerning, given the increasing popularity of vitamin supplements and energy drinks containing taurine. Environmentally induced epigenetic changes may also occur in the gametes and be transmitted to subsequent generations, thus contributing to the epigenetic inheritance of diabetes risk [reviewed in (125)] (126–128). In support of this, impaired glucose homeostasis in either parent has been shown to change the metabolism in the offspring, concomitant with specific epigenetic changes (111, 129, 130).
In line with the above discussion, intrauterine growth restriction (IUGR) leads to impaired pancreatic development, reduced functional beta cell mass, and higher risk for diabetes (131, 132). A large number of studies show that the DNA methylation signatures important for beta cell function and identity are disrupted in models of maternal malnutrition and IUGR, ultimately affecting beta cell homeostasis (14). Several studies have demonstrated that IUGR and maternal dietary variation alter the methylation patterns that govern the expression of transcription factors involved in beta cell identity and function, such as Hnf4a and Pdx1; ultimately leading to impaired functional beta cell mass and eventually diabetes (109, 133, 134). Genome-wide profiling of DNA methylation in islets from control and IUGR rats points to dysregulation of the islet methylome preceding diabetes onset. The genomic regions showing altered DNA methylation in IUGR islets include genes involved in beta cell proliferation, insulin secretion, islet vascularization, and cell death, correlating with changes in the corresponding transcripts and reduced functional beta cell mass (135). Similarly, overnutrition in mouse pups leads to islet dysfunction, corresponding with extensive alterations in the islet DNA methylome at weaning; many of these methylation changes occur at genes involved in islet function and control of endocrine identity (93).
Imprinted genes represent a key group of genes relevant to islet homeostasis and diabetes pathogenesis, that are developmentally regulated by DNA methylation (136, 137). These genes display differential allelic regulation of transcription based on parental origin. The DNA methylation patterns of imprinted genes are highly guarded during early embryonic development, to control the gene dosage of important regulators of growth and differentiation (138). Environmentally induced perturbations in the DNA methylation patterning of imprinted genes can therefore not only cause disease risk transmission, but also dictate the pattern of epigenetic inheritance. In line with this, disturbances in methylation of imprinted genes have been shown to relate to the inheritance of diabetes risk (139). For example, mutations in the imprinted locus KCNQ1, which encodes a potassium channel essential for beta cell function, have been shown to impart diabetes susceptibility when inherited maternally, due to altered DNA methylation in early development (136). Similarly, the methylation patterns of imprinted DLK1-MEG3 locus are altered in islets from subjects with T2D, leading to repression of several maternally expressed miRNA. This in turn leads to upregulation of miRNA targets such as IAPP and TP53INP1, that are associated with beta cell dysfunction and cell death, thus contributing to T2D pathogenesis (137).The significance of imprinting control in islet homeostasis and metabolic health is further emphasized by imprinting disorders such as the Beckwith-Wiedemann Syndrome (BWS) and Transient Neonatal Diabetes Mellitus (TNDM) [reviewed in (11)]. In BWS, imprinting defects lead to loss of expression of cell-cycle inhibitor CDKN1C (p57Kip2), resulting in unchecked beta cell expansion and excessive beta cell mass, which causes hyperinsulinemia, and hypoglycemia (140). In TNDM, germline loss of maternal allele of the cell-cycle regulator PLAGL1 results in its overexpression, which leads to transient beta cell dysfunction and plays a role in TNDM. Environmental changes, such as intrauterine hyperglycemia can also alter DNA methylation of imprinted genes to cause islet dysfunction and increase diabetes risk, as shown in mice for the Igf2/H19 locus in islets (141).
These studies collectively show that DNA methylation patterns of several key regulators essential for the fully functional beta cell phenotype are dysregulated in response to adverse early developmental exposures. An abnormal nutritional milieu during fetal development and early growth may not only impede beta cell growth, but can also negatively impact the epigenetic program that dictates the mature beta cell phenotype. This can not only result in a deficit of functional beta cell mass, but also predispose the offspring to diabetes in adult life (14). Occurrence of such environmentally induced epigenetic defects in the beta cell program in the germline can potentially result in their transmission to subsequent generations, thereby presenting a mechanism for the epigenetic inheritance of beta cell dysfunction and diabetes risk (125).
Dysregulation of Islet DNA Methylation Landscape in Diabetes
People with a family history of diabetes have a much higher risk of disease development (142–146). However, genetic linkage can account for only a small percentage of diabetes cases associated with family history. Furthermore, the incidence of both type 1- and type 2-diabetes (T1D and T2D) has increased over the past half-century at a rate that cannot be explained by genetic factors alone, and has been attributed to environmental factors such as altered nutrition and a sedentary lifestyle. Collectively, this suggests the involvement of epigenetic mechanisms that mediate the effect of environment on gene expression and cellular phenotype (145–148). As elaborated earlier, the involvement of epigenetic factors in diabetes risk is further underscored by studies on the effect of fetal environment (107–110), the critical role of epigenetic mechanisms such as DNA methylation in beta cell homeostasis (31, 64, 96, 97, 149, 150), and a direct link between metabolism and the epigenome (151–153). Changes in the cellular DNA methylation landscape, either due to single nucleotide polymorphisms (SNPs) that add or remove CpG sites, or in response to environmental factors, can alter the function of different metabolic tissues, including islets, and potentially contribute to diabetes risk and pathogenesis [reviewed in (9, 124, 154)].
Changes in DNA methylation patterns have been shown to directly contribute to beta cell defects in type 2 diabetes (T2D) (Tables 1 and 2). Several studies have demonstrated that promoters of genes important for beta cell identity and function such as INS, PDX1, PPARGC1A and GLP1R are hypermethylated in human islets from donors with T2D compared to islets from non-diabetic donors (134, 155, 156, 158), resulting in their decreased expression and the consequent impairment of beta cell identity and insulin secretion. In fact, such epigenetic changes can occur preceding T2D, as illustrated by a recent study in diabetic mice, that identified DNA methylation changes associated with differential expression of several genes. Notably, the differentially methylated and expressed genes were enriched in pathways associated with insulin secretion (161). Similarly, islets from obese mice show an upregulation of Dnmt3a, which leads to increased Nkx6.1 promoter methylation, reduced Nkx6.1 and insulin expression, and consequently, impaired islet function (162).

Table 1 DNA methylation studies in islets from T2D subjects, techniques utilized, and the key findings.
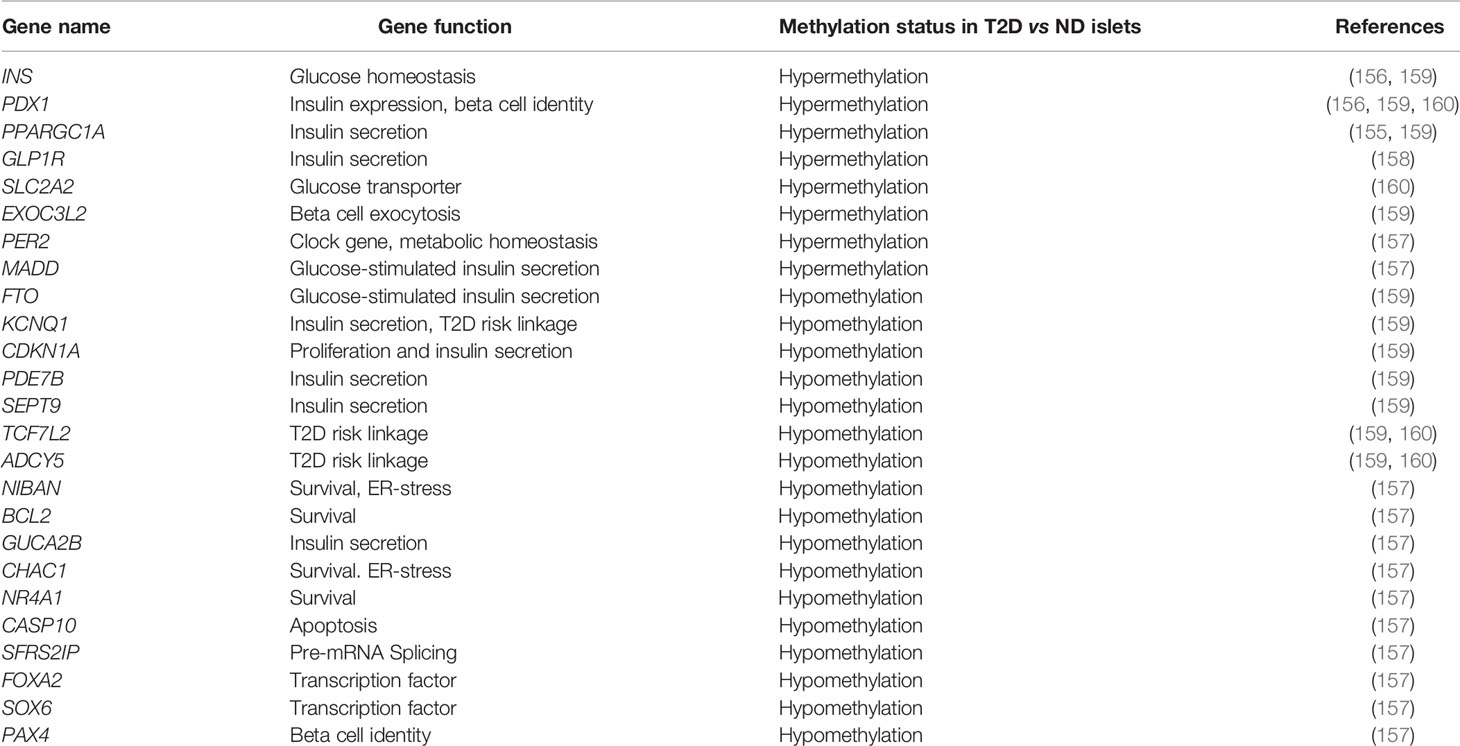
Table 2 DNA methylation status of the key genes related to beta cell homeostasis in human islets in the context of T2D.
Genome-wide profiling of changes in islet DNA methylation patterns in T2D using increasingly sophisticated methods has demonstrated that aberrant DNA methylation of transcriptional programs involved in beta cell function, survival and adaptation contributes to beta cell dysfunction in diabetes (Table 1). One of the first such studies identified 276 differentially methylated CpG sites related to promoters of 254 genes in islets from subjects with T2D, mapping to pathways involved in beta cell function, survival, and stress-response (157). Several of these differentially methylated regions (DMRs) translated in altered gene expression, with majority of them showing an inverse correlation, such that reduced methylation was coincident with increased expression and vice versa. Functional analysis of some of these candidates in human islets using RNAi based knockdown or exposure to stress stimuli underscored the relevance of these genes to dysregulation of beta cell homeostasis in T2D. These analyses, along with functional annotation, revealed epigenetic dysregulation of genes related to pathways such as transcriptional control (FOXA2, PAX4, and SOX6), ER-stress, function, and survival (NIBAN, BCL2, PER2, GUCA2B, CHAC1, MADD, NR4A1, CASP10, and SFRS2IP), in diabetic islets (157). A subsequent genome-wide DNA methylation profiling study using increased depth of mapping identified 853 genes displaying altered DNA methylation in T2D islets including key genes associated with T2D risk, namely TCF7L2, FTO and KCNQ1, highlighting the functional relevance of these DNA methylation changes. Similar to the prior study, several of the differentially methylated genes (e.g. CDKN1A, PDE7B, SEPT9 and EXOC3L2) were differentially expressed (159), and relevant to islet function. More recently, whole-genome bisulfite sequencing has been used to generate an unbiased, single base resolution map of DNA methylation changes in diabetic islets. Such analysis led to the identification of 25,820 DMRs in islets from subjects with T2D, including regions known to regulate islet function, e.g., PDX1, TCF7L2, and ADCY5 (160). Among the DMR associated genes, 457 candidates exhibited concomitant changes in gene expression, and included genes such as SLC2A2, PDX1, and GLP1R that regulate islet function. The DMRs in T2D islets were found to be enriched in binding sites for transcription factors relevant to beta cell function such as FOXA2, NEUROD1, MAFA, and PDX1 (160). This suggests that transcription factors regulate the human islet gene expression program in coordination with DNA methylation patterning.
Alterations in DNA methylation have also been shown to contribute to T1D pathogenesis (163). T1D is characterized by autoimmune destruction of the insulin producing beta cells, and both genetic as well as non-genetic factors contribute to T1D susceptibility (164–166). Several genes associated with T1D susceptibility have been identified, with ~50% of the T1D risk heritability contributed by the MHC locus alone (167), suggesting a major contribution of genetic factors. However, the high average concordance rate of T1D between monozygotic twins (~ 50%) and the rapid rate of increase in T1D incidence over the past few decades suggest the involvement of epigenetic factors. Several studies on blood samples from monozygotic twins discordant for T1D have demonstrated the relevance of altered DNA methylation in immune cells to T1D pathogenesis, with significant methylation changes at genes of immediate relevance to T1D pathogenesis such as HLA genes, INS, IL-2RB, CD226, NFKB1A, TNF, INS-IGF2, CLEC16A, and GAD2 (163, 168, 169). While majority of the studies on DNA methylation differences in T1D have understandably focused on immune cells, a few studies have also demonstrated DNA methylation changes in islets in the context of autoimmune beta cell destruction in the NOD (non-obese diabetic) mice. For instance, one study on disease progression in the NOD mice showed that the inflammation leads to increased expression of Dnmt enzymes, resulting in altered DNA methylation at the INS locus and consequent reduction in insulin expression (170). Beta cell loss during T1D progression is accompanied by the release of unmethylated INS DNA in blood in humans (171–173), and similar release of unmethylated Ins1 and Amylin DNA fragments in the sera of NOD mice (171, 174). Recent data shows that an inflammatory milieu can alter DNA methylation in human islets via TET2 dependent DNA demethylation, concomitant with increased expression of inflammatory and immune pathways genes (175). In line with this, emerging evidence suggests that Tet2 may regulate beta cell response to inflammation in T1D (176). Future studies in islets from auto-antibody positive, non-diabetic cases as well as T1D with residual beta cells in the pancreas may help determine if islet DNA methylation patterns are altered in human islet autoimmunity, and whether they directly contribute to beta cell dysfunction and cell-death in T1D.
While there is abundant evidence that the epigenetic programs that regulate functional beta cell mass are disrupted in diabetic islets, the extent of their causal relationship to disease pathogenesis is not completely clear. One can argue that the DNA methylation changes observed in T2D islets maybe secondary to hyperglycemia. However, the functional analysis of T2D associated DMRs, as well as the overlap of several of these DMRs with regions associated with T2D risk point to causality. Diabetes risk factors such as obesity, aging, and hyperglycemia alter the methylation patterns of T2D associated islet DMRs in non-diabetic subjects, suggesting that epigenetic dysregulation occurs prior to diabetes onset (159). This observation further supports the causal role for such epigenetic changes. While majority of the genetic variations associated with T2D have been shown to be related to islet function (177), they only amount to a minor fraction T2D heritability. It is likely that a combination of genetic and epigenetic factors contribute to diabetes risk and susceptibility, given that DNA methylation changes can be introduced due to genetic variations at even 1-2 CpG sites (178). This also appears to be the case for T1D, as demonstrated by a variant in the imprinted DLK1-MEG3 locus on chromosome 14q32.2, which predisposes to paternally inherited T1D via altered imprinting control (179).
Loss of beta-cells in both T1D and T2D is preceded by progressive beta-cell failure due to impaired beta-cell identity, function, and survival. Functional defects in both scenarios involve beta cell de-differentiation and a recapitulation of the functionally immature phenotype. It is noteworthy that several of the T2D associated DMRs overlap with DMRs associated with endocrine differentiation of TET-deficient stem cells (55), suggesting that a dysregulated methylome may contributes to beta-cell de-differentiation. It is likely that some of these epigenetic changes initiate as an adaptation in response to increased islet workload, or, in case of T1D, to also evade immune assault. However, sustained environmental challenges such as cellular stress, inflammation, altered metabolism, and the inability to meet insulin demand can drive further dysregulation of the islet epigenome and lead to beta cell failure. Altered DNA methylation can also trigger DNA damage and make beta cells prone to apoptosis. This model is supported by studies showing that sustained oxidative stress or hyperglycemia drives a change in the islet transcriptional program from an adaptive mode to stress- response mode (180). In line with this, human islets exposed to lipotoxic stress, a T2D risk factor, exhibit reduced expression of DNMT3A and DNMT1, and increased expression of GADD45A, potentially explaining the mechanistic basis of CpG demethylation observed in T2D islets (159). Collectively, this discussion points to a causal role for islet DNA methylation dysregulation in diabetes risk and pathogenesis, and also underscore the combinatorial role of genetic and epigenetic factors in this process.
Harnessing DNA Methylation for Diabetes Biomarkers and Therapies
Both type 1 and type 2 diabetes are complex disorders that impact multiple organs. A significant number of patients with diabetes develop serious health complications such as retinopathy, nephropathy, neuropathy, as well as cardiovascular disease (181). Loss of functional beta cell mass often begins long before disease diagnosis, and diabetes manifests after more than 50% beta cell mass is already lost (182). Therefore, early detection of beta cell defects may provide opportunities for disease prevention in people at higher risk, as well as inform the development of approaches for monitoring disease progression and personalized patient care. Currently, no established biomarkers exist for the assessment of beta cell loss preceding disease diagnosis. Detection of circulating cell-free DNA (cfDNA) has emerged as a non-invasive and practical tool for the development of blood-based biomarkers. CfDNA fragments are released into the blood due to cell-death, necrosis, or active secretion, and therefore potentially reflect disease related tissue changes. CfDNA is highly stable, and DNA methylation signatures of cfDNA fragments correspond to their tissue-of-origin (183, 184). Development of a comprehensive reference DNA methylation atlas of various human tissues now allows the robust and accurate determination of the tissue-of-origin of cfDNA in human plasma, in healthy and disease conditions (184). As discussed earlier, beta cell death is accompanied by the release of DNA fragments carrying beta cell specific methylation signatures in circulation, and can be detected by assaying for such signatures in the cfDNA. DNA methylation patterns of circulating cfDNA can, therefore, potentially serve as a biomarker to detect beta cell loss in diabetes.
Beta cell specific methylation patterns of human INSULIN (INS) gene have been widely used to detect beta cell death in the context of T1D (171–173, 185), and islet transplantation (172). The premise of these assays is that the circulating unmethylated INS promoter DNA fragments are exclusively of beta cell origin and indicate recent beta cell death (Table 3). Assays based on this principle have been used to assess beta cell death during T1D progression. A study focusing on a small cohort of at-risk subjects showed that the subjects who progressed to T1D displayed a modest increase in the circulating unmethylated INS DNA, corresponding to reduced insulin secretion in an oral glucose tolerance test (172). This study also reported that the half-life of circulating unmethylated INS DNA is only ~2 hrs, suggesting that such assays may only reflect acute beta cell death. Using this assay, this study showed that there is detectable beta cell death in the pre-diabetic period associated with reduced insulin secretion, followed by a dramatic increase in the peri-diagnosis period (172). A recent report described the development of a multiplex ultrasensitive assay that can detect as little as one beta cell genome equivalent. This assay was able to detect beta cell demise in several clinical contexts, such as in islet transplant recipients shortly after transplantation, and in patients with KATP congenital hyperinsulinism. However, unlike prior assays, this approach couldn’t detect an elevation of beta cell derived cfDNA in patients at risk for T1D (autoantibody-positive), and those with recent-onset or long-standing T1D (186). This could be due to several reasons. It is likely that beta cell death in T1D occurs in bursts, such that there may have been no ongoing cell death at the time of sampling. Reduced beta cell mass in pre- and early-T1D due to beta cell de-differentiation could also contribute to this. Longitudinal testing and development of additional markers is required to resolve this. Overall, while DNA methylation analysis of cfDNA holds a lot of promise, several variables can impact the outcome of such assays, including the rate of disease progression.
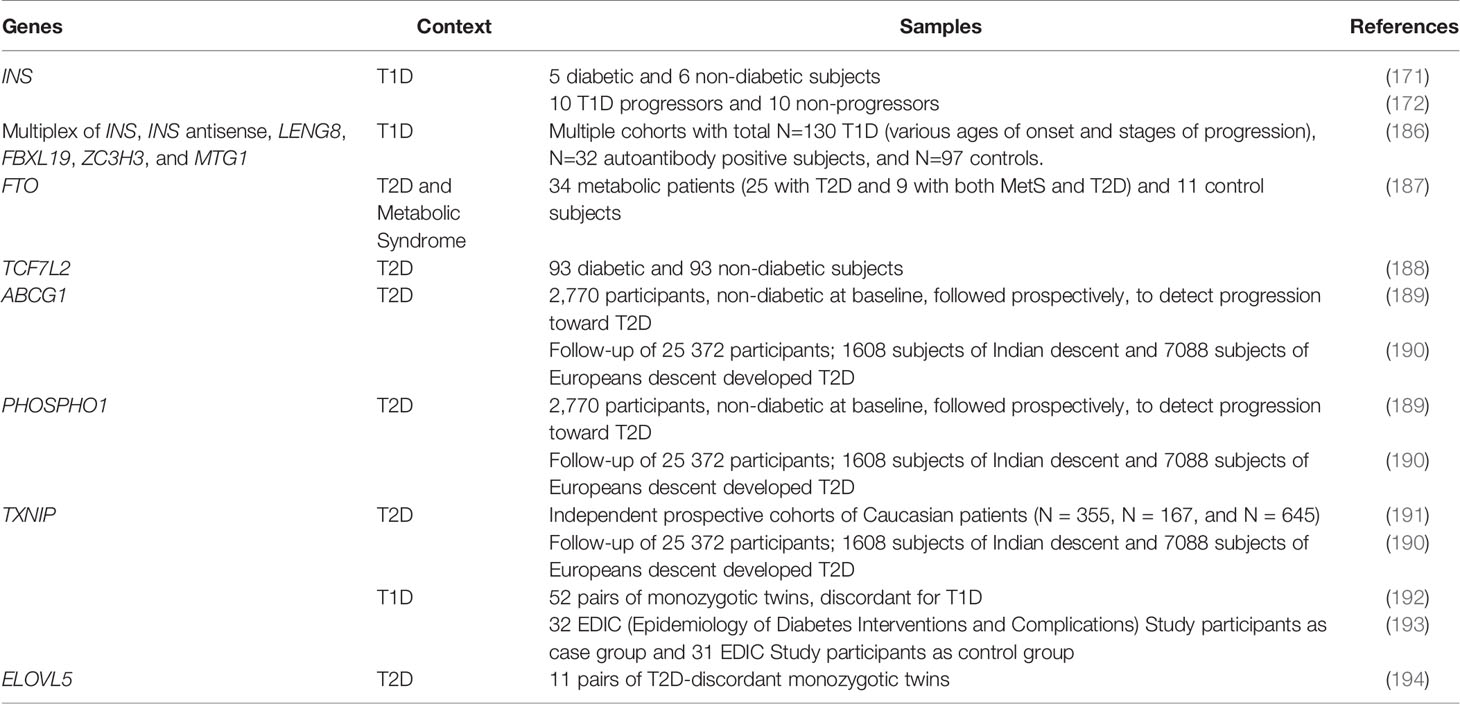
Table 3 Blood-based biomarker candidates T1D and T2D.
While biomarkers based on islet specific DNA methylation signatures in cfDNA have primarily focused on T1D, this approach may also be relevant to T2D (195, 196). Recent data demonstrate that the DNA methylation changes associated with T2D in beta cells and peripheral insulin sensitive tissues are reliably captured in the circulating DNA, supporting the suitability of cfDNA for developing T2D biomarkers (195, 196). Meta-analyses of multiple, highly powered genome-wide DNA methylation profiling studies comparing blood samples diabetic and non-diabetic subjects have identified several potential candidate loci, for which the T2D associated DNA methylation changes in blood are consistent across different ethnic populations (reviewed in (197). These include genes associated with T2D risk such as FTO and TCF7L2 (187, 188), as well as genes involved in beta cell function and glucose homeostasis such as ABCG1, PHOSPHO1, and TXNIP (189–191). Notably, changes in TXNIP methylation in the peripheral blood have also been found to be associated with T1D and its complications (192, 193). In addition to identification of functionally relevant T2D associated DMRs such as those listed above, cross-comparison of DNA methylation profiling studies in multiple metabolic tissues can unravel T2D associated DMRs that are conserved across tissues. This is exemplified by the ELOVL5, which displays conserved T2D associated DMRs in islets and adipose tissue (194). Collectively, these functionally relevant candidates present as robust candidates for blood-based biomarkers for T2D (Table 3). Aging is an important “environmental” factor associated with increased risk of T2D, and leads to DNA methylation changes that dictate reduced beta cell replication and islet function with age (65). Genome-wide methylation profiling in non-diabetic donors (ages 26-74) showed that several age-related DNA methylation changes seen in human islets are conserved in blood samples, including some that associate with islet function and T2D. These data suggest that blood-based epigenetic biomarkers can be good predictors of islet function and T2D risk with age (196). DNA methylation patterns can also be leveraged to determine the differentiation status of stem cell derived products. For instance, promoter hypermethylation of disallowed genes has been used as marker of functional maturity during beta cell differentiation (198). As discussed earlier, the global levels of 5hmC progressively increase during the differentiation and maturation of beta cells. Therefore, in addition to locus specific DNA methylation patterns, global 5hmC content can also serve as a reliable index of beta cell differentiation and functional maturity (56).
Given the specificity and importance of DNA methylation patterning in beta cell homeostasis, and its dysregulation in diabetic islets, this epigenetic module has emerged as an attractive therapeutic target. Approaches that target DNA methylation have been successfully used for cancer therapeutics. For example, DNA methyltransferase inhibitors 5-aza-deoxycytidine (5azadC; Decitabine) and 5-aza-cytidine (5azaC; Azacitidine; Vidaza) are approved for use as anti-tumor agents (199). In a related approach, altering the availability of the methyl donor S-adenosyl methionine (SAM) has been proposed as an intervention in some clinical contexts such as depression, osteoarthritis, and liver disease (200). These agents, however, have a variety of documented harmful side-effects (199, 200). Vitamin B12 (folic acid), another methyl donor and a co-factor in SAM synthesis, is a widely used nutritional supplement, and has been effectively used to prevent neural tube defects (201). However, excessive folate intake during gestation and neonatal life can have obesogenic effects (202), and there are concerns regarding its potential oncogenic effects (201). Overall, the main challenge with all of these approaches is their generalized effect on DNA methylation, which can have deleterious off-target effects. While this currently precludes the use of these drugs for diabetes therapy, 5-aza-dC has recently been used in vitro to improve the efficiency of beta cell differentiation from human induced pluripotent stem cells (iPSCs) cells derived from subjects with T1D, which are otherwise known to differentiate poorly (203). DNA demethylation caused by 5-aza-dC treatment increases PDX1 expression during T1D-iPSC differentiation, thereby improving the differentiation efficiency (203). Thus, while global targeting of DNA methylation may likely find in vitro use for improving cell therapies, there is a need for developing highly specific targeting approaches that can restore the DNA methylation patterns that are disrupted in diabetes and promote a healthy islet phenotype.
Recent advances in genome editing using molecular tools such as CRISPR/Cas9 and TALEN now enable us to modify the DNA methylation patterns at specific regions in the genome, to either remedy disease specific epigenetic changes or introduce epigenetic patterns with potential therapeutic benefits. Locus specific epigenetic engineering has been used to target Dnmt or Tet enzymes to drive methylation or demethylation of specific genes (204). In a recent study, targeted DNA demethylation of the imprinted cell-cycle inhibitor gene CDKN1C was recently used to stimulate the expansion of adult human beta cells that typically do not replicate. Using a TALEN dependent targeting of TET1, Kaestner and colleagues simulated the DNA methylation patterns associated with beta cell hyper-proliferation in Beckwith Wiedemann Syndrome (BWS), where hypomethylation of the CDKN1C locus leads to reduced p57Kip2 and beta-cell hyper-proliferation. Using this approach, they were able to achieve the demethylation of CDKN1C locus and consequent reduction of p57Kip2 levels in human islets, and successfully induce replication of adult human beta cells (205). In another recent proof of principle study, CRISPR/Cas9 based targeting of Dnmt3a was used to drive the DNA methylation and repression of alpha cell-fate determinant gene Arx in mouse embryonic pancreatic progenitors, to promote beta cell lineage (63). Targeted epigenetic editing of developmentally regulated genes, therefore, has the potential to improve existing stem cell differentiation protocols by enhancing beta cell specification and functional maturation. Strategies involving the combinatorial, stage-specific epigenetic targeting of key genes that determine beta cell specification, functional maturation, and expansion, may soon become prevalent. Another potential approach in driving stage-specific epigenetic changes may be to mimic the environmental factors unique to a given developmental stage. It is increasingly being recognized that appropriate developmental niche is critical for the success of in vitro stem cell differentiation protocols (206). In this context, cellular disaggregation and reaggregation of immature beta cell clusters has been shown to promote DNA methylation changes associated with functional maturation (198). This process, intended to mirror the stage-specific changes in cell-cell interaction, is therefore able to induce physiologically relevant epigenetic changes. By combining environmental cues (e.g. nutrient availability) that simulate an in vivo scenario with epigenetic targeting and/or small molecule epigenetic modifiers, future work could dramatically improve the quality of stem cell derived islet products. Data generated from genome-wide DNA methylation profiling studies along with mechanistic work on the dysregulation of DNA methylation of key regulatory pathways (such as the imprinted DLK1-MEG3 locus in T2D), is likely to inspire the development of novel therapeutic approaches for augmenting beta cell function and mass. Given the specificity of epigenetic engineering, such approaches may only have minimal off-target effects, and are thus likely to emerge as powerful therapeutic tools.
Summary
The epigenome serves as a mechanism to adapt the behavior of a cell in response to changes in its environment, by interpreting the genome in a context specific manner and facilitating a transcriptional response accordingly. Methylation of the cytosine within the CpG residues in DNA is one of the most thoroughly studied epigenetic modifications, and one that can be faithfully inherited from parent to daughter cell during cell-division. DNA methylation is indispensable for shaping stage- and cell-type specific transcriptional programs, starting from early embryonic differentiation events to the maintenance of fully differentiated cellular phenotypes (9). While it is clear that the cellular environment shapes the methylome, the precise mechanisms underlying this plasticity are still not well understood. For instance, to what extent does 5hmC contribute to the demethylation of DNA versus serving as an independent regulatory module? What is the regulatory role, if any, of oxidized intermediates such 5fC and 5caC? Some of the work discussed in this review provides strong evidence that DNA methylation does not operate in isolation, but rather requires a coordinated regulatory interaction with other epigenetic modules (207, 208). Recent work, such as studies discussed in this review, also appears to suggest that cell-type and stage-specific transcription factors, including pioneer factors, play an important role in governing the spatio-temporal specificity of DNA methylation patterning. However, our knowledge of such coordinated interaction between cellular environment, signaling pathways, transcriptional programs, and multiple layers of epigenetic regulation is still in its infancy. Studies focusing on these aspects will provide a clearer understanding of the mechanistic underpinnings of gene-environment interaction, and identify sensitive temporal windows of methylome plasticity, genomic regions that are more- or less- vulnerable to methylation changes during a cell’s response to its environment, and the functional implications of such epigenomic flexibility.
The dynamic regulation and appropriate propagation of DNA methylation patterns is essential for the establishment and maintenance of functional beta cell mass, and accumulating evidence suggests that perturbations in the beta cell methylome are associated with beta cell pathologies and may contribute to disease development. Studies using mouse genetics, rodent models of metabolic disease, and stem-cell models of beta cell differentiation have been instrumental in shaping our current understanding of the critical role DNA methylation plays in the regulation of beta cell homeostasis. Environmental factors such as fetal growth impairment, altered nutrition, cellular-stress, aging etc. have been shown to predispose to diabetes, and can cause changes in DNA methylation patterns that regulate functional beta cell mass (14). Whether such changes in the methylome actively drive beta cell dysfunction, or merely reflect the metabolic dysregulation, or both, remains to be seen. While canonically DNA methylation has been viewed as a rather stable mark, emerging evidence shows that highly acute physiologically relevant changes in the methylome can and do occur in many tissue types (175, 209). While in vitro data suggests that acute changes in DNA methylation can occur in the islets (175), it is not yet clear whether similar changes occur in vivo. It is likely that different components of the cellular methylome respond differently to environmental stimuli, and such variations may depend on the genomic location. Whole-genome level mapping of DNA methylation patterns has provided a wealth of knowledge on the specific genes and pathways involved in the epigenetic control of beta cell homeostasis, during beta cell differentiation and in the context of diabetes pathogenesis. In addition, such data is likely to support the development of novel and accurate biomarkers for predicting diabetes risk and monitoring disease progression. The recent recognition of the plasticity of DNA methylation marks, combined with the advances in genome editing techniques, now offers the possibilities of targeted manipulation to remedy disease specific DNA methylation patterns or introduce patterns with therapeutic benefit (13). Future development of methods for cell specific delivery of epigenetic targeting modalities is likely to foster the development of highly precise therapeutic interventions for diabetes.
Author Contributions
Both NP and SD contributed to the conception of this review, critical review of the literature, drafting and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work in Dhawan laboratory is supported by grants from the National Institutes of Health (R01DK120523), the Wanek Family Project to Cure Type 1 Diabetes at City of Hope, and Human Islet Research Network (NIH) UC4 DK104162 (to SD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the members of Dhawan Lab for helpful discussions, and to Dr. Arthur Riggs, City of Hope, for his pioneering studies on DNA methylation and his continued support of our research.
References
1. Allis CD, Jenuwein T. The Molecular Hallmarks of Epigenetic Control. Nat Rev Genet (2016) 17(8):487–500. doi: 10.1038/nrg.2016.59
2. Waddington CH. Canalization of Development and the Inheritance of Acquired Characters. Nature (1942) 150:563–5. doi: 10.1038/150563a0
3. Russo VE, Martienssen RA, Riggs AD. Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor Lab Press (1996) 1–692.
5. Holliday R, Pugh JE. DNA Modification Mechanisms and Gene Activity During Development. Science (1975) 187(4173):226–32. doi: 10.1126/science.1111098
6. Griffith JS, Mahler HR. DNA Ticketing Theory of Memory. Nature (1969) 223(5206):580–2. doi: 10.1038/223580a0
7. Bestor TH, Edwards JR, Boulard M. Notes on the Role of Dynamic DNA Methylation in Mammalian Development. Proc Natl Acad Sci USA (2015) 112(22):6796–9. doi: 10.1073/pnas.1415301111
8. Greenberg MVC, Bourc’his D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat Rev Mol Cell Biol (2019) 20(10):590–607. doi: 10.1038/s41580-019-0159-6
9. Avrahami D, Kaestner KH. The Dynamic Methylome of Islets in Health and Disease. Mol Metab (2019) 27S:S25–32. doi: 10.1016/j.molmet.2019.06.007
10. Pastor WA, Aravind L, Rao A. Tetonic Shift: Biological Roles of TET Proteins in DNA Demethylation and Transcription. Nat Rev Mol Cell Biol (2013) 14(6):341–56. doi: 10.1038/nrm3589
11. Golson ML, Kaestner KH. Epigenetics in Formation, Function, and Failure of the Endocrine Pancreas. Mol Metab (2017) 6(9):1066–76. doi: 10.1016/j.molmet.2017.05.015
12. Hunter CS, Stein RW. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet beta-Cells in Type 2 Diabetes. Front Genet (2017) 8:35. doi: 10.3389/fgene.2017.00035
13. Dhawan S, Natarajan R. Epigenetics and Type 2 Diabetes Risk. Curr Diabetes Rep (2019) 19(8):47. doi: 10.1007/s11892-019-1168-8
14. Bansal A, Simmons RA. Epigenetics and Developmental Origins of Diabetes: Correlation or Causation? Am J Physiol Endocrinol Metab (2018) 315(1):E15–28. doi: 10.1152/ajpendo.00424.2017
15. Rosen ED, Kaestner KH, Natarajan R, Patti ME, Sallari R, Sander M, et al. Epigenetics and Epigenomics: Implications for Diabetes and Obesity. Diabetes (2018) 67(10):1923–31. doi: 10.2337/db18-0537
16. Jones PA. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat Rev Genet (2012) 13(7):484–92. doi: 10.1038/nrg3230
17. Patil V, Ward RL, Hesson LB. The Evidence for Functional non-CpG Methylation in Mammalian Cells. Epigenetics (2014) 9(6):823–8. doi: 10.4161/epi.28741
18. Goll MG, Bestor TH. Eukaryotic Cytosine Methyltransferases. Annu Rev Biochem (2005) 74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721
19. Lyko F. The DNA Methyltransferase Family: A Versatile Toolkit for Epigenetic Regulation. Nat Rev Genet (2018) 19(2):81–92. doi: 10.1038/nrg.2017.80
20. Kaneda M, Sado T, Hata K, Okano M, Tsujimoto N, Li E, et al. Role of De Novo DNA Methyltransferases in Initiation of Genomic Imprinting and X-chromosome Inactivation. Cold Spring Harb Symp Quant Biol (2004) 69:125–9. doi: 10.1101/sqb.2004.69.125
21. Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b are Essential for De Novo Methylation and Mammalian Development. Cell (1999) 99(3):247–57. doi: 10.1016/S0092-8674(00)81656-6
22. Lei H, Oh SP, Okano M, Juttermann R, Goss KA, Jaenisch R, et al. De Novo DNA Cytosine Methyltransferase Activities in Mouse Embryonic Stem Cells. Development (1996) 122(10):3195–205. doi: 10.1242/dev.122.10.3195
23. Li E, Bestor TH, Jaenisch R. Targeted Mutation of the DNA Methyltransferase Gene Results in Embryonic Lethality. Cell (1992) 69(6):915–26. doi: 10.1016/0092-8674(92)90611-F
24. Leonhardt H, Page AW, Weier HU, Bestor TH. A Targeting Sequence Directs DNA Methyltransferase to Sites of DNA Replication in Mammalian Nuclei. Cell (1992) 71(5):865–73. doi: 10.1016/0092-8674(92)90561-P
25. Fatemi M, Hermann A, Pradhan S, Jeltsch A. The Activity of the Murine DNA Methyltransferase Dnmt1 is Controlled by Interaction of the Catalytic Domain With the N-terminal Part of the Enzyme Leading to an Allosteric Activation of the Enzyme After Binding to Methylated DNA. J Mol Biol (2001) 309(5):1189–99. doi: 10.1006/jmbi.2001.4709
26. Geiman TM, Robertson KD. Chromatin Remodeling, Histone Modifications, and DNA Methylation-How Does it All Fit Together? J Cell Biochem (2002) 87(2):117–25. doi: 10.1002/jcb.10286
27. Berger SL. The Complex Language of Chromatin Regulation During Transcription. Nature (2007) 447(7143):407–12. doi: 10.1038/nature05915
28. Deaton AM, Bird A. Cpg Islands and the Regulation of Transcription. Genes Dev (2011) 25(10):1010–22. doi: 10.1101/gad.2037511
29. Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, et al. Orphan CpG Islands Identify Numerous Conserved Promoters in the Mammalian Genome. PloS Genet (2010) 6(9):e1001134. doi: 10.1371/journal.pgen.1001134
30. Bogdanovic O, Veenstra GJ. DNA Methylation and methyl-CpG Binding Proteins: Developmental Requirements and Function. Chromosoma (2009) 118(5):549–65. doi: 10.1007/s00412-009-0221-9
31. Papizan JB, Singer RA, Tschen SI, Dhawan S, Friel JM, Hipkens SB, et al. Nkx2.2 Repressor Complex Regulates Islet Beta-Cell Specification and Prevents Beta-to-Alpha-Cell Reprogramming. Genes Dev (2011) 25(21):2291–305. doi: 10.1101/gad.173039.111
32. Zhu H, Wang G, Qian J. Transcription Factors as Readers and Effectors of DNA Methylation. Nat Rev Genet (2016) 17(9):551–65. doi: 10.1038/nrg.2016.83
33. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of Cytosine Methylation on DNA Binding Specificities of Human Transcription Factors. Science (2017) 356(6337):1–15. doi: 10.1126/science.aaj2239
34. Wu X, Zhang Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat Rev Genet (2017) 18(9):517–34. doi: 10.1038/nrg.2017.33
35. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science (2009) 324(5929):930–5. doi: 10.1126/science.1170116
36. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet Proteins can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science (2011) 333(6047):1300–3. doi: 10.1126/science.1210597
37. Bayraktar G, Kreutz MR. The Role of Activity-Dependent Dna Demethylation in the Adult Brain and in Neurological Disorders. Front Mol Neurosci (2018) 11:169. doi: 10.3389/fnmol.2018.00169
38. Surani MA, Hayashi K, Hajkova P. Genetic and Epigenetic Regulators of Pluripotency. Cell (2007) 128(4):747–62. doi: 10.1016/j.cell.2007.02.010
39. Cedar H, Bergman Y. Programming of DNA Methylation Patterns. Annu Rev Biochem (2012) 81:97–117. doi: 10.1146/annurev-biochem-052610-091920
40. Messerschmidt DM, Knowles BB, Solter D. DNA Methylation Dynamics During Epigenetic Reprogramming in the Germline and Preimplantation Embryos. Genes Dev (2014) 28(8):812–28. doi: 10.1101/gad.234294.113
41. Dhawan S, Georgia S, Bhushan A. Formation and Regeneration of the Endocrine Pancreas. Curr Opin Cell Biol (2007) 19(6):634–45. doi: 10.1016/j.ceb.2007.09.015
42. Shih HP, Wang A, Sander M. Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annu Rev Cell Dev Biol (2013) 29:81–105. doi: 10.1146/annurev-cellbio-101512-122405
43. Bastidas-Ponce A, Scheibner K, Lickert H, Bakhti M. Cellular and Molecular Mechanisms Coordinating Pancreas Development. Development (2017) 144(16):2873–88. doi: 10.1242/dev.140756
44. Zorn AM, Wells JM. Vertebrate Endoderm Development and Organ Formation. Annu Rev Cell Dev Biol (2009) 25:221–51. doi: 10.1146/annurev.cellbio.042308.113344
45. Gittes GK. Developmental Biology of the Pancreas: A Comprehensive Review. Dev Biol (2009) 326(1):4–35. doi: 10.1016/j.ydbio.2008.10.024
46. Larsen HL, Grapin-Botton A. The Molecular and Morphogenetic Basis of Pancreas Organogenesis. Semin Cell Dev Biol (2017) 66:51–68. doi: 10.1016/j.semcdb.2017.01.005
47. Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, et al. DNA Methylation Directs Functional Maturation of Pancreatic Beta Cells. J Clin Invest (2015) 125(7):2851–60. doi: 10.1172/JCI79956
48. Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, Charlton J, et al. Circadian Entrainment Triggers Maturation of Human in Vitro Islets. Cell Stem Cell (2020) 26(1):108–22.e10. doi: 10.1016/j.stem.2019.11.011
49. Wang A, Yue F, Li Y, Xie R, Harper T, Patel NA, et al. Epigenetic Priming of Enhancers Predicts Developmental Competence of hESC-derived Endodermal Lineage Intermediates. Cell Stem Cell (2015) 16(4):386–99. doi: 10.1016/j.stem.2015.02.013
50. Lee K, Cho H, Rickert RW, Li QV, Pulecio J, Leslie CS, et al. Foxa2 Is Required for Enhancer Priming During Pancreatic Differentiation. Cell Rep (2019) 28(2):382–93.e7. doi: 10.1016/j.celrep.2019.06.034
51. Zaret KS, Carroll JS. Pioneer Transcription Factors: Establishing Competence for Gene Expression. Genes Dev (2011) 25(21):2227–41. doi: 10.1101/gad.176826.111
52. Bahar Halpern K, Vana T, Walker MD. Paradoxical Role of DNA Methylation in Activation of FoxA2 Gene Expression During Endoderm Development. J Biol Chem (2014) 289(34):23882–92. doi: 10.1074/jbc.M114.573469
53. Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, et al. MicroRNA-26a Targets Ten Eleven Translocation Enzymes and is Regulated During Pancreatic Cell Differentiation. Proc Natl Acad Sci USA (2013) 110(44):17892–7. doi: 10.1073/pnas.1317397110
54. Li J, Wu X, Zhou Y, Lee M, Guo L, Han W, et al. Decoding the Dynamic DNA Methylation and Hydroxymethylation Landscapes in Endodermal Lineage Intermediates During Pancreatic Differentiation of Hesc. Nucleic Acids Res (2018) 46(6):2883–900. doi: 10.1093/nar/gky063
55. Wu X, Li J, Lee M, Lan Q, Li J, Huang Y, et al. TET1 Dioxygenase is Required for FOXA2-associated Chromatin Remodeling in Pancreatic Beta-Cell Differentiation. BioRxiv (2020) 1–47. doi: 10.1101/2020.05.20.107532
56. Gurlo T, Georgia S, Dhawan S. Dna Hydroxymethylation Regulates Beta-Cell Maturation and Expansion. Diabetes (2018) 67:(Supplement 1): 50–OR. doi: 10.2337/db18-50-OR
57. Koh KP, Rao A. DNA Methylation and Methylcytosine Oxidation in Cell Fate Decisions. Curr Opin Cell Biol (2013) 25(2):152–61. doi: 10.1016/j.ceb.2013.02.014
58. Anderson RM, Bosch JA, Goll MG, Hesselson D, Dong PD, Shin D, et al. Loss of Dnmt1 Catalytic Activity Reveals Multiple Roles for DNA Methylation During Pancreas Development and Regeneration. Dev Biol (2009) 334(1):213–23. doi: 10.1016/j.ydbio.2009.07.017
59. Georgia S, Kanji M, Bhushan A. DNMT1 Represses p53 to Maintain Progenitor Cell Survival During Pancreatic Organogenesis. Genes Dev (2013) 27(4):372–7. doi: 10.1101/gad.207001.112
60. Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC. Replication-Independent Chromatin Loading of Dnmt1 During G2 and M Phases. EMBO Rep (2004) 5(12):1181–6. doi: 10.1038/sj.embor.7400295
61. Ma Y, Jacobs SB, Jackson-Grusby L, Mastrangelo MA, Torres-Betancourt JA, Jaenisch R, et al. Dna CpG Hypomethylation Induces Heterochromatin Reorganization Involving the Histone Variant macroH2A. J Cell Sci (2005) 118(Pt 8):1607–16. doi: 10.1242/jcs.02291
62. Neiman D, Moss J, Hecht M, Magenheim J, Piyanzin S, Shapiro AMJ, et al. Islet Cells Share Promoter Hypomethylation Independently of Expression, But Exhibit Cell-Type-Specific Methylation in Enhancers. Proc Natl Acad Sci USA (2017) 114(51):13525–30. doi: 10.1073/pnas.1713736114
63. Liu J, Banerjee A, Herring CA, Attalla J, Hu R, Xu Y, et al. Neurog3-Independent Methylation is the Earliest Detectable Mark Distinguishing Pancreatic Progenitor Identity. Dev Cell (2019) 48(1):49–63.e7. doi: 10.1016/j.devcel.2018.11.048
64. Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic Beta Cell Identity is Maintained by DNA Methylation-Mediated Repression of Arx. Dev Cell (2011) 20(4):419–29. doi: 10.1016/j.devcel.2011.03.012
65. Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, et al. Aging-Dependent Demethylation of Regulatory Elements Correlates With Chromatin State and Improved Beta Cell Function. Cell Metab (2015) 22(4):619–32. doi: 10.1016/j.cmet.2015.07.025
66. Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 Gene is Essential for Differentiation of Insulin-Producing Beta Cells in the Mammalian Pancreas. Nature (1997) 386(6623):399–402. doi: 10.1038/386399a0
67. Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, et al. Opposing Actions of Arx and Pax4 in Endocrine Pancreas Development. Genes Dev (2003) 17(20):2591–603. doi: 10.1101/gad.269003
68. Collombat P, Hecksher-Sorensen J, Broccoli V, Krull J, Ponte I, Mundiger T, et al. The Simultaneous Loss of Arx and Pax4 Genes Promotes a Somatostatin-Producing Cell Fate Specification At the Expense of the Alpha- and Beta-Cell Lineages in the Mouse Endocrine Pancreas. Development (2005) 132(13):2969–80. doi: 10.1242/dev.01870
69. Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of Beta-Cell Mass in the Growing Rat Pancreas. Estimation With a Simple Mathematical Model. Diabetes (1995) 44(3):249–56. doi: 10.2337/diabetes.44.3.249
70. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 Expression Causes Insulin-Deficient Diabetes and Cdk4 Activation Results in Beta-Islet Cell Hyperplasia. Nat Genet (1999) 22(1):44–52. doi: 10.1038/8751
71. Georgia S, Bhushan A. Beta Cell Replication is the Primary Mechanism for Maintaining Postnatal Beta Cell Mass. J Clin Invest (2004) 114(7):963–8. doi: 10.1172/JCI22098
72. Kulkarni RN. New Insights Into the Roles of Insulin/IGF-I in the Development and Maintenance of Beta-Cell Mass. Rev Endocr Metab Disord (2005) 6(3):199–210. doi: 10.1007/s11154-005-3051-y
73. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-Cell Replication is the Primary Mechanism Subserving the Postnatal Expansion of Beta-Cell Mass in Humans. Diabetes (2008) 57(6):1584–94. doi: 10.2337/db07-1369
74. Asplund K. Dynamics of Insulin Release From the Foetal and Neonatal Rat Pancreas. Eur J Clin Invest (1973) 3(4):338–44. doi: 10.1111/j.1365-2362.1973.tb00360.x
75. Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional Beta-Cell Maturation is Marked by an Increased Glucose Threshold and by Expression of Urocortin 3. Nat Biotechnol (2012) 30(3):261–4. doi: 10.1038/nbt.2141
76. German MS. Glucose Sensing in Pancreatic Islet Beta Cells: The Key Role of Glucokinase and the Glycolytic Intermediates. Proc Natl Acad Sci USA (1993) 90(5):1781–5. doi: 10.1073/pnas.90.5.1781
77. Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, et al. Metabolic Fate of Glucose in Purified Islet Cells. Glucose-regulated Anaplerosis in Beta Cells. J Biol Chem (1997) 272(30):18572–9. doi: 10.1074/jbc.272.30.18572
78. Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, et al. Rat Neonatal Beta Cells Lack the Specialised Metabolic Phenotype of Mature Beta Cells. Diabetologia (2011) 54(3):594–604. doi: 10.1007/s00125-010-2036-x
79. Zeng C, Mulas F, Sui Y, Guan T, Miller N, Tan Y, et al. Pseudotemporal Ordering of Single Cells Reveals Metabolic Control of Postnatal Beta Cell Proliferation. Cell Metab (2017) 25(5):1160–75.e11. doi: 10.1016/j.cmet.2017.04.014
80. Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, et al. Replication Confers Beta Cell Immaturity. Nat Commun (2018) 9(1):485. doi: 10.1038/s41467-018-02939-0
81. Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, et al. Weaning Triggers a Maturation Step of Pancreatic Beta Cells. Dev Cell (2015) 32(5):535–45. doi: 10.1016/j.devcel.2015.01.002
82. Rodnoi P, Rajkumar M, Moin ASM, Georgia SK, Butler AE, Dhawan S. Neuropeptide Y Expression Marks Partially Differentiated Beta Cells in Mice and Humans. JCI Insight (2017) 2(12):1–14. doi: 10.1172/jci.insight.94005
83. Fox JE, Seeberger K, Dai XQ, Lyon J, Spigelman AF, Kolic J, et al. Functional Plasticity of the Human Infant Beta-Cell Exocytotic Phenotype. Endocrinology (2013) 154(4):1392–9. doi: 10.1210/en.2012-1934
84. Lemaire K, Thorrez L, Schuit F. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu Rev Nutr (2016) 36:45–71. doi: 10.1146/annurev-nutr-071715-050808
85. Henning SJ. Postnatal Development: Coordination of Feeding, Digestion, and Metabolism. Am J Physiol (1981) 241(3):G199–214. doi: 10.1152/ajpgi.1981.241.3.G199
86. Jaafar R, Tran S, Shah AN, Sun G, Valdearcos M, Marchetti P, et al. mTORC1 to AMPK Switching Underlies Beta-Cell Metabolic Plasticity During Maturation and Diabetes. J Clin Invest (2019) 129(10):4124–37. doi: 10.1172/JCI127021
87. Ni Q, Gu Y, Xie Y, Yin Q, Zhang H, Nie A, et al. Raptor Regulates Functional Maturation of Murine Beta Cells. Nat Commun (2017) 8:15755. doi: 10.1038/ncomms15755
88. Sharma U, Rando OJ. Metabolic Inputs Into the Epigenome. Cell Metab (2017) 25(3):544–58. doi: 10.1016/j.cmet.2017.02.003
89. Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-Dependent Decline in Beta-Cell Proliferation Restricts the Capacity of Beta-Cell Regeneration in Mice. Diabetes (2009) 58(6):1312–20. doi: 10.2337/db08-1651
90. Rankin MM, Kushner JA. Adaptive Beta-Cell Proliferation is Severely Restricted With Advanced Age. Diabetes (2009) 58(6):1365–72. doi: 10.2337/db08-1198
91. Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, et al. DNA Methylation Aging Clocks: Challenges and Recommendations. Genome Biol (2019) 20(1):249. doi: 10.1186/s13059-019-1824-y
92. Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, et al. Aging Effects on DNA Methylation Modules in Human Brain and Blood Tissue. Genome Biol (2012) 13(10):R97. doi: 10.1186/gb-2012-13-10-r97
93. Li G, Petkova TD, Laritsky E, Kessler N, Baker MS, Zhu S, et al. Early Postnatal Overnutrition Accelerates Aging-Associated Epigenetic Drift in Pancreatic Islets. Environ Epigenet (2019) 5(3):dvz015. doi: 10.1093/eep/dvz015
94. Lemaire K, Granvik M, Schraenen A, Goyvaerts L, Van Lommel L, Gomez-Ruiz A, et al. How Stable is Repression of Disallowed Genes in Pancreatic Islets in Response to Metabolic Stress? PloS One (2017) 12(8):e0181651. doi: 10.1371/journal.pone.0181651
95. Luo C, Hajkova P, Ecker JR. Dynamic DNA Methylation: In the Right Place At the Right Time. Science (2018) 361(6409):1336–40. doi: 10.1126/science.aat6806
96. Dhawan S, Tschen SI, Bhushan A. Bmi-1 Regulates the Ink4a/Arf Locus to Control Pancreatic Beta-Cell Proliferation. Genes Dev (2009) 23(8):906–11. doi: 10.1101/gad.1742609
97. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb Protein Ezh2 Regulates Pancreatic Beta-Cell Ink4a/Arf Expression and Regeneration in Diabetes Mellitus. Genes Dev (2009) 23(8):975–85. doi: 10.1101/gad.1742509
98. Lu TT, Heyne S, Dror E, Casas E, Leonhardt L, Boenke T, et al. The Polycomb-Dependent Epigenome Controls Beta Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab (2018) 27(6):1294–308. doi: 10.1016/j.cmet.2018.04.013
99. Aguayo-Mazzucato C. Functional Changes in Beta Cells During Ageing and Senescence. Diabetologia (2020) 63(10):2022–9. doi: 10.1007/s00125-020-05185-6
100. Goto T, Monk M. Regulation of X-chromosome Inactivation in Development in Mice and Humans. Microbiol Mol Biol Rev (1998) 62(2):362–78. doi: 10.1128/MMBR.62.2.362-378.1998
101. Clausen JO, Borch-Johnsen K, Ibsen H, Bergman RN, Hougaard P, Winther K, et al. Insulin Sensitivity Index, Acute Insulin Response, and Glucose Effectiveness in a Population-Based Sample of 380 Young Healthy Caucasians. Analysis of the Impact of Gender, Body Fat, Physical Fitness, and Life-Style Factors. J Clin Invest (1996) 98(5):1195–209. doi: 10.1172/JCI118903
102. Basu R, Dalla Man C, Campioni M, Basu A, Klee G, Toffolo G, et al. Effects of Age and Sex on Postprandial Glucose Metabolism: Differences in Glucose Turnover, Insulin Secretion, Insulin Action, and Hepatic Insulin Extraction. Diabetes (2006) 55(7):2001–14. doi: 10.2337/db05-1692
103. Hall E, Volkov P, Dayeh T, Esguerra JL, Salo S, Eliasson L, et al. Sex Differences in the Genome-Wide DNA Methylation Pattern and Impact on Gene Expression, microRNA Levels and Insulin Secretion in Human Pancreatic Islets. Genome Biol (2014) 15(12):522. doi: 10.1186/s13059-014-0522-z
104. Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, et al. Converting Adult Pancreatic Islet Alpha Cells Into Beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab (2017) 25(3):622–34. doi: 10.1016/j.cmet.2017.01.009
105. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic Beta Cell Dedifferentiation as a Mechanism of Diabetic Beta Cell Failure. Cell (2012) 150(6):1223–34. doi: 10.1016/j.cell.2012.07.029
106. Kodani N, Nakae J, Kobayashi M, Kikuchi O, Kitamura T, Itoh H. Fcor-Foxo1 Axis Regulates Alpha-Cell Mass Through Repression of Arx Expression. iScience (2020) 23(1):100798. doi: 10.1016/j.isci.2019.100798
107. Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, et al. Fetal and Infant Growth and Impaired Glucose Tolerance At Age 64. BMJ (1991) 303(6809):1019–22. doi: 10.1136/bmj.303.6809.1019
108. Rich-Edwards JW, Colditz GA, Stampfer MJ, Willett WC, Gillman MW, Hennekens CH, et al. Birthweight and the Risk for Type 2 Diabetes Mellitus in Adult Women. Ann Intern Med (1999) 130(4 Pt 1):278–84. doi: 10.7326/0003-4819-130-4_Part_1-199902160-00005
109. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of Type 2 Diabetes Following Intrauterine Growth Retardation in Rats is Associated With Progressive Epigenetic Silencing of Pdx1. J Clin Invest (2008) 118(6):2316–24. doi: 10.1172/JCI33655
110. Jimenez-Chillaron JC, Hernandez-Valencia M, Lightner A, Faucette RR, Reamer C, Przybyla R, et al. Reductions in Caloric Intake and Early Postnatal Growth Prevent Glucose Intolerance and Obesity Associated With Low Birthweight. Diabetologia (2006) 49(8):1974–84. doi: 10.1007/s00125-006-0311-7
111. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent Epigenetic Differences Associated With Prenatal Exposure to Famine in Humans. Proc Natl Acad Sci USA (2008) 105(44):17046–9. doi: 10.1073/pnas.0806560105
112. Hales CN, Barker DJ. The Thrifty Phenotype Hypothesis. Br Med Bull (2001) 60:5–20. doi: 10.1093/bmb/60.1.5
113. Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, et al. High Prevalence of Type 2 Diabetes and Pre-Diabetes in Adult Offspring of Women With Gestational Diabetes Mellitus or Type 1 Diabetes: The Role of Intrauterine Hyperglycemia. Diabetes Care (2008) 31(2):340–6. doi: 10.2337/dc07-1596
114. Holder T, Giannini C, Santoro N, Pierpont B, Shaw M, Duran E, et al. A Low Disposition Index in Adolescent Offspring of Mothers With Gestational Diabetes: A Risk Marker for the Development of Impaired Glucose Tolerance in Youth. Diabetologia (2014) 57(11):2413–20. doi: 10.1007/s00125-014-3345-2
115. Aerts L, van Assche FA. Rat Foetal Endocrine Pancreas in Experimental Diabetes. J Endocrinol (1977) 73(2):339–46. doi: 10.1677/joe.0.0730339
116. Aerts L, Vercruysse L, Van Assche FA. The Endocrine Pancreas in Virgin and Pregnant Offspring of Diabetic Pregnant Rats. Diabetes Res Clin Pract (1997) 38(1):9–19. doi: 10.1016/S0168-8227(97)00080-6
117. Elsakr JM, Dunn JC, Tennant K, Zhao SK, Kroeten K, Pasek RC, et al. Maternal Western-style Diet Affects Offspring Islet Composition and Function in a non-Human Primate Model of Maternal Over-Nutrition. Mol Metab (2019) 25:73–82. doi: 10.1016/j.molmet.2019.03.010
118. Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab (2017) 25(1):27–42. doi: 10.1016/j.cmet.2016.08.009
119. Kintaka Y, Wada N, Shioda S, Nakamura S, Yamazaki Y, Mochizuki K. Excessive Folic Acid Supplementation in Pregnant Mice Impairs Insulin Secretion and Induces the Expression of Genes Associated With Fatty Liver in Their Offspring. Heliyon (2020) 6(4):e03597. doi: 10.1016/j.heliyon.2020.e03597
120. Henderson AM, Tai DC, Aleliunas RE, Aljaadi AM, Glier MB, Xu EE, et al. Maternal Folic Acid Supplementation With Vitamin B12 Deficiency During Pregnancy and Lactation Affects the Metabolic Health of Adult Female Offspring But is Dependent on Offspring Diet. FASEB J (2018) 32(9):5039–50. doi: 10.1096/fj.201701503RR
121. Davis CD, Uthus EO. DNA Methylation, Cancer Susceptibility, and Nutrient Interactions. Exp Biol Med (Maywood) (2004) 229(10):988–95. doi: 10.1177/153537020422901002
122. Vanderkruk B, Hoffman BG. Metabolism as a Central Regulator of Beta-Cell Chromatin State. FEBS J (2020) 1–11. doi: 10.1111/febs.15562
123. Pereda J, Perez S, Escobar J, Arduini A, Asensi M, Serviddio G, et al. Obese Rats Exhibit High Levels of Fat Necrosis and Isoprostanes in Taurocholate-Induced Acute Pancreatitis. PloS One (2012) 7(9):e44383. doi: 10.1371/journal.pone.0044383
124. Ling C, Groop L. Epigenetics: A Molecular Link Between Environmental Factors and Type 2 Diabetes. Diabetes (2009) 58(12):2718–25. doi: 10.2337/db09-1003
125. Sales VM, Ferguson-Smith AC, Patti ME. Epigenetic Mechanisms of Transmission of Metabolic Disease Across Generations. Cell Metab (2017) 25(3):559–71. doi: 10.1016/j.cmet.2017.02.016
126. Martinez D, Pentinat T, Ribo S, Daviaud C, Bloks VW, Cebria J, et al. In Utero Undernutrition in Male Mice Programs Liver Lipid Metabolism in the Second-Generation Offspring Involving Altered Lxra DNA Methylation. Cell Metab (2014) 19(6):941–51. doi: 10.1016/j.cmet.2014.03.026
127. de Castro Barbosa T, Ingerslev LR, Alm PS, Versteyhe S, Massart J, Rasmussen M, et al. High-Fat Diet Reprograms the Epigenome of Rat Spermatozoa and Transgenerationally Affects Metabolism of the Offspring. Mol Metab (2016) 5(3):184–97. doi: 10.1016/j.molmet.2015.12.002
128. Donkin I, Versteyhe S, Ingerslev LR, Qian K, Mechta M, Nordkap L, et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab (2016) 23(2):369–78. doi: 10.1016/j.cmet.2015.11.004
129. Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally Induced Transgenerational Environmental Reprogramming of Metabolic Gene Expression in Mammals. Cell (2010) 143(7):1084–96. doi: 10.1016/j.cell.2010.12.008
130. Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic High-Fat Diet in Fathers Programs Beta-Cell Dysfunction in Female Rat Offspring. Nature (2010) 467(7318):963–6. doi: 10.1038/nature09491
131. Van Assche FA, De Prins F, Aerts L, Verjans M. The Endocrine Pancreas in Small-for-Dates Infants. Br J Obstet Gynaecol (1977) 84(10):751–3. doi: 10.1111/j.1471-0528.1977.tb12486.x
132. Nicolini U, Hubinont C, Santolaya J, Fisk NM, Rodeck CH. Effects of Fetal Intravenous Glucose Challenge in Normal and Growth Retarded Fetuses. Horm Metab Res (1990) 22(8):426–30. doi: 10.1055/s-2007-1004939
133. Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y, et al. Maternal Diet and Aging Alter the Epigenetic Control of a Promoter-Enhancer Interaction At the Hnf4a Gene in Rat Pancreatic Islets. Proc Natl Acad Sci USA (2011) 108(13):5449–54. doi: 10.1073/pnas.1019007108
134. Yang BT, Dayeh TA, Volkov PA, Kirkpatrick CL, Malmgren S, Jing X, et al. Increased DNA Methylation and Decreased Expression of PDX-1 in Pancreatic Islets From Patients With Type 2 Diabetes. Mol Endocrinol (2012) 26(7):1203–12. doi: 10.1210/me.2012-1004
135. Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM. Experimental Intrauterine Growth Restriction Induces Alterations in DNA Methylation and Gene Expression in Pancreatic Islets of Rats. J Biol Chem (2010) 285(20):15111–8. doi: 10.1074/jbc.M109.095133
136. Travers ME, Mackay DJ, Dekker Nitert M, Morris AP, Lindgren CM, Berry A, et al. Insights Into the Molecular Mechanism for Type 2 Diabetes Susceptibility At the KCNQ1 Locus From Temporal Changes in Imprinting Status in Human Islets. Diabetes (2013) 62(3):987–92. doi: 10.2337/db12-0819
137. Kameswaran V, Bramswig NC, McKenna LB, Penn M, Schug J, Hand NJ, et al. Epigenetic Regulation of the DLK1-MEG3 microRNA Cluster in Human Type 2 Diabetic Islets. Cell Metab (2014) 19(1):135–45. doi: 10.1016/j.cmet.2013.11.016
138. Lucifero D, Mertineit C, Clarke HJ, Bestor TH, Trasler JM. Methylation Dynamics of Imprinted Genes in Mouse Germ Cells. Genomics (2002) 79(4):530–8. doi: 10.1006/geno.2002.6732
139. Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental Origin of Sequence Variants Associated With Complex Diseases. Nature (2009) 462(7275):868–74. doi: 10.1038/nature08625
140. Avrahami D, Li C, Yu M, Jiao Y, Zhang J, Naji A, et al. Targeting the Cell Cycle Inhibitor p57Kip2 Promotes Adult Human Beta Cell Replication. J Clin Invest (2014) 124(2):670–4. doi: 10.1172/JCI69519
141. Ding GL, Wang FF, Shu J, Tian S, Jiang Y, Zhang D, et al. Transgenerational Glucose Intolerance With Igf2/H19 Epigenetic Alterations in Mouse Islet Induced by Intrauterine Hyperglycemia. Diabetes (2012) 61(5):1133–42. doi: 10.2337/db11-1314
142. Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The Genetic Architecture of Type 2 Diabetes. Nature (2016) 536(7614):41–7. doi: 10.1038/ng.2383
143. Meigs JB, Cupples LA, Wilson PW. Parental Transmission of Type 2 Diabetes: The Framingham Offspring Study. Diabetes (2000) 49(12):2201–7. doi: 10.2337/diabetes.49.12.2201
144. Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, et al. Large-Scale Association Analysis Provides Insights Into the Genetic Architecture and Pathophysiology of Type 2 Diabetes. Nat Genet (2012) 44(9):981–90. doi: 10.1038/ng.2383
145. Gilbert ER, Liu D. Epigenetics: The Missing Link to Understanding Beta-Cell Dysfunction in the Pathogenesis of Type 2 Diabetes. Epigenetics (2012) 7(8):841–52. doi: 10.4161/epi.21238
146. Huypens P, Sass S, Wu M, Dyckhoff D, Tschop M, Theis F, et al. Epigenetic Germline Inheritance of Diet-Induced Obesity and Insulin Resistance. Nat Genet (2016) 48(5):497–9. doi: 10.1038/ng.3527
147. MacFarlane AJ, Strom A, Scott FW. Epigenetics: Deciphering How Environmental Factors may Modify Autoimmune Type 1 Diabetes. Mamm Genome (2009) 20(9-10):624–32. doi: 10.1007/s00335-009-9213-6
148. Multhaup ML, Seldin MM, Jaffe AE, Lei X, Kirchner H, Mondal P, et al. Mouse-Human Experimental Epigenetic Analysis Unmasks Dietary Targets and Genetic Liability for Diabetic Phenotypes. Cell Metab (2015) 21(1):138–49. doi: 10.1016/j.cmet.2014.12.014
149. Arnes L, Sussel L. Epigenetic Modifications and Long Noncoding RNAs Influence Pancreas Development and Function. Trends Genet (2015) 31(6):290–9. doi: 10.1016/j.tig.2015.02.008
150. Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, et al. Menin Regulates Pancreatic Islet Growth by Promoting Histone Methylation and Expression of Genes Encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci USA (2005) 102(41):14659–64. doi: 10.1073/pnas.0503484102
151. Keating ST, El-Osta A. Epigenetics and Metabolism. Circ Res (2015) 116(4):715–36. doi: 10.1161/CIRCRESAHA.116.303936
152. Lu C, Thompson CB. Metabolic Regulation of Epigenetics. Cell Metab (2012) 16(1):9–17. doi: 10.1016/j.cmet.2012.06.001
153. Sassone-Corsi P. Physiology. When Metabolism and Epigenetics Converge. Science (2013) 339(6116):148–50. doi: 10.1126/science.1233423
154. Davegardh C, Garcia-Calzon S, Bacos K, Ling C. DNA Methylation in the Pathogenesis of Type 2 Diabetes in Humans. Mol Metab (2018) 14:12–25. doi: 10.1016/j.molmet.2018.01.022
155. Ling C, Del Guerra S, Lupi R, Ronn T, Granhall C, Luthman H, et al. Epigenetic Regulation of PPARGC1A in Human Type 2 Diabetic Islets and Effect on Insulin Secretion. Diabetologia (2008) 51(4):615–22. doi: 10.1007/s00125-007-0916-5
156. Yang BT, Dayeh TA, Kirkpatrick CL, Taneera J, Kumar R, Groop L, et al. Insulin Promoter DNA Methylation Correlates Negatively With Insulin Gene Expression and Positively With HbA(1c) Levels in Human Pancreatic Islets. Diabetologia (2011) 54(2):360–7. doi: 10.1007/s00125-010-1967-6
157. Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, et al. DNA Methylation Profiling Identifies Epigenetic Dysregulation in Pancreatic Islets From Type 2 Diabetic Patients. EMBO J (2012) 31(6):1405–26. doi: 10.1038/emboj.2011.503
158. Hall E, Dayeh T, Kirkpatrick CL, Wollheim CB, Dekker Nitert M, Ling C. DNA Methylation of the Glucagon-Like Peptide 1 Receptor (GLP1R) in Human Pancreatic Islets. BMC Med Genet (2013) 14:76. doi: 10.1186/1471-2350-14-76
159. Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, et al. Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets From Type 2 Diabetic and non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion. PloS Genet (2014) 10(3):e1004160. doi: 10.1371/journal.pgen.1004160
160. Volkov P, Bacos K, Ofori JK, Esguerra JL, Eliasson L, Ronn T, et al. Whole-Genome Bisulfite Sequencing of Human Pancreatic Islets Reveals Novel Differentially Methylated Regions in Type 2 Diabetes Pathogenesis. Diabetes (2017) 66(4):1074–85. doi: 10.2337/db16-0996
161. Ouni M, Saussenthaler S, Eichelmann F, Jahnert M, Stadion M, Wittenbecher C, et al. Epigenetic Changes in Islets of Langerhans Preceding the Onset of Diabetes. Diabetes (2020) 69(11):2503–17. doi: 10.2337/db20-0204
162. Zhang FF, Liu YH, Wang DW, Liu TS, Yang Y, Guo JM, et al. Obesity-Induced Reduced Expression of the Lncrna ROIT Impairs Insulin Transcription by Downregulation of Nkx6. 1 Methylation Diabetol (2020) 63(4):811–24. doi: 10.1007/s00125-020-05090-y
163. Stefan M, Zhang W, Concepcion E, Yi Z, Tomer Y. DNA Methylation Profiles in Type 1 Diabetes Twins Point to Strong Epigenetic Effects on Etiology. J Autoimmun (2014) 50:33–7. doi: 10.1016/j.jaut.2013.10.001
164. Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, et al. Heterogeneity of Type I Diabetes: Analysis of Monozygotic Twins in Great Britain and the United States. Diabetologia (2001) 44(3):354–62. doi: 10.1007/s001250051626
165. Jerram ST, Dang MN, Leslie RD. The Role of Epigenetics in Type 1 Diabetes. Curr Diabetes Rep (2017) 17(10):89. doi: 10.1007/s11892-017-0916-x
166. Cerna M. Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus. Int J Mol Sci (2019) 21(1):1–17. doi: 10.3390/ijms21010036
167. Jahromi MM, Eisenbarth GS. Genetic Determinants of Type 1 Diabetes Across Populations. Ann N Y Acad Sci (2006) 1079:289–99. doi: 10.1196/annals.1375.044
168. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of Type 1 Diabetes-Associated DNA Methylation Variable Positions That Precede Disease Diagnosis. PloS Genet (2011) 7(9):e1002300. doi: 10.1371/journal.pgen.1002300
169. Elboudwarej E, Cole M, Briggs FB, Fouts A, Fain PR, Quach H, et al. Hypomethylation Within Gene Promoter Regions and Type 1 Diabetes in Discordant Monozygotic Twins. J Autoimmun (2016) 68:23–9. doi: 10.1016/j.jaut.2015.12.003
170. Rui J, Deng S, Lebastchi J, Clark PL, Usmani-Brown S, Herold KC. Methylation of Insulin DNA in Response to Proinflammatory Cytokines During the Progression of Autoimmune Diabetes in NOD Mice. Diabetologia (2016) 59(5):1021–9. doi: 10.1007/s00125-016-3897-4
171. Akirav EM, Lebastchi J, Galvan EM, Henegariu O, Akirav M, Ablamunits V, et al. Detection of Beta Cell Death in Diabetes Using Differentially Methylated Circulating DNA. Proc Natl Acad Sci USA (2011) 108(47):19018–23. doi: 10.1073/pnas.1111008108
172. Herold KC, Usmani-Brown S, Ghazi T, Lebastchi J, Beam CA, Bellin MD, et al. Beta Cell Death and Dysfunction During Type 1 Diabetes Development in At-Risk Individuals. J Clin Invest (2015) 125(3):1163–73. doi: 10.1172/JCI78142
173. Fisher MM, Watkins RA, Blum J, Evans-Molina C, Chalasani N, DiMeglio LA, et al. Elevations in Circulating Methylated and Unmethylated Preproinsulin DNA in New-Onset Type 1 Diabetes. Diabetes (2015) 64(11):3867–72. doi: 10.2337/db15-0430
174. Olsen JA, Kenna LA, Spelios MG, Hessner MJ, Akirav EM. Circulating Differentially Methylated Amylin DNA as a Biomarker of Beta-Cell Loss in Type 1 Diabetes. PloS One (2016) 11(4):e0152662. doi: 10.1371/journal.pone.0152662
175. Stefan-Lifshitz M, Karakose E, Cui L, Ettela A, Yi Z, Zhang W, et al. Epigenetic Modulation of Beta Cells by Interferon-Alpha Via PNPT1/mir-26a/TET2 Triggers Autoimmune Diabetes. JCI Insight (2019) 4(5):1–21. doi: 10.1172/jci.insight.126663
176. Rui J, Deng S, Ponath G, Kursawe R, Lawlor N, Sumida T, et al. Tet2 Controls β Cells Responses to Inflammation in Type 1 Diabetes. BioRxiv (2020) 1–49. doi: 10.1101/2020.09.01.278028
177. Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, et al. Reduced Insulin Exocytosis in Human Pancreatic Beta-Cells With Gene Variants Linked to Type 2 Diabetes. Diabetes (2012) 61(7):1726–33. doi: 10.2337/db11-1516
178. Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, et al. Genome-Wide Survey Reveals Predisposing Diabetes Type 2-Related DNA Methylation Variations in Human Peripheral Blood. Hum Mol Genet (2012) 21(2):371–83. doi: 10.1093/hmg/ddr472
179. Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The Imprinted DLK1-MEG3 Gene Region on Chromosome 14q32.2 Alters Susceptibility to Type 1 Diabetes. Nat Genet (2010) 42(1):68–71. doi: 10.1038/ng.493
180. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of Specific Beta Cell Transcription Factors in Type 2 Diabetes. J Clin Invest (2013) 123(8):3305–16. doi: 10.1172/JCI65390
181. Reddy MA, Zhang E, Natarajan R. Epigenetic Mechanisms in Diabetic Complications and Metabolic Memory. Diabetologia (2015) 58(3):443–55. doi: 10.1007/s00125-014-3462-y
182. Meier JJ, Breuer TG, Bonadonna RC, Tannapfel A, Uhl W, Schmidt WE, et al. Pancreatic Diabetes Manifests When Beta Cell Area Declines by Approximately 65% in Humans. Diabetologia (2012) 55(5):1346–54. doi: 10.1007/s00125-012-2466-8
183. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-Free DNA Comprises an In Vivo Nucleosome Footprint That Informs its Tissues-Of-Origin. Cell (2016) 164(1-2):57–68. doi: 10.1016/j.cell.2015.11.050
184. Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive Human Cell-Type Methylation Atlas Reveals Origins of Circulating Cell-Free DNA in Health and Disease. Nat Commun (2018) 9(1):5068. doi: 10.1038/s41467-018-07466-6
185. Husseiny MI, Kaye A, Zebadua E, Kandeel F, Ferreri K. Tissue-Specific Methylation of Human Insulin Gene and PCR Assay for Monitoring Beta Cell Death. PloS One (2014) 9(4):e94591. doi: 10.1371/journal.pone.0094591
186. Neiman D, Gillis D, Piyanzin S, Cohen D, Fridlich O, Moss J, et al. Multiplexing DNA Methylation Markers to Detect Circulating Cell-Free DNA Derived From Human Pancreatic Beta Cells. JCI Insight (2020) 5(14):1–15. doi: 10.1172/jci.insight.136579
187. van Otterdijk SD, Binder AM, Szarc Vel Szic K, Schwald J, Michels KB. DNA Methylation of Candidate Genes in Peripheral Blood From Patients With Type 2 Diabetes or the Metabolic Syndrome. PloS One (2017) 12(7):e0180955. doi: 10.1371/journal.pone.0180955
188. Canivell S, Ruano EG, Siso-Almirall A, Kostov B, Gonzalez-de Paz L, Fernandez-Rebollo E, et al. Differential Methylation of TCF7L2 Promoter in Peripheral Blood DNA in Newly Diagnosed, Drug-Naive Patients With Type 2 Diabetes. PloS One (2014) 9(6):e99310. doi: 10.1371/journal.pone.0099310
189. Dayeh T, Tuomi T, Almgren P, Perfilyev A, Jansson PA, de Mello VD, et al. DNA Methylation of Loci Within ABCG1 and PHOSPHO1 in Blood DNA is Associated With Future Type 2 Diabetes Risk. Epigenetics (2016) 11(7):482–8. doi: 10.1080/15592294.2016.1178418
190. Chambers JC, Loh M, Lehne B, Drong A, Kriebel J, Motta V, et al. Epigenome-Wide Association of DNA Methylation Markers in Peripheral Blood From Indian Asians and Europeans With Incident Type 2 Diabetes: A Nested Case-Control Study. Lancet Diabetes Endocrinol (2015) 3(7):526–34. doi: 10.1016/S2213-8587(15)00127-8
191. Soriano-Tarraga C, Jimenez-Conde J, Giralt-Steinhauer E, Mola-Caminal M, Vivanco-Hidalgo RM, Ois A, et al. Epigenome-Wide Association Study Identifies TXNIP Gene Associated With Type 2 Diabetes Mellitus and Sustained Hyperglycemia. Hum Mol Genet (2016) 25(3):609–19. doi: 10.1093/hmg/ddv493
192. Paul DS, Teschendorff AE, Dang MA, Lowe R, Hawa MI, Ecker S, et al. Increased DNA Methylation Variability in Type 1 Diabetes Across Three Immune Effector Cell Types. Nat Commun (2016) 7:13555. doi: 10.1080/15592294.2016.1178418
193. Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, Schones DE, et al. Epigenomic Profiling Reveals an Association Between Persistence of DNA Methylation and Metabolic Memory in the DCCT/EDIC Type 1 Diabetes Cohort. Proc Natl Acad Sci USA (2016) 113(21):E3002–11. doi: 10.1073/pnas.1603712113
194. Hwang JY, Lee HJ, Go MJ, Jang HB, Choi NH, Bae JB, et al. Genome-Wide Methylation Analysis Identifies ELOVL5 as an Epigenetic Biomarker for the Risk of Type 2 Diabetes Mellitus. Sci Rep (2018) 8(1):14862. doi: 10.1038/s41598-018-33238-9
195. Ronn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, et al. Impact of Age, BMI and HbA1c Levels on the Genome-Wide DNA Methylation and mRNA Expression Patterns in Human Adipose Tissue and Identification of Epigenetic Biomarkers in Blood. Hum Mol Genet (2015) 24(13):3792–813. doi: 10.1093/hmg/ddv124
196. Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, et al. Blood-Based Biomarkers of Age-Associated Epigenetic Changes in Human Islets Associate With Insulin Secretion and Diabetes. Nat Commun (2016) 7:11089. doi: 10.1038/ncomms11089
197. Willmer T, Johnson R, Louw J, Pheiffer C. Blood-Based DNA Methylation Biomarkers for Type 2 Diabetes: Potential for Clinical Applications. Front Endocrinol (Lausanne) (2018) 9:744. doi: 10.3389/fendo.2018.00744
198. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating Endocrine Cell Clustering in Culture Promotes Maturation of Human Stem-Cell-Derived Beta Cells. Nat Cell Biol (2019) 21(2):263–74. doi: 10.1038/s41556-018-0271-4
199. Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The Timeline of Epigenetic Drug Discovery: From Reality to Dreams. Clin Epigenet (2019) 11(1):174. doi: 10.1186/s13148-019-0776-0
200. Bottiglieri T. S-Adenosyl-L-methionine (Same): From the Bench to the Bedside–Molecular Basis of a Pleiotrophic Molecule. Am J Clin Nutr (2002) 76(5):1151S–7S. doi: 10.1093/ajcn/76.5.1151S
201. Strickland KC, Krupenko NI, Krupenko SA. Molecular Mechanisms Underlying the Potentially Adverse Effects of Folate. Clin Chem Lab Med (2013) 51(3):607–16. doi: 10.1515/cclm-2012-0561
202. Cho CE, Sanchez-Hernandez D, Reza-Lopez SA, Huot PS, Kim YI, Anderson GH. High Folate Gestational and Post-Weaning Diets Alter Hypothalamic Feeding Pathways by DNA Methylation in Wistar Rat Offspring. Epigenetics (2013) 8(7):710–9. doi: 10.4161/epi.24948
203. Manzar GS, Kim EM, Zavazava N. Demethylation of Induced Pluripotent Stem Cells From Type 1 Diabetic Patients Enhances Differentiation Into Functional Pancreatic Beta Cells. J Biol Chem (2017) 292(34):14066–79. doi: 10.1074/jbc.M117.784280
204. Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA Methylation in the Mammalian Genome. Cell (2016) 167(1):233–47. e17. doi: 10.1016/j.cell.2016.08.056
205. Ou K, Yu M, Moss NG, Wang YJ, Wang AW, Nguyen SC, et al. Targeted Demethylation At the CDKN1C/p57 Locus Induces Human Beta Cell Replication. J Clin Invest (2019) 129(1):209–14. doi: 10.1172/JCI99170
206. Sneddon JB, Tang Q, Stock P, Bluestone JA, Roy S, Desai T, et al. Stem Cell Therapies for Treating Diabetes: Progress and Remaining Challenges. Cell Stem Cell (2018) 22(6):810–23. doi: 10.1016/j.stem.2018.05.016
207. Cedar H, Bergman Y. Linking DNA Methylation and Histone Modification: Patterns and Paradigms. Nat Rev Genet (2009) 10(5):295–304. doi: 10.1038/nrg2540
208. Rose NR, Klose RJ. Understanding the Relationship Between DNA Methylation and Histone Lysine Methylation. Biochim Biophys Acta (2014) 1839(12):1362–72. doi: 10.1016/j.bbagrm.2014.02.007
Keywords: diabetes, beta cells, insulin, DNA methylation, epigenetics
Citation: Parveen N and Dhawan S (2021) DNA Methylation Patterning and the Regulation of Beta Cell Homeostasis. Front. Endocrinol. 12:651258. doi: 10.3389/fendo.2021.651258
Received: 09 January 2021; Accepted: 21 April 2021;
Published: 07 May 2021.
Edited by:
Magalie A. Ravier, INSERM U1191 Institut de Génomique Fonctionnelle (IGF), FranceReviewed by:
Jeffery Sivert Tessem, Brigham Young University, United StatesAmna Khamis, UMR8199 Génomique intégrative et modélisation des maladies métaboliques, France
Christine Doucette, University of Manitoba, Canada
Copyright © 2021 Parveen and Dhawan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangeeta Dhawan, sdhawan@coh.org