Oligomeric Aromatic Oxides (OAO) Production From Sugarcane Bagasse Lignin by Acid-Catalyzed Solvothermal Liquefaction in Methanol
 Yi Cheng
Yi Cheng Shujuan Yang2
Shujuan Yang2 - 1College of Chemical Engineering, Beijing University of Chemical Technology, Beijing, China
- 2Department of Forest Products Chemical Processing, Beijing Forestry University, Beijing, China
- 3Department of Market and Development, Ningxia Pantai Energy Technology Co., Ltd, Yinchuan, China
Extracted alkali lignin (AL) and organosolv lignin (OL) from sugarcane bagasse were acid-catalyzed liquefied in methanol with the aim of producing oligomeric aromatic oxides. Acids were screened for their effects on the output of oligomeric aromatic oxides from alkali lignin liquefaction. Based on the highest amount of lignin conversion, the order of catalytic efficiency was: p-toluenesulfonic acid (TsOH) > CCl3COOH (TCA) > KHSO4 > AlCl3 > H3O40PW12 > H2SO4. The most alkali lignin conversion was 86.2 wt% when catalyzed by p-toluenesulfonic acid. Optimized liquefaction temperatures indicated that AL liquefaction optimum temperature was 250°C and OL was 175°C. GPC characterized AL, OL and resultant products implied that TsOH could degrade both lignins to about 780 g/mol of molecular weight. HSQC-NMR and GC-MS observations suggested that simultaneous vinyl ether cleavage and intermediate stabilization of phenolic hydroxyl group etherification at high temperatures achieved AL liquefaction. Acidolysis of β-ether linkages at mild temperatures was the mechanism of OL liquefaction.
Introduction
Lignin is the most abundant natural aromatic polymer. It consists of methoxylated phenylpropane units, cross-linked by carbon-carbon and ether bonds (Vanholme et al., 2010). This renewable resource is of interest for producing aromatic chemicals (Zakzeski et al., 2013; Ragauskas et al., 2014). Annually, the global paper and pulping industry have over 50 million tons of technical lignin (Lora and Glasser, 2002). Technical lignin is frequently considered a low-value solid fuel and is used to balance production energy needs (Gosselink et al., 2004). In the past two decades, many works on technical lignin valorization have focused on aromatic monomer conversions, like BTX (benzene, toluene, and xylene) and phenol; or as materials (Zakzeski et al., 2013; Upton and Kasko, 2016; Chen et al., 2020). Only small amounts of reports proposed oligomeric lignin (Mousavioun and Doherty, 2010; Yuan et al., 2010; Saito et al., 2014). Unlike polysaccharide, lignin was hardly completely degraded to monomers due to heterogeneous linkages (Jasiukaitytė et al., 2012). The lignin degradation process inevitably generated oligomers (Joffres et al., 2014). Researchers have recognized this problem and conquered further cracking by developing efficiency catalysts or harsh conditions (Tang et al., 2010; Jongerius et al., 2013; Yoshikawa et al., 2014; Kloekhorst et al., 2015). However, confronting the current petroleum market, monomeric chemicals produced from technical lignin by extreme conditions have no obvious economic advantages. Valorizations of dimmer or trimmer lignin, which can use easy-conducted conditions, are potential technical lignin sustainable utilization approaches.
The alcohol-gasoline additive is a potential commercial chemical from lignin liquefaction due to its physical and chemical properties. Alcohol-gasoline is the current and near-term alternative transportation fuel used in spark-ignition engines (Siwale et al., 2014). One obstacle encountered with using this alternative fuel is its tendency to phase separation due to alcohol’s hydrophilicity (Lojkásek et al., 1992). Demirbaş (2000a) has reported a method of biomass glycerin liquefaction for the alcohol-gasoline additive production. The biomass-derived additive proposed to lower the phase separation temperature. The solubility of the final products in gasoline was 1.96% by weight. The octane number increased by 8% when 10% of ethanol was added to gasoline. Wang et al. (2016) argue that aromatic ethers in gasoline are advantageous for improving anti-knock properties. In the upgrading of phenolic-oil by etherification, phenols conversed with methanol to aromatic ethers. Hence, lignin might be a potential resource for gasoline additive production.
Nevertheless, the lignin-derived additive must meet the requirements of solubility in alcohol and chemical stability by degradation. Hydrolytic cleavages of ether linkages were the mechanism of high molecular weight degradation rather than stable bonds of aryl-aryl bonds and C-C bonds between aromatic lignin units (Lin et al., 1997; Lin et al., 2005). Most ether linkages of alkali lignin (AL) are veratryl glycerol-β-guaiacyl ether and organosolv lignin (OL)are guaiacyl glycerol-β-guaiacyl ether (Prinsen et al., 2013; Yang et al., 2013). Mineral acids, Lewis acids, zeolites, and organic acids, have been proved for the degradation of lignin and the related model compounds (Li et al., 2015). Jia et al. (2010) carried out the cleavage of β-O-4 bonds of guaiacyl glycerol-β-guaiacyl ether and nonphenolic veratryl glycerol-β-guaiacyl ether with a catalytic amount of hydrochloric acid. Both ethers degraded to guaiacol in the acid-catalyzed reaction. In acidolysis degradation, the first step is a dehydration reaction, yielding enol aryl ether, different from quinone methide intermediate in the base-catalyzed hydrolysis. The enol aryl ether intermediate rapidly hydrolyzed to guaiacol and α-ketocarbinol (Sarkanen and Hoo, 1981). The reaction rate is usually faster in alcohol than in water due to solvent effects (Miller et al., 1999).
This study aims to produce lignin-derived oligomeric aromatic oxides (OAO) to facilitate alcohol-gasoline additive. The critical problem is discussing the probability of technical lignin conversion. Sugarcane bagasse, which is the main byproduct of the sugar industry, is a potential feedstock for biorefinery because of its availability and low cost. Herein, it was used sugarcane bagasse as feedstock for the AL and OL extraction. Acids, including organic acids, mineral acids, and Lewis acids, were screened for lignin degradation in methanol. Organic solvents have been screened for Kraft lignin fractionation and found that ethyl acetate dissolved only lignin oligomers. In this work, smaller lignin was extracted by ethyl acetate according to the previous study (Wang et al., 2010). Gel permeation chromatography, heteronuclear single quantum coherence spectroscopy-nuclear magnetic resonance spectroscopy (HSQC-NMR), and gas chromatography-mass spectrometry determined the structural changes in lignin observed during the conversion process.
Experimental
Materials
Sugarcane bagasse was kindly provided by Guitang Corporation (Guangxi, China), was water-rinsed and dried at 60°C for 12 h. The dried sugarcane bagasse was grounded to 0.1 mm particles before lignin extraction. The raw material components were 44.63% cellulose, 25.76% hemicelluloses, 22.20% lignin, and 1.45% ash.
Methanol (AR) and ethyl acetate (AR) were purchased from Beijing Chemical Factory and used without further purification. The acids (purchased from Sinopharm Chemical Reagent Co., Ltd.) included TsOH (p-toluenesulfonic acid, AR), (trichloroacetic acid) TCA (CCl3COOH, AR), KHSO4 (AR), H2SO4 (98%, AR), AlCl3 (AR), and H3O40PW12 (AR).
Lignin Extraction
AL extraction: the soda delignification process added 10 g of sugarcane bagasse powders, 100 ml of pH = 11.5 NaOH solution, and 0.5 g anthraquinone. After sealing, the reactor was loaded into a stainless steel tank for rotary stirring, and the reactor was heated at an average of 5°C/min to reach the desired temperature of 160°C. After 1 h of delignification, the reactor was rapidly cooled in water. The insoluble residues were filtrated and washed with 200 ml of water. Delignification solutions and washing water was combined and concentrated to 50 ml and dropped to 500 ml pH = 2 HCl solutions. Obtained AL was acid-solution insoluble parts that were freeze-dried for 48 h.
OL extraction: the organosolv delignification process added 10 g of sugarcane bagasse powders and 100 ml of ethanol and pH = 8.5 NaOH solution mixture (50/50, v/v). After sealing, the reactor was loaded into a stainless steel tank for rotary stirring, and heated at an average of 5°C/min to reach the desired temperature of 140°C for 1 h. After delignification, the reactor was rapidly cooled in water. The insoluble residues were collected by filtration and washed with 200 ml ethanol-water mixture (50:50%, v/v). Delignification solutions and washing solutions were combined and concentrated to 50 ml and dropped to 500 ml pH = 2 HCl solutions. The obtained OL was an acid-solution with insoluble parts that was freeze-dried for 48 h.
Milled wood lignin (MWL), which represents the native lignin released by aqueous dioxane, was prepared according to the typical procedure (Wen et al., 2012). The sugarcane bagasse powders were dried at 60°C in an oven for 24 h before ball milling with a planetary ball mill (FritschGMBH, Idar-Oberstein, Germany). The milling bowl was composed of zirconium dioxide (500 ml) and contained 25 zirconium dioxide balls (1 cm diameter). The milling was conducted at room temperature under N2 atmosphere with a milling frequency of 500 rpm. In the case of overheating, 10 min of ball milling followed 10 min breaks. Typically, 10 g of sugarcane bagasse powders we added into the milling bowl and milled for 5 h. The milled powders were extracted with dioxane/water (96:4, v/v, 50 ml) in the dark, and the extracted solution was collected by centrifugation and concentration, as well as precipitation. MWL was obtained by dropped concentrated solution to pH = 2 HCl solution before freeze-dried for 48 h. Further purification was non-performed to preserve structural features of the isolated MWL, although some carbohydrates remained in the MWL.
Lignin Conversion and Separation
Hydrothermal autoclaves (25 ml) were used as reactors and conducted each group of batch mode tests. Every experiment loaded 10 ml methanol, 0.5 g lignin samples (AL or OL), and 0.05 g acid catalysts in each reactor. Heated reactor to desired reaction temperatures (150°C–250°C) and retained for 30–150 min without any extra gases. After the reaction, the reactors were rapidly quenched in a water bath to cool down. The undissolved residues were dried for 24 h after the slurry centrifugation. The determination of lignin conversion was 100% minus the ratio of undissolved residues. The liquid part was directly analyzed by gas chromatography for monomers detection after being filtered. The obtained soluble lignin was dropped into 90 ml pH = 2 HCl solutions to remove the residual acids. Acid precipitates were dried at room temperature and later dissolved in the ethyl acetate for OAO determination. Herein, OAO yields were product selectivity determined by the amounts of soluble conversed lignin in ethyl acetate.
Characterization of Lignin and Products
The monomer quantification and qualification analyses were detected by gas chromatography-flame ionization detector (GC-FID) (Agilent 7980A) and identified by gas chromatography-mass spectrometry (Shimadzu QP-2010), respectively. The column in the equipment was HP-5MS capillary column (30 m × 0.25 mm × 0.25 mm), and the temperature program was from 50°C to 270°C at 10°C/min with 2 min for the initial time. N-hexadecane was added as an internal standard for quantification. The identification of the compounds was achieved by comparing the mass spectra obtained with those contained in the system’s database (NIST02).
1H-13C HSQC NMR spectra were obtained by using a Bruker-500 MHz NMR. About 80 mg of the lignin samples were dissolved in 0.5 ml DMSO-d6. The quantitative analysis of the HSQC cross-signal intensities was performed according to published methods [16, 17]: the spectral widths were 5,000 and 13,200 Hz for the 1H and 13C dimensions, respectively. The number of collected complex points was 2,048 for the 1H dimension, with a recycle delay of 1 s. The number of transients was 32, and 256-time increments were recorded in the 13C dimension. Before Fourier transformation, the data matrices were zero-filled up to 1,024 points in the 13C dimension. In the aromatic region, the ratio of S/G units can be obtained by integrating the C2, 6-H2, 6 correlations of syringyl (S), guaiacyl (G), and aromatic units. The assignments of signals were according to reports (Wen et al., 2013; Yang et al., 2014).
The weight average (Mw), number average (Mn) molecular weight, and polydispersity index (PDI) were determined by using a Gel permeation chromatography instrument (Waters 2695e, UV detector at 280 nm, PL-gel 10 mm Mixed-Bn 7.5 mm ID column in a Perkin-Elmer instrument) at a column temperature of 35°C and THF as the eluent at a flow rate of 1 ml/min. Liner monodisperse polystyrene standards were employed for the molecular weight calibration curve.
Results and Discussion
Effects of Acids on Alkali Lignin Conversion
The results of the AL liquefaction at 250°C, both with and without acids, are given in Figure 1. Firstly, AL liquefaction was conducted with the aim of catalyst screening in methanol solvent. It was observed that 49.4 wt% of the lignin was converted in the absence of acid at 250°C in methanol solvent, similar to the literature shown (Singh et al., 2014). The lignin conversion increased significantly upon p-toluenesulfonic acid (TsOH) into the system, with 86.2 wt% of lignin conversion and 64.7 wt% of OAO yield. Among the selected catalysts, TCA showed the best product selectivity, with 71.3 wt% of OAO. Herein, TsOH performed better than TCA in lignin conversion, whereas the OAO yields with TsOH catalysts were less than that of with TCA catalysts.
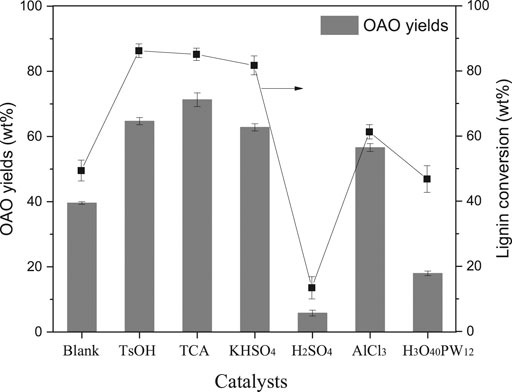
FIGURE 1. Effects of catalyst on alkali lignin conversion and oligomeric aromatic oxides (OAO) yielded when the reactions were conducted at 250°C for 60 min with 10 wt% catalysts loading.
Two mineral acids with sulfate of H2SO4 and KHSO4 were compared for lignin conversion and performed differently. The H2SO4 has been proved to be a significant catalyst in wood liquefaction in phenol and polyol solvents when used for phenolic resin and polyurethane synthesis (Wen et al., 2018). Results of AL conversion in methanol solvent with sulfate compounds have been rarely reported previously. In this work, lignin conversion catalyzed by H2SO4 performed unsatisfied results, in which lignin conversion was 13.4 wt% and only obtained 5.8 wt% of OAO. Conversely, KHSO4 outputted acceptable results: 81.7 wt% of lignin conversion and 62.8 wt% of OAO yields. In this acid catalysis process, acid can simultaneously promote both de- and re-polymerization. High hydrogen ion concentration might significantly condensate the liquefied phenolic product (Yao et al., 2018). In this work, the acid dissociation constant (pKa) in water consulted acidity comparison in AL liquefaction. In the water solvent, the pKa1 of H2SO4 was −2.00, and the pKa2 of was +1.99, which was much more significant than pKa1. The pKa of KHSO4 was +1.99 if assumed that potassium ions can completely ionize. It concluded that the acidity of KHSO4 was weaker than that of H2SO4. The dissociative proton might re-polymerize the lignin fragments rather than break linkages of lignin units. It can hence be concluded that mild acid was optimal for AL degradation. It should point out that the pKa of TsOH was −1.34. The TsOH was also confirmed to be a useful catalyst in lignocellulose liquefaction in mild conditions (Demirbaş, 2000b). Therefore, a significant difference in this system’s performance implied that the acidolysis mechanism was not the unique answer of AL liquefaction in methanol.
The catalytic activities of AlCl3 and H3O40PW12 were intermediate between that of organic acids and sulfuric acid, with AlCl3 outperforming H3O40PW12. Lignin conversions catalyzed by AlCl3 and H3O40PW12 were 61.3 and 46.8 wt%, respectively. The OAO yields from AlCl3 and H3O40PW12 catalysis were 56.6 and 18 wt%, respectively. The acid strengths of these two acids were considered responsible for lignin degradation. The higher valence metal increased the number of acidic centers, thereby increasing the catalyst’s acidic strength (Shu et al., 2015). Lewis acids might drive unpredicted reactions in specific systems. For example, aqueous AlCl3 can catalyze delignification reactions (Shen et al., 2016), and H3O40PW12 can oxidize carbonyl groups of lignin (Voitl and Philipp, 2010). In this work, Lewis acids were not applicable unless employed under controlled conditions.
Effect on Reaction Temperatures on Alkali Lignin and Organosolv Lignin Conversion
Figure 2 gives the results of the effects of reaction temperature on lignin conversion. It compared AL and OL liquefactions at different reaction temperatures by using the TsOH catalyst. AL and OL performed different results in the same range of reaction temperatures. Figures 2A illustrated that 81.8 wt% of AL conversion and 8.2 wt% of OAO yield when conducted the reaction at 150°C. Figures 2B gave the results of 96.9 wt% of AL conversion and 87.6 wt% of OAO yield when the liquefaction was completed at the same reaction temperature. With the growth of reaction temperatures, both two lignin conversions achieved peak values at 225°C. The highest lignin conversion of AL was 86.1 wt% and of OL was 98.3 wt%. Both two peak values later decreased when heated at 250°C. Notably, OL conversion was always higher than that of AL.
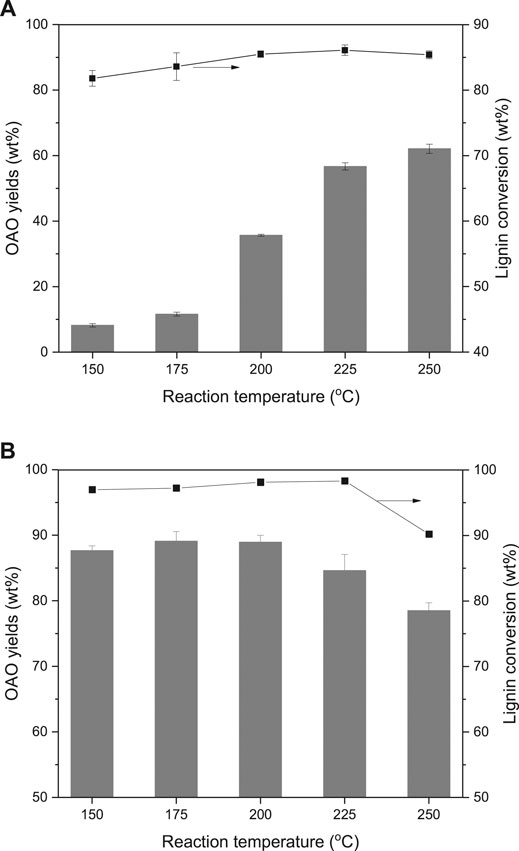
FIGURE 2. Effects of reaction temperature on alkali lignin (A) and organosolv lignin (B) conversion and oligomeric aromatic oxides (OAO) yields when TsOH was the catalyst with 10 wt% loading and kept for 60 min.
It can be observed that with the growth reaction, the temperature performed little influence on AL liquefaction but increased OAO yields. It indicated that the temperature was the key to the advanced degradation of lignin. The OAO yield was from 8.2 to 35.7 wt% when the reaction temperature increased from 150 to 200°C. While it grew from 200 to 250°C, the OAO yield was from 35.7 to 62.1 wt%. Conversely, OAO yields of OL performed little change when increased reaction temperature from 150 to 200°C. The peak value of 89.1 wt% emerged at 175°C. This value was closed to 87.6 wt% at 150°C and 88.9 wt% at 200°C. These results indicated that AL and OL were undergoing two different degradation routes. With the growth of the reaction temperature, OAO yields of OL were decreased to 78.5 wt% when conducted reaction at 250°C. These results implied that condensation of OL derived OAO occurred at high temperature. The OAO yields of AL kept growth at high temperature, which indicated that OAO of AL was more stable than that of OL.
Molecular Weight Analysis of Lignin and Liquefaction Products
Table 1 demonstrates the molecular weight results of milled wood lignin, extracted lignin (AL and OL), and OAO (OAO-AL and OAO-OL). It can note that alkali delignification and organosolv delignification processes decreased the weight molecular weight of 5,644 g/mol (MWL) to 1,692 g/mol (AL) and 1,378 g/mol (OL), respectively. The molecular weight of OL was lower than that of AL. The TsOH catalyzed the liquefaction process decreased AL’s molecular weight from 1,692 g/mol to 785 g/mol (Mw). It was observed that the OL-OAO molecular weight was 788 g/mol (Mw) in OL liquefaction, which was close to AL-OAO molecular weight. These results indicated that a molecular weight of about 780 g/mol was the limit of lignin liquefaction by the TsOH catalyst. Further degradation depended on C-C linkages cleavage rather than β-ether linkages cleavage at high temperatures. Group characterizations should provide more information on lignin degradation by TsOH catalysis.
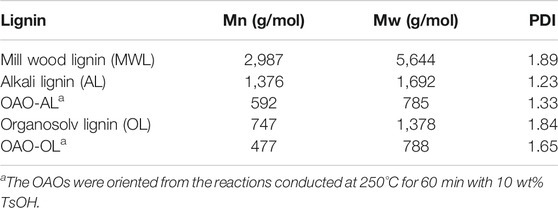
TABLE 1. Weight-average molecular weights (Mw), number-average (Mn) molecular weights, and polydispersity (Mw/Mn) of milled wood lignin, extracted lignin (AL and OL), and oligomeric aromatic oxides (OAO-AL and OAO-OL).
Heteronuclear Single Quantum Coherence Spectroscopy-Nuclear Magnetic Resonance Spectroscopy Analysis of Lignin and Liquefaction Products
Figure 3 illustrated the side chain HSQC-NMR spectra of native lignin, extracted lignin, and their liquefaction products. HSQC-NMR clarified the structural determination of lignin as well as a lignin-carbohydrate linkage to unveil the structural change. The NMR signal assignments of spectra were assigned according to references (Wen et al., 2012; Wen et al., 2013; Yang et al., 2014). OL performed signals on δC/δH 72.0/4.85 ppm and 59.9/3.80 ppm, which were assigned to Cα-Hα and Cγ-Hγ of β-O-4 linkage, respectively. The spectrum performed calls on δC/δH 83.9/4.29 ppm and 85.8/4.10 ppm, which were given to Cβ-Hβ of β-O-4 (G/H) and β-O-4(S), respectively. Signals of resinol (β-β) and phenylcoumaran (β-5) were not obviously in the spectrum. It observed a few amounts of β-O-4(S) linkages in the scope of OL-OAO. These results implied that OL liquefaction was undergoing the mechanism of β-O-4 linkage acidolysis, which could be clarified by many reports on native lignin liquefaction (Li et al., 2015). Also, residue signals of β-O-4(S) linkages confirmed that β-O-4(S) connections were more stable than that of β-O-4(G) in the acidolysis process. In the AL spectrum, 2-O-Ac-X and xylan signals emerged, and of β-O-4 linkage were disappeared. Calls assigned to xylan identified in AL, with its C2-H2, C3-H3, and C4-H4 correlations (X2, X3, and X4) at δC/δH 72.5/3.05, 73.6/3.25, and 75.3/3.56 ppm, respectively. It found correlations of β-D-xylopyranoside units (X1) at δC/δH 101.5/4.30 ppm. Correlation of C2-H2 at δC/δH 73.5/4.64 ppm suggested that the AL has a 2-O-Ac-X linkage. The observations of the AL-OAO spectrum indicated methoxy and aliphatic hydroxyl groups existed, and no native lignin connection after liquefaction. It implied that AL liquefaction was achieved by dehydrated ether linkages cracking rather than typical β-O-4 linkages acidolysis, which should be confirmed by further deduction.
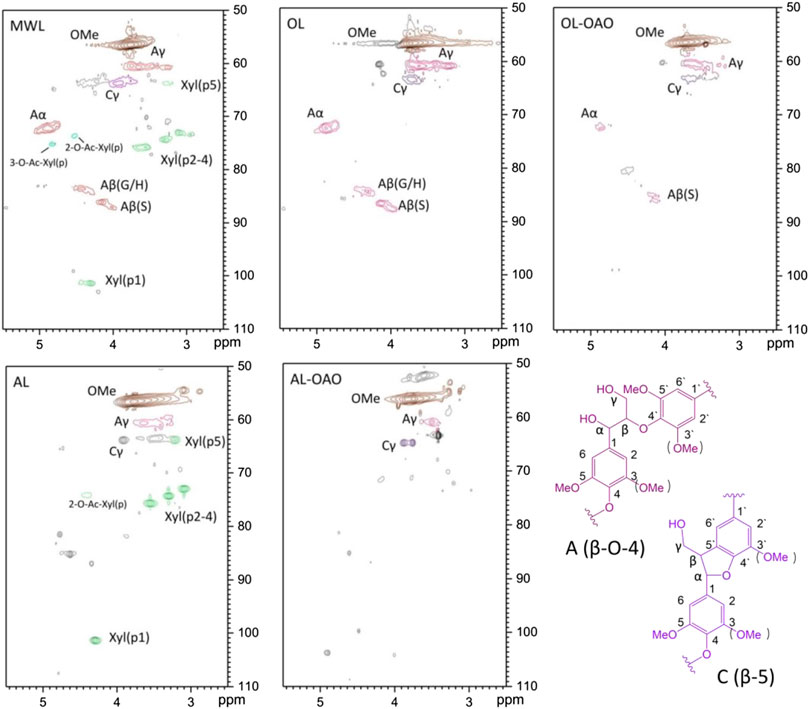
FIGURE 3. HSQC-NMR spectra of side regions resulted from MWL, organosolv lignin (AL), alkali lignin (OL), and oligomeric aromatic oxides obtained from AL (AL-OAO) and OL (OL-OAO). (The OAO was received by the reactions conducted at 250°C for 60 min with 10 wt% TsOH).
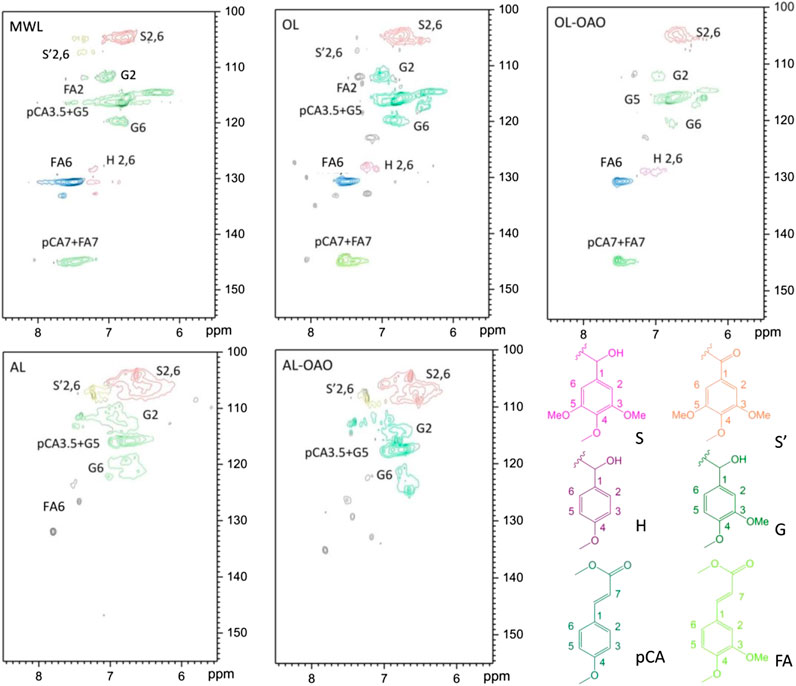
FIGURE 4. HSQC-NMR spectra of aromatic regions resulted from MWL, organosolv lignin (AL), alkali lignin (OL), and oligomeric aromatic oxides obtained from AL (AL-OAO) and OL (OL-OAO). (The OAO was received by the reactions conducted at 250°C for 60 min with 10°wt% TsOH).
Figure 5 performs the HSQC-NMR spectra of the aromatic region. It observed the significant correlation for the S2, 6 at δC/δH 103.8/6.69 ppm in the aromatic region spectrum of OL, and correlation for the oxidized S units (S’) was at δC/δH 106.1/7.28 ppm. The H2, 6 was at δC/δH 127.8/7.17 ppm, and the pCA2, 6 emerged at δC/δH 130.2/7.48 ppm. It also showed that the signals of pCA3, 5 at δC/δH 115.8/6.83 ppm, overlapped with G5. Also, C-H correlations of G2 and G6 emerged at δC/δH 110.7/7.35 ppm and 118.8/6.77 ppm, and the pCA7 correlation was at δC/δH 144.8/7.51. The signals observed were highly consistent with the data for p-coumarate units. The FA7 correlation coincides with that of pCA7 at δC/δH 144.8/7.51 ppm. After liquefaction, signals of S’ had disappeared, which might be due to TsOH catalyzed α-ketone. Remained calls of S, G, H, pCA, and FA indicated that the aromatic structures remained in the liquefaction process. In AL, signals assigned to H and pCA cannot found on the spectrum. After liquefaction, S, S’ and G’ indications were still existing in AL-OAO because S units and G units were relatively stable compared to that of H units. Accordingly, acid-catalyzation kept the liquefaction of most aromatic structures. Methoxy groups (G-type and S-type) existed in degraded lignin, which allowed OAO to be employed as a methanol gasoline anti-knock additive. (Demirbaş, 2000a).
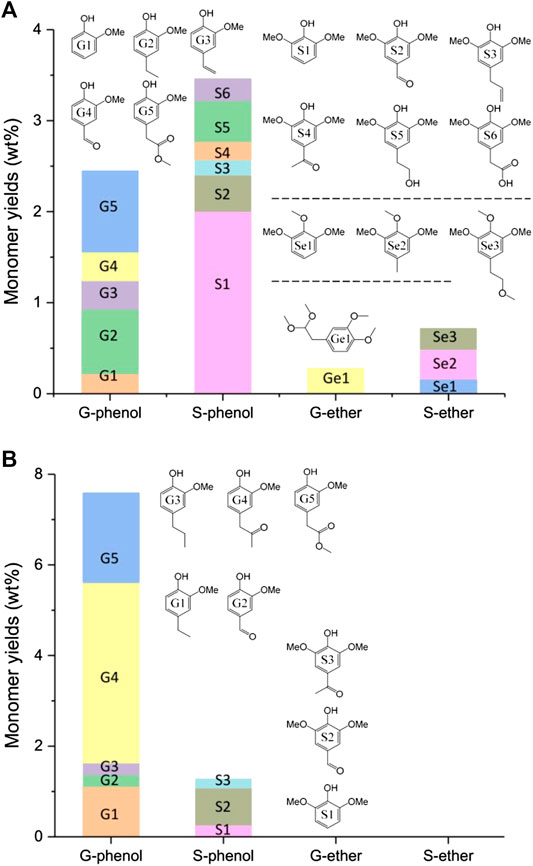
FIGURE 5. Monomers analysis of liquefaction products obtained from alkali lignin (A) and organosolv lignin (B) when TsOH was the catalyst with 10 wt% loading and kept for 60 min in each reaction.
Monomer Analysis of Lignin Conversion Products
Figures 5A B, illustrate the monomers detected by AL and OL liquefaction when catalyzed by TsOH, respectively. Lignin liquefaction generated small amounts of aromatic monomers, and these monomers provided evidence of lignin structure change from low molecular weight lignin change. The most prevalent compounds were monomeric phenolic compounds and their derivatives, including phenolic species substituted with methoxyl and ether groups. According to para and ortho substitutions, these aromatics could be divided into four groups: G-phenol, S-phenol, G-ether, and S-ether. The four group products obtained from AL liquefaction were 2.45 wt% of G-phenol, 3.46 wt% of S-phenol, 0.28 wt% of G-ether, and 0.49 wt% of S-ether. The monomers’ total amounts were 6.91 wt%, and phenolic compounds were higher than those of ether groups.
Lee et al. (2015) has classified the products from Kraft lignin ethanol-water mixture liquefaction: direct crack products, secondary crack products, and hydrolysis or esterification products. Herein, G2, 3, and 4 and S2, 3, 4, 5, and 6 in Figures 5A were the direct crack products of AL liquefaction due to their non-ester para-substituents of phenolic hydroxyl groups. Guaiacol and 2, 6-dimethoxy phenol were secondary crack products. Ether and G5 were the etherification and esterification products. Phenolic hydroxyl groups of lignin could undergo O-alkylation in the process of lignin degradation (Sad et al., 2008; Goodarznia and Smith, 2010; Sad et al., 2011). The resulting aromatic ether resisted condensation as the ether group decreased ortho sites activity with formaldehyde in the liquefaction process (Huang et al., 2014; Huang et al., 2015). Secondary crack and etherification products of 2, 6-dimethoxy phenol, and aromatic ethers were more stable than direct crack products at high temperatures.
Figures 5B gave the Monomers of OL liquefaction. The monomers obtained from OL liquefaction were only phenolic compounds. The total amounts of the monomers were 8.87 wt%, and G-phenol was the primary product. The G1, 2, 3, and 4, and S2 and 3 were direct crack products of AL liquefaction. The 2, 6-dimethoxy phenol was secondary crack products, and G5 was the esterification product. No ether monomer emerged after liquefaction. As discussed in Figure 2, optimum conditions for OL-OAO production were at 175°C. This reaction temperature may not meet the requirements of intermediates secondary crack. Additionally, the amounts of 2, 6-dimethoxy phenol from OL liquefaction (0.25 wt%) were lower than AL liquefaction (2 wt%). These results suggested that direct crack was the primary reaction of OL liquefaction. Stable products at high temperatures of aromatic ethers did not exist in the final products.
Accordingly, it suggested that TsOH acts as different roles in two lignin liquefaction processes in this study. As demonstrated in Figure 6, TsOH was the β-ether linkage acidolysis catalyst in the process of OL liquefaction (Duan et al., 2018). The primary step was a dehydration reaction, and the rate-determining step for β-ether hydrolysis, by yielding enol aryl ether. Enol aryl ether intermediate hydrolyzed to guaiacol and unstable α-ketocarbinol (Jia et al., 2010). The unstable intermediate gradually converted to a mixture of ketones called Hibbert’s ketones. Hence, it can be found that are several monomeric ketones in OL liquefaction products. In the process of AL extraction, β-ether linkage cleaved by the conversion of the phenolate unit into the corresponding quinone methide intermediate and further transformed to alkali-stable vinyl ether. Vinyl ether was more stable than native β-ether due to conjugated alkene (Parthasarathi et al., 2015). In AL liquefaction, the TsOH acted as two roles of acidolysis catalyst and etherification catalyst. AL first degraded by C-O bonds cleaved and generated a phenolic hydroxyl group simultaneously. Phenolic groups can link with formaldehyde generated from Cγ hydroxyl groups’ cleavage at high temperatures (Huang et al., 2015). TsOH catalyzed phenolic groups O-alkylation. The formation of undissolved parts and liquid resulted from simultaneous de- and re-polymerization in the process of AL liquefaction at high temperatures.
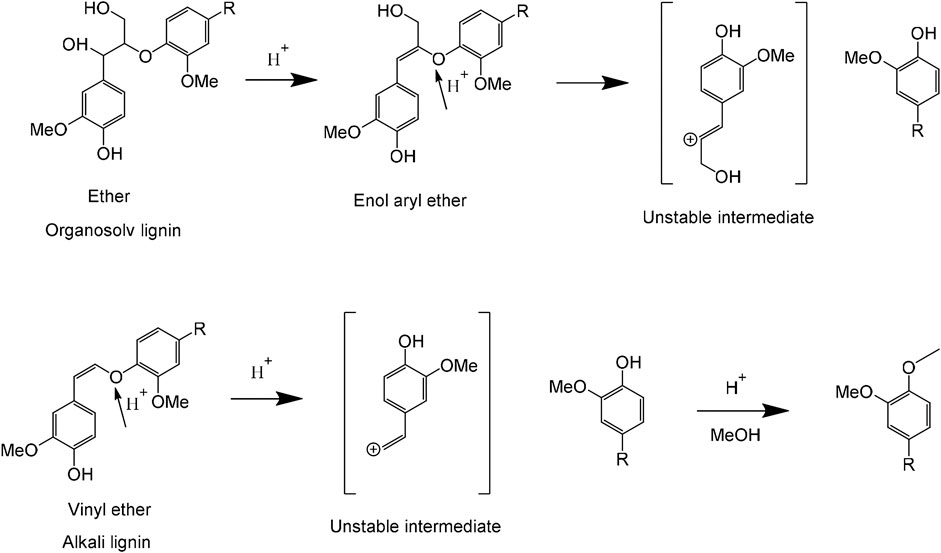
FIGURE 6. Liquefaction mechanisms of alkali lignin and organosolv lignin catalyzed by p-toluenesulfonic acid (TsOH) in methanol solvent.
Conclusion
Sugarcane bagasse lignin was liquefied for the production of OAO, which will be used as a methanol gasoline additive. The evaluation of acid performance was based on AL conversion; and gave the following order of catalytic efficiency: organic acid > inorganic acid salt > Lewis acid > sulfuric acid. The optimum reaction temperature of AL and OL were 250°C and 175°C, respectively. Lignin conversion and OAO yield of OL conversion were higher than that of AL. HSQC-NMR and GC/MS results showed that the liquefaction mechanisms were different, although they used the same catalyst of TsOH. OL liquefaction was by native ether linkages acidolysis. AL liquefaction was by simultaneous degradation and stabilization of aromatic etherification.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
YC conceptualized the work, designed the experiment, and wrote the manuscript. SY conducted lignin liquefaction experiments, and LJ completed the characterizations and analyzed the data. HS discussed the results and tested OAO feasibility. All authors have approved the manuscript and agreed with submission to Frontiers in Energy Research.
Funding
This work was supported by the Guangxi Key Laboratory of Chemistry and Engineering of Forest Products (GXFC1503).
Conflict of Interest
Author HS was employed by company Ningxia Pantai Energy Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Chen, J., Fan, X., Zhang, L., Chen, X., Sun, S., and Sun, R. (2020). Research progress in lignin‐based slow/controlled release fertilizer. ChemSusChem 13, 4356–4366. doi:10.1002/cssc.202000455
Demirbaş, A. (2000a). Conversion of biomass using glycerin to liquid fuel for blending gasoline as alternative engine fuel. Energ. Convers. Manage. 41, 1741–1748. doi:10.1016/S0196-8904(00)00015-7
Demirbaş, A. (2000b). Mechanism of liquefaction and pyrolysis reactions of biomass. Energ. Convers. Manage. 41, 633–646. doi:10.1016/S0196-8904(99)00130-2
Duan, D., Wang, Y., Ruan, R., Tayier, M., Dai, L., Zhao, Y., et al. (2018). Comparative study on various alcohols solvolysis of organosolv lignin using microwave energy: physicochemical and morphological properties. Chem. Eng. Process. 126, 38–44. doi:10.1016/j.cep.2017.10.023
Goodarznia, S., and Smith, K. J. (2010). Properties of alkali-promoted Cu–MgO catalysts and their activity for methanol decomposition and C2-oxygenate formation. J. Mol. Catal. A 320, 1–13. doi:10.1016/j.molcata.2010.01.002
Gosselink, R. J. A., Jong, E. D., Guran, B., and Abächerli, A. (2004). Co-ordination network for lignin-standardisation, production and applications adapted to market requirements (EUROLIGNIN). Ind. Crop. Prod. 20, 121–129. doi:10.1016/j.indcrop.2004.04.015
Huang, X., Tamás, I. K., Boot, M. D., and Hensen, E. J. M. (2014). Catalytic depolymerization of lignin in supercritical ethanol. ChemSusChem 7, 2276–2288. doi:10.1002/cssc.201402094
Huang, X., Tamás, I. K., Boot, M. D., and Hensen, E. J. M. (2015). Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 17, 4941–4950. doi:10.1039/c5gc01120e
Jasiukaitytė, E., Kunaver, M., and Crestini, C. (2012). Lignin structural changes during liquefaction in acidified ethylene glycol. J. Wood Chem. Technol. 32, 342–360. doi:10.1080/02773813.2012.698690
Jia, S., Cox, B. J., Guo, X., Zhang, Z. C., and Ekerdt, J. G. (2010). Cleaving the β-O-4 bonds of lignin model compounds in an acidic ionic liquid, 1-H-3-methylimidazolium chloride: an optional strategy for the degradation of lignin. Chemsuschem 3, 1078–1084. doi:10.1002/cssc.201000112
Joffres, B., Lorentz, C., Vidalie, M., Laurenti, D., Quoineaud, A. A., Charon, N., et al. (2014). Catalytic hydroconversion of a wheat straw soda lignin: characterization of the products and the lignin residue. Appl. Catal. B-Environ. 145, 167–176. doi:10.1016/j.apcatb.2013.01.039
Jongerius, A. L., Bruijnincx, P. C. A., and Weckhuysen, B. M. (2013). Liquid-phase reforming and hydrodeoxygenation as a two-step route to aromatics from lignin. Green Chem. 15, 3049–3056. doi:10.1039/c3gc41150h
Kloekhorst, A., Shen, Y., Yie, Y., Fang, M., and Heeres, H. J. (2015). Catalytic hydrodeoxygenation and hydrocracking of alcell lignin in alcohol/formic acid mixtures using a Ru/C catalyst. Biomass Bioenerg. 80, 147–161. doi:10.1016/j.biombioe.2015.04.039
Lee, H. S., Jae, J., Ha, J. M., and Dong, J. S. (2015). Hydro- and solvothermolysis of kraft lignin for maximizing production of monomeric aromatic chemicals. Bioresour. Technol. 203, 142–149. doi:10.1016/j.biortech.2015.12.022
Li, C., Zhao, X., Wang, A., Huber, G. W., and Zhang, T. (2015). Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 115, 11559–11624. doi:10.1021/acs.chemrev.5b00155
Lin, L., Yao, Y., and Shiraishi, N. (2005). Liquefaction mechanism of β-O-4 lignin model compound in the presence of phenol under acid catalysis. Part 2. Reaction behaviour and pathways. Holzforschung 55, 617–624. doi:10.1515/HF.2001.102
Lin, L., Yao, Y., Yoshioka, M., and Shiraishi, N. (1997). Liquefaction mechanism of lignin in the presence of phenol at elevated temperature without catalysts. Studies on ß-O-4 lignin model compound. II. Reaction pathway. Holzforschung 51, 325–332. doi:10.1515/hfsg.1997.51.4.325
Lojkásek, M., Růžička, V., and Kohoutová, A. (1992). Solubility of water in blends of gasoline, methanol and a solubilizer. Fluid Phase Equilibr. 71, 113–123. doi:10.1016/0378-3812(92)85008-V
Lora, J. H., and Glasser, W. G. (2002). Recent industrial applications of lignin: a sustainable alternative to nonrenewable materials. J. Polym. Environ. 10, 39–48. doi:10.1023/A:1021070006895
Miller, J. E., Evans, L., Littlewolf, A., and Trudell, D. E. (1999). Batch microreactor studies of lignin and lignin model compound depolymerization by bases in alcohol solvents. Fuel 78, 1363–1366. doi:10.1016/S0016-2361(99)00072-1
Mousavioun, P., and Doherty, W. O. S. (2010). Chemical and thermal properties of fractionated bagasse soda lignin. Ind. Crop. Prod. 31, 52–58. doi:10.1016/j.indcrop.2009.09.001
Parthasarathi, R., Romero, R. A., Redondo, A., and Gnanakaran, S. (2015). Theoretical study of the remarkably diverse linkages in lignin. J. Phys. Chem. Lett. 2, 2660–2666. doi:10.1021/jz201201q
Prinsen, P., Rencoret, J., Gutiérrez, A., Liitiä, T., Tamminen, T., Colodette, J. L., et al. (2013). Modification of the lignin structure during alkaline delignification of eucalyptus wood by kraft, soda-aq, and soda-O2 cooking. Ind. Eng. Chem. Res. 52, 15702–15712. doi:10.1021/ie401364d
Ragauskas, A., Beckham, G., Biddy, M., Chandra, R., Chen, F., Davis, M., et al. (2014). Lignin valorization: improving lignin processing in the biorefnery. Science 344, 709–718. doi:10.1126/science.1246843
Sad, M. E., Neurock, M., and Iglesia, E. (2011). Formation of C-C and C-O bonds and oxygen removal in reactions of alkanediols, alkanols, and alkanals on copper catalysts. J. Am. Chem. Soc. 133, 20384–20398. doi:10.1021/ja207551f
Sad, M. E., Padró, C. L., and Apesteguía, C. R. (2008). Synthesis of cresols by alkylation of phenol with methanol on solid acids. Catal. Today 133, 720–728. doi:10.1016/j.cattod.2007.12.074
Saito, T., Perkins, J. H., Vautard, F., Meyer, H. M., Messman, J. M., Tolnai, B., et al. (2014). Methanol fractionation of softwood kraft lignin: impact on the lignin properties. Chemsuschem 7, 221–228. doi:10.1002/cssc.201300509
Sarkanen, K. V., and Hoo, L. H. (1981). Kinetics of hydrolysis of erythro-guaiacylglycerol beta-(2-methoxyphenyl) ether and its veratryl analogue using HCl and aluminum chloride as catalysts. J. Wood Chem. Technol. 1, 11–27. doi:10.1080/02773818108085091
Shen, X. J., Wang, B., Huang, P. L., Wen, J. L., and Sun, R. C. (2016). Understanding the structural changes and depolymerization of Eucalyptus lignin under mild conditions in aqueous AlCl3. RSC Advance. 6, 45315–45325. doi:10.1039/C6RA08945C
Shu, R., Long, J., Yuan, Z., Zhang, Q., Wang, T., Wang, C., et al. (2015). Efficient and product-controlled depolymerization of lignin oriented by metal chloride cooperated with Pd/c. Bioresour. Technol. 179, 84–90. doi:10.1016/j.biortech.2014.12.021
Singh, R., Prakash, A., Dhiman, S. K., Balagurumurthy, B., and Bhaskar, T. (2014). Hydrothermal conversion of lignin to substituted phenols and aromatic ethers. Bioresour. Technol. 165, 319–322. doi:10.1016/j.biortech.2014.02.076
Siwale, L., Kristóf, L., Bereczky, A., Mbarawa, M., and Kolesnikov, A. (2014). Performance, combustion and emission characteristics of n-butanol additive in methanol-gasoline blend fired in a naturally-aspirated spark ignition engine. Fuel Process. Technol. 118, 318–326. doi:10.1016/j.fuproc.2013.10.007
Tang, Z., Zhang, Y., and Guo, Q. (2010). Catalytic hydrocracking of pyrolytic lignin to liquid fuel in supercritical ethanol. Ind. Eng. Chem. Res. 49, 2040–2046. doi:10.1021/ie9015842
Upton, B. M., and Kasko, A. M. (2016). Strategies for the conversion of lignin to high-value polymeric materials: review and perspective. Chem. Rev. 116, 2275–2306. doi:10.1021/acs.chemrev.5b00345
Vanholme, R., Demedts, B., and Morreel, K. (2010). Lignin biosynthesis and structure. Plant Physiol. 153, 895–905. doi:10.1104/pp.110.155119
Voitl, T., and Philipp, R. (2010). Oxidation of lignin using aqueous polyoxometalates in the presence of alcohols. ChemSusChem 1, 763–769. doi:10.1002/cssc.200800050
Wang, K., Xu, F., and Sun, R. C. (2010). Molecular characteristics of Kraft-AQ pulping lignin fractionated by sequential organic solvent extraction. Int. J. Mol. Sci. 11, 2988–3001. doi:10.3390/ijms11082988
Wang, Z., Dang, D., Lin, W., and Song, W. (2016). Catalytic upgrading of phenolic oil by etherification with methanol. Chem. Eng. Technol. 39, 1797–1803. doi:10.1002/ceat.201600252
Wen, J., Anuj, K., and Stergios, A. (2018). Liquefaction of lignocellulosic materials and its applications in wood adhesives-a review. Ind. Crop. Prod. 124, 325–342. doi:10.1016/j.indcrop.2018.07.053
Wen, J. L., Sun, S. L., Xue, B. L., and Sun, R. C. (2013). Recent advances in characterization of lignin polymer by solution-state nuclear magnetic resonance (NMR) methodology. Materials 6, 359–391. doi:10.3390/ma6010359
Wen, J. L., Xue, B. L., Xu, F., and Sun, R. C. (2012). Unveiling the structural heterogeneity of bamboo lignin by in situ HSQC NMR technique. Bioenerg. Res. 5, 886–903. doi:10.1007/s12155-012-9203-5
Yang, H., Zheng, X., Yao, L., and Xie, Y. (2013). Structural changes of lignin in the soda-aq pulping process studied using the carbon-13 tracer method. Bioresources 9, 176–190. doi:10.15376/biores.9.1.176-190
Yang, S., Wen, J. L., Yuan, T. Q., and Sun, R. C. (2014). Characterization and phenolation of biorefinery technical lignins for lignin–phenol–formaldehyde resin adhesive synthesis. Rsc Advance. 4, 57996–58004. doi:10.1039/c4ra09595b
Yao, L., Yang, H., Yoo, C. G., Meng, X., Pu, Y., Hao, N., et al. (2018). Characteristics of lignin fractions from dilute acid pretreated switchgrass and their effect on cellobiohydrolase from trichoderma longibrachiatum. Front. Energy Res. 6, 1. doi:10.3389/fenrg.2018.00001
Yoshikawa, T., Shinohara, S., Yagi, T., Ryumon, N., Nakasaka, Y., Tago, T., et al. (2014). Production of phenols from lignin-derived slurry liquid using iron oxide catalyst. Appl. Catal. B Environ. 146, 289–297. doi:10.1016/j.apcatb.2013.03.010
Yuan, Z., Cheng, S., Leitch, M., and Xu, C. (2010). Hydrolytic degradation of alkaline lignin in hot-compressed water and ethanol. Bioresour. Technol. 101, 9308–9313. doi:10.1016/j.biortech.2010.06.140
Keywords: sugarcane bagasse, alkali lignin, liquefaction, etherification, organic acids
Citation: Cheng Y, Yang S, Ji L and Shi H (2020) Oligomeric Aromatic Oxides (OAO) Production From Sugarcane Bagasse Lignin by Acid-Catalyzed Solvothermal Liquefaction in Methanol. Front. Energy Res. 8:608415. doi: 10.3389/fenrg.2020.608415
Received: 20 September 2020; Accepted: 14 October 2020;
Published: 26 November 2020.
Edited by:
Shaolong Sun, South China Agricultural University, ChinaReviewed by:
Xiaojun Shen, Haidian District, ChinaLing-Ping Xiao, Dalian Polytechnic University, China
Copyright © 2020 Cheng, Yang, Ji and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Cheng, yicheng@mail.buct.edu.cn