 Erika Kague
Erika Kague Carolina Medina-Gomez
Carolina Medina-Gomez Simeon A. Boyadjiev3
Simeon A. Boyadjiev3 Fernando Rivadeneira
Fernando Rivadeneira- 1School of Physiology, Pharmacology and Neuroscience, Biomedical Sciences, University of Bristol, Bristol, United Kingdom
- 2Department of Internal Medicine, Erasmus Medical Center (MC), University Medical Center Rotterdam, Rotterdam, Netherlands
- 3Department of Pediatrics, University of California, Davis, Sacramento, CA, United States
- 4Department of Oral and Maxillofacial Surgery, Erasmus Medical Center (MC), University Medical Center Rotterdam, Rotterdam, Netherlands
Osteoporosis is the most prevalent bone condition in the ageing population. This systemic disease is characterized by microarchitectural deterioration of bone, leading to increased fracture risk. In the past 15 years, genome-wide association studies (GWAS), have pinpointed hundreds of loci associated with bone mineral density (BMD), helping elucidate the underlying molecular mechanisms and genetic architecture of fracture risk. However, the challenge remains in pinpointing causative genes driving GWAS signals as a pivotal step to drawing the translational therapeutic roadmap. Recently, a skull BMD-GWAS uncovered an intriguing intersection with craniosynostosis, a congenital anomaly due to premature suture fusion in the skull. Here, we recapitulate the genetic contribution to both osteoporosis and craniosynostosis, describing the biological underpinnings of this overlap and using zebrafish models to leverage the functional investigation of genes associated with skull development and systemic skeletal homeostasis.
Introduction
Osteoporosis is characterized by low bone mass, microarchitectural deterioration, and impairment of bone quality, predisposing individuals to fractures (1). Although fragility fractures caused by osteoporosis may not lead to immediate death, they have a big impact on the quality of life of patients and bring on a considerable burden to the health and economic systems. Fractures cause substantial pain, disability, loss of independence and are associated with a 20% mortality rate within one-year post-fracture (2).
Osteoporosis is the most prevalent metabolic bone disease, with estimates that 1 in 3 women and 1 in 5 men over the age of 50 will experience fragility fractures in their remaining lifetimes. Therefore, it is a major health problem as the aging population increases worldwide. In the United States, the direct medical costs of osteoporosis-related fractures alone exceed $20 billion annually (3). More than 500,000 fractures due to osteoporosis happen every year in the UK alone, representing treatment costs of over £4 billion per year (1). In Europe, osteoporosis causes 4.3 million fragility fractures per year, costing the health care systems more than 56 billion euros each year (The International Osteoporosis Foundation, SCOPE 2021). Given such alarming numbers, the World Health Organization (WHO) considers osteoporosis a leading health problem worldwide, with a burden only exceeded by cardiovascular diseases. However, despite the severe consequences of the increment of fracture number in elderly patients worldwide and the availability of diverse therapeutic options, there is still limited and in some instances, inadequate adherence of patients to treatments (described as the “gap in osteoporosis treatment”). This is a multifactorial phenomenon where limitations to long-term therapies, response to treatment and fear of adverse events contribute importantly. Altogether there is room for the identification of new biologic pathways and factors that constitute new therapeutic targets, which begins through gaining a better understanding of the underlying molecular mechanisms.
The causes of osteoporosis are attributed to a complex interplay between genetic and environmental factors. For instance, physical activity, smoking, alcohol use, and diet (calcium and protein intake) have been shown to influence the risk of osteoporotic fractures (4). Yet, twin and family studies have demonstrated that osteoporosis is highly heritable, and genetic factors are solid contributors to the risk of disease. Indeed, a wide range of osteoporosis-related phenotypes have been robustly associated with genetic factors, including markers of bone turnover, skeletal dimensions, bone growth, bone mineral density (BMD), and fracture risk itself (5–8). Among these, BMD measured using dual-energy X-ray absorptiometry (DXA), which is used to diagnose osteoporosis and assess fracture risk (9), has the highest heritability (ranging between 50% to 80%) (10). Therefore, most genetic studies in osteoporosis have focused on the BMD phenotype.
Over the years, genome wide-association studies (GWAS) have uncovered over 500 genomic loci associated with BMD. Beyond the contribution to our understanding of molecular mechanisms in bone biology these findings might frame the discovery of new therapeutic targets for osteoporosis (11, 12). In fact, the success rate during a drug’s development is doubled for drug targets with human genetic support (13). Nevertheless, GWASs have not yet contributed to translational therapeutic approaches for bone conditions. This is highly attributed to mapping the association signals and identifying the causal genes. Bone diseases with a known genetic cause help interpret a gene’s function, as learned from many cases of monogenic disorders (14). However, functional evidence from animal models provides crucial causality evidence in most cases. Great initiatives in mouse models, such as the “Origins of Bone and Cartilage Disease” (OBCD) and the “International Mouse Phenotyping Consortium” (IMPC), have helped to functional annotate GWAS candidate genes (12, 15). But, given the large number of BMD-associated loci, mouse models still fall behind in providing high-throughput evidence of implication for the numerous genes within an associated locus harmonically, rapidly, and longitudinally.
BMD is a widely available measurement of bone strength, resulting in a large yield of associated loci that have helped characterize the genetic architecture of osteoporosis during the last decade. In addition, is an excellent trait suitable for the identification of a wide spectrum of bone biology, relevant to many other bone conditions, such as osteogenesis imperfecta, sclerosteosis, and Paget’s disease of bone (16, 17), with diverse, overlapping loci. Recently, an exciting overlap in associated signals between BMD and craniosynostosis has become evident. Craniosynostosis is a developmental condition that affects the skull shape and volume as a consequence of premature fusion of the calvarial sutures (18). In this review, we will briefly discuss the genetics of osteoporosis, its overlap with craniosynostosis, and will explore the implications of this intriguing intersection. Furthermore, we will introduce zebrafish as a model organism that allows versatile functional studies aimed at the identification of causal genes from GWAS, and specifically to understand the observed overlap between these two diseases.
A brief walkthrough of 15 years of GWAS of osteoporosis
GWAS for osteoporosis mainly focused on investigating the effect of genetic factors on BMD, the strongest predictor of fracture risk; therefore this review will focus on BMD. Through the years, BMD GWAS have dramatically increased the number of participant and cohorts, resulting in the development of better methodologies for harmonizing data, expansion of reference panels for imputation, and cutting-edge tools for statistical analysis. More importantly, GWAS have progressively delineated genes to be prioritized for functional studies, so their biological function in the disease can be identified. In Figure 1 we provide an overview of the GWAS for BMD.
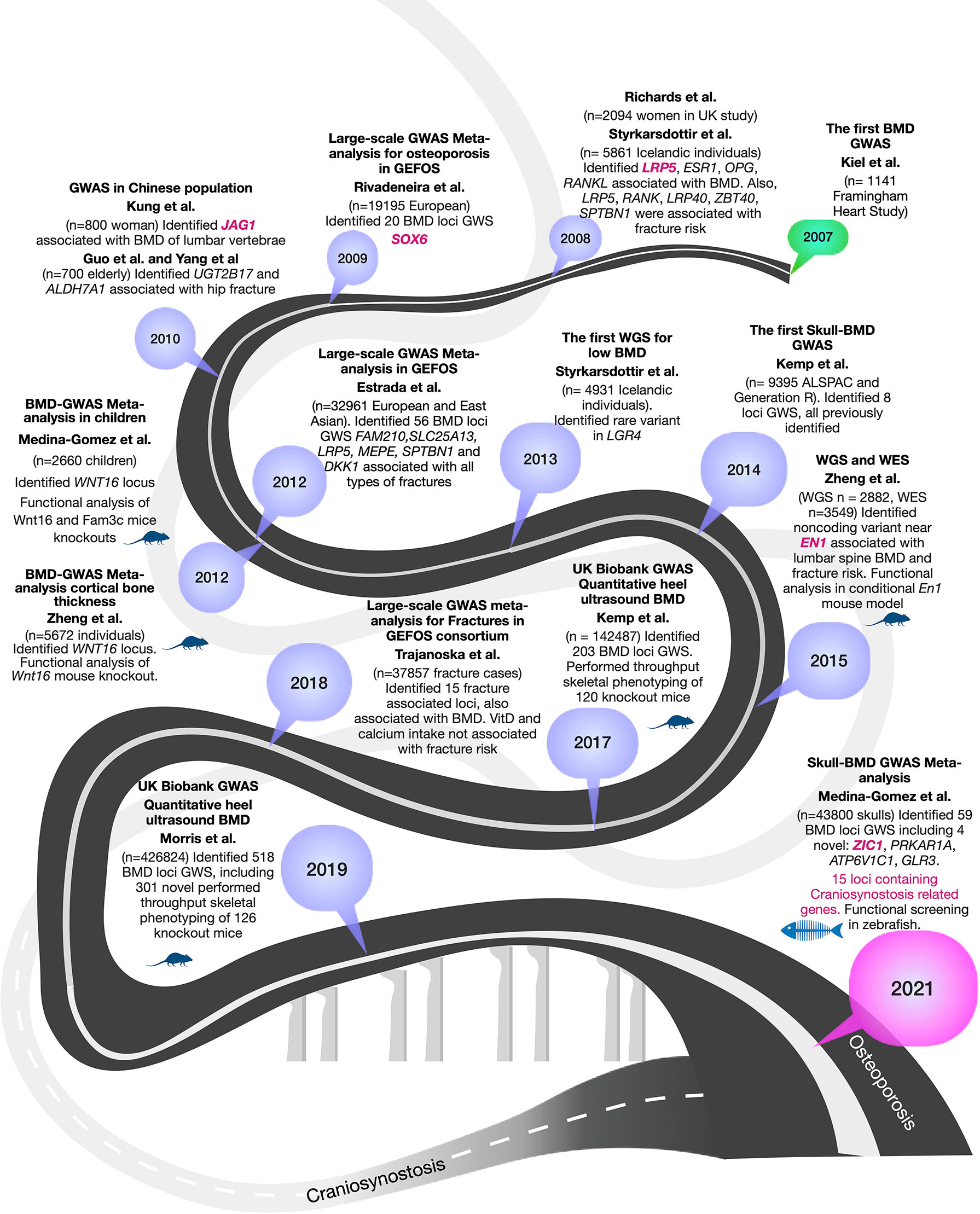
Figure 1 Summary of 15 years of GWASs for BMD to the current encounter with craniosynostosis. BMD-GWAS timeline is represented in a dark grey roadmap. Mouse and fish symbols indicate studies with functional work performed in mice and zebrafish, respectively. Genes highlighted in magenta are known craniosynostosis genes. A light grey roadmap represents the path by the genetics of craniosynostosis that finally meets with that of osteoporosis in 2021.
The first BMD-GWAS was conducted in 2007 by Kiel et al. (19), including 1141 individuals from the Framingham Heart Study, but given the small sample size, none of the scrutinized SNPs reached genome-wide significance (GWS) (P< 5 x 10-8) (19). In 2008, two GWAS were published. Richards et al. (20) included 2094 women from the Twins UK study and carried out replication in 6463 participants from the Rotterdam Study (n=4081) and other European cohorts, identifying variants in TNFRSF11B and LRP5 associated at GWS level (20). The second study, published by Styrkarsdottir et al. (21), was performed on 5861 Icelandic subjects and identified TNFSF11 (RANKL), TNFRSF11B (OPG), ESR1, ZBTB40, and TNFRSF11A (RANK). The authors also reported association of SPTBN1, LRP4, and RANKL with osteoporotic fractures (21). Since then, the greatest leaps in discoveries have evolved from extensive collaborative studies leading to the identification of hundreds of loci and SNPs associated with osteoporosis traits.
In 2009, Rivadeneira et al. (22) published the first large collaborative study of the Genetic Factors for Osteoporosis Consortium (GEFOS-1) cohort, including 19,195 European subjects. It identified 20 loci associated with BMD, of which 13 were novel (GPR177, SPTBN1, CTNNB1, MEPE, MEF2C, STARD3NL, FLJ42280, LRP4. DCDC5, SOX6, FOXL1, HDAC5, CRHR1). The meta-analysis also confirmed seven known BMD loci at GWS (SBTB40, ESR1, TNFRSF11B, LRP5, SP7, TNFSF11, TNFSF11A) (22). The second GEFOS study was published not long after three small GWAS in Chinese populations identified variants in JAG1 (study of n= 800 women), UGT2B17 (n=700 elderly, comprising 350 cases with a history of hip osteoporotic fracture and 350 healthy controls), and ALDH7A1 (study of n= 700 elderly) (23–25), but the latter two were not replicated by subsequent studies. In 2012, the second large GEFOS-2 meta-analysis increased to 32,961 participants of Europeans and East Asia backgrounds (26). This study by Estrada et al. (26) identified 56 BMD GWS loci, including variants in 14 loci associated with all types of fractures (including FAM210, SLC25A13, LRP5, MEPE, SPTBN1, and DKK1) (26). In 2012, two other independent GWAS identified a prominent signal in WNT16 (Wnt family member 16). Medina-Gomez et al. (27) analyzed 2,660 children of different ethnicities identifying WNT16 locus influencing bone accrual (27). Zheng et al. (28) analyzed cortical bone thickness on 5,878 European subjects and identified an association with WNT16. Both groups validated their results with functional studies in homozygous mutant mice for Wnt16-/-, which resulted in 10% lower BMD, 27% thinner cortical bones at the femur midshaft and 43%-61% reduction of bone strength (28), therefore showing evidence of gene causality for this respective locus. Additional functional studies of WNT16 locus were previously reviewed (29). Of note, rare coding variants in CPED1 mapping in the same locus, appears to be also involved in regulating BMD variation (28).
With the advancements of next-generation sequencing technologies and prices for sequencing dropping down, it became realistic to move forward with the identification of rare variants (minor allele frequency, MAF<1%) through the incorporation of Whole-Genome (WGS) and Exome (WES) sequencing (reviewed by (29)). In 2013, the first WGS was performed by Styrkarsdottir et al. (30) for BMD in an Icelandic population comprising 4,931 individuals, which identifying a rare nonsense mutation (c.376C >T) within LGR4 associated with low BMD and fracture risk (30). In 2015, Zheng et al. (31) used WGS [n = 2,882 from UK10K (sequenced 10K people to identify rare genetic variants in health and disease) (32)], WES, deep imputation of genotypes samples employing a combined UK10K/1000 Genomes reference (n=26,534), and de novo replication genotyping (n = 20,271). This effort resulted in the identification of a less frequent non-coding variant near a novel locus, harboring EN1 (encoding a homeobox protein engrailed 1). Conditional mouse En1 knockout presented reduced BMD and increased skull bone resorption (31). Further, non-coding variants mapping to CPED1 were also identified, suggesting this gene in close vicinity of WNT16, is also involved in regulating BMD variation. In 2015, Styrkarsdottir et al. (17) performed GWAS using sequence variants found through WGS of 2,636 Icelanders, resulting in identifying two rare variants in the COL1A2, a gene known for to underlie many Osteogenesis Imperfecta cases, in association with low BMD and osteoporotic fractures. The carriers of these mutations did not show phenotypic signs of osteogenesis imperfecta but had low BMD (17). Collectively, these WGS-baswed studies underscore the importance of rare variants of larger effects in coding and non-coding regions that may not be identified through low powered GWAS. Despite the successful examples described here, the research community was already aware of the steep increase in sample size needed to adequately survey rare variation.
The emergence of large biobanks presented an unprecedented opportunity to assess the contribution of rare genetic variation to osteoporosis-related phenotypes within the so-called spectrum of “mega-GWAS” that gave rise to the two largest efforts in the osteoporosis field, studying estimated BMD (eBMD) measured with quantitative ultrasound of the heel. First, in 2017, Kemp et al. (15) undertook a GWAS in 142,487 individuals from the UK Biobank and identified 203 loci associated with eBMD, from which 153 were novel loci. This work used an exciting approach, among others, for gene prioritization. Genes within 1MB of any lead SNPs at the associated eBMD loci were checked for implication in knock-out mice models part of the International Mouse Phenotyping Consortium (IMPC) and the International Knockout Mouse Consortium (IKMC). In total, 120 genes had knockout mice available that presented abnormal bone structure after high-throughput skeletal phenotyping was undertaken by the Origins of Bone and Cartilage Disease (OBCD) consortium (15). In 2019, the largest GWAS to date on the genetics of osteoporosis was published as an extension of the eBMD analysis in the UK Biobank, involving now 426,824 participants (12). Morris et al. identified 515 loci, of which 301 were novel. This work involved skeletal phenotyping of 126 knockout mice, representing the largest GWAS with the largest functional evidence from mice.
This study dissected the contribution of independent lead SNPs per locus, showing that only 4.6% were rare (MAF<1%), whereas 9.3% were low frequency (1%>MAF ≤ 5%), and 86.1% were common (MAF>5%). Rare variants explained 0.8% of the variance in eBMD, whereas common variants explained 17.8%. Yet, it is important to mention that imputation of rare variants is challenging, and sequencing is essential to truly understand the impact of rare variations on human health. For instance, recently, the release of the UKBB exome sequencing revealed a catalog of 8-fold the coding variation accessible in the UKBB through imputation, in which a rare variant in MEPE was strongly associated with low BMD (33).
Beyond BMD, it is essential to mention the largest fracture risk GWAS performed by Trajanoska et al. (34). This GWAS on a set of 37,857 fracture cases and 227,116 controls resulted in the identification of 15 fracture-associated loci (SPTBN1, CTNNB1, RSPO3, ESR1, WNT16/CPED1, SHFM1, STRARD3NL, GRB10/COLBL, FUBP3, MBL2/DKK1, LRP5, RPS6KA5, SOST, FAM210A, ETS2) from which all had been previously associated with BMD. Through two-sample Mendelian randomization, the author assessed different risk factors for fracture; BMD and hand grip strength showed significant evidence for a causal effect; whereas genetically-determined vitamin D levels and calcium intake were not associated with fracture risk. Other relevant GWASs were also published during the mentioned period (35–38).
To date, GWAS for BMD have significantly advanced our knowledge of the complex genetics underpinning osteoporosis, shed light on the genetic causes of fracture risk, and hinted on the molecular mechanisms underlying bone homeostasis. BMD measurements constitute excellent endophenotypes of fracture risk, while also pinpointing diverse aspects of bone biology and homeostasis. For example, significant genetic correlation has been described between BMD loci and those of bone growth and shape (39), osteoarthritis (40, 41), high bone mass disorders (42), rare skeletal disorders (17), Paget’s disease of bone (16, 43), metabolic disorders (i.e., diabetes) (44), etc. Such genetic overlaps indicate that diverse factors and pathways regulate the skeletal mineral content in our bodies. By studying bone conditions of known genetic causes and dissecting their function to maintain BMD, we will learn key elements in bone biology with the potential of translating this knowledge into therapeutic strategies for bone diseases.
Recently, a curious overlap between BMD and craniosynostosis, a condition that affects the cranial bones, has emerged. We may ask ourselves, “what does the skull have to do with osteoporosis?”
Why should we give attention to the skull?
While most GWASs were performed using BMD at the femoral neck and lumbar spine, less attention has been given to the skull. However, the skull has a few attributes that may allow the identification of new genetic factors in bone biology and, surprisingly, also for osteoporosis.
The skull (not including the jaw) is subjected to considerably less mechanical loading than axial or appendicular bones. Studies in mice estimated that the human fibula has a load nearly twice that of the skull bones (45). In vivo studies in humans showed that parietal bones in the skull were subjected to at least ten times lower loads than the appendicular skeleton (46). The skull is subject to a distinctive mechanical forces pattern, loaded radially and tangentially by intracranial pressure and mastication (45). This is also reflected in the morphology of mechanosensing osteocytes in the skull compared to those present in the tibia, as demonstrated by studies in mice. Skull bones have fewer osteocytes with smaller bodies but increased surface area, due to the increased number of cellular processes (47, 48). Skull of rats highly express genes in response to mechanical loading in the limbs (48). These findings suggest distinct molecular adaptations of mechanosensing osteocytes in the skull, which can maintain bone mass despite being subjected to lower mechanical loading than appendicular bones.
Interestingly, the calvaria does not lose bone during disuse or after menopause (49). In contrast to the rest of the body, cranial bone mass does not decline during spaceflight (50). Similarly, prolonged bed rest and spinal cord injury result in bone loss from the appendicular and axial skeleton but not from the skull (51, 52). Therefore, GWAS derived from skull BMD may represent an excellent source for identifying genes associated with yet to be elucidated mechanosensing properties and subjected to fewer environmental influences (i.e., physical activity) than appendicular bones.
Another peculiarity of the skull is its embryonic origin and bone formation modes. The skull is formed from cranial skeletogenic mesenchyme derived from two distinct embryonic sources: mesoderm and neural crest (53). The neural crest cells are multipotent stem cells that, in early development, arise in the dorsal-most aspect of the forming neural tube, from which undergoes an epithelial-to-mesenchymal transition to delaminate from the neuroepithelium. Neural crest cells migrate extensively throughout the early embryo to contribute to important cell types, including the peripheral nervous system, the craniofacial skeleton, and the cardiovascular system (54). Lineage tracing studies in mice, chicken, and zebrafish have demonstrated that the frontal bones are derived from neural crests, while parietal bones originate from the mesoderm (53, 55, 56). Macroscopically, at birth, the skull has movable fibrous regions, providing plasticity required during delivery and later for the growth and development of the head and brain. These regions are composed of dense connective tissue shaped into six fontanelles: anterior (frontal), posterior (occipital), anterolateral (sphenoid 2x) and posterolateral (mastoid 2x); which fuse normally by the age of 12 to 18 months. Although 80% of skull adult size is reached by the age of 3, bone growth continues via membranous ossification.
When it comes to types of ossification, the skull is complex as it is formed both by intramembranous (without a cartilaginous intermediate as seen in the calvaria) and endochondral ossification (temporal and skull-base). Mesenchymal cells within a vascularized area of the connective tissue proliferate and differentiate directly into preosteoblasts and finally into osteoblasts (57). The structure of the flat bones of the skull resembles a sandwich, where cortical bones represent the bread, while the center of the sandwich is the cancellous (trabecular) bone. The amount of cortical bone in the skull exceeds that of trabecular bone. Hence, GWAS on the BMD of the skull may primarily highlight genes associated with cortical bones and highly mechanosensing.
Finally, the skull is often compromised in monogenic skeletal conditions, exemplified by patients with recessive mutations in SOST (encodes sclerostin). In both cases of SOST-related sclerosing bone dysplasia, including sclerosteosis and van Buchem disease, patients display excessive bone formation (hyperostosis) phenotypically, leading to increased bone mass throughout the body, but particularly thickening of the skull bones (58, 59). SOST codifies sclerostin, a secreted glycoprotein predominantly produced by osteocytes and known to inhibit bone formation (60). Remarkably, the identification of SOST led to the development of romosozumab. Although with concerns of cardiovascular complications, this humanized monoclonal antibody inhibits sclerostin and is currently the most effective drug for osteoporosis, approved in Europe in 2019 (61). Calvaria thickening is also observed in cases of pyknodysostosis, a rare autosomal recessive bone dysplasia caused by mutations in CTSK (cathepsin K), characterized by osteosclerosis and short stature. Cathepsin K is among the most attractive targets for anti-osteoporosis drug development, reducing resorption while maintaining bone formation. Cathepsin K inhibitor odanacatib demonstrated high therapeutic efficacy in patients with postmenopausal osteoporosis in Phase III clinical trials. Still, unfortunately, due to cardio-cerebrovascular adverse effects, it was not approved to go into the market (62). More examples will be highlighted throughout the text. Still, it is essential to notice that genetic conditions affecting skull intramembranous bones have been vital in developing potent therapeutics for osteoporosis.
Craniosynostosis is the premature closure of the sutures between the flat bones of the skull
Craniosynostosis is a major structural birth defect characterized by the premature fusion of one or more cranial sutures (18). It happens in 1 in 2,500 liveborn babies, with approximately 80% of children presenting an isolated phenotype (i.e., nonsyndromic craniosynostosis), with the suture fusion being the only defect (63). The remainder of children with craniosynostosis present with syndromic craniosynostosis that includes additional birth defects and developmental delays (64).
The cranial sutures bridge the gap between the cranial bones, allowing to mold the head while passing through the birth canal and are growth sites of the skull. At the suture osteogenic fronts, a balance between proliferation and differentiation ensures the coordinated growth of the calvaria during the first years of life (64) (Figure 2). Within the sutures, a stem cell skeletogenic niche is found, self-renewing and allowing bone modeling, remodeling, regeneration, and healing over an extensive period during the skeletal development (65) (Figure 2). The metopic sutures, located between the frontal bones, are the first to close at approximately nine months of age. The fusion of the remaining sutures are largely completed by 18 months, but complete fusion does not occur until the third decade of life (64). Therefore, the sutures compromised in craniosynostosis comprise a unique paradigm for studying osteoblast differentiation, bone remodeling, and regeneration.
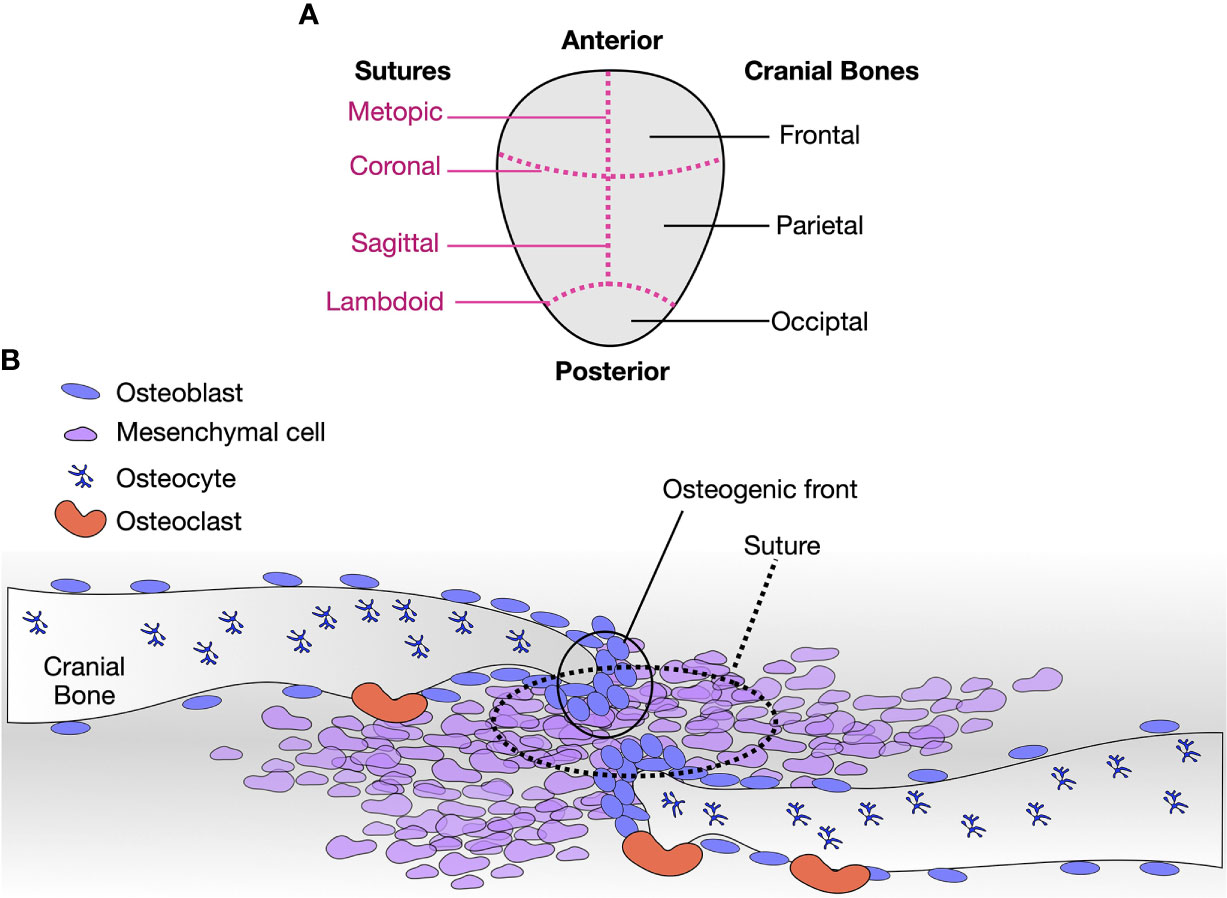
Figure 2 Cranial sutures are paradigms to study osteoblast differentiation, remodeling, and regeneration. (A) Diagram of a human calvaria, with respective cranial bones (right, black) and sutures (left, magenta, dashed lines). (B) Diagram of a suture. Osteoblasts, osteocytes, osteoclasts, and mesenchymal cells are indicated. The tip of each cranial plate is a region of active osteoblasts bone deposits. Osteoblasts lining cells are positioned away from the fronts and along the bone. Osteocytes are embedded in the bone matrix, and osteoclasts resorb bone. At the sutures, pluripotent mesenchymal cells self-renew and eventually enter osteoblastic differentiation to contribute to the osteogenic fronts. As observed with craniosynostosis, minor molecular disturbances at the sutures lead to severe phenotypes.
In the presence of premature closure of the cranial sutures, the brain cannot expand naturally during growth. Thus, affected children may experience ongoing medical complications, such as increased intracranial pressure (66, 67), vision problems (optic neuropathy secondary to elevated intracranial pressure, strabismus, refracture errors, exposure keratopathy from exorbitism) (68), and learning disabilities (69–71). As consequence, children with craniosynostosis require extensive surgical treatment through their first years of life to correct the malformation and allow brain expansion. However, after correction surgeries, re-synostosis is a recurrent problem requiring follow-up surgeries, given the enhanced osteoanabolic activity in the cranial sutures (72). The complexity and number of surgeries needed reflect the healthcare and financial burden caused by craniosynostosis. Some data exist regarding the long-term results of the surgical corrections (73, 74) and developmental outcomes (75, 76).
Despite the high healthcare burden of craniosynostosis, the genetics of craniosynostosis started to be elucidated only in 1993, when Jabs et al. identified a mutation in MSX2 in a patient with Boston-type syndromic craniosynostosis, marking the first gene discovered as causing craniosynostosis (77). Since then, the genetic causes of most syndromic cases have been identified (78, 79). These include mutations in FGFR2 explaining Apert, Crouzon, Pfeiffer, and Jackson-Weiss syndromes; mutations in FGFR3 leading to Muenke syndrome and acanthosis nigricans, in TWIST1, causing Saethre-Chotzen syndrome; mutations in JAG1 leading to Alagille syndrome, ERF, TCF12 causing ERF-related and TCF12-related craniosynostosis, and other mutations associated with Mendelian forms of syndromic craniosynostosis (64, 80–82). Nearly 100 genes have been identified as causative of craniosynostosis (79). Syndromic cases have also been associated with several chromosomal aberrations (78).
Approximately 80% of craniosynostosis cases are non-syndromic and of unknown genetic cause (79, 83, 84). Most children with nonsyndromic craniosynostosis present as sporadic cases, although an estimated 6-8% of families have multiple affected individuals (85). Segregation analyses of multiplex nonsyndromic craniosynostosis families support an autosomal dominant inheritance with 38% penetrance for sagittal nonsyndromic craniosynostosis (86) and 60% penetrance for coronal nonsyndromic craniosynostosis (87), although pedigrees suggestive of autosomal recessive inheritance have also been observed. The etiopathogenesis of nonsyndromic craniosynostosis represents a significant gap in our current knowledge, but genomic technologies have begun to narrow this knowledge gap. In 2012, Justice et al. (88) performed the first GWAS for sagittal nonsyndromic craniosynostosis, using 130 non-Hispanic case-parent trios of European ancestry (88). This study identified novel, strong associations to BMP2 and BBS9 (88). Functional studies using zebrafish demonstrated that rs1884302 in the vicinity of BMP2 acts as an enhancer (89). In 2020, Justice et al. (90) published the second GWAS of nonsyndromic metopic craniosynostosis using 225 non-Hispanic case-parent trios, identifying six GWS SNPs: rs781716 intronic to SPRY3, rs6127972 intronic to BMP7, rs62590971 ~155 kb upstream from TGIF2LX, and rs2522623, rs2573826, and rs2754857 all intronic to PCDH11X (90). A replication study using an independent cohort of 194 unrelated metopic cases and 333 unaffected controls, confirmed rs6127972 mapping nearby a member of the bone morphogenetic protein (BMP) family BMP7. Furthermore, WES studies have identified SMAD6 loss-of-function mutations in 7% of midline nonsyndromic patients (91), mutations in ERF in 5 of 12 nonsyndromic multiplex families (92), and mutations in TCF12 in 10% of unilateral coronal nonsyndromic craniosynostosis and 32% of bicoronal cases (93). More complex presentations are well-characterized by the discovery of a two-loci inheritance resulting in midline craniosynostosis, involving interaction between rare coding variants in SMAD6 and the risk allele of rs1884302 in the vicinity of BMP2, finding that has been replicated (94).
Human fused suture specimens, although important, are a limited resource that does not allow for comprehensive investigations of events leading to suture fusion. Thus, animal models remain critical for investigating the alterations within cranial sutures that precede fusion. Traditionally, mice have been used to study craniofacial development and become fundamental to our current understanding of craniosynostosis. Mouse models have been developed for syndromic forms of craniosynostosis, such as Apert, Crouzon, Muenke, and Saethre–Chotzen syndromes (95, 96), and a limited number for nonsyndromic craniosynostosis (97–100). These models have served as a proof-of-principle for nonsurgical craniosynostosis treatment, as observed from prenatal phenotype rescue of Apert and Crouzon mouse models with small molecules (101, 102) and of midline nonsyndromic craniosynostosis with rapamycin (103). However, these mouse models do not allow visualization of suture formation in vivo and do not serve as rapid screening systems for testing genes in loci identified by GWAS, WGS and/or WES.
Children diagnosed with craniosynostosis are rarely systematically evaluated longitudinally for late-onset morbidities, limiting our knowledge about the late effects mutations in craniosynostosis genes have on our entire skeleton. However, there is evidence that some craniosynostosis genes also regulate whole-body bone homeostasis, such as mutations in LRP5 causing osteoporosis-Pseudoglioma, ALPL causing low serum alkaline phosphatase, and low-BMD, PHEX leading to hypophosphatemia rickets and diminishing bone regenerative capacity (79), etc. Moreover, all known craniosynostosis causative genes are also identified by GWAS for human height (msk.hugeamp.org), suggesting the role of bone growth in the etiology of the condition.
Osteoporosis meets craniosynostosis
In 2014, Kemp et al. (104) published the first GWAS for skull BMD (SK-BMD) in pediatric populations. First, the study estimated the heritability of BMD across skeletal sites. For this, they partitioned total body-BMD into lower-limb, upper-limb, and skull using total-body DXA scans of 4,890 participants recruited by the Avon Longitudinal Study of Parents and their Children (ALSPAC). Interestingly, common SNPs explained a greater proportion of the overall variance of SK-BMD when compared to BMD measured at the appendicular sites. Such difference might reflect the differential exposure of each skeletal site to environmental influences, particularly those acting through the mechanical load, suggesting that skull BMD is less influenced by environmental factors. Next, combining data from ALSPAC with the Generation R Study, they performed a GWAS meta-analysis on 9,395 participants, identifying eight loci associated with SK-BMD, all of which were previously associated with BMD at appendicular regions, suggesting distinct architecture of gene regulation among skeletal regions (104). These regions included TNFR11A1, TNFS11, RSPO3/CENPW, WNT16/CPED1, and WNT4, all genes with essential functions in bone biology and previously associated with osteoporosis. For example, RSPO3 is one of the top genetic determinants for fractures and has been robustly associated with increased BMD (12, 34). Later, functional studies demonstrated RSPO3 expression in osteoblasts, blood cells, adipocytes, and skeletal stem cells (105). RSPO3 expression leads to increased osteoblast proliferation and differentiation. Conditional mice Rspo3 knockouts showed that osteoblasts are the principal source of its expression in bone and a central regulator of trabecular bone mass (105). Lead SNPs in the RSPO3 locus associated with SK-BMD differ from those identified associated at the lumbar spine and femoral neck, pointing to different regulatory elements governing the gene expression across skeletal sites (104). This study suggested that mostly the same genes would be identified from SK-BMD GWASs, but with variations in lead SNPs, and gene regulation.
Recently, we conducted the largest GWAS for SK-BMD (106), including 43,800 participants. By increasing the sample size from the previous work, we identified 59 loci, of which 4 were novel (ZIC1, ATP6V1C1, PRKAR1A, and GLRX3). Indeed, our study demonstrated that most genes associated with SK-BMD also partake in axial skeletal homeostasis, as fifty-five out of fifty-nine SK-BMD loci (93%) co-localized with total-body-BMD loci. RSPO3 locus also displayed independent lead SNP, as previously identified by Kemp et al. (104). We highlighted associated variants mapping to known BMD genes that belong to 1% of most deleterious skeletal variants in the human genome (e.g., LRP5 (107), WNT16 (108), LGR4 (109), CPED1 (27), LRP4 (110)). Association with SOST, which provides the genetic basis of romosozumab, a recently developed osteoporosis medication (61), was also detected. Our skull-BMD study underscored genes with mechanosensing properties, including the novel BMD-associated gene ZIC1, which encodes a zinc-finger transcription factor. A few studies have demonstrated mechano-function properties of ZIC1, including its differential expression in skeletal regions of higher and lower mechanical load, also ZIC1 immunolocalization in the cytoplasm, and translocation to the nucleus of murine osteocytes upon fluid shear and stress experiments (111, 112). While more studies are needed to evaluate ZIC1 function in osteocytes and its function as a mechanosensing gene, its current understanding corroborates the high potential of GWAS for SK-BMD to identify genes with mechanosensing properties.
Additionally, our GWAS for SK-BMD revealed a remarkable overlap between BMD loci and genes associated with craniosynostosis. Fifteen of the 59 SK-BMD loci (25%) harbored craniosynostosis causative genes or family members of causative genes (Figure 1). Among known craniosynostosis causative genes were EN1, IDUA, CSNK1G3, DLX6, SOX6, JAG1, LRP5 (79), and the novel candidate BMD gene ZIC1 (84). Mutations in craniosynostosis genes lead to premature suture fusion frequently due to enhanced osteoblast activity or reduction of the mesenchymal cell pool (64). Because the sutures experience a very fine regulation of osteoblast differentiation, small changes in this process lead to severe craniofacial phenotypes. This implies that the genetic overlap between BMD and craniosynostosis automatically flags the gene’s function partaking in osteogenesis and possibly with osteoanabolic properties. By studying the molecular mechanisms of suture fusion, we may learn shortcuts to decipher a gene’s biological impact on bone homeostasis of other parts of the body. For example, at the sutures, EN1 influences osteoblasts, and it has a non-cell-autonomous function in regulating osteoclast recruitment and activation, affecting the resorption and remodeling, as demonstrated by studies in mice (113). EN1 has been robustly associated with BMD at different skeletal sites and fracture risk (31). Additional functional studies in mice showed En1 expression in osteoblast cells of cortical and of lumbar trabecular bone and in osteocytes of cortical bones. Conditional mice knockout showed lower trabecular bone volume, number, and thickness, reduced femoral cortical thickness and increased osteoclast activity (31). Therefore, this points to similar mechanisms in osteogenesis as in suture fusion. En1 is expressed in the paraxial cephalic mesoderm, forming an important cell population above the mice developing eye that migrate to form the coronal suture (114). Zic1 is expressed in the same cellular domain as En1 (84) and is homologous to the Drosophila gene odd-parred, which is required to activate embryonic engrailed expression (115). The novel BMD gene, ZIC1, may regulate EN1 in bone development and homeostasis. Gain-of-function mutations in ZIC1 cause coronal craniosynostosis and learning disability. In mice, Zic1-/- display cerebellar hypoplasia and vertebral fusions, similar to Gli3 knockout (Gli-Kruppel family member 3), which is highly important for mesenchymal cell patency in the sutures (116, 117). The joint involvement of ZIC1, EN1, and GLI3 during bone formation and homeostasis has yet to be determined.
The association of noncoding variants in the 3’ region of BMP2 resulting in nonsyndromic sagittal craniosynostosis points to another interesting intersection with osteoporosis. Craniosynostosis occurs due to accelerated osteoblast differentiation with increased BMD implying overexpression of BMP2. Since osteoporosis involves opposite processes (i.e., decreased osteoblast differentiation and activity), the mechanisms leading to low BMD would involve downregulation of BMP2. Indeed, Styrkarsdottir et al. (118), reported three BMP2 variants, a missense polymorphism, and two anonymous single nucleotide polymorphism haplotypes, to be associated with osteoporosis in the Icelandic patients (118).
Because the sutures are a source of pluripotent mesenchymal cells with regenerative capacity (119), identified BMD genes that are expressed in the suture mesenchyme and involved in craniosynostosis may be exciting targets as osteoinductive in bone regeneration. This is supported by the Notch ligand JAG1, where mutations in the gene lead to Alagille syndrome and craniosynostosis, and was one of the first genes found associated with BMD and osteoporotic fractures by GWAS in 2010 (23, 82). Studies in mice have shown that Jag1 is expressed in the suture mesenchyme, functioning along Twist1 in maintaining suture patency, and downregulation of Jag1 leads to enhanced osteoblast differentiation (82). Mice studies also showed that Jag1 is required for normal trabecular bone formation, leading to downregulation of osteoblast differentiation genes in trabecular bone and osteopenic phenotype (120). Remarkably, Jag1 has emerged as a potent osteoinductive protein that positively regulates post-traumatic bone healing in the last decades. Intraoperative delivery of Jag1 with collagen sponges at the fractures has rendered exceptional and promising healing results without ectopic bone formation, observed with BMPs (121). This bond opens opportunities to explore the osteoinductive potential of other craniosynostosis genes for therapeutic approaches in fracture healing.
Nevertheless, several questions remain unanswered, such as the primordial question of whether craniosynostosis genes are indeed causal in BMD, or whether skull BMD genes are involved in skull development and/or craniosynostosis. For this, functional studies in animal models that allow a longitudinal analysis of skull formation and skeletal homeostasis will provide valuable insight.
Zebrafish for functional studies of craniosynostosis and osteoporosis genetics
Mice have been the traditional animal models for functional experiments in the osteoporosis and craniosynostosis fields (15, 84, 92, 105). However, zebrafish (Danio rerio) have attracted recent interest in the functional validation of genetic findings from human studies. But not only that, they are becoming preferable models for higher throughput rapid phenotypic and genetic screening, especially for the scrutiny of genes identified by GWAS. Zebrafish are freshwater fish of relatively simple maintenance and low cost, of external fertilization and rapid development, that weekly generates over 200 individuals per cross (122). The zebrafish community has made available an arsenal of genetic tools (i.e., reporter lines, site-directed mutagenesis, reverse and forward genetic screenings, etc.) and phenotypic tools for skeletal studies in zebrafish (123, 124). Despite zebrafish having evolutionarily diverged from humans around 450 million years ago, about 80% of disease-causing genes in humans have an ortholog in zebrafish (125). During evolution, zebrafish passed through additional whole-genome duplication, which resulted in gene duplicates (orthologs to mouse/human genes). In many cases, orthologs functionally balance each other, permitting to phenotypically study of individual orthologs in zebrafish that are embryonic lethal in mouse (126, 127). Also, while mutations in many skeletal genes are lethal in mice, zebrafish carrying mutations in the same genes may survive to adulthood, due to the supportive buoyancy in the aquatic environment (128). The molecular pathways and types of ossification (endochondral and intramembranous) are conserved between zebrafish and humans, with zebrafish displaying osteoblasts, osteoclasts, osteocytes, and chondrocytes (129, 130). For these many reasons, zebrafish have contributed to an increasing number of models for human skeletal diseases over the years (reviewed by Kague et al. (29)).
Adult zebrafish models to study osteoporosis have only been developed recently, alongside studies characterizing skeletal changes during aging (123, 131, 132). Recently, we provided strong support for osteoporosis in aging zebrafish, showing that aged zebrafish spines display increased susceptibility to fractures and have bone quality deterioration (increased cortical porosity and tendency towards reduction of BMD), reminiscent of the osteoporosis (Figures 3A–C) (123). While aging zebrafish studies allow us to learn how to recognize critical hallmarks of skeletal architecture deterioration, varying from micro to nanoscale, under genetic manipulation, young zebrafish provide consistent and compelling models for osteoporosis. Mutant zebrafish are attractive models for functional studies referent to adult skeletal maintenance, with BMD changes easily detected by standard techniques (Figure 3D). This has been demonstrated with several mutants, including zebrafish sp7-/- and lrp5-/- among others (123, 133). By using high-resolution 3D imaging (<0.1um, synchrotron radiation), osteocyte lacunar profile can also be obtained (123, 134).
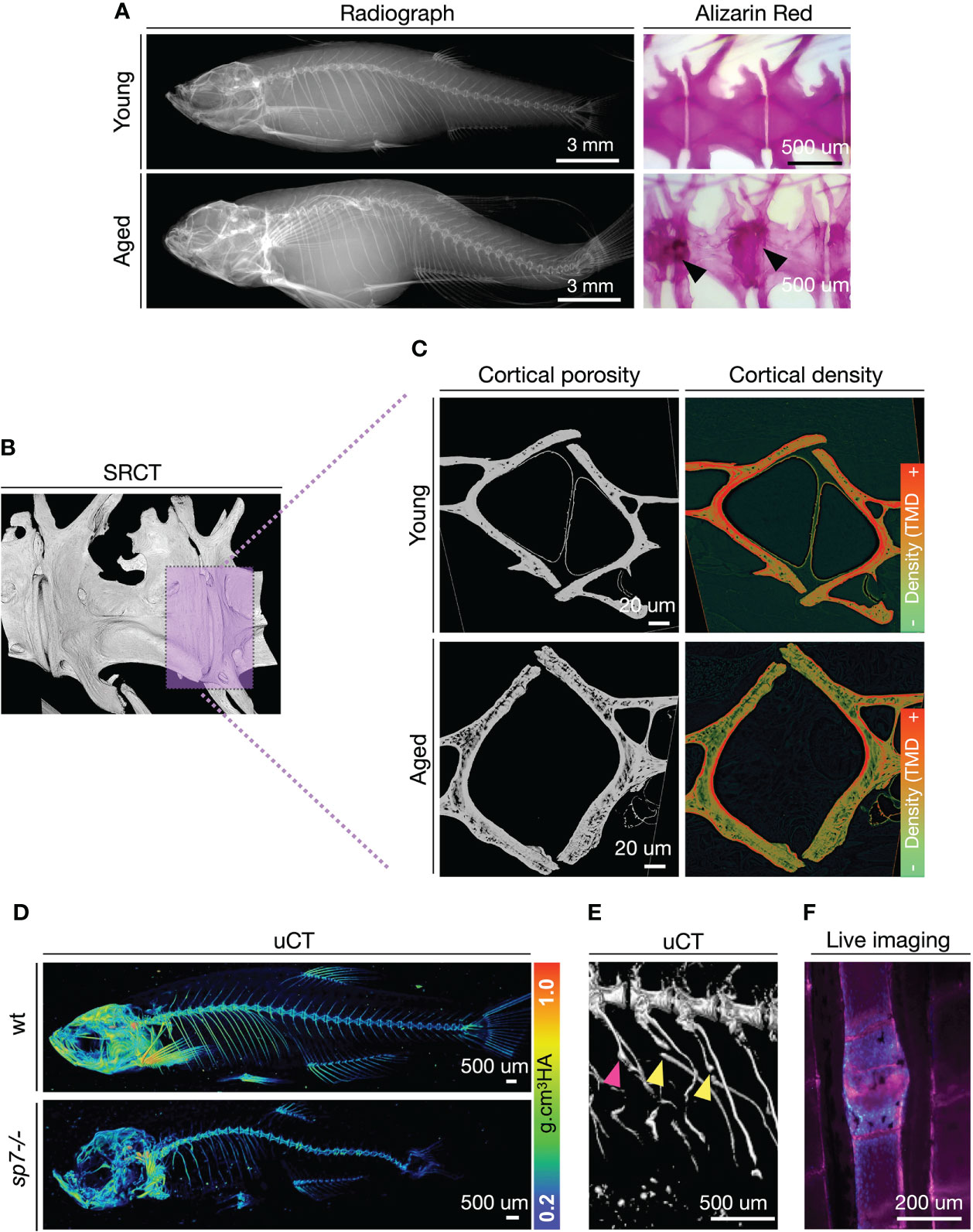
Figure 3 Zebrafish develop bone microarchitecture deterioration during aging, and young zebrafish mutants are used to study osteoporosis-associated genes. (A) Radiographs and Alizarin Red Staining of young (1 year = ~30 human years) and aged (3 years = ~80 human years). Alizarin red staining from Kague et al. (123). Note bone sclerosis in endplates of aged spines (arrowhead). (B) Synchrotron radiation µCT of a region of interest in the zebrafish spine. The magenta box shows a selected region for a digital cross-section. (C) Digital cross-sections of the endplate region of the zebrafish vertebral column to show cortical porosity (greyscale) and density (colored by pixel grey intensity). TMD, Tissue Mineral Density. Regions of higher densities are shown in red, while lower densities are in green. (D) Micro-computed tomography (µCT) of young (3 months old) wild-type (wt) and sp7-/- mutant with values of tissue mineral density (cortical density). µCT images from Kague et al. (123). (E) Fractures are observed in ribs of sp7-/- mutants. Fractures (yellow arrowhead), calluses (magenta arrowhead). (F) Example of a fracture in the zebrafish caudal fin imaged from live fish. Calcified bone was stained with Alizarin Red (magenta) and osteoblasts (blue) are labeled with Tg(sp7:gfp) reporter line. Scale bar is represented in each image.
While tools for the prediction of fractures risk in zebrafish have not been developed yet, genetically modified zebrafish offer alternative ways to study fracture occurrence, prediction, and fracture healing (135, 136). Due to buoyance in the aquatic environment, fish are subjected to a lower mechanical load than terrestrial species. However, swimming behavior through a viscous medium plays an important factor in mechanical resistance to the zebrafish skeleton. This is confirmed by recent findings of vertebral compression fractures in zebrafish modeling osteogenesis imperfecta (137). Also, zebrafish subjected to an exercise regime increase bone formation at the vertebral endplates (138), while those subjected to restricted mobility develop osteopenia (139). Fragility fractures are often observed in ribs and fins (Figures 3E, F) (128, 140). The zebrafish bony fin rays, known by fish experts as lepidotrichia, are regions of interest to monitor bone fractures. Aged zebrafish show increased numbers of spontaneous fin fractures, detected by the presence of calluses (Figure 3F) (141). Calluses are also observed prematurely in young zebrafish carrying a loss-of-function mutation in wnt16 or bmp1a1 (136, 141). Moreover, a fracture can be induced by applying pressure on fish fins, and fracture healing can be monitored longitudinally in vivo.
One of the fascinating advantages of zebrafish is their transparency, which prevails from early development to the formation of the main skeleton during juvenile stages. This permits in vivo cell traceability while the skeleton is formed using reporter lines labeling specific cell types (i.e., osteoblasts, osteoclasts, and chondrocytes) (14, 126). Zebrafish cranial bones and sutures are homologous to those in mammals, representing the only vertebrates enabling non-invasive in vivo visualization of cranial bones growth and suture formation (55, 128), therefore of great interest for models of craniosynostosis (89, 142, 143). Zebrafish develop the same sutures as humans and mice (55, 128) (Figures 4A, B). In 2012, we mapped the contribution of neural crest cells to the whole zebrafish adult skeleton and the skull, taking advantage of the cre-lox system, where cre was expressed in neural crest cells through an enhancer for sox10 and used to permanently label neural crest derivatives (55). We demonstrated that the anterior part of the frontal bones was neural crest-derived and the remaining posterior part, including the coronal suture, was of mesoderm origin (55). Therefore, while the coronal suture in mice is of neural crest-mesoderm origin, our experiments demonstrated that the coronal suture is formed by mesoderm-mesoderm origin in zebrafish.
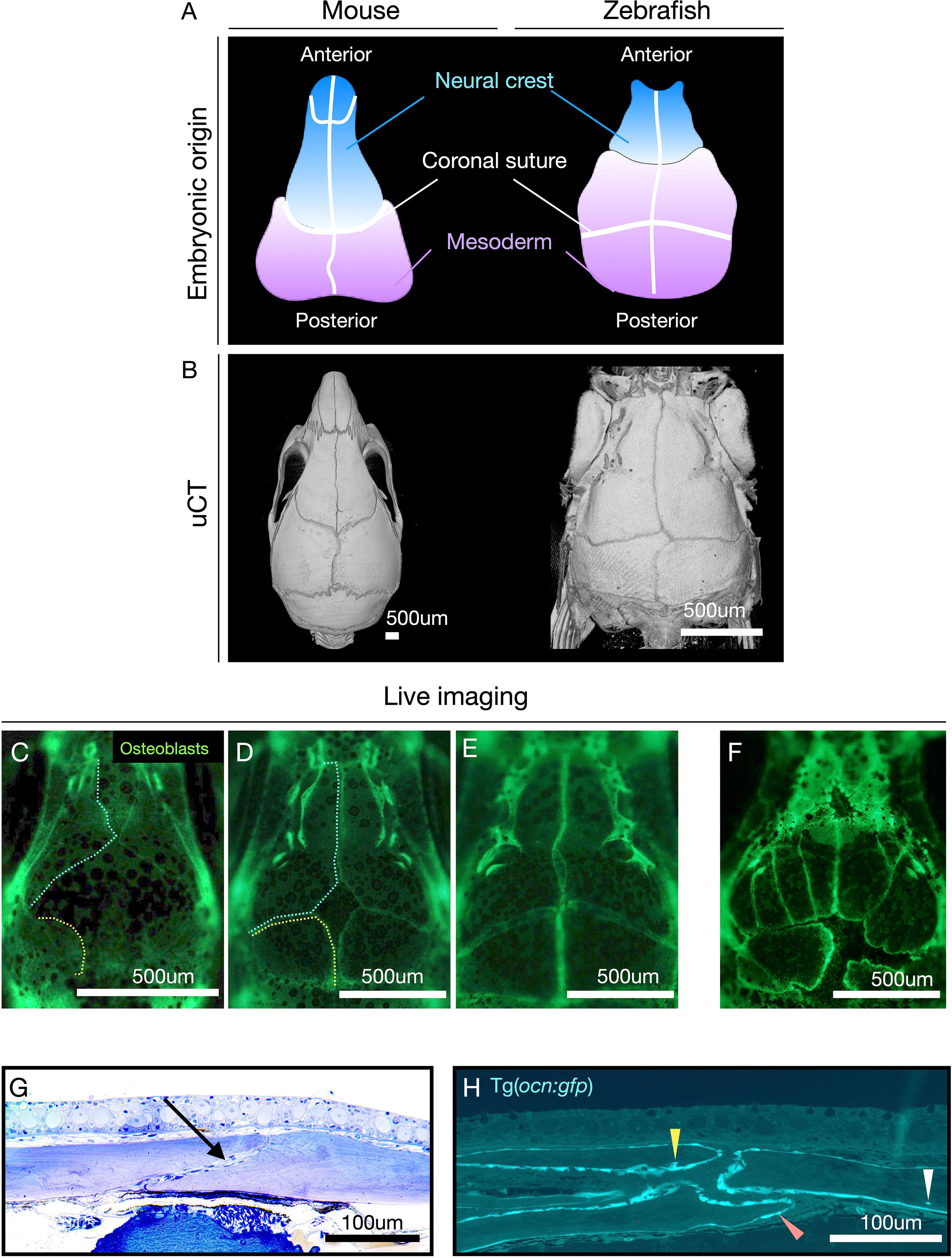
Figure 4 Zebrafish have the same sutures as mammals with the advantage of allowing the visualization of suture formation in vivo. (A) Diagram showing the contribution of neural crest (blue) and mesoderm (lilac) cells to the mouse and zebrafish calvariae (top panel). Note: coronal suture in zebrafish is formed by mesoderm/mesoderm while in mice by neural crest/mesoderm. (B) µCT of calvaria of mice and zebrafish. (C–E) Live imaging of wild-type (wt) zebrafish calvariae during suture formation using Tg(Runx2:egfp) reporter line labeling osteoblasts. Note: osteogenic fronts of the frontal bones (cyan dashed line) and the parietal bones (yellow dashed line). Bone growth is initiated from lateral condensations and progress towards the middle of the calvaria to totally cover the brain. (F) Live imaging of sp7-/- as an example of abnormal suture patterning, and ectopic sutures. (G) Histological semi-thin section of adult male zebrafish, at the coronal suture, stained with Toluidine Blue. Note: in contrast to humans bones overlap at the suture. The coronal suture is indicated with an arrow. (H) Semi-thin plastic section at the sagittal suture of a 3-month-old zebrafish carrying the reporter line Tg(ocn:gfp) labeling osteoblasts and osteocytes (expressing osteocalcin) (according to protocol (144)). Osteoblasts (orange arrowhead), terminal osteoblasts being immersed in bone matrix (yellow arrowhead), osteocyte embedded in bone (white arrowhead). Images (G, H) were kindly provided by the laboratory of Dr. P. Eckhard Witten. Scale bars are indicated in each image.
Additionally, the zebrafish sutures do not fuse during their lifetime, and the two cranial flat bones remain overlapping (Figures 4A, G, H), highlighting a difference from humans (128). However, these differences are insufficient to prevent craniosynostosis in zebrafish genetic models. Craniosynostosis was first modeled in zebrafish through loss-of-function mutations in cyp26b1, reproducing coronal craniosynostosis observed in human patients (143). Later, Teng et al. (142) demonstrated that bi-coronal synostosis could be reproduced in zebrafish when twist1b and tcf12 were simultaneously knockout, similarly to results in mice knockouts (142). The group also showed that mutations in these genes led to the reduction of osteoprogenitor cells at the suture mesenchyme culminating in the coronal synostosis (142).
Advantageously, given the transparency of zebrafish and the superficial position of the cranial bones, the sutures can be studied longitudinally as they are formed (Figures 4C–F). We were the first to show in vivo formation of the calvaria and the formation of ectopic sutures in zebrafish lacking the important osteogenesis gene sp7 (Figure 4F) (128). By using transgenic lines labeling osteoblasts, it is simple to detect the osteogenic fronts of the sutures, sites of dynamic bone growth, and renewal (Figures 4C–F, H). Genes associated with maintenance of suture pattern when mutated in zebrafish often result in the formation of ectopic sutures, as seen in sp7 (128), twist1a, twist1b (142), fgfr3 (145), and bmp7 (unpublished data). Another peculiarity of mutations in craniosynostosis genes in zebrafish is their association with morphological changes in suture position and differences in calvariae bone growth rates (142, 145). The ossification centers, formed laterally in the skull next to the eyes, as in mice, are essential regions of mesenchymal condensation and initial bone formation. The genes associated with these steps in osteogenesis timely regulate bone condensation and growth rate. Delays during condensation lead to fontanelles, ectopic bones, and changes in suture pattern (Figure 4F) (128). These phenotypic changes serve as hallmarks indicating a gene’s function in suture homeostasis and bone growth. Therefore, they can be easily detected during phenotypic screenings.
Remarkably, zebrafish models have become of great interest to pinpoint causal genes in GWAS loci. The main reason for its suitability is the unbeatable speed to test individual or multiple genes in zebrafish. The CRISPR/Cas9 technology efficiently operates in zebrafish, permitting a reliable recapitulation of complex mutant phenotypes in the first generation (mosaic G0s, commonly known as crispants), as demonstrated for genes associated with osteogenesis imperfecta and osteoporosis (124, 133). Crispants involved with osteogenesis imperfecta (PLOD2) and osteoporosis (LRP5) were able to reproduce similar phenotypic characteristics, such as low BMD, like those of knockout mutants (124, 133). Zebrafish crispants represent powerful tools for rapid functional annotation of GWAS-identified variants, able to add a new layer for gene prioritization based on animal findings. Altogether, among the advantages that zebrafish models can offer to the functional evaluation of genetic associations, they represent the ultimate optimum to study the genetic overlap between osteoporosis and craniosynostosis.
Our SK-BMD is the first study that has combined adult zebrafish to provide functional support for GWAS findings in BMD (106). Taking advantage of crispants and the transparency of zebrafish, we tested selected genes harbored in novel identified BMD loci, ZIC1, ATP6V1C1, and PRKAR1A. These genes were targeted by CRISPR/Cas9 to cause loss-of-function mutations and analyzed longitudinally under a fluorescent microscope to visualize cranial suture formation in vivo. Invariably, we detected suture mis-patterning, ectopic sutures, and abnormal bone growth associated with zic1 crispants. Similar results were found for atp6v1c1; while for prkar1a, only abnormal calvarial bone growth was detected. These results suggest that ATP6V1C1 is a BMD-associated gene of potential interest as causative of craniosynostosis and should be further investigated in unresolved forms of the condition. Moreover, micro-computed tomography revealed BMD changes supporting the role of these three genes in bone homeostasis and control of bone mass. These results constitute an excellent starting point, exemplifying the great potential that SK-BMD GWAS and functional validation in zebrafish will bring to the fields of osteoporosis and craniosynostosis.
Future perspectives
The genetic overlap between osteoporosis and craniosynostosis only became apparent recently through the GWAS for SK-BMD. However, this intersection actually dates back to the first successful GWAS in 2008 reporting variants mapping to LRP5 (21), then in 2010 JAG1 was reported (23), and more recently, the discovery of EN1 in 2015 (31) (Figure 1). Whether this overlap is merely a coincidence is unlikely. Some craniosynostosis genes are essential for the critical balance between bone formation and resorption, a process that is mirrored by measurements of BMD (64, 79). One can conclude that the pool of genes involved in overall bone function is very similar across distinct skeletal locations and despite different embryonic origin (104). GWAS on SK-BMD have confirmed that genes involved in bone homeostasis of the skull are indeed largely the same as those regulating the appendicular skeleton, including genes identified using more distant phenotypes like from the heel GWAS (12, 15). This way, genes associated with craniosynostosis have also been associated with fracture risk and trabecular bone maintenance, as is the case for EN1 (31). Despite the large similarity among the genes contributing to BMD of the skull and of BMD measured at other skeletal sites, SK-BMD studies illustrate distinctive regulatory mechanisms in the same genes and associated genetic networks (104, 106). Deciphering these regulatory mechanisms could help us understand (among other mechanisms) temporospatial shifts in gene expression among distinct skeletal sites and types of ossification.
Further, the prioritization of BMD genes associated with craniosynostosis for functional studies may highlight genes with osteoinductive and osteoanabolic properties. It will be essential to prove that identified craniosynostosis genes and family members are causal genes in BMD and understand why not all craniosynostosis genes are being identified through BMD-GWAS and whether they would belong to a different class of “bone-active” genes. Additionally, it will be interesting to validate the osteoinductive potential of craniosynostosis genes for the treatment of fracture healing and regeneration, especially for genes already known to act in the suture mesenchymal cell pool.
From a clinical perspective, the intersection between BMD-GWAS and craniosynostosis also alludes to the possibility of identifying new craniosynostosis genes, that could underlie the presentation of non-syndromic cases in which 80% of the genetic etiology remains unknown. To date, craniosynostosis GWASs are limited by sample size (typically in the hundreds). This is in contrast to the large numbers achieved by the SK-BMD GWAS, (with more than 50K samples and a proportional increase in the yield of identified loci). We advocate that clinicians in the field of craniosynostosis draw close attention to searching mutations in SK-BMD genes among patients whose genetic causes remain unsolved.
In conclusion, zebrafish now constitute the preferred model organism to validate genes overlapping craniosynostosis and osteoporosis, helping to elucidate the genetic overlap between the conditions. Such pleiotropy studies hold the potential to enhance our knowledge about the underlying biology of diverse skeletal conditions and potentially identify therapeutic targets. A glimpse into the near future depicts a considerable increase in the incorporation of zebrafish models for identifying causal genes and providing in-depth mechanistic evaluations with multiple translational opportunities to improve the care of patients with skeletal conditions. Similarly, researchers in the zebrafish field will incorporate more comprehensive genomic evaluations and innovative artificial intelligence approaches to speed phenotypic analyses. Last but not least, the implementation of cohesive platforms to access functional data from studies performed on zebrafish and standardization of methodologies to assess bone phenotypes in zebrafish will become a reality through multidisciplinary collaboration.
Author contributions
EK: Conceptualization, drafting, writing, visualization, and editing. All authors have read and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
EK was supported by Versus Arthritis, grant #21937. SB: National Institute of Health-NIDCR grants R01 DE-16886 and R03 DE-031061.
Acknowledgments
We acknowledge Dr. P. Eckhard Witten (University of Gent, Belgium) and his laboratory for providing images for Figure 4. We thank Dr John Kemp for proofreading the manuscript and for sharing critics. EK Thank Dr. Chrissy Hammond for her support. This article is based upon work from COST Action GEMSTONE CA18139 supported by COST (European Cooperation in Science and Technology) www.cost.eu.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BMD, Bone mineral density; BMP, bone morphogenic protein; DXA, dual-energy X-ray absorptiometry; MAF, minor allele frequency; GEFOS, Genetic Factors for Osteoporosis; GOF, Gain-of-function; GWAS, Genome-Wide Association Studies; IMPC, International Mouse Phenotyping Consortium; IKMC, International Knockout Mouse Consortium; OBCD, Origins of Bone and Cartilage Diseases; SK-BMD, skull bone mineral density; WES, Whole-exome sequencing; WGS, Whole-genome sequencing; WHO, World Health Organization.
References
1. Clynes MA, Harvey NC, Curtis EM, Fuggle NR, Dennison EM, Cooper C. The epidemiology of osteoporosis. Br Med Bull (2020) 133:105–17. doi: 10.1093/bmb/ldaa005
2. Lindsay R, Silverman SL, Cooper C, Hanley DA, Barton I, Broy SB, et al. Risk of new vertebral fracture in the year following a fracture. JAMA (2001) 285:320–3. doi: 10.1001/jama.285.3.320
3. Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res (2007) 22:465–75. doi: 10.1359/jbmr.061113
4. Krall EA, Dawson-Hughes B. Heritable and life-style determinants of bone mineral density. J Bone Miner Res (1993) 8:1–9. doi: 10.1002/jbmr.5650080102
5. Slemenda CW, Turner CH, Peacock M, Christian JC, Sorbel J, Hui SL, et al. The genetics of proximal femur geometry, distribution of bone mass and bone mineral density. Osteoporos Int (1996) 6:178–82. doi: 10.1007/BF01623944
6. Arden NK, Baker J, Hogg C, Baan K, Spector TD. The heritability of bone mineral density, ultrasound of the calcaneus and hip axis length: a study of postmenopausal twins. J Bone Miner Res (1996) 11:530–4. doi: 10.1002/jbmr.5650110414
7. Koller DL, Liu G, Econs MJ, Hui SL, Morin PA, Joslyn G, et al. Genome screen for quantitative trait loci underlying normal variation in femoral structure. J Bone Miner Res (2001) 16:985–91. doi: 10.1359/jbmr.2001.16.6.985
8. Flicker L, Faulkner KG, Hopper JL, Green RM, Kaymacki B, Nowson CA, et al. Determinants of hip axis length in women aged 10-89 years: a twin study. Bone (1996) 18:41–5. doi: 10.1016/8756-3282(95)00418-1
9. Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, Richards JB. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat Rev Genet (2018) 19:110–24. doi: 10.1038/nrg.2017.101
10. Zhu X, Zheng H. Factors influencing peak bone mass gain. Front Med (2021) 15:53–69. doi: 10.1007/s11684-020-0748-y
11. Medina-Gomez C, Kemp JP, Dimou NL, Kreiner E, Chesi A, Zemel BS, et al. Bivariate genome-wide association meta-analysis of pediatric musculoskeletal traits reveals pleiotropic effects at the SREBF1/TOM1L2 locus. Nat Commun (2017) 8:121. doi: 10.1038/s41467-017-00108-3
12. Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet (2019) 51:258–66. doi: 10.1038/s41588-018-0302-x
13. Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet (2015) 47:856–60. doi: 10.1038/ng.3314
14. Rauner M, Foessl I, Formosa MM, Kague E, Prijatelj V, Alonso Lopez N, et al. Perspective of the GEMSTONE Consortium on Current and Future Approaches to Functional Validation for Skeletal Genetic Disease Using Cellular, Molecular and Animal-Modeling Techniques. Front Endocrinol (Lausanne) (2021) 12:731217. doi: 10.3389/fendo.2021.731217
15. Kemp JP, Morris JA, Medina-Gomez C, Forgetta V, Warrington NM, Youlten SE, et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat Genet (2017) 49:1468–75. doi: 10.1038/ng.3949
16. Albagha OM, Wani SE, Visconti MR, Alonso N, Goodman K, Brandi ML, et al. Genetic Determinants of Paget's Disease, Genome-wide association identifies three new susceptibility loci for Paget's disease of bone. Nat Genet (2011) 43:685–9. doi: 10.1038/ng.845
17. Styrkarsdottir U, Thorleifsson G, Eiriksdottir B, Gudjonsson SA, Ingvarsson T, Center JR, et al. Two Rare Mutations in the COL1A2 Gene Associate With Low Bone Mineral Density and Fractures in Iceland. J Bone Miner Res (2016) 31:173–9. doi: 10.1002/jbmr.2604
18. Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. Am J Med Genet A 146A (2008), 984–91. doi: 10.1002/ajmg.a.32208
19. Kiel DP, Demissie S, Dupuis J, Lunetta KL, Murabito JM, Karasik D. Genome-wide association with bone mass and geometry in the Framingham Heart Study. BMC Med Genet (2007) 8 Suppl 1:S14. doi: 10.1186/1471-2350-8-S1-S14
20. Richards JB, Rivadeneira F, Inouye M, Pastinen TM, Soranzo N, Wilson SG, et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet (2008) 371:1505–12. doi: 10.1016/S0140-6736(08)60599-1
21. Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, et al. Multiple genetic loci for bone mineral density and fractures. N Engl J Med (2008) 358:2355–65. doi: 10.1056/NEJMoa0801197
22. Rivadeneira F, Styrkarsdottir U, Estrada K, Halldorsson BV, Hsu YH, Richards JB, et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet (2009) 41:1199–206. doi: 10.1038/ng.446
23. Kung AW, Xiao SM, Cherny S, Li GH, Gao Y, Tso G, et al. Association of JAG1 with bone mineral density and osteoporotic fractures: a genome-wide association study and follow-up replication studies. Am J Hum Genet (2010) 86:229–39. doi: 10.1016/j.ajhg.2009.12.014
24. Yang TL, Chen XD, Guo Y, Lei SF, Wang JT, Zhou Q, et al. Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis. Am J Hum Genet (2008) 83:663–74. doi: 10.1016/j.ajhg.2008.10.006
25. Guo Y, Tan LJ, Lei SF, Yang TL, Chen XD, Zhang F, et al. Genome-wide association study identifies ALDH7A1 as a novel susceptibility gene for osteoporosis. PloS Genet (2010) 6:e1000806. doi: 10.1371/journal.pgen.1000806
26. Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet (2012) 44:491–501. doi: 10.1038/ng.2249
27. Medina-Gomez C, Kemp JP, Estrada K, Eriksson J, Liu J, Reppe S, et al. Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PloS Genet (2012) 8:e1002718. doi: 10.1371/journal.pgen.1002718
28. Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PloS Genet (2012) 8:e1002745. doi: 10.1371/journal.pgen.1002745
29. Kague E, Karasik D. Functional Validation of Osteoporosis Genetic Findings Using Small Fish Models. Genes (Basel) (2022) 13(2):279. doi: 10.3390/genes13020279
30. Styrkarsdottir U, Thorleifsson G, Sulem P, Gudbjartsson DF, Sigurdsson A, Jonasdottir A, et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature (2013) 497:517–20. doi: 10.1038/nature12124
31. Zheng HF, Forgetta V, Hsu YH, Estrada K, Rosello-Diez A, Leo PJ, et al. Whole-genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature (2015) 526:112–7. doi: 10.1038/nature14878
32. Consortium UK, Walter K, Min JL, Huang J, Crooks L, Memari Y, et al. The UK10K project identifies rare variants in health and disease. Nature (2015) 526:82–90. doi: 10.1038/nature14962
33. Backman JD, Li AH, Marcketta A, Sun D, Mbatchou J, Kessler MD, et al. and XXXM. A.R. Ferreira Exome sequencing Anal 454,787 UK Biobank participants. Nat (2021) 599:628–34. doi: 10.1038/s41586-021-04103-z
34. Trajanoska K, Morris JA, Oei L, Zheng HF, Evans DM, Kiel DP, et al. G.G. consortium, and t. the 23andMe research, Assessment of the genetic and clinical determinants of fracture risk: genome wide association and mendelian randomisation study. BMJ (2018) 362:k3225.
35. Timpson NJ, Tobias JH, Richards JB, Soranzo N, Duncan EL, Sims AM, et al. Common variants in the region around Osterix are associated with bone mineral density and growth in childhood. Hum Mol Genet (2009) 18:1510–7. doi: 10.1093/hmg/ddp052
36. Duncan EL, Danoy P, Kemp JP, Leo PJ, McCloskey E, Nicholson GC, et al. Genome-wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PloS Genet (2011) 7:e1001372. doi: 10.1371/journal.pgen.1001372
37. Paternoster L, Lorentzon M, Lehtimaki T, Eriksson J, Kahonen M, Raitakari O, et al. Genetic determinants of trabecular and cortical volumetric bone mineral densities and bone microstructure. PloS Genet (2013) 9:e1003247. doi: 10.1371/journal.pgen.1003247
38. Paternoster L, Lorentzon M, Vandenput L, Karlsson MK, Ljunggren O, Kindmark A, et al. Genome-wide association meta-analysis of cortical bone mineral density unravels allelic heterogeneity at the RANKL locus and potential pleiotropic effects on bone. PloS Genet (2010) 6:e1001217. doi: 10.1371/journal.pgen.1001217
39. Styrkarsdottir U, Stefansson OA, Gunnarsdottir K, Thorleifsson G, Lund SH, Stefansdottir L, et al. GWAS of bone size yields twelve loci that also affect height, BMD, osteoarthritis or fractures. Nat Commun (2019) 10:2054. doi: 10.1038/s41467-019-09860-0
40. Boer CG, Hatzikotoulas K, Southam L, Stefansdottir L, Zhang Y, Coutinho de Almeida R, et al. Zeggini, Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell (2021) 184:4784–4818 e17. doi: 10.1016/j.cell.2021.11.003
41. Tachmazidou I, Hatzikotoulas K, Southam L, Esparza-Gordillo J, Haberland V, Zheng J, et al. Zeggini, Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat Genet (2019) 51:230–6. doi: 10.1038/s41588-018-0327-1
42. Gregson CL, Newell F, Leo PJ, Clark GR, Paternoster L, Marshall M, et al. Genome-wide association study of extreme high bone mass: Contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Bone (2018) 114:62–71. doi: 10.1016/j.bone.2018.06.001
43. Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget's disease of bone. Nat Genet (2010) 42:520–4. doi: 10.1038/ng.562
44. Xue A, Wu Y, Zhu Z, Zhang F, Kemper KE, Zheng Z, et al. Yang, Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun (2018) 9:2941. doi: 10.1038/s41467-018-04951-w
45. Lambert KL. The weight-bearing function of the fibula. A strain gauge study. J Bone Joint Surg Am (1971) 53:507–13. doi: 10.2106/00004623-197153030-00007
46. Hillam RA, Goodship AE, Skerry TM. Peak strain magnitudes and rates in the tibia exceed greatly those in the skull: An in vivo study in a human subject. J Biomech (2015) 48:3292–8. doi: 10.1016/j.jbiomech.2015.06.021
47. Himeno-Ando A, Izumi Y, Yamaguchi A, Iimura T. Structural differences in the osteocyte network between the calvaria and long bone revealed by three-dimensional fluorescence morphometry, possibly reflecting distinct mechano-adaptations and sensitivities. Biochem Biophys Res Commun (2012) 417:765–70. doi: 10.1016/j.bbrc.2011.12.031
48. Rawlinson SC, McKay IJ, Ghuman M, Wellmann C, Ryan P, Prajaneh S, et al. Adult rat bones maintain distinct regionalized expression of markers associated with their development. PloS One (2009) 4:e8358. doi: 10.1371/journal.pone.0008358
49. Gallagher JC, Goldgar D, Moy A. Total bone calcium in normal women: effect of age and menopause status. J Bone Miner Res (1987) 2:491–6. doi: 10.1002/jbmr.5650020605
50. Vico L, Alexandre C. [Adaptation of the skeleton to microgravity]. Rev Rhum Ed Fr (1994) 61:853–7.
51. Leblanc AD, Schneider VS, Evans HJ, Engelbretson DA, Krebs JM. Bone mineral loss and recovery after 17 weeks of bed rest. J Bone Miner Res (1990) 5:843–50. doi: 10.1002/jbmr.5650050807
52. Garland DE, Stewart CA, Adkins RH, Hu SS, Rosen C, Liotta FJ, et al. Osteoporosis after spinal cord injury. J Orthop Res (1992) 10:371–8. doi: 10.1002/jor.1100100309
53. Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol (2002) 241:106–16. doi: 10.1006/dbio.2001.0487
54. Martik ML, Bronner ME. Riding the crest to get a head: neural crest evolution in vertebrates. Nat Rev Neurosci (2021) 22:616–26. doi: 10.1038/s41583-021-00503-2
55. Kague E, Gallagher M, Burke S, Parsons M, Franz-Odendaal T, Fisher S. Skeletogenic fate of zebrafish cranial and trunk neural crest. PloS One (2012) 7:e47394. doi: 10.1371/journal.pone.0047394
56. Evans DJ, Noden DM. Spatial relations between avian craniofacial neural crest and paraxial mesoderm cells. Dev Dyn (2006) 235:1310–25. doi: 10.1002/dvdy.20663
57. Franz-Odendaal TA. Induction and patterning of intramembranous bone. Front Biosci (Landmark Ed) (2011) 16:2734–46. doi: 10.2741/3882
58. Van Hul W, Balemans W, Van Hul E, Dikkers FG, Obee H, Stokroos RJ, et al. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am J Hum Genet (1998) 62:391–9. doi: 10.1086/301721
59. Gregson CL, Duncan EL. The Genetic Architecture of High Bone Mass. Front Endocrinol (Lausanne) (2020) 11:595653. doi: 10.3389/fendo.2020.595653
60. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med (2013) 19:179–92. doi: 10.1038/nm.3074
61. Kobayakawa T, Miyazaki A, Saito M, Suzuki T, Takahashi J, Nakamura Y. Denosumab versus romosozumab for postmenopausal osteoporosis treatment. Sci Rep (2021) 11:11801. doi: 10.1038/s41598-021-91248-6
62. McClung MR, O'Donoghue ML, Papapoulos SE, Bone H, Langdahl B, Saag KG, et al. Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol (2019) 7:899–911. doi: 10.1016/S2213-8587(19)30346-8
63. Ko JM. Genetic Syndromes Associated with Craniosynostosis. J Korean Neurosurg Soc (2016) 59:187–91. doi: 10.3340/jkns.2016.59.3.187
64. Lattanzi W, Barba M, Di Pietro L, Boyadjiev SA. Genetic advances in craniosynostosis. Am J Med Genet A (2017) 173:1406–29. doi: 10.1002/ajmg.a.38159
65. Lattanzi W, Parolisi R, Barba M, Bonfanti L. Osteogenic and Neurogenic Stem Cells in Their Own Place: Unraveling Differences and Similarities Between Niches. Front Cell Neurosci (2015) 9:455. doi: 10.3389/fncel.2015.00455
66. Thompson DN, Harkness W, Jones B, Gonsalez S, Andar U, Hayward R. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst (1995) 11:269–75. doi: 10.1007/BF00301758
67. Tamburrini G, Caldarelli M, Massimi L, Santini P, Di Rocco C. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Childs Nerv Syst (2005) 21:913–21. doi: 10.1007/s00381-004-1117-x
68. Gupta PC, Foster J, Crowe S, Papay FA, Luciano M, Traboulsi EI. Ophthalmologic findings in patients with nonsyndromic plagiocephaly. J Craniofac Surg (2003) 14:529–32. doi: 10.1097/00001665-200307000-00026
69. Shipster C, Hearst D, Somerville A, Stackhouse J, Hayward R, Wade A. Speech, language, and cognitive development in children with isolated sagittal synostosis. Dev Med Child Neurol (2003) 45:34–43. doi: 10.1111/j.1469-8749.2003.tb00857.x
70. Kapp-Simon KA. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac J (1998) 35:197–203. doi: 10.1597/1545-1569_1998_035_0197_mdaldi_2.3.co_2
71. Altarac M, Saroha E. Lifetime prevalence of learning disability among US children. Pediatrics (2007) 119 Suppl 1:S77–83. doi: 10.1542/peds.2006-2089L
72. Foster KA, Frim DM, McKinnon M. Recurrence of synostosis following surgical repair of craniosynostosis. Plast Reconstr Surg (2008) 121:70e–6e. doi: 10.1097/01.prs.0000299393.36063.de
73. Natghian H, Song M, Jayamohan J, Johnson D, Magdum S, Richards P, et al. Long-Term Results in Isolated Metopic Synostosis: The Oxford Experience over 22 Years. Plast Reconstr Surg (2018) 142:509e–15e. doi: 10.1097/PRS.0000000000004768
74. Salokorpi N, Savolainen T, Sinikumpu JJ, Ylikontiola L, Sandor GK, Pirttiniemi P, et al. Outcomes of 40 Nonsyndromic Sagittal Craniosynostosis Patients as Adults: A Case-Control Study With 26 Years of Postoperative Follow-up. Oper Neurosurg (Hagerstown) (2018) 16(1):1–8. doi: 10.1093/ons/opy047
75. Bellew M, Chumas P. Long-term developmental follow-up in children with nonsyndromic craniosynostosis. J Neurosurg Pediatr (2015) 16:445–51. doi: 10.3171/2015.3.PEDS14567
76. Speltz ML, Collett BR, Wallace ER, Starr JR, Cradock MM, Buono L, et al. Intellectual and academic functioning of school-age children with single-suture craniosynostosis. Pediatrics (2015) 135:e615–23. doi: 10.1542/peds.2014-1634
77. Jabs EW, Muller U, Li X, Ma L, Luo W, Haworth IS, et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell (1993) 75:443–50. doi: 10.1016/0092-8674(93)90379-5
78. Passos-Bueno MR, Sertie AL, Jehee FS, Fanganiello R, Yeh E. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations Front Oral Biol (2008) 12:107–43. doi: 10.1159/000115035
79. Goos JAC, Mathijssen IMJ. Genetic Causes of Craniosynostosis: An Update. Mol Syndromol (2019) 10:6–23. doi: 10.1159/000492266
80. Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr (2017) 29:622–8. doi: 10.1097/MOP.0000000000000542
81. Twigg SR, Wilkie AO. A Genetic-Pathophysiological Framework for Craniosynostosis. Am J Hum Genet (2015) 97:359–77. doi: 10.1016/j.ajhg.2015.07.006
82. Yen HY, Ting MC, Maxson RE. Jagged1 functions downstream of Twist1 in the specification of the coronal suture and the formation of a boundary between osteogenic and non-osteogenic cells. Dev Biol (2010) 347:258–70. doi: 10.1016/j.ydbio.2010.08.010
83. Sasaki E, Byrne AT, Murray DJ, Reardon W. Caput membranaceum: A novel clinical presentation of ZIC1 related skull malformation and craniosynostosis. Am J Med Genet A (2020) 182:2994–8. doi: 10.1002/ajmg.a.61882
84. Twigg SR, Forecki J, Goos JA, Richardson IC, Hoogeboom AJ, Ouweland AMvd, et al. Gain-of-Function Mutations in ZIC1 Are Associated with Coronal Craniosynostosis and Learning Disability. Am J Hum Genet (2015) 97:378–88. doi: 10.1016/j.ajhg.2015.07.007
85. Boyadjiev SA, International Craniosynostosis C. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res (2007) 10:129–37. doi: 10.1111/j.1601-6343.2007.00393.x
86. Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of scaphocephaly. Am J Med Genet (1996) 62:282–5. doi: 10.1002/(SICI)1096-8628(19960329)62:3<282::AID-AJMG15>3.0.CO;2-G
87. Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet (1995) 55:500–4. doi: 10.1002/ajmg.1320550422
88. Justice CM, Yagnik G, Kim Y, Peter I, Jabs EW, Erazo M, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet (2012) 44:1360–4. doi: 10.1038/ng.2463
89. Justice CM, Kim J, Kim SD, Kim K, Yagnik G, Cuellar A, et al. A variant associated with sagittal nonsyndromic craniosynostosis alters the regulatory function of a non-coding element. Am J Med Genet A (2017) 173:2893–7. doi: 10.1002/ajmg.a.38392
90. Justice CM, Cuellar A, Bala K, Sabourin JA, Cunningham ML, Crawford K, et al. A genome-wide association study implicates the BMP7 locus as a risk factor for nonsyndromic metopic craniosynostosis. Hum Genet (2020) 139:1077–90. doi: 10.1007/s00439-020-02157-z
91. Timberlake AT, Furey CG, Choi J, Nelson-Williams C, Yale Center for Genome A, Loring E, et al. De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis. Proc Natl Acad Sci U.S.A. (2017) 114:E7341-E7347. doi: 10.1073/pnas.1709255114
92. Twigg SR, Vorgia E, McGowan SJ, Peraki I, Fenwick AL, Sharma VP, et al. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet (2013) 45:308–13. doi: 10.1038/ng.2539
93. Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JA, Hoogeboom AJ, et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet (2013) 45:304–7. doi: 10.1038/ng.2531
94. Calpena E, Cuellar A, Bala K, Swagemakers SMA, Koelling N, McGowan SJ, et al. SMAD6 variants in craniosynostosis: genotype and phenotype evaluation. Genet Med (2020) 22:1498–506. doi: 10.1038/s41436-020-0817-2
95. Holmes G. The role of vertebrate models in understanding craniosynostosis. Childs Nerv Syst (2012) 28:1471–81. doi: 10.1007/s00381-012-1844-3
96. Carver EA, Oram KF, Gridley T. Craniosynostosis in Twist heterozygous mice: a model for Saethre-Chotzen syndrome. Anat Rec (2002) 268:90–2. doi: 10.1002/ar.10124
97. Yu HM, Jerchow B, Sheu TJ, Liu B, Costantini F, Puzas JE, et al. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development (2005) 132:1995–2005. doi: 10.1242/dev.01786
98. Zhang X, Kuroda S, Carpenter D, Nishimura I, Soo C, Moats R, et al. Craniosynostosis in transgenic mice overexpressing Nell-1. J Clin Invest (2002) 110:861–70. doi: 10.1172/JCI15375
99. Komatsu Y, Yu PB, Kamiya N, Pan H, Fukuda T, Scott GJ, et al. Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J Bone Miner Res (2013) 28:1422–33. doi: 10.1002/jbmr.1857
100. Ting MC, Wu NL, Roybal PG, Sun J, Liu L, Yen Y, et al. EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development (2009) 136:855–64. doi: 10.1242/dev.028605
101. Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet (2007) 39:1145–50. doi: 10.1038/ng2096
102. Eswarakumar VP, Ozcan F, Lew ED, Bae JH, Tome F, Booth CJ, et al. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci U.S.A. (2006) 103:18603–8.
103. Kramer K, Yang J, Swanson WB, Hayano S, Toda M, Pan H, et al. Rapamycin rescues BMP mediated midline craniosynostosis phenotype through reduction of mTOR signaling in a mouse model. Genesis (2018) 56:e23220. doi: 10.1002/dvg.23220
104. Kemp JP, Medina-Gomez C, Estrada K, St Pourcain B, Heppe DH, Warrington NM, et al. Phenotypic dissection of bone mineral density reveals skeletal site specificity and facilitates the identification of novel loci in the genetic regulation of bone mass attainment. PloS Genet (2014) 10:e1004423. doi: 10.1371/journal.pgen.1004423
105. Nilsson KH, Henning P, El Shahawy M, Nethander M, Andersen TL, Ejersted C, et al. RSPO3 is important for trabecular bone and fracture risk in mice and humans. Nat Commun (2021) 12:4923. doi: 10.1038/s41467-021-25124-2
106. Carolina Medina-Gomez VP, Mullin BH, Chesi A, Kemp JP, Shochat-Carvalho C, Trajanoska P, et al. Genome Wide Association Metanalysis Of Skull Bone Mineral Density Identifies Loci Relevant For Osteoporosis And Craniosynostosis. MedRxiv (2021). doi: 10.1101/2021.11.01.21265592
107. Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, et al. Lrp5 functions in bone to regulate bone mass. Nat Med (2011) 17:684–91. doi: 10.1038/nm.2388
108. Gori F, Lerner U, Ohlsson C, Baron R. A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. Bonekey Rep (2015) 4:669. doi: 10.1038/bonekey.2015.36
109. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med (2016) 22:539–46. doi: 10.1038/nm.4076
110. Xiong L, Jung JU, Wu H, Xia WF, Pan JX, Shen C, et al. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc Natl Acad Sci U.S.A. (2015) 112:3487–92.
111. Kalogeropoulos M, Varanasi SS, Olstad OK, Sanderson P, Gautvik VT, Reppe S, et al. Zic1 transcription factor in bone: neural developmental protein regulates mechanotransduction in osteocytes. FASEB J (2010) 24:2893–903. doi: 10.1096/fj.09-148908
112. Datta HK, Kringen MK, Tuck SP, Salpingidou G, Olstad OK, Gautvik KM, et al. Mechanical-Stress-Related Epigenetic Regulation of ZIC1 Transcription Factor in the Etiology of Postmenopausal Osteoporosis. Int J Mol Sci 23 (2022). doi: 10.3390/ijms23062957
113. Deckelbaum RA, Majithia A, Booker T, Henderson JE, Loomis CA. The homeoprotein engrailed 1 has pleiotropic functions in calvarial intramembranous bone formation and remodeling. Development (2006) 133:63–74. doi: 10.1242/dev.02171
114. Deckelbaum RA, Holmes G, Zhao Z, Tong C, Basilico C, Loomis CA. Regulation of cranial morphogenesis and cell fate at the neural crest-mesoderm boundary by engrailed 1. Development (2012) 139:1346–58. doi: 10.1242/dev.076729
115. Benedyk MJ, Mullen JR, DiNardo S. odd-paired: a zinc finger pair-rule protein required for the timely activation of engrailed and wingless in Drosophila embryos. Genes Dev (1994) 8:105–17. doi: 10.1101/gad.8.1.105
116. Aruga J, Mizugishi K, Koseki H, Imai K, Balling R, Noda T, et al. Zic1 regulates the patterning of vertebral arches in cooperation with Gli3. Mech Dev (1999) 89:141–50. doi: 10.1016/S0925-4773(99)00220-8
117. Tanimoto Y, Veistinen L, Alakurtti K, Takatalo M, Rice DP. Prevention of premature fusion of calvarial suture in GLI-Kruppel family member 3 (Gli3)-deficient mice by removing one allele of Runt-related transcription factor 2 (Runx2). J Biol Chem (2012) 287:21429–38. doi: 10.1074/jbc.M112.362145
118. Styrkarsdottir U, Cazier JB, Kong A, Rolfsson O, Larsen H, Bjarnadottir E, et al. Linkage of osteoporosis to chromosome 20p12 and association to BMP2. PloS Biol (2003) 1:E69. doi: 10.1371/journal.pbio.0000069
119. Doro DH, Grigoriadis AE, Liu KJ. Calvarial Suture-Derived Stem Cells and Their Contribution to Cranial Bone Repair. Front Physiol (2017) 8:956. doi: 10.3389/fphys.2017.00956
120. Youngstrom DW, Dishowitz MI, Bales CB, Carr E, Mutyaba PL, Kozloff KM, et al. Jagged1 expression by osteoblast-lineage cells regulates trabecular bone mass and periosteal expansion in mice. Bone (2016) 91:64–74. doi: 10.1016/j.bone.2016.07.006
121. Youngstrom DW, Senos R, Zondervan RL, Brodeur JD, Lints AR, Young DR, et al. Intraoperative delivery of the Notch ligand Jagged-1 regenerates appendicular and craniofacial bone defects. NPJ Regener Med (2017) 2:32. doi: 10.1038/s41536-017-0037-9
122. Patton EE, Zon LI, Langenau DM. Zebrafish disease models in drug discovery: from preclinical modelling to clinical trials. Nat Rev Drug Discovery (2021) 20:611–28. doi: 10.1038/s41573-021-00210-8
123. Kague E, Turci F, Newman E, Yang Y, Brown KR, Aglan MS, et al. 3D assessment of intervertebral disc degeneration in zebrafish identifies changes in bone density that prime disc disease. Bone Res (2021) 9:39. doi: 10.1038/s41413-021-00156-y
124. Watson CJ, Monstad-Rios AT, Bhimani RM, Gistelinck C, Willaert A, Coucke P, et al. Phenomics-Based Quantification of CRISPR-Induced Mosaicism in Zebrafish. Cell Syst (2020) 10:275–286 e5. doi: 10.1016/j.cels.2020.02.007
125. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature (2013) 496:498–503. doi: 10.1038/nature12111
126. Kwon RY, Watson CJ, Karasik D. Using zebrafish to study skeletal genomics. Bone (2019) 126:37–50. doi: 10.1016/j.bone.2019.02.009
127. Busse B, Galloway JL, Gray RS, Harris MP, Kwon RY. Zebrafish: An Emerging Model for Orthopedic Research. J Orthop Res (2020) 38:925–36. doi: 10.1002/jor.24539
128. Kague E, Roy P, Asselin G, Hu G, Simonet J, Stanley A, et al. Osterix/Sp7 limits cranial bone initiation sites and is required for formation of sutures. Dev Biol (2016) 413:160–72. doi: 10.1016/j.ydbio.2016.03.011
129. Witten PE, Huysseune A. A comparative view on mechanisms and functions of skeletal remodelling in teleost fish, with special emphasis on osteoclasts and their function. Biol Rev Camb Philos Soc (2009) 84:315–46. doi: 10.1111/j.1469-185X.2009.00077.x
130. Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn (2006) 235:176–90. doi: 10.1002/dvdy.20603
131. Hayes AJ, Reynolds S, Nowell MA, Meakin LB, Habicher J, Ledin J, et al. Spinal deformity in aged zebrafish is accompanied by degenerative changes to their vertebrae that resemble osteoarthritis. PloS One (2013) 8:e75787. doi: 10.1371/journal.pone.0075787
132. Monma Y, Shimada Y, Nakayama H, Zang L, Nishimura N, Tanaka T. Aging-associated microstructural deterioration of vertebra in zebrafish. Bone Rep (2019) 11:100215. doi: 10.1016/j.bonr.2019.100215
133. Bek JW, Shochat C, De Clercq A, De Saffel H, Boel A, Metz J, et al. Lrp5 Mutant and Crispant Zebrafish Faithfully Model Human Osteoporosis, Establishing the Zebrafish as a Platform for CRISPR-Based Functional Screening of Osteoporosis Candidate Genes. J Bone Miner Res (2021) 36:1749–64. doi: 10.1002/jbmr.4327
134. Fiedler IAK, Schmidt FN, Wölfel EM, Plumeyer C, Milovanovic P, Gioia R, et al. Severely Impaired Bone Material Quality in Chihuahua Zebrafish Resembles Classical Dominant Human Osteogenesis Imperfecta. J Bone Mineral Res (2018) 33:1489–99. doi: 10.1002/jbmr.3445
135. Foessl I, Bassett JHD, Bjornerem A, Busse B, Calado A, Chavassieux P, et al. Bone Phenotyping Approaches in Human, Mice and Zebrafish - Expert Overview of the EU Cost Action GEMSTONE ("GEnomics of MusculoSkeletal traits TranslatiOnal NEtwork"). Front Endocrinol (Lausanne) (2021) 12:1–32. doi: 10.3389/fendo.2021.720728
136. Tomecka MJ, Ethiraj LP, Sanchez LM, Roehl HH, Carney TJ. Clinical pathologies of bone fracture modelled in zebrafish. Dis Model Mech 12 (2019). doi: 10.1242/dmm.037630
137. Cotti S, Huysseune A, Larionova D, Koppe W, Forlino A, Witten PE. Compression Fractures and Partial Phenotype Rescue With a Low Phosphorus Diet in the Chihuahua Zebrafish Osteogenesis Imperfecta Model. Front Endocrinol (Lausanne) (2022) 13:851879. doi: 10.3389/fendo.2022.851879
138. Suniaga S, Rolvien T, Vom Scheidt A, Fiedler IAK, Bale HA, Huysseune A, et al. Increased mechanical loading through controlled swimming exercise induces bone formation and mineralization in adult zebrafish. Sci Rep (2018) 8:3646. doi: 10.1038/s41598-018-21776-1
139. Khajuria DK, Karasik D. Novel model of restricted mobility induced osteopenia in zebrafish. J Fish Biol (2021) 98:1031–8. doi: 10.1111/jfb.14369
140. Fisher S, Jagadeeswaran P, Halpern ME. Radiographic analysis of zebrafish skeletal defects. Dev Biol (2003) 264:64–76. doi: 10.1016/S0012-1606(03)00399-3
141. McGowan LM, Kague E, Vorster A, Newham E, Cross S, Hammond CL Wnt16 Elicits a Protective Effect Against Fractures and Supports Bone Repair in Zebrafish. JBMR Plus (2021) 5:e10461. doi: 10.1002/jbm4.10461
142. Teng CS, Ting MC, Farmer DT, Brockop M, Maxson RE, Crump JG. Altered bone growth dynamics prefigure craniosynostosis in a zebrafish model of Saethre-Chotzen syndrome. Elife 7 (2018) 7:e37024. doi: 10.7554/eLife.37024
143. Laue K, Pogoda HM, Daniel PB, van Haeringen A, Alanay Y, von Ameln S, et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet (2011) 89:595–606. doi: 10.1016/j.ajhg.2011.09.015
144. Dambroise E, Ktorza I, Brombin A, Abdessalem G, Edouard J, Luka M, et al. Fgfr3 is a positive regulator of osteoblast expansion and differentiation during zebrafish skull vault development. J Bone Miner Res (2020) 35(9):1782–97. doi: 10.1002/jbmr.4042
Keywords: craniosynostosis, zebrafish, fractures, bone mineral density, genome-wide association studies, osteoporosis
Citation: Kague E, Medina-Gomez C, Boyadjiev SA and Rivadeneira F (2022) The genetic overlap between osteoporosis and craniosynostosis. Front. Endocrinol. 13:1020821. doi: 10.3389/fendo.2022.1020821
Received: 16 August 2022; Accepted: 08 September 2022;
Published: 26 September 2022.
Edited by:
Melanie Haffner-Luntzer, University of Ulm, GermanyReviewed by:
David Karasik, Bar-Ilan University, IsraelHao Zhang, Shanghai Jiao Tong University, China
Hanna Taipaleenmäki, Ludwig Maximilian University of Munich, Germany
Copyright © 2022 Kague, Medina-Gomez, Boyadjiev and Rivadeneira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erika Kague, erika.kague@bristol.ac.uk