Exploring the Diversity and Metabolic Profiles of Bacterial Communities Associated With Antarctic Sponges (Terra Nova Bay, Ross Sea)
 Maria Papale1
Maria Papale1  Carmen Rizzo2
Carmen Rizzo2  Renato Fani3 Marco Bertolino4 Gabriele Costa4 Andreu Paytuví-Gallart5
Renato Fani3 Marco Bertolino4 Gabriele Costa4 Andreu Paytuví-Gallart5  Stefano Schiaparelli4,6 Luigi Michaud7 Maurizio Azzaro1
Stefano Schiaparelli4,6 Luigi Michaud7 Maurizio Azzaro1  Angelina Lo Giudice1*
Angelina Lo Giudice1*- 1Institute of Polar Sciences, National Research Council (CNR-ISP), Messina, Italy
- 2Stazione Zoologica Anton Dohrn, National Institute of Biology, Department BIOTECH, Villa Pace, Messina, Italy
- 3Department of Biology, University of Florence, Florence, Italy
- 4Department of Earth, Environment and Life Sciences (DISTAV), University of Genoa, Genoa, Italy
- 5Sequentia Biotech S.L., Barcelona, Spain
- 6Italian National Antarctic Museum (MNA, Section of Genoa), University of Genoa, Genoa, Italy
- 7Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, Messina, Italy
Complex cell-to-cell interactions (including the production of antibiotics and the quorum sensing phenomenon) occur between benthic marine organisms and bacteria, leading to the establishment of synergistic interactions, especially in extreme and harsh environments, such as Antarctica. Despite this, current data concerning the composition, host- and site-relatedness, and biotechnological values of the bacterial community associated with Antarctic sponges are limited to few works, resulting in a still fragmented and incomplete knowledge. In this context, a total of 11 sponge species (belonging to Demospongiae and Hexactinellida) from the Terra Nova Bay area (Ross Sea) were explored for the associated bacterial diversity by the ION Torrent sequencing. An additional predictive functional analysis on 16S rRNA gene data was performed to unravel metabolic and biotechnological potentials of the associated bacterial communities. Data obtained highlighted the predominance of Proteobacteria, mainly affiliated to Alpha- and Gammaproteobacteria. Retrieved phyla were similarly distributed across samples, with dissimilarities encountered for the sponge Haliclona (Rhizoniera) dancoi (Topsent, 1901). Functional prediction results suggested that the associated bacterial community may be involved in the biosynthesis of antibiotics, quorum sensing, and degradation of aromatic compounds.
Introduction
Symbiosis is one of the most fascinating phenomena occurring in nature, bringing relevant implications from ecological, evolutionistic, and bioprospecting perspectives (Li et al., 2014). Marine sponges are the most ancient multicellular invertebrates, peculiar representatives of the benthic communities, and very sensitive indicator organisms (Glasl et al., 2019). Symbiotic relationships involving marine sponges possess a great evolutionary and ecological significance. These peculiar interactions are affected by a diverse set of environmental parameters and require the establishment of complex signal communication systems and fine regulating processes between the host and its symbionts, as well as within the symbiotic community itself (Mohamed et al., 2008; Mangano et al., 2009, 2018). Several studies have contributed to the prokaryotic diversity characterization of sponge-associated assemblages. They have been mainly aimed at establishing the taxonomical specificity of sponge core microbiome and/or the possible vertical or horizontal transmission processes involved in the acquisition and maintenance of ancient symbionts (Taylor et al., 2007; Webster et al., 2010). However, greater efforts are available for the exploration of sponges inhabiting more accessible study areas, mainly tropical and temperate habitats, while studies performed in polar regions remain very scant (Cárdenas et al., 2014; Lo Giudice and Rizzo, 2018; Lo Giudice et al., 2019a).
Even though sponges dominate vast areas of the Antarctic shelves, the sponge–bacteria association has been seldom explored (Savoca et al., 2019 and references therein). Recently, interesting evolutionary and ecological insights have been pointed out, and they highlight the great versatility of Antarctic sponges as hosts of a wide range of microorganisms, with Proteobacteria and Bacteroidetes dominance within the whole prokaryotic community (Webster et al., 2004; Rodríguez-Marconi et al., 2015; Cárdenas et al., 2018). Rodríguez-Marconi et al. (2015) also emphasized differences occurring in the taxonomical structure of sponge-associated bacterial assemblages in Antarctic and tropical-temperate regions, by suggesting strong peculiarity and specificity for Antarctic sponges. Indeed, in contrast with some studies reporting the high stability of sponge-associated bacterial assemblages (Cárdenas et al., 2014; Erwin et al., 2015), others detected important shifts under thermal stress (Lemoine et al., 2007; Webster et al., 2008; Fan et al., 2013) or under a combination of stressful factors (Pineda et al., 2016, 2017). Beside culture-independent methods, the cultivable-based approach should not be neglected, as a complementary tool to the molecular approach, gaining useful information in an applicative purview and a better comprehension of metabolic and physiological processes, also in Antarctic sponges (Papaleo et al., 2012; Savoca et al., 2019).
Contrary to bacterial diversity, the metabolic functional aspects of prokaryotic symbionts of Antarctic sponges (from the Antarctic Peninsula) have been only recently explored by Steinert et al. (2019), highlighting the possible role of symbionts in the biosynthesis of secondary metabolites alongside their involvement in the biodegradation of xenobiotics. The predicted functional evaluation of sponge-associated bacterial communities is a new challenge of sponge symbioses, especially in extreme environments whose peculiar conditions might be responsible for the development of unique adaptation strategies. This approach could be a time-reducing tool useful to support the discovery of novel metabolic mechanisms of remarkable interest in a bioprospecting outlook (Sipkema, 2016; Steinert et al., 2019).
In line with these considerations, this work was aimed at expanding our current knowledge on the taxonomic composition of the whole bacterial communities associated with Antarctic sponges, by coupling these new information with a predictive profiling approach applied on 16S rRNA data. The study contributes to provide interesting insights on the diversity, biotechnological potential, and ecological role of bacterial sponge symbionts in the Terra Nova Bay area, one of the most studied in terms of sponge biodiversity in the Ross Sea (Ghiglione et al., 2018 and references therein).
Materials and Methods
Sampling Area
Sampling activities were carried out in the Terra Nova Bay area (hereafter TNB). TNB is often ice-free during polar summer months and span for about 64 km along the coast of Victoria Land, being comprised between Cape Washington and the Drygalski Ice Tongue. It represents an important site for long-term research on the marine ecology of benthic communities (e.g., Piazza et al., 2019, 2020), supporting rich and complex sponge and anthozoan communities, alongside loosely structured and low diversity assemblages, often coexisting in mosaics (e.g., Sarà et al., 1992; Cattaneo-Vietti et al., 1997; Gambi et al., 1997; Cerrano et al., 2009).
Collection of Sponge Specimens
Sponge specimens (n = 12) were collected from four sites (namely, A, B, C, and D) at Terra Nova Bay (Ross Sea, Antarctica) during the XXIX Italian Expedition to Antarctica. Sampling depths ranged between 30 and 271 m (Table 1). Sponge specimen collection (by dredge, SCUBA diver, or remotely operated underwater vehicle, ROV) was authorized by the PRNA project, conformably to the Antarctic Treaty legislation and the SCAR Code of Conduct for the Use of Animals for Scientific Purposes in Antarctica (ATCM XXXIV, 2011). To minimize the risk of environmental contamination (especially in the case of specimens collected by dredging), dissected specimens were rinsed with 0.2 μm filtered seawater (Mangano et al., 2009; Steinert et al., 2019). Briefly, organisms were immediately washed at least three times with filter-sterilized natural seawater to remove transient and loosely attached bacteria and/or debris. Specimens were then placed into individual sterile plastic bags containing filter-sterilized natural seawater and transported directly to the laboratory at 4°C for microbiological processing (within 2 h after sampling). Fragments of each specimen were then preserved in 70% ethanol for taxonomic identification and at –20°C for DNA extraction. Sponge fragments and glass slides of spicules used for morphological identification are curated at the Italian National Antarctic Museum (MNA, Section of Genoa, Italy). The MNA voucher codes of the specimens here studied are reported in Table 1.
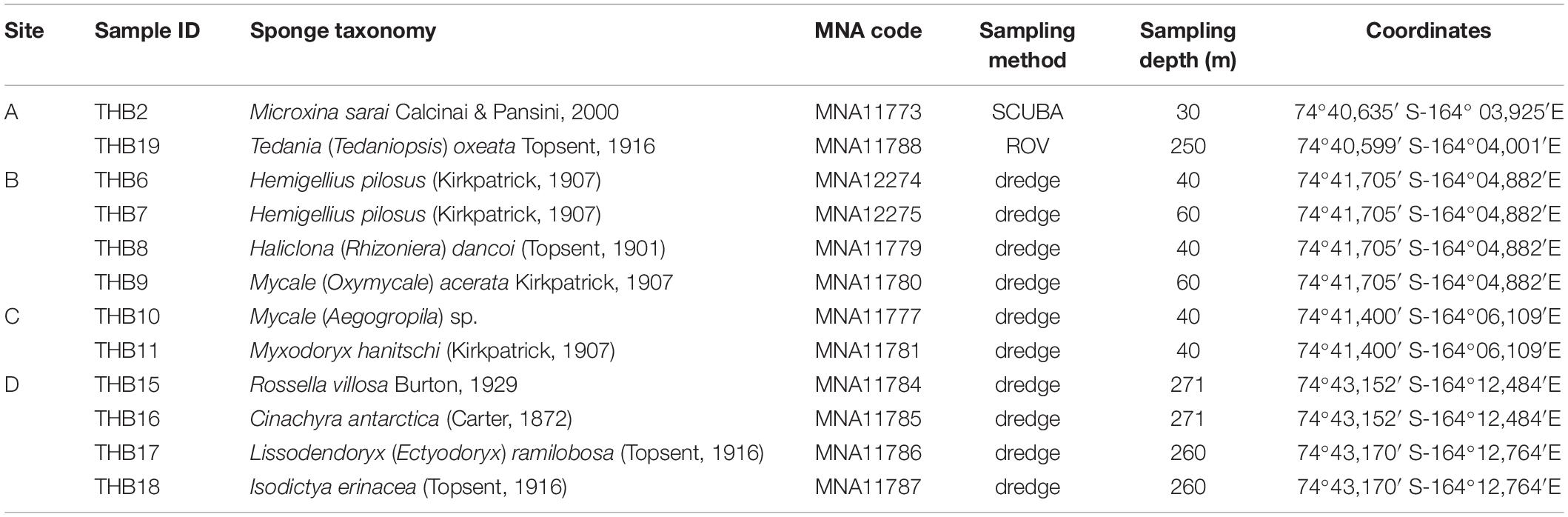
Table 1. Sample identifiers, sponge identification, sampling site and depth, and coordinates.
Sponge Taxonomy
Skeletal architecture was examined by light microscopy. Hand-cut sections of the ectosome and choanosome were made following Hooper (2000). Spicule complement was analyzed according to Rützler (1978). Taxonomic decisions are in agreement with the Systema Porifera (Hooper and van Soest, 2002), the revision of demosponge classification of Morrow and Cárdenas (2015), and the World Porifera Database (WPD) (Van Soest et al., 2018).
Bacterial Community Diversity and Composition
DNA Extraction
Portions of sponge tissues (between 1 and 1.5 g) were reduced to pulp by using sterile pestles. Obtained pulps were used for DNA extraction by employing the PowerSoil DNA extraction kit (MoBio Laboratories, Carlsbad, CA, United States) according to the manufacturer’s instructions. DNA concentration and purity were quantified by using a NanoDrop ND-1000 UV-vis Spectrophotometer (NanoDrop Technologies, United States).
Amplification of 16S rRNA Genes by ION Torrent Sequencing
Extracted DNA was used to amplify the V1–V2 region of 16S rRNA gene of Bacteria (primers 27f 5′-AGAGTTTGATCCTGGCTCAG-3′ and 338 5′-GCT GCC TCC CGT AGG AGT-3′). PCR amplification was carried out under the conditions described by Conte et al. (2018). Briefly, in order to reduce bias in massive sequencing, a two-step PCR protocol was applied: the first step consisted of a conventional PCR, and then amplicons were used as a template for the second PCR with barcoded primers for Ion Torrent sequencing. PCR products were purified using the AgencourtAMPure XP kit (Beckman Coulter, Inc.), according to the manufacturer’s instructions, and then quantified using the Qubit dsDNA HS Assay Kit with Qubit Fluorometer 2.0 (Invitrogen, Thermo Fisher Scientific). Each purified product (20 ng) was pooled for emulsion PCR with Ion PGM Template OT2 400 Kit. Sequencing was performed on an Ion Torrent Personal Genome MachineTM, using the Ion PGM Sequencing 400 Kit and the Ion 314TM chip (all Ion Torrent reagents by Thermo Fischer Scientific) following the manufacturer’s protocols. All steps during amplification and sequencing were checked using negative controls. Ion Torrent sequence data obtained from this study have been registered as an NCBI BioProject PRJNA594262.
Post-run Analysis
At the quality check and trimming steps, read portions with a Phred quality score less than 20 per base and four or more consecutive low-quality base calls were removed using Trimmomatic (sliding window 4:20) (Bolger et al., 2014). Then, denoising was carried out with DADA2 with the denoise-single command (using –p-trim-left 20 –p-trunc-len 0 –i-demultiplexed-seqs). Quality trimming resulted in approximately 338,091 high-quality reads for 12 samples with an average of 28,174 reads (65%) per sample. All samples were included in the downstream quantitative analyses. Sequences were taxonomically classified in QIIME2 (version 2019.4) (Bolyen et al., 2019) by SILVA reference files (SILVA release 132 full-length sequences and taxonomy references) using classify-consensus-blast. Sequences were clustered into OTUs and taxonomically assigned at 97% identity. The resulting OTU table was normalized to the lowest number of reads among the samples (24,980) for the downstream diversity and quantitative analyses.
Predictive Functional Profiling
Metagenomes from 16S data were predicted by QIIME2 with the PICRUSt2 tool (version 2.1.2), which uses evolutionary modeling and a reference genome database (Douglas et al., 2019). The Hidden state prediction method with the mp (Maximum Parsimony) approach was used. To specify how distantly a sequence needs to be placed in the reference phylogeny, before it is excluded, the command –p-max-nsti (cutoff 2) was applied. The accuracy of metagenome predictions was tested trough the Nearest Sequenced Taxon Index (NSTI). The accuracy prediction is related to the presence of closely representative bacterial genomes. The lower values reveal a closer mean relationship. The data obtained by PICRUSt2 were analyzed by Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2017).
Statistical Analyses
Shannon diversity index (H’) for each sample was calculated by QIIME2 software based on the total clustered OTU number. Bacterial community structure and predicted expressed pathways were normalized by Trimmed Mean of M-values (TTM) normalization using R package HTSFilter (Bioconductor 3.10) (Rau et al., 2013). The abundance of all retrieved OTUs was clustered in a heat map using R pheatmap package (v 1.0.12). Abundance results were used to perform statistical analyses using PRIMER v6 for Windows (PRIMER-E Ltd, Plymouth, United Kingdom). Data were analyzed for eventual differences/similarities among the bacterial communities, by considering the sampling sites (A to D) as factors. The Bray–Curtis similarity matrices were used to perform the cluster analysis. Non-metric multidimensional scaling analysis (nMDS) was computed on Bray–Curtis similarity matrices obtained from OTU relative abundances after a log(x+1) transformation.
Results
Sponge Taxonomy
We collected in total 12 sponge specimens and identified 11 species belonging to two classes: Demospongiae (10) and Hexactinellida (1). Demospongiae were represented by three orders: Haplosclerida [i.e., Hemigellius pilosus (Kirkpatrick, 1907), Microxina sarai Calcinai & Pansini, 2000, and Haliclona (Rhizoniera) dancoi (Topsent, 1901)], Poecilosclerida (i.e., Isodictya erinacea (Topsent, 1916), Lissodendoryx (Ectyodoryx) ramilobosa (Topsent, 1916), Mycale spp., Myxodoryx hanitschi (Kirkpatrick, 1907), and Tedania (Tedaniopsis) oxeata Topsent, 1916), and Tetractinellida [i.e., Cinachyra antarctica (Carter, 1872)]; instead, the class Hexactinellida was represented by the sole Lyssacinosida order (i.e., Rossella villosa Burton, 1929) (Table 1).
Diversity of Sponge-Associated Bacterial Communities
Analyzed bacterial sequences were resolved in a total of 177 OTUs. On average, 40 distinct OTUs were found per sample, with the lowest value (21 OTUs) that was observed for the sponges THB6 and THB7, both classified as H. pilosus (Table 2). The nMDS analysis computed on data from the relative abundances of retrieved OTUs is shown in Figure 1. Most sponge samples clustered in a larger group, which included a subcluster composed of H. (R.) dancoi_THB8 and R. villosa_THB15. The sponges H. pilosus_THB6 and THB7 grouped together, as well as L. (E.) ramibolosa_THB17 that clustered separately from the rest.
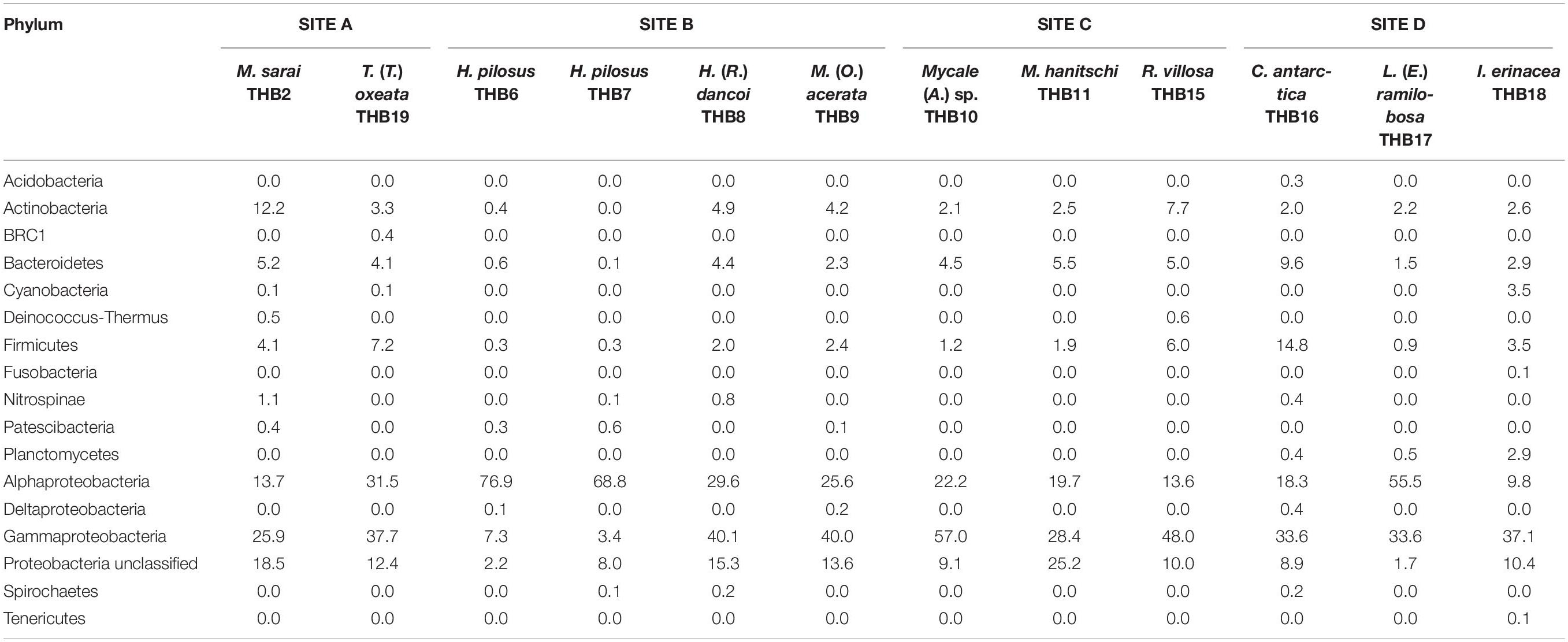
Table 2. Relative abundance values (%) of bacterial phyla retrieved in sponge samples.
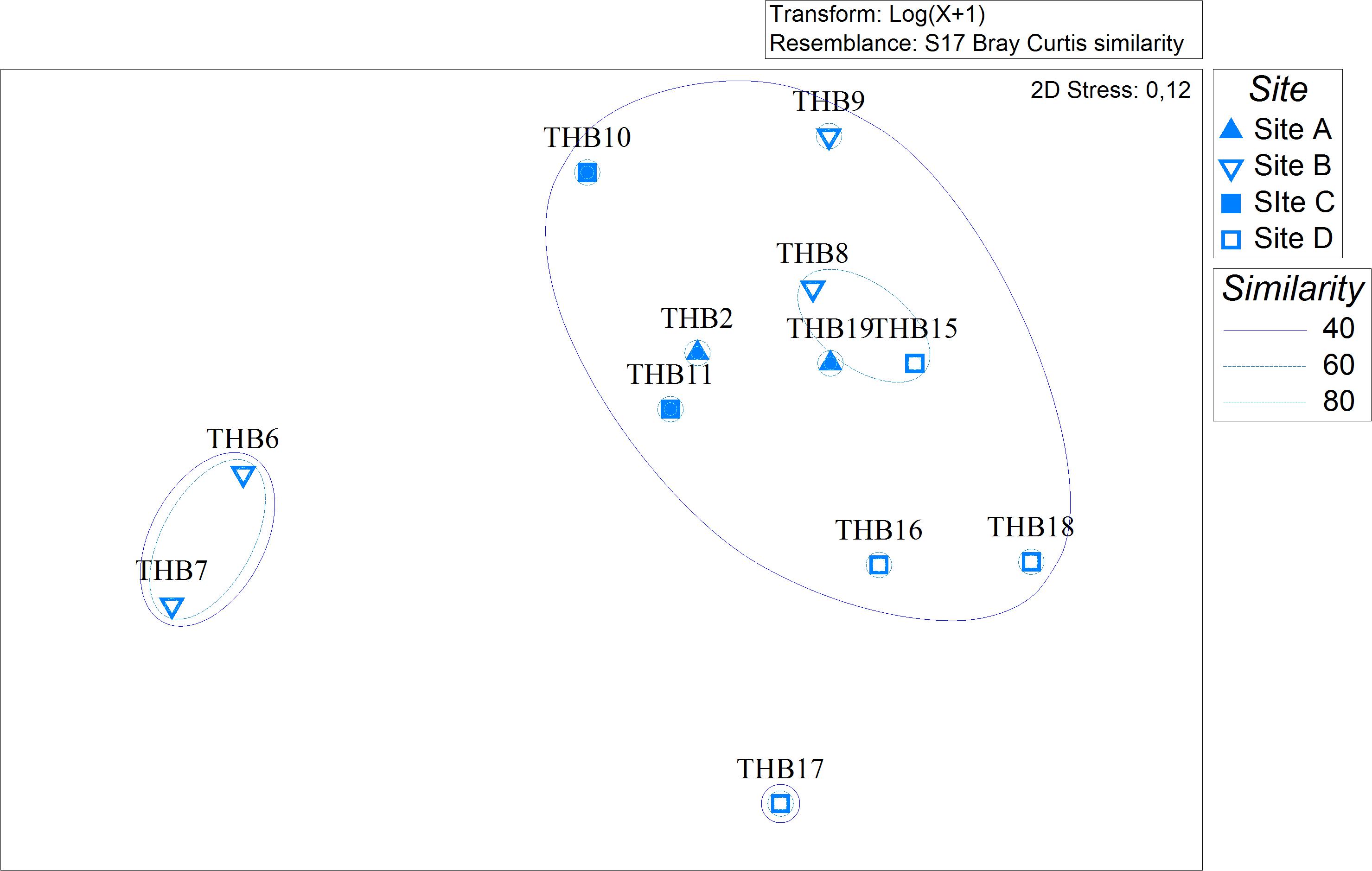
Figure 1. Non-metric multidimensional scaling analysis (nMDS) computed on Bray-Curtis similarity values obtained for relative abundances of bacterial OTUs retrieved in sponge samples.
Overall, OTUs were distributed in 16 bacterial phyla, differently distributed among samples (Figure 2 and Table 2). Proteobacteria dominated in all samples (range 57.3–90.8% of the total community) and were particularly abundant within the bacterial community associated with L. (E.) ramilobosa_THB17 (90.8% of sequences for this sponge). Minor contributes were provided by Bacteroidetes, Actinobacteria, and Firmicutes, accounting on average for 7.3, 7.1, and 6.5% of the total community, respectively. Bacteroidetes were mainly represented in C. antarctica_THB16 (9.6%), while Actinobacteria presented a higher relative abundance in M. sarai_THB2 (12.2%). Firmicutes resulted considerably abundant in C. antarctica_THB16 (14.8%), whereas they ranged from 0.3 to 7.2% in all other sponge samples. The relative abundances of the remaining phyla (i.e., Acidobacteria, BRC1, Cyanobacteria, Deinococcus-Thermus, Fusobacteria, Nitrospinae, Patescibacteria, Planctomycetes, Spirochaetes, Tenericutes), among which there were representatives of some typical sponge-associated bacteria, were mainly less than ≤1% each, and they were generally not shared among all sponge specimens (Table 2). Within Proteobacteria, Alpha- and Gammaproteobacteria were well represented. Alphaproteobacteria were particularly abundant within the bacterial community associated with H. pilosus (samples THB6 and THB7) and L. (E.) ramilobosa_THB 17. Gammaproteobacteria mainly dominated the bacterial communities associated with Mycale (A.) sp._THB10 and R. villosa_THB15. Cyanobacteria were quite absent in all sponge samples.
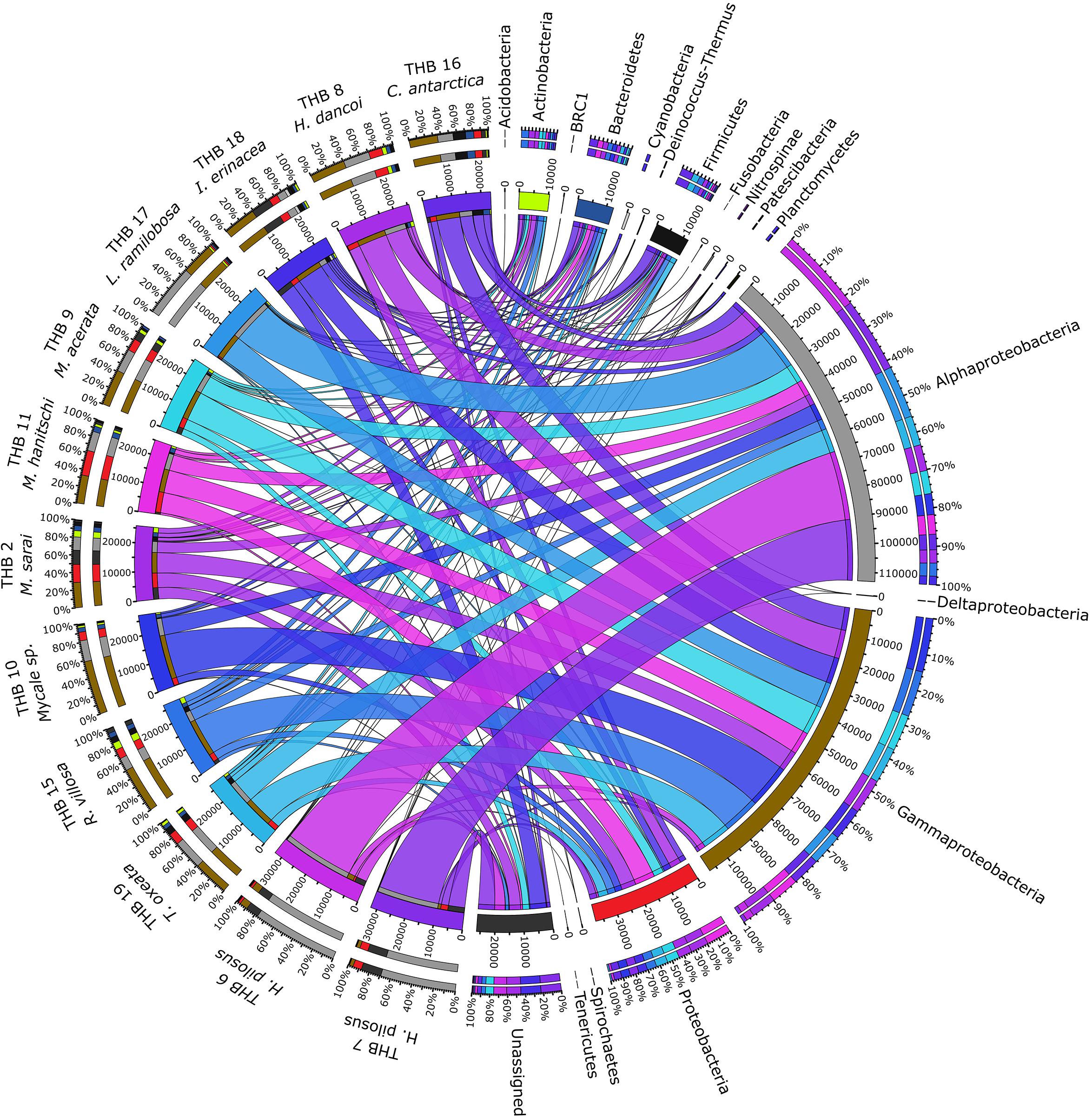
Figure 2. The figure shows the connection among main phyla found in association with the different analyzed sponges. The sponges analyzed are shown on the right of the image, with the color scale from blue to purple (the colors of the ribbons inside the figure). Main phyla found in different colors are shown on the left (colors from top to bottom: orange, yellow, sea blue, silver, gray, black, light green, dark blue, blue, ash, light gray, teal, brown, red, dark green, light green and dark gray). Ribbon size in the figure codifies cell value associated with Sponge/phyla segment pair. Finally, the histograms present under the different sponge species report the percentage distribution of the different phyla inside the sponge.
The 70% of the total high-quality bacterial sequences were classified at genus level, by resulting in a total of 157 genera (from 0.01 to 38.2% of total sequences) (heatmaps in Figure 3). In line with their marked dominance, a high number of retrieved genera were affiliated with Alpha- and Gammaproteobacteria. In detail, among Alphaproteobacteria, a total of 46 genera were detected, with the predominance of Methylobacterium (up to 38.2%) that occurred at high percentages in almost all samples (exceptions were H. pilosus_THB6 and THB7). The genus Sphingomonas showed a similar distribution among samples, with the highest relative abundance (15.9%) in H. (R.) dancoi_THB8. Conversely, the genera Thalassobius and Amylibacter resulted dominant in H. pilosus_THB6 and THB7 (19.9 and 7.2% in THB6, respectively; 16.3 and 6.3% in THB7, respectively). The genus Bradyrhizobium was exclusively detected in M. hanitschi_THB11 (20.5% of total sequences). Among Gammaproteobacteria, the genus Methylobacter was retrieved in L. (E.) ramilobosa_THB17 (9%), while Erwinia representatives were detected at a high percentage (on average 7.1%) in almost all samples (exceptions were H. pilosus_THB6 and THB7). All the remaining alpha- and gammaproteobacterial genera occurred at percentages <3%. With respect to Bacteroidetes, the genus Prevotella was particularly abundant (up to 12.1%) and ubiquitous, while Polaribacter members were observed almost only in M. hanitschi_THB11 (4.0%). With respect to Gram-positives, among Actinobacteria, the genus Cutibacterium occurred in all samples, ranging from 0.3 and 7.8%, while Rhodococcus members were present only in H. (R.) dancoi_THB8 (3.7%). Finally, a total of 23 genera were detected among Firmicutes, each occurring at <2% in all samples. Exception was the genus Paenibacillus, which was well represented in C. antarctica_THB16 and T. (T.) oxeata_THB19 (17.1 and 4%, respectively). The bacterial community composition (with respect to both phyla and genera) is also shown as interactive Krona charts in Supplementary Material S1: i.e., SiteA.html, SiteB.html, SiteC.html, SiteD.html (Krona charts are in HTML format and need to use a web browser to be opened).
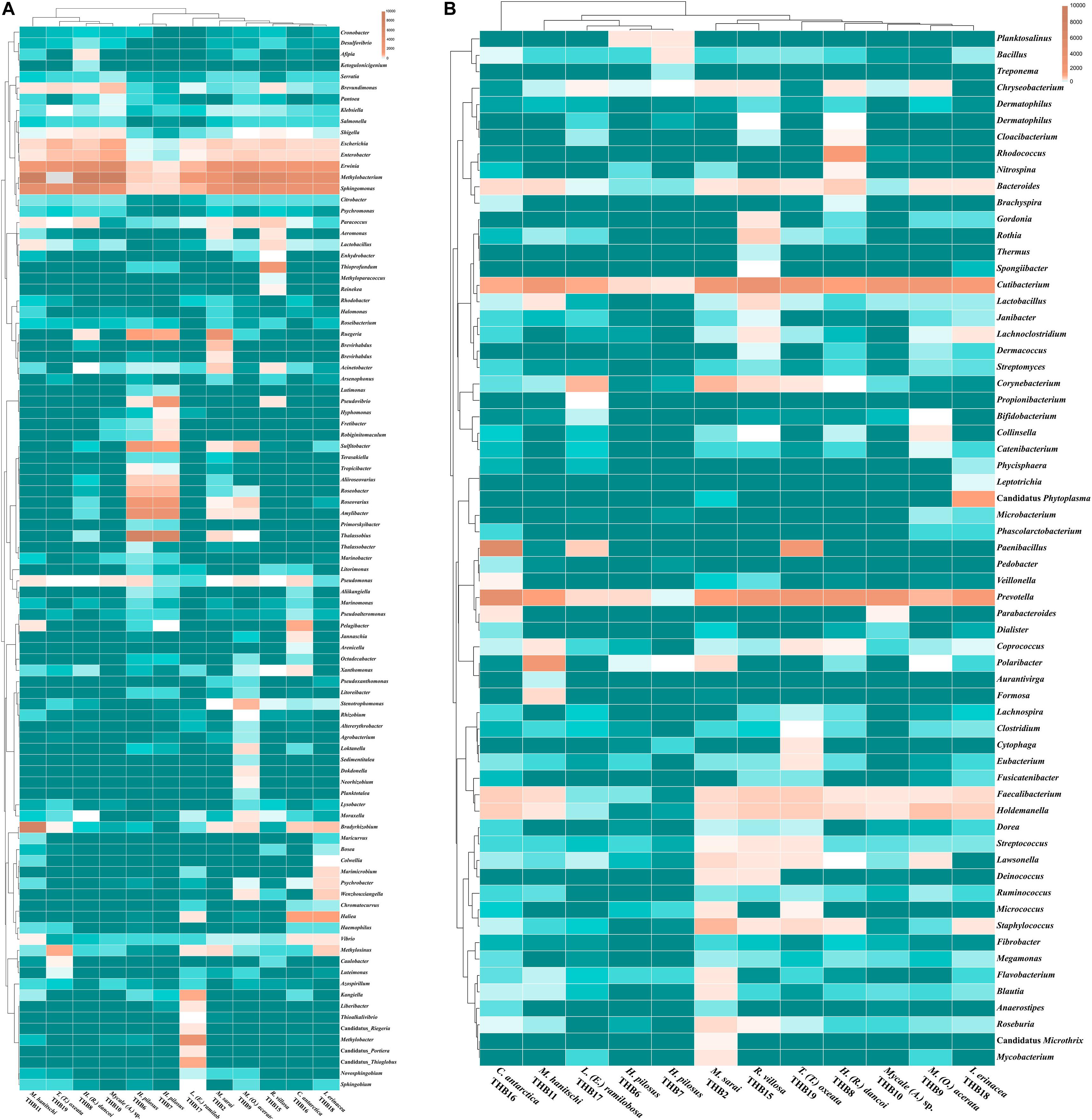
Figure 3. Heatmaps showing the genera distribution and clustering based on relative abundances detected in sponge samples. Color blocks represent the relative abundance of genera. More dark indicates a higher relative abundance. (A) Proteobacterial genera. (B) Genera belonging to other phyla or classes.
Shannon diversity values (ranging from 3.1 and 5.5) suggested a very different bacterial community composition among samples (Table 3). The cluster analysis of the total retrieved genera in all samples pointed out a larger group of samples composed of Mycale sp._THB10, T. (T.) oxeata_THB19, H. (R.) dancoi_THB8, M. (O.) acerata_THB9, R. villosa_THB15, and I. erinacea_THB18. On the other side, samples H. pilosus_THB6 and THB7 clearly separated from the rest, whereas L. (E.) ramibolosa_THB17, C. antarctica_THB16, M. sarai_THB2, and M. hanitschi_THB11 were more related to the larger group, even if they appeared separated one to each other. Instead, the analysis generated from results of all genera retrieved within Proteobacteria showed a larger group constituted of M. sarai_THB2, M. (O.) acerata_THB9, R. villosa_THB15, C. antarctica_THB16, L. (E.) ramibolosa_THB17 and I. erinacea_THB18. H. pilosus_THB6, and THB7 clearly separated from the rest, whereas H. (R.) dancoi_THB8, Mycale (A.) sp._THB10, M. hanitschi_THB11, and T. (T.) oxeata_THB19 generated a second group of samples (Figure 3A). The clustering obtained from all genera not related to Proteobacteria (i.e., Actinobacteria, Bacteroidetes, Deinococcus-Thermus, Firmicutes, Fusobacterial, Nitrospinae, Planctomycete, Spirochaetes, and Tenericutes) underlined a different situation in which C. antarctica_THB16 was completely separated from the other samples. Two main groups were observed. The first one was composed by M. hanitschi_THB11, L. (E.) ramibolosa_THB17, H. pilosus_THB6, and THB7 (these latter were closely related). The second group was composed by M. sarai_THB2, R. villosa_THB15, H. (R.) dancoi_THB8, M. (O.) acerata_THB9, Mycale (A.) sp._THB10, I. erinacea_THB18, and T. (T.) oxeata_THB19 (Figure 3B).
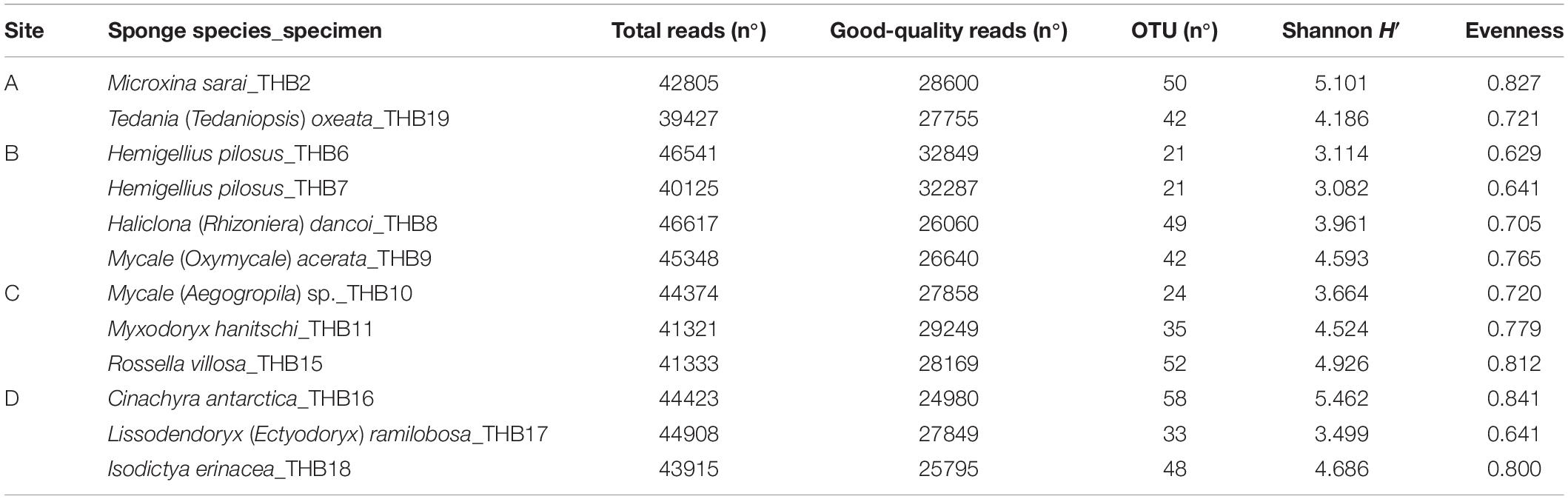
Table 3. Results of the next-generation sequencing analysis and retrieved diversity associated with Antarctic sponge samples.
Predictive Functional Profiling
A total of 5,523 predicted KEGG Orthologs (KOs; i.e., sets of homologous sequences) were present in the analyzed samples. Overall, 752 KEGG pathways resulting in 70 different molecular functions were found. Results relative to biological processes (>2%) predicted for the whole bacterial community associated with sponges are reported in Figure 4. In detail, the “biosynthesis of secondary metabolites” was the most represented KEGG pathway (49.9%), followed by the ATP-binding cassette transporters (ABC transporters; one of the largest and possibly one of the oldest gene families) and the “two-component system” (TCS), consisting of two proteins in most bacteria, a sensor histidine kinase and a response regulator involved in chemical signal transmission and reception (22.8 and 14.1%, respectively). The “biosynthesis of antibiotics” (4.5%), “quorum sensing” (3.2%), “methane metabolism” (3.2%), and “degradation of aromatic compounds” (2%) pathways were less represented. A detailed list of the biological processes, with the corresponding molecular functions, exhibited by the individual sponge-associated bacterial communities, is reported in Table 4 and detailed below.
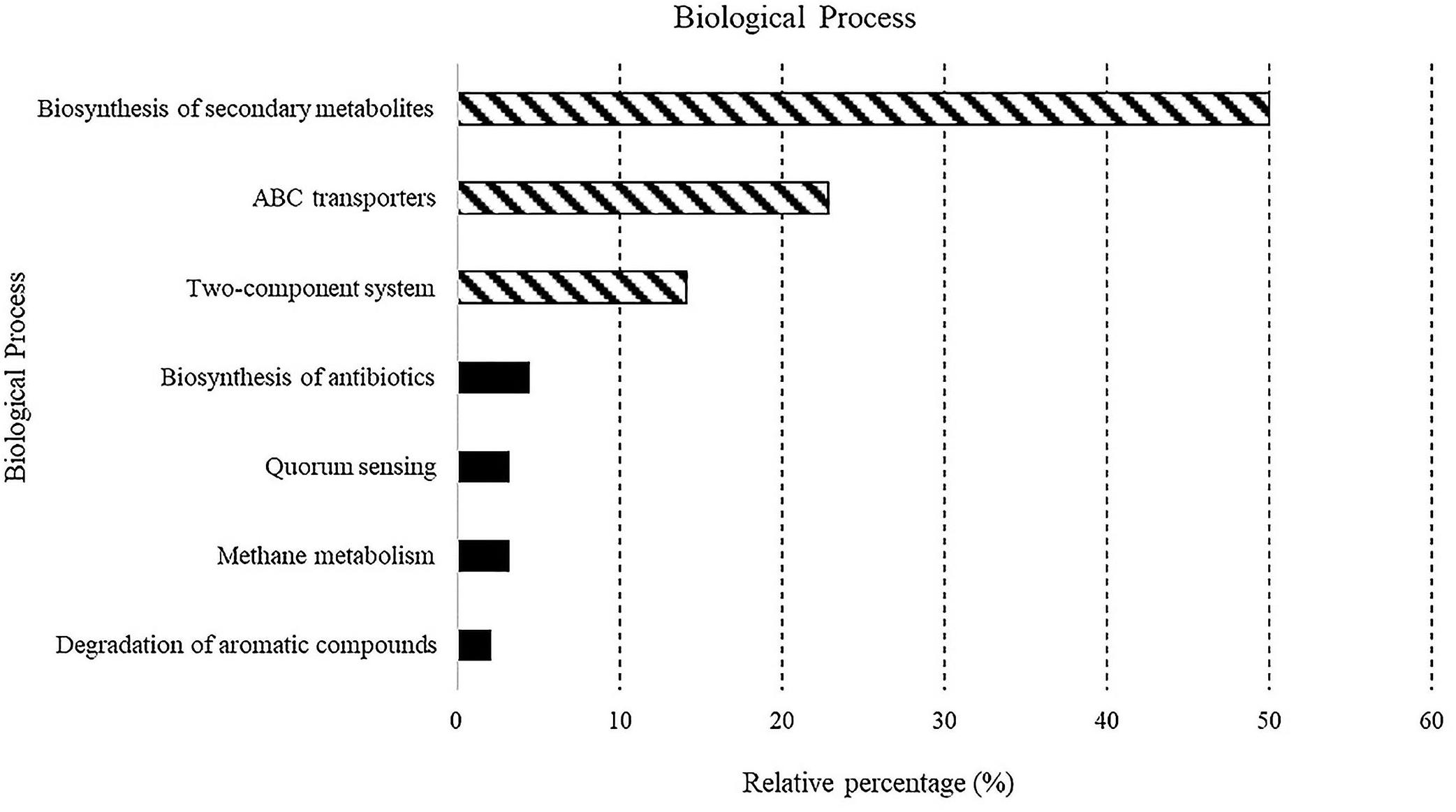
Figure 4. Predicted biological processes expressed as percentages in all samples.

Table 4. Sample identifiers, sponge identification, sampling site and depth, and coordinates.
Antibiotic Biosynthesis
Myo-inositol-1(or 4)-monophosphatase (KO01093) and glucose-1-phosphate thymidylyltransferase (KO00973) molecular functions, both associated with streptomycin biosynthesis, were observed in all samples (range 3.4–6.4% and 2.4–3.4%, respectively) (Supplementary Figure S1). The former was particularly evident in H. pilosus_THB6 and THB7 (5.9 and 6.4%, respectively) and M. hanitschi_THB11 (3.9%). Other pathways retrieved in all samples were correlated with the staurosporine biosynthesis (i.e., glutamate 5-kinase, KO00931; UTP-glucose-1-phosphate uridylyltransferase, KO00963; range 2.5–3.4 and 2.1–3.4%, respectively).
Degradation Pathways
The potential ability to degrade aromatic compounds was highlighted by the occurrence of two specific functions (i.e., 3-phenylpropionate/trans-cinnamate dioxygenase ferredoxin reductase component, KO00529, and 4-carboxymuconolactone decarboxylase, K01607; range 1.01–2.8 and 1.6–3.5%, respectively) that are related to the degradation of toluene, benzaldehyde, benzyl alcohol, benzoate, and other water-insoluble aromatic hydrocarbons (Supplementary Figure S2).
Methane Metabolism
Molecular functions related to methane metabolism (Supplementary Figure S3) were also observed in all samples. Among the most abundant processes, there were K00123 (formate dehydrogenase major subunit) and K00831 (phosphoserine aminotransferase) (ranged between 2.2–5.3 and 1.6–3.3%, respectively). They are involved in the oxidation of carbon compounds, such as methanol and methane. Otherwise, the KO14083 (trimethylamine-corrinoid protein co-methyltransferase), involved in methanogenesis from trimethylamine, was mainly abundant in the bacterial community from C. antartica_THB16 and L. (E.) ramilobosa_THB17 (6.4 and 5.4%, respectively).
Quorum Sensing
Molecular functions associated with the quorum sensing phenomenon (Supplementary Figure S4) were generally retrieved at very high percentages. Among the most abundant processes, a number of KOs were associated with sensing proteins, such as Qrr, LuxQ, LuxO, LuxU, CqsS, CAI-1, LitR/HapR, LuxP, AI-2, LuxN/AinR, and AI- (e.g., branched-chain amino acid transport system permease protein, K01998; peptide/nickel transport system ATP-binding protein, K02032; preprotein translocase subunit SecA, K03070; signal recognition particle subunit SRP54; K03106; range 5.7–9.3, 8.5–14.9, 2.2–3.4, and 2.2–3.4%, respectively). The acyl homoserine lactone synthase (KO13060) was mainly expressed by the bacterial communities associated with H. pilosus_THB6 and THB7 (1.4 and 1.3%, respectively).
Discussion
Previous microbiological studies on sponges from the Terra Nova Bay (Ross Sea) area were mainly focused on the phylogenetic affiliation, on the ecological roles and biotechnological potential of the cultivable bacterial fraction (e.g., Mangano et al., 2009, 2014, 2018; Papaleo et al., 2012; Caruso et al., 2018; Savoca et al., 2019), and, at a lesser extent, on the diversity and ecology of associated microeukaryotes, specifically diatoms (e.g., Cerrano et al., 2000, 2004a,b; Totti et al., 2005) and, in even more rare cases, on the associated macrofauna (e.g., Schiaparelli et al., 2003). In this context, we aimed at exploring more in depth the diversity and predictive functional profiles of the whole bacterial communities associated with 11 Antarctic sponge species, within the classes Demospongiae and Hexactinellida, commonly found in the Terra Nova Bay. In particular, in consideration of the growing interest in Antarctic resources, the predictive analysis on the bacterial metabolic potentials was performed to evaluate the potential exploitability in biotechnological fields of bacteria associated with Antarctic sponges. Data obtained in this study add further elements to the current knowledge, which still remains rather scarce and fragmentary (also in terms of biogeography), on the Antarctic sponges–bacteria association (Webster et al., 2004; Rodríguez-Marconi et al., 2015; Cárdenas et al., 2018). It is noteworthy that Antarctica must be preserved and, also by virtue of the best intentions, great care must be taken toward it and its living and non-living resources. In this regard, in order to keep sampling as less invasive as possible, sponge specimens belonging to different species (sufficiently representative for an explorative purpose) were collected from four different sites within the sampling area.
Phylogenetic Affiliation of Sponge-Associated Bacterial Communities
Taxonomic affiliation identified a diversified bacterial communities hosted by the analyzed sponges (overall, 177 distinct OTUs were retrieved), with the number of OTUs hosted per species that ranged between 21 (H. pilosus_THB6 and THB7) and 58 (C. antarctica_THB16). Such numbers of bacterial OTUs are lower than those reported by other authors for Antarctic sponges. In fact, Steinert et al. (2019) and Rodríguez-Marconi et al. (2015) yielded average values of 142 and 1,601 (this latter including Bacteria and Archaea) distinct OTUs per sponge sample, respectively. However, in line with previous results by Cárdenas et al. (2018) and Rodríguez-Marconi et al. (2015), a high diversity value (H’ values >3) was calculated for our samples. These two aspects taken in consideration together suggested a rather uniform distribution of OTUs among the analyzed sponge samples.
Overall, a strong dominance of Proteobacteria, with considerable abundance of both Alpha- and Gammaproteobacteria, characterized the communities of sponge-associated bacteria. Bacteroidetes, Actinobacteria, and Firmicutes were less represented than Proteobacteria, even if they were retrieved in almost all samples. Finally, members of other phyla (i.e., Acidobacteria, Nitrospinae, and Cyanobacteria) were sporadically detected in a smaller number of sponges. Such findings are in line with previous investigations (e.g., Webster et al., 2004; Savoca et al., 2019; Steinert et al., 2019) reporting a high abundance of associated Proteobacteria (in addition to Actinobacteria and Bacteroidetes), including cultivable members, in cold-water sponges, and with general observations on marine sponges worldwide (Hentschel et al., 2012; Thomas et al., 2016; Moitinho-Silva et al., 2017). The bacterial communities associated with the analyzed sponges were affiliated to 157 different known genera, which differed in both number and frequency among sponges. In particular, Proteobacteria genera strongly contributed to the diversification of sponges at the genus level. The bacterial communities associated with Antarctic sponges analyzed in this study included members involved in methane biochemical processes, such as Methylobacterium and Methylobacter (among Alpha- and Gammaproteobacteria, respectively). Some Methylobacterium affiliates are able to grow on one-carbon compounds (such as methanol, methylamine, formaldehyde, and formate). They are frequently found in association with plants (Green, 2006), but recently Methylobacterium sequences were also retrieved in sponges from temperate environments (Najafi et al., 2018). Methylobacter is a methanotroph frequently detected in numerous freshwater and marine habitats (Smith et al., 2018). Interestingly, in line with Steinert et al. (2019), the predictive analysis (see below) highlighted the presence of pathways involved in the methane metabolism. Bacterial members involved in the nitrogen cycle, i.e., Bradyrhizobium spp., were also found, even if only in M. hanitschi_THB11. Members in such genus are well-known typical symbionts of leguminous plant, but they were recently detected also in the microbiome of sponges from the Persian Gulf (Najafi et al., 2018). Among Alphaproteobacteria, the genus Sphingomonas was present at similar percentages in almost all sponges. Sphingomonas members are frequently found in marine matrices and play important ecological roles in the marine environment (Cavicchioli et al., 1999). Its occurrence was already reported in association with the Antarctic marine sponges Haliclona virens (Topsent, 1908), Tedania sp., and Lissodendoryx (Ectyodoryx) nobilis (Ridley and Dendy, 1886) from the Terra Nova Bay area (i.e., the Adelie Cove) (Savoca et al., 2019). Finally, among Bacteroidetes, we observed the occurrence of Polaribacter members, especially in M. hanitschi_THB11. Such cold-adapted copiotrophic bacteria are frequently found in marine environments of polar regions, also in association with Myxodoryx hanitschi from Antarctica (Webster et al., 2004; Savoca et al., 2019).
Results obtained for sponges collected from the same site highlighted that the bacterial communities associated with M. sarai_THB2 and T. (T.) oxeata_THB19 from site A (OTU-sharing 88.7%), and H. pilosus_THB6 and THB7 from site B were similar at the OTU level (OTU-sharing 55.5%). Interestingly, the specimens THB6 and THB7, both affiliated to H. pilosus, showed a high similarity in terms of genera, with a sharing percentage of 98% mainly due to the relative abundances of the genera Acinetobacter and Enterobacter, and hosted bacterial communities different from those observed for the other sponges. Such differences may be mainly dependent on the lower abundances of Gammaproteobacteria (below 10% of total sequences) and the higher abundances of Alphaproteobacteria (approx. 70%) than other sponge samples. The cultivable community associated with H. pilosus from the Terra Nova Bay was previously characterized by Mangano et al. (2014), who reported on the predominance of Gammaproteobacteria. Such discrepancy is probably dependent on the r-strategy adopted by members of this phylogenetic group that, being able to rapidly grow on nutrient-rich media (such as marine agar) and successfully competing under heterotrophic conditions, are generally well represented in culture collection (Lo Giudice et al., 2012). Further, if compared with the other samples, members of the genera Thalassobius, Amylibacter, Roseovarius, and Sulphitobacter were better represented within the bacterial communities of H. pilosus_THB6 and THB7, whereas other genera (such as Cutibacterium, Sphingomonas, and Erwinia) were absent or less represented. In particular, Thalassobius spp. are chemoorganotrophic marine bacteria showing complex ionic requirements to grow (Pujalte et al., 2018).
The similarity observed at the OTU (29.4%) and genus (47.7%) levels for Mycale (O.) acerata_THB9 and Mycale (A.) sp._THB10, collected from two different sites (site B and site C, respectively), mainly derived from the relative abundances of Bacteroidetes and Chryseobacterium spp., suggesting a possible influence of sponge genus features in shaping the associated bacterial community. In this regard, the microbiota associated with Mycale spp. was previously characterized by Webster et al. (2004) and Cárdenas et al. (2018) using culture-independent techniques. In particular, Cárdenas et al. (2018) explored the microbial communities associated with the Mycale (Aegogroipila) magellanica and Mycale (Oxymycale) acerata sampled from distant areas. Results revealed a high similarity (total OTU-sharing, 74%) in the community composition, with the dominance of Proteobacteria and Bacteroidetes. Our Mycale spp. [i.e., M. (O.) acerata_THB9 and Mycale (A.) sp._THB10] shared the 29.5 and 47.7% of OTUs and genera, respectively. The predominance of Proteobacteria observed by Cárdenas et al. (2018) in Mycale spp. sponges was confirmed by the present study. Conversely, Bacteroidetes were less represented, whereas an increased presence of Firmicutes was observed. Differently from Cárdenas et al. (2018), Gammaproteobacteria were more abundant than Alphaproteobacteria in M. (O.) acerata_THB9.
Predictive Functional Profiling
To date, the functional analysis of Antarctic sponge-associated prokaryotic communities has been rarely performed (Steinert et al., 2019). It should be noted that findings on the functional patterns are predictive hypotheses and they do not necessary reflect the real microbial processes inside the sponges. However, this approach provides more insight into the potential role that microbes play in this critical host association. From a functional viewpoint, it was very interesting that a very high percentage of predicted pathways was associated with the biosynthesis of secondary metabolites. This finding might be related to the production of bioactive compounds playing a role in the inhibition of microorganisms, which could act as competitors for essential nutrients. The nature of these bioactive compounds is still unknown, but some of them might be volatile organic compounds (VOCs), as previously suggested for bacteria associated with Haliclonissa verrucosa Burton, 1932, Lissodendoryx (E.) nobilis, and Anoxycalyx (Scolymastra) joubini (Topsent, 1916) from the Terra Nova Bay (Romoli et al., 2011, 2014; Papaleo et al., 2012, 2013). Such results highlighted the important role of sponge/benthic invertebrate symbiotic bacteria, also from polar areas, in a bioprospecting scenario as producers of bioactive natural products (Lo Giudice and Rizzo, 2018; Rizzo and Lo Giudice, 2018). Several pathways involved in the production of antibiotics were predicted in this study. Among them, molecular functions correlated with the biosynthesis of streptomycin (expressed at particularly high extent by the bacterial communities associated with H. pilosus_THB6 and THB7 and M. hanitschi_THB11) and staurosporine. In particular, molecular processes at the basis of streptomycin biosynthesis were also evidenced by Steinert et al. (2019), who suggested that the presence of such predicted pathways might indicate a host-specific antimicrobial activity in the interested sponges.
The second highly predicted processes were the two-component systems and ABC transporter. This finding is not surprising since bacteria inhabiting such harsh environments should sense the presence of nutrients, even at low concentrations, to growth and proliferate under extreme conditions.
Quorum sensing was firstly reported in the present study as a prediction for bacterial communities associated with Antarctic sponges. Cell-to-cell communication occurs within the dense bacterial communities associated with sponges (Taylor et al., 2004). Quorum sensing is among environmental sensing systems involved in regulating the bacterial symbiotic colonization of metazoan organisms, including population density, social activities, and physiological processes (such as the formation of biofilm for bacterial adhesion to surfaces; Mohamed et al., 2008). Our results reinforced previous observations by Mangano et al. (2018) on 211 cultivable Gram-negative bacteria isolated from Terra Nova Bay sponges (i.e., Lissodendoryx (Ectyodoryx) nobilis, Phorbas glaberrimus, and Myxodoryx hanitschi) and able to produce N-Acyl homoserine lactones (AHLs), signal molecules involved in the QS mechanism. The AHL producer mainly belonged to bacterial genera that are known to be involved in surface colonization by biofilm synthesis.
Interestingly, biological processes related to the degradation of xenobiotics (such as toluene, benzaldehyde, benzyl alcohol, benzoate, and other hydrophobic compounds), particularly aromatic substances, were also predicted. Such compounds are well-known environmental contaminants, which could accumulate in marine filter-feeding organism tissues, thus probably stimulating the development of specific bacterial symbiotic communities with the ability to utilize them as a source of carbon and energy (Mangano et al., 2018; Rizzo and Lo Giudice, 2018; Lo Giudice et al., 2019b). The occurrence of molecular pathways involved in pollutant degradation represents a starting step for exploring more in depth the possible implications in bioremediation processes of Antarctic sponge-associated bacteria. Further insights are necessary in order to understand if the predicted degradation pathway may be an indication of the local pollution level and/or if such capacities are included in the detoxifying system owned by prokaryotes within the sponge-holobiont, as previously suggested (Steinert et al., 2019 and therein references).
Concluding Remarks
In conclusion, the application of a combined approach, which included the analysis of both diversity and predictive functional metabolic profile of Antarctic sponge-associated bacterial communities, furnished novel and interesting insights on the symbiotic communities hosted by benthic filter-feeding invertebrates living in polar areas, and allowed individuating a number of their putative ecological roles and biotechnological potentialities. In terms of diversity, our Antarctic sponges seemed to be attributable to low microbial abundance (LMA) sponges, which commonly host bacterial communities dominated by few taxonomic groups (mainly Proteobacteria and Bacteroidetes). This assumption is strengthened, as it was previously reported for sponges of Antarctic areas different from the Terra Nova Bay, by the absence of the Poribacter group, which are instead normally associated with high microbial abundance (HMA) sponges. Furthermore, our results confirm the so far documented absence of such taxon in the Antarctic environments. Further investigation should be performed to establish at which extent the surrounding environment (e.g., seawater and sediment) influences the composition of the bacterial communities associated with Antarctic sponges, as well as the processes involved in the selection of true symbiotic (instead of potentially transient) bacteria. At this regard, more research efforts should be addressed to the quorum sensing phenomenon, which is probably strictly involved in the adhesion (and the subsequent colonization) of symbiotic bacteria to the sponge surfaces through the formation of biofilm. Finally, on the basis of the predictive metabolic profiling, the involvement of associated bacteria in possible host defensive strategies (including the biodegradation of toxic compounds and the biosynthesis of metabolites inhibiting sponge pathogens or biofouling) is not to be excluded.
Data Availability Statement
The datasets generated for this study can be found in NCBI BioProject, NCBI Accession No. PRJNA594262.
Author Contributions
AL and RF designed and conceived the study. LM, MP, and SS collected samples during the XXIX Italian Expedition to Antarctica. MB and GC classified the sponge samples. MP and CR undertook the laboratory work and sequencing, and generated data. AP-G and MP performed bioinformatics analysis. AL, MP, CR, and MA analyzed and interpreted the data and wrote the first draft of the manuscript. All authors contributed to manuscript corrections and improved the final version.
Funding
This research was supported by grants from the PNRA (Programma Nazionale di Ricerche in Antartide) and Italian Ministry of Education and Research (Research Projects PNRA 2013/AZ1.04 and PNRA16_00020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all of the staff at Mario Zucchelli Station for logistical help and support and the Sequentia biotech staff (Spain) for supporting the data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00268/full#supplementary-material
MATERIAL S1 | Supplementary Krona charts: SiteA.html, Kronachart relative to the bacterial community composition in samples THB2 (M. sarai) and THB19 [T. (T.) oxeata]; SiteB.html, Kronachart relative to the bacterial community composition in samples THB6, THB7 (both H. pilosus), THB8 [H. (R.) dancoi] and THB9 [M. (O.) acerata]; SiteC.html; Kronachart relative to the bacterial community composition in samples THB10 (Mycale (A.) sp.) and THB11 (M. hanitschi); SiteD.html, Kronachart relative to the bacterial community composition in samples THB15 (R. villosa), THB16 (C. antarctica), THB17 (L. (E.) ramilobosa) and THB18 (I. erinacea).
References
ATCM XXXIV (2011). “SCAR’s code of conduct for the use of animals for scientific purposes in antarctica,” in Proceedings of the IP53, Agenda Item CEP 8c, presented by SCAR during XXXIV Antarctic Treaty Consultative Meeting, Buenos Aires.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Cárdenas, C. A., Bell, J. J., Davy, S. K., Hoggard, M., and Taylor, M. W. (2014). Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiol. Ecol. 88, 516–527. doi: 10.1111/1574-6941.12317
Cárdenas, C. A., González-aravena, M., Font, A., Hestetun, J. T., Hajdu, E., Trefault, N., et al. (2018). High similarity in the microbiota of cold-water sponges of the genus Mycale from two different geographical areas. Peer J. 6:e4965.
Caruso, C., Rizzo, C., Mangano, S., Poli, A., Di Donato, P., Finore, I., et al. (2018). Production and biotechnological potentialities of extracellular polymeric substances from sponge-associated Antarctic bacteria. Appl. Environ. Microbiol. 84:e01624-17.
Cattaneo-Vietti, R., Chiantore, M., and Albertelli, G. (1997). The population structure and ecology of the antarctic scallop, adamussiumcolbeckiin terra nova bay (Ross Sea, Antarctica). Sci. Mar. 61, 15–24.
Cavicchioli, R., Fegatella, F., Ostrowski, M., Eguchi, M., and Gottschal, J. (1999). Sphingomonads from marine environments. J. Ind. Microbiol. Biotechnol. 23, 268–272. doi: 10.1038/sj.jim.2900732
Cerrano, C., Arillo, A., Bavestrello, G., Calcinai, B., Cattaneo-Vietti, R., Penna, A., et al. (2000). Diatom invasion in the Antarctic hexactinellid sponge Scolymastra joubini. Polar Biol. 23, 441–444. doi: 10.1007/s003000050466
Cerrano, C., Bertolino, M., Valisano, L., Bavestrello, G., and Calcinai, B. (2009). Epibiotic demosponges of the Antarctic scallop, Adamussium colbecki (Smith, 1902) and the seaurchins Ctenocidaris perrieri Koehler, 1912 in Ross Sea, Antarctica. Polar Biol. 32, 1067–1076. doi: 10.1007/s00300-009-0606-5
Cerrano, C., Calcinai, B., Cucchiari, E., Di Camillo, C., Nigro, M., Regoli, F., et al. (2004a). Are diatoms a food source for Antarctic sponges? Chem. Ecol. 20, 57–64. doi: 10.1080/02757540310001629198
Cerrano, C., Calcinai, B., Cucchiari, E., Di Camillo, C., Totti, C., and Bavestrello, G. (2004b). The diversity of relationships between Antarctic sponges and diatoms: the case of Mycale acerata Kirkpatrick, 1907 (Porifera. Demospongiae). Polar Biol. 27, 231–237. doi: 10.1007/s00300-003-0581-1
Conte, A., Papale, M., Amalfitano, S., Mikkonen, A., Rizzo, C., De Domenico, E., et al. (2018). Bacterial community structure along the subtidal sandy sediment belt of a high Arctic fjord (Kongsfjorden. Svalbard Islands). Sci. Total Environ. 61, 203–211. doi: 10.1016/j.scitotenv.2017.11.077
Douglas, G. M., Maffei, V. J., Zaneveld, J., Yurgel, S. N., Brown, J. N., Taylor, C. M., et al. (2019). PICRUSt2: an improved and extensible approach for metagenome inference. bioRxiv [Preprint]. doi: 10.1101/672295
Erwin, P. M., Coma, R., López-Sendino, P., Serrano, E., and Ribes, M. (2015). Stable symbionts across the HMA-LMA dichotomy: low seasonal and interannual variation in sponge associated bacteria from taxonomically diverse hosts. FEMS Microbiol. Ecol. 91:fiv11.
Fan, L., Liu, M., Simister, R., Webster, N. S., and Thomas, T. (2013). Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 7, 991–1002. doi: 10.1038/ismej.2012.165
Gambi, M. C., Castelli, A., and Guizzardi, M. (1997). Polychaete populations of the shallow soft bottoms off Terra Nova Bay (Ross Sea. Antarctica): distribution, diversity and biomass. Polar Biol. 17, 199–210. doi: 10.1007/s003000050123
Ghiglione, C., Alvaro, M. C., Cecchetto, M., Canese, S., Downey, R., Guzzi, A., et al. (2018). Porifera collection of the Italian National Antarctic Museum (MNA), with an updated checklist from Terra Nova Bay (Ross Sea). ZooKeys 758, 137–156. doi: 10.3897/zookeys.758.23485
Glasl, B., Bourne, D. G., Frade, P. R., Thomas, T., Schaffelke, B., and Webster, N. S. (2019). Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome 7:94.
Green, P. N. (2006). “Methylobacterium,” in The Prokaryotes, 3rd Edn, Vol. 5, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 257–265.
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 10, 641–654. doi: 10.1038/nrmicro2839
Hooper, J. N., and van Soest, R. W. (2002). Systema Porifera. A Guide to the Classification of Sponges. Boston, MA: Springer, 94. doi: 10.1007/978-1-4615-0747-5
Hooper, J. N. A. (2000). ‘Sponguide’. Guide to Sponge Collection and Identification. Available online at: https://www.academia.edu/34258606/SPONGE_GUIDE_GUIDE_TO_SPONGE_COLLECTION_AND_IDENTIFICATION_Version_August_2000
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, 353–361.
Lemoine, N., Buell, N., Hill, A., and Hill, M. (2007). “Assessing the utility of sponge microbial symbiont communities as models to study global climate change: a case study with Halichondria bowerbanki,” in Porifera Research: Biodiversity, Innovation and Sustainability, eds M. R. Custodio, G. Lobo-Hajdu, E. Hajdu, and G. Muricy (Rio de Janeiro: Museu Nacional), 419–425.
Li, Z. Y., Wang, Y. Z., He, L. M., and Zheng, H. J. (2014). Metabolic profiles of prokaryotic and eukaryotic communities in deep-seasponge Lamellomorpha sp. indicated by metagenomics. Sci. Rep. 4:3895.
Lo Giudice, A., Azzaro, M., and Schiaparelli, S. (2019a). “Microbial symbionts of Antarctic marine benthic invertebrates,” in The Ecological Role of Micro-organisms in the Antarctic Environment, ed. S. Castro-Sowinski (Berlin: Springer Polar Sciences), 277–296. doi: 10.1007/978-3-030-02786-5_13
Lo Giudice, A., Caruso, G., Rizzo, C., Papale, M., and Azzaro, M. (2019b). Bacterial communities versus anthropogenic disturbances in the Antarctic coastal marine environment. Environ. Sustain. 2, 297–310. doi: 10.1007/s42398-019-00064-2
Lo Giudice, A., Caruso, C., Mangano, S., Bruni, V., De Domenico, M., and Michaud, L. (2012). Marine bacterioplankton diversity and community composition in an Antarctic coastal environment. Microb. Ecol. 63, 210–223. doi: 10.1007/s00248-011-9904-x
Lo Giudice, A., and Rizzo, C. (2018). Bacteria associated with marine benthic invertebrates from polar environments: unexplored frontiers for biodiscovery? Diversity 10:80. doi: 10.3390/d10030080
Mangano, S., Caruso, C., Michaud, L., and Lo Giudice, A. (2018). First evidence of quorum sensing activity in bacteria associated with Antarctic sponges. Polar Biol. 41, 1435–1445. doi: 10.1007/s00300-018-2296-3
Mangano, S., Michaud, L., Caruso, C., Brilli, M., Bruni, V., Fani, R., et al. (2009). Antagonistic interactions among psychrotrophic cultivable bacteria isolated from Antarctic sponges: a preliminary analysis. Res. Microbiol. 160, 27–37. doi: 10.1016/j.resmic.2008.09.013
Mangano, S., Michaud, L., Caruso, C., and Lo Giudice, A. (2014). Metal and antibiotic-resistance in psychrotrophic bacteria associated with the Antarctic sponge Hemigellius pilosus (Kirkpatrick, 1907). Polar Biol. 37, 227–235. doi: 10.1007/s00300-013-1426-1
Mohamed, N. M., Cicirelli, E. M., Kan, J., Chen, F., Fuqua, C., and Hill, R. T. (2008). Diversity and quorum-sensing signal production of Proteobacteria associated with marine sponges. Environ. Microbiol. 10, 75–86.
Moitinho-Silva, L., Steinert, G., Nielsen, S., Hardoim, C. C. P., Wu, Y.-C., McCormack, G. P., et al. (2017). Predicting the HMA-LMA status in marine sponges by machine learning. Front. Microbiol. 8:752. doi: 10.3389/fmicb.2017.00752
Morrow, C., and Cárdenas, P. (2015). Proposal for a revised classification of the Demospongiae (Porifera). Front. Zool. 12:7. doi: 10.1186/s12983-015-0099-8
Najafi, A., Moradinasab, M., and Nabipour, I. (2018). First record of microbiomes of sponges collected from the Persian Gulf, using tag pyrosequencing. Front. Microbiol. 9:1500. doi: 10.3389/fmicb.2018.01500
Papaleo, M. C., Fondi, M., Maida, I., Perrin, E., Lo Giudice, A., Michaud, L., et al. (2012). Sponge-associated microbial Antarctic communities exhibiting antimicrobial activity against Burkholderia cepacia complex bacteria. Biotechnol. Adv. 30, 272–293. doi: 10.1016/j.biotechadv.2011.06.011
Papaleo, M. C., Romoli, R., Bartolucci, G., Maida, I., Perrin, E., Fondi, M., et al. (2013). Bioactive volatile organic compounds from Antarctic (sponges) bacteria. New Biotechnol. 30, 824–838. doi: 10.1016/j.nbt.2013.03.011
Piazza, P., Cummings, V., Guzzi, A., Hawes, I., Lohrer, A., Marini, S., et al. (2019). Underwater photogrammetry in Antarctica: long-term observations in benthic ecosystems and legacy data rescue. Polar Biol. 42, 1061–1079. doi: 10.1007/s00300-019-02480-w
Piazza, P., Gattone, S. A., Guzzi, A., and Schiaparelli, S. (2020). Towards a robust baseline for long term monitoring of Antarctic coastal benthos. Hydrobiologia 847, 1753–1771. doi: 10.1007/s10750-020-04177-2
Pineda, M.-C., Strehlow, B., Duckworth, A., Doyle, J., Jones, R., and Webster, N. S. (2016). Effects of light attenuation on the sponge holobiont- implications for dredging management. Sci. Rep. 6:39038.
Pineda, M.-C., Strehlow, B., Sternel, M., Duckworth, A., Jones, R., and Webster, N. S. (2017). Effects of suspended sediments on the sponge holobiont with implications for dredging management. Sci. Rep. 7:4925.
Pujalte, M. J., Lucena, T., Rodrigo-Torres, L., and Arahal, D. R. (2018). Comparative genomics of Thalassobiusincluding the description of Thalassobius activus sp. nov., and Thalassobius autumnalis sp. nov. Front. Microbiol. 8:2645. doi: 10.3389/fmicb.2017.02645
Rau, A., Gallopin, M., Celeux, G., and Jaffrezic, F. (2013). Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 29, 2146–2152. doi: 10.1093/bioinformatics/btt350
Rizzo, C., and Lo Giudice, A. (2018). Marine invertebrates: underexplored sources of bacteria producing biologically active molecules. Diversity 10: 52. doi: 10.3390/d10030052
Rodríguez-Marconi, S., De la Iglesia, R., Díez, B., Fonseca, C. A., Hajdu, E., and Trefault, N. (2015). Characterization of bacterial, archaeal and eukaryote symbionts from Antarctic sponges reveals a high diversity at a three-domain level and a particular signature for this ecosystem. PLoS One 10:e0138837. doi: 10.1371/journal.pone.0138837
Romoli, R., Papaleo, M. C., De Pascale, D., Tutino, M. L., Michaud, L., Lo Giudice, A., et al. (2011). Characterization of the volatile profile of Antarctic bacteria by using solid-phase microextraction - gas chromatography mass spectrometry. J. Mass Spectr. 46, 1051–1059. doi: 10.1002/jms.1987
Romoli, R., Papaleo, M. C., de Pascale, D., Tutino, M. L., Michaud, L., Lo Giudice, A., et al. (2014). GC-MS volatolomic approach to study the antimicrobial activity of the Antarctic bacterium Pseudoalteromonas sp. TB41. Metabolomics 10, 42–51. doi: 10.1007/s11306-013-0549-2
Rützler, K. (1978). “Sponges in coral reefs,” in Coral Reefs: Research Methods, Monographs on Oceanographic Methodology, eds D. R. Stoddart and R. E. Johannes (Paris: Unesco), 299–313.
Sarà, M., Balduzzi, A., Barbieri, M., Bavestrello, G., and Burlando, B. (1992). Biogeographic traits and checklist of Antarctic demosponges. Polar Biol. 12, 559–585.
Savoca, S., Lo Giudice, A., Papale, M., Mangano, S., Caruso, C., Spanò, N., et al. (2019). Antarctic sponges from the Terra Nova Bay (Ross Sea) host a diversified bacterial community. Sci. Rep. 9:16135.
Schiaparelli, S., Albertelli, G., and Cattaneo-Vietti, R. (2003). The epibiotic assembly on the sponge Haliclona dancoi (Topsent, 1901) at Terra Nova Bay (Antarctica. Ross Sea). Pol. Biol. 26, 342–347. doi: 10.1007/s00300-003-0481-4
Sipkema, D. (2016). Marine biotechnology: diving deeper for drugs. Microb. Biotechnol. 10, 7–8. doi: 10.1111/1751-7915.12410
Smith, G. J., Angle, J. C., Solden, L. –M., Borton, M. A., Morin, T. H., Daly, R. A., et al. (2018). Members of the genus Methylobacter are inferred to account for the majority of aerobic methane oxidation in oxic soils from a freshwater wetland. mBio 9:e00815-18.
Steinert, G., Wemheuer, B., Janussen, D., Erpenbeck, D., Daniel, R., Simon, M., et al. (2019). Prokaryotic diversity and community patterns in Antarctic continental shelf sponges. Front. Mar. Sci. 6:297. doi: 10.3389/fmars.2019.00297
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Boil. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Taylor, M. W., Schupp, P. J., Baille, H. J., Charlton, T. S., de Nys, R., Kjelleberg, S., et al. (2004). Evidence for acyl homoserine lactone signal production in bacteria associated with marine sponges. Appl. Environ. Microbiol. 70, 4387–4389. doi: 10.1128/aem.70.7.4387-4389.2004
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870.
Totti, C., Calcinai, B., Cerrano, C., Camillo, C., Romagnoli, T., and Bavestrello, G. (2005). Diatom assemblages associated with Sphaerotylus antarcticus (Porifera: Demospongiae). J. Mar. Biol. Associat. U. K. 85, 795–800. doi: 10.1017/s0025315405011720
Van Soest, R. W. M., Boury-Esnault, N., Hooper, J. N. A., Rützler, K., de Voogd, N. J., Alvarez, B., et al. (2018). World Porifera Database. Available from: http://www.marinespecies.org/porifera/ (accessed 10 May 2019).
Webster, N., Taylor, M. W., Behnam, F., Lücker, S., Rattei, T., Whalan, S., et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12, 2070–2082.
Webster, N. S., Cobb, R. E., and Negri, A. P. (2008). Temperature thresholds for bacterial symbiosis with a sponge. ISME J. 2, 830–842. doi: 10.1038/ismej.2008.42
Keywords: bacterial diversity, NGS, predictive metabolic profiling, sponge-bacteria association, Antarctica
Citation: Papale M, Rizzo C, Fani R, Bertolino M, Costa G, Paytuví-Gallart A, Schiaparelli S, Michaud L, Azzaro M and Lo Giudice A (2020) Exploring the Diversity and Metabolic Profiles of Bacterial Communities Associated With Antarctic Sponges (Terra Nova Bay, Ross Sea). Front. Ecol. Evol. 8:268. doi: 10.3389/fevo.2020.00268
Received: 11 January 2020; Accepted: 28 July 2020;
Published: 02 September 2020.
Edited by:
Michele Kiyoko Nishiguchi, New Mexico State University, United StatesReviewed by:
Robert W. Thacker, Stony Brook University, United StatesAlison Gould, California Academy of Sciences, United States
Copyright © 2020 Papale, Rizzo, Fani, Bertolino, Costa, Paytuví-Gallart, Schiaparelli, Michaud, Azzaro and Lo Giudice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelina Lo Giudice, angelina.logiudice@cnr.it