 Konrad Patyra1,2†Kristiina Makkonen1,2†Maria Haanpää3,4Sinikka Karppinen5Liisa Viikari5
Konrad Patyra1,2†Kristiina Makkonen1,2†Maria Haanpää3,4Sinikka Karppinen5Liisa Viikari5 Jorma Toppari1,2,5
Jorma Toppari1,2,5 Mary Pat Reeve6
Mary Pat Reeve6 Jukka Kero1,2,5*
Jukka Kero1,2,5*- 1Research Centre for Integrative Physiology and Pharmacology, Institute of Biomedicine, University of Turku, Turku, Finland
- 2Turku Center for Disease Modeling, University of Turku, Turku, Finland
- 3Department of Genomics and Clinical Genetics, Turku University Hospital, Turku, Finland
- 4Department of Genetics, University of Turku, Turku, Finland
- 5Department of Pediatrics, Turku University Hospital, Turku, Finland
- 6Institute for Molecular Medicine Finland, HiLIFE, University of Helsinki, Helsinki, Finland
Background: Central hypothyroidism (CeH) is a rare condition affecting approximately 1:16 000- 100 000 individuals. Congenital forms can harm normal development if not detected and treated promptly. Clinical and biochemical diagnosis, especially of isolated CeH, can be challenging. Cases are not usually detected in neonatal screening, which, in most countries, is focused on detection of the more prevalent primary hypothyroidism. Until now, five genetic causes for isolated CeH have been identified. Here we aimed to identify the genetic cause in two brothers with impaired growth diagnosed with CeH at the age of 5 years. We further evaluated the candidate gene variants in a large genetic database.
Methods: Clinical and biochemical characterization together with targeted next-generation sequencing (NGS) was used to identify the genetic cause in a family of two brothers presenting with CeH. Screening of insulin receptor substrate 4 (IRS4) variants was carried out in the FinnGen database.
Results: A novel monoallelic frameshift mutation c.1712_1713insT, p.Gly572Trp fs*32 in the X-linked IRS4 gene was identified by NGS analysis in both affected males and confirmed using Sanger sequencing. Their mother was an unaffected carrier. In addition to the declined growth at presentation, central hypothyroidism and blunted TRH test, no other phenotypic alterations were found. Diagnostic tests included head MRI, thyroid imaging, bone age, and laboratory tests for thyroid autoantibodies, glucose, insulin and glycosylated hemoglobin levels. Examination of the IRS4 locus in FinnGen (R5) database revealed the strongest associations to a rare Finnish haplotype associated with thyroid disorders (p = 1.3e-7) and hypothyroidism (p = 8.3e-7).
Conclusions: Here, we identified a novel frameshift mutation in an X-linked IRS4 gene in two brothers with isolated CeH. Furthermore, we demonstrate an association of IRS4 gene locus to a general thyroid disease risk in the FinnGen database. Our findings confirm the role of IRS4 in isolated central hypothyroidism.
Introduction
Central hypothyroidism (CeH) is defined as a reduced thyroid hormone secretion from an otherwise functional thyroid gland due to diminished stimulation of the gland. Reduced stimulation can result either from an impaired secretion of thyroid stimulating hormone (TSH) from the anterior pituitary, defective secretion or action of hypothalamic thyrotropin releasing hormone (TRH), or both (1).
Central hypothyroidism is rarely an isolated defect, but most often appears congenitally as a part of panhypopituitarism affecting also gonadotropin, adrenocorticotrophic hormone (ACTH) or growth hormone secretion. Panhypopituitarism can be potentially life-threatening, primarily because of severe hypoglycemia. The exact prevalence of CeH is unknown, but the presentation of genetic forms shows a peak in childhood, whereas other forms due to pituitary lesions are typical later in adulthood (2). Estimations of CeH prevalence vary between 1:16 000-100 000 individuals (3, 4). Overall, CeH has been reported to be equally distributed in both sexes. However, known X-linked forms suggest a male predominance (2). In addition to the genetic etiology of isolated central hypothyroidism, impairing only TSH secretion, CeH has been described in patients with pituitary tumors, trauma, radiation therapy, diabetes mellitus or it may be idiopathic (5). Genetic defects in pituitary transcription factors can lead to CeH, but are usually associated also with other hormonal defects (1). Isolated TSH deficiency covers approximately 20% of all CeH cases (2). Inherited isolated CeH has been demonstrated to be caused by mutations in thyroid releasing hormone receptor (TRHR), thyroid stimulating hormone beta subunit (TSHB), immunoglobulin superfamily member 1 (IGSF1), transducin (beta)-like 1X-linked (TBL1X) and more recently, in insulin receptor substrate 4 (IRS4) genes (6). Among these genes TRHR mutations has been shown to lead to blunted TSH response to TRH, growth retardation and obesity during childhood (7). Furthermore, TSHB mutations are characterized by neonatal onset of low TSH, high glycoprotein hormone alpha-subunit levels and pituitary hyperplasia, which is reversible with thyroxine treatment (8). Mutations in both IGSF1 and TBL1X can lead to X-linked isolated CeH, but IGSF1 mutations are also associated with low PRL, variable GH deficiency, metabolic syndrome, and postpubertal macroorchidism (9). In addition, the mutations in the TBL1X can also lead to impaired hearing (10).
Here we describe a genetic, biochemical and clinical characterization of a family with two brothers diagnosed with CeH at the age of 5 years and shown to carry a novel frameshift mutation in the IRS4 gene. Furthermore, we evaluate the overall occurrence of IRS4 variants and their association to other clinical phenotypes in a large national genetic database.
Materials and Methods
Study Participants
The study participants were recruited to the study by a pediatric endocrinologist, and they and their parents signed a written consent. The Ethics Committee of the Hospital District of Southwest Finland approved the study (108/180/2010).
The clinical examinations were performed by a pediatrician (SK) and thyroid ultrasound, head MRI and bone age by the pediatric radiologists. Laboratory tests were done at the Turku University Hospital Laboratory, except for the IGF-1 test, which was performed in the Islab-laboratory (Kuopio, Finland). Umbilical serum TSH (uS-TSH), serum TSH, free T4 (fT4), insulin and cortisol concentrations were determined with the Cobas e801 immunoassay analyzer (Roche Diagnostics, Rotkreuz, Switzerland). Serum cholesterol (HDL cholesterol, LDL cholesterol and triglycerides) were determined with the Cobas c702 chemistry analyzer (Roche Diagnostics). Serum glycosylated hemoglobin (HbA1c) was determined with the Cobas c501 immunoturbidimetric assay (Roche Diagnostics). Serum IGF-1 concentrations were determined with the Liaison XL chemiluminescence analyzer (DiaSorin S.p.A, Saluggia, Italy). Growth data were collected from the hospital records based on measurements performed at the visits using stabilized and calibrated scale and wall mounted Harpenden Stadiometer (Holtain Limited, Crosswell, Crymych, Pembs., UK) with ±0.1 cm precision.
TRH Stimulation Test
At the start of the TRH stimulation test, non-fasting serum TSH concentrations were measured. A bolus of 7 μg/kg of TRH (Ferring Pharmaceuticals, Saint-Prex, Switzerland) was given intravenously, and subsequently the serum TSH concentrations were measured at 20 and 60 minutes.
Genetic Analysis
Genetic analyses were performed on DNA extracted from peripheral blood. Amplification of target region was performed with PCR using AmpliTaq Gold 360 (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol in the Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA, USA). Primers used are listed in the Supplemental Table 2. For the index patient an NGS-based targeted panel was performed with Sophia Genetics custom clinical exome solution (Sophia Genetics, Boston, MA, USA) and Illumina sequencing (Illumina, San Diego, CA, USA), including 4400 known disease-causing genes. NGS libraries were prepared using a hybrid capture method according to the manufacturer´s protocol (Sophia Genetics CCE_A_v1). DNA was sequenced with NextSeq sequencer (Illumina, San Diego, CA, USA) using 2x151bp paired-end technique. The identified variation was visually inspected using the Integrative Genomics Viewer (11). Bioinformatic analysis and annotation was focused on the following candidate gene panel for CeH: HESX1, IGSF1, IRS4, LEPR, LHX3, LHX4, OTX2, POU1F1, PROP1, SOX3, TBL1X, TRHR and TSHB.
Confirmation of the IRS4 mutation and its segregation was tested with PCR and Sanger sequencing. The primer sequences and PCR conditions are listed in the Supplementary Materials. Sequencing reactions were performed by using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequencing was performed with the ABI3500xl Dx (Applied Biosystems) and chromatograms were analyzed using Sequencher v5. (Gene Codes Corporation, MI, USA). The alignment of WT and mutated IRS4 sequences was performed using the Clustal O (1.2.4) multiple sequence alignment tool (12).
IRS4 Variant Analysis in the FinnGen Database
The FinnGen project has been approved by the Ethical Review Board of the Hospital District of Helsinki and Uusimaa with the protocol Nr. HUS/990/2017. The FinnGen data release 5 was used. Detailed information about of the different releases is described on the FinnGen’s website1. Release 5 comprises of data from 218 792 Finnish participants with disease endpoints2 constructed from national registries using International Classification of Diseases (ICD), Social Insurance Institute (KELA) drug reimbursement and ATC codes linked with DNA data.
Results
Clinical Characteristics
The two male subjects were referred to the endocrinologist due to mild growth retardation and normal TSH, but low serum fT4 concentrations. They were both diagnosed with CeH between the age of 5 – 6 years. The index case (patient #1) and his brother (patient #2) had normal serum TSH and low fT4 values prior the diagnosis (Figures 1A, B). Both cases had blunted TSH response (ΔTSH, <1.6) to TRH stimulation (Figure 1D), no interfering antibodies in TSH or TH assays, and the TG or TPO antibody tests were negative (Figure 1A, table). Secretion defects or dysfunction of other pituitary hormones were excluded (Supplemental Table 1). Both MRI of the head and ultrasound evaluation of the thyroid were normal in both cases at the time of diagnosis. Patient #1 had linear growth until the age of 2 years, after which his height standard deviation scores (SDS) decreased from –1 to –2.4 SD between the age 2 and 5 years (Figure 1E). His brother (#2) had similar growth retardation prior to the CeH diagnosis. Both had delayed bone age (2.0 - 2.3 and 0.7 years behind the calendar age) at the time of diagnosis. There were no significant alterations in other biochemical or metabolic variables measured including glucose, glycosylated hemoglobin (HbA1c), insulin, insulin-like growth factor-1, cholesterol or cortisol levels (Supplemental Table 1). The thyroxine replacement was started promptly after CeH diagnosis and fT4 levels returned to normal. Furthermore, the serum TSH concentrations, although within the normal range at diagnosis, decreased significantly in both cases after thyroxine supplementation. A small increase in growth velocity was also noted after the initiation of thyroxine treatment (Figure 1E).
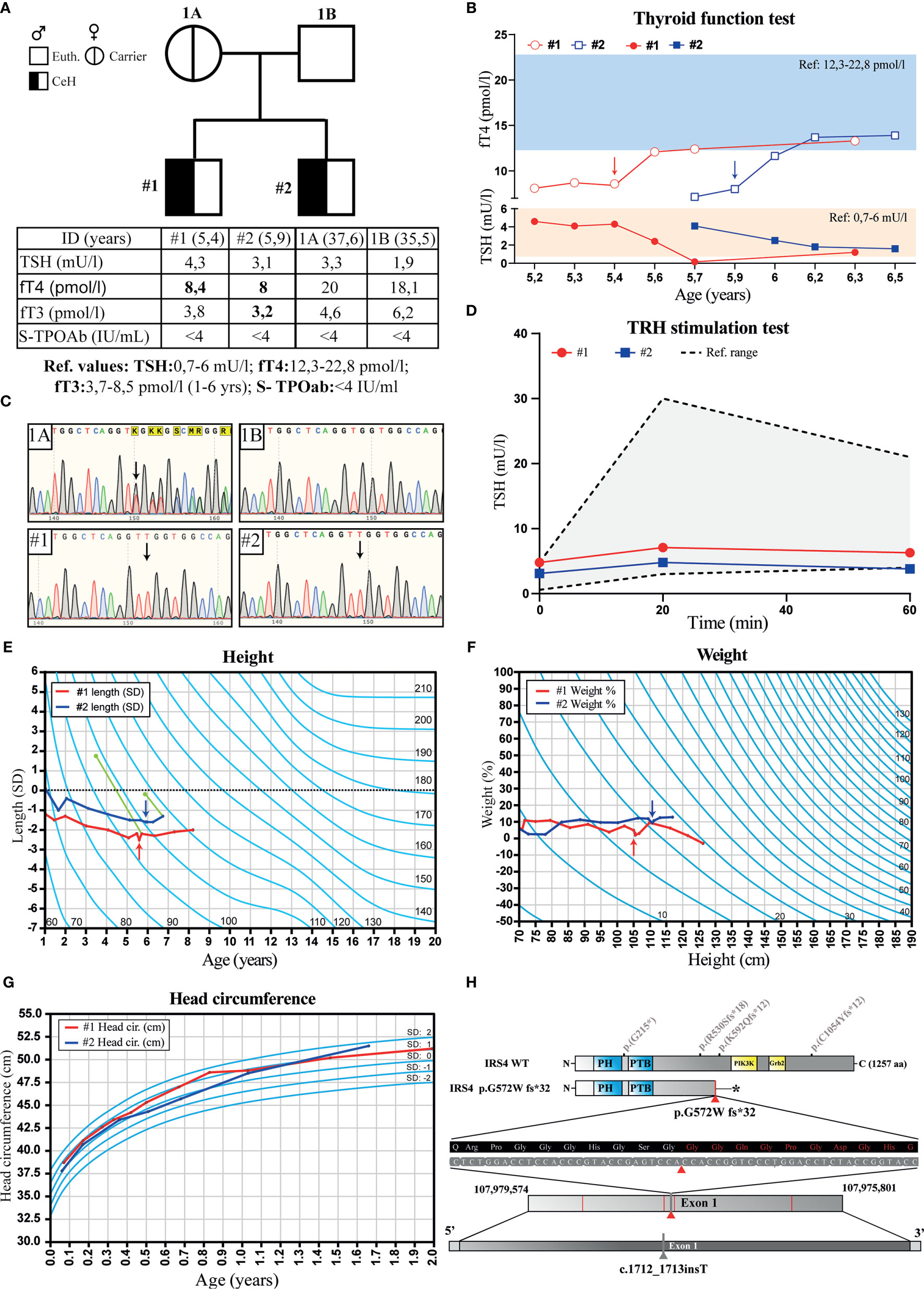
Figure 1 Pedigree, thyroid function tests, growth charts and chromatograms of the family with two brothers diagnosed with isolated central hypothyroidism (CeH). (A) Pedigree, serum TSH, fT4, fT3 and TPO antibody (S-TPOAb) concentrations in parents and their offspring. Bolded values indicate concentration below the reference range. (B) Follow-up graph of the serum TSH and fT4 concentrations in the affected cases before and after the thyroxin replacement therapy. Blue and orange rectangles show fT4 and TSH reference ranges, respectively. (C) Chromatograms of the IRS4 sequence flanking the mutation in mother (1A), father (1B), and the two patients #1 and #2 presenting CeH. Black arrows show the position of thymidine insertion. (D) TSH response in TRH stimulation test of the affected cases, gray area indicates a range of normal TSH response. (E) Growth (height SD) and (F) weight (%) curves of the affected siblings: #1 (red) and #2 (blue). Arrows indicate the time of CeH diagnosis and the start of thyroxine treatment. The green dots and lines show bone age determined at the indicated calendar age using the Tanner-Whitehouse method, and dotted line shows the expected length calculated from the parents´ heights. (G) Head circumference in affected siblings carrying IRS4 mutation during the first 2 years. Light blue lines show SD values. (H) Location of the IRS4 frameshift mutation detected in this study (marked with red triangle), premature stop codon (indicated with star) previously published (6) IRS4 mutations (marked with red lines). The location of IRS4 gene shown is based on the GRCh37.p13 primary assembly.
Both brothers were born at term after a normal pregnancy and uncomplicated birth with >9 APGAR-scores. However, patient #1 received phototherapy for prolonged jaundice as a newborn. The birth weight, height and head circumferences were within average Finnish standards. Patients #1 and #2 had normal umbilical serum TSH levels (patient #1: 11 mU/l and patient #2: 6.6 mU/l) measured at birth as a part of the CH screening program. Both brothers reached all developmental milestones, had normal weight gain and head growth pattern during the first two years (Figures 1F, G), normal weight gain and development and no further diagnoses. Both parents were healthy, had thyroid function tests (TSH, fT4, fT3) in normal range, and negative TPO and Tg antibody tests, when measured at the recruitment visit. There was no positive family history for thyroid disease.
Genetic Findings
A novel frameshift variant was detected in hemizygous state in exon 1 of the IRS4 gene located on the X chromosome (X:107977861; NM_003604.2) of the affected male proband (Figure 1H). The detected variant had a 1bp insertion (c.1712_1713insT), which leads to a frameshift mutation (p.Gly572Trp fs*32, amino acid sequence listed in Supplemental Figure 1), premature stop codon and strongly truncated protein (Figure 1H). This variant most likely degrades through nonsense-mediated decay and is not present in the gnomAD and dbSNP databases. Confirmation and co-segregation of the mutant in the family was performed using Sanger sequencing. The same variant was also found in the DNA of the affected brother and the mother, who was a healthy carrier (Figures 1A, C). No other pathogenic variants associated with CeH were detected in the clinical exome.
Exploring IRS4 Mutations in the FinnGen Study
To explore the role of IRS4 in both hypothyroidism and across the medical spectrum, we used the large FinnGen population study which in release 5 had integrated genome-wide genotyping and extensive medical history data from 218 792 Finns. We searched for any rare loss-of-function (LoF) or damaging missense mutations (Table 1) and found no LoFs. Of note, the only rare predicted damaging missense variant observed (rs766893547, p.Arg8His) was carried by two female congenital hypothyroidism cases (an enrichment odds ratio of 6.7 compared to all females, p = 0.03). Furthermore, another variant (rs1801164) had significant association to renal failure (Supplemental Figure 2).
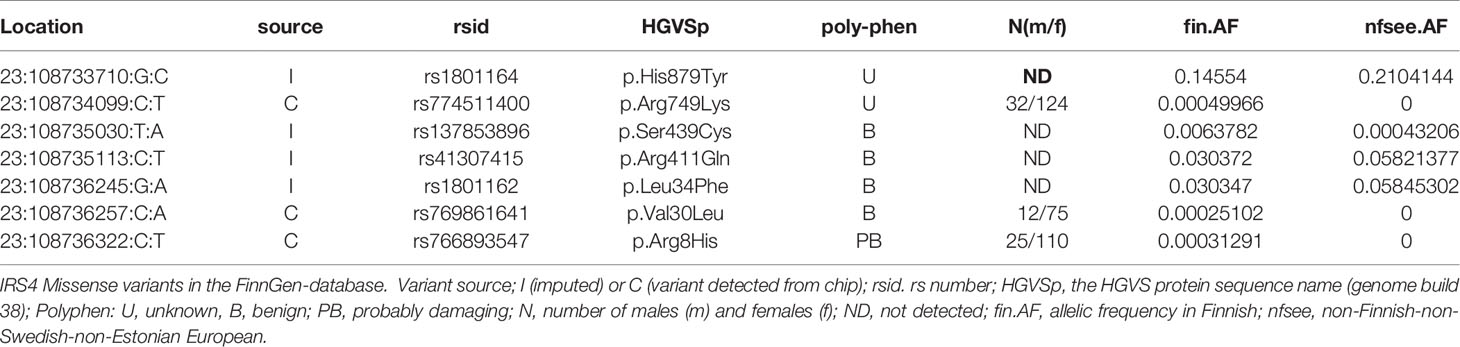
Table 1 IRS4 missense variants in the FinnGen-database.
We then surveyed the IRS4 locus to see if any medical phenotypes might be associated to this genomic region. This was done by evaluating the DNA variants at the IRS4 locus in FinnGen participants and comparing those to disease phenotypes2 obtained from national registries linked with the DNA data. Across all 2925 phenotypes studied in the FinnGen project, the strongest associations discovered were with thyroid disorders (p = 1.3x10-7) and hypothyroidism (p = 8.3x10-7), which were associated to a rare Finnish-enriched haplotype (tagged by SNP rs1452561670 20 kb downstream of IRS4) (Supplemental Figure 2). This haplotype spans a roughly 500 kb interval within which IRS4 is the only documented protein-coding gene.
Discussion
Isolated central hypothyroidism (CeH) in children is rare and challenging to diagnose clinically. It has a multifactorial etiology and a small proportion of cases that are due to gene mutations. Therefore, the etiology for several cases remains unsolved. Currently, mutations in five different genes have been shown to associate with CeH. In this study, we describe a genetic, clinical and biochemical characterization of a family with two brothers presenting CeH at the age of 5 years. Furthermore, an evaluation of candidate gene variants for isolated CeH and their putative association to other phenotypes in a national genetic database was performed. In the present study, both affected cases with a novel frameshift mutation in an X-linked IRS4 gene were shown to have normal umbilical TSH measured at newborn screening and normal psychomotor development. However, their growth started to slowly decline 2-3 years prior to the diagnosis of CeH, which prompted the investigations. The TSH response was blunted in the TRH-test and TSH levels declined from mid-normal to low- normal after start of the thyroxine replacement. The mother was a healthy carrier with normal TSH and also fT4 concentrations at the upper normal range. The data from this family supports the pathogenic role of IRS4 in isolated central hypothyroidism and the cases showed no obvious additional phenotypes. Furthermore, the candidate gene analysis using the FinnGen database showed an association between the IRS4 locus and thyroxine purchases.
So far, only one study has reported the association of CeH with pathogenic mutations in IRS4 (6). Heinen et al. described five families and seven affected male patients with four different mutations in the IRS4 gene. Most of those patients were detected at birth via neonatal screening which can identify both primary and CeH (13). Unfortunately, CeH is not usually detected in congenital hypothyroidism (CH) screening programmes, which are generally targeted to detect the more prevalent primary CH using TSH-based screening (4, 14). However, in some countries with T4-based screening programmes CeH can be detected at birth and potential neurodevelopmental disabilities can be therefore prevented. The importance of such screening was recently demonstrated in a study by Lanting et al. (15), which showed a relatively high (1 in 16 404) occurrence of CeH. Moreover, this finding is also supported by the fact that the clinical recognition of isolated CeH can be challenging without the classic symptoms, such as hypoglycemia, jaundice and micropenis observed typically in panhypothyroidism (2). Thus the debate continues whether the screening should also detect central CH continues (15, 16). In Finland, TSH-based screening is used and the CeH cases have not been routinely detected (17). The two cases described in our study, show that the impaired IRS4 function does not necessary lead to severe CH, as the boys´ development, growth and head growth for the first 2 years was normal. Similarly, in the study from Heinen et al. (6) they report a male case diagnosed with CeH at the age of 12, originally evaluated because of short stature and delayed tooth eruption but otherwise normal development. This suggests that a significant degree of compensation can occur either in the regulation of the growth during the infancy and childhood phase or at the thyroid axis level. In fact, the fT3 levels in our patients were normal or only slightly below the reference range, and normal among the patients described in the previous study (6). In the previously described IRS4 mutation cases the TSH response to the TRH-test was blunted in six of seven male IRS4 mutation carriers, similar to that seen in our patients. The value of the TRH-test in the diagnosis of CeH with multiple etiologies has been shown to be controversial (18, 19), and the response can be affected by multiple factors such as initial TSH level, age, weight or nutritional state (20–22). However, the blunted TSH response seen in our CeH cases and the previously described IRS4 mutation carriers suggest that the IRS4 is needed for the proper signaling of the TRH. The IRS4 gene codes for a cytoplasmic protein which interacts with tyrosine kinase receptors and mediates their signaling (23). It has been shown to be expressed in the hypothalamus, but also has been found in several other tissues including the pituitary, thyroid and ovaries (10). IRS4 knock-out mice exhibit mild metabolic differences including lower blood glucose levels, impaired glucose tolerance and decreased fertility (24). Additionally, IRS4 female knockout mice have decreased Tshb mRNA expression in the pituitary, but no altered serum TSH or thyroid hormone concentrations. In contrast, the lack of IRS4 function in humans impairs TSH pulsatile secretion but the detailed mechanisms remain unclear. Impaired TSH pulsatile secretion may be linked to leptin action (6) that has been shown to participate in TRH response (25) and its action is partially mediated via IRS proteins.
A limitation of our study is the lack of functional test of the mutation pathogenicity. However, IRS4 mutation segregation in the family, the frameshift alteration leading to premature stop codon strongly support the pathogenicity of the mutation. Since the fine regulation of thyroid function via hypothalamus and pituitary seems to differ between human and mice (6) further functional studies, for example using induced human pluripotent pituitary cell lines, are warranted to elucidate the detailed mechanisms.
Using FinnGen data we could not identify new LoF mutations. However, one rare predicted damaging missense variant (rs766893547, p.Arg8His) was present in two females with a congenital hypothyroidism diagnosis. Furthermore, an association of the variant rs1452561670 to the thyroid endpoint category was observed. Although these associations include cases with central isolated hypothyroidism (ICD code E23.05), they may also indicate primary thyroid diseases.
To our knowledge, general association of the IRS4 variants to thyroid diseases has not been previously described in GWAS studies or genetic databases. However, our observation of a Finnish-enriched haplotype spanning IRS4 would not be detectable outside of a large Finnish study. This haplotype spans a roughly 500 kb interval within which IRS4 is the only documented protein-coding gene.
In addition to the association to thyroid endpoint category in FinnGen one IRS4 missense variant (rs1801164) had a significant association to renal failure. However, no renal phenotypes were described in humans with IRS4 frameshift mutations in the previous publication (6). Furthermore, in our study the creatinine values were normal and there was no sign of kidney dysfunction in the individuals with IRS4 mutation and the family history was negative for any kidney diseases. Therefore, the present clinical findings do not support this database association at least in young individuals.
In summary, our data from patients with a novel frameshift mutation in IRS4 gene together with the observed association between the rare IRS4 haplotype and thyroid disease risk supports the pathogenic role of IRS4 in isolated central hypothyroidism.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ebi.ac.uk/eva/, PRJEB45041.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of the Hospital District of Southwest Finland (108/180/2010). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The study was supported by the Finnish Cultural and Sigrid Juselius (JK) Foundation (KP), Finnish Pediatric Foundation (JK) and a grant from Turku University graduate school (KM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Minna Toivonen, Pia Pohjola and Matilda Kuusi for the help with laboratory and genetic analysis, and Andreina Kero for the proofreading this manuscript and comments on its content. Furthermore, we want to acknowledge the participants and investigators of FinnGen study. Following biobanks are acknowledged for collecting the FinnGen project samples: Auria Biobank (https://www.auria.fi/biopankki), THL Biobank (https://thl.fi/fi/web/thl-biopankki), Helsinki Biobank (https://www.helsinginbiopankki.fi/fi/etusivu), Biobank Borealis of Northern Finland (https://www.oulu.fi/university/node/38474), Finnish Clinical Biobank Tampere (https://www.tays.fi/en-US/Research_and_development/Finnish_Clinical_Biobank_Tampere), Biobank of Eastern Finland (https://itasuomenbiopankki.fi), Central Finland Biobank (https://www.ksshp.fi/fi-FI/Potilaalle/Biopankki), Finnish Red Cross Blood Service Biobank (https://www.veripalvelu.fi/verenluovutus/biopankkitoiminta) and Terveystalo Biobank (https://www.terveystalo.com/fi/Yritystietoa/Terveystalo-Biopankki/Biopankki/). All Finnish Biobanks are members of BBMRI.fi infrastructure (www.bbmri.fi). The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and by twelve industry partners (AbbVie Inc, AstraZeneca UK Ltd, Biogen MA Inc, Celgene Corporation, Celgene International II Sàrl, Genentech Inc, Merck Sharp & Dohme Corp, Pfizer Inc., GlaxoSmithKline, Sanofi, Maze Therapeutics Inc., Janssen Biotech Inc, Novartis AG).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.658137/full#supplementary-material
Footnotes
- ^ https://finngen.gitbook.io/documentation/
- ^ https://www.finngen.fi/en/researchers/clinical-endpoints
References
1. Persani L, Cangiano B, Bonomi M. The Diagnosis and Management of Central Hypothyroidism in 2018. Endocr Connect (2019) 8:R44–54. doi: 10.1530/EC-18-0515
2. Naafs JC, Verkerk PH, Fliers E, van Trotsenburg ASP, Zwaveling-Soonawala N. Clinical and Genetic Characteristics of Dutch Children With Central Congenital Hypothyroidism, Early Detected by Neonatal Screening. Eur J Endocrinol (2020) 183:627–36. doi: 10.1530/EJE-20-0833
3. Hanna CE, Krainz PL, Skeels MR, Miyahira RS, Sesser DE, LaFranchi SH. Detection of Congenital Hypopituitary Hypothyroidism: Ten-Year Experience in the Northwest Regional Screening Program. J Pediatr (1986) 109:959–64. doi: 10.1016/S0022-3476(86)80276-1
4. Ford G, Lafranchi SH. Screening for Congenital Hypothyroidism: A Worldwide View of Strategies. Best Pract Res Clin Endocrinol Metab (2014) 28:175–87. doi: 10.1016/j.beem.2013.05.008
5. Beck-Peccoz P, Rodari G, Giavoli C, Lania A. Central Hypothyroidism - a Neglected Thyroid Disorder. Nat Rev Endocrinol (2017) 13:588–98. doi: 10.1038/nrendo.2017.47
6. Heinen CA, De Vries EM, Alders M, Bikker H, Zwaveling-Soonawala N, Van Den Akker ELT, et al. Mutations in IRS4 are Associated With Central Hypothyroidism. J Med Genet (2018) 55:693–700. doi: 10.1136/jmedgenet-2017-105113
7. Collu R, Tang J, Castagné J, Lagacé G, Masson N, Huot C, et al. A Novel Mechanism for Isolated Central Hypothyroidism: Inactivating Mutations in the Thyrotropin-Releasing Hormone Receptor Gene. J Clin Endocrinol Metab (1997) 82:1561–5. doi: 10.1210/jc.82.5.1561
8. Hayashizaki Y, Hiraoka Y, Endo Y, Matsubara K. Thyroid-Stimulating Hormone (TSH) Deficiency Caused by a Single Base Substitution in the CAGYC Region of the β-Subunit. EMBO J (1989) 8:2291–6. doi: 10.1002/j.1460-2075.1989.tb08355.x
9. Sun Y, Bak B, Schoenmakers N, Van Trotsenburg ASP, Oostdijk W, Voshol P, et al. Loss-of-Function Mutations in IGSF1 Cause an X-Linked Syndrome of Central Hypothyroidism and Testicular Enlargement. Nat Genet (2012) 44:1375–81. doi: 10.1038/ng.2453
10. Heinen CA, Losekoot M, Sun Y, Watson PJ, Fairall L, Joustra SD, et al. Mutations in TBL1X are Associated With Central Hypothyroidism. J Clin Endocrinol Metab (2016) 101:4564–73. doi: 10.1210/jc.2016-2531
11. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief Bioinform (2013) 14:178–92. doi: 10.1093/bib/bbs017
12. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol Syst Biol (2011) 7(539):1–6. doi: 10.1038/msb.2011.75
13. Kempers MJE, Van Der Sluijs Veer L, Nijhuis-van Der Sanden MWG, Kooistra L, Wiedijk BM, Faber I, et al. Intellectual and Motor Development of Young Adults With Congenital Hypothyroidism Diagnosed by Neonatal Screening. J Clin Endocrinol Metab (2006) 91:418–24. doi: 10.1210/jc.2005-1209
14. Cherella CE, Wassner AJ. Update on Congenital Hypothyroidism. Curr Opin Endocrinol Diabetes Obes (2020) 27:63–9. doi: 10.1097/MED.0000000000000520
15. Lanting CI, Van Tijn DA, Loeber JG, Vulsma T, De Vijlder JJM, Verkerk PH. Clinical Effectiveness and Cost-Effectiveness of the Use of the Thyroxine/Thyroxine-Binding Globulin Ratio to Detect Congenital Hypothyroidism of Thyroidal and Central Origin in a Neonatal Screening Program. Pediatrics (2005) 116:168–73. doi: 10.1542/peds.2004-2162
16. Price A, Weetman AP. Screening for Central Hypothyroidism is Unjustified. Br Med J (2001) 322:798. doi: 10.1136/bmj.322.7289.798
17. Virtanen M, Perheentupa J, Mäenpää J, Pitänen L, Pikkarainen J. Finnish National Screening for Hypothyroidism. Eur J Pediatr (1984) 143:2–5. doi: 10.1007/bf00442737
18. Mehta A, Hindmarsh PC, Stanhope RG, Brain CE, Preece MA, Dattani MT. Is the Thyrotropin-Releasing Hormone Test Necessary in the Diagnosis of Central Hypothyroidism in Children. J Clin Endocrinol Metab (2003) 88:5696–703. doi: 10.1210/jc.2003-030943
19. Van Tijn DA, De Vijlder JJM, Vulsma T. Role of the Thyrotropin-Releasing Hormone Stimulation Test in Diagnosis of Congenital Central Hypothyroidism in Infants. J Clin Endocrinol Metab (2008) 93:410–9. doi: 10.1210/jc.2006-2656
20. Aizawa T, Koizumi Y, Yamada T, Tawata M, Nagata H, Izumiyama T, et al. Difference in Pituitary-Thyroid Feedback Regulation in Hypothyroid Patients, Depending on the Severity of Hypothyroidism. J Clin Endocrinol Metab (1978) 47:560–5. doi: 10.1210/jcem-47-3-560
21. Leslie RDG, Isaacs AJ, Gomez J, Raggatt PR, Bayliss R. Hypothalamo-Pituitary-Thyroid Function in Anorexia Nervosa: Influence of Weight Gain. Br Med J (1978) 2:526–8. doi: 10.1136/bmj.2.6136.526
22. Rijks J, Penders B, Dorenbos E, Straetemans S, Gerver WJ, Vreugdenhil A. Pituitary Response to Thyrotropin Releasing Hormone in Children With Overweight and Obesity. Sci Rep (2016) 6:1–6. doi: 10.1038/srep31032
23. Wauman J, De Smet AS, Dominiek Catteeuw DB, Tavernier J. Insulin Receptor Substrate 4 Couples the Leptin Receptor to Multiple Signaling Pathways. Mol Endocrinol (2008) 22:965–77. doi: 10.1210/me.2007-0414
24. Fantin VR, Wang Q, Lienhard GE, Keller SR. Mice Lacking Insulin Receptor Substrate 4 Exhibit Mild Defects in Growth, Reproduction, and Glucose Homeostasis. Am J Physiol - Endocrinol Metab (2000) 278:127–33. doi: 10.1152/ajpendo.2000.278.1.e127
Keywords: insulin receptor substrate 4, IRS4, central hypothyroidism, thyroid disorders, genetic screening, FinnGen, congenital hypothyroidism
Citation: Patyra K, Makkonen K, Haanpää M, Karppinen S, Viikari L, Toppari J, Reeve MP and Kero J (2021) Screening for Mutations in Isolated Central Hypothyroidism Reveals a Novel Mutation in Insulin Receptor Substrate 4 . Front. Endocrinol. 12:658137. doi: 10.3389/fendo.2021.658137
Received: 25 January 2021; Accepted: 31 March 2021;
Published: 21 May 2021.
Edited by:
Cintia E. Citterio, CONICET Institute of Immunology, Genetics and Metabolism (INIGEM), ArgentinaReviewed by:
Arturo Hernandez, Maine Medical Center, United StatesPaul Van Trotsenburg, Amsterdam University Medical Centers, Netherlands
Hector Manuel Targovnik, University of Buenos Aires, Argentina
Copyright © 2021 Patyra, Makkonen, Haanpää, Karppinen, Viikari, Toppari, Reeve and Kero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jukka Kero, jukka.kero@utu.fi
†These authors have contributed equally to this work