 Xiangyu Ke
Xiangyu Ke Zhirui Lin1
Zhirui Lin1 Guowei Li
Guowei Li- 1Centre of Clinical Epidemiology and Methodology, Guangdong Second Provincial General Hospital, Guangzhou, China
- 2Department of Cardiology, Guangdong Second Provincial General Hospital, Guangzhou, China
- 3Department of Endocrinology, Guangdong Second Provincial General Hospital, Guangzhou, China
- 4Department of Nutrition and Food Hygiene, School of Public Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 5Department of Pulmonary and Critical Care Medicine, Guangdong Second Provincial General Hospital, Guangzhou, China
- 6Department of Health Research Methods, Evidence, and Impact, McMaster University, Hamilton, ON, Canada
The global burden of diabetes mellitus and its complications are currently increasing. Diabetic cardiomyopathy (DCM) is the main cause of diabetes mellitus associated morbidity and mortality; therefore, a comprehensive understanding of DCM development is required for more effective treatment. A disorder of epigenetic posttranscriptional modification of histones in chromatin has been reported to be associated with the pathology of DCM. Recent studies have implicated that histone deacetylases could regulate cardiovascular and metabolic diseases in cellular processes including cardiac fibrosis, hypertrophy, oxidative stress and inflammation. Therefore in this review, we summarized the roles of histone deacetylases in the pathogenesis of DCM, aiming to provide insights into exploring potential preventative and therapeutic strategies of DCM.
Introduction
Diabetes mellitus is a metabolic disease characterized by hyperglycemia. With the improvement of living standard, the incidence of diabetes mellitus continues to rise across the world (1). Diabetes mellitus causes long-term damage to multiple organs, ultimately leading to severe complications. Moreover, diabetes mellitus affects the heart through various mechanisms including metabolic disorder, microvascular injury, cardiac autonomic dysfunction, and a maladaptive immune response (2). Diabetic cardiomyopathy (DCM) is a common and severe complication of diabetes mellitus and increases the risk of heart failure with heavy social and economic burden worldwide (3, 4). DCM is a clinical problem which is present in both type 1 and 2 diabetes (5). People with diabetes (30%) always have more than twice the risk of developing heart failure as compared to people without diabetes (23%). Emerging evidence disclosed that 19-26% of diabetic patients were prone to develop heart failure (6, 7). DCM is characterized by left ventricular hypertrophy, cardiac fibrosis, chronic inflammation in the absence of hypertension, coronary, and other heart diseases in diabetic patients (4, 8, 9), ranging from left ventricular fibrosis and diastolic cardiac dysfunction in the early stage, to severe diastolic heart failure with normal ejection fraction (HFpEF, EF<40%) and ultimately to systolic dysfunction accompanied by heart failure with reduced ejection fraction (HFrEF, EF<50%) (10, 11). Importantly, DCM also increases the risks for the development of extensive focal myocardial necrosis, shock and sudden death. Recent studies have highlighted that a complex interplay between genes and environment may significantly contribute to pathogenesis of microvascular complications associated with diabetes (12). Several potential mechanisms that may contribute to the pathogenesis of the DCM have been proposed, including cardiac structural abnormalities, metabolic disturbances, mitochondrial damage, oxidative stress, autophagy/mitophagy defect, apoptosis, systemic inflammation, epigenetic modification, dampened coronary flow reserve, coronary microvascular disease (microangiopathy), and endothelial impairment (5, 13–17).
A concerted definition of epigenetic trait, “stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence”, was reached at a Cold Spring Harbor meeting (18–20). There are three major epigenetic modifications: histone modifications, DNA methylation and microRNAs (21). Histone acetylation which is the best-characterized histone modifications and is controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Histone deacetylases (HDACs) are a family of enzymes that are important regulators of epigenetic gene modification (22). Accumulating evidence has implicated that HDACs are associated with many cardiovascular diseases (CVDs) (19) including hypertension (23, 24), DCM (25, 26), myocardial infarction (27–29) and atrial fibrillation (30). HDACs have been implicated in numerous cellular processes relevant to DCM, which include cardiac fibrosis, hypertrophy, inflammation and oxidative stress (31). However, only quite a few publications have reported the potential impact of HDACs on DCM. In this review, we comprehensively reviewed the roles of HDACs in cellular processes relevant to DCM, aiming to discuss the implication of HDACs in the pathogenesis of DCM and provide insights into exploring potential preventative and therapeutic strategies of DCM.
Epigenetics in DCM
Epigenetic mechanisms such as histone modification, DNA methylation and microRNA changes may play an important role in the development of DCM (32–34). Previous studies suggested that factors possibly implicated in the pathogenesis of DCM include cardiac fibrosis, cardiac hypertrophy, oxidative stress, and inflammation, which may contribute to alterations in the pathogenic gene expression by epigenetic mechanisms to initiate the pathogenic changes in the target cells and organs (35).
Reversible modifications of histones indicate covalent posttranslational modification of histone proteins, including methylation, acetylation, phosphorylation, ubiquitination and sumoylation (21). Histone modification, especially histone acetylation, is a major epigenetic mechanism regulating gene expression. Histone acetyltransferases (HATs) and histone deacetylases (HDACs), which catalyze histone acetylation resulting in gene transcriptional activation and remove acetyl groups resulting in gene silencing respectively, are the major players in maintaining the equilibrium of histone acetylation (8, 36).
To date, there have been 18 HDACs reported that can be divided into four categories based on their sequence identity and catalytic activity (37). Descriptions of the classification, cellular localization, substrates and main biological functions of HDACs are shown in Table 1. Brief summary of the four class HDACs are also mentioned below.
● Class I HDACs contain HDAC1, HDAC2, HDAC3 and HDAC8.
● Class II HDACs are subdivided into Class IIa (HDAC 4, 5, 7 and 9) and Class IIb (HDAC 6 and 10) (81, 82).
● Class III HDACs are called sirtuins sharing sequence homology with the yeast Sirt 2 protein, which contains seven sirtuin members, namely, SIRT1-SIRT7 (83). This highly conserved class of proteins thereby controls a range of different biological processes (84, 85).
● Class IV HDACs include a solitary member HDAC 11, which shares sequence homology with the yeast RPD 3 and HAD 1 proteins.
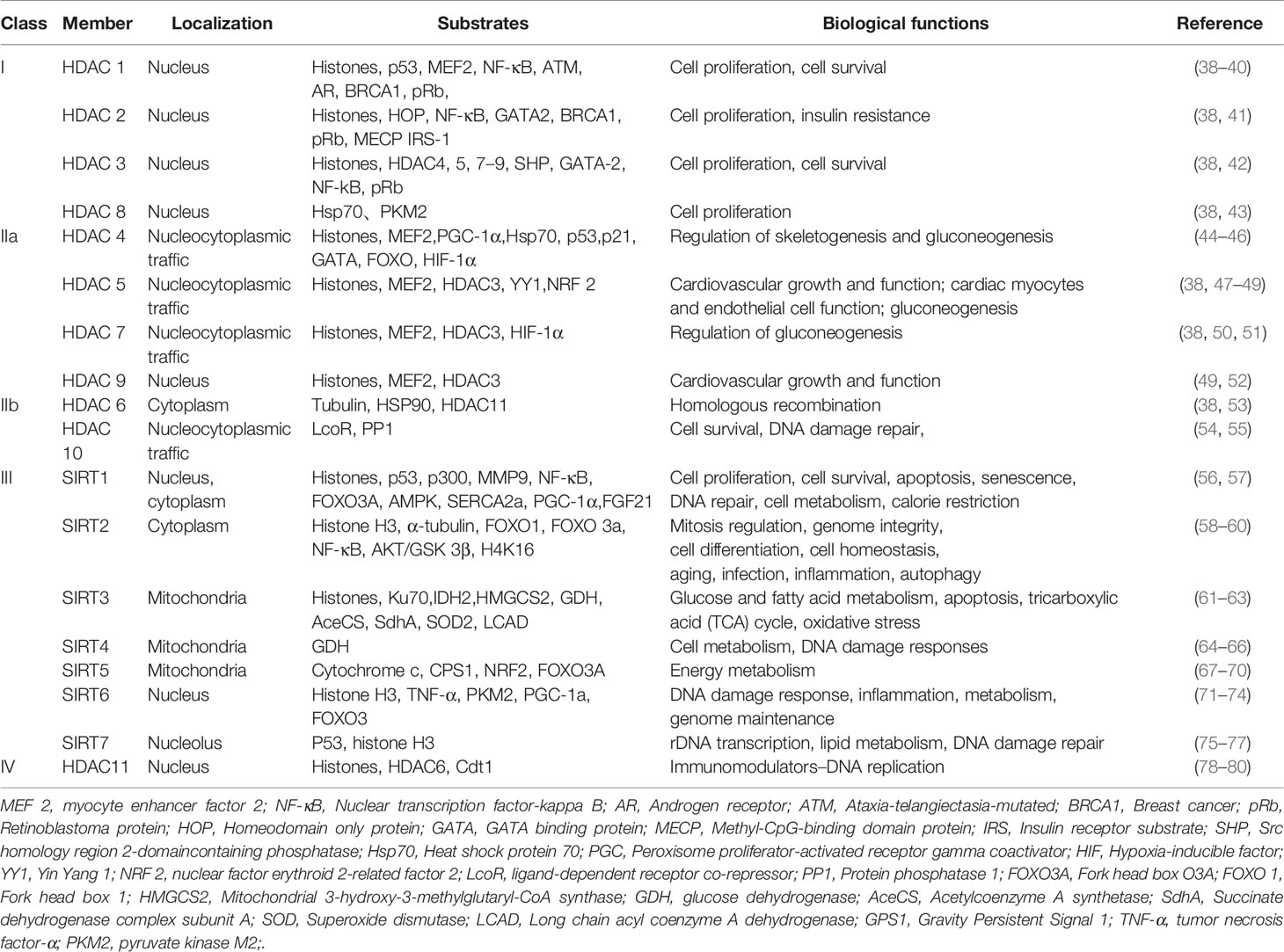
Table 1 The HDAC family: classification, cellular localization and substrates.
HDACs, Cardiac Fibrosis and DCM
Cardiac fibrosis is a hallmark of DCM and is caused by excessive matrix (ECM) proteins accumulation including collagen I and collagen II. Fibrosis increases the passive stiffness of the myocardium and impairs relaxation and diastolic dysfunction (86). Elevated perivascular and intermyofibrillar fibrosis has been observed in human myocardial samples in the absence of coronary heart disease and hypertension (87, 88). This further illustrates the presence of myocardial fibrosis in diabetic cardiomyopathy.
HDACs are emerging as crucial regulators of cardiac fibrosis, although the cellular mechanisms by which HDACs regulate cardiac fibrosis have not been fully understood (89). Current studies provide insufficient evidence for the role of HDACs in DCM; however sizable explorations have reported that HDACs were dysregulated in cardiac fibrosis (29, 90). For instance, SIRT6 knockout mice presented cardiac fibrosis and dysfunction with cardiomyocyte hypertrophy and increased apoptosis (90). Recently, SIRT1, as a protein regulator, has attracted widespread attention because of its salutary effect in DCM (56). One study found that SIRT1 alleviated cardiac fibrosis in the development of DCM. Specifically, bakuchiol (BAK) alleviated cardiac fibrosis in DCM via SIRT1-induced inhibition of ROS generation. Moreover, the TGF-β1/Smad3 signaling pathway played a key role in mediating ROS generation to pathologic fibrosis (91). Likewise, recent researches indicated that Class IIa HDACs could also own profibrotic functions (92–95). Zhang et al. found that the overexpression of activated HDAC4 exacerbated cardiac dysfunction and interstitial fibrosis in the model of myocardial infarction (29). Another study showed that HDAC 4 knockdown blocked cardiac fibrosis by inhibiting the expression of α-SMA and the phosphorylation of ERK (96). It is probably that HDAC4 is adverse to the development of DCM, but whether it will exacerbate cardiac fibrosis in DCM needs to be further explored.
HDACs inhibitors have been reported to be efficacious in rodent models of heart failure. By blocking pathological cardiac hypertrophy and fibrosis, HDACs inhibitors can improve cardiac function (97, 98). For example, MPT0E014 (a Class I/IIb HDAC inhibitor) attenuated cardiac fibrosis with heart failure induced by isoproterenol administration in rats (99). It was associated with downregulation of Ang II type I receptor (AT1R) and transforming growth factor-β (TGF-β) (99). HDACs inhibitors also have been reported to show protective effects on the diabetic heart (25, 26). Xu et al. found that selective inhibitor RGFP966 of HDAC3 ameliorated diabetes-induced fibrosis and deterred the development of DCM by obstructing the enhanced phosphorylated ERK1/2, and upregulating dual specificity phosphatase 5 (DUSP5) expression through increased acetylated histone H3 on the primer region of DUSP5 gene (26). Likewise, Chen et al. reported that the protective effects of HDACs inhibitor (sodium butyrate) in the diabetic myocardium were closely related to mitigating apoptosis, stimulating angiogenesis and increased SOD1 (25). Thus, their findings indicated that HDACs inhibitor had the potential to alleviate cardiac fibrosis and prevent the development of DCM (25).
HDACs, Cardiac Hypertrophy and DCM
Cardiac hypertrophy is defined as an increase in heart mass through growth of individual cardiomyocytes rather than an increment in cardiomyocyte. Physiological and pathological hypertrophy are two types of hypertrophy. Cardiac hypertrophy as a risk factor for heart failure, is a compensatory response that occurs as a result of hemodynamic overload (100–102). The process of hypertrophic cardiac remodeling is the response to the pathological insults, and ultimately cause impaired cardiac function (103, 104). Diabetic patients with impaired cardiac function are prone to the development of DCM. According to the Strong Heart study and the Cardiovascular Health study, they found that cardiac hypertrophy was often accompanied by cardiac systolic and/or diastolic function, suggesting a link between DM and cardiac hypertrophy (103, 105). In addition, diabetes-induced cardiac hypertrophy has been obtained from animal studies, which showed the increased ratio of heart weight to body weight (HW/BW) and cardiomyocyte size and upregulated hypertrophic gene expression (106).
Evidence for the role of HDACs in DCM is limited, but numerous studies revealed that HDACs contribute to the cardiac hypertrophy. Class I HDACs are generally identified pro-hypertrophic. Class I selective HDAC inhibitors have been reported to be efficacious agents, which could block cardiac hypertrophy induced by angiotensin II infusion and aortic banding (107). Similar results have been reported in other studies, that HDAC inhibitors (trichostatin A or scriptaid) ameliorated the cardiac hypertrophy induced by aortic banding (108). All this amount of evidence indirectly supported that HDACs contribute to the cardiac hypertrophy. Further studies disclosed that sodium butyrate which was a specific HADC inhibitor reduced heart/tibia ratio and areas of cardiomyocytes, suggesting that sodium butyrate lessened cardiac hypertrophy in the diabetic mice model. Adult mice were injected intraperitoneally with streptozotocin (STZ, 200mg/kg) to establish the diabetic mice model. In addition, sodium butyrate lessened cardiac hypertrophy which was associated with reducing interstitial fibrosis, relieving the apoptosis and stimulating angiogenesis in STZ-injected diabetic mice. These results have demonstrated that HDAC inhibitor reduced cardiac hypertrophy which in turn preventing diabetic mice from progressing to DCM (25). In contrast, class IIa HDACs have been identified as negative regulators of cardiac hypertrophy, by suppressing hypertrophic gene transcription (93, 109). Taken together, these evidences indirectly proved that HDACs dulled the occurrence of DCM by regulating cardiac hypertrophy.
Based on Bagul’s study, SIRT1 activation by resveratrol led to deacetylation of both NF-κB-p65 and H3. SIRT1 activation decreased binding of NFκB-p65 to DNA, and lessened cardiac hypertrophy and oxidative stress, thereby ultimately blunting the development of DCM (110). The aforementioned study also suggested that oxidative stress may affect the physiology of the diabetic heart. There is some evidence showing the effect of oxidative stress on cardiac abnormalities including cardiac hypertrophy (110). These results suggest that HDACs are regulators of cardiac hypertrophy in the development of DCM. Figure 1 depicted the role of HDACs regulated cardiac fibrosis and hypertrophy, and thus alleviating DCM. HDAC inhibitors reduced cardiac fibrosis and hypertrophy.
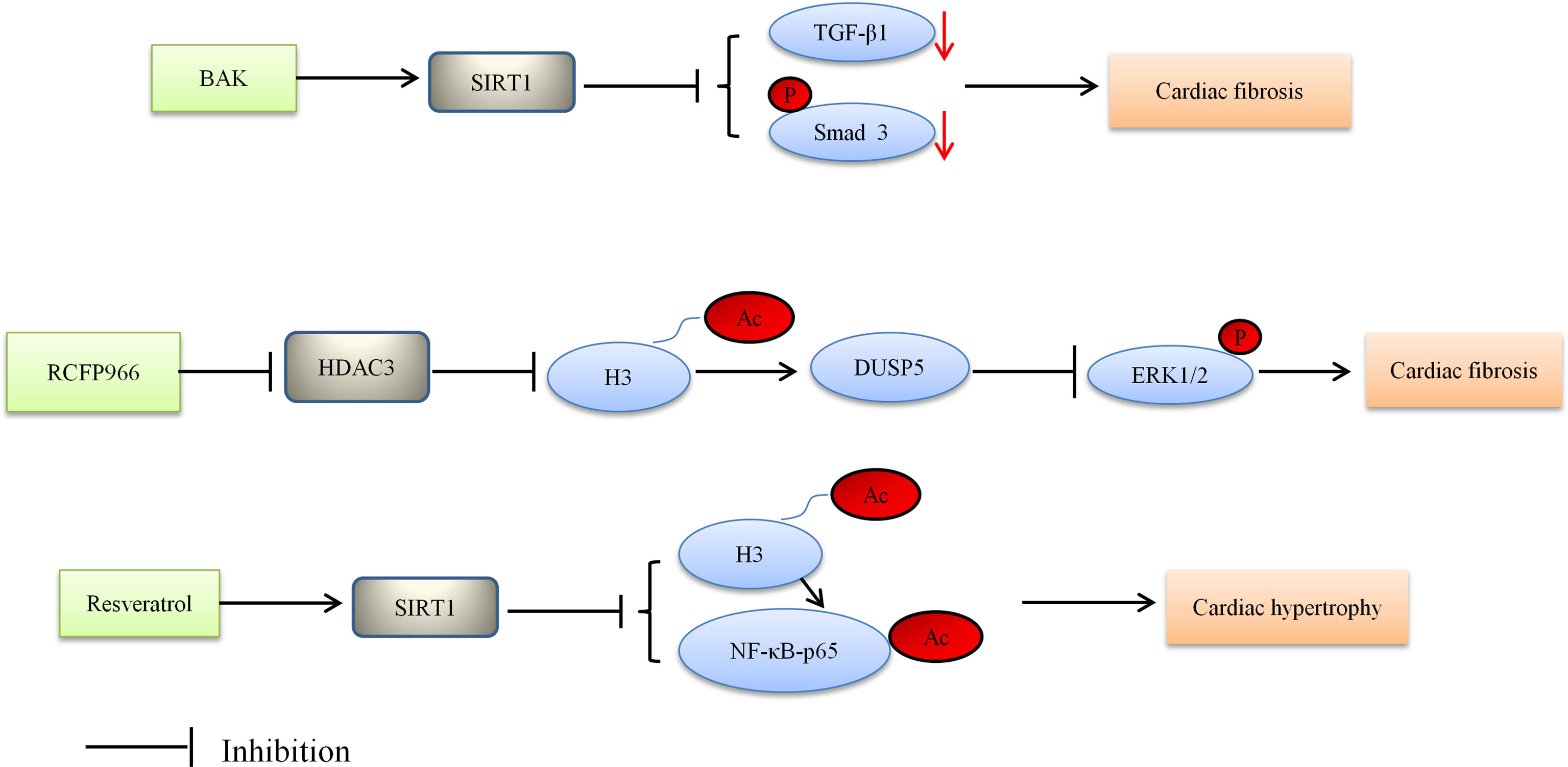
Figure 1 Proposed model depicting regulation and the role of HDACs in cardiac fibrosis and hypertrophy, and thus alleviating DCM. BAK, bakuchiol; Ac, acetylation; P, phosphorylation.
HDACs, Oxidative Stress and DCM
Oxidative stress is widely considered to be one of the main contributors in the development and progression of diabetic cardiovascular complications, including DCM (36, 111). Under the conditions of DCM, the antioxidant factors such as superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) are sharply decreased in heart tissue, while the production of reactive oxygen species (ROS) is remarkably increased, which are responsible for cellular oxidative stress (112, 113). Excessive production of ROS can directly damage proteins, lipid membranes and DNA, oxidize lipids into harmful lipid peroxides, and increase the generation of reactive nitrogen species (RNS) (8, 36, 114, 115). Excessive generation of ROS can cause as well as activate several cellular stress-signaling and inflammatory pathways (116). Previous studies have reported that oxidative stress increased in human diabetic hearts (117).
HDACs have recently been reported to play a part in the pathological process of DCM, including oxidative stress. SIRT1 is the first member to be discovered in sirtuins and still the most studied one, especially as a potential target to treat cardiovascular diseases (56). SIRT1 mitigates oxidative stress and improves DCM via SIRT1/NF-κB-p65, SIRT1/FOXO1, SIRT1/NRF2 pathways. Resveratrol which was a polyphenolic compound used to be the potential prevention or therapy for DCM. Pankaj et al. found that SIRT1 activation by resveratrol led to deacetylation of both NF-κB-p65 at K310 and histone 3 at K9, thereby decreasing binding of NF-κB-p65 to DNA (110). The mice experiment showed that resveratrol ameliorated oxidative stress in diabetic mouse hearts depending on regulation of autophagic flux. Activation of SIRT1 led to deacetylation of FOXO1 and increased the transcriptional activity of FOXO1, ultimately enhanced the autophagy flux and protected diabetes-induced cardiac injury (118). Similarly, in Ren’s study, it was shown that curcumin treatment could alleviate DCM by modulating apoptosis and oxidative stress via the SIRT1-FOXO1 pathway (119). Given the oxidative stress seems a critical cause for the development of DCM, other anti-oxidative approaches have also been used to treat DCM, such as allisartan isoproxil, BAK and tetrahydrocurcumin (THC).
However, these anti-oxidative approaches respectively activated different SIRT1 signaling pathways (120–122). Previous researches have reported that BAK (a bioactive monoterpene phenol) and Allisartan isoproxil (a new nonpeptide angiotensin II receptor blocker [ARB] precursor drug) play crucial role in DCM by attenuating myocardial oxidative damage via activating the SIRT1/NRF 2 signaling pathway (91, 123). Furthermore, another research by Li et al. found that THC treatment could alleviate DCM by attenuating oxidative stress via activating the SIRT1 pathway (120). However, the SIRT1 pathway in Li’s study was not identical to those previously mentioned. SIRT1 activation by THC led to deacetylation of Ac-SOD2, while leading to produce SOD which is a vital molecule in maintaining ROS homeostasis. Thus, generation of ROS was reduced by THC via enhancing the SIRT1 pathway. These studies suggested that SIRT1 attenuates oxidative stress and improves DCM via SIRT1/NF-κB-p65, SIRT1/FOXO1, SIRT1/NRF2 pathways.
Among the seven different sirtuins, SIRT3 as a major protein deacetylase in mitochondria is involved in mediating cellular redox status, mitochondrial energetic and apoptosis (61). In Song’s study, it was demonstrated that SIRT3 deficiency increased ROS accumulation, aggravated hyperglycemia-induced mitochondrial damage, accelerated necroptosis, possibly activated the NLRP3 inflammasome, and ultimately exacerbated development of DCM in the mice (124). Elabela, another endogenous ligand of APJ, has been known as peptides. SIRT3 which is a downstream of APJ has been shown to protect DCM from oxidative stress-mediated cellular injury. Evidence from Li’s study indicated that the protective effects of Elabela in DCM are regulated by inhibition of oxidative stress via FOXO3a deacetylation (125).
Taken together, HDACs potentially contribute to the pathogenesis of DCM, and they are also considered potential therapeutic target in DCM. Figure 2 depicted the role of HDACs regulation oxidative stress and alleviating DCM.
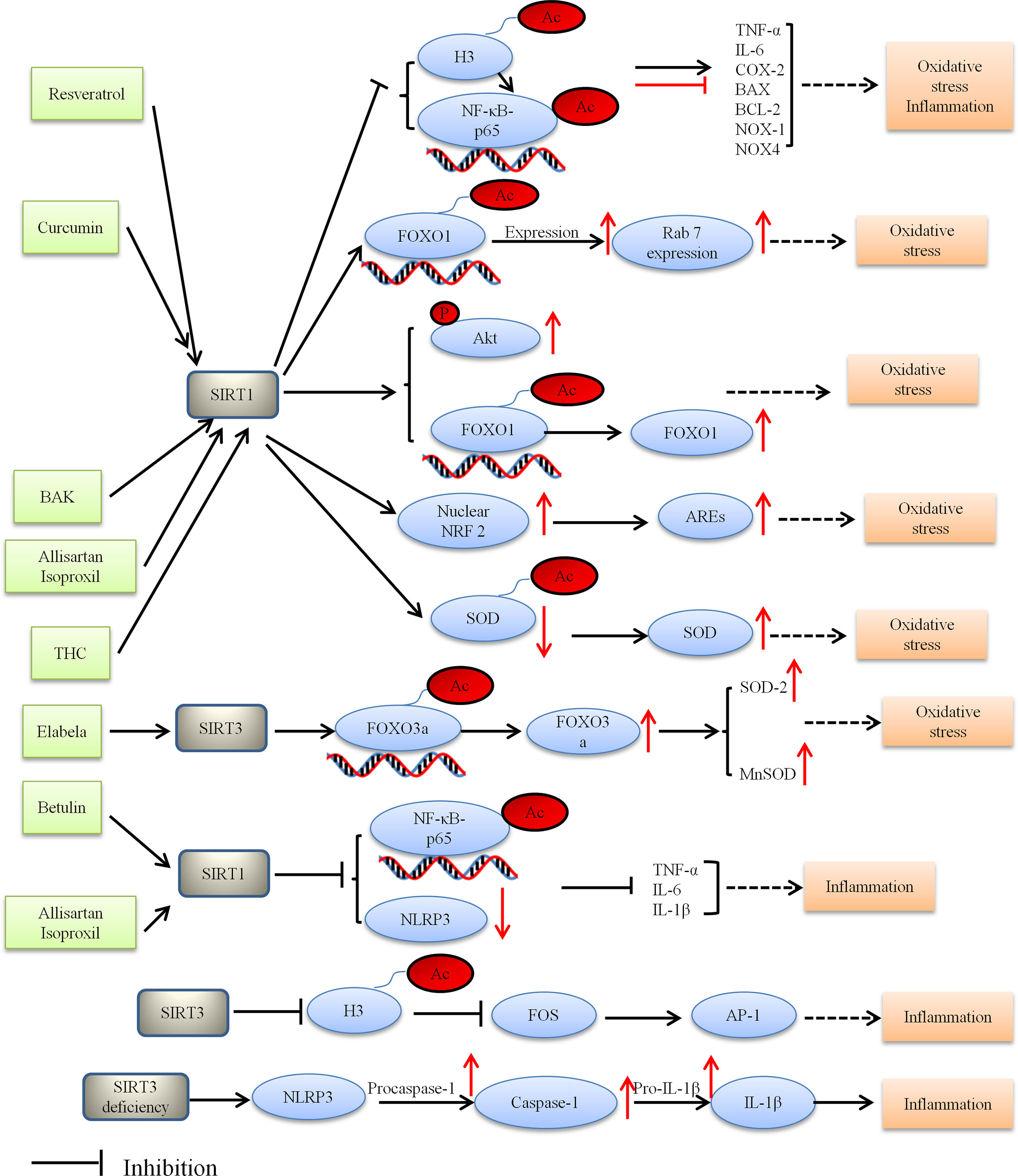
Figure 2 Proposed model depicting regulation and the role of HDACs in oxidative stress and inflammation, and thus alleviating DCM. BAK, bakuchiol; THC, tetrahydrocumin, AREs, antioxidant-responsive elements; Ac, acetylation; P, phosphorylation.
HDACs, Inflammation and DCM
Diabetic mellitus is a pro-inflammatory state (36, 126, 127), and many studies have revealed that cytokine of tissue concentrations increase in various diabetic mouse models, suggesting that inflammation plays an important role in the development of DCM. These studies demonstrated intramyocardial inflammation in DCM including increased inflammatory cells (infiltration of macrophages and leucocytes) and increased expression of inflammatory cytokines [leptin, TNF-α, TGF-β1, intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 (ICAM-1 and VCAM-1), interleukin1 beta (IL-1β), interleukin 6 (IL-6), and interleukin 18 (IL-18)] (121, 122, 128–131).
Miao et al. revealed HDACs are associated with inflammation under diabetic conditions. High glucose can activate NF-κB and increase the expression of inflammation cytokines. In vivo experiment demonstrated that recruitment of NF-κB, histone acetyltransferases (HATs) and histone acetylation at the promoters of inflammatory genes was increased under diabetic conditions, which indicated that HDACs were associated with inflammation (132). The role of NF-κB in regulating inflammatory gene expression is well manifested (127, 132, 133). Likewise, NF-κB (a transcription factor) can be modified by HATs and HDACs leading to the upregulation of inflammatory genes (127). SIRT1 directly interacts with Rela/p65 and NF-κB subunits and inhibits NF-κB by deacetylating Rela/p65 at lysine 310, resulting in the nuclear translocation of NF-κB dependent on IκBa (134). Thereby, it decreased the expression of proinflammatory genes. Betulin and allisartan isoproxil alleviated DCM by attenuating inflammation via SIRT1/NF-κB pathway, however there are still differences between them (110, 123, 134, 135). NLRP3 inflammasome is a member of the NLRP superfamily. Inflammasomes are a group of protein complexes involved in inflammation, immunity and metabolic abnormalities of various diseases (135). In Wen’s study, it was demonstrated that Betulin (triterpene compound) plays anti-inflammation effect in the development of DCM by SIRT1 simultaneously activated NF-κB and NLRP3 (134). Moreover, Jin et al. reported that allisartan isoproxil alleviated DCM by attenuating diabetes-induced inflammation via the SIRT1/NF-κB pathway (123). From what has been discussed above, we draw the conclusion that SIRT1 reduced the inflammatory response and thus alleviated DCM. Taken together, SIRT1 is considered to be a potential intervention target in DCM (3, 136).
Several studies have uncovered multiple important links between the SIRT3 and inflammatory processes. SIRT3 knockout mice showed inflammation and cardiac fibrosis due to upregulation of AP-1 activity. SIRT3 inhibited FOS by deacetylating histone 3 at lysine K27. Palomer et al. found that SIRT3 regulated the proinflammatory and profibrotic responses of cardiac cells via the FOS/AP-1 pathway (137). Song et al. demonstrated that SIRT3 deficiency raised the expression of inflammation-related proteins containing NLRP3, caspase 1 p20, and interleukin-1β both in vitro and in vivo. Moreover, SIRT3 deficiency affected the development of DCM via the NLRP3 inflammasome (124). These results suggest that SIRT3 can be a molecular intervention target for the prevention and treatment of DCM. Figure 2 depicted the role of HDACs regulation inflammatory and alleviating DCM.
HDACIs and DCM
HDACs may be promising drug targets owing to their function in cell proliferation, cell cycle regulation, apoptosis, differentiation, metabolism, protein trafficking and DNA repair. HDAC inhibitors (HDACIs) are chemical compounds that block Zn2+-dependent HDAC enzymes involved in epigenetic modifications which regulate histone acetylation state. HDACIs have been approved by the US Food and Drug Administration (FDA) for clinical use, particularly for cancer treatment (138). Moreover, emerging studies indicated that epigenetic regulation of histone acetylation state may also own a potential for clinical application in the treatment of cardiovascular disease (139).
Currently, five HDACIs that are structurally divided into hydroxamic acid derivates (e.g., trichostatin A), short chain fatty (aliphatic) acids (e.g., sodium butyrate), cyclic peptides, benzamides and sirtuin inhibitors have been approved and are being used globally (140–142). While currently available HDACIs are largely non-selective (pan- HDACIs), the effects of HDACIs are usually studied by examining changes in bulk histone acetylation or the therapeutic effects observed in experimental model or in clinical trials (38). Table 2 summarizes some progress of HDACIs related to treatment of cardiomyopathy regarding their anti-fibrotic and anti-hypertrophy effects. Therefore, more well-designed studies exploring the potential of selective HDACIs for DCM treatment is needed.
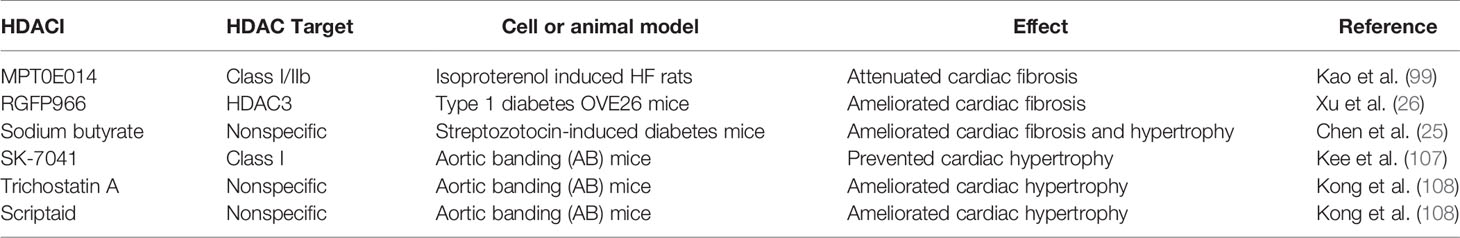
Table 2 Evidence showing the potential effect of inhibitors of HDACs in cardiomyopathy.
Future Research Direction
The current evidence indicates that HDACs are involved in several biological pathways relevant to the pathogenesis of DCM as presented in Figures 1, 2. However, more research is required to better understand the roles of HDACs in the pathogenesis of DCM and the mechanism that regulate them, and address the curative potential in treating DCM. For instance, some HDACs outside of the nucleus can also be post-translationally modified, which can subsequently alter the protein function. Exploring the proteins outside of the nucleus would therefore be a worthwhile endeavor to further understand the potential role of HDACs in DCM. HDACs do not solely remove acetyl residues from proteins but can also remove other acyl modifications, which provide better understanding of the mechanism. Likewise, assessing the relationship between HDACs and diabetes in heart failure may offer important insight into novel mechanisms for DCM. Nevertheless the currently available evidence provides a strong rationale for continuing preclinical studies and initiating clinical trials, with the ultimate purpose of testing the clinical utility of HDACs in DCM.
Conclusion
Increasing evidence from in vitro and in vivo revealed that HDACs plays a critical role in the pathogenesis of DCM, suggesting that HDACs could be molecular intervention targets for the prevention and treatment of DCM. However, more endeavors are needed to further understand the roles of HDACs in the pathogenesis of DCM and the mechanism that regulate them. A comprehensive understanding of the mechanism of HDACs may provide a novel option for the prevention and treatment of DCM.
Author Contributions
XK, XJ and GL prepared the initial draft of the manuscript. ZL, ZY, ML, BC and CJ edited the manuscript for intellectual content. Figures and Tables were created by XK and GL. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Science and Technology Program of Guangzhou (Grant sponsor: GL; Grant no.202002030252), the Science Foundation of Guangdong Second Provincial General Hospital (Grant sponsor: GL; Grant no.YY2018-002), and Doctoral workstation foundation of Guangdong Second Provincial general Hospital (Grant sponsor: XK; Grant no. 2021BSGZ008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Ruoting Wang for her editing and polishing the manuscript.
References
1. A. American Diabetes. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care (2012) 35(Suppl 1):S64–71. doi: 10.2337/dc12-s064
2. Lee WS, Kim J. Diabetic Cardiomyopathy: Where We Are and Where We Are Going. Korean J Intern Med (2017) 32(3):404–21. doi: 10.3904/kjim.2016.208
3. Luo W, Jin Y, Wu G, Zhu W, Qian Y, Zhang Y, et al. Blockage of ROS and MAPKs-Mediated Inflammation via Restoring SIRT1 by a New Compound LF10 Prevents Type 1 Diabetic Cardiomyopathy. Toxicol Appl Pharmacol (2019) 370:24–35. doi: 10.1016/j.taap.2019.03.005
4. Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, et al. Diabetic Cardiomyopathy - A Comprehensive Updated Review. Prog Cardiovasc Dis (2019) 62(4):315–26. doi: 10.1016/j.pcad.2019.03.003
5. Wold LE, Ceylan-Isik AF, Ren J. Oxidative Stress and Stress Signaling: Menace of Diabetic Cardiomyopathy. Acta Pharmacol Sin (2005) 26(8):908–17. doi: 10.1111/j.1745-7254.2005.00146.x
6. Dewanjee S, Vallamkondu J, Kalra RS, John A, Reddy PH, Kandimalla R. Autophagy in the Diabetic Heart: A Potential Pharmacotherapeutic Target in Diabetic Cardiomyopathy. Ageing Res Rev (2021) 68:101338. doi: 10.1016/j.arr.2021.101338
7. Kenny HC, Abel ED. Heart Failure in Type 2 Diabetes Mellitus. Circ Res (2019) 124(1):121–41. doi: 10.1161/CIRCRESAHA.118.311371
8. Hu X, Bai T, Xu Z, Liu Q, Zheng Y, Cai L. Pathophysiological Fundamentals of Diabetic Cardiomyopathy. Compr Physiol (2017) 7(2):693–711. doi: 10.1002/cphy.c160021
9. Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic Cardiomyopathy: Insights Into Pathogenesis, Diagnostic Challenges, and Therapeutic Options. Am J Med (2008) 121(9):748–57. doi: 10.1016/j.amjmed.2008.03.046
10. Schwinger RHG. Pathophysiology of Heart Failure. Cardiovasc Diagn Ther (2021) 11(1):263–76. doi: 10.21037/cdt-20-302
11. Jia G, Whaley-Connell A, Sowers JR. Diabetic Cardiomyopathy: A Hyperglycaemia- and Insulin-Resistance-Induced Heart Disease. Diabetologia (2018) 61(1):21–8. doi: 10.1007/s00125-017-4390-4
12. Khullar M, Cheema BS, Raut SK. Emerging Evidence of Epigenetic Modifications in Vascular Complication of Diabetes. Front Endocrinol (Lausanne) (2017) 8:237. doi: 10.3389/fendo.2017.00237
13. Ren J, Wu NN, Wang S, Sowers JR, Zhang Y. Obesity Cardiomyopathy: Evidence, Mechanisms and Therapeutic Implications. Physiol Rev (2021). doi: 10.1152/physrev.00030.2020
14. Gutierrez-Cuevas J, Sandoval-Rodriguez A, Meza-Rios A, Monroy-Ramirez HC, Galicia-Moreno M, Garcia-Banuelos J, et al. Molecular Mechanisms of Obesity-Linked Cardiac Dysfunction: An Up-Date on Current Knowledge. Cells (2021) 10(3):629. doi: 10.3390/cells10030629
15. Lu L, Ma J, Sun M, Wang X, Gao E, Lu L, et al. Melatonin Ameliorates MI-Induced Cardiac Remodeling and Apoptosis Through a JNK/p53-Dependent Mechanism in Diabetes Mellitus. Oxid Med Cell Longev (2020) 2020:1535201. doi: 10.1155/2020/1535201
16. Bi Y, Zhang Y, Ren J. Phosphoinositide 3-Kinase Therapy in Diabetic Cardiomyopathy: Unravelling an Enigma. Am J Physiol Heart Circ Physiol (2020) 318(5):H1029–31. doi: 10.1152/ajpheart.00160.2020
17. Jia G, Hill MA, Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res (2018) 122(4):624–38. doi: 10.1161/CIRCRESAHA.117.311586
18. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An Operational Definition of Epigenetics. Genes Dev (2009) 23(7):781–3. doi: 10.1101/gad.1787609
19. Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J, et al. Dysregulation of Histone Acetyltransferases and Deacetylases in Cardiovascular Diseases. Oxid Med Cell Longev (2014) 2014:641979. doi: 10.1155/2014/641979
20. Zhang Y, Ren J. Epigenetics and Obesity Cardiomyopathy: From Pathophysiology to Prevention and Management. Pharmacol Ther (2016) 161:52–66. doi: 10.1016/j.pharmthera.2016.03.005
21. Lyu X, Hu M, Peng J, Zhang X, Sanders YY. HDAC Inhibitors as Antifibrotic Drugs in Cardiac and Pulmonary Fibrosis. Ther Adv Chronic Dis (2019) 10:2040622319862697. doi: 10.1177/2040622319862697
22. Bagchi RA, Weeks KL. Histone Deacetylases in Cardiovascular and Metabolic Diseases. J Mol Cell Cardiol (2019) 130:151–9. doi: 10.1016/j.yjmcc.2019.04.003
23. Lee HA, Lee DY, Cho HM, Kim SY, Iwasaki Y, Kim IK. Histone Deacetylase Inhibition Attenuates Transcriptional Activity of Mineralocorticoid Receptor Through Its Acetylation and Prevents Development of Hypertension. Circ Res (2013) 112(7):1004–12. doi: 10.1161/CIRCRESAHA.113.301071
24. Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, et al. Epigenetic Modulation of the Renal Beta-Adrenergic-WNK4 Pathway in Salt-Sensitive Hypertension. Nat Med (2011) 17(5):573–80. doi: 10.1038/nm.2337
25. Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, et al. Histone Deacetylase (HDAC) Inhibition Improves Myocardial Function and Prevents Cardiac Remodeling in Diabetic Mice. Cardiovasc Diabetol (2015) 14:99. doi: 10.1186/s12933-015-0262-8
26. Xu Z, Tong Q, Zhang Z, Wang S, Zheng Y, Liu Q, et al. Inhibition of HDAC3 Prevents Diabetic Cardiomyopathy in OVE26 Mice via Epigenetic Regulation of DUSP5-ERK1/2 Pathway. Clin Sci (Lond) (2017) 131(15):1841–57. doi: 10.1042/CS20170064
27. Zhang L, Qin X, Zhao Y, Fast L, Zhuang S, Liu P, et al. Inhibition of Histone Deacetylases Preserves Myocardial Performance and Prevents Cardiac Remodeling Through Stimulation of Endogenous Angiomyogenesis. J Pharmacol Exp Ther (2012) 341(1):285–93. doi: 10.1124/jpet.111.189910
28. Thal MA, Krishnamurthy P, Mackie AR, Hoxha E, Lambers E, Verma S, et al. Enhanced Angiogenic and Cardiomyocyte Differentiation Capacity of Epigenetically Reprogrammed Mouse and Human Endothelial Progenitor Cells Augments Their Efficacy for Ischemic Myocardial Repair. Circ Res (2012) 111(2):180–90. doi: 10.1161/CIRCRESAHA.112.270462
29. Zhang LX, Du J, Zhao YT, Wang J, Zhang S, Dubielecka PM, et al. Transgenic Overexpression of Active HDAC4 in the Heart Attenuates Cardiac Function and Exacerbates Remodeling in Infarcted Myocardium. J Appl Physiol (1985) (2018) 125(6):1968–78. doi: 10.1152/japplphysiol.00006.2018
30. Seki M, LaCanna R, Powers JC, Vrakas C, Liu F, Berretta R, et al. Class I Histone Deacetylase Inhibition for the Treatment of Sustained Atrial Fibrillation. J Pharmacol Exp Ther (2016) 358(3):441–9. doi: 10.1124/jpet.116.234591
31. Weeks KL. HDAC Inhibitors and Cardioprotection: Homing in on a Mechanism of Action. EBioMedicine (2019) 40:21–2. doi: 10.1016/j.ebiom.2019.01.015
32. Asrih M, Steffens S. Emerging Role of Epigenetics and miRNA in Diabetic Cardiomyopathy. Cardiovasc Pathol (2013) 22(2):117–25. doi: 10.1016/j.carpath.2012.07.004
33. Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, et al. Hyperglycemia Induces a Dynamic Cooperativity of Histone Methylase and Demethylase Enzymes Associated With Gene-Activating Epigenetic Marks That Coexist on the Lysine Tail. Diabetes (2009) 58(5):1229–36. doi: 10.2337/db08-1666
34. Nikoshkov A, Sunkari V, Savu O, Forsberg E, Catrina SB, Brismar K. Epigenetic DNA Methylation in the Promoters of the Igf1 Receptor and Insulin Receptor Genes in Db/Db Mice. Epigenetics (2011) 6(4):405–9. doi: 10.4161/epi.6.4.14791
35. Reddy MA, Zhang E, Natarajan R. Epigenetic Mechanisms in Diabetic Complications and Metabolic Memory. Diabetologia (2015) 58(3):443–55. doi: 10.1007/s00125-014-3462-y
36. Bugger H, Abel ED. Molecular Mechanisms of Diabetic Cardiomyopathy. Diabetologia (2014) 57(4):660–71. doi: 10.1007/s00125-014-3171-6
37. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone Deacetylases as Regulators of Inflammation and Immunity. Trends Immunol (2011) 32(7):335–43. doi: 10.1016/j.it.2011.04.001
38. Wang X, Wei X, Pang Q, Yi F. Histone Deacetylases and Their Inhibitors: Molecular Mechanisms and Therapeutic Implications in Diabetes Mellitus. Acta Pharm Sin B (2012) 2(4):387–95. doi: 10.1016/j.apsb.2012.06.005
39. Zhang LZ, Yang JE, Luo YW, Liu FT, Yuan YF, Zhuang SM. A P53/lnc-Ip53 Negative Feedback Loop Regulates Tumor Growth and Chemoresistance. Adv Sci (Weinh) (2020) 7(21):2001364. doi: 10.1002/advs.202001364
40. Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE, et al. A Role for Histone Deacetylase Activity in HDAC1-Mediated Transcriptional Repression. Proc Natl Acad Sci USA (1998) 95(7):3519–24. doi: 10.1073/pnas.95.7.3519
41. Tang W, Zhou W, Xiang L, Wu X, Zhang P, Wang J, et al. The P300/YY1/miR-500a-5p/HDAC2 Signalling Axis Regulates Cell Proliferation in Human Colorectal Cancer. Nat Commun (2019) 10(1):663. doi: 10.1038/s41467-018-08225-3
42. Ozawa Y, Towatari M, Tsuzuki S, Hayakawa F, Maeda T, Miyata Y, et al. Histone Deacetylase 3 Associates With and Represses the Transcription Factor GATA-2. Blood (2001) 98(7):2116–23. doi: 10.1182/blood.v98.7.2116
43. Zhang R, Shen M, Wu C, Chen Y, Lu J, Li J, et al. HDAC8-Dependent Deacetylation of PKM2 Directs Nuclear Localization and Glycolysis to Promote Proliferation in Hepatocellular Carcinoma. Cell Death Dis (2020) 11(12):1036. doi: 10.1038/s41419-020-03212-3
44. Luo L, Martin SC, Parkington J, Cadena SM, Zhu J, Ibebunjo C, et al. HDAC4 Controls Muscle Homeostasis Through Deacetylation of Myosin Heavy Chain, PGC-1alpha, and Hsc70. Cell Rep (2019) 29(3):749–63.e12. doi: 10.1016/j.celrep.2019.09.023
45. Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 Deacetylase Associates With and Represses the MEF2 Transcription Factor. EMBO J (1999) 18(18):5099–107. doi: 10.1093/emboj/18.18.5099
46. Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone Deacetylase 4 Controls Chondrocyte Hypertrophy During Skeletogenesis. Cell (2004) 119(4):555–66. doi: 10.1016/j.cell.2004.10.024
47. Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-Dependent Activation of the MEF2 Transcription Factor by Dissociation From Histone Deacetylases. Proc Natl Acad Sci USA (2000) 97(8):4070–5. doi: 10.1073/pnas.080064097
48. Berger I, Bieniossek C, Schaffitzel C, Hassler M, Santelli E, Richmond TJ. Direct Interaction of Ca2+/calmodulin Inhibits Histone Deacetylase 5 Repressor Core Binding to Myocyte Enhancer Factor 2. J Biol Chem (2003) 278(20):17625–35. doi: 10.1074/jbc.M301646200
49. Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone Deacetylases 5 and 9 Govern Responsiveness of the Heart to a Subset of Stress Signals and Play Redundant Roles in Heart Development. Mol Cell Biol (2004) 24(19):8467–76. doi: 10.1128/MCB.24.19.8467-8476.2004
50. Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, Khochbin S. Mechanism for Nucleocytoplasmic Shuttling of Histone Deacetylase 7. J Biol Chem (2001) 276(50):47496–507. doi: 10.1074/jbc.M107631200
51. Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM, Muscat GE. A Dynamic Role for HDAC7 in MEF2-Mediated Muscle Differentiation. J Biol Chem (2001) 276(20):17007–13. doi: 10.1074/jbc.M101508200
52. Mahlknecht U, Schnittger S, Will J, Cicek N, Hoelzer D. Chromosomal Organization and Localization of the Human Histone Deacetylase 9 Gene (HDAC9). Biochem Biophys Res Commun (2002) 293(1):182–91. doi: 10.1016/S0006-291X(02)00193-6
53. Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 Is a Microtubule-Associated Deacetylase. Nature (2002) 417(6887):455–8. doi: 10.1038/417455a
54. Kao HY, Lee CH, Komarov A, Han CC, Evans RM. Isolation and Characterization of Mammalian HDAC10, a Novel Histone Deacetylase. J Biol Chem (2002) 277(1):187–93. doi: 10.1074/jbc.M108931200
55. Tong JJ, Liu J, Bertos NR, Yang XJ. Identification of HDAC10, A Novel Class II Human Histone Deacetylase Containing a Leucine-Rich Domain. Nucleic Acids Res (2002) 30(5):1114–23. doi: 10.1093/nar/30.5.1114
56. Karbasforooshan H, Karimi G. The Role of SIRT1 in Diabetic Cardiomyopathy. BioMed Pharmacother (2017) 90:386–92. doi: 10.1016/j.biopha.2017.03.056
57. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. Hsir2(SIRT1) Functions as an NAD-Dependent P53 Deacetylase. Cell (2001) 107(2):149–59. doi: 10.1016/s0092-8674(01)00527-x
58. Wang Y, Yang J, Hong T, Chen X, Cui L. SIRT2: Controversy and Multiple Roles in Disease and Physiology. Ageing Res Rev (2019) 55:100961. doi: 10.1016/j.arr.2019.100961
59. Wang F, Tong Q. SIRT2 Suppresses Adipocyte Differentiation by Deacetylating FOXO1 and Enhancing FOXO1’s Repressive Interaction With PPARgamma. Mol Biol Cell (2009) 20(3):801–8. doi: 10.1091/mbc.E08-06-0647
60. North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol Cell (2003) 11(2):437–44. doi: 10.1016/s1097-2765(03)00038-8
61. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A Role for the Mitochondrial Deacetylase Sirt3 in Regulating Energy Homeostasis. Proc Natl Acad Sci USA (2008) 105(38):14447–52. doi: 10.1073/pnas.0803790105
62. Sun W, Liu C, Chen Q, Liu N, Yan Y, Liu B. SIRT3: A New Regulator of Cardiovascular Diseases. Oxid Med Cell Longev (2018) 2018:7293861. doi: 10.1155/2018/7293861
63. Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of Succinate Dehydrogenase Activity by SIRT3 in Mammalian Mitochondria. Biochemistry (2010) 49(2):304–11. doi: 10.1021/bi901627u
64. Min Z, Gao J, Yu Y. The Roles of Mitochondrial SIRT4 in Cellular Metabolism. Front Endocrinol (Lausanne) (2018) 9:783. doi: 10.3389/fendo.2018.00783
65. Zaganjor E, Vyas S, Haigis MC. SIRT4 Is a Regulator of Insulin Secretion. Cell Chem Biol (2017) 24(6):656–8. doi: 10.1016/j.chembiol.2017.06.002
66. Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S, et al. SIRT4 Regulates Fatty Acid Oxidation and Mitochondrial Gene Expression in Liver and Muscle Cells. J Biol Chem (2010) 285(42):31995–2002. doi: 10.1074/jbc.M110.124164
67. Kumar S, Lombard DB. Functions of the Sirtuin Deacylase SIRT5 in Normal Physiology and Pathobiology. Crit Rev Biochem Mol Biol (2018) 53(3):311–34. doi: 10.1080/10409238.2018.1458071
68. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Science (2011) 334(6057):806–9. doi: 10.1126/science.1207861
69. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 Regulates the Mitochondrial Lysine Succinylome and Metabolic Networks. Cell Metab (2013) 18(6):920–33. doi: 10.1016/j.cmet.2013.11.013
70. Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, et al. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol Cell (2013) 50(6):919–30. doi: 10.1016/j.molcel.2013.06.001
71. Tasselli L, Zheng W, Chua KF. SIRT6: Novel Mechanisms and Links to Aging and Disease. Trends Endocrinol Metab (2017) 28(3):168–85. doi: 10.1016/j.tem.2016.10.002
72. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 Is a Histone H3 Lysine 9 Deacetylase That Modulates Telomeric Chromatin. Nature (2008) 452(7186):492–6. doi: 10.1038/nature06736
73. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-KappaB-Dependent Gene Expression and Organismal Life Span. Cell (2009) 136(1):62–74. doi: 10.1016/j.cell.2008.10.052
74. Dominy JE Jr., Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, et al. The Deacetylase Sirt6 Activates the Acetyltransferase GCN5 and Suppresses Hepatic Gluconeogenesis. Mol Cell (2012) 48(6):900–13. doi: 10.1016/j.molcel.2012.09.030
75. Vazquez BN, Thackray JK, Serrano L. Sirtuins and DNA Damage Repair: SIRT7 Comes to Play. Nucleus (2017) 8(2):107–15. doi: 10.1080/19491034.2016.1264552
76. Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 Homolog SIRT7 Is an Activator of RNA Polymerase I Transcription. Genes Dev (2006) 20(9):1075–80. doi: 10.1101/gad.1399706
77. Grob A, Roussel P, Wright JE, McStay B, Hernandez-Verdun D, Sirri V. Involvement of SIRT7 in Resumption of rDNA Transcription at the Exit From Mitosis. J Cell Sci (2009) 122(Pt 4):489–98. doi: 10.1242/jcs.042382
78. Liu SS, Wu F, Jin YM, Chang WQ, Xu TM. HDAC11: A Rising Star in Epigenetics. BioMed Pharmacother (2020) 131:110607. doi: 10.1016/j.biopha.2020.110607
79. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The Histone Deacetylase HDAC11 Regulates the Expression of Interleukin 10 and Immune Tolerance. Nat Immunol (2009) 10(1):92–100. doi: 10.1038/ni.1673
80. Cheng F, Lienlaf M, Perez-Villarroel P, Wang HW, Lee C, Woan K, et al. Divergent Roles of Histone Deacetylase 6 (HDAC6) and Histone Deacetylase 11 (HDAC11) on the Transcriptional Regulation of IL10 in Antigen Presenting Cells. Mol Immunol (2014) 60(1):44–53. doi: 10.1016/j.molimm.2014.02.019
81. Yang XJ, Gregoire S. Class II Histone Deacetylases: From Sequence to Function, Regulation, and Clinical Implication. Mol Cell Biol (2005) 25(8):2873–84. doi: 10.1128/MCB.25.8.2873-2884.2005
82. Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the Hidden Catalytic Activity of Vertebrate Class IIa Histone Deacetylases. Proc Natl Acad Sci USA (2007) 104(44):17335–40. doi: 10.1073/pnas.0706487104
83. Kane AE, Sinclair DA. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ Res (2018) 123(7):868–85. doi: 10.1161/CIRCRESAHA.118.312498
84. Grunstein M. Histone Acetylation in Chromatin Structure and Transcription. Nature (1997) 389(6649):349–52. doi: 10.1038/38664
85. Ziegler N, Raichur S, Brunner B, Hemmann U, Stolte M, Schwahn U, et al. Liver-Specific Knockdown of Class IIa HDACs Has Limited Efficacy on Glucose Metabolism But Entails Severe Organ Side Effects in Mice. Front Endocrinol (Lausanne) (2020) 11:598. doi: 10.3389/fendo.2020.00598
86. Moreo A, Ambrosio G, De Chiara B, Pu M, Tran T, Mauri F, et al. Influence of Myocardial Fibrosis on Left Ventricular Diastolic Function: Noninvasive Assessment by Cardiac Magnetic Resonance and Echo. Circ Cardiovasc Imaging (2009) 2(6):437–43. doi: 10.1161/CIRCIMAGING.108.838367
87. Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, et al. Evidence for Cardiomyopathy in Familial Diabetes Mellitus. J Clin Invest (1977) 60(4):884–99. doi: 10.1172/JCI108843
88. Shimizu M, Umeda K, Sugihara N, Yoshio H, Ino H, Takeda R, et al. Collagen Remodelling in Myocardia of Patients With Diabetes. J Clin Pathol (1993) 46(1):32–6. doi: 10.1136/jcp.46.1.32
89. Schuetze KB, McKinsey TA, Long CS. Targeting Cardiac Fibroblasts to Treat Fibrosis of the Heart: Focus on HDACs. J Mol Cell Cardiol (2014) 70:100–7. doi: 10.1016/j.yjmcc.2014.02.015
90. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, et al. The Sirtuin SIRT6 Blocks IGF-Akt Signaling and Development of Cardiac Hypertrophy by Targeting C-Jun. Nat Med (2012) 18(11):1643–50. doi: 10.1038/nm.2961
91. Ma W, Guo W, Shang F, Li Y, Li W, Liu J, et al. Bakuchiol Alleviates Hyperglycemia-Induced Diabetic Cardiomyopathy by Reducing Myocardial Oxidative Stress via Activating the SIRT1/Nrf2 Signaling Pathway. Oxid Med Cell Longev (2020) 2020:3732718. doi: 10.1155/2020/3732718
92. Ellmers LJ, Scott NJ, Piuhola J, Maeda N, Smithies O, Frampton CM, et al. Npr1-Regulated Gene Pathways Contributing to Cardiac Hypertrophy and Fibrosis. J Mol Endocrinol (2007) 38(1-2):245–57. doi: 10.1677/jme.1.02138
93. Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II Histone Deacetylases Act as Signal-Responsive Repressors of Cardiac Hypertrophy. Cell (2002) 110(4):479–88. doi: 10.1016/s0092-8674(02)00861-9
94. Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM Kinase II Selectively Signals to Histone Deacetylase 4 During Cardiomyocyte Hypertrophy. J Clin Invest (2006) 116(7):1853–64. doi: 10.1172/JCI27438
95. Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, et al. Protein Kinases C and D Mediate Agonist-Dependent Cardiac Hypertrophy Through Nuclear Export of Histone Deacetylase 5. Mol Cell Biol (2004) 24(19):8374–85. doi: 10.1128/MCB.24.19.8374-8385.2004
96. Zhang Y, Gao F, Tang Y, Xiao J, Li C, Ouyang Y, et al. Valproic Acid Regulates Ang II-Induced Pericyte-Myofibroblast Trans-Differentiation via MAPK/ERK Pathway. Am J Transl Res (2018) 10(7):1976–89.
97. Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, et al. Class I HDACs Regulate Angiotensin II-Dependent Cardiac Fibrosis via Fibroblasts and Circulating Fibrocytes. J Mol Cell Cardiol (2014) 67:112–25. doi: 10.1016/j.yjmcc.2013.12.013
98. Bush EW, McKinsey TA. Protein Acetylation in the Cardiorenal Axis: The Promise of Histone Deacetylase Inhibitors. Circ Res (2010) 106(2):272–84. doi: 10.1161/CIRCRESAHA.109.209338
99. Kao YH, Liou JP, Chung CC, Lien GS, Kuo CC, Chen SA, et al. Histone Deacetylase Inhibition Improved Cardiac Functions With Direct Antifibrotic Activity in Heart Failure. Int J Cardiol (2013) 168(4):4178–83. doi: 10.1016/j.ijcard.2013.07.111
100. Johnson EJ, Dieter BP, Marsh SA. Evidence for Distinct Effects of Exercise in Different Cardiac Hypertrophic Disorders. Life Sci (2015) 123:100–6. doi: 10.1016/j.lfs.2015.01.007
101. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic Implications of Echocardiographically Determined Left Ventricular Mass in the Framingham Heart Study. N Engl J Med (1990) 322(22):1561–6. doi: 10.1056/NEJM199005313222203
102. Lorell BH, Carabello BA. Left Ventricular Hypertrophy: Pathogenesis, Detection, and Prognosis. Circulation (2000) 102(4):470–9. doi: 10.1161/01.cir.102.4.470
103. Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, et al. Impact of Diabetes on Cardiac Structure and Function: The Strong Heart Study. Circulation (2000) 101(19):2271–6. doi: 10.1161/01.cir.101.19.2271
104. Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of Glucose Intolerance and Insulin Resistance on Cardiac Structure and Function: Sex-Related Differences in the Framingham Heart Study. Circulation (2003) 107(3):448–54. doi: 10.1161/01.cir.0000045671.62860.98
105. Lee M, Gardin JM, Lynch JC, Smith VE, Tracy RP, Savage PJ, et al. Diabetes Mellitus and Echocardiographic Left Ventricular Function in Free-Living Elderly Men and Women: The Cardiovascular Health Study. Am Heart J (1997) 133(1):36–43. doi: 10.1016/s0002-8703(97)70245-x
106. Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic Cardiomyopathy: Mechanisms and New Treatment Strategies Targeting Antioxidant Signaling Pathways. Pharmacol Ther (2014) 142(3):375–415. doi: 10.1016/j.pharmthera.2014.01.003
107. Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, et al. Inhibition of Histone Deacetylation Blocks Cardiac Hypertrophy Induced by Angiotensin II Infusion and Aortic Banding. Circulation (2006) 113(1):51–9. doi: 10.1161/CIRCULATIONAHA.105.559724
108. Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, et al. Suppression of Class I and II Histone Deacetylases Blunts Pressure-Overload Cardiac Hypertrophy. Circulation (2006) 113(22):2579–88. doi: 10.1161/CIRCULATIONAHA.106.625467
109. Kee HJ, Kook H. Roles and Targets of Class I and IIa Histone Deacetylases in Cardiac Hypertrophy. J BioMed Biotechnol (2011) 2011:928326. doi: 10.1155/2011/928326
110. Bagul PK, Deepthi N, Sultana R, Banerjee SK. Resveratrol Ameliorates Cardiac Oxidative Stress in Diabetes Through Deacetylation of NFkB-P65 and Histone 3. J Nutr Biochem (2015) 26(11):1298–307. doi: 10.1016/j.jnutbio.2015.06.006
111. Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, et al. Diabetic Cardiovascular Disease Induced by Oxidative Stress. Int J Mol Sci (2015) 16(10):25234–63. doi: 10.3390/ijms161025234
112. Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive Oxygen Species Signalling in the Diabetic Heart: Emerging Prospect for Therapeutic Targeting. Heart (2018) 104(4):293–9. doi: 10.1136/heartjnl-2017-311448
113. Kowluru RA, Mishra M. Oxidative Stress, Mitochondrial Damage and Diabetic Retinopathy. Biochim Biophys Acta (2015) 1852(11):2474–83. doi: 10.1016/j.bbadis.2015.08.001
114. Wallace DC. Mitochondrial Genetics: A Paradigm for Aging and Degenerative Diseases? Science (1992) 256(5057):628–32. doi: 10.1126/science.1533953
115. Bugger H, Bode C. The Vulnerable Myocardium. Diabetic Cardiomyopathy Hamostaseologie (2015) 35(1):17–24. doi: 10.5482/HAMO-14-09-0038
116. Madamanchi NR, Runge MS. Mitochondrial Dysfunction in Atherosclerosis. Circ Res (2007) 100(4):460–73. doi: 10.1161/01.RES.0000258450.44413.96
117. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J Am Coll Cardiol (2009) 54(20):1891–8. doi: 10.1016/j.jacc.2009.07.031
118. Wang B, Yang Q, Sun YY, Xing YF, Wang YB, Lu XT, et al. Resveratrol-Enhanced Autophagic Flux Ameliorates Myocardial Oxidative Stress Injury in Diabetic Mice. J Cell Mol Med (2014) 18(8):1599–611. doi: 10.1111/jcmm.12312
119. Ren BC, Zhang YF, Liu SS, Cheng XJ, Yang X, Cui XG, et al. Curcumin Alleviates Oxidative Stress and Inhibits Apoptosis in Diabetic Cardiomyopathy via Sirt1-Foxo1 and PI3K-Akt Signalling Pathways. J Cell Mol Med (2020) 24(21):12355–67. doi: 10.1111/jcmm.15725
120. Li K, Zhai M, Jiang L, Song F, Zhang B, Li J, et al. Tetrahydrocurcumin Ameliorates Diabetic Cardiomyopathy by Attenuating High Glucose-Induced Oxidative Stress and Fibrosis via Activating the SIRT1 Pathway. Oxid Med Cell Longev (2019) 2019:6746907. doi: 10.1155/2019/6746907
121. Westermann D, Van Linthout S, Dhayat S, Dhayat N, Escher F, Bucker-Gartner C, et al. Cardioprotective and Anti-Inflammatory Effects of Interleukin Converting Enzyme Inhibition in Experimental Diabetic Cardiomyopathy. Diabetes (2007) 56(7):1834–41. doi: 10.2337/db06-1662
122. Rajesh M, Batkai S, Kechrid M, Mukhopadhyay P, Lee WS, Horvath B, et al. Cannabinoid 1 Receptor Promotes Cardiac Dysfunction, Oxidative Stress, Inflammation, and Fibrosis in Diabetic Cardiomyopathy. Diabetes (2012) 61(3):716–27. doi: 10.2337/db11-0477
123. Jin Q, Zhu Q, Wang K, Chen M, Li X. Allisartan Isoproxil Attenuates Oxidative Stress and Inflammation Through the SIRT1/Nrf2/NF−κb Signalling Pathway in Diabetic Cardiomyopathy Rats. Mol Med Rep (2021) 23(3):215. doi: 10.3892/mmr.2021.11854
124. Song S, Ding Y, Dai GL, Zhang Y, Xu MT, Shen JR, et al. Sirtuin 3 Deficiency Exacerbates Diabetic Cardiomyopathy via Necroptosis Enhancement and NLRP3 Activation. Acta Pharmacol Sin (2020) 42(2):230–41. doi: 10.1038/s41401-020-0490-7
125. Li C, Miao X, Wang S, Liu Y, Sun J, Liu Q, et al. Elabela may Regulate SIRT3-Mediated Inhibition of Oxidative Stress Through Foxo3a Deacetylation Preventing Diabetic-Induced Myocardial Injury. J Cell Mol Med (2021) 25(1):323–32. doi: 10.1111/jcmm.16052
126. Diamant M, Lamb HJ, Smit JW, de Roos A, Heine RJ. Diabetic Cardiomyopathy in Uncomplicated Type 2 Diabetes Is Associated With the Metabolic Syndrome and Systemic Inflammation. Diabetologia (2005) 48(8):1669–70. doi: 10.1007/s00125-005-1821-4
127. Reddy MA, Natarajan R. Epigenetic Mechanisms in Diabetic Vascular Complications. Cardiovasc Res (2011) 90(3):421–9. doi: 10.1093/cvr/cvr024
128. Westermann D, Rutschow S, Jager S, Linderer A, Anker S, Riad A, et al. Contributions of Inflammation and Cardiac Matrix Metalloproteinase Activity to Cardiac Failure in Diabetic Cardiomyopathy: The Role of Angiotensin Type 1 Receptor Antagonism. Diabetes (2007) 56(3):641–6. doi: 10.2337/db06-1163
129. Tschope C, Walther T, Escher F, Spillmann F, Du J, Altmann C, et al. Transgenic Activation of the Kallikrein-Kinin System Inhibits Intramyocardial Inflammation, Endothelial Dysfunction and Oxidative Stress in Experimental Diabetic Cardiomyopathy. FASEB J (2005) 19(14):2057–9. doi: 10.1096/fj.05-4095fje
130. Westermann D, Rutschow S, Van Linthout S, Linderer A, Bucker-Gartner C, Sobirey M, et al. Inhibition of P38 Mitogen-Activated Protein Kinase Attenuates Left Ventricular Dysfunction by Mediating Pro-Inflammatory Cardiac Cytokine Levels in a Mouse Model of Diabetes Mellitus. Diabetologia (2006) 49(10):2507–13. doi: 10.1007/s00125-006-0385-2
131. Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, Pacher P. Interplay of Oxidative, Nitrosative/Nitrative Stress, Inflammation, Cell Death and Autophagy in Diabetic Cardiomyopathy. Biochim Biophys Acta (2015) 1852(2):232–42. doi: 10.1016/j.bbadis.2014.06.030
132. Miao F, Gonzalo IG, Lanting L, Natarajan R. In Vivo Chromatin Remodeling Events Leading to Inflammatory Gene Transcription Under Diabetic Conditions. J Biol Chem (2004) 279(17):18091–7. doi: 10.1074/jbc.M311786200
133. Barnes PJ, Karin M. Nuclear Factor-Kappab: A Pivotal Transcription Factor in Chronic Inflammatory Diseases. N Engl J Med (1997) 336(15):1066–71. doi: 10.1056/NEJM199704103361506
134. Wen Y, Geng L, Zhou L, Pei X, Yang Z, Ding Z. Betulin Alleviates on Myocardial Inflammation in Diabetes Mice via Regulating Siti1/NLRP3/NF-kappaB Pathway. Int Immunopharmacol (2020) 85:106653. doi: 10.1016/j.intimp.2020.106653
135. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J Immunol (2013) 191(10):5230–8. doi: 10.4049/jimmunol.1301490
136. Ying Y, Jiang C, Zhang M, Jin J, Ge S, Wang X. Phloretin Protects Against Cardiac Damage and Remodeling via Restoring SIRT1 and Anti-Inflammatory Effects in the Streptozotocin-Induced Diabetic Mouse Model. Aging (Albany NY) (2019) 11(9):2822–35. doi: 10.18632/aging.101954
137. Palomer X, Roman-Azcona MS, Pizarro-Delgado J, Planavila A, Villarroya F, Valenzuela-Alcaraz B, et al. SIRT3-Mediated Inhibition of FOS Through Histone H3 Deacetylation Prevents Cardiac Fibrosis and Inflammation. Signal Transduct Target Ther (2020) 5(1):14. doi: 10.1038/s41392-020-0114-1
138. Qin HT, Li HQ, Liu F. Selective Histone Deacetylase Small Molecule Inhibitors: Recent Progress and Perspectives. Expert Opin Ther Pat (2017) 27(5):621–36. doi: 10.1080/13543776.2017.1276565
139. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action. J Pharmacol Exp Ther (2007) 321(3):892–901. doi: 10.1124/jpet.107.120188
140. Marks PA. Histone Deacetylase Inhibitors: A Chemical Genetics Approach to Understanding Cellular Functions. Biochim Biophys Acta (2010) 1799(10-12):717–25. doi: 10.1016/j.bbagrm.2010.05.008
141. Liu N, Zhuang S. Treatment of Chronic Kidney Diseases With Histone Deacetylase Inhibitors. Front Physiol (2015) 6:121. doi: 10.3389/fphys.2015.00121
Keywords: diabetic cardiomyopathy, histone deacetylases, cardiac fibrosis, cardiac hypertrophy, oxidative stress, inflammation
Citation: Ke X, Lin Z, Ye Z, Leng M, Chen B, Jiang C, Jiang X and Li G (2021) Histone Deacetylases in the Pathogenesis of Diabetic Cardiomyopathy. Front. Endocrinol. 12:679655. doi: 10.3389/fendo.2021.679655
Received: 12 March 2021; Accepted: 06 July 2021;
Published: 22 July 2021.
Edited by:
Khalid Siddiqui, King Saud University, Saudi ArabiaReviewed by:
Kyle Stephan McCommis, Saint Louis University, United StatesJun Ren, University of Washington, United States
Adam R. Wende, University of Alabama at Birmingham, United States
Copyright © 2021 Ke, Lin, Ye, Leng, Chen, Jiang, Jiang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyun Jiang, tgccem@hotmail.com; Guowei Li, lig28@mcmaster.ca