 Victoria N. Tedjawirja
Victoria N. Tedjawirja Max Nieuwdorp
Max Nieuwdorp Kak Khee Yeung1
Kak Khee Yeung1- 1Department of Surgery, Amsterdam University Medical Center (UMC), University of Amsterdam, Amsterdam Cardiovascular Sciences, Amsterdam, Netherlands
- 2Departments of Internal and Vascular Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 3Department of Medical Biochemistry, Amsterdam UMC, University of Amsterdam, Amsterdam Cardiovascular Sciences, Amsterdam, Netherlands
An abdominal aortic aneurysm (AAA) is a dilatation of the abdominal aorta, which can potentially be fatal due to exsanguination following rupture. Although AAA is less prevalent in women, women with AAA have a more severe AAA progression compared to men as reflected by enhanced aneurysm growth rates and a higher rupture risk. Women are diagnosed with AAA at an older age than men, and in line with increased osteoporosis and cardiovascular events, the delayed AAA onset has been attributed to the reduction of the protective effect of oestrogens during the menopausal transition. However, new insights have shown that a high follicle stimulating hormone (FSH) level during menopause may also play a key role in those diseases. In this report we hypothesize that FSH may aggravate AAA development and progression in postmenopausal women via a direct and/or indirect role, promoting aorta pathology. Since FSH receptors (FSHR) are reported on many other cell types than granulosa cells in the ovaries, it is feasible that FSH stimulation of FSHR-bearing cells such as aortic endothelial cells or inflammatory cells, could promote AAA formation directly. Indirectly, AAA progression may be influenced by an FSH-mediated increase in osteoporosis, which is associated with aortic calcification. Also, an FSH-mediated decrease in cholesterol uptake by the liver and an increase in cholesterol biosynthesis will increase the cholesterol level in the circulation, and subsequently promote aortic atherosclerosis and inflammation. Lastly, FSH-induced adipogenesis may lead to obesity-mediated dysfunction of the microvasculature of the aorta and/or modulation of the periaortic adipose tissue. Thus the long term increased plasma FSH levels during the menopausal transition may contribute to enhanced AAA disease in menopausal women and could be a potential novel target for treatment to lower AAA-related events in women.
Introduction
An abdominal aortic aneurysm (AAA) is a dilatation of the abdominal aorta with a diameter of ≥ 3 cm, which, after further aortic dilatation, poses a risk for aortic rupture and subsequent death (1). Although the prevalence of AAA is higher in men than in women, the condition in women is more deleterious as they are at higher risk of rupture than men at the same aortic diameter and have increased AAA growth rates than men (2–7). While the higher rupture risk at equal aortic diameters may be explained by the fact that women are in general smaller than men, which suggests a proportionally greater enlargement, this does not explain the enhanced AAA growth rates in women. Therefore, the exact cause of this morbidity in women is not yet fully understood. The hormonal changes during the menopausal transition have been a subject of interest as a possible explanation since women present themselves with AAA at an older age than men (8). However, the role of sex hormones in the progression of AAA in women is inconclusive as described in a review (9) and will be elaborated on further below after briefly addressing current management and characteristics of the pathogenesis of AAA. As hormone replacement therapy in postmenopausal women was not clearly negatively associated with AAA (10, 11), this led to the exploration of an additional (hormonal) factor that may be at play. The current report hence shows the development of AAA in postmenopausal women from another hormonal perspective, leading to our hypothesis that follicle stimulating hormone (FSH) may enhance AAA onset or progression, via different mechanisms as outlined below.
Contemporary Surgical and Pharmacologic Treatment Options for AAA
Elective surgical AAA repair to prevent rupture is currently indicated at an aortic diameter of more than 5.0 cm in women and more than 5.5 cm in men (12). In the acute setting, patients with ruptured AAA also require surgery. The observation that women have higher mortality rates than men after both elective and ruptured AAA repair is worrisome (13, 14). Female sex has been associated with mortality and it has been suggested that it may be a proxy of risk factors that are specific to women (15).
Prior research of ex vivo cultures of human AAA tissue, animal AAA models and clinical trials in patients investigated various pharmacological agents as an intervention, which were aimed at inhibiting AAA progression. Outcomes for effectiveness of these drugs include the growth of AAA diameter in mm, AAA characteristics on histology level, or the measurement of AAA-related markers, such as the proteases matrix metalloproteinase 2 (MMP-2) and/or 9 (MMP-9) (16–18). Suggested possible future roles of those pharmacological agents were to be used as prevention in patients at risk for AAA, patients with a small AAA diameter or after surgical repair as additional support (19). Despite extensive AAA research, currently no established drugs are available that can diminish or stabilize AAA growth (19–22), besides cardiovascular risk management that may improve long-term survival of AAA patients (23). Interestingly, diabetes has been reported to be inversely associated with AAA (24, 25). An explanation for this observation from a pharmacological point of view is that diabetes medication, such as metformin, may have played a role through its vascular anti-inflammatory effects and have an effect on other mechanisms that are involved in AAA formation (26–29). As such, in part based on a large retrospective AAA study with 13,834 AAA patients (30), there are currently three trials running using metformin (NCT04500756, NCT04224051, NCT03507413). However, in the large retrospective AAA study, there were only 0.6% women with an AAA and a sub-analysis of the association of metformin prescription with the protection of AAA in women was not shown (30). Since it is not yet clear if metformin is also associated with lower risk of AAA in women, further research to find a potential target for pharmacological treatment is needed, with perhaps a different target in women.
AAA Histopathology and Pathogenesis
The understanding of the mechanisms involved in AAA pathogenesis has been expanding. Many complex processes have been identified to contribute to the remodelling and weakening of the aortic wall, leading to AAA (1, 22). On a histopathological level, AAA is characterized by smooth muscle cell (SMC) apoptosis, accumulation of inflammatory cells, extracellular matrix (ECM) degradation and oxidative stress (1, 31). However, it seems that not only the abdominal aorta is affected in patients with AAA but also the rest of the cardiovascular system. A study using individuals with asymptomatic AAA from the UK Small Aneurysm Trial showed an association between AAA diameter and cardiovascular and all-cause mortality before aneurysm surgery or rupture and after surgical repair, suggesting that AAA diameter is a marker for progressive cardiovascular disease (CVD) (32). Another study showed that patients with small AAA had a high prevalence of CVD and that patients with small AAA were at increased risk for CVD death (33). Indeed, atherosclerosis in itself is a major contributor to CVD. Although AAA share some common risk factors with atherosclerosis, including advanced age, smoking and hyperlipidaemia (34–36), there was some uncertainty if atherosclerosis has a causal role in AAA or if both conditions coexist (36–38). The current view is that the mechanisms for both pathologies are different, yet that certain risk factors may overlap (36–38). Furthermore, while atherosclerosis does not always precede AAA, it could contribute to AAA through the effects of chronic inflammation (36, 39). A study comparing AAA tissue with atherosclerotic wall samples from the same region showed that AAA distinguishes from atherosclerotic disease by enhanced expression and activation of inflammatory transcription factors (40). This finding is corroborated in a recent study that compared AAA patients with age and sex-matched atherosclerotic patients, and demonstrated that inflammation and calcification of the aorta was greater in patients with AAA (41). On a histological level, the evidently highly inflamed aorta in AAA shows massive infiltration of inflammatory cells in the outer SMC layers (media) and external collagen layer (adventitia). These inflammatory cells can form lymphoid follicles and some are united into lymph node-like structures (42). During this process, microvessels in the medial and adventitial layer, also known as the vasa vasorum, are surrounded by T-cells, dendritic cells, macrophages and to a lesser extent B-cells (42), revealing that the inflammatory cells in AAA are likely recruited from the vasa vasorum.
Hormones in Relation With AAA
Sex Steroid Hormones: Oestrogen and Testosterone
There is an increasing number of studies on hormonal differences in AAA as a potential explanation for the worse morbidity in women compared to men. Many studies have investigated the effects of both endogenous and exogenous oestrogen. The cellular mechanisms of oestrogen in modulating anti-inflammatory and vasoprotective processes in response to vascular injury have been identified in multiple laboratory studies (43, 44). As plasma levels of oestrogen diminish around the menopause (45), AAA has been proposed to be attributed to the decline of the protective effect of oestrogen. In support of this theory, an observational study found that women with earlier menopause were characterized by larger AAA diameters (46). The oestrogen effect has also been demonstrated in various animal AAA models (47–50). Two experimental AAA studies that compared ovariectomized mice with a sham operation group showed specifically an increased expression of MMP-9 in AAA tissue in the ovariectomized group (47, 48). Whereas one study also showed an increase in MMP-2 and the other study found no difference (47, 48). Both are proteases that have been identified to promote the development of AAA (51, 52). The effect of oestrogen supplementation in older female rats however seems to be lost (43, 53). This was demonstrated in a study in which older female rats and young female rats underwent balloon injury of the carotid artery and were treated with either vehicle or oestrogen. Upon injury, oestrogen did not reduce, but actually enhanced neointima formation in older female rats while it did reduce neointima formation in young female rats. Furthermore, oestrogen did not have an inhibitory effect on leukocyte infiltration and mRNA expression of inflammatory mediators in injured arteries as is seen in young female rats (53). The timing hypothesis has been proposed to explain this discrepancy in older versus younger subjects. This hypothesis consists of the protective effect of oestrogen/oestrogen and progestin on atherosclerosis when given early after the onset of menopause in women, which is lost when initiated long after the menopause (54). This hypothesis may explain why hormone replacement therapy (HRT) in older postmenopausal women is associated with no benefit or even a slightly higher risk of AAA, whereas HRT was reported to have an inverse association with AAA in women who had a mean age of 43.5 years (10, 11, 55, 56). This complex and inconclusive effect of HRT was also seen in the setting of CVD in postmenopausal women (57–59), which have led to the question if there may be an additional factor that could have contributed to the differences observed after the menopause.
While oestrogen receptors are considered to be protective transcription factors in vascular disease (60), a single administration of testosterone in female neonatal mice increased the AAA incidence compared to mice administered vehicle as neonates in the angiotensin-II induced aneurysm/dissection model (61). In addition, the authors reported that the external diameters of the abdominal aortas and aneurysm pathology of female mice administered testosterone as neonates were similar to those observed previously in adult male mice by the same study group (61). Testosterone is also an essential hormone in women and circulating testosterone plasma levels were slightly lower than those of oestrogen in premenopausal women in a cross-sectional study (62). In a prospective longitudinal study, the total testosterone plasma levels were annually measured in women who underwent menopause and the study reported that the total testosterone levels were unchanged across the menopausal transition (63). Although interestingly, Burger et al. (63) reported higher pre-final menstrual period testosterone levels compared to the levels reported in premenopausal women by Skiba et al. (62). Could the timing of drawing blood partly explain this difference? As oestrogen levels decrease during/after the menopause, the ratio of testosterone to oestrogen increases (64, 65). Testosterone activates the androgen receptor, a transcription factor similar to the oestrogen receptors, that is expressed in many different cell types, including in vasculature and immune cells (66, 67). While some researchers found that pharmacological blockade of the androgen receptor or deficiency of the receptor in male mice attenuated AAA formation (68) and castration of male mice with established AAA promoted aneurysm stabilization (69), others find the opposite (70). Also in humans there is controversy as to the beneficial or detrimental roles that testosterone (replacement therapy) plays in vascular disease (60, 71–74). Since testosterone replacement therapy is provided nowadays in women and men for various reasons (72, 75), for example in postmenopausal women for treating hypoactive sexual desire disorder (76), its effect on AAA development may become evident in the future. So far there is only an association of low testosterone levels in men with AAA (77). A more elaborate overview of sex hormones in AAA is given in a review by Makrygiannis et al. (78). Taking these findings together, the inconclusive effect of oestrogen and the as-yet unidentified role of testosterone on AAA in postmenopausal women have led to the exploration of the potential impact of other hormones.
Gonadotropins: Follicle Stimulating Hormone and Luteinizing Hormone
Upon a reduction in oestrogen production in the menopausal transition, there is a gradual increase in follicle stimulating hormone (FSH) and luteinizing hormone (LH) levels. FSH and LH are hormones produced and released by the anterior pituitary gland. They are gonadotropic hormones because they stimulate the function of ovaries in females and testis in males with the purpose of reproduction. In women, the hormones activate the ovaries to produce oestrogen, androgen and progesterone to stimulate follicular growth, maturation and ovulation (79, 80). In men, both gonadotropins are necessary for spermatogenesis. FSH and LH stimulate the production of inhibin and testosterone by the Sertoli and Leydig cells, respectively (81). During the menopausal transition period in women, when the follicle reservoir in the ovaries is diminishing, plasma FSH and LH levels become elevated to maintain follicle development and compensate the decrease in oestrogen production (82). Once the ovaries are completely depleted of follicles, the negative feedback by oestradiol on the hypothalamus and/or pituitary gland is missing, which leads to excess gonadotropin production (83). Whilst FSH and LH are mostly known for their gonadal functions, FSH and LH receptors (FSHR and LHR) are also found in numerous cell types unrelated to sexual development (84, 85), which suggests there are roles for FSH and LH beyond its gonadal function in women. These FSH and LH receptors may become extensively activated upon chronic high FSH and LH blood levels seen during the menopause. In this respect, women with Turner Syndrome, a genetic condition due to a chromosomal abnormality where (part of) one of the X chromosomes is missing, is partly characterized by hypergonadotropic hypogonadism (86). These patients are at risk to develop aortic dilatations and associated dissections in the ascending and descending thoracic aorta (87, 88), and multiple other comorbidities including hypertension, increased risk of bone fracture, impaired glucose tolerance, increased total fat mass, and a range of congenital heart diseases and autoimmune diseases (86). Would it be possible that FSH and LH are not innocent bystanders, but are active participants in aneurysm formation? Indeed, high plasma LH has been associated with increased ischaemic heart disease and AAA in older men (77, 89). Contributing to CVD, a study with ovariectomized female mice showed that administration of LH promoted atherosclerosis (90). For the interested reader, a number of extragonadal sites of LHR expression is summarized in a review on the extragonadal effects of LH and chorionic gonadotropin (CG) (85), including the (foetal) adrenal gland and kidneys, and other parts of the reproductive tract such as the cervix, oviduct, endometrium and myometrium (91, 92). However, as contemporary studies demonstrated a possible role for FSH in conditions which were once thought to be solely a result of the diminished protective effect of oestrogen in women during the menopausal transition (84), we similarly focused in the current report on the potential role of the extragonadal FSHR in AAA. The previously described potential role of FSH with respect to postmenopausal osteoporosis and cardiovascular disease can be appreciated in an excellent review by Zhu et al. (84). Furthermore, for the interested reader, we refer to a recently published chapter that has been dedicated to various aspects of FSH in fertility that extends to the potential actions beyond fertility (93). We summarized some important studies on the extragonadal FSHR in Supplemental Table 1 which may be relevant for the current report. Although Chrusciel et al. reviewed some discrepancies found in FSH research, suggested to be partly based on species-specific findings and/or the use of poorly validated antibodies (94), with the current knowledge, there are signs that there is a role for FSH in various diseases in postmenopausal women.
Hypothesis: FSH Enhances the Onset or Progression of AAA
FSH is a glycoprotein polypeptide hormone and can exert its effects on cells expressing the FSH receptor (FSHR) on their cell membrane. FSH primarily stimulates signalling in the granulosa cells in ovaries and Sertoli cells in testis (84). Some extragonadal sites with FSHR expression have been identified that may contribute to disease, upon chronic stimulation. The most profound examples are postmenopausal CVD and osteoporosis, which were thought to be primarily related to reduced oestrogen levels (95, 96). New insights however reveal that FSH can contribute to the development of both conditions (84). Therefore, we believe that the elevation of circulating FSH for a number of years spanning the menopausal transition may also have consequences for the development of other diseases affecting postmenopausal women. An example of extragonadal FSHR expression is in stem cells in bone marrow in mice (97). Upon stimulation with FSH in women, those stem cells were shown to be mobilized to the circulation (98), which thereby reach all organs. We hereby hypothesize that there may be direct and/or indirect effects of FSH that contribute to AAA development or progression. First, a summary of known mechanisms promoting AAA development is provided in Figure 1. The potential effects of FSH on AAA are described in the following paragraphs and summarized graphically in Figure 2.
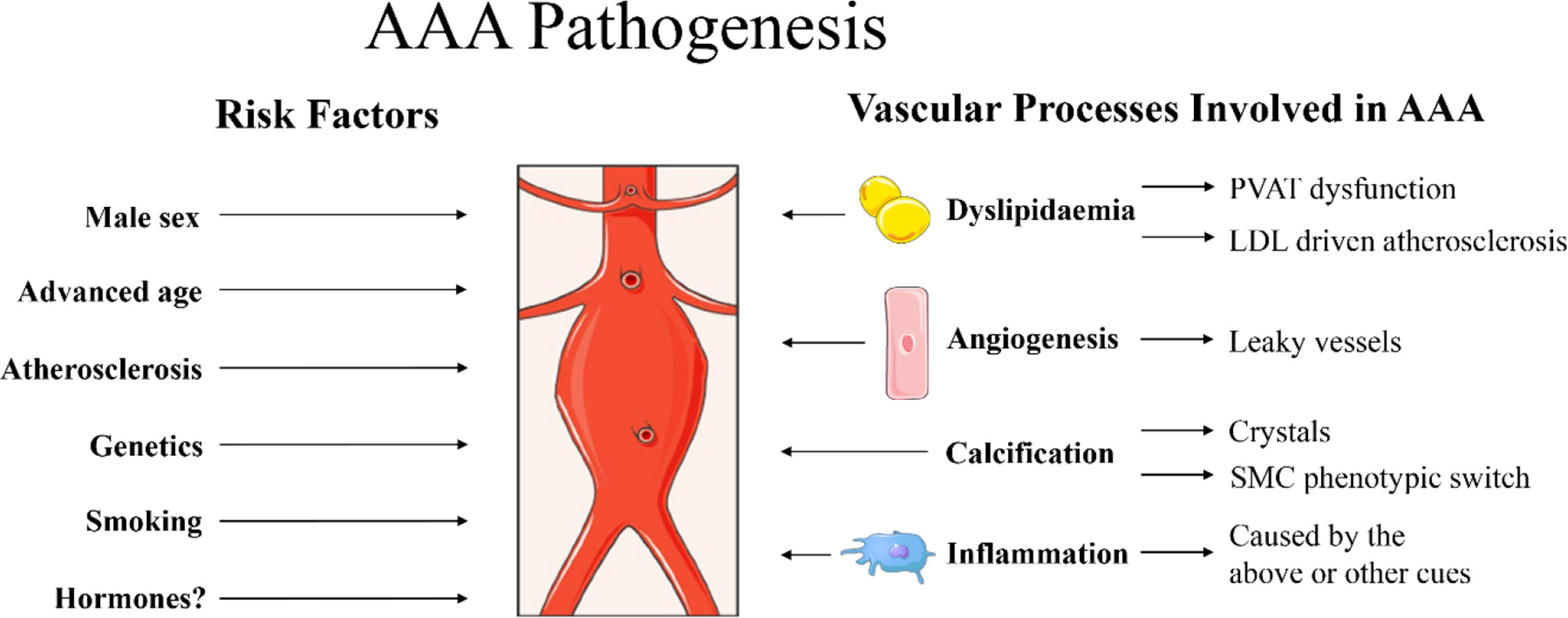
Figure 1 Summary of risk factors for abdominal aortic aneurysm (AAA) pathogenesis as described in the current report. Male sex, advanced age, atherosclerosis, genetics and smoking are identified clinical risk factors for AAA (1). Hormones are also suggested to play a role, although the exact mechanisms are still under investigation. Specific processes at play in the vascular wall that may promote AAA include dyslipidaemia, promoting dysfunctional perivascular adipose tissue (PVAT) (99, 100) and low-density lipoprotein (LDL) driven atherosclerosis (39). Furthermore, angiogenesis [newly formed angiogenic vessels with increased permeability that can result in vascular leakage (101, 102)], calcification (41) [with crystals causing damage to the vascular wall and/or vascular mineralization through smooth muscle cell (SMC) phenotypic switch into osteoblast-like cells (103)] and massive inflammation (1) are also observed in aneurysmal tissue. Images were used from Smart Servier Medical Art.
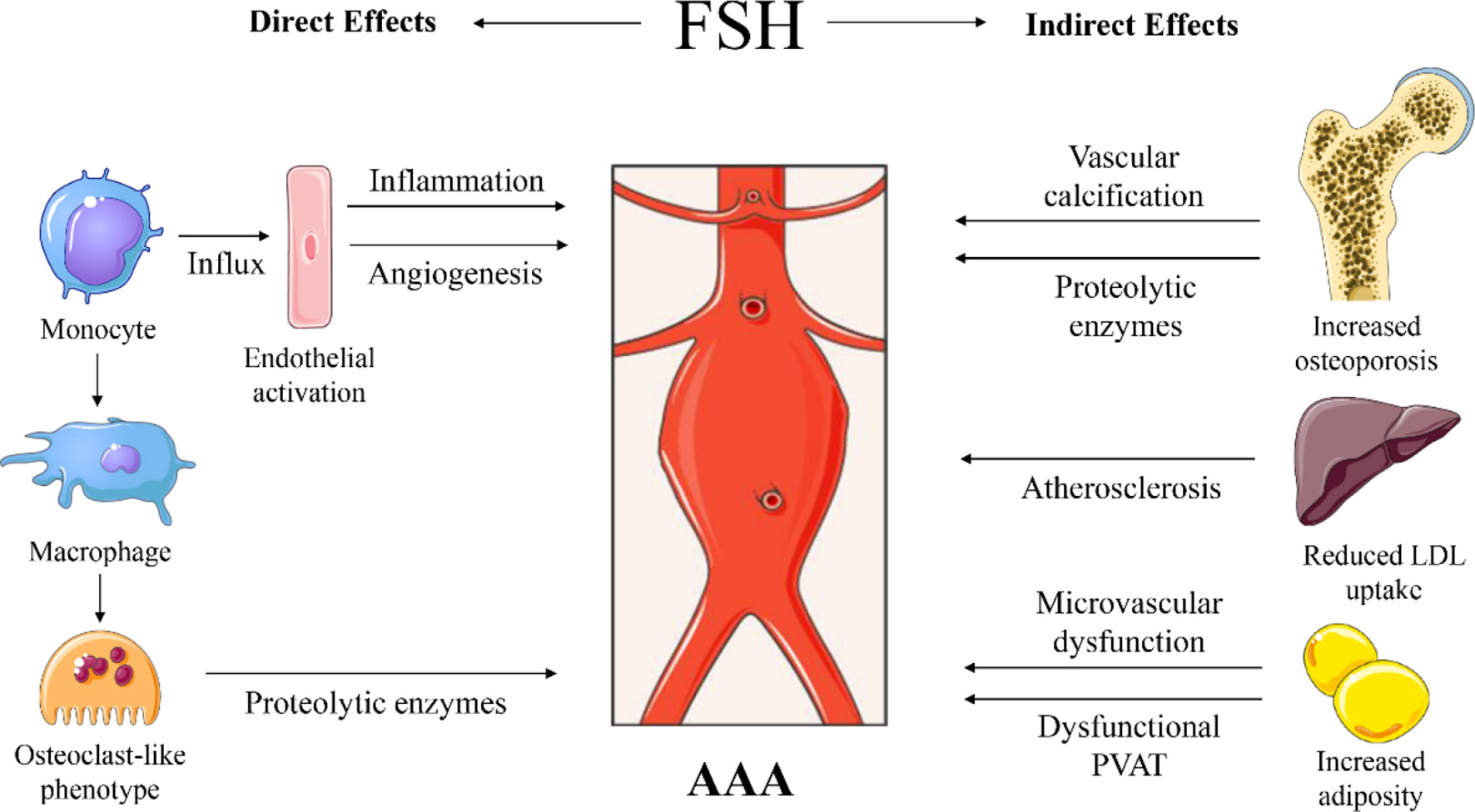
Figure 2 Proposed sites of action and mechanisms of follicle stimulating hormone (FSH) on abdominal aortic aneurysm (AAA) development. Direct effects through increased influx of monocytes in the vascular wall, through FSH-mediated endothelial cell activation and angiogenesis. FSH can stimulate macrophages to become osteoclast-like cells that will produce proteolytic enzymes. Indirect effects through FSH-induced osteoporosis associated with vascular calcification and potential systemic release of proteolytic enzymes. FSH reduces low-density lipoprotein (LDL) cholesterol uptake by the liver and increases cholesterol biosynthesis, causing increased LDL in the circulation, contributing to atherosclerosis. FSH induces adipogenesis, thus adiposity with potential microvascular dysfunction and impaired perivascular adipose tissue (PVAT) function. Images were used from Smart Servier Medical Art.
The Direct Effects of FSH on the Vascular Wall
Macrophages
Macrophages are inflammatory cells that can play a pro-inflammatory or anti-inflammatory role and as such can regulate tissue injury and repair (104). In AAA, there are many different processes ongoing that macrophages are also thought to be involved in, such as ECM remodelling, inflammation and oxidative stress (105). Dependent on (micro)environmental cues, macrophages can adopt different phenotypes. As such, if disturbed, an imbalance in the identity and function of the macrophage can result in (vascular) disease (106, 107). Interestingly, macrophages are involved in vascular calcification (108). In AAA and atherosclerosis calcification is often observed, and in atherosclerotic plaques the contribution of macrophages to vascular calcification is proposed to be via disrupted signalling in macrophages that impairs their osteoclast-like activity, and/or via macrophage-derived cytokines that induce osteogenic differentiation and mineralization of SMC (109, 110). During SMC calcification in the aortic root in mice, oxidative stress induced the expression of receptor activator of nuclear factor-κB ligand (RANKL) via runt-related transcription factor 2 (Runx2; a transcription factor associated with osteoblast differentiation) in SMC. This upregulation increased bone marrow derived monocyte migration and differentiation into tartrate-resistant acid phosphatase (TRAP) positive osteoclast-like cells (111). This finding suggests a direct role for osteoclast-like cells in vascular calcification. Possibly, there is also a role for osteoclastogenesis in AAA (112, 113). AAA often contain advanced stages of atherosclerosis with calcification. Due to the abundance of macrophages in atherosclerotic areas in AAA, these cells can be a potential local source of osteoclast-like cells (112, 114). The role of osteoclast-like cells in AAA has been demonstrated in an interesting study using human aortic tissue, the murine RAW 264.7 macrophage cell line, and two aneurysm mouse models using calcium chloride (CaCl2) and angiotensin II (113). The authors showed that the majority of the macrophages in human AAA tissue was differentiated into osteoclast-like cells (TRAP positive) and that they produced significantly greater MMP-9 activity compared to the undifferentiated macrophages (113). Furthermore, the authors showed that Tumour Necrosis Factor alpha (TNFα) in the presence of calcium phosphate enhanced macrophages to differentiate into osteoclast-like cells (TRAP-positive cells) in vitro (113). This suggests that the deposition of calcium phosphate is a prerequisite for the formation of osteoclast-like cells from macrophages via TNFα in AAA.
The macrophages in the aortic wall in AAA are mostly derived from peripheral blood monocytes (105), a cell-type on which the FSHR has been identified previously (115). A study on bone loss, using mice, an in silico model and in vitro data, suggested that stimulation of macrophages with FSH resulted in increased TNFα expression that increased the osteoclast precursor pool (116). FSH could possibly have the same effect in the vascular wall since vascular calcifications in atherosclerotic plaques resemble bone calcifications as the organic matrix and calcium phosphate mineral atomic interface has been suggested to be similar in both conditions (117). Thus if we would extrapolate the findings in the setting of bone loss to the (calcified) vascular wall, FSH stimulation could promote macrophage differentiation into osteoclast-like cells via TNFα, and subsequently contributed to ECM degradation in AAA.
Next to changing the macrophage phenotype to an osteoclast-like cell, can FSH also stimulate the newly formed osteoclast-like cells directly to become more proteolytic? Indeed, FSH stimulation of RANKL-induced osteoclast-like cells (derived from RAW264.7 macrophages) induced a dose-dependent increase in mRNA expression of RANK, TRAP, MMP-9 and cathepsin K (118). However, RANKL was not upregulated in end stage aneurysmal tissue (113), suggesting that osteoclastogenesis in AAA may not be RANKL-induced or may have occurred at an earlier disease stage.
These reports may indicate a role for FSH in the transdifferentiation of macrophages into osteoclast-like cells in the calcified aorta through TNFα. Once activated, an excessive production of proteases such as MMP-9 and cathepsin K, similar as in bone, can potentially degrade the aortic ECM, leading to AAA progression. Both MMP-9 and cathepsin K are higher in human AAA tissue compared to the atherosclerotic aorta (119). Evidence of the extrapolation of the effects of FSH on macrophages and osteoclast-like cells from an in vitro setting into one in vivo in human AAA however requires further investigation.
Endothelial Cells
In addition to FSHR expression on monocytes and osteoclasts, FSHR has been found on endothelial cells. Endothelial cells form the inner lining of the vasculature and are responsible for a proper barrier function and influx of inflammatory cells, amongst many other functions (120). FSH stimulation was observed to promote vascular endothelial adhesion molecule-1 (VCAM-1) expression on endothelial cells, which is a protein involved in monocyte influx through the endothelial cell layer into the tissue (121). This finding suggests a functional FSHR on endothelial cells and may promote the pro-inflammatory influx of cells into the aortic vessel wall. Furthermore, FSH was shown to induce a proangiogenic response similarly effective as vascular endothelial growth factor in cultured endothelial cells (122). These findings were further substantiated by showing that the FSHR is present on endothelial cells in angiogenic blood vessels in different types of tumours (123). In the aorta, the microvascular bed responsible for ample nutrient and oxygen delivery is the vasa vasorum. Upon vascular wall damage, neovascularisation in the arterial wall in AAA is common and seems to be an ongoing process to promote cellular survival in areas of decreased nutrient and oxygen supply (124, 125). Inflammatory cues are also triggers for neovascularisation as is observed in atherosclerotic plaques (101). Newly formed angiogenic vessels have an increased permeability that results in vascular leakage and enhanced inflammatory cells migration into the tissue (101), that may promote the inflammatory status of the AAA. This could possibly contribute to a vicious cycle of angiogenesis and inflammation in AAA. Angiogenesis in AAA has mainly been observed in the SMC-rich medial layers (126) and a correlation between medial neovascularisation and inflammatory infiltration in AAA has been observed (102). As such, angiogenesis should be considered a sign of disease progression as increased gene expression of angiogenic factors was observed at the edge of ruptured AAA compared to other sites within the ruptured AAA (102). In line with the angiogenic factors at the edge in ruptured AAA, the density of microvessels in the medial layer of AAA was significantly increased and the microvessels had smaller diameters (102). Taken together, if FSH contributes to increased VCAM-1 expression and enhanced angiogenesis, this may lead to increased inflammation in the aortic media and subsequent AAA progression.
Indirect Effects of FSH on AAA
Osteoporosis and Vascular Calcification
Osteoporosis is characterized by decreased bone mass density (BMD) and discrepancies in the rate of bone resorption (osteoclasts) and formation (osteoblasts), favouring resorption (127). Although seemingly paradoxical, osteoporosis has been associated with arterial calcification in epidemiological studies (128). Vascular calcification is characterized by the pathological deposition of calcium phosphate crystals (129). The observation that both conditions coexist suggest a common mechanism and there are a number of theories on the development of vascular calcification (129, 130). While vascular calcification was believed to be a passive process, comprising the precipitation of calcium and phosphate (131), more recently, it is considered to be an active process, consisting of a regulated process between the induction of osteogenesis and loss of inhibitors of mineralization (131). Changes in blood and urinary calcium have been observed in disease states where bone turnover is increased such as in hyperparathyroidism (132), but could perhaps to a lesser extent also be applicable in osteoporosis. Elevated calcium and phosphate levels could exacerbate vascular calcification (129, 133–135). The accumulating evidence around the mechanisms of osteoporosis and vascular calcification is too extensive to cover in the current work. Although the exact and highly complex mechanisms are as-yet to be determined, possibly the systemic imbalance of bone minerals and associated regulatory factors in the process of osteoporosis can be associated with vascular calcification.
As such, aortic calcification has been seen in relation with postmenopausal osteoporosis. In postmenopausal women, lower metacarpal bone mass and density was associated with a higher degree of abdominal aortic calcification (136), even when adjusted for age (137). Another study suggested a possible role of hormonal factors that are unique to women, showing that after adjustment for age and other confounders, a significant association between vascular calcification of the abdominal aorta and lower bone mass was seen in women and not in men (138). This potential relationship with a hormonal aspect coincides with an older case-control study, showing that the prevalence of aortic calcification was higher in men than in women <65 years, yet after the age of 65, women had more vascular calcification than men (139). With a change in prevalence between the sexes after the age of 65 years, the hormonal alterations during the menopause are of interest. Although the exact mechanism is still under investigation, perhaps there is a role for FSH linking osteoporosis and aortic calcification in postmenopausal women as a shared mechanism. For postmenopausal osteoporosis treatment, denosumab acts by interrupting RANKL-RANK signalling in osteoclasts (140). A role for FSH in osteoporosis may be through involvement of RANKL-induced osteoclastogenesis as at perimenopausal concentrations (50 mIU/ml) FSH can induce increased RANK expression in peripheral blood mononuclear cells (141). In line hereof, FSH treatment of murine bone marrow cells that were differentiated with RANKL and CSF-1 into osteoclasts, resulted in increased osteoclastic differentiation. Subsequently, when antibodies were used against FSH, the FSH-induced osteoclast formation was inhibited. The authors also compared the formation of TRAP+ osteoclasts after RANKL treatment between FSHR-/-, FSHR+/- and wild type mice. In mice devoid of the FSHR, the formation of osteoclasts was significantly attenuated (142). Given the presence of the FSHR on murine and human osteoclasts, a direct role for FSH may be suggested (115, 142, 143). Thus the question arises how the consequences of enhanced osteoporosis and aortic calcification could be responsible for AAA progression.
Interestingly, an elevated degree of calcification of the aortic wall was seen in symptomatic and ruptured AAA patients compared to asymptomatic AAA patients (144), which suggests that aortic calcification is associated with a severe AAA phenotype. Although another study does not reinforce this theory (145), in a murine AAA model, promoting aortic medial calcification with calcium phosphate enhanced AAA severity (146). The phenotypic switch of SMC into osteoblast-like cells has been identified as an important factor in this matter in relation to AAA (103). An in vitro and in vivo study showed that under calcifying conditions, aortic SMC lost their SMC markers SM22α and SM α-actin, and gained osteogenic markers (alkaline phosphatase, osteocalcin and osteopontin) (147). Calcification consists of macrocalcification (≥ 50 µm crystals) and microcalcification (< 50 µm crystals), in which the latter has been seen to be more prevalent in mouse and human AAA and is suggested to associate positively with AAA formation (148). More specifically, SMC microcalcification mediated via Runx2 seems to precede AAA and its presence can further enhance inflammatory cytokines, MMPs or activation of the NLR family pyrin domain-containing 3 (NLRP3) inflammasome pathway (148). A potential mechanism of AAA development in the calcified aorta is that the calcium crystals can physically damage the aorta, which attracts inflammatory cells. Otherwise, the calcification cues may induce macrophages phenotypic switching into the previously mentioned osteoclast-like cells that can produce various proteolytic enzymes, as an association between calcification and elastic lamina degradation in the aortic ECM has been seen in a murine model (149). A role for osteoclasts was demonstrated in a murine AAA model with calcium phosphate. When bisphosphonate (a drug to treat osteoporosis) was administered to bind to calcium phosphate, this resulted in the inhibition of vascular osteoclastogenesis (113). Could FSH possibly also affect these osteoclasts? Although there are signs that aortic calcification may be an important preceding factor, how this may exactly affect aneurysm development remains to be elucidated.
Alternatively, during bone resorption as in osteoporosis, osteoclast markers cathepsin K and TRAP can be found in the circulation (150). The higher production of these proteins during osteoporosis is reflected in the finding that postmenopausal women with osteoporosis have higher circulating levels of cathepsin K (151, 152). Cathepsin K is a known protease which is involved in AAA (119) and may not only be produced locally by activated (osteoclast-like) macrophages, but could possibly also come from the circulation.
Lipid Metabolism: Cholesterol Accumulation and Adipogenesis
Another indirect effect of FSH may be via the liver. The liver plays a key role in lipid homeostasis, by taking up cholesterol from the circulation, and to be disposed of via bile and faeces (153). The FSHR is present on human hepatocytes and it has been reported that FSH attenuated endocytosis of cholesterol-rich low density lipoprotein particles (LDL) by reducing the LDL receptor (LDLR) expression in liver tissue (154). The downregulation of LDLR expression consequently reduces LDL uptake from the circulation. This increases serum LDL levels and subsequently promotes LDL accumulation in the arteries enhancing arterial inflammation (154). Furthermore, FSH regulated cholesterol biosynthesis by inducing 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and sterol regulatory element-binding protein 2 (SREBP-2) expression in murine hepatocytes and human HepG2 cells, further enhancing cholesterol blood levels (155). Both processes contribute to atherosclerosis, which may affect AAA development. The atherosclerotic plaques will become more advanced, promoting vascular inflammation and calcification (156, 157).
Adipose tissue has also been reported to be FSH sensitive. White adipocytes function as storage for lipids (triglycerides), while brown adipocytes burn these lipids to generate heat (158). In ovariectomized mice that have increased FSH plasma levels, blocking the FSHR with an FSH antibody resulted in reduced adiposity and increased production of thermogenic adipose tissue (159). The localization of the FSHR on adipocytes implicated a direct role for FSH in adipogenesis (159). If these findings were to be extrapolated to humans, FSH could enhance the accumulation of body fat, which is associated with CVD and metabolic disease in postmenopausal women (160). If FSH contributes to body fat accumulation leading to obesity, then microvascular dysfunction throughout the body may be present as obesity is associated with microvascular dysfunction, including endothelial dysfunction (161–163). Several mechanisms have been identified that can modulate the microvasculature; in part this is affected through visceral adipose tissue derived inflammatory adipokines (161, 164). These changes in the microvasculature can result in impaired tissue oxygenation and low grade inflammation, although the exact mechanism and impact of obesity on the microcirculation can differ between organs (161). As such, possibly (the endothelial cells of) the vasa vasorum oxygenating the aorta can similarly be affected.
Furthermore, changes in lipid accumulation or lipid serum levels can also affect perivascular adipose tissue (PVAT) (161, 162). PVAT is adipose tissue surrounding arteries and is critical to maintain the normal functional status of the vasculature, partially through secretion of paracrine factors (165). However, dysfunctional PVAT including obese or aged PVAT can induce abnormal changes and vascular pathology (165). The alterations in PVAT induced by obesity may thus have consequences. In obese rats with metabolic syndrome, PVAT mass was increased and PVAT-derived cytokine leptin contributed to SMC phenotypic switching in vascular remodelling (99). A potential direct link through PVAT and AAA development was suggested in the angiotensin II-induced AAA mouse model. Under obese conditions, secretion of platelet-derived growth factor-D (PDGF-D) by PVAT contributed to AAA formation in these mice through adventitial fibrosis and inflammation (100). In humans, a relationship between the alteration in PVAT and AAA has also been suggested (166–168). This was reflected in the findings that in AAA patients compared to controls, PVAT had a higher density on CT-scans (differences in PVAT quantity) and an increased pro-inflammatory and MMP gene expression profile (166, 167). Moreover, in human AAA tissue there is an increase in adventitial adipocyte clusters and higher expression of adipogenic transcription factors (169). Interestingly, AAA-derived mesenchymal cells showed enhanced adipogenic potential in culture, compared to control aortic tissue-derived cells (169). Lastly, the significant enrichment of adipogenesis in ruptured AAA supports an association between the extent of PVAT remodelling and rupture (169). Thus, the extragonadal effects of FSH leading to increased cholesterol levels and adiposity in postmenopausal women could have indirectly led to enhanced atherosclerosis with arterial inflammation and calcification, dysfunctional vasa vasorum and PVAT, all potentially affecting AAA development.
Discussion
The current report highlights the potential extragonadal roles of FSH in AAA progression, which may in part explain the severe course of AAA pathogenesis in postmenopausal women. Improving our understanding of the mechanisms underlying AAA development in women should lead to personalized treatment in women in the future. While oestrogen has been believed to protect premenopausal women from AAA, HRT supplementation in postmenopausal women was not associated with protection against AAA. This could in part be explained by the “timing hypothesis theory”, as discussed earlier. However, there may be an additional hormonal factor that contributes to the more severe AAA phenotype in postmenopausal women, which is in line with newly gained insights of the role of FSH in postmenopausal osteoporosis and CVD. Our hypothesis is that the chronically high FSH plasma levels during the menopausal transition may directly and/or indirectly promote AAA severity. A direct role for FSH in the calcified aorta may be in triggering the macrophages to differentiate into osteoclast-like cells, which through their proteolytic activity can degrade the ECM of the aorta. Secondly, FSH can induce endothelial cells to become angiogenic and to express adhesion molecules that attract inflammatory cells (121, 122). For the indirect role, FSH is a newly identified key player in postmenopausal osteoporosis (84), which is associated with aortic calcification. Since vascular calcification is associated with enhanced AAA development, osteoporosis may have an indirect role in promoting AAA progression through aortic calcification. If and how osteoporosis and vascular calcification are causally related has yet to be determined. Furthermore, FSH can downregulate the expression of the LDL receptor and increase cholesterol synthesis in the liver, thereby enhancing circulating LDL which can accumulate in the aortic wall and increase atherosclerotic burden (154). Although there is no causal relation between atherosclerosis and the development of AAA, both conditions carry the same risk factors (34, 35). As atherosclerosis is a risk factor for AAA, this may aggravate AAA development by promoting aortic inflammation. Lastly, FSH can stimulate adipogenesis (159), which may induce adiposity with subsequent increased risk of microvascular dysfunction, potentially affecting the vasa vasorum. Furthermore, obesity has been associated with dysfunctional PVAT, which can secrete disease-promoting factors that may affect the aneurysmal vascular wall.
The current report focused on the hormonal impact on AAA progression in women. However, there is also evidence that hormonal factors play a role in the development of AAA in men. As the condition primarily affects men, testosterone has been thought to attribute to AAA as demonstrated in murine studies with male mice (68, 69), and even in female mice administered testosterone as neonates (61). However, a recent case-control study comparing 65-year old men with and without AAA found that higher oestrogen, higher progesterone and lower testosterone levels were associated with AAA (170). The association of low testosterone levels with AAA is in line with an earlier clinical and murine study (70, 77). This discrepancy in findings reflects the complexity of characterizing AAA development. As male sex is a known risk factor for AAA, this could be easily explained by hormonal differences between women and men. However, a genetic component may also be involved, demonstrated in an angiotensin II-induced AAA mouse model using phenotypic female mice with either sex chromosome XY or XX (171). The authors showed that sex chromosomes influence the gene expression profiles of abdominal aortas. In XY mice specifically, an increase in the expression of inflammatory pathway genes was observed. Interestingly, female XY mice exposed to testosterone had an increased rupture risk of AAA compared to female XX mice (171), suggesting that testosterone may have a different impact in men than in women.
Furthermore, as lifestyle aspect, smoking is an important risk factor for AAA, which is reported to be more prevalent in men across all age groups (172). However, for women who do smoke, the impact on AAA risk seems to be larger than for men (173). A complex interplay of various factors including hormonal, genetic and lifestyle elements seem to contribute to the differences in the development of AAA in men compared to women. However, there may still be other (as-yet unknown) factors that play a role in the sex-disparity of AAA.
Currently, no studies on the presence of the FSHR in aortic tissue and/or the potential effects of FSH on AAA have been conducted and there are some aspects to take into consideration when anticipating this line of research. While many researchers confirmed the presence of FSHR in extragonadal tissues on both protein and mRNA level, there are also studies that could not detect the FSHR as outlined in Supplemental Table 1. As the extragonadal FSHR may have a different splice variant (115), this may have challenged the detection of FSHR mRNA with primers that include exon 9 in one study (174). Alternatively, another study that could not detect the FSHR, used male osteoclasts (175). Could male sex have influenced this issue possibly? Furthermore, differences between experimental designs also affect results, reflecting the complexity of assessing the hormonal impact. For example, while transgenic mice with high FSH levels and normal LH levels showed an increase in bone mass, without detection of FSHR in bone (174), another study using FSHR-/- mice with high FSH levels demonstrated normal bone mass, and partial FSH deficiency increased bone mass (143).
As a first step in the investigation of a possible direct effect of FSH on the aorta, the mRNA expression or localization of the FSHR in aortic tissue should be assessed, to reveal if there are differences in FSHR gene expression or density on cells in the aorta and/or number of cells positive for FSHR, between AAA and non-diseased aortic tissue. Furthermore, the various cell types present in the aorta could be stimulated in vitro with/without FSH to determine the effect of FSH on those cells by measuring the differences in gene expression of e.g. inflammatory/osteoblast/bone resorption markers, depending on the cell type. The indirect role of FSH on AAA may be more difficult to capture. For example, in a mouse model of AAA with female mice, it would be interesting to explore if FSH-induced osteoporosis resulted in larger AAA diameters with enhanced AAA histopathology characteristics and microcalcification compared to female mice without osteoporosis. In humans, one may investigate if postmenopausal women with osteoporosis have larger AAA diameters than postmenopausal women without osteoporosis, indicating a severe AAA phenotype. Some challenges in detecting the FSHR and its effects on extragonadal cells have been elaborated on thoroughly in a previous review by Chrusciel et al. (94). Nevertheless, the role for FSH in AAA pathogenesis seems worth exploring by further fundamental and clinical research following the above pathways, as an attractive potential target to reduce the AAA-related morbidity in women.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
VT, MN, RB, and VW contributed to the concept of the work. VT wrote the manuscript with support from VW who supervised the writing process. All authors contributed to the article and approved the submitted version.
Funding
VT is funded by the AMC Foundation. MN is supported by a ZONMW VICI grant (number 09150182010020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.726107/full#supplementary-material
References
1. Kuivaniemi H, Ryer EJ, Elmore JR, Tromp G. Understanding the Pathogenesis of Abdominal Aortic Aneurysms. Expert Rev Cardiovasc Ther (2015) 13(9):975–87. doi: 10.1586/14779072.2015.1074861
2. Brown PM, Zelt DT, Sobolev B. The Risk of Rupture in Untreated Aneurysms: The Impact of Size, Gender, and Expansion Rate. J Vasc Surg (2003) 37(2):280–4. doi: 10.1067/mva.2003.119
3. Powell JT, Brady AR, Brown LC, Fowkes FG, Greenhalgh RM, Ruckley CV, et al. Long-Term Outcomes of Immediate Repair Compared With Surveillance of Small Abdominal Aortic Aneurysms. New Engl J Med (2002) 346(19):1445–52. doi: 10.1056/NEJMoa013527
4. Norman PE, Powell JT. Abdominal Aortic Aneurysm: The Prognosis in Women is Worse Than in Men. Circulation (2007) 115(22):2865–9. doi: 10.1161/CIRCULATIONAHA.106.671859
5. Skibba AA, Evans JR, Hopkins SP, Yoon HR, Katras T, Kalbfleisch JH, et al. Reconsidering Gender Relative to Risk of Rupture in the Contemporary Management of Abdominal Aortic Aneurysms. J Vasc Surg (2015) 62(6):1429–36. doi: 10.1016/j.jvs.2015.07.079
6. Mofidi R, Goldie VJ, Kelman J, Dawson AR, Murie JA, Chalmers RT. Influence of Sex on Expansion Rate of Abdominal Aortic Aneurysms. Br J Surg (2007) 94(3):310–4. doi: 10.1002/bjs.5573
7. Solberg S, Singh K, Wilsgaard T, Jacobsen BK. Increased Growth Rate of Abdominal Aortic Aneurysms in Women. Tromsø Study Eur J Vasc Endovasc Surg (2005) 29(2):145–9. doi: 10.1016/j.ejvs.2004.11.015
8. Egorova NN, Vouyouka AG, McKinsey JF, Faries PL, Kent KC, Moskowitz AJ, et al. Effect of Gender on Long-Term Survival After Abdominal Aortic Aneurysm Repair Based on Results From the Medicare National Database. J Vasc Surg (2011) 54(1):1–12.e6; discussion 1-2. doi: 10.1016/j.jvs.2010.12.049
9. Villard C, Hultgren R. Abdominal Aortic Aneurysm: Sex Differences. Maturitas (2018) 109:63–9. doi: 10.1016/j.maturitas.2017.12.012
10. Lederle FA, Larson JC, Margolis KL, Allison MA, Freiberg MS, Cochrane BB, et al. Abdominal Aortic Aneurysm Events in the Women’s Health Initiative: Cohort Study. BMJ (2008) 337:a1724. doi: 10.1136/bmj.a1724
11. Hsia J, Criqui MH, Herrington DM, Manson JE, Wu L, Heckbert SR, et al. Conjugated Equine Estrogens and Peripheral Arterial Disease Risk: The Women’s Health Initiative. Am Heart J (2006) 152(1):170–6. doi: 10.1016/j.ahj.2005.09.005
12. Wanhainen A, Verzini F, Van Herzeele I, Allaire E, Bown M, Cohnert T, et al. Editor’s Choice - European Society for Vascular Surgery (ESVS) 2019 Clinical Practice Guidelines on the Management of Abdominal Aorto-Iliac Artery Aneurysms. Eur J Vasc Endovasc Surg (2019) 57(1):8–93. doi: 10.1016/j.ejvs.2018.09.020
13. Sidloff DA, Saratzis A, Sweeting MJ, Michaels J, Powell JT, Thompson SG, et al. Sex Differences in Mortality After Abdominal Aortic Aneurysm Repair in the UK. Br J Surg (2017) 104(12):1656–64. doi: 10.1002/bjs.10600
14. Aber A, Tong TS, Chilcott J, Thokala P, Maheswaran R, Thomas SM, et al. Sex Differences in National Rates of Repair of Emergency Abdominal Aortic Aneurysm. Br J Surg (2019) 106(1):82–9. doi: 10.1002/bjs.11006
15. Indrakusuma R, Jalalzadeh H, Vahl AC, Koelemay MJW, Balm R. Editor’s Choice - Sex Related Differences in Peri-Operative Mortality After Elective Repair of an Asymptomatic Abdominal Aortic Aneurysm in the Netherlands: A Retrospective Analysis of 2013 to 2018. Eur J Vasc Endovasc Surg (2019) 58(6):813–20. doi: 10.1016/j.ejvs.2019.05.017
16. Nagashima H, Aoka Y, Sakomura Y, Uto K, Sakuta A, Aomi S, et al. Matrix Metalloproteinase 2 is Suppressed by Trapidil, a CD40-CD40 Ligand Pathway Inhibitor, in Human Abdominal Aortic Aneurysm Wall. J Vasc Surg (2004) 39(2):447–53. doi: 10.1016/j.jvs.2003.07.005
17. Mosorin M, Juvonen J, Biancari F, Satta J, Surcel H-M, Leinonen M, et al. Use of Doxycycline to Decrease the Growth Rate of Abdominal Aortic Aneurysms: A Randomized, Double-Blind, Placebo-Controlled Pilot Study. J Vasc Surg (2001) 34(4):606–10. doi: 10.1067/mva.2001.117891
18. Xiong F, Zhao J, Zeng G, Huang B, Yuan D, Yang Y. Inhibition of AAA in a Rat Model by Treatment With ACEI Perindopril. J Surg Res (2014) 189(1):166–73. doi: 10.1016/j.jss.2014.01.057
19. Yoshimura K, Morikage N, Nishino-Fujimoto S, Furutani A, Shirasawa B, Hamano K. Current Status and Perspectives on Pharmacologic Therapy for Abdominal Aortic Aneurysm. Curr Drug Targets (2018) 19(11):1265–75. doi: 10.2174/1389450119666171227223331
20. Kokje VB, Hamming JF, Lindeman JH. Editor’s Choice - Pharmaceutical Management of Small Abdominal Aortic Aneurysms: A Systematic Review of the Clinical Evidence. Eur J Vasc Endovasc Surg (2015) 50(6):702–13. doi: 10.1016/j.ejvs.2015.08.010
21. Golledge J. Abdominal Aortic Aneurysm: Update on Pathogenesis and Medical Treatments. Nat Rev Cardiol (2019) 16(4):225–42. doi: 10.1038/s41569-018-0114-9
22. Miyake T, Morishita R. Pharmacological Treatment of Abdominal Aortic Aneurysm. Cardiovasc Res (2009) 83(3):436–43. doi: 10.1093/cvr/cvp155
23. Bahia SS, Vidal-Diez A, Seshasai SR, Shpitser I, Brownrigg JR, Patterson BO, et al. Cardiovascular Risk Prevention and All-Cause Mortality in Primary Care Patients With an Abdominal Aortic Aneurysm. Br J Surg (2016) 103(12):1626–33. doi: 10.1002/bjs.10269
24. De Rango P, Cao P, Cieri E, Parlani G, Lenti M, Simonte G, et al. Effects of Diabetes on Small Aortic Aneurysms Under Surveillance According to a Subgroup Analysis From a Randomized Trial. J Vasc Surg (2012) 56(6):1555–63. doi: 10.1016/j.jvs.2012.05.078
25. Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Diabetes and Aortic Aneurysm: Current State of the Art. Cardiovasc Res (2018) 114(13):1702–13. doi: 10.1093/cvr/cvy174
26. Hinchliffe RJ. Metformin and Abdominal Aortic Aneurysm. Eur J Vasc Endovascular Surg (2017) 54(6):679–80. doi: 10.1016/j.ejvs.2017.08.016
27. Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin Inhibits Proinflammatory Responses and Nuclear factor-kappaB in Human Vascular Wall Cells. Arterioscler Thromb Vasc Biol (2006) 26(3):611–7. doi: 10.1161/01.ATV.0000201938.78044.75
28. Golledge J, Moxon J, Pinchbeck J, Anderson G, Rowbotham S, Jenkins J, et al. Association Between Metformin Prescription and Growth Rates of Abdominal Aortic Aneurysms. Br J Surg (2017) 104(11):1486–93. doi: 10.1002/bjs.10587
29. Raffort J, Hassen-Khodja R, Jean-Baptiste E, Lareyre F. Relationship Between Metformin and Abdominal Aortic Aneurysm. J Vasc Surg (2020) 71(3):1056–62. doi: 10.1016/j.jvs.2019.08.270
30. Itoga NK, Rothenberg KA, Suarez P, Ho TV, Mell MW, Xu B, et al. Metformin Prescription Status and Abdominal Aortic Aneurysm Disease Progression in the U.S. Veteran Population. J Vasc Surg (2019) 69(3):710–6.e3. doi: 10.1016/j.jvs.2018.06.194
31. Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and Epidemiology of Abdominal Aortic Aneurysms. Nat Rev Cardiol (2011) 8(2):92–102. doi: 10.1038/nrcardio.2010.180
32. Brady AR, Fowkes FG, Thompson SG, Powell JT. Aortic Aneurysm Diameter and Risk of Cardiovascular Mortality. Arterioscler Thromb Vasc Biol (2001) 21(7):1203–7. doi: 10.1161/hq0701.091999
33. Bath MF, Gokani VJ, Sidloff DA, Jones LR, Choke E, Sayers RD, et al. Systematic Review of Cardiovascular Disease and Cardiovascular Death in Patients With a Small Abdominal Aortic Aneurysm. Br J Surg (2015) 102(8):866–72. doi: 10.1002/bjs.9837
34. Kent KC, Zwolak RM, Egorova NN, Riles TS, Manganaro A, Moskowitz AJ, et al. Analysis of Risk Factors for Abdominal Aortic Aneurysm in a Cohort of More Than 3 Million Individuals. J Vasc Surg (2010) 52(3):539–48. doi: 10.1016/j.jvs.2010.05.090
35. Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int J Prev Med (2014) 5(8):927–46.
36. Toghill BJ, Saratzis A, Bown MJ. Abdominal Aortic Aneurysm-an Independent Disease to Atherosclerosis? Cardiovasc Pathol (2017) 27:71–5. doi: 10.1016/j.carpath.2017.01.008
37. Johnsen SH, Forsdahl SH, Singh K, Jacobsen BK. Atherosclerosis in Abdominal Aortic Aneurysms: A Causal Event or a Process Running in Parallel? The Tromsø Study. Arterioscler Thromb Vasc Biol (2010) 30(6):1263–8. doi: 10.1161/ATVBAHA.110.203588
38. Peshkova IO, Schaefer G, Koltsova EK. Atherosclerosis and Aortic Aneurysm - is Inflammation a Common Denominator? FEBS J (2016) 283(9):1636–52. doi: 10.1111/febs.13634
39. Golledge J, Norman PE. Atherosclerosis and Abdominal Aortic Aneurysm: Cause, Response, or Common Risk Factors? Arterioscler Thromb Vasc Biol (2010) 30(6):1075–7. doi: 10.1161/ATVBAHA.110.206573
40. Lindeman JH, Abdul-Hussien H, Schaapherder AF, Van Bockel JH, Von der Thüsen JH, Roelen DL, et al. Enhanced Expression and Activation of Pro-Inflammatory Transcription Factors Distinguish Aneurysmal From Atherosclerotic Aorta: IL-6- and IL-8-Dominated Inflammatory Responses Prevail in the Human Aneurysm. Clin Sci (Lond) (2008) 114(11):687–97. doi: 10.1042/CS20070352
41. Joshi NV, Elkhawad M, Forsythe RO, McBride OMB, Rajani NK, Tarkin JM, et al. Greater Aortic Inflammation and Calcification in Abdominal Aortic Aneurysmal Disease Than Atherosclerosis: A Prospective Matched Cohort Study. Open Heart (2020) 7(1):e001141. doi: 10.1136/openhrt-2019-001141
42. Bobryshev YV, Lord RS. Vascular-Associated Lymphoid Tissue (VALT) Involvement in Aortic Aneurysm. Atherosclerosis (2001) 154(1):15–21. doi: 10.1016/S0021-9150(00)00441-X
43. Xing D, Nozell S, Chen YF, Hage F, Oparil S. Estrogen and Mechanisms of Vascular Protection. Arterioscler Thromb Vasc Biol (2009) 29(3):289–95. doi: 10.1161/ATVBAHA.108.182279
44. Arnal JF, Douin-Echinard V, Brouchet L, Tremollières F, Laurell H, Lenfant F, et al. Understanding the Oestrogen Action in Experimental and Clinical Atherosclerosis. Fundam Clin Pharmacol (2006) 20(6):539–48. doi: 10.1111/j.1472-8206.2006.00445.x
45. Burger HG, Dudley EC, Hopper JL, Groome N, Guthrie JR, Green A, et al. Prospectively Measured Levels of Serum Follicle-Stimulating Hormone, Estradiol, and the Dimeric Inhibins During the Menopausal Transition in a Population-Based Cohort of Women. J Clin Endocrinol Metab (1999) 84(11):4025–30. doi: 10.1210/jcem.84.11.6158
46. Villard C, Swedenborg J, Eriksson P, Hultgren R. Reproductive History in Women With Abdominal Aortic Aneurysms. J Vasc Surg (2011) 54(2):341–5, 5.e1-2. doi: 10.1016/j.jvs.2010.12.069
47. Wu XF, Zhang J, Paskauskas S, Xin SJ, Duan ZQ. The Role of Estrogen in the Formation of Experimental Abdominal Aortic Aneurysm. Am J Surg (2009) 197(1):49–54. doi: 10.1016/j.amjsurg.2007.11.022
48. Miyamoto C, Kugo H, Hashimoto K, Moriyama T, Zaima N. Ovariectomy Increases the Incidence and Diameter of Abdominal Aortic Aneurysm in a Hypoperfusion-Induced Abdominal Aortic Aneurysm Animal Model. Sci Rep (2019) 9(1):18330. doi: 10.1038/s41598-019-54829-0
49. Thatcher SE, Zhang X, Woody S, Wang Y, Alsiraj Y, Charnigo R, et al. Exogenous 17-β Estradiol Administration Blunts Progression of Established Angiotensin II-Induced Abdominal Aortic Aneurysms in Female Ovariectomized Mice. Biol Sex Dif (2015) 6(1):12. doi: 10.1186/s13293-015-0030-1
50. Martin-McNulty B, Tham DM, da Cunha V, Ho JJ, Wilson DW, Rutledge JC, et al. 17 Beta-Estradiol Attenuates Development of Angiotensin II-Induced Aortic Abdominal Aneurysm in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol (2003) 23(9):1627–32. doi: 10.1161/01.ATV.0000085842.20866.6A
51. Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix Metalloproteinases 2 and 9 Work in Concert to Produce Aortic Aneurysms. J Clin Invest (2002) 110(5):625–32. doi: 10.1172/JCI0215334
52. Maguire EM, Pearce SWA, Xiao R, Oo AY, Xiao Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharmaceuticals (Basel) (2019) 12(3):118. doi: 10.3390/ph12030118
53. Miller AP, Xing D, Feng W, Fintel M, Chen YF, Oparil S. Aged Rats Lose Vasoprotective and Anti-Inflammatory Actions of Estrogen in Injured Arteries. Menopause (2007) 14(2):251–60. doi: 10.1097/01.gme.0000235366.39726.f6
54. Clarkson TB, Meléndez GC, Appt SE. Timing Hypothesis for Postmenopausal Hormone Therapy: Its Origin, Current Status, and Future. Menopause (2013) 20(3):342–53. doi: 10.1097/gme.0b013e3182843aad
55. Hsia J, Criqui MH, Rodabough RJ, Langer RD, Resnick HE, Phillips LS, et al. Estrogen Plus Progestin and the Risk of Peripheral Arterial Disease: The Women’s Health Initiative. Circulation (2004) 109(5):620–6. doi: 10.1161/01.CIR.0000115309.63979.92
56. Iribarren C, Darbinian JA, Go AS, Fireman BH, Lee CD, Grey DP. Traditional and Novel Risk Factors for Clinically Diagnosed Abdominal Aortic Aneurysm: The Kaiser Multiphasic Health Checkup Cohort Study. Ann Epidemiol (2007) 17(9):669–78. doi: 10.1016/j.annepidem.2007.02.004
57. Mosca L, Collins P, Herrington DM, Mendelsohn ME, Pasternak RC, Robertson RM, et al. Hormone Replacement Therapy and Cardiovascular Disease: A Statement for Healthcare Professionals From the American Heart Association. Circulation (2001) 104(4):499–503. doi: 10.1161/hc2901.092200
58. Boardman HM, Hartley L, Eisinga A, Main C, Roqué i Figuls M, Bonfill Cosp X, et al. Hormone Therapy for Preventing Cardiovascular Disease in Post-Menopausal Women. Cochrane Database Syst Rev (2015) 3):Cd002229. doi: 10.1002/14651858.CD002229.pub4
59. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The Protective Role of Estrogen and Estrogen Receptors in Cardiovascular Disease and the Controversial Use of Estrogen Therapy. Biol Sex Differ (2017) 8(1):33. doi: 10.1186/s13293-017-0152-8
60. Kurakula K, Hamers AA, de Waard V, de Vries CJ. Nuclear Receptors in Atherosclerosis: A Superfamily With Many ’Goodfellas’. Mol Cell Endocrinol (2013) 368(1-2):71–84. doi: 10.1016/j.mce.2012.05.014
61. Zhang X, Thatcher SE, Rateri DL, Bruemmer D, Charnigo R, Daugherty A, et al. Transient Exposure of Neonatal Female Mice to Testosterone Abrogates the Sexual Dimorphism of Abdominal Aortic Aneurysms. Circ Res (2012) 110(11):e73–85. doi: 10.1161/CIRCRESAHA.111.253880
62. Skiba MA, Bell RJ, Islam RM, Handelsman DJ, Desai R, Davis SR. Androgens During the Reproductive Years: What Is Normal for Women? J Clin Endocrinol Metab (2019) 104(11):5382–92. doi: 10.1210/jc.2019-01357
63. Burger HG, Dudley EC, Cui J, Dennerstein L, Hopper JL. A Prospective Longitudinal Study of Serum Testosterone, Dehydroepiandrosterone Sulfate, and Sex Hormone-Binding Globulin Levels Through the Menopause Transition1. J Clin Endocrinol Metab (2000) 85(8):2832–8. doi: 10.1210/jcem.85.8.6740
64. Morley JE, Perry HM 3rd. Androgens and Women at the Menopause and Beyond. J Gerontol A Biol Sci Med Sci (2003) 58(5):M409–16. doi: 10.1093/gerona/58.5.M409
65. Torréns JI, Sutton-Tyrrell K, Zhao X, Matthews K, Brockwell S, Sowers M, et al. Relative Androgen Excess During the Menopausal Transition Predicts Incident Metabolic Syndrome in Midlife Women: Study of Women’s Health Across the Nation. Menopause (2009) 16(2):257–64. doi: 10.1097/gme.0b013e318185e249
66. Davey RA, Grossmann M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin Biochem Rev (2016) 37(1):3–15.
67. Liu PY, Death AK, Handelsman DJ. Androgens and Cardiovascular Disease. Endocrine Rev (2003) 24(3):313–40. doi: 10.1210/er.2003-0005
68. Davis JP, Salmon M, Pope NH, Lu G, Su G, Meher A, et al. Pharmacologic Blockade and Genetic Deletion of Androgen Receptor Attenuates Aortic Aneurysm Formation. J Vasc Surg (2016) 63(6):1602–12.e2. doi: 10.1016/j.jvs.2015.11.038
69. Zhang X, Thatcher S, Wu C, Daugherty A, Cassis LA. Castration of Male Mice Prevents the Progression of Established Angiotensin II-Induced Abdominal Aortic Aneurysms. J Vasc Surg (2015) 61(3):767–76. doi: 10.1016/j.jvs.2013.11.004
70. Son BK, Kojima T, Ogawa S, Akishita M. Testosterone Inhibits Aneurysm Formation and Vascular Inflammation in Male Mice. J Endocrinol (2019) 241(3):307–17. doi: 10.1530/JOE-18-0646
71. Glaser R, Dimitrakakis C. Testosterone Therapy in Women: Myths and Misconceptions. Maturitas (2013) 74(3):230–4. doi: 10.1016/j.maturitas.2013.01.003
72. Tsametis CP, Isidori AM. Testosterone Replacement Therapy: For Whom, When and How? Metabolism (2018) 86:69–78. doi: 10.1016/j.metabol.2018.03.007
73. Kelly DM, Jones TH. Testosterone: A Vascular Hormone in Health and Disease. J Endocrinol (2013) 217(3):R47–71. doi: 10.1530/JOE-12-0582
74. Vigen R, O’Donnell CI, Barón AE, Grunwald GK, Maddox TM, Bradley SM, et al. Association of Testosterone Therapy With Mortality, Myocardial Infarction, and Stroke in Men With Low Testosterone Levels. Jama (2013) 310(17):1829–36. doi: 10.1001/jama.2013.280386
75. Panzer C, Guay A. Testosterone Replacement Therapy in Naturally and Surgically Menopausal Women. J Sex Med (2009) 6(1):8–18; quiz 9-20. doi: 10.1111/j.1743-6109.2008.01128.x
76. Davis SR, Baber R, Panay N, Bitzer J, Perez SC, Islam RM, et al. Global Consensus Position Statement on the Use of Testosterone Therapy for Women. J Clin Endocrinol Metab (2019) 104(10):4660–6. doi: 10.1210/jc.2019-01603
77. Yeap BB, Hyde Z, Norman PE, Chubb SA, Golledge J. Associations of Total Testosterone, Sex Hormone-Binding Globulin, Calculated Free Testosterone, and Luteinizing Hormone With Prevalence of Abdominal Aortic Aneurysm in Older Men. J Clin Endocrinol Metab (2010) 95(3):1123–30. doi: 10.1210/jc.2009-1696
78. Makrygiannis G, Courtois A, Drion P, Defraigne JO, Kuivaniemi H, Sakalihasan N. Sex Differences in Abdominal Aortic Aneurysm: The Role of Sex Hormones. Ann Vasc Surg (2014) 28(8):1946–58. doi: 10.1016/j.avsg.2014.07.008
79. Raju GA, Chavan R, Deenadayal M, Gunasheela D, Gutgutia R, Haripriya G, et al. Luteinizing Hormone and Follicle Stimulating Hormone Synergy: A Review of Role in Controlled Ovarian Hyper-Stimulation. J Hum Reprod Sci (2013) 6(4):227–34. doi: 10.4103/0974-1208.126285
80. Jamnongjit M, Hammes SR. Ovarian Steroids: The Good, the Bad, and the Signals That Raise Them. Cell Cycle (2006) 5(11):1178–83. doi: 10.4161/cc.5.11.2803
81. Ramaswamy S, Weinbauer GF. Endocrine Control of Spermatogenesis: Role of FSH and LH/ Testosterone. Spermatogenesis (2014) 4(2):e996025. doi: 10.1080/21565562.2014.996025
82. Hall JE. Endocrinology of the Menopause. Endocrinol Metab Clin North Am (2015) 44(3):485–96. doi: 10.1016/j.ecl.2015.05.010
83. Shaw ND, Histed SN, Srouji SS, Yang J, Lee H, Hall JE. Estrogen Negative Feedback on Gonadotropin Secretion: Evidence for a Direct Pituitary Effect in Women. J Clin Endocrinol Metab (2010) 95(4):1955–61. doi: 10.1210/jc.2009-2108
84. Zhu D, Li X, Macrae VE, Simoncini T, Fu X. Extragonadal Effects of Follicle-Stimulating Hormone on Osteoporosis and Cardiovascular Disease in Women During Menopausal Transition. Trends Endocrinol Metab: TEM (2018) 29(8):571–80. doi: 10.1016/j.tem.2018.06.001
85. Pakarainen T, Ahtiainen P, Zhang F-P, Rulli S, Poutanen M, Huhtaniemi I. Extragonadal LH/hCG Action—Not Yet Time to Rewrite Textbooks. Mol Cell Endocrinol (2007) 269(1):9–16. doi: 10.1016/j.mce.2006.10.019
86. Gravholt CH, Viuff MH, Brun S, Stochholm K, Andersen NH. Turner Syndrome: Mechanisms and Management. Nat Rev Endocrinol (2019) 15(10):601–14. doi: 10.1038/s41574-019-0224-4
87. Silberbach M, Roos-Hesselink JW, Andersen NH, Braverman AC, Brown N, Collins RT, et al. Cardiovascular Health in Turner Syndrome: A Scientific Statement From the American Heart Association. Circ Genom Precis Med (2018) 11(10):e000048. doi: 10.1161/HCG.0000000000000048
88. Duijnhouwer AL, Bons LR, Timmers H, van Kimmenade RRL, Snoeren M, Timmermans J, et al. Aortic Dilatation and Outcome in Women With Turner Syndrome. Heart (2019) 105(9):693–700. doi: 10.1136/heartjnl-2018-313716
89. Zoë H, Paul EN, Leon F, Graeme JH, Kieran AM, Osvaldo PA, et al. Elevated LH Predicts Ischaemic Heart Disease Events in Older Men: The Health in Men Study. Eur J Endocrinol (2011) 164(4):569–77. doi: 10.1530/EJE-10-1063
90. Meng X, Li X, Xu X, Li P, Chen Y, Fu X, et al. Elevated Luteinizing Hormone Contributes to Atherosclerosis Formation by Inhibiting Nitric Oxide Synthesis via PI3K/Akt Pathway. Vascul Pharmacol (2019) 121:106582. doi: 10.1016/j.vph.2019.106582
91. Fields MJ, Shemesh M. Extragonadal Luteinizing Hormone Receptors in the Reproductive Tract of Domestic Animals. Biol Reprod (2004) 71(5):1412–8. doi: 10.1095/biolreprod.104.027201
92. Apaja PM, Aatsinki JT, Rajaniemi HJ, Petäjä-Repo UE. Expression of the Mature Luteinizing Hormone Receptor in Rodent Urogenital and Adrenal Tissues Is Developmentally Regulated at a Posttranslational Level. Endocrinology (2005) 146(8):3224–32. doi: 10.1210/en.2005-0073
93. Simoni M, Huhtaniemi I, Casarini L, Santi D. Follicle-Stimulating Hormone: Fertility and Beyond. Lausanne: Frontiers Media (2019).
94. Chrusciel M, Ponikwicka-Tyszko D, Wolczynski S, Huhtaniemi I, Rahman NA. Extragonadal FSHR Expression and Function—Is It Real? Front Endocrinol (2019) 10(32). doi: 10.3389/fendo.2019.00032
95. El Khoudary SR, Wildman RP, Matthews K, Thurston RC, Bromberger JT, Sutton-Tyrrell K. Endogenous Sex Hormones Impact the Progression of Subclinical Atherosclerosis in Women During the Menopausal Transition. Atherosclerosis (2012) 225(1):180–6. doi: 10.1016/j.atherosclerosis.2012.07.025
96. Ji MX, Yu Q. Primary Osteoporosis in Postmenopausal Women. Chronic Dis Transl Med (2015) 1(1):9–13. doi: 10.1016/j.cdtm.2015.02.006
97. Shaikh A, Bhartiya D, Kapoor S, Nimkar H. Delineating the Effects of 5-Fluorouracil and Follicle-Stimulating Hormone on Mouse Bone Marrow Stem/Progenitor Cells. Stem Cell Res Ther (2016) 7(1):59. doi: 10.1186/s13287-016-0311-6
98. Zbucka-Kretowska M, Eljaszewicz A, Lipinska D, Grubczak K, Rusak M, Mrugacz G, et al. Effective Mobilization of Very Small Embryonic-Like Stem Cells and Hematopoietic Stem/Progenitor Cells But Not Endothelial Progenitor Cells by Follicle-Stimulating Hormone Therapy. Stem Cells Int (2016) 2016:8530207. doi: 10.1155/2016/8530207
99. Li H, Wang YP, Zhang LN, Tian G. Perivascular Adipose Tissue-Derived Leptin Promotes Vascular Smooth Muscle Cell Phenotypic Switching via P38 Mitogen-Activated Protein Kinase in Metabolic Syndrome Rats. Exp Biol Med (Maywood) (2014) 239(8):954–65. doi: 10.1177/1535370214527903
100. Zhang ZB, Ruan CC, Lin JR, Xu L, Chen XH, Du YN, et al. Perivascular Adipose Tissue-Derived PDGF-D Contributes to Aortic Aneurysm Formation During Obesity. Diabetes (2018) 67(8):1549–60. doi: 10.2337/db18-0098
101. Sedding DG, Boyle EC, Demandt JAF, Sluimer JC, Dutzmann J, Haverich A, et al. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front Immunol (2018) 9:706. doi: 10.3389/fimmu.2018.00706
102. Choke E, Thompson MM, Dawson J, Wilson WR, Sayed S, Loftus IM, et al. Abdominal Aortic Aneurysm Rupture is Associated With Increased Medial Neovascularization and Overexpression of Proangiogenic Cytokines. Arterioscler Thromb Vasc Biol (2006) 26(9):2077–82. doi: 10.1161/01.ATV.0000234944.22509.f9
103. Petsophonsakul P, Furmanik M, Forsythe R, Dweck M, Schurink GW, Natour E, et al. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler Thromb Vasc Biol (2019) 39(7):1351–68. doi: 10.1161/ATVBAHA.119.312787
104. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
105. Raffort J, Lareyre F, Clement M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and Macrophages in Abdominal Aortic Aneurysm. Nat Rev Cardiol (2017) 14(8):457–71. doi: 10.1038/nrcardio.2017.52
106. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in Atherosclerosis: A Dynamic Balance. Nat Rev Immunol (2013) 13(10):709–21. doi: 10.1038/nri3520
107. Udalova IA, Mantovani A, Feldmann M. Macrophage Heterogeneity in the Context of Rheumatoid Arthritis. Nat Rev Rheumatol (2016) 12(8):472–85. doi: 10.1038/nrrheum.2016.91
108. Li Y, Sun Z, Zhang L, Yan J, Shao C, Jing L, et al. Role of Macrophages in the Progression and Regression of Vascular Calcification. Front Pharmacol (2020) 11:661. doi: 10.3389/fphar.2020.00661
109. Chinetti-Gbaguidi G, Daoudi M, Rosa M, Vinod M, Louvet L, Copin C, et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ Res (2017) 121(1):19–30. doi: 10.1161/CIRCRESAHA.116.310262
110. Tintut Y, Patel J, Parhami F, Demer LL. Tumor Necrosis Factor-Alpha Promotes In Vitro Calcification of Vascular Cells via the cAMP Pathway. Circulation (2000) 102(21):2636–42. doi: 10.1161/01.CIR.102.21.2636
111. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, et al. Runx2-Upregulated Receptor Activator of Nuclear Factor κb Ligand in Calcifying Smooth Muscle Cells Promotes Migration and Osteoclastic Differentiation of Macrophages. Arterioscler Thromb Vasc Biol (2011) 31(6):1387–96. doi: 10.1161/ATVBAHA.110.222547
112. Yamanouchi D, Takei Y, Komori K. Balanced Mineralization in the Arterial System: Possible Role of Osteoclastogenesis/Osteoblastogenesis in Abdominal Aortic Aneurysm and Stenotic Disease. Circ J (2012) 76(12):2732–7. doi: 10.1253/circj.CJ-12-1240
113. Takei Y, Tanaka T, Kent KC, Yamanouchi D. Osteoclastogenic Differentiation of Macrophages in the Development of Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol (2016) 36(9):1962–71. doi: 10.1161/ATVBAHA.116.307715
114. Wu M, Rementer C, Giachelli CM. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcified Tissue Int (2013) 93(4):365–73. doi: 10.1007/s00223-013-9712-z
115. Robinson LJ, Tourkova I, Wang Y, Sharrow AC, Landau MS, Yaroslavskiy BB, et al. FSH-Receptor Isoforms and FSH-Dependent Gene Transcription in Human Monocytes and Osteoclasts. Biochem Biophys Res Commun (2010) 394(1):12–7. doi: 10.1016/j.bbrc.2010.02.112
116. Iqbal J, Sun L, Kumar TR, Blair HC, Zaidi M. Follicle-Stimulating Hormone Stimulates TNF Production From Immune Cells to Enhance Osteoblast and Osteoclast Formation. Proc Natl Acad Sci USA (2006) 103(40):14925–30. doi: 10.1073/pnas.0606805103
117. Duer MJ, Friscić T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, et al. Mineral Surface in Calcified Plaque is Like That of Bone: Further Evidence for Regulated Mineralization. Arterioscler Thromb Vasc Biol (2008) 28(11):2030–4. doi: 10.1161/ATVBAHA.108.172387
118. Wang J, Zhang W, Yu C, Zhang X, Zhang H, Guan Q, et al. Follicle-Stimulating Hormone Increases the Risk of Postmenopausal Osteoporosis by Stimulating Osteoclast Differentiation. PloS One (2015) 10(8):e0134986. doi: 10.1371/journal.pone.0134986
119. Abdul-Hussien H, Hanemaaijer R, Kleemann R, Verhaaren BF, van Bockel JH, Lindeman JH. The Pathophysiology of Abdominal Aortic Aneurysm Growth: Corresponding and Discordant Inflammatory and Proteolytic Processes in Abdominal Aortic and Popliteal Artery Aneurysms. J Vasc Surg (2010) 51(6):1479–87. doi: 10.1016/j.jvs.2010.01.057
120. Michiels C. Endothelial Cell Functions. J Cell Physiol (2003) 196(3):430–43. doi: 10.1002/jcp.10333
121. Li X, Chen W, Li P, Wei J, Cheng Y, Liu P, et al. Follicular Stimulating Hormone Accelerates Atherogenesis by Increasing Endothelial VCAM-1 Expression. Theranostics (2017) 7(19):4671–88. doi: 10.7150/thno.21216
122. Stilley JA, Guan R, Duffy DM, Segaloff DL. Signaling Through FSH Receptors on Human Umbilical Vein Endothelial Cells Promotes Angiogenesis. J Clin Endocrinol Metab (2014) 99(5):E813–20. doi: 10.1210/jc.2013-3186
123. Radu A, Pichon C, Camparo P, Antoine M, Allory Y, Couvelard A, et al. Expression of Follicle-Stimulating Hormone Receptor in Tumor Blood Vessels. N Engl J Med (2010) 363(17):1621–30. doi: 10.1056/NEJMoa1001283
124. Thompson MM, Jones L, Nasim A, Sayers RD, Bell PR. Angiogenesis in Abdominal Aortic Aneurysms. Eur J Vasc Endovasc Surg (1996) 11(4):464–9. doi: 10.1016/S1078-5884(96)80183-3
125. Paik DC, Fu C, Bhattacharya J, David Tilson M. Ongoing Angiogenesis in Blood Vessels of the Abdominal Aortic Aneurysm. Exp Mol Med (2004) 36(6):524–33. doi: 10.1038/emm.2004.67
126. Vijaynagar B, Bown MJ, Sayers RD, Choke E. Potential Role for Anti-Angiogenic Therapy in Abdominal Aortic Aneurysms. Eur J Clin Invest (2013) 43(7):758–65. doi: 10.1111/eci.12103
127. Lane JM, Russell L, Khan SN. Osteoporosis. Clin Orthop Relat Res (2000) 372):139–50. doi: 10.1097/00003086-200003000-00016
128. Hofbauer LC, Brueck CC, Shanahan CM, Schoppet M, Dobnig H. Vascular Calcification and Osteoporosis–From Clinical Observation Towards Molecular Understanding. Osteoporos Int (2007) 18(3):251–9. doi: 10.1007/s00198-006-0282-z
129. Giachelli CM. The Emerging Role of Phosphate in Vascular Calcification. Kidney Int (2009) 75(9):890–7. doi: 10.1038/ki.2008.644
130. Cozzolino M, Ciceri P, Galassi A, Mangano M, Carugo S, Capelli I, et al. The Key Role of Phosphate on Vascular Calcification. Toxins (Basel) (2019) 11(4):213. doi: 10.3390/toxins11040213
131. Johnson RC, Leopold JA, Loscalzo J. Vascular Calcification: Pathobiological Mechanisms and Clinical Implications. Circ Res (2006) 99(10):1044–59. doi: 10.1161/01.RES.0000249379.55535.21
132. Shaker JL, Deftos L. Calcium and Phosphate Homeostasis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, editors. Endotext. South Dartmouth (MA: MDText.com, Inc.Copyright © (2000). p. 2000–21.
133. Lee SJ, Lee I-K, Jeon J-H. Vascular Calcification—New Insights Into Its Mechanism. Int J Mol Sci (2020) 21(8):2685. doi: 10.3390/ijms21082685
134. Karwowski W, Naumnik B, Szczepański M, Myśliwiec M. The Mechanism of Vascular Calcification - a Systematic Review. Med Sci Monit (2012) 18(1):Ra1–11. doi: 10.12659/MSM.882181
135. Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J Am Soc Nephrol (2004) 15(11):2857–67. doi: 10.1097/01.ASN.0000141960.01035.28
136. Hak AE, Pols HA, van Hemert AM, Hofman A, Witteman JC. Progression of Aortic Calcification is Associated With Metacarpal Bone Loss During Menopause: A Population-Based Longitudinal Study. Arterioscler Thromb Vasc Biol (2000) 20(8):1926–31. doi: 10.1161/01.ATV.20.8.1926
137. Lampropoulos CE, Kalamara P, Konsta M, Papaioannou I, Papadima E, Antoniou Z, et al. Osteoporosis and Vascular Calcification in Postmenopausal Women: A Cross-Sectional Study. Climacteric (2016) 19(3):303–7. doi: 10.3109/13697137.2016.1164134
138. Kiel DP, Kauppila LI, Cupples LA, Hannan MT, O’Donnell CJ, Wilson PWF. Bone Loss and the Progression of Abdominal Aortic Calcification Over a 25 Year Period: The Framingham Heart Study. Calcified Tissue Int (2001) 68(5):271–6. doi: 10.1007/BF02390833
139. Witteman JC, Kok FJ, van Saase JL, Valkenburg HA. Aortic Calcification as a Predictor of Cardiovascular Mortality. Lancet (1986) 2(8516):1120–2. doi: 10.1016/S0140-6736(86)90530-1
140. Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, et al. Denosumab for Prevention of Fractures in Postmenopausal Women With Osteoporosis. N Engl J Med (2009) 361(8):756–65. doi: 10.1056/NEJMoa0809493
141. Cannon JG, Kraj B, Sloan G. Follicle-Stimulating Hormone Promotes RANK Expression on Human Monocytes. Cytokine (2011) 53(2):141–4. doi: 10.1016/j.cyto.2010.11.011
142. Zhu LL, Tourkova I, Yuen T, Robinson LJ, Bian Z, Zaidi M, et al. Blocking FSH Action Attenuates Osteoclastogenesis. Biochem Biophys Res Commun (2012) 422(1):54–8. doi: 10.1016/j.bbrc.2012.04.104
143. Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, et al. FSH Directly Regulates Bone Mass. Cell (2006) 125(2):247–60. doi: 10.1016/j.cell.2006.01.051
144. Buijs RVC, Willems TP, Tio RA, Boersma HH, Tielliu IFJ, Slart RHJA, et al. Calcification as a Risk Factor for Rupture of Abdominal Aortic Aneurysm. Eur J Vasc Endovascular Surg (2013) 46(5):542–8. doi: 10.1016/j.ejvs.2013.09.006
145. Hendy K, Gunnarsson R, Cronin O, Golledge J. Infra-Renal Abdominal Aortic Calcification Volume Does Not Predict Small Abdominal Aortic Aneurysm Growth. Atherosclerosis (2015) 243(1):334–8. doi: 10.1016/j.atherosclerosis.2015.07.027
146. Yamanouchi D, Morgan S, Stair C, Seedial S, Lengfeld J, Kent KC, et al. Accelerated Aneurysmal Dilation Associated With Apoptosis and Inflammation in a Newly Developed Calcium Phosphate Rodent Abdominal Aortic Aneurysm Model. J Vasc Surg (2012) 56(2):455–61. doi: 10.1016/j.jvs.2012.01.038
147. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth Muscle Cell Phenotypic Transition Associated With Calcification: Upregulation of Cbfa1 and Downregulation of Smooth Muscle Lineage Markers. Circ Res (2001) 89(12):1147–54. doi: 10.1161/hh2401.101070
148. Li Z, Zhao Z, Cai Z, Sun Y, Li L, Yao F, et al. Runx2 (Runt-Related Transcription Factor 2)-Mediated Microcalcification Is a Novel Pathological Characteristic and Potential Mediator of Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol (2020) 40(5):1352–69. doi: 10.1161/ATVBAHA.119.314113
149. Basalyga DM, Simionescu DT, Xiong W, Baxter BT, Starcher BC, Vyavahare NR. Elastin Degradation and Calcification in an Abdominal Aorta Injury Model: Role of Matrix Metalloproteinases. Circulation (2004) 110(22):3480–7. doi: 10.1161/01.CIR.0000148367.08413.E9
150. Shetty S, Kapoor N, Bondu JD, Thomas N, Paul TV. Bone Turnover Markers: Emerging Tool in the Management of Osteoporosis. Indian J Endocrinol Metab (2016) 20(6):846–52. doi: 10.4103/2230-8210.192914
151. Muñoz-Torres M, Reyes-García R, Mezquita-Raya P, Fernández-García D, Alonso G, Luna Jde D, et al. Serum Cathepsin K as a Marker of Bone Metabolism in Postmenopausal Women Treated With Alendronate. Maturitas (2009) 64(3):188–92. doi: 10.1016/j.maturitas.2009.09.011
152. Meier C, Meinhardt U, Greenfield JR, De Winter J, Nguyen TV, Dunstan CR, et al. Serum Cathepsin K Concentrations Reflect Osteoclastic Activity in Women With Postmenopausal Osteoporosis and Patients With Paget’s Disease. Clin Lab (2006) 52(1-2):1–10.
153. Vrins CL. From Blood to Gut: Direct Secretion of Cholesterol via Transintestinal Cholesterol Efflux. World J Gastroenterol (2010) 16(47):5953–7. doi: 10.3748/wjg.v16.i47.5953
154. Song Y, Wang ES, Xing LL, Shi S, Qu F, Zhang D, et al. Follicle-Stimulating Hormone Induces Postmenopausal Dyslipidemia Through Inhibiting Hepatic Cholesterol Metabolism. J Clin Endocrinol Metab (2016) 101(1):254–63. doi: 10.1210/jc.2015-2724
155. Guo Y, Zhao M, Bo T, Ma S, Yuan Z, Chen W, et al. Blocking FSH Inhibits Hepatic Cholesterol Biosynthesis and Reduces Serum Cholesterol. Cell Res (2019) 29(2):151–66. doi: 10.1038/s41422-018-0123-6
156. Shioi A, Ikari Y. Plaque Calcification During Atherosclerosis Progression and Regression. J Atheroscler Thromb (2018) 25(4):294–303. doi: 10.5551/jat.RV17020
157. New SE, Aikawa E. Molecular Imaging Insights Into Early Inflammatory Stages of Arterial and Aortic Valve Calcification. Circ Res (2011) 108(11):1381–91. doi: 10.1161/CIRCRESAHA.110.234146
158. Rosell M, Kaforou M, Frontini A, Okolo A, Chan YW, Nikolopoulou E, et al. Brown and White Adipose Tissues: Intrinsic Differences in Gene Expression and Response to Cold Exposure in Mice. Am J Physiol Endocrinol Metab (2014) 306(8):E945–64. doi: 10.1152/ajpendo.00473.2013
159. Liu P, Ji Y, Yuen T, Rendina-Ruedy E, DeMambro VE, Dhawan S, et al. Blocking FSH Induces Thermogenic Adipose Tissue and Reduces Body Fat. Nature (2017) 546(7656):107–12. doi: 10.1038/nature22342
160. Tchernof A, Poehlman ET. Effects of the Menopause Transition on Body Fatness and Body Fat Distribution. Obes Res (1998) 6(3):246–54. doi: 10.1002/j.1550-8528.1998.tb00344.x
161. Sorop O, Olver TD, van de Wouw J, Heinonen I, van Duin RW, Duncker DJ, et al. The Microcirculation: A Key Player in Obesity-Associated Cardiovascular Disease. Cardiovasc Res (2017) 113(9):1035–45. doi: 10.1093/cvr/cvx093
162. Virdis A, Masi S, Colucci R, Chiriacò M, Uliana M, Puxeddu I, et al. Microvascular Endothelial Dysfunction in Patients With Obesity. Curr Hypertens Rep (2019) 21(4):32. doi: 10.1007/s11906-019-0930-2
163. de Jongh RT, Serné EH RGIJ, de Vries G, Stehouwer CD. Impaired Microvascular Function in Obesity: Implications for Obesity-Associated Microangiopathy, Hypertension, and Insulin Resistance. Circulation (2004) 109(21):2529–35. doi: 10.1161/01.CIR.0000129772.26647.6F
164. Levy BI, Schiffrin EL, Mourad JJ, Agostini D, Vicaut E, Safar ME, et al. Impaired Tissue Perfusion: A Pathology Common to Hypertension, Obesity, and Diabetes Mellitus. Circulation (2008) 118(9):968–76. doi: 10.1161/CIRCULATIONAHA.107.763730
165. Chang L, Garcia-Barrio MT, Chen YE. Perivascular Adipose Tissue Regulates Vascular Function by Targeting Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol (2020) 40(5):1094–109. doi: 10.1161/ATVBAHA.120.312464
166. Dias-Neto M, Meekel JP, van Schaik TG, Hoozemans J, Sousa-Nunes F, Henriques-Coelho T, et al. High Density of Periaortic Adipose Tissue in Abdominal Aortic Aneurysm. Eur J Vasc Endovasc Surg (2018) 56(5):663–71. doi: 10.1016/j.ejvs.2018.07.008
167. Meekel JP, Dias-Neto M, Bogunovic N, Conceição G, Sousa-Mendes C, Stoll GR, et al. Inflammatory Gene Expression of Human Perivascular Adipose Tissue in Abdominal Aortic Aneurysms. Eur J Vasc Endovasc Surg (2021) 61(6):1008–16. doi: 10.1016/j.jvssci.2020.11.011
168. Folkesson M, Vorkapic E, Gulbins E, Japtok L, Kleuser B, Welander M, et al. Inflammatory Cells, Ceramides, and Expression of Proteases in Perivascular Adipose Tissue Adjacent to Human Abdominal Aortic Aneurysms. J Vasc Surg (2017) 65(4):1171–9.e1. doi: 10.1016/j.jvs.2015.12.056
169. Doderer SA, Gäbel G, Kokje VBC, Northoff BH, Holdt LM, Hamming JF, et al. Adventitial Adipogenic Degeneration is an Unidentified Contributor to Aortic Wall Weakening in the Abdominal Aortic Aneurysm. J Vasc Surg (2018) 67(6):1891–900.e4. doi: 10.1016/j.jvs.2017.05.088
170. Villard C, Roy J, Bogdanovic M, Eriksson P, Hultgren R. Sex Hormones in Men With Abdominal Aortic Aneurysm. J Vasc Surg (2021) S0741-5214(21):00995-2. doi: 10.1016/j.jvs.2021.06.020
171. Alsiraj Y, Thatcher SE, Charnigo R, Chen K, Blalock E, Daugherty A, et al. Female Mice With an XY Sex Chromosome Complement Develop Severe Angiotensin II-Induced Abdominal Aortic Aneurysms. Circulation (2017) 135(4):379–91. doi: 10.1161/CIRCULATIONAHA.116.023789
172. Syamlal G, Mazurek JM, Dube SR. Gender Differences in Smoking Among U.S. Working Adults. Am J Prev Med (2014) 47(4):467–75. doi: 10.1016/j.amepre.2014.06.013
173. Stackelberg O, Björck M, Larsson SC, Orsini N, Wolk A. Sex Differences in the Association Between Smoking and Abdominal Aortic Aneurysm. Br J Surg (2014) 101(10):1230–7. doi: 10.1002/bjs.9526
174. Allan CM, Kalak R, Dunstan CR, McTavish KJ, Zhou H, Handelsman DJ, et al. Follicle-Stimulating Hormone Increases Bone Mass in Female Mice. Proc Natl Acad Sci USA (2010) 107(52):22629–34. doi: 10.1073/pnas.1012141108
Keywords: follicle stimulating hormone, abdominal aortic aneurysm, menopause, women, osteoporosis, macrophages, atherosclerosis, adiposity
Citation: Tedjawirja VN, Nieuwdorp M, Yeung KK, Balm R and de Waard V (2021) A Novel Hypothesis: A Role for Follicle Stimulating Hormone in Abdominal Aortic Aneurysm Development in Postmenopausal Women. Front. Endocrinol. 12:726107. doi: 10.3389/fendo.2021.726107
Received: 17 June 2021; Accepted: 02 September 2021;
Published: 13 October 2021.
Edited by:
Andrzej Bartke, Southern Illinois University School of Medicine, United StatesReviewed by:
Bu Yeap, University of Western Australia, AustraliaDeepa Bhartiya, Indian Council of Medical Research (ICMR), India
Christina Villard, Karolinska Institutet (KI), Sweden
Copyright © 2021 Tedjawirja, Nieuwdorp, Yeung, Balm and de Waard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria N. Tedjawirja, v.n.tedjawirja@amsterdamumc.nl