 Raghad Alhuthil1
Raghad Alhuthil1 Afaf Alsagheir1,2*
Afaf Alsagheir1,2* Maha Almslam1Jana Raed2Farah Barakat2
Maha Almslam1Jana Raed2Farah Barakat2 Sarah Murad3
Sarah Murad3 Bassam Bin-Abbas2
Bassam Bin-Abbas2- 1Department of Pediatrics, King Faisal Specialist Hospital & Research Centre, Riyadh, Saudi Arabia
- 2College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 3Department of Radiology, King Faisal Specialist Hospital and Research Centre, Jeddah, Saudi Arabia
Background: 3M syndrome (3MS) is a very rare autosomal recessive disorder characterized by short stature, distinctive facial features, and skeletal abnormalities. The condition is frequently underdiagnosed due to its nonspecific symptoms and normal neurocognitive development. Few reports exist on its clinical course and response to growth hormone (GH) therapy. Therefore, this study aims to describe the clinical features of Saudi patients with 3MS and to investigate the effects of growth hormone therapy on growth.
Methods: We conducted a retrospective case series of 14 Saudi patients from 11 families with genetically confirmed 3MS at King Faisal Specialist Hospital and Research Centre in Riyadh.
Results: The mean age at diagnosis was 5.4 years. Consanguinity was present in 79% of cases. The most frequently affected gene was CUL7 (57% of cases), followed by OBSL1 and CCDC8. All variants were predominantly homozygous and classified as pathogenic or likely pathogenic. Clinical abnormalities included growth retardation, dental abnormalities, spinal abnormalities, and a characteristic facial appearance. GH therapy was administered to 10 children; 5 demonstrated a measurable improvement in growth velocity, while 5 did not respond or discontinued treatment. IGF-1 was within/low-normal in most tested cases, with two elevated results.
Conclusion: Our study highlights the extensive phenotypic variability of 3MS and underscores the predominantly autosomal recessive inheritance pattern in this population. GH therapy may provide a growth benefit in select cases, although resistance and poor response remain a challenge. Genetic testing is crucial for accurate diagnosis, individualized management, and appropriate family counseling.
1 Introduction
3M syndrome (3MS) is a rare autosomal recessive disorder characterized by short stature, distinctive facial features, and skeletal abnormalities (1). It is inherited in accordance with an autosomal recessive pattern (1), and is considered very uncommon, with approximately 200 cases reported worldwide. The actual prevalence, however, might be higher because many cases might go unnoticed due to normal cognitive development (2, 3).
Individuals diagnosed with 3MS experience profound prenatal growth retardation, attributed to fetal growth delays, leading to a diminished birth weight (1). The growth impediments persist beyond birth, manifesting as a consistent pattern of delayed growth throughout childhood and adolescence, culminating in a stature significantly below the average. Many distinctive physical characteristics associated with this condition are congenital in nature. Craniofacial anomalies commonly observed encompass a disproportionately elongated and narrow head, a prominently broad forehead, and a triangular facial appearance marked by a hypoplastic midface, pointed chin, extended philtrum, noticeable mouth, depressed nasal bridge, fleshy-tipped upturned nose, large ears, and full lips (1, 4–6).
While skeletal anomalies are not apparent at birth, they gradually manifest, including delayed bone maturation, elongated and slender tubular bones, and heightened vertebral bodies (1, 4). Some individuals exhibit joint hypermobility and an increased susceptibility to hip dislocation (1, 5). Anomalous spinal curvature, such as kyphoscoliosis or hyperlordosis, leading to back pain, is also documented in this disorder (1, 5).
Additional physical abnormalities identified in certain children consist of an unusually short and broad neck and thorax, square shoulders, flared shoulder blades, atypical curvature of the 5th finger, and prominent heels (1, 5, 6).
Three different genes have been involved in the disease so far, with mutations in CUL7, OBSL1 and CCDC8 (1). The CUL7 gene, initially documented in 2005 (7), is responsible for 77.5% of genetically confirmed cases, with a specific mutation identified in exon 24 for Maghreb families (7–9).
Consequently, there is limited data on this syndrome in the existing literature. Therefore, the purpose of this study is to describe the clinical characteristics of 14 Saudi patients who have 3MS and investigate the impact of growth hormone (GH) therapy on their growth.
2 Methodology
This retrospective case series research involved 14 cases of 3MS who are currently receiving care at endocrinology clinics at King Faisal Specialist Hospital and Research Centre (KFSHRC) in Riyadh, Saudi Arabia. Data retrieval occurred from November 2023 to January 2025 from our database and included both pediatric and adult patients. Individuals without available genetic testing data were excluded. The study documented patients’ demographics, medical history, presentations, management and investigative results. Approval for this study was obtained from the Office of Research Affairs at King Faisal Specialist Hospital and Research Centre (reference number: 2245444).
The diagnostic criteria for 3MS include proportionate short stature, characteristic facial and skeletal features, with confirmation typically achieved by identifying pathogenic variants in the CUL7, OBSL1, or CCDC8 genes.
Genetic testing was conducted as part of routine clinical practice. Following patient consent, DNA was extracted from peripheral blood samples, and whole-exome sequencing was carried out at the Molecular Diagnostic Laboratory of the Clinical Genomic Department, Center for Genomic Medicine at KFSHRC.
Clinical improvement with GH therapy is defined as a significant and sustained increase in height velocity (≥2 cm/year above baseline) and/or an improvement in height SDS (≥0.3–0.5 within one year), without adverse effects, indicating partial restoration of growth potential.
This research was performed according to the guidelines of the Declaration of Helsinki and approved by the Office of Research Affairs in King Faisal Specialist Hospital and Research Centre (Reference number: 2231134). This was a retrospective study; therefore, informed consent was not required.
3 Results
Table 1 presents clinical and genetic characteristics of 14 individuals (10 females, 4 males) from 11 families diagnosed with 3MS. The mean current age of the cases was approximately 11.2 years, with a range of 4 to 39 years. The average age at diagnosis (excluding intrauterine cases) was 5.4 years. The most commonly affected gene was CUL7 (8/14 cases), followed by OBSL1 (5/14), and CCDC8 (2/14). One subject had no gene documented. Most variants were homozygous (13/14), with one heterozygous. Consanguinity was reported in 11/14 (78.6%) cases, and intrauterine growth restriction (IUGR) was present in all cases. Most mutations were classified as pathogenic (P) or likely pathogenic (LP), with three variants of uncertain significance (VUS) noted.
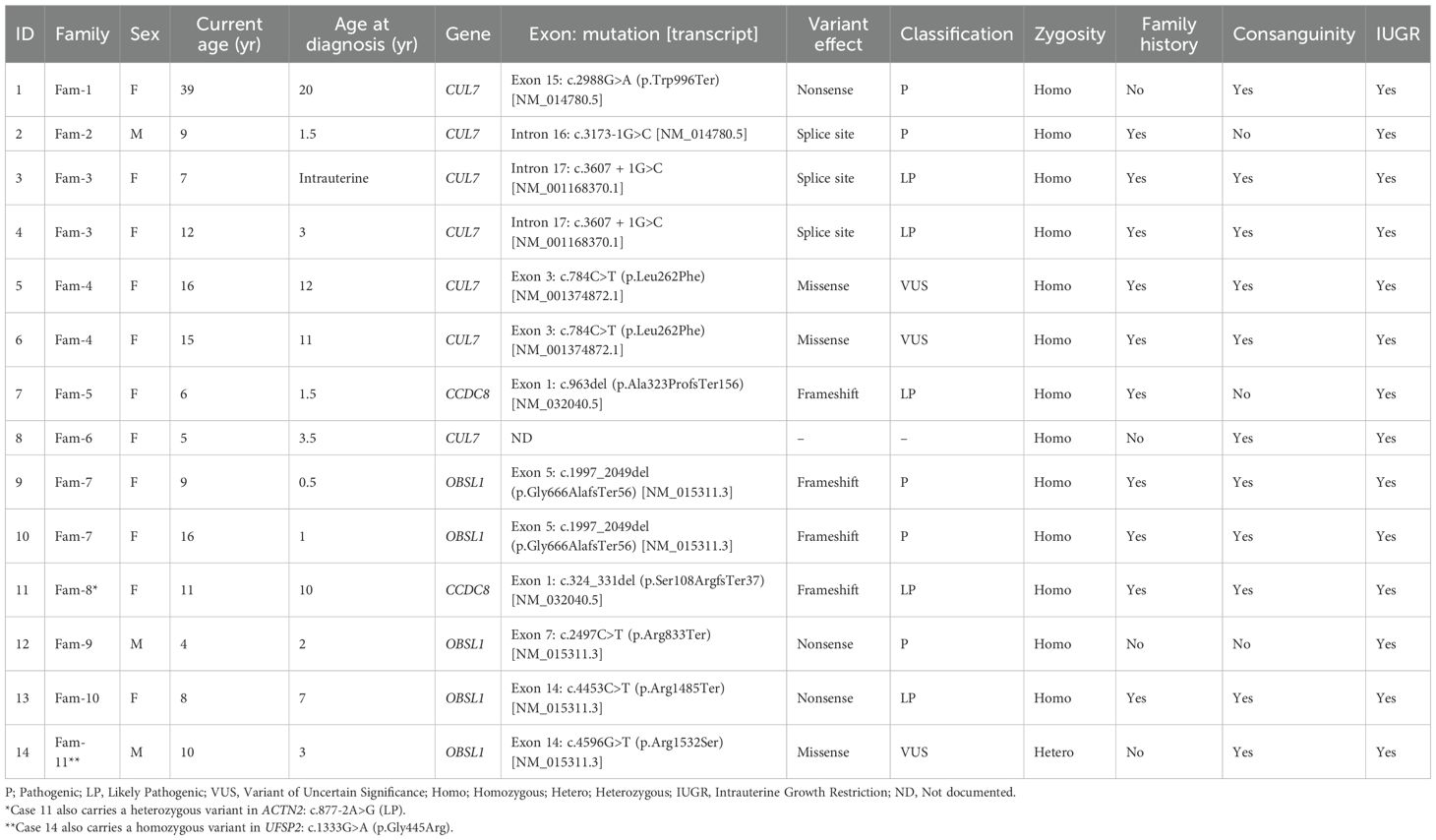
Table 1. Clinical and genetic characteristics of study subjects (n=14).
At baseline, patients had significantly reduced height with a mean SDS of -3.9. Short stature with dysmorphic/skeletal features was universal, with frequent orthopedic issues (e.g., scoliosis, skeletal dysplasia) and occasional renal involvement (hydronephrosis, nephrotic-range proteinuria). Bone age was usually age-appropriate when assessed; one child had mildly advanced bone age, and one had very low BMD (Z -4.2). IGF-1 was within/low-normal in most tested cases, with two elevated results (Table 2).
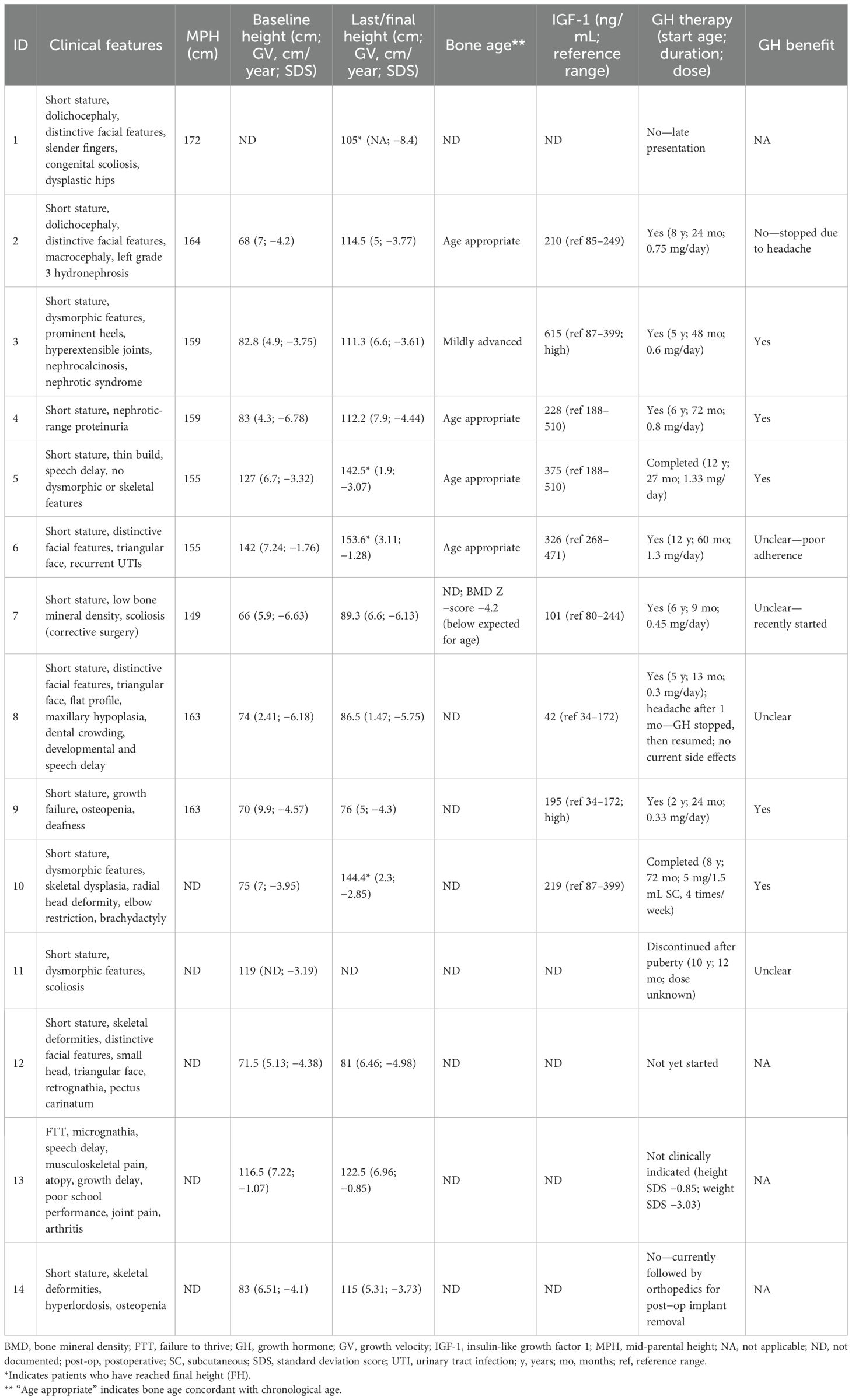
Table 2. Presentations, growth data, and management outcomes.
GH therapy was initiated in 10/14 (71.4%): five showed a clear clinical benefit, one stopped due to headaches (no benefit), and four had indeterminate benefit (recent start/poor adherence/insufficient data). Among patients with paired height SDS, most improved over time (median ΔSDS ≈ +0.43 in GH-treated vs ≈ +0.22 in non-treated; small numbers), with the largest gain of +2.34 SDS in a long-treated child. Four patients had reached final height (including one untreated adult at 105 cm); others remain under follow-up, with GH not started or not indicated in selected cases (Table 2).
Figure 1 shows hand radiographs from Case 1 revealing slender tubular phalanges, a characteristic skeletal feature of 3MS. Figure 2 includes spinal imaging from Cases 1 and 6. Case 1 exhibits lumbar hyperlordosis, thoracolumbar scoliosis, and rib abnormalities, while Case 6 shows tall lumbar vertebral bodies and scoliosis, further supporting skeletal involvement. Figure 3 illustrates pelvic radiographs from multiple cases. Cases 8 and 9 exhibit narrow triangular pelvis, while Case 1 demonstrates severe hip dysplasia with pseudoacetabuli and displaced femoral heads, reflecting the heterogeneity in pelvic morphology among affected patients. Figure 4 shows frontal facial radiographs. A triangular face was noted in Cases 2, 6, and 8, with additional craniofacial anomalies in Case 8, including maxillary hypoplasia and dental crowding, features consistent with 3MS-related dysmorphism.
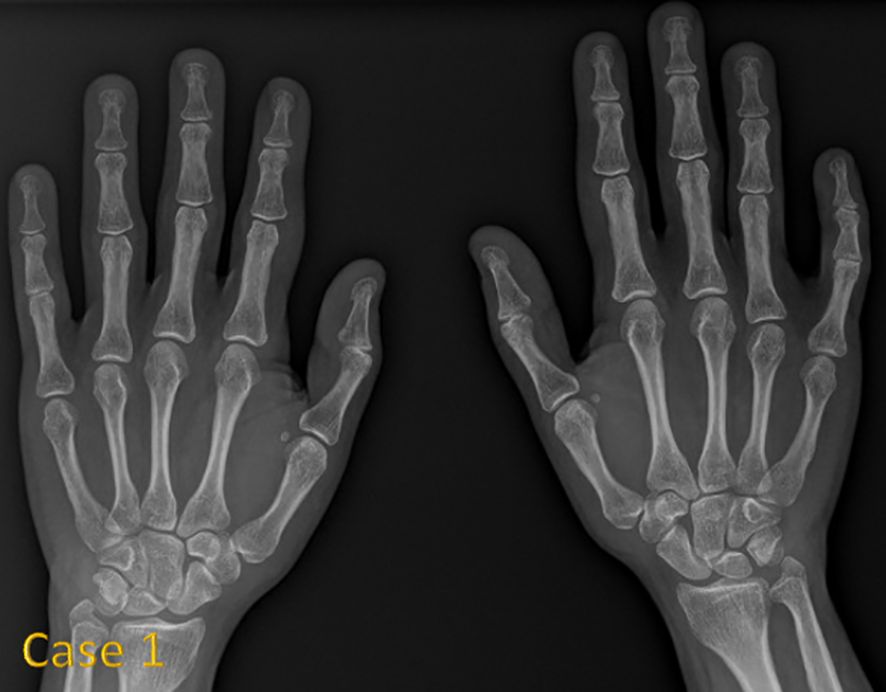
Figure 1. Anteroposterior radiograph of both hands in case 1 reveals slender tubular phalanges.
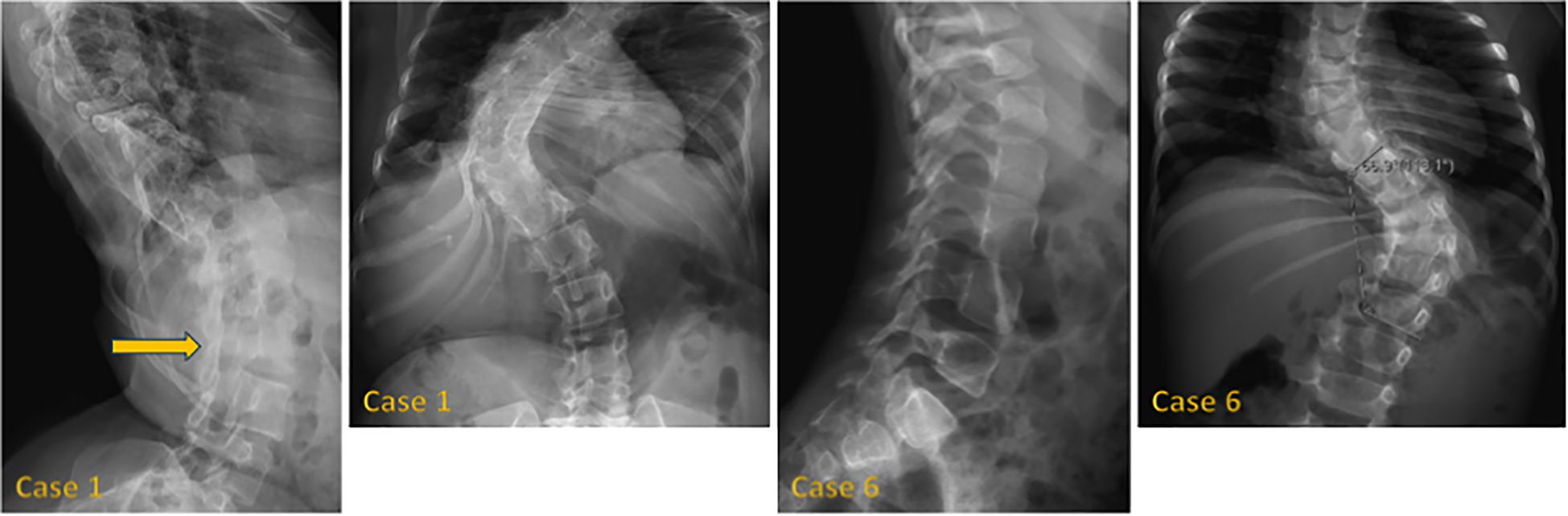
Figure 2. Lateral radiographs of case 1 reveal lumbar hyperlordosis (arrow), while frontal radiographs show thoracolumbar scoliosis and abnormal rib morphology. In case 6, lateral radiographs demonstrate tall lumbar vertebral bodies, and frontal radiographs display thoracolumbar scoliosis.
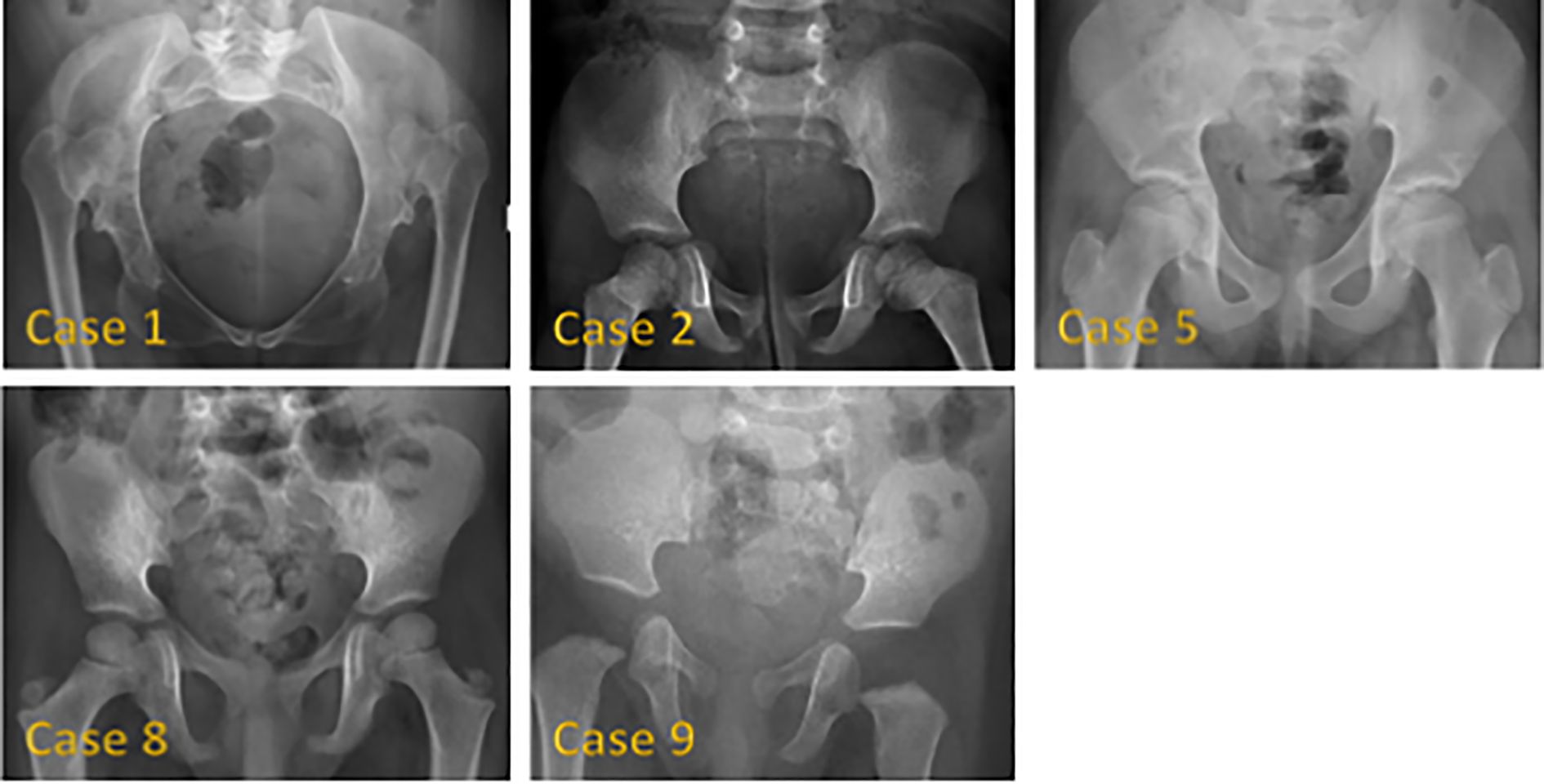
Figure 3. Pelvic radiographs of 5 patients with 3M syndrome. No obvious abnormality was found in Case 2. The pelvic measurements of Cases 5, 8 and 9 are smaller than their peers. Cases 8 and 9 showed a narrow triangular pelvis. Cases 1 and 5 showed a wide pelvis. Case 1 also showed severely dysplastic hip joints, pseudoacetabili formation, and superiorly displaced bilateral femoral heads.
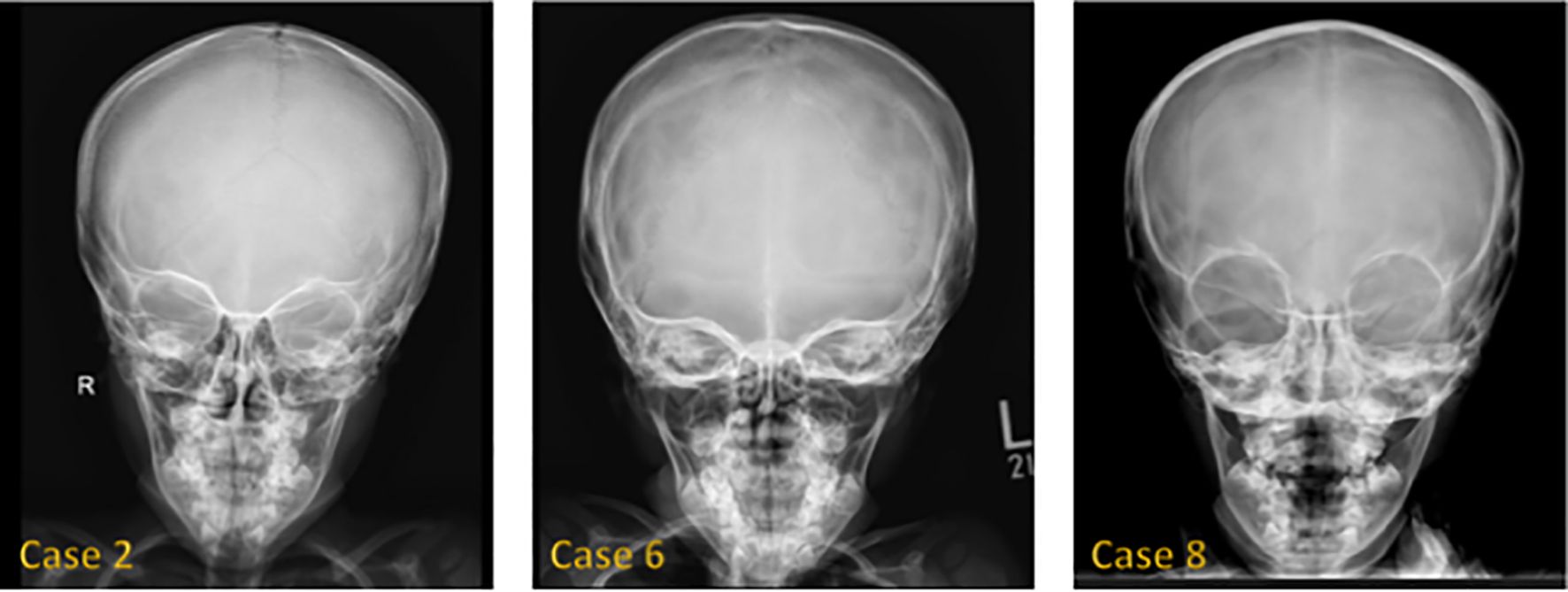
Figure 4. Frontal face radiograph showed a triangular configuration of the face in Cases 2, 6, and 8. Case 8 also showed maxillary hypoplasia, teeth malocclusion, and teeth crowding.
4 Discussion
This case series aims to describe the clinical characteristics of 14 Saudi patients who have 3MS, investigate the impact of growth hormone therapy on their growth. Additionally, it provides a review of the recent literature of this condition (3, 10–15, 18, 21–31).
The clinical manifestations observed in this series align with classical 3MS characteristics (10–13) (Table 3). Common features included IUGR, postnatal short stature, and a triangular face, frequently accompanied by dental abnormalities, pectus abnormalities, spinal abnormalities, hyperlordosis, and other skeletal abnormalities, such as scoliosis, lordosis, and slender tubular phalanges. One patient (Case 6) was described with recurrent infections, a feature previously documented in Turkey by Ceylan et al. (2023) in a patient with severe combined immunodeficiency (SCID) and 3MS (14). Less frequently described abnormalities, such as macrocephaly, acanthosis nigricans, voracious appetite, and obstructive sleep apnea, were also reported by Yang & Patni (2020) in a patient with a CUL7 mutation (15).
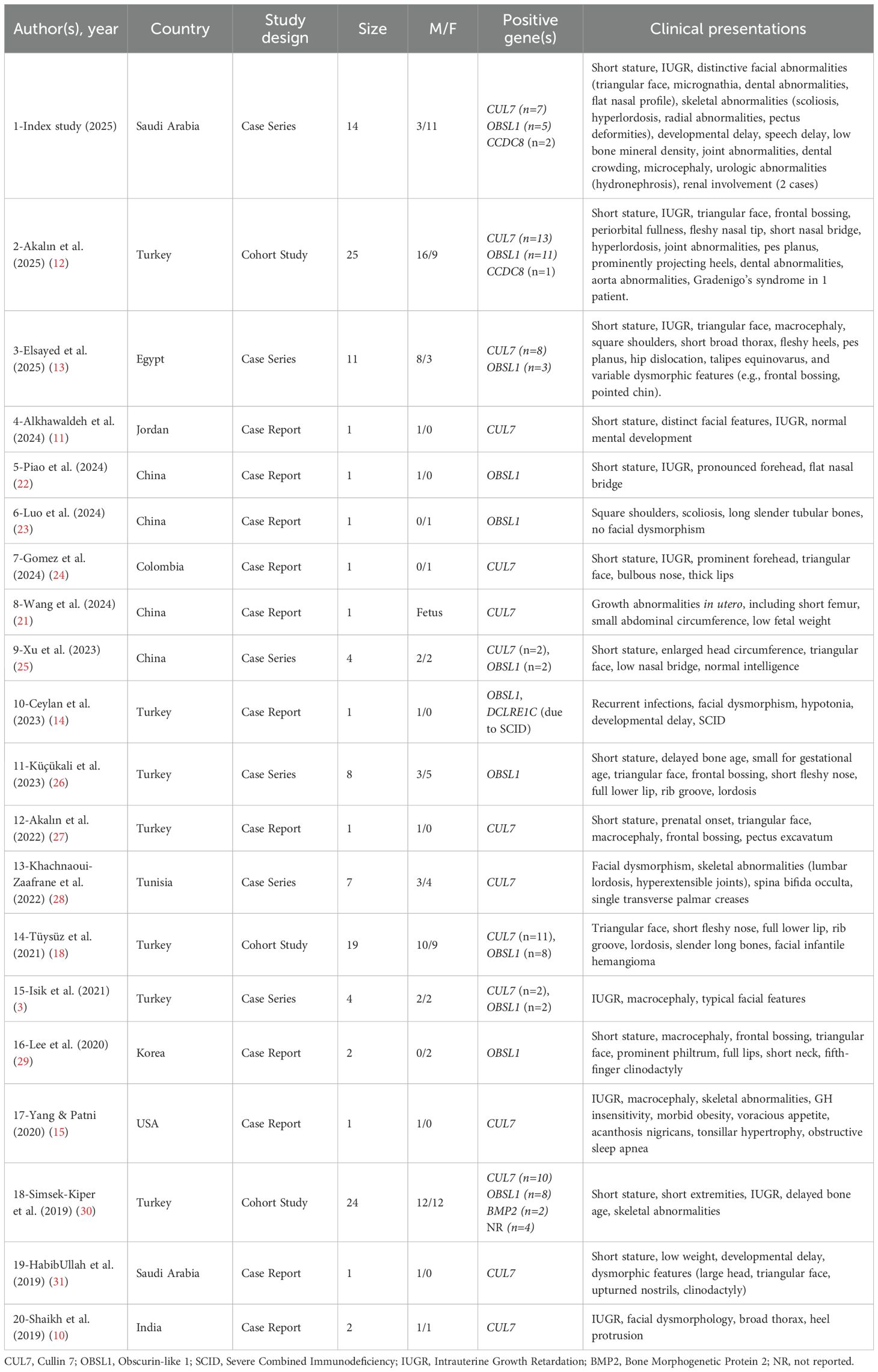
Table 3. Summary of case studies on 3M syndrome (2019–2025): clinical and genetic findings from 20 studies (n = 129 patients).
Genetically, we identified variants predominantly in CUL7, OBSL1, and CCDC8, reflecting autosomal recessive inheritance, a pattern frequently described in 3MS cases (Table 3). The most frequently affected gene was CUL7, followed by OBSL1, while CCDC8 variants were rare, which is consistent with the findings by Akalın et al. (2025) (12). Consanguinity was present in nearly 79% of cases, which may explain the predominantly homozygous variants in this population. CUL7 mutations identified in cases 1 and 2 — c.2988G>A (p.Trp996Ter) and c.3173-1G>C, respectively — have previously been described in 3MS cases (16, 17).
In mechanistic context, the three canonical 3MS genes form an interrelated “3M complex” that participates in cytoskeletal integrity, mitotic progression, and ubiquitin–proteasome–mediated protein turnover (2, 9, 12, 13, 20). CUL7 encodes a scaffold of a cullin-RING E3 ubiquitin ligase that, together with adaptor proteins, regulates turnover of signaling intermediates, including components of insulin/IGF pathways (2, 7, 9, 20). Experimental work indicates that perturbations of CUL7 can dysregulate IRS-1 handling and downstream PI3K–AKT signaling, contributing to impaired chondrocyte proliferation and skeletal growth despite ostensibly intact upstream GH stimulation (2, 9, 13, 25). OBSL1 (a cytoskeletal adaptor) and CCDC8 (a coiled-coil protein with roles in cell division and genome surveillance) interact with CUL7; loss of function across any of these partners disrupts the complex, with convergent effects on growth-factor signaling and endochondral ossification (2, 9, 13, 20). Clinically, this biology predicts GH insensitivity or post-receptor resistance, a pattern we observed—several children had normal or even elevated IGF-1 while linear growth remained suboptimal (18, 19, 26, 29).
As for management, of the ten reported cases where GH therapy was administered, the response was highly variable. Five cases demonstrated a notable improvement, while the other five showed no clear response or discontinued therapy due to factors like puberty or poor compliance. This variability is consistent with previous reports; for instance, Tüysüz et al. (2021) described analogous cases where GH treatment improved growth outcomes in only a subset of children with 3MS (18). Similarly, Altun et al. (2025) observed modest but statistically significant improvements in annual growth velocity (from 5.3 to 6.1 cm/year) in eight pediatric cases, despite evidence of underlying GH resistance indicated by elevated IGF-1 levels in some patients (19). While final height in these studies remained significantly below average, the stabilization or slight improvement in height SDS suggests a potential, albeit limited, benefit from GH therapy (19).
The challenges of this treatment are further highlighted across other studies. In one study, three out of four children exhibited significant growth acceleration with recombinant human GH (rhGH), though their long-term outcomes remain under observation (32). Another study reported that five out of seven patients discontinued GH treatment due to an insufficient growth response, underscoring the difficulty in sustaining effective therapy over time (Küçükali et al., 2023) (26). The genetic background of 3MS, particularly mutations in the CUL7 and OBSL1 genes, is thought to influence this variable response and contribute to the observed GH resistance (Xu et al., 2023) (25). In cases of suspected resistance, recombinant human IGF-1 (rhIGF-1) has been trialed as an alternative; however, one such report noted it did not provide substantial height benefits and was associated with side effects like obesity and acanthosis (Yang & Patni, 2020) (15). To potentially enhance outcomes, combination therapies have been explored, with one report of two siblings showing height gains following a regimen of GH and a gonadotropin-releasing hormone (GnRH) agonist (Lee et al., 2020) (29).
Beyond pharmacological interventions, the comprehensive management of 3MS includes surgical interventions for skeletal and joint abnormalities, as well as adaptive measures for persistent short stature. Furthermore, genetic counseling is recommended for affected families to explain the autosomal recessive inheritance pattern, discuss the 25% recurrence risk, and review options such as preimplantation genetic testing and prenatal ultrasound for early diagnosis (20, 21).
This study has a few limitations. Since it included only a small number of patients, the results may not apply to all individuals with 3MS. Also, because the data were collected by looking back at medical records, there’s a chance some details were missed or not recorded consistently. Finally, the study took place at just one specialized hospital, so the findings might not represent how 3MS is diagnosed or treated in other hospitals or regions.
In conclusion, this study highlights the clinical variability of 3MS, predominantly homozygous variants in CUL7, OBSL1, and CCDC8, and the potential role of GH therapy in improving growth outcomes in some cases. Nevertheless, the response to treatment is heterogeneous and underscores the necessity for individualized management plans and ongoing follow-up. Furthermore, genomic testing and proper genetic counseling are crucial for guiding future family planning and disease management.
Author contributions
RA: Data curation, Methodology, Writing – original draft. AA: Conceptualization, Supervision, Writing – review & editing. MA: Data curation, Writing – original draft. JR: Data curation, Writing – original draft. FB: Data curation, Writing – original draft. SM: Data curation, Methodology, Writing – original draft. BB-A: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Meazza C, Lausch E, Pagani S, Bozzola E, Calcaterra V, Superti-Furga A, et al. 3-M syndrome associated with growth hormone deficiency: 18-year follow-up of a patient. Ital J Pediatr. (2013) 39:21. doi: 10.1186/1824-7288-39-21
2. Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu-Amero SN, et al. Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin Endocrinol (Oxf). (2012) 77:335–42. doi: 10.1111/j.1365-2265.2012.04428.x
3. Isik E, Arican D, Atik T, Ooi JE, Darcan S, Ozen S, et al. A rare cause of syndromic short stature: 3M syndrome in three families. Am J Med Genet A. (2021) 185:461–8. doi: 10.1002/ajmg.a.61989
4. Winter RM, Baraitser M, Grant DB, Preece MA, and Hall CM. The 3-M syndrome. J Med Genet. (1984) 21:124–8. doi: 10.1136/jmg.21.2.124
5. Guven A and Cebeci AN. 3-M syndrome: A report of four cases in two families. J Clin Res Pediatr Endocrinol. (2011) 3:154–9. doi: 10.4274/jcrpe.v3i3.30
6. Miller JD, McKusick VA, Malvaux P, Temtamy S, and Salinas C. The 3-M syndrome: A heritable low birthweight dwarfism. Birth Defects Orig Artic Ser. (1975) 11:39–47.
7. Huber C, Dias-Santagata D, Glaser A, O’Sullivan J, Brauner R, Wu K, et al. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. (2005) 37:1119–24. doi: 10.1038/ng1628
8. Huber C, Delezoide AL, Guimiot F, Baumann C, Malan V, Le Merrer M, et al. A large-scale mutation search reveals genetic heterogeneity in 3M syndrome. Eur J Hum Genet. (2009) 17:395–400. doi: 10.1038/ejhg.2008.200
9. Hanson D, Murray PG, Coulson T, Sud A, Omokanye A, Stratta E, et al. Mutations in CUL7, OBSL1 and CCDC8 in 3-M syndrome lead to disordered growth factor signalling. J Mol Endocrinol. (2012) 49:267. doi: 10.1530/JME-12-0034
10. Shaikh S, Shettigar SK, Kumar S, Kantharia S, Kurva J, and Cherian S. Novel mutation in Cul7 gene in a family diagnosed with 3M syndrome. J Genet. (2019) 98:1–4. doi: 10.1007/s12041-019-1057-6
11. Alkhawaldeh A, Alsayed AR, Daghash R, Albaramiki J, Shibli D, Abudahab S, et al. The first study of 3-M syndrome in Jordan and Literature Review. Pharm Pract (Granada). (2024) 22:2908. doi: 10.18549/PharmPract.2024.1.2908
12. Akalın A, Özalkak Ş, Yıldırım R, Karakaya AA, Kolbaşı B, Durmuşalioğlu EA, et al. Clinical and molecular spectrum along with genotype–phenotype correlation of 25 patients diagnosed with 3M syndrome: a study from Turkey. Eur J Pediatr. (2025) 184:68. doi: 10.1007/s00431-024-05855-2
13. Elsayed S, Elmakkawy GA, Abdelrazek IM, Fawzy DA, Kim J, Song Y, et al. An update on 3M syndrome: review of clinical and molecular aspects and report of additional families. Am J Med Genet Part A. (2025) 197:e64068. doi: 10.1002/ajmg.a.64068
14. Ceylan A, Tekdemir IE, Kocak N, Chinn IK, Orange JS, and Artac H. Case report: Artemis deficiency and 3M syndrome—coexistence of two distinct genetic disorders. Front Pediatr. (2023) 11:1211254. doi: 10.3389/fped.2023.1211254
15. Yang M and Patni N. Effect of recombinant human insulin-like growth factor 1 therapy in a child with 3-M syndrome-1 with CUL7 gene mutation. J Pediatr Endocrinol Metab. (2020) 33:1609–12. doi: 10.1515/jpem-2020-0278
16. Holder-Espinasse M, Irving M, and Cormier-Daire V. Clinical utility gene card for: 3M syndrome. Eur J Hum Genet. (2011) 19:1017. doi: 10.1038/ejhg.2011.32
17. Lugli L, Bertucci E, Mazza V, Elmakky A, Ferrari F, Neuhaus C, et al. Pre-and post-natal growth in two sisters with 3-M syndrome. Eur J Med Genet. (2016) 59:232–6. doi: 10.1016/j.ejmg.2016.01.009
18. Tüysüz B, Turan H, Gezdirici A, Alkaya DU, Kasap B, Yeşil G, et al. Natural history of facial and skeletal features from neonatal period to adulthood in a 3M syndrome cohort with biallelic CUL7 or OBSL1 variants. Eur J Med Genet. (2021) 64:104346. doi: 10.1016/j.ejmg.2021.104346
19. Altun I, Bayramoğlu E, Karakas H, Ucar M, Velioğlu HG, Guney A, et al. Treatment responce of the growth hormone in 3M syndrome: a single center experience. In: Endocrine abstracts, vol. 110. United Kingdom: Bioscientifica (2025). doi: 10.1530/endoabs.110.EP733
20. Akilapa R, Irving M, and Holder-Espinasse M. Three M syndrome. In: Adam MP, Feldman J, and Mirzaa GM, editors. GeneReviews®. University of Washington, Seattle (WA (2002). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1481/.
21. Wang X, He Y, Wang X, Kong X, Lin Y, Yao Y, et al. Prenatal diagnosis and preimplantation genetics testing of 3M syndrome in a Chinese family with novel biallelic variants of CUL7. Mol Genet Genomic Med. (2024) 12:e2284. doi: 10.1002/mgg3.2284
22. Piao Y, Li R, Wang Y, Chen C, and Sang Y. Novel OBSL1 variant in a Chinese patient with 3M syndrome: The c. 458dupG mutation may be a potential hotspot mutation in the Chinese population. J Clin Res Pediatr Endocrinol. (2024) 16:501. doi: 10.4274/jcrpe.galenos.2024.2023-11-6
23. Luo MR, Dai SM, Li Y, Wang Q, Liu H, Gao P, et al. 3M syndrome patient with a novel mutation: A case report. World J Clin cases. (2024) 12:1454. doi: 10.12998/wjcc.v12.i8.1454
24. Gomez SI, Polanco AM, Valencia GM, Hernández JB, Torres AC, and Espinosa NA. Síndrome 3-M (Miller, McKusick, Malvaux): otra causa de talla baja genética. Rev Colomb Endocrinol Diabetes Metab. (2024) 11:438–44. doi: 10.53853/encr.11.3.900
25. Xu N, Liu K, Yang Y, Li X, and Zhong Y. Chinese patients with 3M syndrome: clinical manifestations and two novel pathogenic variants. Front Genet. (2023) 14:1164936. doi: 10.3389/fgene.2023.1164936
26. Küçükali GK, Keskin M, Aycan Z, Savaş-Erdeve Ş, and Çetinkaya S. 3M syndrome: Evaluating the clinical and laboratory features and the response of the growth hormone treatment: Single center experience. Eur J Med Genet. (2023) 66:104828. doi: 10.1016/j.ejmg.2023.104828
27. Akalın A, Simsek-Kiper PÖ, Taşkıran E, Utine GE, and Boduroğlu K. Typical face, developmental delay, and hearing loss in a patient with 3M syndrome: the co-occurrence of two rare conditions. Mol Syndromol. (2022) 13:537–42. doi: 10.1159/000524703
28. Khachnaoui-Zaafrane K, Ouertani I, Zanati A, Kandara H, Maazoul F, and Mrad R. 3M syndrome: A Tunisian seven-cases series. Eur J Med Genet. (2022) 65:104448. doi: 10.1016/j.ejmg.2022.104448
29. Lee IK, Lim HH, and Kim YM. The effect of combined growth hormone and a gonadotropin-release hormone agonist therapy on height in korean 3-M syndrome siblings. Yonsei Med J. (2020) 61:981–5. doi: 10.3349/ymj.2020.61.11.981
30. Simsek-Kiper PO, Taskiran E, Kosukcu C, Arslan UE, Cormier-Daire V, Gonc N, et al. Further expanding the mutational spectrum and investigation of genotype–phenotype correlation in 3M syndrome. Am J Med Genet A. (2019) 179:1157–72. doi: 10.1002/ajmg.a.61154
31. HabibUllah H, Al-Baradie R, and Bashir S. 3-M syndrome: A local case report. Am J Case Rep. (2019) 20:36–8. doi: 10.12659/AJCR.912736
Keywords: 3M syndrome, growth hormone therapy, rare hereditary disorder, CUL7, OBSL1, CCDC8
Citation: Alhuthil R, Alsagheir A, Almslam M, Raed J, Barakat F, Murad S and Bin-Abbas B (2025) 3M syndrome in Saudi Arabia: a case series study and literature review. Front. Endocrinol. 16:1666468. doi: 10.3389/fendo.2025.1666468
Received: 15 July 2025; Accepted: 29 October 2025;
Published: 11 November 2025.
Edited by:
Alberto Falchetti, Santa Maria della Misericordia, ItalyReviewed by:
Ruijin Xie, Affiliated Hospital of Jiangnan University, ChinaGülin Karacan Küçükali, Dr Sami Ulus Child Health and Diseases Training and Research Hospital, Türkiye
Copyright © 2025 Alhuthil, Alsagheir, Almslam, Raed, Barakat, Murad and Bin-Abbas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afaf Alsagheir, QVNhZ2hlaXJAa2ZzaHJjLmVkdS5zYQ==