 Balamurugan Dhayalan
Balamurugan Dhayalan Deepak Chatterjee
Deepak Chatterjee Yen-Shan Chen
Yen-Shan Chen Michael A. Weiss
Michael A. Weiss- Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, United States
Insight into folding mechanisms of proinsulin has been provided by analysis of dominant diabetes-associated mutations in the human insulin gene (INS). Such mutations cause pancreatic β-cell dysfunction due to toxic misfolding of a mutant proinsulin and impairment in trans of wild-type insulin secretion. Anticipated by the “Akita” mouse (a classical model of monogenic diabetes mellitus; DM), this syndrome illustrates the paradigm endoreticulum (ER) stress leading to intracellular proteotoxicity. Diverse clinical mutations directly or indirectly perturb native disulfide pairing leading to protein misfolding and aberrant aggregation. Although most introduce or remove a cysteine (Cys; leading in either case to an unpaired thiol group), non-Cys-related mutations identify key determinants of folding efficiency. Studies of such mutations suggest that the hormone’s evolution has been constrained not only by structure-function relationships, but also by the susceptibility of its single-chain precursor to impaired foldability. An intriguing hypothesis posits that INS overexpression in response to peripheral insulin resistance likewise leads to chronic ER stress and β-cell dysfunction in the natural history of non-syndromic Type 2 DM. Cryptic contributions of conserved residues to folding efficiency, as uncovered by rare genetic variants, define molecular links between biophysical principles and the emerging paradigm of Darwinian medicine: Biosynthesis of proinsulin at the edge of non-foldability provides a key determinant of “diabesity” as a pandemic disease of civilization.
Introduction
The Centennial of insulin’s discovery (1) coincides with renewed interest in cellular mechanisms of biosynthesis. The mature hormone is the post-translational product of a single-chain precursor, proinsulin (2, 3). Diverse dominant mutations in the human insulin gene (INS) have been identified associated with diabetes mellitus (DM) (4–10). Such mutations impair oxidative folding of nascent proinsulin in the endoplasmic reticulum (ER) of pancreatic β-cells (11, 12). Originally identified as a monogenic cause of permanent neonatal-onset DM (7, 13–15), this syndrome (designated mutant INS-gene-induced diabetes of youth; MIDY) can also present in childhood (16) or adolescence (17) (maturity-onset diabetes of the young; MODY). Such variation in onset is ascribed to mutation-specific differences in extent of perturbed folding (12, 18). The spectrum of phenotypes may also reflect polygenic differences in how the β-cell responds to chronic ER stress (19, 20).
MIDY patients are heterozygous. Although one wild-type (WT) insulin allele would ordinarily be sufficient to maintain metabolic homeostasis, studies of the Akita mouse [a corresponding mouse model (21–23)] first demonstrated biochemical dominance: misfolding of the variant proinsulin impairs wild-type (WT) biosynthesis (24, 25). Analogous biochemical interference occurs in β-cell lines (11, 12, 18). ER stress leads to distorted organelle architecture, impaired glucose-stimulated β-cell secretion and eventual cell death (26, 27). Discovery of the mutant proinsulin syndrome has stimulated renewed interest in structural mechanisms of disulfide pairing (28–33) as a critical step in the biosynthesis of insulin (2, 3, 34). The central importance of such mechanisms—both in β-cells and as a general model for oxidative protein folding—have motivated extensive cell-based and animal studies (35–39). Together, these efforts have deepened the biophysical understanding of classical structure-functional relationships in the insulin molecule (9, 10, 19, 40) in relation to cellular mechanisms of biosynthesis (4, 10, 41, 42).
The goal of this review is to provide a structural perspective on INS mutations in human proinsulin [for clinical background and history of discovery, see (43)]. A starting point is provided by a general biophysical paradigm: that key interactions in intermediate stages of protein folding often foreshadow spatial relationships in the native state (44–46). Accordingly and in the reverse direction, we will regard the classical crystal structure of insulin (47) as a framework for interpreting folding mechanisms. Given this context, we will restrict our attention to mutations in (or adjoining) the well-organized insulin moiety of proinsulin (48) (Figure 1A and Table 1). Whereas traditional structure-activity relationships (SAR) pertain to receptor binding (9), contributions of the same residues to folding efficiency may be inapparent once the native structure is reached. The growing MIDY/MODY database of INS mutations (Figure 1B and Table S1) may be exploited to decipher this hidden layer of meaning. As a seeming paradox in Darwinian medicine (49, 50), the biophysical non-robustness of proinsulin biosynthesis suggests that the hormone has evolved to the precarious edge of foldability (40, 51, 52). We envisage that foundational principles of protein folding, structure and stability will be found to rationalize the distribution of MIDY/MODY mutations and broad spectrum of clinical presentations.
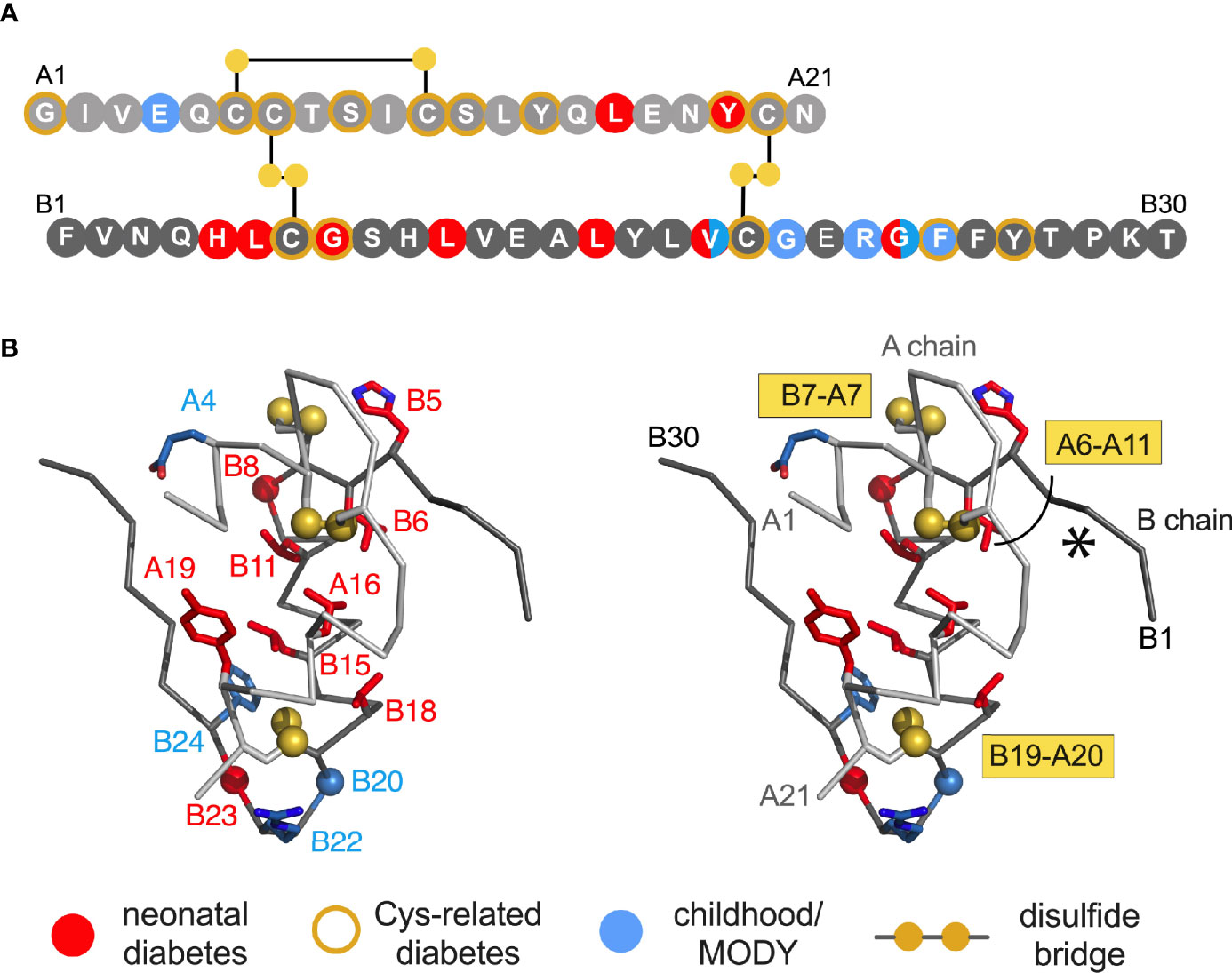
Figure 1 Clinical mutations in INS gene. (A) Sequence of insulin showing positions of clinical mutations. Residues are labelled by standard single letter code (bold white). The A chain is shown as light gray circles (upper sequence), and B chain as dark gray circles (lower sequence). Color code: neonatal- or delayed onset is indicated by filled red or blue circles, respectively. Sites of Cys-related mutations are highlighted by gold borders (Tables 1A, B). Disulfide bridges are indicated by filled gold circles connected by black lines. (B) Stereo view of insulin monomer (Cα-trace ribbon model; PDB entry 4INS) (47). Non-Cys-related mutations are highlighted as in (A); side chains are shown in red as labeled (Tables 1C, D). The Cα atoms of GlyB8, GlyB20 and GlyB23 are respectively shown as red, blue and red spheres (one-third Van der Waals radii), and sulfur atoms likewise as gold spheres. The A- and B chain ribbons are shown in light and dark gray, respectively. For clarity, symbols are also defined at bottom.

Table 1 Sites of clinical mutations in proinsulina,b.
Studies of Insulin Biosynthesis
Essential background is provided by the molecular biology of the insulin gene (53–57). In brief, INS encodes a single-chain precursor polypeptide, designated preproinsulin. Its signal peptide is cleaved on ER translocation. The translocated polypeptide (reduced proinsulin) contains a C domain between B- and A domains (thus connecting ThrB30 to GlyA1) (58). Folding in the ER requires specific pairing of three disulfide bridges (cystines B7-A7, B19-A20 and A6-A11). These bridges (gold spheres in Figure 1B) stabilize the protein and its receptor-binding surface (31, 59–67). Heteronuclear NMR studies of proinsulin (as an engineered monomer) have defined a folded insulin core with flexible C domain (48) as suggested by earlier studies (68–73). The contribution of each disulfide bridge to structure, stability, and activity have been extensively investigated (31, 59–61, 63–67). Together, these bridges provide both interior struts (cystines B19-A20 and A6-A11) and an external staple between chains (A7-B7). Mispairing of insulin’s cysteines (to form disulfide isomers) markedly impairs stability and activity (74–76).
Processing of proinsulin by prohormone convertases (PC1/3 and PC2) liberates the mature hormone (3, 77). Such conversion, occurring in the Golgi apparatus (GA) and immature secretory granules (78), ensures hormonal activity as proinsulin binds more weakly than insulin to the insulin receptor (IR) (79). Insulin and C-peptide are stored within glucose-regulated secretory granules (80) with microcrystalline assembly of Zn2+-stabilized hexamers (81–83). Evolution of such assembly foreshadowed its pharmacologic exploitation in clinical formulations (84). The marked susceptibility of the Zn2+-free insulin monomer (the active form of the hormone) to fibrillation complicated its manufacture and clinical use in the immediate decades after its discovery in 1921, thus recapitulating evolutionary constraints faced by β-cells due to the implicit threat of toxic misfolding (34, 85). This perspective has been reinforced by studies of a mouse model lacking the β-cell zinc transporter (86). Although key to the stable pharmaceutical formulation of “first-generation” rapid-acting insulin analogs [lispro and aspart (87), otherwise exhibiting heightened susceptibility to fibrillation (88)], in β-cells such assembly occurs only after exit from the ER and so cannot mitigate toxic misfolding of proinsulin variants.
Unlike native biosynthesis, chemical synthesis of insulin has traditionally employed isolated A- and B-chain peptides (89). The success of insulin chain combination implies that chemical information required for folding is contained within A- and B-chain sequences (90, 91). Hundreds of analogues have been prepared by this protocol, facilitating pharmaceutical innovation (87, 92). Despite the general robustness of insulin chain combination, synthesis of certain analogues has been confounded by low yields (30, 93–99). In selected cases such limitations have been overcome through the use of proinsulin or foreshortened single-chain synthetic intermediates [“mini-proinsulins” (100–103)]. Chemical protein synthesis via native ligation of peptide segments (104, 105) has also enabled synthetic access to the proinsulin molecule (106). In addition to their practical utility, such synthetic advances promise to provide insight into structural mechanisms of disulfide pairing (31, 95, 107–109). Sites of mutation among MIDY patients in large measure coincide with past difficulties in synthetic efforts.
Oxidative Folding Mechanisms
An historic foundation for studies of MIDY mutations in proinsulin has been provided by basic studies of protein folding over the past sixty years. Whereas studies of isolated peptides motifs and model globular domains were often designed to circumvent the complexity of disulfide pairing (28, 29, 110), oxidative protein folding has provided an attractive opportunity to define intermediates investigated by chemical trapping of partial folds (111). An extensive literature pertains to such disulfide-rich globular proteins as bovine pancreatic trypsin inhibitor (44, 112–114), hen egg white lysozyme (115–118) and α-lactalbumin (119–122). Insights from these model proteins and their application to proinsulin and homologous polypeptides underlie efforts to interpret INS mutations associated with toxic misfolding.
Chemical-trapping studies of proinsulin and homologous proteins have provided evidence for preferential accumulation of one- and two-disulfide intermediates (28, 29, 123, 124). These intermediates define a series of partial folds and corresponding trajectories on successive free-energy landscapes [“landscape maturation”; Figure 2A). The landscapes (maturing from shallow to steep; left to right in Figure 2A)] are each associated with (a) stepwise stabilization on successive disulfide pairing and (b) a corresponding ensemble of dynamic trajectories constrained by the bridges. Chemical-trapping studies are thus consistent with both multiple folding trajectories on funnel-shaped landscapes and a preferred sequence of specific disulfide intermediates (125) in general accordance with biophysical principles (126–128).
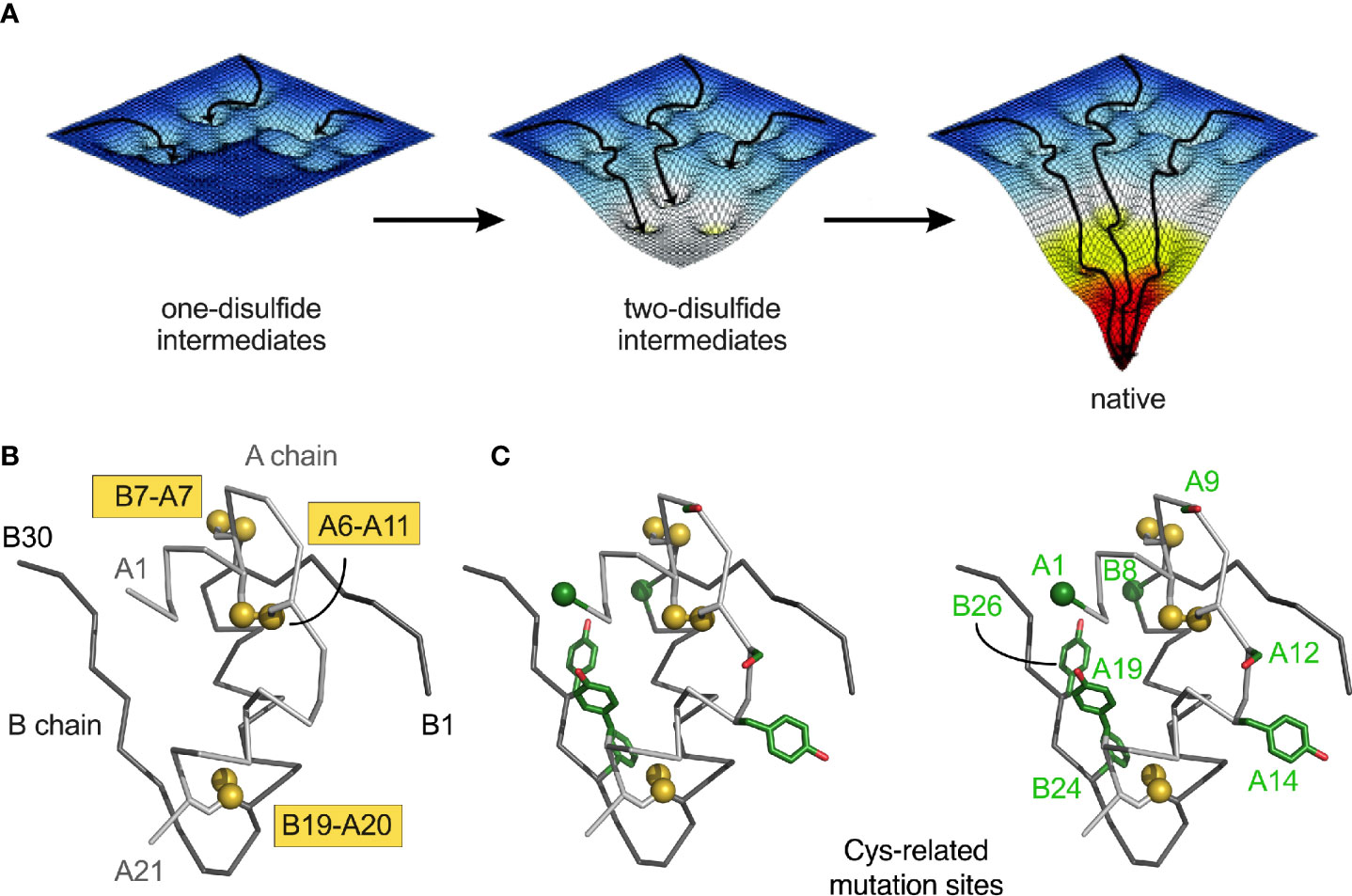
Figure 2 Energy landscape paradigm. (A) Landscape maturation: successive disulfide pairing enables a sequence of folding trajectories on ever-steeper funnel-shaped free-energy landscapes. (B) Ribbon model of insulin showing the three native disulfide bonds (yellow boxes). Coordinates were obtained from PDB entry 4INS (47). The A- and B chains are shown in light- and dark gray, respectively. (C) Stereo view of insulin with Cys substitutions highlighted in green (Table 1B). Side chains are shown as sticks; Cys-related sulfur atom and alpha-carbons of GlyA1 and GlyB8 represented as spheres (one-third Van der Waals radii).
Physiological interpretation of proinsulin refolding studies has been limited by its aggregation near neutral pH [thereby imposing a technical requirement for pH > 9) (29, 129)]. This limitation has been circumvented through the use of mini-proinsulin and IGF-I as more tractable models (28, 29, 59, 62, 108, 110, 123, 130). A structural pathway was proposed based on spectroscopic studies of equilibrium models (31, 59–61, 63–67, 131); this scheme highlights initial formation of cystine A20-B19 within a hydrophobic cluster of conserved side chains between the C-terminal A-chain α-helix and central B-chain α-helix (75, 76, 95). Because in the refolding of mini-proinsulin and IGF-I the A20-B19 disulfide bridge is the first to form (as the only one-disulfide intermediate to accumulate) (28, 29, 110), its pairing defines a biophysical milestone, formation of a specific folding nucleus (31, 131, 132). The predominance of cystine A20-B19 among populated intermediates motivated design of equilibrium models based on pairwise Ala- or Ser substitution of the other cystines (31, 59–67). Such analogues exhibit reduced α-helix content with native-like structure near cystine B19-A20 (31). Mutations in the putative B19-A20-related folding nucleus impair insulin chain combination, biosynthetic expression, and secretion of single-chain precursors in yeast (63, 97, 132–134).
1H-NMR spectra of one- and two-disulfide analogues exhibit progressive chemical-shift dispersion with successive disulfide pairing. These data are in accordance with stepwise structural stabilization in the landscape paradigm illustrated above (59, 131). Despite the predominance of A20-B19 pairing as an initial step, folding subsequently proceeds in parallel via multiple channels. Mini-proinsulin, for example, can rapidly form cystine A7-B7 or slowly undergo pairing of A6-A11. Although it is not apparent that pairing of cysteines distant in the sequence (such as A7 and B7) should be favored relative to pairing of nearby cysteines (A6 and A11), pairwise substitution of cystine A7-B7 (by Ser) destabilizes insulin more markedly than does pairwise substitution of A6-A11 (132). These findings suggest that nascent structure in the one-disulfide [B19-A21] intermediate either more effectively aligns CysA7 and CysB7 or more significantly impairs pairing of CysA6 and CysA11. These on-pathway two-disulfide intermediate may interconvert with non-native disulfide isomers as off-pathway kinetic traps. The danger posed by such traps has been highlighted in studies of IGF-I and its non-native disulfide isomers (28, 135). Relative isomer stabilities (as probed in a mini-IGF model) are specified by N-terminal residues in the B domain (136, 137). Although the refolding of proinsulin is more stringent, related non-native disulfide isomers (76) may readily be generated by disulfide exchange on addition of a chemical denaturant (75). Corresponding insulin isomers are molten globules whose stability and cooperativity are marginal (76).
Non-native disulfide isomers of proinsulin and related polypeptides have also been observed in transfection studies of mammalian cells (27, 30, 138–140). These studies have exploited electrophoretic mobility differences between native and non-native disulfide isomers in non-denaturing gels [as demonstrated by Arvan, Liu and colleagues (138)]. Less compact structures of non-native states are presumably associated with slower mobilities. Formation of non-native proinsulin isomers has thus been observed on transfection of expression constructs in a variety of mammalian cell lines. Although non-native proinsulin isomers are generally not secreted, mutations can enhance mispairing in the ER (139, 140). Extent of cellular misfolding does not correlate with in vitro thermodynamic stability, suggesting that the ER machinery does not evaluate free energies of unfolding (ΔGu) as a criterion of quality-control.
Studies of proinsulin variants containing N-terminal substitutions or deletions suggest that contributions of specific side chains to foldability may not be apparent in the native state (141). The substituted side chains may perturb the relative stabilities or kinetic accessibility of disulfide intermediates, for example, disproportionately to effects on the native state, once achieved. Such residues may also contribute to interactions of the nascent polypeptide with ER chaperones and its oxidative machinery (142). Indeed, the ER of β-cells may contain a lineage-specific set of chaperones and foldases required for proinsulin biosynthesis. Defining such a β-cell-specific “ER proteome” defines a key frontier of current research. Cell-type-specific differences in ER proteomes are likely to underlie the inefficient folding and secretion of proinsulin in the majority of human cell lines (143).
Foreshortened “mini-proinsulins” (144) can misfold in yeast to form a metastable disulfide isomer as the predominant secretion product. Such quantitative misfolding indicates that the ER folding machinery of a eukaryotic cell can selectively direct folding into a non-ground-state conformation. Characterizing this alternative pairing scheme and assessing its structural resemblance to the native fold would be of broad interest. Because the aberrant protein is not degraded prior to ER trafficking (i.e., it passes ER quality-control checkpoints), such analogues provide models of “stealth” misfolding, in turn leading to secretion of a protein caught in a kinetic trap. As described in the following two sections, clinical mutations in proinsulin conversely exemplify “non-stealth” misfolding leading to activation of the unfolded protein response (UPR) (145–150).
Monogenic Diabetes and the INS Gene
The majority of INS mutations cause permanent neonatal-onset DM (Figure 1 and Table S1) (14). Because impaired β-cell function develops prior to maturation of the immune system, the patients present with auto-antigen-negative DM. Similar phenotypes may be caused by mutations in other genes (151), most frequently a heterozygous activating mutation in the β-cell voltage-gated potassium channel (either KCNJ11 or ABCC8, respectively encoding its Kir6.2 and Sur1 subunits) (152, 153). The resulting diabetic phenotype in this genetic background may be transient or permanent. It is important to recognize this subset of neonates or toddlers as in favorable cases they can successfully be treated with oral agents that inhibit the channel (sulfonylureas) rather than by insulin injections (151).
Dominant INS mutations are the second most common genetic cause of permanent neonatal DM (7, 13, 14, 16). Such mutations occur in each region of preproinsulin: its signal peptide, B-, C- and A domains (Table S1) (9, 10). The majority result in the addition or removal of a cysteine, leading in either case to an odd number of potential pairing sites (Figure 1A). Mutations have been found at each of insulin’s six canonical cysteines, generally associated with neonatal onset (Figure 2B, Table 1A). An additional cysteine may be introduced at various positions in the insulin moiety (Figure 2C and Table 1B). The resulting odd number of thiol groups leads in general to misfolding and aggregation (11, 12, 18). Even in this context structure may matter, as it is possible that some sites of Cys introduction lead more readily to aberrant intra- or intermolecular disulfide pairing than others, depending on the conformational properties of oxidative folding intermediates and their interactions surfaces. Such biophysical variability would be expected to be associated with differences in ER stress and hence age of DM onset.
Among human MIDY mutations is the same “Akita” substitution (CysA7→Tyr) as in the Ins2 gene of the Mody4 mouse (21–23); this dominant murine substitution has thus been characterized as a model of the human syndrome (25–27). The variant murine proinsulin in vitro undergoes partial unfolding with increased aggregation (154). Analogous perturbations were found in human insulin- and proinsulin analogues lacking cystine A7-B7 (66, 132). Heterozygous expression of related Ins2 allele CysA6→Ser in the mouse also causes DM (155).
Identification of identical human and murine mutations at position A7 suggests that the mechanisms of neonatal DM have shared pathogenetic features independent of species (21–23, 25–27). Although β-cell degeneration in the Akita mouse remains incompletely understood, early defects have been observed in the folding and trafficking of both wild-type and variant proinsulins. These defects are associated with elevated markers of ER stress, electron-dense deposits in abnormal ER and GA, and mitochondria swelling—together leading to a progressive decline in β-cell mass (25–27). Evidence for the clinical relevance of these findings has been obtained by the construction of innovative fluorescent proinsulin fusion proteins and their use in cell lines and transgenic mice to detect subcellular localization and aggregation (35–38).
Deciphering Determinants of Foldability
The Akita variant is representative of a mutant proinsulin with an odd number of cysteines. However, a distinct subset of MIDY- or MODY-associated mutations does not involve cysteine (Table 1C). Although widely scattered in the sequence, these mutations occur more often in the B domain than in the A domain—and not at all in the C domain. Because the variant proinsulins retain the six canonical cysteines and yet pair inefficiently, such mutations are of special biophysical interest. A structural overview is provided in Figures 3–5 as described in turn.
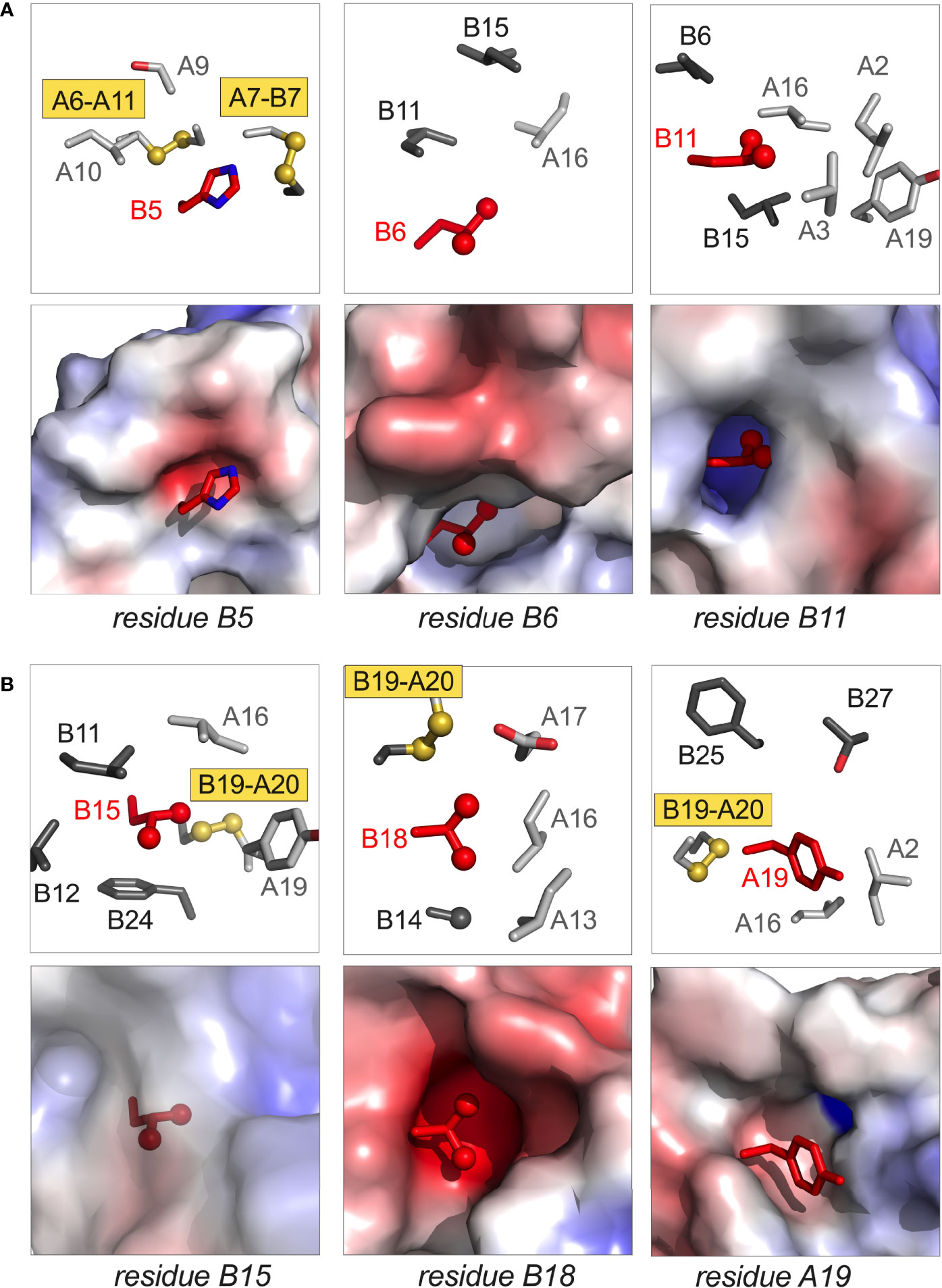
Figure 3 Structural sites of neonatal-onset mutations. (A) Spatial environments of residues B5, B6, and B11; (B) spatial environments of residues B15, B18 and A19. In each panel the highlighted side chain is shown in red; in each pair of images, stick models are shown in upper panels whereas electrostatic surfaces (calculated in absence of indicated side chain) are shown in the lower panels. In stick models, side chains belonging to the A- and B chains are respectively shown in light and dark gray; Cys-related sulfur atoms (gold) and aliphatic methyl groups (red) are represented as spheres (one-third Van der Waals radii). Coordinates were obtained from PDB entry 4INS (47).
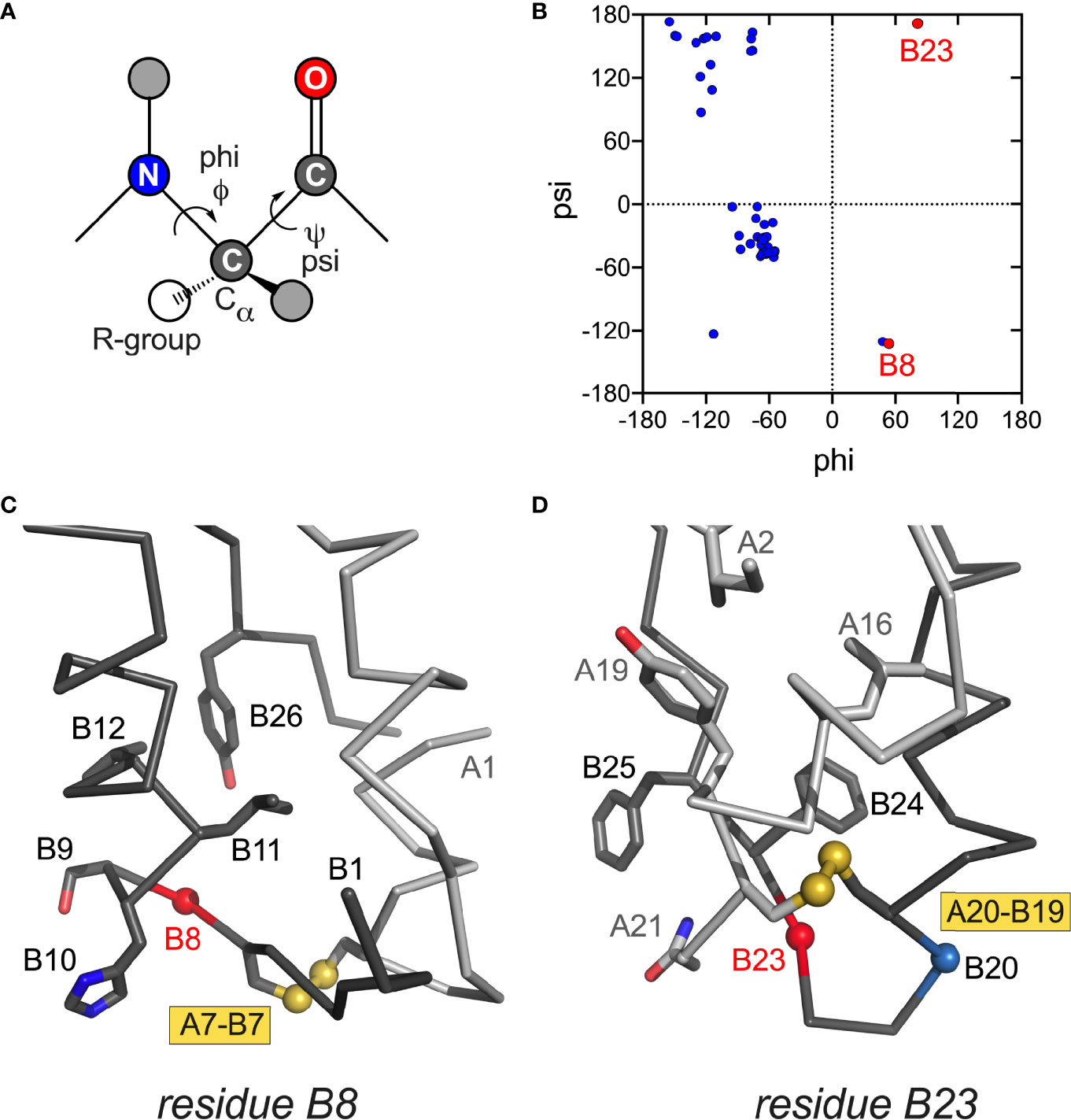
Figure 4 Conformations and structural environments of conserved glycines at positions B8 and B23. (A) Main-chain dihedral angles phi (φ) and psi (ψ) in a peptide. (B) Ramachandran plot of crystallographic insulin T state (PDB entry 4INS; φ/ψ angles generated using PyMOL and plotted using GraphPad Prism software). Residues B8 and B23 are as labeled (red). In canonical T state GlyB8 and GlyB23 lie within β-turns with positive φ–angles, thereby residing on the right side of Ramachandran plane in regions unfavorable or “forbidden” for L-amino acids. In R state GlyB8 residues in α-helix and so on the left side of Ramachandran plot (not shown). (C) Canonical environment of GlyB8 in the T-state β-turn; key nearby side chains are shown. (D) Canonical environment of GlyB23 in B20-B23 β-turn, shared by T- and R states. Residue B23 is near the side chain of AsnA21, and the positive B23 φ angle enables formation of an inter-chain hydrogen bond (A21 side-chain carboxamide NH … O=C B23). Nearby side chains are shown. α-Carbon traces of the A- and B chains are shown in light- and dark gray, respectively. Coordinates were obtained from PDB entry 4INS (47).
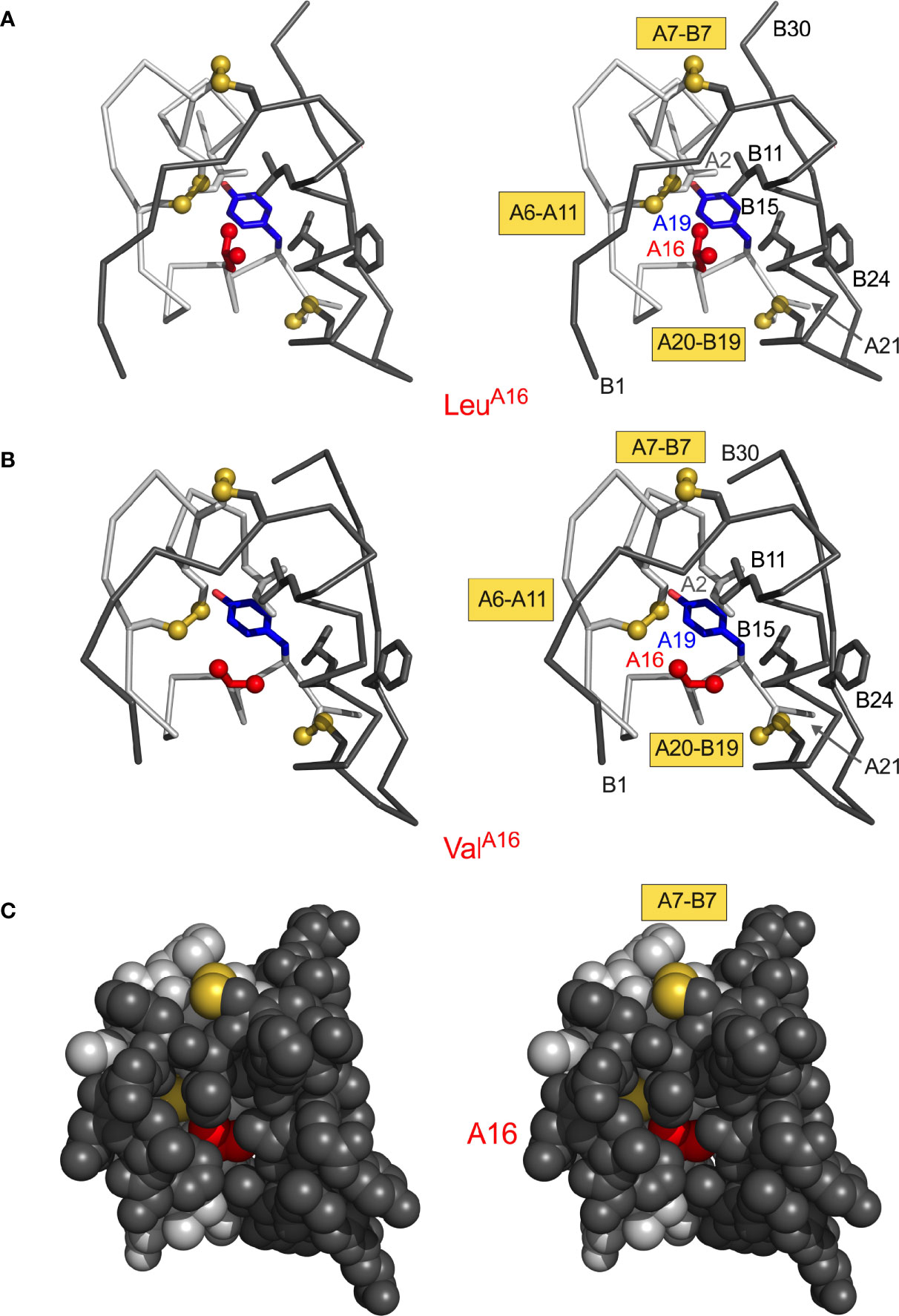
Figure 5 Structural environment of conserved position A16. (A) LeuA16 packs in core of insulin monomer: ribbon model (stereo pair) showing LeuA16 (red) in relation to TyrA19 (blue) and internal side chains IleA2 (light gray), LeuB11 (dark gray) and LeuB15 (dark gray). A- and B-chain ribbons are shown in light- and dark gray, respectively; disulfide bridges are shown as gold spheres. Molecular coordinates were obtained from PDB entry 4INS (47). (B) Corresponding ribbon model (same orientation) of “non-foldable” analogue ValA16 (PDB entry 3GKY) (156). Structural similarities highlight cryptic folding defect. (C) Stereo space-filling model showing limited exposure of internal ValA16 side chain (red) between B-chain surface (dark gray, overlying surface) and A-chain surface (light gray). The solvent-exposed A7–B7 disulfide bridge is shown in gold (top); internal cystine side chains A6–A11 and A20–B19 are not visible.
Structural relationships in insulin were examined using the monomer derived from a representative wild-type T6 zinc insulin hexamer [PDB entry 4INS (47)]. NMR studies have shown that the conformation of an engineered insulin monomer in solution closely resembles the T-state protomer in a zinc insulin hexamer as characterized by X-ray crystallography (60, 157–159). Short- and medium-range NOEs are consistent with spatial relationships in the T state (159). Although positions of C-terminal B-chain residues (B25-B30) are generally less well defined than in crystallographic dimers and hexamers, classical attachment of B24-B28 β-strand to the α-helical core is maintained in solution. The free monomer thus does not exhibit a major change in B-chain conformation [hinge opening of the B20-B23 β-turn (160)]. The present analysis has thus focused on spatial relationships in T-state monomers (as extracted from crystal structures) because of their higher resolution (relative to NMR ensembles) and likely pertinence to proinsulin (48).
N-terminal segment. In NMR-derived structures of insulin as a Zn2+-free monomer (60, 157, 158), residues B1-B6 are extended (asterisk in Figure 1B); B7-B10 comprise a β-turn adjoining the central α-helix. Similar features occur in the crystallographic T-state protomers within Zn2+ hexamers (47, 161). The N-terminal five residues favor A7-B7 disulfide pairing in vitro (136, 137) and to overall efficiency of proinsulin folding in cell lines (141). These residues are dispensable for receptor binding (162). Although PheB1 has not been identified to date as a site of clinical mutation, studies of des-B1 analogues nonetheless suggest that its loose T-state-specific packing against a nonpolar A-chain surface (principally the otherwise exposed side chain of LeuA13) contributes to disulfide specification (12). Sites of clinical mutation (HisB5, LeuB6 and GlyB8; broadly conserved among vertebrate insulins) have been well characterized (30, 96, 98, 99).
i. Position B5. In the native state HisB5 packs within an inter-chain crevice, making one or more hydrogen bonds to carbonyl oxygens in the A chain (Figure 3A, left). Clinical mutations are Asp, Gln and Tyr (Table 1C); in mammalian cell culture substitution of HisB5 by Asp blocks the folding and secretion of human proinsulin (30). Although some substitutions impair chain combination (30), ArgB5 (found in non-mammalian insulins) is well tolerated. We imagine that HisB5 and ArgB5 form analogous inter-chain hydrogen bonds in the course of disulfide pairing; this hypothesis is in accordance with the respective crystal structures of WT and ArgB5-insulin (99). AlaB5-insulin (as an engineered monomer) exhibits decreased stability (30), presumably due to the absence of these hydrogen bonds and to a cavity penalty (163, 164). Its solution structure is nonetheless similar to the parent HisB5 monomer (30), suggesting that critical perturbations in an oxidative folding intermediate can be inapparent in the native state, once reached.
ii. Position B6. LeuB6 inserts into a deep inter-chain cavity bounded by the invariant side chains of LeuB11, LeuB15 and LeuA16 (Figure 3A, middle). Neonatal-onset mutations are Arg, Gln, Pro and Val (Table 1C). Each variant would be expected to be destabilizing in this environment: Arg and Gln via insertion of charged or polar functions into a nonpolar cavity, Pro and Val via introduction of packing defects. Substitution of the branched and nonpolar side chain of LeuB6 by the linear non-polar side chain Met by contrast leads to MODY (Table 1D). Delay in clinical onset presumably follows the structural biology: we envision that MetB6 can be accommodated within the B6-related cavity but with less optimal packing interactions.
iii. Position B8. Special structural principles pertain to position B8. Neonatal-onset mutations are Arg, Ser and Val (Table 1C; also Cys in Table 1B). In an insulin or proinsulin monomer in solution (48, 60) GlyB8 exhibits a positive ϕ dihedral angle [as in the crystallographic T state (47)] and so occupies a position in the Ramachandran plane ordinarily forbidden to L-amino acids (Figures 4A, B). In a protein-folding intermediate an L-amino-acid side chain at B8 would presumably change the orientation of CysB7 and so impair its pairing with CysA7 (Figure 4C) (98). The side chain itself would be expected to project into solvent.
Kent, Weiss and colleagues described synthetic studies of human proinsulin variants containing L-Ala or D-Ala at B8 (109). Such protein diastereomers exhibited L-specific impairment of specific disulfide pairing; D-AlaB8 was well tolerated, presumably due to its enforcement of a positive ϕ angle favorable to [B7-A7] pairing. These findings corroborated prior studies of mini-proinsulin analogues (134, 165) and insulin chain combination (96). In the latter stereospecific B-chain libraries were exploited to demonstrate that L-substitution at B8 generally impair chain combination whereas yield was generally enhanced by D-substitutions (96). Together, these studies rationalize the invariance of GlyB8 among vertebrate insulins and insulin-related polypeptides and the diversity of clinical mutations at this site. Interestingly, SerB8-insulin (but not AlaB8-insulin) exhibits substantial biological activity despite its reduced foldability (109). Indeed, its solution structure retain native-like features. Decreased thermodynamic stability was nonetheless observed, presumably due to an unfavorable local main-chain conformation on the right side of the Ramachandran plot (166).
Central α-helix. Nascent α-helical structure in the B chain has been observed in one- and two-disulfide analogues containing the key [A20-B19] disulfide bridge (31, 59, 63, 66, 67, 132). Neonatal-onset mutations have been identified at positions B11, B15 and B18 (Table 1C) as described in turn.
(i, ii) Helicogenic residues B11 and B15. LeuB11 and LeuB15 each contribute to segmental α-helical propensity (167, 168) and to the nascent clustering of nonpolar residues (31, 131). We imagine that mutations at these sites (Pro or Gln at B11, Pro or Val at B15; Table 1C) would impede nascent α-helix formation and in turn initial [B19-A20] disulfide pairing. In the mature structure the B11 side chain is buried within a cavity abutting the nonpolar inner surface of the A chain (Figure 3A, right) whereas the B15 side chain packs within a shallower neighboring inter-chain crevice delimited by CysB19 and PheB24 (Figure 3B, left). Should native disulfide pairing be achieved, we would expect that that mutations ProB11 and ProB15 would profoundly perturb native structure, stability and self-assembly. GlnB11 and ValB15 would also be destabilizing, but likely less so than Pro. GlnB11 would fit within the B11-related cavity, but its carboxamide group would impose an electrostatic penalty; the smaller, β-branched side chain of ValB15 would be predicted to attenuate segmental α-helical propensity (167, 168) and impose a cavity penalty (163).
The importance of LeuB11 and LeuB15 to folding efficiency was first demonstrated in a model organism. Ala substitutions at these positions (although compatible with α-helix) were found to hinder secretion of mini-proinsulin in S. cerevisiae (134). Insulin chain combination was likewise impaired by interchange of LeuB11 and ValB12, presumably due to perturbed long-range packing (93). Native spacing between CysB7 and CysB19—and hence length of the central B-chain α-helix—are also likely to influence the efficiency of disulfide pairing as a complex MIDY mutation combines a point mutation with deletion with an adjacent residue: LeuB15-TyrB16 are replaced by His (43), leaving an even number of residues between the B-chain cysteines.
(iii) Non-helicogenic residue B18. ValB18 packs near cystine [B19-A20] in a solvent-exposed inter-chain crevice. This environment is polar on one side (due to GluA17) and non-polar on other sides (due to the cystine, AlaB14, LeuA13 and LeuA16). Although the β-branched side chain of Val is not in principle favorable within an α-helix (167, 168), its mutation to Gly (also of low helical propensity; Table 1C) would enhance main-chain flexibility and introduce an inter-chain packing defect; each perturbation could reduce efficiency of [B19-A20] disulfide pairing. In the native state 1H-2H exchange studies in D2O have established that the main-chain amide proton of ValB18 is the most highly protected site in insulin (159). Extending this to variant on-pathway folding intermediates, we propose that enhanced segmental conformational fluctuations and decreased thermodynamic stability could each contribute to impaired biosynthesis.
B20-B23 β-Turn. The B chain contains a U-turn between its central α-helix and C-terminal β-strand (B24-B28). This super-secondary motif requires a solvent-exposed β-turn (Gly-Glu-Arg-Gly tetrapeptide motif). Like GlyB8 (above), the flanking glycines each exhibit positive ϕ angles associated with a specific pattern of hydrogen bonds within the turn (47). Discussed more fully in the following section (MODY), mutation of GlyB23 to Val is associated with neonatal-onset DM (Table 1C). Cell-based and biophysical studies of this mutation have demonstrated profound perturbations (97). Qualitative NMR studies suggest that the β-branched side chain leads to transmitted perturbations in the position or conformation of the following B24-B27 segment (12).
A-chain mutations. Studies of peptide models have suggested that initial pairing of cystine [B19-A20] is coupled to nascent α-helical conformations of the A16-A19 segment, coincident with nonlocal hydrophobic collapse of LeuA16 and TyrA19 within a folding nucleus (31, 131). Indeed, substitutions at these sites were found to impair the yield of insulin chain combination (94, 95, 156). In accordance with the above mechanism and such synthetic experience, recent clinical studies have uncovered neonatal-onset MIDY mutations ProA16 and AspA19 (Table 1C).
The structural environments of α-helical residues A16 and A19 are distinctive. Whereas TyrA19 projects from a non-polar crevice (lined in part by cystine [B19-A20]) to expose its para-hydroxyl group (Figure 3B, right), the side chain of LeuA16 is inaccessible to solvent (Figure 5). AspA19 would place a negative charge within the non-polar confines of the core. ProA16 would perturb segmental main-chain conformation and (when modelled in a native-like framework) introduce both side-chain steric clash and a destabilizing cavity. The essential contribution of LeuA16 to protein-folding intermediates has been demonstrated through studies of ValA16-proinsulin and ValA16-insulin (156). Although this substitution is compatible with a native-like crystal structure (essentially identical to WT insulin), ValA16 markedly impairs both insulin chain combination and cellular folding of the variant proinsulin (156). Because ValA16-insulin also exhibits high biological activity (156), the evolutionary invariance of Leu at this position presumably reflects its cryptic yet key contribution to folding efficiency.
MIDY mutations have not been identified in the N-terminal A-domain α-helix (residues A1-A8). Their absence may simply reflect incomplete sampling of patients to date; however, it is also possible that non-cysteine residues in this segment are tangential in the mechanism of disulfide pairing. Indeed, successful combination of variant A chains containing Gly at positions A1-A2, A1-A4 or A1-A4 (in each case with WT B chain S-sulfonate) provided evidence that an N-terminal A-chain α-helical conformation is not required for native disulfide pairing (95). Such dispensability is in accord with a putative structural pathway in which segmental folding of this α-helix is a late event.
From MIDY to MODY
INS mutations may also be associated with onset of DM in childhood or adolescence (Table 1D) (169–171); diagnoses may be carried as auto-antibody-negative presumed Type 1 DM or Type 2 DM. Substitution of ValB18 (Figure 3B, center) by Ala (172) was identified as a MODY allele (DM onset <25 years of age, autoantigen negative) in a three-generation Italian pedigree (three siblings, the parent and presumed grandfather) (172). Unlike MIDY patients with neonatal onset, birth weights were normal. The Ala mutation at position B18 would be expected to enhance segmental α-helical propensity (167, 168), but introduce a destabilizing cavity (163, 164) adjacent to the critical [B19-A20] disulfide bridge. Unlike the perturbations introduced by GlyB18 (above), these effects would offset to yield, rationalizing a mild net impairment of initial disulfide pairing.
Four additional MODY mutations occur within the B20-B23 β-turn and its aromatic anchor at B24: GlyB20→Arg, ArgB22→Gln, GlyB23→Asp and PheB24→Ser (Figure 6). Although the mechanism by which GlnB22 causes MODY is not apparent, L-amino-acid substitutions of GlyB20 or GlyB23 would be expected to alter their respective ϕ dihedral angles. It has previously been reported that Ala substitutions impair the expression of mini-proinsulin in S. cerevisiae and impede chain combination, whereas efficient disulfide pairing in vitro can be rescued by D-Ala substitutions (97). That B23 mutations may cause either neonatal onset (ValB23) or delayed onset (AspB23) suggests that details of side-chain chemistry influence folding efficiency.
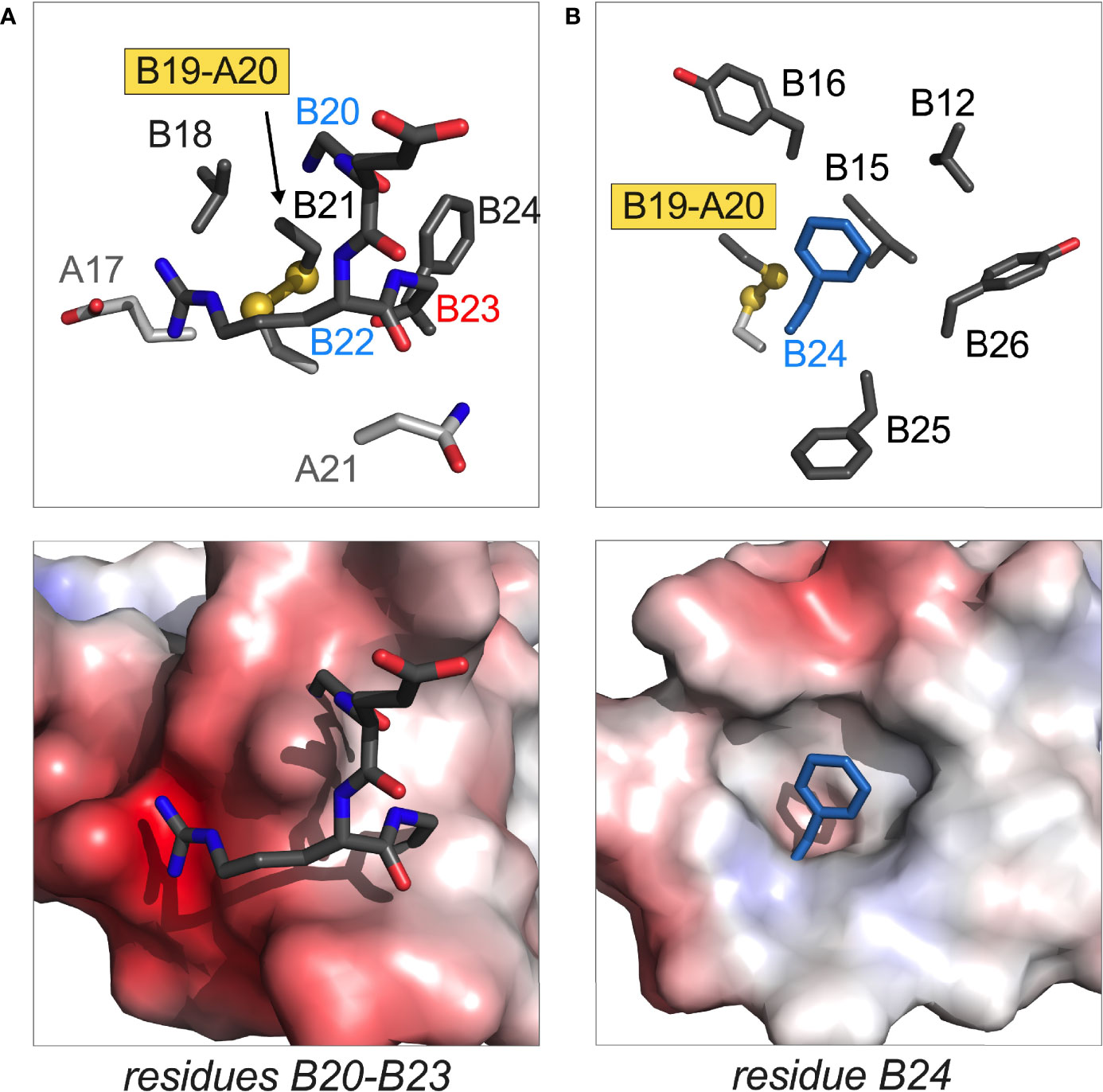
Figure 6 Structural sites of MODY mutations. (A) Residues B20 and B22 in B-chain B20-B23 β-turn; (B) residue B24 anchoring this β-turn and adjoining B24-B28 β-strand. In each pair of images, stick models are in upper panel and electrostatic surfaces in lower panel. The latter highlights the groove or cavity occupied by the designated structural element; blue and red surfaces are coded by positive or negative electrostatic potential. In stick models main-chain atoms in A- or B main chains are shown in light or dark gray, respectively. Disulfide bridges are shown as balls and sticks with sulfur atoms in gold (one-third Van der Waals radii). The side chain of PheB24 in (B) is shown as dark blue stick (not related to electrostatic potential). Coordinates were obtained from PDB entry 4INS (47).
SerB24 (originally designated insulin Los Angeles) is associated with variable genetic penetrance with hyperinsulinemia. The latter finding indicates that SerB24-proinsulin can in fact fold in the β-cell ER, undergo proper trafficking and processing to mature SerB24-insulin (173). In cell culture the variant proinsulin nonetheless induces ER stress, albeit at a level below MIDY variants (12). In vivo mutational induction of mild or moderate ER stress can presumably cause (depending on other genetic risk alleles and environmental factors) slow but progressive loss of β-cell mass (174, 175) as in the Akita mouse (24, 176).
The final MODY-associated mutation occurs on the surface of the A domain: GluA4→Lys (Table 1D and Figure 7A). That this substitution should perturb the folding of proinsulin seems surprising given (a) the absence of structural constraints at this position in insulin and (b) the broad tolerance of insulin chain combination to substitutions within the N-terminal A-chain α-helix (95). We speculate that LysA4 introduces a subtle perturbation in proinsulin through electrostatic repulsion of the dibasic element at the CA junction (red box in Figure 7B). In particular, nascent α-helical structure in the A1-A8 segment may be stabilized by a salt bridge between “ArgA0” (i.e., the final residue of the C domain [position 89 of preproinsulin]; Figure 7A) and WT GluA4 (Figure 7C). Such an interaction, together with GlyA1, could in essence provide a favorable N-Cap (177), which could overcome the adverse helical propensities of the three β-branched residues in this segment (IleA2, ValA3 and ThrA8). This contribution would not pertain to insulin chain combination due to the absence of ArgA0 (an analogous C-capping salt bridge from GluA4 to the A1 α-amino group would be blocked by its deprotonation at the reaction pH of 10.5).
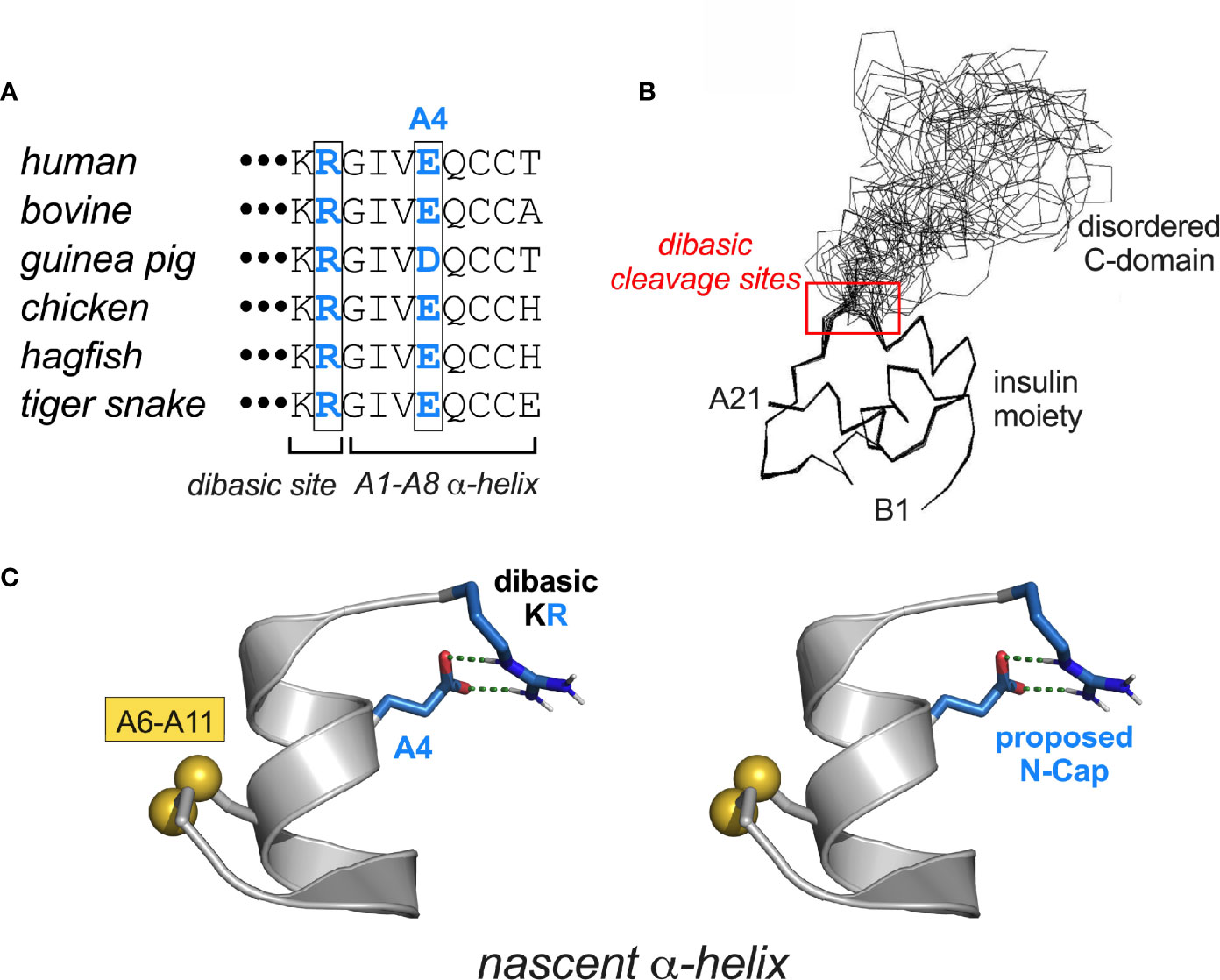
Figure 7 MIDY-related mutations at CA junction. (A) Vertebrate sequence alignment showing conserved KR dibasic site (CA junction) and acidic side chain at residue A4. (B) Solution structure of proinsulin (line drawing) showing the folded insulin moiety and disordered C-domain (48). The dibasic RR (BC junction) and KR sites (CA junction) are within red box. (C) Proposed stabilization of a nascent α-helix in proinsulin folding intermediate by junctional (i, i+4) salt bridge between residues Arg and GluA4 (blue in panels A, C). The putative salt bridge was modeled as an α-helical N-Cap element (177) using PyMOL.
Diversity of INS-Related Disease Mechanisms
For completeness, we note that mutations in the insulin gene that are not associated with impaired folding can nonetheless be associated with adult-onset DM phenotypes of variable penetrance (57) (Table S1). Such heterogeneity is in accord with “Murphy’s Law of genetics”: in a complex pathway or set of mechanisms, what can go wrong will go wrong. For example, insulin variants Wakayama and Chicago (i.e., classical insulinopathies ValA3→Leu and PheB25→Leu respectively) markedly impede receptor binding (173) in association with mutant hyperinsulinemia (178). These mutations directly perturb the hormone-receptor interface (160). A complementary example is provided by diabetes-associated mutation HisB10→Asp, which enhances receptor binding (179). Although AspB10 would introduce a favorable electrostatic interaction at the hormone-receptor interface, in β-cells AspB10-proinsulin exhibits inappropriate sorting to a constitutive granule (180, 181). Unlike glucose-regulated secretory granules, constitutive granules lack prohormone convertases, and so the patients exhibit mutant hyperproinsulinemia. Yet another syndrome is characterized by impaired prohormone processing leading to circulation of a split proinsulin with reduced activity (182).
Evolution at the Edge of Foldability
Protein evolution is generally enjoined by overlapping biological constraints, including biosynthesis, structure, and function (Figure 8A). Particular residues in insulin may thus contribute to one or more critical mechanisms, including nascent foldability in the ER, protection from intra- or extracellular toxic misfolding, trafficking from the ER through the GA to glucose-regulated secretory granules, self-assembly within these granules, disassembly of Zn2+-insulin hexamer in the portal circulation and in turn receptor binding. The stringency of these overlapping constraints rationalizes the limited sequence variation among vertebrate insulins (47).
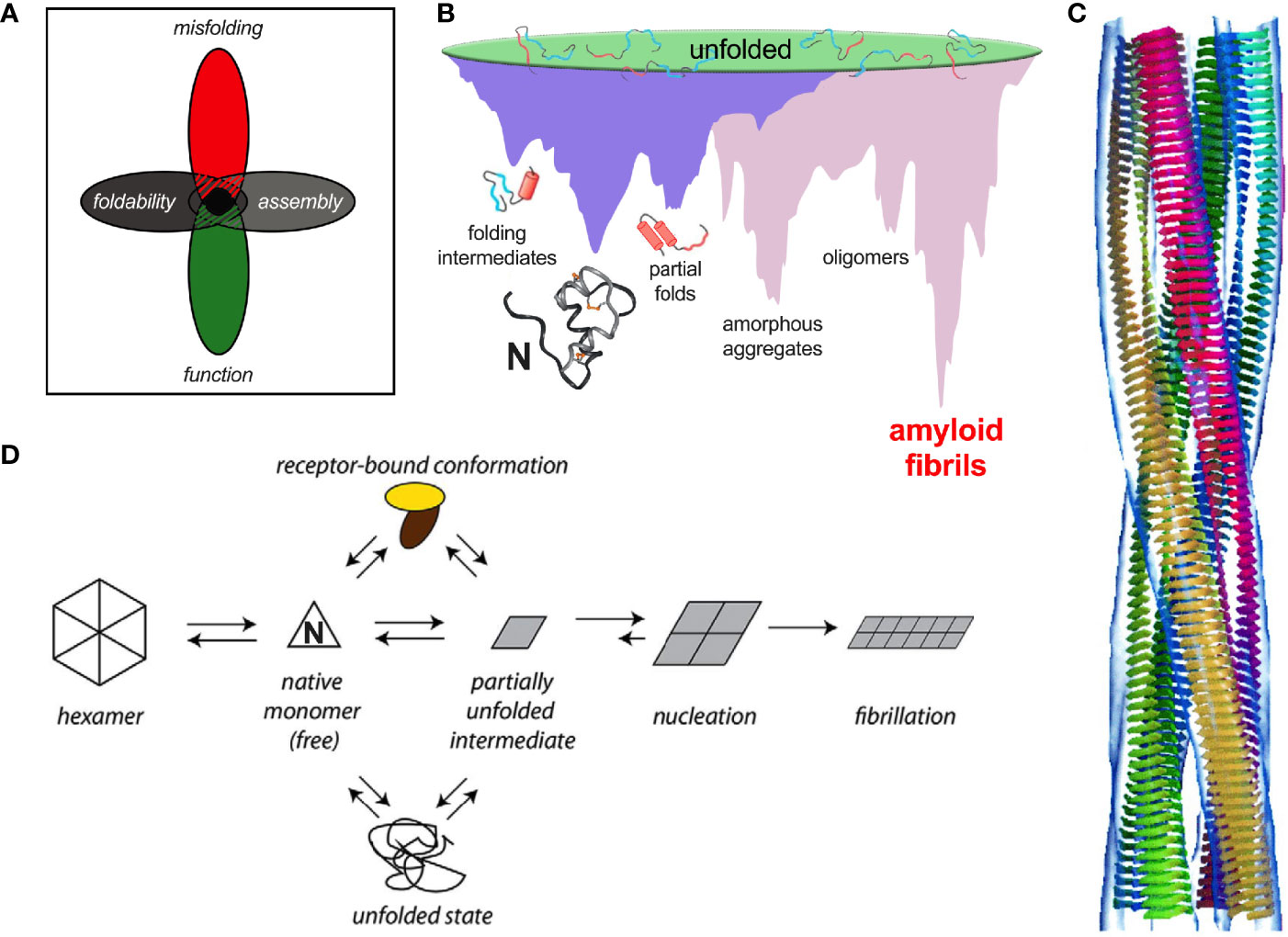
Figure 8 Evolutionary constraints and insulin fibrillation. (A) Venn diagram showing intersection of multiple constraints: function, foldability, misfolding, and assembly (183). (B) Energy landscape of protein folding (purple) and coupled landscape of aggregation (pink) (184). Cytotoxic oligomers may form as off-pathway intermediates en route to amyloid. (C) Models of protofilament packing based on low-resolution cryo-EM images. The image is reproduced from the reference (185), Copyright (2002) National Academy of Sciences. (D) General scheme of insulin fibrillation via a partially unfolded monomeric intermediate (parallelogram at center) (186). The native state (triangle) is protected by classic self-assembly (far left). Disassembly leads to an equilibrium between native and partially folded monomers. The receptor-bound conformation of insulin (top) may also participate in this equilibrium. This partial fold may unfold completely (bottom) as an off-pathway event or aggregate to form an amyloidogenic nucleus en route to a proto-filament (right).
Evolutionary constraints may be coincident or opposing at a given position. An example of a coincident constraint is the concurrent contributions of invariant PheB24 to core packing, dimerization and receptor binding. Opposing constraints call for compromise. An example is provided by GlyB8, invariant as an achiral amino acid free to roam in the Ramachandran plane. Systematic studies of L- or D substitutions have suggested that at B8 kinetic determinants of foldability are at odds with conformational requirements of receptor binding (96, 98). Whereas a positive ϕ dihedral angle (enforced by a D-substitution) facilitates disulfide pairing, a D side chain impedes receptor binding. Conversely, negative dihedral angle (like that of an L-amino acid) impair folding efficiency but may be compatible with receptor binding (96, 98). These opposing requirements presumably underlie the invariance of glycine – the only achiral amino acid – at a site of conformational change. A switch in conformation of GlyB8 between the right side of the Ramachandran plot and the left (respectively corresponding to positive or negative ϕ angles) was anticipated by the classical TR transition among zinc insulin hexamers (187). Although such allostery may pertain only to hexamers (99), the TR transition exemplified the long-range transmission of conformational change (188) —a theme central to the transmembrane propagation of an insulin signal via receptor reorganization (189–191). The examples posed by clinical mutations at B24 and B8 (Table 1C) suggest that premature adoption of the hormone’s receptor-engaged conformation within β-cells (either by proinsulin in the ER or GA or by insulin in secretory granules) may trigger toxic misfolding.
Recent co-crystal and cryo-EM-derived structures of insulin bound to receptor fragments have demonstrated the function of a protective hinge in B chain (160, 189, 190, 192). Mechanisms of hormone-receptor recognition [for review, see (191)], extend to IGF-I as visualized in a landmark series of homologous cryo-EM-derived structures of respective receptor ectodomain complexes (189, 190, 193–195). As predicted based on studies of “anomalous” insulin analogues (157, 196, 197), detachment of the C-terminal β-strand (residues B24-B28) enables both its own binding in a groove between receptor elements L1 and αCT (respectively at the N- and C-terminal ends of the IR α-subunit); the latter element also packs against the N-terminal A-chain α-helix.
Insulin’s B-chain hinge—opened on receptor binding—may represent an evolutionary response to the danger of proteotoxicity. This danger, aggravated by exposure of non-polar surfaces, is intrinsic to the coupled folding/misfolding landscapes wherein the true ground state is defined by β-sheet-rich amyloid (Figure 8B). Models of insulin amyloid as superhelices of protofilaments have been derived at low resolution by cryo-EM (Figure 8C). Studies of insulin fibrils by infrared and Raman spectroscopy have demonstrated a predominance of β-sheet (198–200) in accordance with fibril X-ray diffraction (201–203). Despite the striking biophysical features of fibrils as a universal thermodynamic ground state of polypeptides (Figure 8B) (85), oligomeric intermediates in the pathway of fibrillation pose the greater cytopathic danger (Figure 8D) (204).
Recent evolutionary studies of insulin have highlighted the importance of PheB24, whose conserved aromatic ring plays multiple roles: anchoring the native B-chain β-strand, stabilizing the α-helical core, and contributing to both self-assembly (47) and hinge opening on receptor binding (197). In the open state the aromatic ring binds within a classical nonpolar pocket at the hormone-receptor interface (189, 190, 195). On substitution of PheB24 by Gly, native function is paradoxically retained (157). Comparative studies of “register shift” analogues indicate that an alternative mode of receptor binding supervenes in which PheB25 takes the place of the missing PheB24 (52); residues B20-B24 form a flexible pentaloop rather than an aromatic-anchored β-turn (205). This alternative binding mode is apparently disallowed in evolution due to toxic misfolding of GlyB24-proinsulin (as evidenced impaired folding efficiency, induction of ER stress and impaired secretability in transfected cell models) and possibly by the heightened susceptibility of GlyB24-insulin analogues to fibrillation (52). Evidence for the paradoxical evolution of vertebrate insulins to the edge of foldability has been provided by biophysical studies of a native-like variant, TyrB24-insulin (40). Although providing the C-terminal B-chain β-turn and β-strand with an homologous “aromatic anchor,” TyrB24 is also disallowed due its perturbation of biosynthesis and induction of ER stress. Indeed, of the 20 natural amino acids, only Phe at position B24 enables the efficient biosynthesis of proinsulin (40). We speculate that such marked sensitivity to mutation—signifying the paradoxical non-robustness of an adaptive landscape (40)—will be found at many or most sites associated with neonatal-onset DM (Table 1C).
Because, to our knowledge, clinical mutations that selectively perturb insulin’s hexameric structure and storage in secretory granules have not been described, this review has not focused on these processes. Any such perturbations would be downstream from the major sites of perturbation in the MIDY syndrome: misfolding in the ER and impaired trafficking through the GA. It is possible, however, that processes in the secretory granule are affected by SerB24 and AspB10 in concert with other perturbations. (i) PheB24→Ser. The invariant aromatic ring of PheB24 packs at the dimer interface. Its substitution by SerB24 impairs self-assembly (as monitored by gel-filtration) and leads to accelerated disassembly of the R6 hexamer once formed (40). Receptor binding and biological activity are low. (ii) HisB10→Asp. The conserved imidazole ring of HisB10 coordinates the axial zinc ions at the trimer interface of insulin hexamers (47). Genetic variant AspB10 causes a diabetes syndrome characterized by baseline mutant proinsulinemia due to constitutive secretion (180) as the mutation perturbs specific trafficking to glucose-regulated secretory granules (34, 57). The corresponding substitution in insulin blocks both zinc binding and trimer formation in vitro (206, 207). AspB10-insulin exhibits increased affinity for both IR and IGF-1R with prolonged residence times in association with augmented mitogenic signaling (179, 208–211).
An Evolutionary Hypothesis
Given the ancestral history of metazoan insulin-like proteins over the past 540 million years and its broad radiation among diverse body plans (212–214), why might vertebrate proinsulins be susceptible to misfolding and lacking in mutational robustness? A possible answer is given by the history of the INS gene as traced by the late D.F. Steiner and coworkers (215–218). This seminal study characterized an insulin-like gene encoding an insulin-like protein (ILP) in an extant protochordate (amphioxus; Branchiostoma californiensis) (Figure 9A). The predicted polypeptide precursor pro-ILP contains a C-terminal peptide resembling the D and E domains of vertebrate IGFs, suggesting an intermediate form linking the ancestral proto-insulin gene with modern IGF genes. In accordance with this perspective, ILP is the only INS-like gene in amphioxus; its genome also contains a single gene encoding a putative insulin-IGF receptor (216) and a single gene encoding a putative IGF-binding protein (IGFBP) (220, 221).
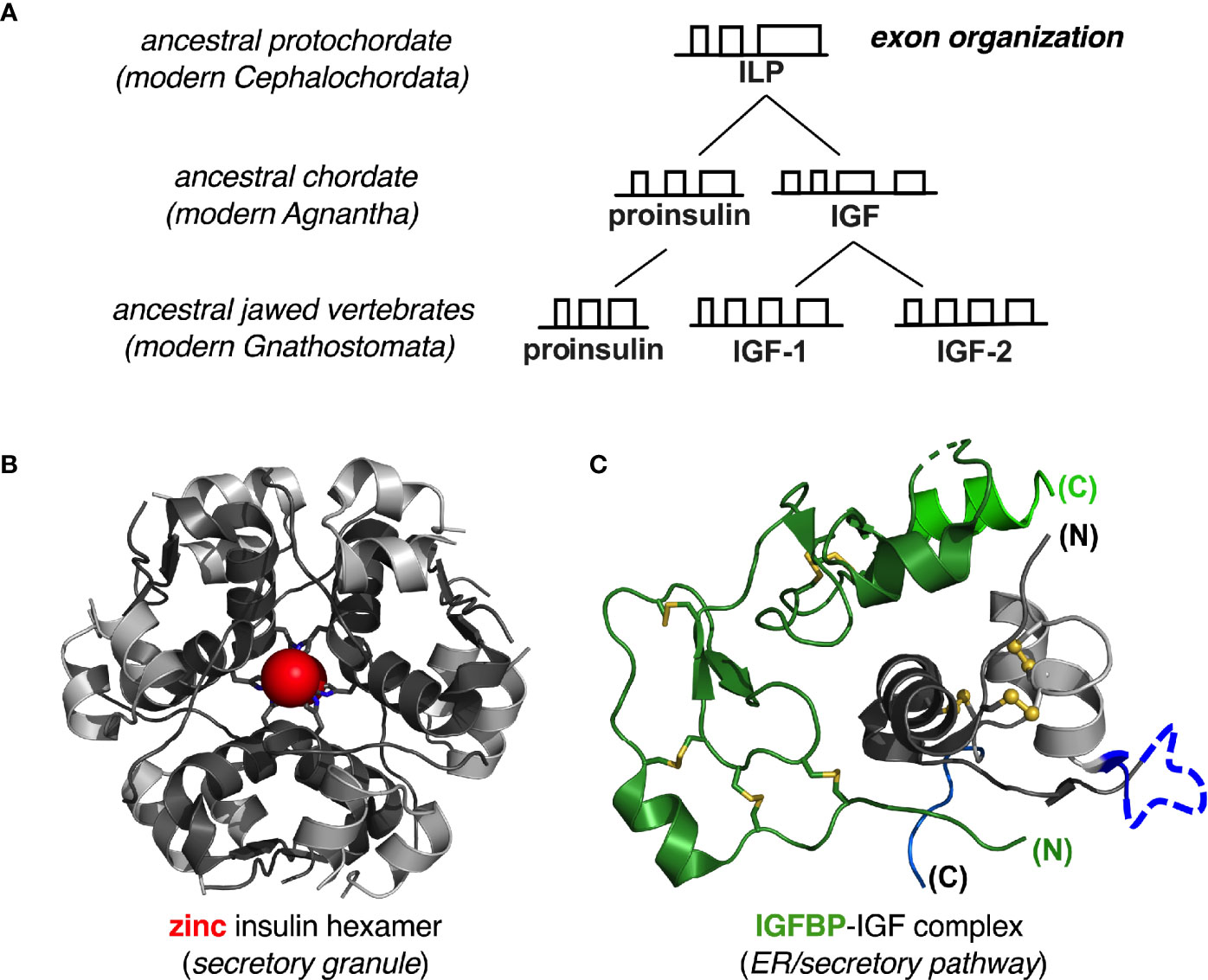
Figure 9 Evolution of insulin-like genes in vertebrates. (A) Steiner and colleagues proposed that a primordial insulin-like gene in protochordates was the common ancestor of vertebrate insulin/IGF factors (215, 217). Amphioxus ILP represents this ancestral gene (as a “living fossil”) prior to gene duplication in early agnathan vertebrates. Further gene duplication results in distinct IGF-I and IGF-II genes found in all gnathosomes (jawed vertebrates). Independent gene duplication events occurred during the evolution of invertebrate insulin-like peptides (not shown). See Supplemental Figure S1A for further details of exonic structure. (B) Proinsulin folds autonomously, and the mature hormone is stabilized by native self-assembly: ribbon model of classical zinc-insulin hexamer. The A- and B chains are shown in light- and dark gray, respectively; the two overlying axial zinc ions are shown as red spheres (enlarged for emphasis at twice Van der Waals radii), each coordinated by three HisB10 side chains (sticks). (C) IGF-I and IGF-II fold together with specific IGF binding proteins: ribbon model of illustrative IGFBP-IGF complex. We hypothesize that heterodimeric folding “rescues” the foldability of the IGFs, whose folding properties would otherwise be ambiguous. Color codes as follows: IGF domain A (light gray), domain B (dark gray), domain C (dark blue) and domain D (light blue); IGFBP N-terminal domain (dark green) and C- terminal domain (light green). Molecular coordinates were respectively obtained from PDB entries 1MSO (zinc insulin hexamer) (186) and 2DSP (IGF-1/IGFBP-4 complex) (219).
ILP was thus proposed to combine the functions of insulin, IGF-I and IGF-II prior to the duplication the proto-insulin gene and specialization of distinct factors (215). Evolutionary changes in intron-exon structures are shown in greater detail in Supplemental Figure S1A. In this scheme conversion of a metazoan proto-insulin gene to ILP would have been effected by a nonsense-to-sense mutation at the end of the A-domain-encoding sequence in the putative proto-insulin gene; conversion of ILP to proto-lGF would have been effected by an upstream shift in the intron donor site into the B-domain-encoding exon. IGF-I and -II genes subsequently evolved from the posited proto-IGF by insertion of an intron into the E-encoding domain followed by gene duplication.
Thus, predating the divergence of insulin and IGFs as distinct gene products, ILP retains framework residues conserved among vertebrate insulins and IGFs, including the six canonical cysteines, LeuB6, LeuB11, LeuB15, ValB18, LeuA16 and TyrA19—hotspots for MIDY mutations (Table 1C). Whereas mammalian insulins contain LeuB17, however, residue B17 in ILP is Phe as in IGFs. Similarly, ILP residue A8 is Tyr, resembling the homologous Phe in IGF-I and IGF-II but unlike AlaA8 or ThrA8 in mammalian insulins (215, 218). ILP would not be expected to undergo insulin-like self-assembly: (a) it lacks a His at position B10 and so would not be expected to coordinate zinc ions; and (b) dimerization would be predicted to be impaired by ILP residues AlaB12 and SerB26 (in place of ValB12 and TyrB26) (183, 222). Representative vertebrate insulin B-chain sequences and IGF-I B-domain sequences are shown in Supplemental Figure S1B.
Given the evolutionary framework established by Steiner and coworkers (215–217), we hypothesize that the primordial insulin/IGF precursor protein folded as a heterodimer in partnership with a proto-IGFBP. Such heterodimeric folding occurs in vertebrate IGF-IGFBP systems (221, 223–226) and appears to compensate for the ambiguous refolding properties of IGFs in vitro (28, 29, 123, 130). We envisage that in heterodimeric folding requirements of foldability are relaxed in each partner (when considered in isolation). Crystal structures of human IGF-I/IGFBP complexes [illustrated in a representative case in Figure 9C (219)] exhibit extensive engagement of IGF surfaces adjoining disulfide bridges and sites of MIDY mutations. This model predicts that the foldability of pro-IGF variants in mammalian cells would be more robust to MIDY-like mutations than is proinsulin—but only in cells co-expressing one or more IGFBPs.
Proinsulin by contrast folds in the ER as an autonomous monomer, aided by chaperonins and oxidoreductases but not, to our knowledge, by specific proinsulin-binding proteins. Native zinc-mediated self-assembly of insulin (Figure 9B) can include proinsulin [which can form corresponding hexamers (69)], but such self-assembly occurs in secretory granules and not in the zinc-poor environment of the ER. Although IGFBPs do not bind to insulin or proinsulin, this model predicts that engineered proinsulin-binding proteins may be designed to enhance the foldability of WT and variant proinsulins. Although such artificial proteins are unlikely to find therapeutic application, they may be of interest as reagents to probe the mechanism of proinsulin folding in vivo, including steps susceptible to misfolding.
Structural Determinants of ER Quality Control
Arvan and colleagues have studied the ER folding of proinsulin in β-cell lines using a systematic set of variants that can form only one or two disulfide bonds; to this end, specific disulfide bridges were removed by pairwise mutagenesis (227). These constructs differed in biosynthetic properties and so provided probes of quality-control determinants. Their results demonstrated that cystines A20-B19 and A7-B7 (but not cystine A6-A11) are critical to enable native folding and ER exit. Prior biophysical studies of an insulin analogue lacking cystine A7-B7 (due to pairwise Ser substitution) demonstrated a more marked decrease in stability and chain-combination efficiency relative to analogous analogues lacking A6-A11 (60, 132). Further studies on single-chain insulin analogues (67) and IGF-1-related peptides and peptide fragments (29, 31, 60) provided evidence for a kinetic pathway in which pairing of cystine A20-B19 provides a required first step to stabilize a native-like molten mini-core (31). Native-like NOEs were observed in such a one-disulfide peptide model even in the absence of stable secondary structure (31). Together, these cell-based and in vitro studies suggest possible structural features that might be sensed by ER quality control: as a general principle, the more destabilizing the disulfide intermediate or isomer destabilizing, the greater the degree of exposed non-polar surfaces and in turn the intervention of detection and degradation by the quality-control machinery.
Whereas variant proinsulin polypeptides without interdomain disulfide bridges cannot be secreted (227), non-native disulfide isomers can accumulate and evade ER quality control (138, 140). Early work from the Arvan laboratory demonstrated secretion of mispaired disulfide isomers in cells using various single-chain insulin constructs (138, 140). Indeed, prior studies of IGF-I revealed that its oxidative refolding in vitro yielded two isoenergetic products (28, 123). Although these had similar α-helical propensities and thermodynamic stabilities, 2D 1H-NMR spectra were remarkable for distinct well-dispersed patterns of chemical shifts, indicative of different three-dimensional structures (28). Unlike IGF-I and its disulfide “swapped” isomer, insulin disulfide isomers are less stable and less well-ordered than is native insulin (75, 76). The respective N-terminal segments of proinsulin and IGF-I contribute to such salient differences in the fidelity of disulfide specification and relative stability (228, 229). The chain asymmetry of non-Cys-related MIDY mutations—more in the B domain than in the A domain (Figure 1)—is consistent with a hierarchical disulfide pathway in which nascent structure in the B domain provides a structural template for folding of the A domain (95).
Mutations that impair the foldability of proinsulin (or efficiency of insulin chain combination (95) can nonetheless be compatible with high activity (108). This lack of correlation suggests that determinants of quality control in the ER differ from determinants of receptor binding. A prominent example is provided by substitution of invariant LeuA16 by Val (156). This cavity-associated mutation (not [yet] seen among MIDY patients) markedly impairs both cellular folding of ValA16-proinsulin and chain combination, and yet substantial biological activity is retained once the native state is reached (94, 156). Similarly, folding of SerB8-proinsulin is significantly reduced in vitro, yet IR affinity is similar to WT insulin (109). A recent study reported that substitution of PheB24 by Tyr (also not [yet] seen among MIDY patients) blocks cellular folding (40) whereas the corresponding two-chain insulin analogue retains substantial activity in a rat model of DM.
Concluding Remarks
We imagine that insulin’s conserved side chains, as exemplified by PheB24, play different roles in the course of a complex conformational “life cycle.” If so, this would represent a marked compression of structural information within a short protein sequence. The present cryo-EM revolution promises to provide snapshots of structures through this life cycle, likely to be extended by solid-state NMR-based models of non-native insulin aggregates and fibrils. As we celebrate the Centennial of insulin’s discovery in Toronto in 1921 (1, 230)—and coincidentally the gold anniversary of its high-resolution crystal structure at Oxford in 1971 (161)—it is remarkable to appreciate how much remains to be discovered in relation to biosynthesis, folding, function and evolution. Further, in a related review article, J. S. Flier and C. R. Kahn have discussed how the discovery of insulin has defined a milestone in the history of molecular medicine that extends beyond the insulin molecule itself (231).
This review has focused on the structural lessons of the mutant proinsulin syndrome (4–6). Patient-derived experiments of nature are providing an opportunity to investigate biophysical principles at the intersection of cell biology and human genetics. As envisioned by classical diffusion-collision and framework models (232), folding of globular proteins (such as proinsulin) represent the coalescence of discrete subdomains (233, 234). Even as funnel-like energy landscapes make possible parallel events in folding (126), the existence of preferred trajectories (235) is implied by disulfide trapping studies of insulin-related polypeptides. Biophysical studies of these trajectories and equilibrium models promise to deepen a structural understanding of MIDY/MODY mutations. Sites of mutation reflect mechanisms of folding or misfolding that may not be apparent in the native state (9, 10, 19, 51). Many of the principles discussed here were foreshadowed in pioneering efforts toward the total chemical synthesis of insulin wherein specific disulfide pairing posed a key challenge to chain combination (89).
Foldability is an evolved property (236), highlighting the general threat of toxic misfolding as a hidden constraint in protein evolution. In the genetics of proteotoxic diseases these principles connect bench to bedside. Critical questions for continuing investigation include: can over-expression of the WT INS gene in response to peripheral insulin resistance likewise tax the folding capacity of the β-cell and induce ER stress analogous to that of the mutant proinsulin syndrome? Might structural mechanisms of misfolding due to MIDY mutations broadly inform a hidden landscape of toxic aggregation awaiting WT biosynthesis? A key frontier in molecular metabolism is thus defined by the role of the UPR and chronic ER stress in the progression of non-canonical Type 2 DM (9, 24, 25). Molecular dissection of how β-cells respond to the challenge of proinsulin overexpression (237) is of compelling translational interest as a strategy to arrest the progression of prediabetes to frank diabetes (9, 38, 238). Structural lessons of the mutant proinsulin syndrome may thus inform UPR-based approaches to mitigate the growing pandemic of diabesity.
Author Contributions
MW oversaw preparation of the manuscript and wrote the first draft. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was supported by current and past grants from the National Institutes of Health (DK040949 and DK0697674) to MW. BD was supported in part by a grant from the Diabetes Research Connection. The authors thank G.I. Bell for summary of clinical mutations, members of the Weiss group for advice; P. Arvan, G.I. Bell, and M. Liu for communication of unpublished results; and Prof. P. Arvan, F. Ismail-Beigi, R. Kaufman, S. B. Kent, M. C. Lawrence, M. Liu, L. Philipson, and N.B. Phillips for helpful discussion. This article, a contribution from the Center for Diabetes and Metabolic Diseases at the Indiana University School of Medicine, is jointly dedicated to the memory of late senior colleagues P.G. Katsoyannis and D.F. Steiner.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2021.754693/full#supplementary-material
Abbreviations
DM, diabetes mellitus; ER, endoplasmic reticulum; GA, Golgi apparatus; MIDY, mutant INS-gene-induced diabetes of youth; MODY, maturity-onset diabetes of the young, and NMR, nuclear magnetic resonance. Amino acids are designated by standard one- and three-letter codes.
References
1. Hegele RA, Maltman GM. Insulin’s Centenary: The Birth of an Idea. Lancet Diabetes Endocrinol (2020) 8(12):971–7. doi: 10.1016/S2213-8587(20)30337-5
2. Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin Biosynthesis: Evidence for a Precursor. Science (1967) 157:697–700. doi: 10.1126/science.157.3789.697
3. Steiner DF, Clark JL, Nolan C, Rubenstein AH, Margoliash E, Aten B, et al. Proinsulin and the Biosynthesis of Insulin. Recent Prog Horm Res (1969) 25:207–82. doi: 10.1016/B978-0-12-571125-8.50008-9
4. Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, et al. Proinsulin Misfolding and Diabetes: Mutant INS Gene-Induced Diabetes of Youth. Trends Endocrinol Metab (2010) 21(11):652–9. doi: 10.1016/j.tem.2010.07.001
5. Greeley SA, Tucker SE, Naylor RN, Bell GI, Philipson LH. Neonatal Diabetes Mellitus: A Model for Personalized Medicine. Trends Endocrinol Metab (2010) 21(8):464–72. doi: 10.1016/j.tem.2010.03.004
6. Greeley SA, Naylor RN, Philipson LH, Bell GI. Neonatal Diabetes: An Expanding List of Genes Allows for Improved Diagnosis and Treatment. Curr Diabetes Rep (2011) 11(6):519–32. doi: 10.1007/s11892-011-0234-7
7. Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin Gene Mutations as a Cause of Permanent Neonatal Diabetes. Proc Natl Acad Sci USA (2007) 104:15040–4. doi: 10.1073/pnas.0707291104
8. Stoy J, Steiner DF, Park SY, Ye H, Philipson LH, Bell GI. Clinical and Molecular Genetics of Neonatal Diabetes Due to Mutations in the Insulin Gene. Rev Endocr Metab Disord (2010) 11(3):205–15. doi: 10.1007/s11154-010-9151-3
9. Liu M, Weiss MA, Arunagiri A, Yong J, Rege N, Sun J, et al. Biosynthesis, Structure, and Folding of the Insulin Precursor Protein. Diabetes Obes Metab (2018) 20:28–50. doi: 10.1111/dom.13378
10. Sun J, Xiong Y, Li X, Haataja L, Chen W, Mir SA, et al. Role of Proinsulin Self-Association in Mutant INS Gene–Induced Diabetes of Youth. Diabetes (2020) 69(5):954–64. doi: 10.2337/db19-1106
11. Park SY, Ye H, Steiner DF, Bell GI. Mutant Proinsulin Proteins Associated With Neonatal Diabetes are Retained in the Endoplasmic Reticulum and Not Efficiently Secreted. Biochem Biophys Res Commun (2010) 391(3):1449–54. doi: 10.1016/j.bbrc.2009.12.090
12. Liu M, Haataja L, Wright J, Wickramasinghe DN, Hua QX, Phillips NB, et al. Mutant INS-Gene Induced Diabetes of Youth: Proinsulin Cysteine Residues Impose Dominant-Negative Inhibition on Nonmutant Proinsulin Transport. PloS One (2010) 5(10):e13333. doi: 10.1371/journal.pone.0013333
13. Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, et al. Seven Mutations in the Human Insulin Gene Linked to Permanent Neonatal/Infancy-Onset Diabetes Mellitus. J Clin Invest (2008) 118:2148–56. doi: 10.1172/JCI33777
14. Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, et al. Insulin Mutation Screening in 1044 Patients With Diabetes: Mutations in the INS Gene Are a Common Cause of Neonatal Diabetes But a Rare Cause of Diabetes Diagnosed in Childhood or Adulthood. Diabetes (2008) 57:1034–42. doi: 10.2337/db07-1405
15. Polak M, Dechaume A, Cave H, Rivtal N, Crosnier H, Sulmont V, et al. Heterozygous Missense Mutations in the Insulin Gene Are Linked to Permanent Diabetes Appearing in the Neonatal Period or in Early Infancy. Diabetes (2008) 57:1115–9. doi: 10.2337/db07-1358
16. Molven A, Ringdal M, Nordbo AM, Raeder H, Stoy J, Lipkind GM, et al. Mutations in the Insulin Gene Can Cause MODY and Autoantibody-Negative Type 1 Diabetes. Diabetes (2008) 57:1131–5. doi: 10.2337/db07-1467
17. Boesgaard TW, Pruhova S, Andersson EA, Cinek O, Obermannova B, Lauenborg J, et al. Further Evidence That Mutations in INS can be a Rare Cause of Maturity-Onset Diabetes of the Young (MODY). BMC Med Genet (2010) 12(11):42. doi: 10.1186/1471-2350-11-42
18. Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, et al. Insulin Gene Mutations Resulting in Early-Onset Diabetes: Marked Differences in Clinical Presentation, Metabolic Status, and Pathogenic Effect Through Endoplasmic Reticulum Retention. Diabetes (2010) 59(3):653–61. doi: 10.2337/db09-1091
19. Weiss MA. Proinsulin and the Genetics of Diabetes Mellitus. J Biol Chem (2009) 284:19159–63. doi: 10.1074/jbc.R109.009936
20. Arunagiri A, Haataja L, Pottekat A, Pamenan F, Kim S, Zeltser LM, et al. Proinsulin Misfolding Is an Early Event in the Progression to Type 2 Diabetes. Elife (2019) 8:e44532. doi: 10.7554/eLife.44532
21. Yoshioka M, Kayo T, Ikeda T, Koizumi A. A Novel Locus, Mody4, Distal to D7Mit189 on Chromosome 7 Determines Early-Onset NIDDM in Nonobese C57BL/6 (Akita) Mutant Mice. Diabetes (1997) 46:887–94. doi: 10.2337/diab.46.5.887
22. Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, et al. A Mutation in the Insulin 2 Gene Induces Diabetes With Severe Pancreatic B-Cell Dysfunction in the Mody Mouse. J Clin Invest (1999) 103:27–37. doi: 10.1172/JCI4431
23. Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted Disruption of the Chop Gene Delays Endoplasmic Reticulum Stress-Mediated Diabetes. J Clin Invest (2002) 109:525–32. doi: 10.1172/JCI0214550
24. Ron D. Proteotoxicity in the Endoplasmic Reticulum: Lessons From the Akita Diabetic Mouse. J Clin Invest (2002) 109:443–5. doi: 10.1172/JCI0215020
25. Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant Negative Pathogenesis by Mutant Proinsulin in the Akita Diabetic Mouse. Diabetes (2003) 52:409–16. doi: 10.2337/diabetes.52.2.409
26. Zuber C, Fan JY, Guhl B, Roth J. Misfolded Proinsulin Accumulates in Expanded Pre-Golgi Intermediates and Endoplasmic Reticulum Subdomains in Pancreatic β Cells of Akita Mice. FASEB J (2004) 18:917–9. doi: 10.1096/fj.03-1210fje
27. Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin Maturation, Misfolding, and Proteotoxicity. Proc Natl Acad Sci USA (2007) 104:15841–6. doi: 10.1073/pnas.0702697104
28. Miller JA, Narhi LO, Hua QX, Rosenfeld R, Arakawa T, Rohde M, et al. Oxidative Refolding of Insulin-Like Growth Factor 1 Yields Two Products of Similar Thermodynamic Stability: A Bifurcating Protein-Folding Pathway. Biochemistry (1993) 32:5203–13. doi: 10.1021/bi00070a032
29. Qiao ZS, Guo ZY, Feng YM. Putative Disulfide-Forming Pathway of Porcine Insulin Precursor During its Refolding In Vitro. Biochemistry (2001) 40:2662–8. doi: 10.1021/bi001613r
30. Hua QX, Liu M, Hu SQ, Jia W, Arvan P, Weiss MA. A Conserved Histidine in Insulin Is Required for the Foldability of Human Proinsulin. Structure and Function of an AlaB5 Analog. J Biol Chem (2006) 281:24889–99. doi: 10.1074/jbc.M602617200
31. Hua QX, Mayer J, Jia W, Zhang J, Weiss MA. The Folding Nucleus of the Insulin Superfamily: A Flexible Peptide Model Foreshadows the Native State. J Biol Chem (2006) 281:28131–42. doi: 10.1074/jbc.M602616200
32. Qiao ZS, Guo ZY, Feng YM. In Vitro Folding/Unfolding of Insulin/Single-Chain Insulin. Protein Pept Lett (2006) 13:423–9. doi: 10.2174/092986606776819583
33. Guo ZY, Qiao ZS, Feng YM. The In Vitro Oxidative Folding of the Insulin Superfamily. Antioxid Redox Signal (2008) 10:127–40. doi: 10.1089/ars.2007.1860
34. Dodson G, Steiner D. The Role of Assembly in Insulin’s Biosynthesis. Curr Opin Struct Biol (1998) 8:189–94. doi: 10.1016/S0959-440X(98)80037-7
35. Hodish I, Liu M, Rajpal G, Larkin D, Holz RW, Adams A, et al. Misfolded Proinsulin Affects Bystander Proinsulin in Neonatal Diabetes. J Biol Chem (2010) 285(1):685–94. doi: 10.1074/jbc.M109.038042
36. Hodish I, Absood A, Liu L, Liu M, Haataja L, Larkin D, et al. In Vivo Misfolding of Proinsulin Below the Threshold of Frank Diabetes. Diabetes (2011) 60(8):2092–101. doi: 10.2337/db10-1671
37. Haataja L, Snapp E, Wright J, Liu M, Hardy AB, Wheeler MB, et al. Proinsulin Intermolecular Interactions During Secretory Trafficking in Pancreatic β Cells. J Biol Chem (2013) 288(3):1896–906. doi: 10.1074/jbc.M112.420018
38. Absood A, Gandomani B, Zaki A, Nasta V, Michail A, Habib PM, et al. Insulin Therapy for Pre-Hyperglycemic β-Cell Endoplasmic Reticulum Crowding. PloS One (2013) 8(2):e54351. doi: 10.1371/journal.pone.0054351
39. Austin AL, Gatward LFD, Cnop M, Santos G, Andersson D, Sharp S, et al. The KINGS Ins2+/G32S Mouse: A Novel Model of β-Cell Endoplasmic Reticulum Stress and Human Diabetes. Diabetes (2020) 69(12):2667–77. doi: 10.2337/db20-0570
40. Rege NK, Liu M, Yang Y, Dhayalan B, Wickramasinghe NP, Chen Y-S, et al. Evolution of Insulin at the Edge of Foldability and Its Medical Implications. Proc Natl Acad Sci USA (2020) 117(47):29618–28. doi: 10.1073/pnas.2010908117
41. Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F, et al. INS-Gene Mutations: From Genetics and Beta Cell Biology to Clinical Disease. Mol Aspects Med (2015) 42:3–18. doi: 10.1016/j.mam.2014.12.001
42. Stoy J, Olsen J, Park SY, Gregersen S, Hjorringgaard CU, Bell GI. In Vivo Measurement and Biological Characterisation of the Diabetes-Associated Mutant Insulin P.R46Q (GlnB22-Insulin). Diabetologia (2017) 60:1423–31. doi: 10.1007/s00125-017-4295-2
43. Støy J, De Franco E, Ye H, Park S-Y, Bell GI, Hattersley AT. In Celebration of a Century With Insulin–Update of Insulin Gene Mutations in Diabetes. Mol Metab (2021), 101280.doi: 10.1016/j.molmet.2021.101280
44. Weissman JS, Kim PS. Reexamination of the Folding of BPTI: Predominance of Native Intermediates. Science (1991) 253:1386–93. doi: 10.1126/science.1716783
45. Peng ZY, Wu LC, Schulman BA, Kim PS. Does the Molten Globule Have a Native-Like Tertiary Fold. Philos Trans R Soc Lond B Biol Sci (1995) 348:43–7. doi: 10.1098/rstb.1995.0044
46. Dobson CM, Karplus M. The Fundamentals of Protein Folding: Bringing Together Theory and Experiment. Curr Opin Struct Biol (1999) 9:92–101. doi: 10.1016/S0959-440X(99)80012-8
47. Baker EN, Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, et al. The Structure of 2Zn Pig Insulin Crystals at 1.5 Å Resolution. Philos Trans R Soc Lond B Biol Sci (1988) 319:369–456. doi: 10.1098/rstb.1988.0058
48. Yang Y, Hua QX, Liu J, Shimizu EH, Choquette MH, Mackin RB, et al. Solution Structure of Proinsulin: Connecting Domain Flexibility and Prohormone Processing. J Biol Chem (2010) 285:7847–51. doi: 10.1074/jbc.C109.084921
49. Nesse RM, Williams GC. Why We Get Sick: The New Science of Darwinian Medicine: Vintage. New York: Times Books (2012).
50. Watve M, Diwekar-Joshi M. What to Expect From an Evolutionary Hypothesis for a Human Disease: The Case of Type 2 Diabetes. Homo (2016) 67(5):349–68. doi: 10.1016/j.jchb.2016.07.001
51. Weiss MA. Diabetes Mellitus Due to the Toxic Misfolding of Proinsulin Variants. FEBS Lett (2013) 587(13):1942–50. doi: 10.1016/j.febslet.2013.04.044
52. Rege NK, Liu M, Dhayalan B, Chen Y-S, Smith NA, Rahimi L, et al. “Register-Shift” Insulin Analogs Uncover Constraints of Proteotoxicity in Protein Evolution. J Biol Chem (2020) 295(10):3080–98. doi: 10.1074/jbc.RA119.011389
53. Bell GI, Pictet RL, Rutter WJ, Cordell B, Tischer E, Goodman HM. Sequence of the Human Insulin Gene. Nature (1980) 284(5751):26–32. doi: 10.1038/284026a0
54. Steiner DF, Chan SJ, Welsh JM, Kwok SC. Structure and Evolution of the Insulin Gene. Annu Rev Genet (1985) 19:463–84. doi: 10.1146/annurev.ge.19.120185.002335
55. Steiner DF, Chan SJ. An Overview of Insulin Evolution. Horm Metab Res (1988) 20:443–4. doi: 10.1055/s-2007-1010855
57. Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH. Lessons Learned From Molecular Biology of Insulin-Gene Mutations. Diabetes Care (1990) 13:600–9. doi: 10.2337/diacare.13.6.600
58. Steiner DF. Evidence for a Precursor in the Biosynthesis of Insulin. Trans N Y Acad Sci (1967) 30:60–8. doi: 10.1111/j.2164-0947.1967.tb02452.x
59. Narhi LO, Hua QX, Arakawa T, Fox GM, Tsai L, Rosenfeld R, et al. Role of Native Disulfide Bonds in the Structure and Activity of Insulin-Like Growth Factor 1: Genetic Models of Protein-Folding Intermediates. Biochemistry (1993) 32:5214–21. doi: 10.1021/bi00070a033
60. Hua QX, Hu SQ, Frank BH, Jia W, Chu YC, Wang SH, et al. Mapping the Functional Surface of Insulin by Design: Structure and Function of a Novel A-Chain Analogue. J Mol Biol (1996) 264:390–403. doi: 10.1006/jmbi.1996.0648
61. Dai Y, Tang JG. Characteristic, Activity and Conformational Studies of [A6-Ser, A11-Ser]-Insulin. Biochim Biophys Acta (1996) 1296:63–8. doi: 10.1016/0167-4838(96)00054-4
62. Hober S, Uhlen M, Nilsson B. Disulfide Exchange Folding of Disulfide Mutants of Insulin-Like Growth Factor I In Vitro. Biochemistry (1997) 36:4616–22. doi: 10.1021/bi9611265
63. Weiss MA, Hua QX, Jia W, Chu YC, Wang RY, Katsoyannis PG. Hierarchiacal Protein “Un-Design”: Insulin’s Intrachain Disulfide Bridge Tethers a Recognition a-Helix. Biochemistry (2000) 39:15429–40. doi: 10.1021/bi001905s
64. Guo ZY, Feng YM. Effects of Cysteine to Serine Substitutions in the Two Inter-Chain Disulfide Bonds of Insulin. Biol Chem (2001) 382:443–8. doi: 10.1515/BC.2001.054
65. Feng Y, Liu D, Wang J. Native-Like Partially Folded Conformations and Folding Process Revealed in the N-Terminal Large Fragments of Staphylococcal Nuclease: A Study by NMR Spectroscopy. J Mol Biol (2003) 330:821–37. doi: 10.1016/S0022-2836(03)00660-0
66. Jia XY, Guo ZY, Wang Y, Xu Y, Duan SS, Feng YM. Peptide Models of Four Possible Insulin Folding Intermediates With Two Disulfides. Protein Sci (2003) 12:2412–9. doi: 10.1110/ps.0389303
67. Yan H, Guo ZY, Gong XW, Xi D, Feng YM. A Peptide Model of Insulin Folding Intermediate With One Disulfide. Protein Sci (2003) 12:768–75. doi: 10.1110/ps.0237203
68. Frank BH, Veros AJ. Physical Studies on Proinsulin-Association Behavior and Conformation in Solution. Biochem Biophys Res Commun (1968) 32(2):155–60. doi: 10.1016/0006-291X(68)90362-8
69. Pekar AH, Frank BH. Conformation of Proinsulin. A Comparison of Insulin and Proinsulin Self-Association at Neutral Ph. Biochemistry (1972) 11:4013–6. doi: 10.1021/bi00772a001
70. Frank BH, Pekar AH, Veros AJ. Insulin and Proinsulin Conformation in Solution. Diabetes (1972) 21:486–91. doi: 10.2337/diab.21.2.S486
71. Snell CR, Smyth DG. Proinsulin: A Proposed Three-Dimensional Structure. J Biol Chem (1975) 250:6291–5. doi: 10.1016/S0021-9258(19)41065-X
72. Brems DN, Brown PL, Heckenlaible LA, Frank BH. Equilibrium Denaturation of Insulin and Proinsulin. Biochemistry (1990) 29:9289–93. doi: 10.1021/bi00491a026
73. Weiss MA, Frank BH, Khait I, Pekar A, Heiney R, Shoelson SE, et al. NMR and Photo-CIDNP Studies of Human Proinsulin and Prohormone Processing Intermediates With Application to Endopeptidase Recognition. Biochemistry (1990) 29:8389–401. doi: 10.1021/bi00488a028
74. Sieber PS, Eisler K, Kamber B, Riniker B, Rittel W, Marki F, et al. Synthesis and Biological Activity of Two Disulphide Bond Isomers of Human Insulin: [A7-A11, A6-B7-Cystine]- and [A6-A7, A11-B7-Cystine] Insulin (Human). Hoppe Seylers Z Physiol Chem (1978) 359:113–23. doi: 10.1515/bchm.1978.359.1.113
75. Hua QX, Gozani SN, Chance RE, Hoffmann JA, Frank BH, Weiss MA. Structure of a Protein in a Kinetic Trap. Nat Struct Biol (1995) 2:129–38. doi: 10.1038/nsb0295-129
76. Hua QX, Jia W, Frank BH, Phillips NB, Weiss MA. A Protein Caught in a Kinetic Trap: Structures and Stabilities of Insulin Disulfide Isomers. Biochemistry (2002) 41:14700–15. doi: 10.1021/bi0202981
77. Galloway JA, Hooper SA, Spradlin CT, Howey DC, Frank BH, Bowsher RR, et al. Biosynthetic Human Proinsulin. Review of Chemistry, In Vitro and In Vivo Receptor Binding, Animal and Human Pharmacology Studies, and Clinical Trial Experience. Diabetes Care (1992) 15:666–92. doi: 10.2337/diacare.15.5.666
78. Huang XF, Arvan P. Formation of the Insulin-Containing Secretory Granule Core Occurs Within Immature β-Granules. J Biol Chem (1994) 269:20838–44. doi: 10.1016/S0021-9258(17)31898-7
79. Steiner DF. The Proprotein Convertases. Curr Opin Chem Biol (1998) 2:31–9. doi: 10.1016/S1367-5931(98)80033-1
80. Huang XF, Arvan P. Intracellular Transport of Proinsulin in Pancreatic β-Cells. Structural Maturation Probed by Disulfide Accessibility. J Biol Chem (1995) 270:20417–23. doi: 10.1074/jbc.270.35.20417
81. Greider MH, Howell SL, Lacy PE. Isolation and Properties of Secretory Granules From Rat Islets of Langerhans. II. Ultrastructure of the β Granule. J Cell Biol (1969) 41:162–6. doi: 10.1083/jcb.41.1.162
82. Lange RH. Crystalline Islet B-Granules in the Grass Snake (Natrix Natrix (L.)): Tilting Experiments in the Electron Microscope. J Ultrastruct Res (1974) 46:301–7. doi: 10.1016/S0022-5320(74)80064-X
83. Michael J, Carroll R, Swift HH, Steiner DF. Studies on the Molecular Organization of Rat Insulin Secretory Granules. J Biol Chem (1987) 262:16531–5. doi: 10.1016/S0021-9258(18)49288-5
84. Brange J, Langkjaer L. Insulin Formation and Delivery. In: Sanders LM, Hendren RW, editors. Protein Delivery: Physical Systems. Pharmaceutical Biotechnology. 10. New York, NY: Plenum Press, New York (1997). p. 343–410.
86. Lemaire K, Ravier MA, Schraenen A, Creemers JW, Van de Plas R, Granvik M, et al. Insulin Crystallization Depends on Zinc Transporter ZnT8 Expression, But is Not Required for Normal Glucose Homeostasis in Mice. Proc Natl Acad Sci USA (2009) 106(35):14872–7. doi: 10.1073/pnas.0906587106
88. Woods RJ, Alarcon J, McVey E, Pettis RJ. Intrinsic Fibrillation of Fast-Acting Insulin Analogs. J Diabetes Sci Technol (2012) 6(2):265–76. doi: 10.1177/193229681200600209
89. Katsoyannis PG. Synthesis of Insulin. Science (1966) 154:1509–14. doi: 10.1126/science.154.3756.1509
90. Tang JG, Tsou CL. The Insulin A and B Chains Contain Structural Information for the Formation of the Native Molecule. Studies With Protein Disulphide- Isomerase. Biochem J (1990) 268:429–35. doi: 10.1042/bj2680429
91. Wang CC, Tsou CL. The Insulin A and B Chains Contain Sufficient Structural Information to Form the Native Molecule. Trends Biochem Sci (1991) 16:279–81. doi: 10.1016/0968-0004(91)90114-B
92. Brange J. The New Era of Biotech Insulin Analogues. Diabetologia (1997) 40:S48–53. doi: 10.1007/s001250051400
93. Hu SQ, Burke GT, Schwartz GP, Ferderigos N, Ross JB, Katsoyannis PG. Steric Requirements at Position B12 for High Biological Activity in Insulin. Biochemistry (1993) 32:2631–5. doi: 10.1021/bi00061a022
94. Weiss MA, Nakagawa SH, Jia W, Xu B, Hua QX, Chu YC, et al. Protein Structure and the Spandrels of San Marco: Insulin’s Receptor-Binding Surface is Buttressed by an Invariant Leucine Essential for Protein Stability. Biochemistry (2002) 41:809–19. doi: 10.1021/bi011839+
95. Hua QX, Chu YC, Jia W, Phillips NB, Wang RY, Katsoyannis PG, et al. Mechanism of Insulin Chain Combination. Asymmetric Roles of A-Chain α-Helices in Disulfide Pairing. J Biol Chem (2002) 277:43443–53. doi: 10.1074/jbc.M206107200
96. Nakagawa SH, Zhao M, Hua QX, Hu SQ, Wan ZL, Jia W, et al. Chiral Mutagenesis of Insulin. Foldability and Function are Inversely Regulated by a Stereospecific Switch in the B Chain. Biochemistry (2005) 44:4984–99. doi: 10.1021/bi048025o
97. Nakagawa SH, Hua QX, Hu SQ, Jia W, Wang S, Katsoyannis PG, et al. Chiral Mutagenesis of Insulin. Contribution of the B20-B23 β-Turn to Activity and Stability. J Biol Chem (2006) 281:22386–96. doi: 10.1074/jbc.M603547200
98. Hua QX, Nakagawa SH, Hu SQ, Jia W, Wang S, Weiss MA. Toward the Active Conformation of Insulin. Stereospecific Modulation of a Structural Switch in the B Chain. J Biol Chem (2006) 281:24900–9. doi: 10.1074/jbc.M602691200
99. Wan Z, Huang K, Whittaker J, Weiss MA. The Structure of a Mutant Insulin Uncouples Receptor Binding From Protein Allostery. An Electrostatic Block to the TR Transition. J Biol Chem (2008) 283:21198–210. doi: 10.1074/jbc.M800235200
100. Tofteng AP, Jensen KJ, Schäffer L, Hoeg-Jensen T. Total Synthesis of Desb30 Insulin Analogues by Biomimetic Folding of Single-Chain Precursors. ChemBioChem (2008) 9(18):2989–96. doi: 10.1002/cbic.200800430
101. Sohma Y, Kent SB. Biomimetic Synthesis of Lispro Insulin via a Chemically Synthesized “Mini-Proinsulin” Prepared by Oxime-Forming Ligation. J Am Chem Soc (2009) 131:16313–8. doi: 10.1021/ja9052398
102. Sohma Y, Hua QX, Whittaker J, Weiss MA, Kent SB. Design and Folding of [Glua4(Oβthrb30)]Insulin (“Ester Insulin”): A Minimal Proinsulin Surrogate That can be Chemically Converted Into Human Insulin. Angew Chem Int Ed Engl (2010) 49:5489–93. doi: 10.1002/anie.201001151
103. Zaykov AN, Mayer JP, Gelfanov VM, Dimarchi RD. Chemical Synthesis of Insulin Analogs Through a Novel Precursor. ACS Chem Biol (2014) 9:683–91. doi: 10.1021/cb400792s
104. Kochendoerfer GG, Kent SB. Chemical Protein Synthesis. Curr Opin Chem Biol (1999) 3:665–71. doi: 10.1016/S1367-5931(99)00024-1
105. Kent S. Chemical Protein Synthesis: Inventing Synthetic Methods to Decipher How Proteins Work. Biorg Med Chem (2017) 25(18):4926–37. doi: 10.1016/j.bmc.2017.06.020
106. Luisier S, Avital-Shmilovici M, Weiss MA, Kent SB. Total Chemical Synthesis of Human Proinsulin. Chem Commun (Camb) (2010) 46(43):8177–9. doi: 10.1039/c0cc03141k
107. Sohma Y, Pentelute BL, Whittaker J, Hua QX, Whittaker LJ, Weiss MA, et al. Comparative Properties of Insulin-Like Growth Factor 1 (IGF-1) and [Gly7D-Ala]IGF-1 Prepared by Total Chemical Synthesis. Angew Chem Int Ed Engl (2008) 47:1102–6. doi: 10.1002/anie.200703521
108. Sohma Y, Hua QX, Liu M, Phillips NB, Hu SQ, Whittaker J, et al. Contribution of Residue B5 to the Folding and Function of Insulin and IGF-I: Constraints and Fine-Tuning in the Evolution of a Protein Family. J Biol Chem (2010) 285:5040–55. doi: 10.1074/jbc.M109.062992
109. Avital-Shmilovici M, Whittaker J, Weiss MA, Kent SB. Deciphering a Molecular Mechanism of Neonatal Diabetes Mellitus by the Chemical Synthesis of a Protein Diastereomer,[D-AlaB8] Human Proinsulin. J Biol Chem (2014) 289(34):23683–92. doi: 10.1074/jbc.M114.572040
110. Milner SJ, Carver JA, Ballard FJ, Francis GL. Probing the Disulfide Folding Pathway of Insulin-Like Growth Factor-I. Biotechnol Bioeng (1999) 62:693–703. doi: 10.1002/(SICI)1097-0290(19990320)62:6<693::AID-BIT8>3.0.CO;2-S
111. Baldwin TO, Ziegler MM, Chaffotte AF, Goldberg ME. Contribution of Folding Steps Involving the Individual Subunits of Bacterial Luciferase to the Assembly of the Active Heterodimeric Enzyme. J Biol Chem (1993) 268:10766–72. doi: 10.1016/S0021-9258(18)82051-8
112. Creighton TE. Protein Folding Pathways Determined Using Disulphide Bonds. Bioessays (1992) 14:195–9. doi: 10.1002/bies.950140310
113. Ascenzi P, Bocedi A, Bolognesi M, Spallarossa A, Coletta M, Cristofaro RD, et al. The Bovine Basic Pancreatic Trypsin Inhibitor (Kunitz Inhibitor): A Milestone Protein. Curr Protein Pept Sci (2003) 4(3):231–51. doi: 10.2174/1389203033487180
114. Donovan AJ, Dowle J, Yang Y, Weiss MA, Kent SB. Total Synthesis of Bovine Pancreatic Trypsin Inhibitor and the Protein Diastereomer [Gly37D-Ala] BPTI Using Boc Chemistry Solid Phase Peptide Synthesis. Pept Sci (2020) 112(4):e24166. doi: 10.1002/pep2.24166
115. Kato S, Okamura M, Shimamoto N, Utiyama H. Spectral Evidence for a Rapidly Formed Structural Intermediate in the Refolding Kinetics of Hen Egg-White Lysozyme. Biochemistry (1981) 20(5):1080–5. doi: 10.1021/bi00508a006
116. Dobson CM, Evans PA, Radford SE. Understanding How Proteins Fold: The Lysozyme Story So Far. Trends Biochem Sci (1994) 19(1):31–7. doi: 10.1016/0968-0004(94)90171-6
117. De Bernardez Clark E, Hevehan D, Szela S, Maachupalli-Reddy J. Oxidative Renaturation of Hen Egg-White Lysozyme. Folding vs Aggregation. Biotechnol Prog (1998) 14(1):47–54. doi: 10.1021/bp970123w
118. Durek T, Torbeev VY, Kent SB. Convergent Chemical Synthesis and High-Resolution X-Ray Structure of Human Lysozyme. Proc Natl Acad Sci USA (2007) 104(12):4846–51. doi: 10.1073/pnas.0610630104
119. Nozaka M, Kuwajima K, Nitta K, Sugai S. Detection and Characterization of the Intermediate on the Folding Pathway of Human. Alpha.-Lactalbumin. Biochemistry (1978) 17(18):3753–8. doi: 10.1021/bi00611a013
120. Kuwajima K, Hiraoka Y, Ikeguchi M, Sugai S. Comparison of the Transient Folding Intermediates in Lysozyme and. Alpha.-Lactalbumin. Biochemistry (1985) 24(4):874–81. doi: 10.1021/bi00325a010
121. Baum J, Dobson CM, Evans PA, Hanley C. Characterization of a Partly Folded Protein by NMR Methods: Studies on the Molten Globule State of Guinea Pig. Alpha.-Lactalbumin. Biochemistry (1989) 28(1):7–13. doi: 10.1021/bi00427a002
122. Ewbank JJ, Creighton TE. Structural Characterization of the Disulfide Folding Intermediates of Bovine. Alpha.-Lactalbumin. Biochemistry (1993) 32(14):3694–707. doi: 10.1021/bi00065a023
123. Hober S, Forsberg G, Palm G, Hartmanis M, Nilsson B. Disulfide Exchange Folding of Insulin-Like Growth Factor I. Biochemistry (1992) 31:1749–56. doi: 10.1021/bi00121a024
124. Huang Y, Liang Z, Feng Y. The Relationship Between the Connecting Peptide of Recombined Single Chain Insulin and its Biological Function. Sci China C Life Sci (2001) 44(6):593–600. doi: 10.1007/BF02879353
125. Creighton TE. Protein Folding Coupled to Disulphide Bond Formation. Biol Chem (1997) 378:731–44. doi: 10.1515/bchm.1997.378.8.731
126. Dill KA, Chan HS. From Levinthal to Pathways to Funnels. Nat Struct Biol (1997) 4:10–9. doi: 10.1038/nsb0197-10
127. Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of Protein Folding: The Energy Landscape Perspective. Annu Rev Phys Chem (1997) 48:545–600. doi: 10.1146/annurev.physchem.48.1.545
128. Oliveberg M, Wolynes PG. The Experimental Survey of Protein-Folding Energy Landscapes. Q Rev Biophys (2005) 38(3):245–88. doi: 10.1017/S0033583506004185
129. Qiao ZS, Min CY, Hua QX, Weiss MA, Feng YM. In Vitro Refolding of Human Proinsulin. Kinetic Intermediates, Putative Disulfide-Forming Pathway, Folding Initiation Site, and Protential Role of C-Peptide in Folding Process. J Biol Chem (2003) 278:17800–9. doi: 10.1074/jbc.M300906200
130. Hober S, Hansson A, Uhlen M, Nilsson B. Folding of Insulin-Like Growth Factor I is Thermodynamically Controlled by Insulin-Like Growth Factor Binding Protein. Biochemistry (1994) 33:6758–61. doi: 10.1021/bi00188a003
131. Hua QX, Narhi L, Jia W, Arakawa T, Rosenfeld R, Hawkins N, et al. Native and non-Native Structure in a Protein-Folding Intermediate: Spectroscopic Studies of Partially Reduced IGF-I and an Engineered Alanine Model. J Mol Biol (1996) 259:297–313. doi: 10.1006/jmbi.1996.0320
132. Hua QX, Nakagawa SH, Jia W, Hu SQ, Chu YC, Katsoyannis PG, et al. Hierarchical Protein Folding: Asymmetric Unfolding of an Insulin Analogue Lacking the A7-B7 Interchain Disulfide Bridge. Biochemistry (2001) 40:12299–311. doi: 10.1021/bi011021o
133. Chu YC, Burke GT, Chanley JD, Katsoyannis PG. Possible Involvement of the A20-A21 Peptide Bond in the Expression of the Biological Activity of Insulin. 2. [21-Asparagine Diethylamide-A]insulin. Biochemistry (1987) 26:6972–5. doi: 10.1021/bi00396a017
134. Kristensen C, Kjeldsen T, Wiberg FC, Schaffer L, Hach M, Havelund S, et al. Alanine Scanning Mutagenesis of Insulin. J Biol Chem (1997) 272:12978–83. doi: 10.1074/jbc.272.20.12978
135. Gill R, Verma C, Wallach B, Urso B, Pitts J, Wollmer A, et al. Modelling of the Disulphide-Swapped Isomer of Human Insulin-Like Growth Factor-1: Implications for Receptor Binding. Protein Eng (1999) 12:297–303. doi: 10.1093/protein/12.4.297
136. Chen Y, You Y, Jin R, Guo ZY, Feng YM. Sequences of B-Chain/Domain 1-10/1-9 of Insulin and Insulin-Like Growth Factor 1 Determine Their Different Folding Behavior. Biochemistry (2004) 43:9225–33. doi: 10.1021/bi049710y
137. Huang QL, Zhao J, Tang YH, Shao SQ, Xu GJ, Feng YM. The Sequence Determinant Causing Different Folding Behaviors of Insulin and Insulin-Like Growth Factor-1. Biochemistry (2007) 46:218–24. doi: 10.1021/bi0616798
138. Liu M, Ramos-Castaneda J, Arvan P. Role of the Connecting Peptide in Insulin Biosynthesis. J Biol Chem (2003) 278:14798–805. doi: 10.1074/jbc.M212070200
139. Zhang BY, Liu M, Arvan P. Behavior in the Eukaryotic Secretory Pathway of Insulin-Containing Fusion Proteins and Single-Chain Insulins Bearing Various B-Chain Mutations. J Biol Chem (2003) 278:3687–93. doi: 10.1074/jbc.M209474200
140. Liu M, Li Y, Cavener D, Arvan P. Proinsulin Disulfide Maturation and Misfolding in the Endoplasmic Reticulum. J Biol Chem (2005) 280:13209–12. doi: 10.1074/jbc.C400475200
141. Liu M, Hua Q-X, Hu S-Q, Jia W, Yang Y, Saith SE, et al. Deciphering the Hidden Informational Content of Protein Sequences: Foldability of Proinsulin Hinges on a Flexible Arm That Is Dispensable in the Mature Hormone. J Biol Chem (2010) 285:30989–1001. doi: 10.1074/jbc.M110.152645
142. Noiva R. Protein Disulfide Isomerase: The Multifunctional Redox Chaperone of the Endoplasmic Reticulum. Semin Cell Dev Biol (1999) 10:481–93. doi: 10.1006/scdb.1999.0319
143. Zhu YL, Abdo A, Gesmonde JF, Zawalich KC, Zawalich W, Dannies PS. Aggregation and Lack of Secretion of Most Newly Synthesized Proinsulin in Non-β-Cell Lines. Endocrinology (2004) 145:3840–9. doi: 10.1210/en.2003-1512
144. Markussen J. Comparative Reduction/Oxidation Studies With Single Chain Des-(B30) Insulin and Porcine Proinsulin. Int J Pept Protein Res (1985) 25:431–4. doi: 10.1111/j.1399-3011.1985.tb02197.x
145. Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and Translational Control in the Mammalian Unfolded Protein Response. Annu Rev Cell Dev Biol (2002) 18:575–99. doi: 10.1146/annurev.cellbio.18.011402.160624
146. Rutkowski DT, Kaufman RJ. A Trip to the ER: Coping With Stress. Trends Cell Biol (2004) 14(1):20–8. doi: 10.1016/j.tcb.2003.11.001
147. Marciniak SJ, Ron D. Endoplasmic Reticulum Stress Signaling in Disease. Physiol Rev (2006) 86(4):1133–49. doi: 10.1152/physrev.00015.2006
148. Scheuner D, Kaufman RJ. The Unfolded Protein Response: A Pathway That Links Insulin Demand With β-Cell Failure and Diabetes. Endocr Rev (2008) 29(3):317–33. doi: 10.1210/er.2007-0039
149. Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science (2011) 334(6059):1081–6. doi: 10.1126/science.1209038
150. Han J, Kaufman RJ. Physiological/pathological Ramifications of Transcription Factors in the Unfolded Protein Response. Genes Dev (2017) 31(14):1417–38. doi: 10.1101/gad.297374.117
151. Murphy R, Ellard S, Hattersley AT. Clinical Implications of a Molecular Genetic Classification of Monogenic β-Cell Diabetes. Nat Clin Pract Endocrinol Metab (2008) 4:200–13. doi: 10.1038/ncpendmet0778
152. Slingerland AS, Hattersley AT. Mutations in the Kir6.2 Subunit of the KATP Channel and Permanent Neonatal Diabetes: New Insights and New Treatment. Ann Med (2005) 37:186–95. doi: 10.1080/07853890510007287
153. Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, et al. Activating Mutations in the ABCC8 Gene in Neonatal Diabetes Mellitus. N Engl J Med (2006) 355:456–66. doi: 10.1056/NEJMoa055068
154. Yoshinaga T, Nakatome K, Nozaki J, Naitoh M, Hoseki J, Kubota H, et al. Proinsulin Lacking the A7-B7 Disulfide Bond, Ins2Akita, Tends to Aggregate Due to the Exposed Hydrophobic Surface. Biol Chem (2005) 386:1077–85. doi: 10.1515/BC.2005.124
155. Herbach N, Rahtkolb B, Kemter E, Pichl L, Klaften M, De Angelis MH, et al. Dominant-Negative Effects of a Novel Mutated Ins2 Allele Causes Early-Onset Diabetes and Severe β-Cell Loss in Munich Ins2C95S Mutant Mice. Diabetes (2007) 56:1268–76. doi: 10.2337/db06-0658
156. Liu M, Wan ZL, Chu YC, Aladdin H, Klaproth B, Choquette MH, et al. Crystal Structure of a “Non-Foldable” Insulin: Impaired Folding Efficiency and ER Stress Despite Native Activity. J Biol Chem (2009) 284:35259–72. doi: 10.1074/jbc.M109.046888
157. Hua QX, Shoelson SE, Kochoyan M, Weiss MA. Receptor Binding Redefined by a Structural Switch in a Mutant Human Insulin. Nature (1991) 354:238–41. doi: 10.1038/354238a0
158. Olsen HB, Ludvigsen S, Kaarsholm NC. Solution Structure of an Engineered Insulin Monomer at Neutral Ph. Biochemistry (1996) 35:8836–45. doi: 10.1021/bi960292+
159. Hua QX, Jia W, Weiss MA. Conformational Dynamics of Insulin. Front Endocrin (2011) 2:48. doi: 10.3389/fendo.2011.00048
160. Menting JG, Yang Y, Chan SJ, Phillips NB, Smith BJ, Whittaker J, et al. A Structural Hinge in Insulin Enables its Receptor Engagement. Proc Natl Acad Sci USA (2014) 111(33):1–48. doi: 10.1073/pnas.1412897111
161. Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DC, et al. Atomic Positions in Rhombohedral 2-Zinc Insulin Crystals. Nature (1971) 231:506–11. doi: 10.1038/231506a0
162. Schwartz G, Katsoyannis PG. Synthesis of Des(Tetrapeptide B(1-4)) and Des(Pentapeptide B(1-5) Human Insulins. Two Biologically Active Analogues. Biochemistry (1978) 17:4550–56. doi: 10.1021/bi00614a029
163. Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A Cavity-Containing Mutant of T4 Lysozyme Is Stabilized by Buried Benzene. Nature (1992) 355(6358):371–3. doi: 10.1038/355371a0
164. Xu J, Baase WA, Baldwin E, Matthews BW. The Response of T4 Lysozyme to Large-to-Small Substitutions Within the Core and Its Relation to the Hydrophobic Effect. Protein Sci (1998) 7:158–77. doi: 10.1002/pro.5560070117
165. Guo ZY, Zhang Z, Jia XY, Tang YH, Feng YM. Mutational Analysis of the Absolutely Conserved B8Gly: Consequence on Foldability and Activity of Insulin. Acta Biochim Biophys Sin (Shanghai) (2005) 10:673–9. doi: 10.1111/j.1745-7270.2005.00093.x
166. Anil B, Song B, Tang Y, Raleigh DP. Exploiting the Right Side of the Ramachandran Plot: Substitution of Glycines by D-Alanine can Significantly Increase Protein Stability. J Am Chem Soc (2004) 126(41):13194–5. doi: 10.1021/ja047119i
167. O’Neil KT, DeGrado WF. A Thermodynamic Scale for the Helix-Forming Tendencies of the Commonly Occurring Amino Acids. Science (1990) 250:646–51. doi: 10.1126/science.2237415
168. Chakrabartty A, Kortemme T, Baldwin RL. Helix Propensities of the Amino Acids Measured in Alanine-Based Peptides Without Helix-Stabilizing Side-Chain Interactions. Protein Sci (1994) 3:843–52. doi: 10.1002/pro.5560030514
169. Schober E, Rami B, Grabert M, Thon A, Kapellen T, Reinehr T, et al. Phenotypical Aspects of Maturity-Onset Diabetes of the Young (MODY Diabetes) in Comparison With Type 2 Diabetes Mellitus (T2DM) in Children and Adolescents: Experience From a Large Multicentre Database. Diabetes Med (2009) 26(5):466–73. doi: 10.1111/j.1464-5491.2009.02720.x
170. Shields B, Hicks S, Shepherd M, Colclough K, Hattersley AT, Ellard S. Maturity-Onset Diabetes of the Young (MODY): How Many Cases are We Missing? Diabetologia (2010) 53(12):2504–8. doi: 10.1007/s00125-010-1799-4
171. Thanabalasingham G, Owen KR. Diagnosis and Management of Maturity Onset Diabetes of the Young (MODY). BMJ (2011) 343:d6044. doi: 10.1136/bmj.d6044
172. Johnson SR, McGown I, Oppermann U, Conwell LS, Harris M, Duncan EL. A Novel INS Mutation in a Family With Maturity-Onset Diabetes of the Young: Variable Insulin Secretion and Putative Mechanisms. Pediatr Diabetes (2018) 19(5):905–9. doi: 10.1111/pedi.12679
173. Shoelson S, Haneda M, Blix P, Nanjo A, Sanke T, Inouye K, et al. Three Mutant Insulins in Man. Nature (1983) 302:540–3. doi: 10.1038/302540a0
174. Fonseca SG, Gromada J, Urano F. Endoplasmic Reticulum Stress and Pancreatic β-Cell Death. Trends Endocrinol Metab (2011) 22(7):266–74. doi: 10.1016/j.tem.2011.02.008
175. Thomas SE, Dalton L, Malzer E, Marciniak SJ. Unravelling the Story of Protein Misfolding in Diabetes Mellitus. World J Diabetes (2011) 2(7):114–8. doi: 10.4239/wjd.v2.i7.114
176. Yuan Q, Tang W, Zhang X, Hinson JA, Liu C, Osei K, et al. Proinsulin Atypical Maturation and Disposal Induces Extensive Defects in Mouse Ins2+/Akita β-Cells. PloS One (2012) 7(4):e35098. doi: 10.1371/journal.pone.0035098
177. Doig AJ, Baldwin RL. N- and C-Capping Preferences for All 20 Amino Acids in α-Helical Peptides. Protein Sci (1995) 4:1325–36. doi: 10.1002/pro.5560040708
178. Shoelson SE, Polonsky KS, Zeidler A, Rubenstein AH, Tager HS. Human Insullin B24 (Phe®Ser), Secretion and Metabolic Clearance of the Abnormal Insulin in Man and in a Dog Model. J Clin Invest (1984) 73:1351–8. doi: 10.1172/JCI111338
179. Schwartz GP, Burke GT, Katsoyannis PG. A Superactive Insulin: [B10-Aspartic Acid]Insulin(Human). Proc Natl Acad Sci USA (1987) 84:6408–11. doi: 10.1073/pnas.84.18.6408
180. Carroll RJ, Hammer RE, Chan SJ, Swift HH, Rubenstein AH, Steiner DF. A Mutant Human Proinsulin Is Secreted From Islets of Langerhans in Increased Amounts via an Unregulated Pathway. Proc Natl Acad Sci USA (1988) 85:8943–7. doi: 10.1073/pnas.85.23.8943
181. Gross DJ, Halban PA, Kahn CR, Weir GC, Villa-Komaroff L. Partial Diversion of a Mutant Proinsulin (B10 Aspartic Acid) From the Regulated to the Constitutive Secretory Pathway in Transfected AtT-20 Cells. Proc Natl Acad Sci USA (1989) 86(11):4107–11. doi: 10.1073/pnas.86.11.4107
182. Chan SJ, Seino S, Gruppuso PA, Schwartz R, Steiner DF. A Mutation in the B Chain Coding Region Is Associated With Impaired Proinsulin Conversion in a Family With Hyperproinsulinemia. Proc Natl Acad Sci USA (1987) 84:2194–7. doi: 10.1073/pnas.84.8.2194
183. Pandyarajan V, Phillips NB, Rege NK, Lawrence MC, Whittaker J, Weiss MA. Contribution of TyrB26 to the Function and Stability of Insulin. Structure-Activity Relationships at a Conserved Hormone-Receptor Interface. J Biol Chem (2016) 291(25):12978–90. doi: 10.1074/jbc.M115.708347
184. Hartl FU, Hayer-Hartl M. Converging Concepts of Protein Folding In Vitro and In Vivo. Nat Struct Mol Biol (2009) 16(6):574–81. doi: 10.1038/nsmb.1591
185. Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, Saibil HR. The Protofilament Structure of Insulin Amyloid Fibrils. Proc Natl Acad Sci USA (2002) 99:9196–201. doi: 10.1073/pnas.142459399
186. Nielsen L, Frokjaer S, Brange J, Uversky VN, Fink AL. Probing the Mechanism of Insulin Fibril Formation With Insulin Mutants. Biochemistry (2001) 40:8397–409. doi: 10.1021/bi0105983
187. Brader ML, Dunn MF. Insulin Hexamers: New Conformations and Applications. Trends Biochem Sci (1991) 16:341–5. doi: 10.1016/0968-0004(91)90140-Q
188. Chothia C, Lesk AM, Dodson GG, Hodgkin DC. Transmission of Conformational Change in Insulin. Nature (1983) 302:500–5. doi: 10.1038/302500a0
189. Scapin G, Dandey VP, Zhang Z, Prosise W, Hruza A, Kelly T, et al. Structure of the Insulin Receptor-Insulin Complex by Single-Particle Cryo-EM Analysis. Nature (2018) 556(7699):122–5. doi: 10.1038/nature26153
190. Weis F, Menting JG, Margetts MB, Chan SJ, Xu Y, Tennagels N, et al. The Signalling Conformation of the Insulin Receptor Ectodomain. Nat Commun (2018) 9(1):4420. doi: 10.1038/s41467-018-06826-6
191. Lawrence MC. Understanding Insulin and its Receptor From Their Three-Dimensional Structure. Mol Metab (2021) 101255. doi: 10.1016/j.molmet.2021.101255
192. Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, et al. How Insulin Engages its Primary Binding Site on the Insulin Receptor. Nature (2013) 493(7431):241–5. doi: 10.1038/nature11781
193. Zhang Z, Liang WG, Bailey LJ, Tan YZ, Wei H, Wang A, et al. Ensemble cryoEM Elucidates the Mechanism of Insulin Capture and Degradation by Human Insulin Degrading Enzyme. Elife (2018) 7:e33572. doi: 10.7554/eLife.33572
194. Uchikawa E, Choi E, Shang G, Yu H, Bai X-C. Activation Mechanism of the Insulin Receptor Revealed by Cryo-EM Structure of the Fully Liganded Receptor–Ligand Complex. Elife (2019) 8:e48630. doi: 10.7554/eLife.48630
195. Zhang X, Yu D, Sun J, Wu Y, Gong J, Li X, et al. Visualization of Ligand-Bound Ectodomain Assembly in the Full-Length Human IGF-1 Receptor by Cryo-EM Single-Particle Analysis. Structure (2020) 28:555e561. doi: 10.1016/j.str.2020.03.007
196. Mirmira RG, Nakagawa SH, Tager HS. Importance of the Character and Configuration of Residues B24, B25, and B26 in Insulin-Receptor Interactions. J Biol Chem (1991) 266(3):1428–36. doi: 10.1016/S0021-9258(18)52312-7
197. Hua QX, Xu B, Huang K, Hu SQ, Nakagawa S, Jia W, et al. Enhancing the Activity of Insulin by Stereospecific Unfolding. Conformational Life Cycle of Insulin and its Evolutionary Origins. J Biol Chem (2009) 284:14586–96. doi: 10.1074/jbc.M900085200
198. Bouchard M, Zurdo J, Nettleton EJ, Dobson CM, Robinson CV. Formation of Insulin Amyloid Fibrils Followed by FTIR Simultaneously With CD and Electron Microscopy. Protein Sci (2000) 9:1960–7. doi: 10.1110/ps.9.10.1960
199. Dong J, Wan Z, Popov M, Carey PR, Weiss MA. Insulin Assembly Damps Conformational Fluctuations: Raman Analysis of Amide I Line Widths in Native States and Fibrils. J Mol Biol (2003) 330:431–42. doi: 10.1016/S0022-2836(03)00536-9
200. Sereda V, Sawaya MR, Lednev IK. Structural Organization of Insulin Fibrils Based on Polarized Raman Spectroscopy: Evaluation of Existing Models. J Am Chem Soc (2015) 137(35):11312–20. doi: 10.1021/jacs.5b07535
201. Burke MJ, Rougvie MA. Cross-B Protein Structures. I. Insulin Fibrils. Biochemistry (1972) 11:2435–9. doi: 10.1021/bi00763a008
202. Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, Fraser PE, et al. Instability, Unfolding and Aggregation of Human Lysozyme Variants Underlying Amyloid Fibrillogenesis. Nature (1997) 385:787–93. doi: 10.1038/385787a0
203. Ivanova MI, Sievers SA, Sawaya MR, Wall JS, Eisenberg D. Molecular Basis for Insulin Fibril Assembly. Proc Natl Acad Sci USA (2009) 106(45):18990–5. doi: 10.1073/pnas.0910080106
204. Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, et al. Inherent Toxicity of Aggregates Implies a Common Mechanism for Protein Misfolding Diseases. Nature (2002) 416:507–11. doi: 10.1038/416507a
205. Pandyarajan V, Smith BJ, Phillips NB, Whittaker L, Cox GP, Wickramasinghe N, et al. Aromatic Anchor at an Invariant Hormone-Receptor Interface Function of Insulin Residue B24 With Aplication to Protein Design. J Biol Chem (2014) 289(50):34709–27. doi: 10.1074/jbc.M114.608562
206. Kang S, Creagh FM, Peters JR, Brange J, Volund A, Owens DR. Comparison of Subcutaneous Soluble Human Insulin and Insulin Analogues (AspB9, GluB27; AspB10; AspB28) on Meal-Related Plasma Glucose Excursions in Type I Diabetic Subjects. Diabetes Care (1991) 14:571–7. doi: 10.2337/diacare.14.7.571
207. Shoelson SE, Lu ZX, Parlautan L, Lynch CS, Weiss MA. Mutations at the Dimer, Hexamer, and Receptor-Binding, Surfaces of Insulin Independently Affect Insulin-Insulin and Insulin-Receptor Interactions. Biochemistry (1992) 31:1757–67. doi: 10.1021/bi00121a025
208. Milazzo G, Sciacca L, Papa V, Goldfine ID, Vigneri R. AspB10 Insulin Induction of Increased Mitogenic Responses and Phenotypic Changes in Human Breast Epithelial. Mol Carcinog (1997) 18:19–25. doi: 10.1002/(SICI)1098-2744(199701)18:1<19::AID-MC3>3.0.CO;2-M
209. Hansen BF, Kurtzhals P, Jensen AB, Dejgaard A, Russell-Jones D. Insulin X10 Revisited: A Super-Mitogenic Insulin Analogue. Diabetologia (2011) 54(9):2226–31. doi: 10.1007/s00125-011-2203-8
210. Svendsen AM, Winge SB, Zimmermann M, Lindvig AB, Warzecha CB, Sajid W, et al. Down-Regulation of Cyclin G2 by Insulin, IGF-I (Insulin-Like Growth Factor 1) and X10 (AspB10 Insulin): Role in Mitogenesis. Biochem J (2014) 457(1):69–77. doi: 10.1042/BJ20130490
211. Gallagher EJ, Alikhani N, Tobin-Hess A, Blank J, Buffin NJ, Zelenko Z, et al. Insulin Receptor Phosphorylation by Endogenous Insulin or the Insulin Analog AspB10 Promotes Mammary Tumor Growth Independent of the IGF-I Receptor. Diabetes (2013) 62(10):3553–60. doi: 10.2337/db13-0249
212. Smit A, Van Marle A, Van Elk R, Bogerd J, Van Heerikhuizen H, Geraerts W. Evolutionary Conservation of the Insulin Gene Structure in Invertebrates: Cloning of the Gene Encoding Molluscan Insulin-Related Peptide III From Lymnaea Stagnalis. J Mol Endocrinol (1993) 11(1):103–13. doi: 10.1677/jme.0.0110103
213. Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, et al. Regulation of DAF-2 Receptor Signaling by Human Insulin and Ins-1, a Member of the Unusually Large and Diverse C. Elegans Insulin Gene Family. Genes Dev (2001) 15:672–86. doi: 10.1101/gad.867301
214. Blumenthal S. From Insulin and Insulin-Like Activity to the Insulin Superfamily of Growth-Promoting Peptides: A 20th-Century Odyssey. Perspect Biol Med (2010) 53(4):491–508. doi: 10.1353/pbm.2010.0001
215. Chan SJ, Cao QP, Steiner DF. Evolution of the Insulin Superfamily: Cloning of a Hybrid Insulin/Insulin- Like Growth Factor cDNA From Amphioxus. Proc Natl Acad Sci USA (1990) 87:9319–23. doi: 10.1073/pnas.87.23.9319
216. Pashmforoush M, Chan SJ, Steiner DF. Structure and Expression of the Insulin-Like Peptide Receptor From Amphioxus. Mol Endocrinol (1996) 10:857–66. doi: 10.1210/mend.10.7.8813726
217. Chan SJ, Steiner DF. Insulin Through the Ages: Phylogeny of a Growth Promoting and Metabolic Regulatory Hormone. Integr Comp Biol (2000) 40(2):213–22. doi: 10.1093/icb/40.2.213
218. Guo Z-Y, Shen L, Gu W, Wu A-Z, Ma J-G, Feng Y-M. In Vitro Evolution of Amphioxus Insulin-Like Peptide to Mammalian Insulin. Biochemistry (2002) 41(34):10603–7. doi: 10.1021/bi020223x
219. Sitar T, Popowicz GM, Siwanowicz I, Huber R, Holak TA. Structural Basis for the Inhibition of Insulin-Like Growth Factors by Insulin-Like Growth Factor-Binding Proteins. Proc Natl Acad Sci USA (2006) 103(35):13028–33. doi: 10.1073/pnas.0605652103
220. Zhou J, Xiang J, Zhang S, Duan C. Structural and Functional Analysis of the Amphioxus IGFBP Gene Uncovers Ancient Origin of IGF-Independent Functions. Endocrinology (2013) 154(10):3753–63. doi: 10.1210/en.2013-1201
221. Allard JB, Duan C. IGF-Binding Proteins: Why do They Exist and Why are There So Many? Front Endocrinol (2018) 9:117. doi: 10.3389/fendo.2018.00117
222. Nakagawa SH, Tager HS, Steiner DF. Mutational Analysis of Invariant ValB12 in Insulin: Implications for Receptor Binding. Biochemistry (2000) 39:15826–35. doi: 10.1021/bi001802+
223. Allander SV, Coleman M, Luthman H, Powell DR. Chicken Insulin-Like Growth Factor Binding Protein (IGFBP)-5: Conservation of IGFBP-5 Structure and Expression During Evolution. Comp Biochem Physiol B Biochem Mol Biol (1997) 116(4):477–83. doi: 10.1016/S0305-0491(96)00289-1
224. Collett-Solberg PF, Cohen P. The Role of the Insulin-Like Growth Factor Binding Proteins and the IGFBP Proteases in Modulating IGF Action. Endocrinol Metab Clin North Am (1996) 25(3):591–614. doi: 10.1016/S0889-8529(05)70342-X
225. Hwa V, Oh Y, Rosenfeld R. Insulin-Like Growth Factor Binding Proteins: A Proposed Superfamily. Acta Paediatr (1999) 88:37–45. doi: 10.1111/j.1651-2227.1999.tb14349.x
226. Cheng R, Chang K-M, Wu J-L. Different Temporal Expressions of Tilapia (Oreochromis Mossambicus) Insulin-Like Growth Factor-I and IGF Binding Protein-3 After Growth Hormone Induction. Mar Biotechnol (2002) 4(3):218–25. doi: 10.1007/s10126-002-0014-0
227. Haataja L, Manickam N, Soliman A, Tsai B, Liu M, Arvan P. Disulfide Mispairing During Proinsulin Folding in the Endoplasmic Reticulum. Diabetes (2016) 65(4):1050–60. doi: 10.2337/db15-1345
228. Guo ZY, Shen L, Feng YM, Wang DC. The Different Energetic State of the Intra A-Chain/Domain Disulfide of Insulin and Insulin-Like Growth Factor 1 Is Mainly Controlled by Their B-Chain/Domain. Biochemistry (2002) 40:10585–92. doi: 10.1021/bi020165f
229. Guo ZY, Shen L, Feng YM. The Different Folding Behavior of Insulin and Insulin-Like Growth Factor 1 is Mainly Controlled by Their B-Chain/Domain. Biochemistry (2002) 41:1556–67. doi: 10.1021/bi011166v
230. Bliss M. The Discovery of Insulin: Twenty-Fifth Anniversary Edition. Chicago: University of Chicago Press (2007). p. 310.
231. Flier JS, Kahn CR. Insulin: A Pacesetter for the Shape of Modern Biomedical Science and the Nobel Prize. Mol Metab (2021) 101194. doi: 10.1016/j.molmet.2021.101194
232. Ptitsyn OB. How Does Protein Synthesis Give Rise to the 3D-Structure. FEBS Lett (1991) 285:176–81. doi: 10.1016/0014-5793(91)80799-9
233. Matagne A, Radford SE, Dobson CM. Fast and Slow Tracks in Lysozyme Folding: Insight Into the Role of Domains in the Folding Process. J Mol Biol (1997) 267:1068–74. doi: 10.1006/jmbi.1997.0963
234. Yokota A, Izutani K, Takai M, Kubo Y, Noda Y, Koumoto Y, et al. The Transition State in the Folding-Unfolding Reaction of Four Species of Three-Disulfide Variant of Hen Lysozyme: The Role of Each Disulfide Bridge. J Mol Biol (2000) 295:1275–88. doi: 10.1006/jmbi.1999.3442
235. Lazaridis T, Karplus M. “New View” of Protein Folding Reconciled With the Old Through Multiple Unfolding Simulations. Science (1997) 278:1928–31. doi: 10.1126/science.278.5345.1928
236. Mirny LA, Abkevich VI, Shakhnovich EI. How Evolution Makes Proteins Fold Quickly. Proc Natl Acad Sci USA (1998) 95:4976–81. doi: 10.1073/pnas.95.9.4976
237. Despa F. Endoplasmic Reticulum Overcrowding as a Mechanism of β-Cell Dysfunction in Diabetes. Biophys J (2010) 98(8):1641–8. doi: 10.1016/j.bpj.2009.12.4295
Keywords: protein folding, protein structure, folding efficiency, hormone, metabolism
Citation: Dhayalan B, Chatterjee D, Chen Y-S and Weiss MA (2021) Structural Lessons From the Mutant Proinsulin Syndrome. Front. Endocrinol. 12:754693. doi: 10.3389/fendo.2021.754693
Received: 06 August 2021; Accepted: 13 September 2021;
Published: 30 September 2021.
Edited by:
Pierre De Meyts, Université Catholique de Louvain, BelgiumReviewed by:
Rongpeng Gong, Qinghai University Medical College, ChinaRasheed Ahmad, Dasman Diabetes Institute, Kuwait
Copyright © 2021 Dhayalan, Chatterjee, Chen and Weiss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Weiss, weissma@iu.edu