The Complete Mitochondrial Genome of Two Armored Catfish Populations of the Genus Hypostomus (Siluriformes, Loricariidae, Hypostominae)
 Dinaíza Abadia Rocha-Reis1*
Dinaíza Abadia Rocha-Reis1*  Rubens Pasa1
Rubens Pasa1  Fabiano Bezerra Menegidio2
Fabiano Bezerra Menegidio2  John Seymour Heslop-Harrison3
John Seymour Heslop-Harrison3  Trude Schwarzacher3
Trude Schwarzacher3  Karine Frehner Kavalco1
Karine Frehner Kavalco1- 1Laboratory of Ecological and Evolutionary Genetics, Institute of Biological and Health Sciences, Federal University of Viçosa-Campus Rio Paranaíba, Rio Paranaíba, Brazil
- 2Integrated Nucleus of Biotechnology, University of Mogi das Cruzes, São Paulo, Brazil
- 3Department of Genetics and Genome Biology, University of Leicester, Leicester, United Kingdom
Introduction
Hypostomus Lacépède (1803) is the dominant genus of armored catfish (Siluriformes, Loricariidae, Hypostominae) in Brazilian rivers (Britski, 1972). This group presents a wide interspecific color and morphology variation (Oyakawa et al., 2005), which hinders the identification of some species. Likewise, the existence of various cytogenetic phenotypes, including different chromosomal numbers, karyotype formulas, and location of ribosomal genes (Rocha-Reis et al., 2020), reinforces the need for more appropriate methodologies for species identification.
The Alto Paraná river basin concentrates the largest number of studies on freshwater fish in Brazil (Agostinho et al., 2007). This drainage basin is the habitat of 25 species of the genus Hypostomus (Weber, 2003), suggesting a complex evolutionary history for the taxon in this region. Hypostomus ancistroides Ihering (1911), which has a natural distribution in the basins of the rivers Tietê, Ribeira de Iguape, and Alto Paraná (Fricke et al., 2020), is considered a complex of cryptic species due to the different morphotypes found in its area. In fact, this complex of cryptic species has representatives with small morphological variations among populations, different chromosomal numbers, unique karyotype formulas for each population, polymorphisms related to the number and location of Nucleolus Organizer Regions, ribosomal DNAs, constitutive heterochromatin patterns, and even the (occasional) existence of sex chromosome systems (Rocha-Reis et al., 2020).
A previous study conducted by Rocha-Reis et al. (2018) showed that a population of this complex collected in the Paranapanema river (Alto Paraná basin, Brazil) had highly differentiated traits when compared to other groups. Although morphologically similar to H. ancistroides, the specimens presented distinct karyotypic data, characterized by lower chromosome numbers and the presence of a neo-XY sex chromosome system in which X chromosomes constitute a pair of small acrocentric chromosomes in females with only one chromosome of this type in males (Rocha-Reis et al., 2018). The Y chromosome, on the other hand, is the largest metacentric of the karyotype complement, only present in males.
Species delimitation within a complex is often difficult because lineages might not show large morphological differences, despite being reproductively isolated (Lane, 1997). Consequently, Evolutionary Significant Units (ESU) and species can be both identified using alternative approaches, such as genetic techniques (Rocha-Reis et al., 2018).
The mitochondrial genome (mtDNA) of vertebrates is a circular and closed molecule, with conserved gene content (Billington and Hebert, 1991). It is 16–20 kb in length and consists of a light and a heavy chain with a non-coding control region and 37 genes: 13 protein-coding genes (PCGs), 22 RNA transfer genes (tRNAs), and 2 ribosomal RNA genes (rRNAs) (Boore, 1999; Taanman, 1999; Bernt et al., 2013). Features such as high evolutionary rate, low recombination, and maternal inheritance make mtDNA a powerful molecular marker and a useful tool for biological identification studies, phylogenetic analyses, and population genetics (Harrison, 1989; Caccone et al., 2004; Ma et al., 2012; Hirase et al., 2016).
Here, we describe the complete mitochondrial genome of two Hypostomus populations of the H. ancistroides species complex. In addition, we seek to elucidate the phylogenetic positioning of these populations based on their mitogenomes.
Materials and Methods
Specimen Collection and DNA Extraction
We collected one male specimen of H. ancistroides from the Tietê river basin, near the municipality of Conchas, São Paulo, Brazil (22°59′57.84″S; 48°0′16.09″O), and two individuals of Hypostomus aff. ancistroides, one of each sex, from a stream near the municipality of São Miguel Arcanjo, São Paulo, Brazil (23°54′44.58″S; 47°57′40.50″O). The identification of their sex was performed either through morphological observation, since many species of Hypostomus have sexual dimorphism, or through the presence of gonads. We extracted heart and liver to obtain DNA, which were stored in a freezer at −20°C. We deposited the tissue and DNA samples from individuals under accession numbers in the Tissue, Cell Suspension, and DNA Bank in the Laboratory of Ecological and Evolutionary Genetics, Federal University of Viçosa—Campus Rio Paranaíba. Sampling and euthanasia procedures were conducted in accordance with the Conselho Nacional de Controle de Experimentação Animal (CONCEA).
We extracted the total genomic DNA from liver and heart samples according to the instructions of Invitrogen's DNA extraction and purification kit and checked DNA quality and concentration on a 1% agarose gel, with subsequent measurement using a NanoDrop 2000 spectrophotometer and a Qubit fluorometer.
Library Construction and Sequencing
The Whole Genome Sequencing was performed by Novaseq 6000 (Illumina, San Diego, CA) by the company Novogene, UK. A total amount of 1.0 μg of DNA per sample was used as input material for the DNA sample preparations. Sequencing libraries were generated using NEBNext® DNA Library Prep Kit following the manufacturer's recommendations and indices were added to each sample. The genomic DNA was randomly sheared to 350-bp fragments. These DNA fragments were then end-polished, A-tailed, and ligated with the NEBNext adapter for Illumina sequencing, and further PCR enriched by P5 and indexed P7 oligos. The PCR products were purified (AMPure XP system) and the resulting libraries were analyzed for size distribution through Agilent 2100 Bioanalyzer and quantified using real-time PCR. The qualified libraries were input into Illumina sequencers after pooling according to their effective concentration and expected data volume.
Sequence Assembly and Analysis
For the de novo mitogenome assembly, we used NovoPlasty 3.7 (Dierckxsens et al., 2017) with 2 × 150 raw reads and three distinct kmers (19, 21, and 23).
We annotated the mitochondrial sequences obtained in MitoAnnotator (Iwasaki et al., 2013) present in MitoFish. We performed the analyses as a base composition with Fasta Statistics 1.0.1 (Seemann and Gladman, 2012), available at The Galaxy Project (https://usegalaxy.org/) (Afgan et al., 2018). The complete mitochondrial genomes of the two populations are available at GenBank under accessions MT066232 for H. ancistroides, MT081402 for the male, and MT396945 for the female of Hypostomus aff. ancistroides.
Phylogenetic Analysis
We estimated the phylogenetic relationships of H. ancistroides and H. aff. ancistroides by comparison with three Hypostominae species: Hypostomus francisci (sequence obtained through personal communication with the author, Pereira et al., 2019), Hypostomus plecostomus (NC025584, Liu et al., 2016), and Pterygoplichthys disjunctivus (NC015747, identical to AP012021, Nakatani et al., 2011). Additionally, we included a species of the family Hypoptopomatinae, Hypoptopoma incognitum (NC028072, Moreira et al., 2015), as the outgroup. We used ClustalW (Thompson et al., 1994), implemented by MEGA 7.0.21 software (Kumar et al., 2018), with all the default parameters to align the complete mitogenome sequences.
We performed Bayesian Analysis with the concatenated sequences using the 13 PCGs. Each gene was divided into three partitions, allowing the selection of the best model for each segment individually. The analysis of the model choice was performed in PartitionFinder 2.1.1 (Lanfear et al., 2016) using the Bayesian Information Criterion (BIC), which resulted in five partitions with the following models: SYM + I, HKY + I, GTR + G, HKY + I, and HKY. After 10 million samples of Markov Chain Monte Carlo in MrBayes 3.2.7a (Ronquist et al., 2012), the length of the sampling chain was calculated every 1,000 generations using Tracer 2.6.2 (Rambaut et al., 2018) to estimate the effectiveness of sample size and chain convergence. We visualized the tree in the FigTree 1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/) and edited the images with Inkscape (https://www.inkscape.org) and GNU Image Manipulation Program (GIMP) 2.10.14 (https://www.gimp.org/).
Results and Discussion
Organization of Mitochondrial Genomes
The organization of the mitogenomes of the two populations was very similar to other ostariophysan fishes (Satoh et al., 2016) and, consequently, to other Hypostomus already described: 13 PCGs, 22 tRNA genes, 2 rRNA genes, and a partial control region (D-loop) (Figure 1). However, we found different sizes for the three mitochondrial genomes: H. ancistroides presented 16,826 bp, while the male of H. aff. ancistroides was 16,505 bp and the female was 17,066 bp (Figure 1, Supplementary Table 1). The difference in sizes is due to the control region.
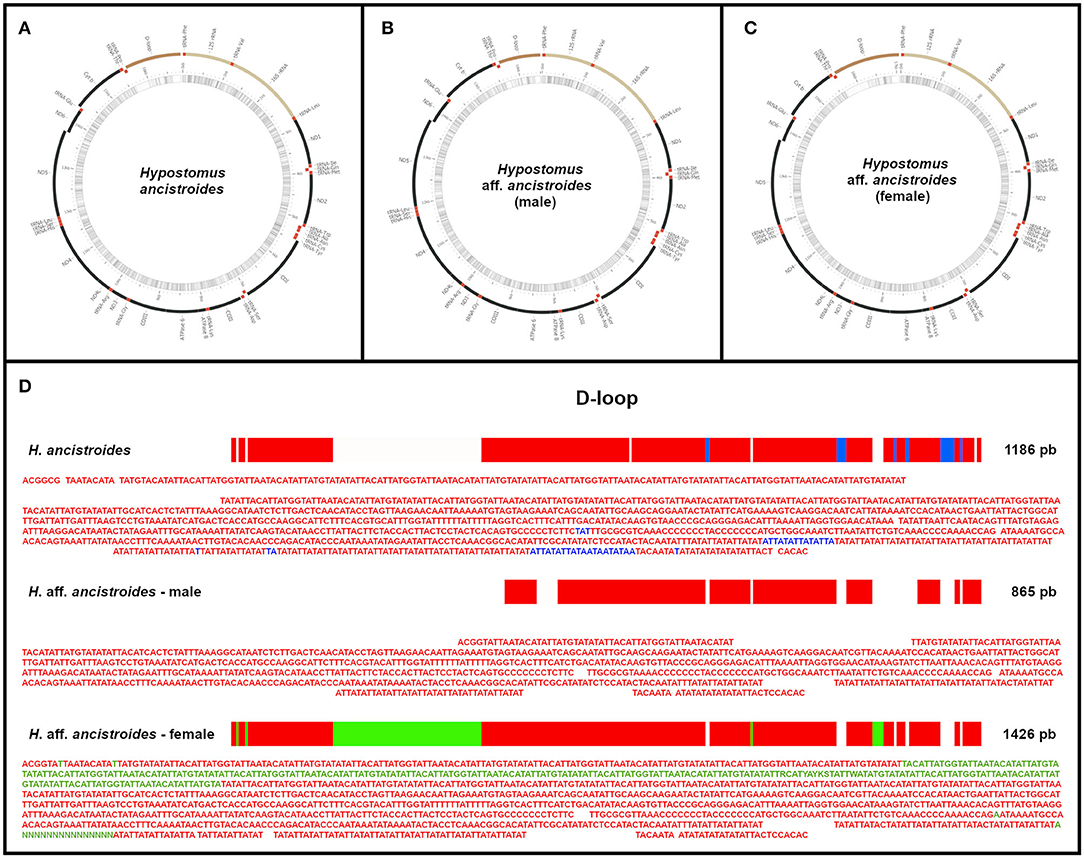
Figure 1. The complete mitochondrial genomes of Hypostomus ancistroides (A) and male (B) and female (C) of Hypostomus aff. ancistroides. The mitogenomes of the two populations have a similar organization, with small variations in the start and stop positions of genes. In (D), a comparison between the control regions (D-loop): homologous bases among individuals are represented in red, bases exclusive to H. ancistroides are in blue, and bases exclusive to the female of H. aff. ancistroides are in green. Blanks represent absent regions.
The base composition of the mitochondrial genome of H. ancistroides was estimated as 31.7% A, 26.6% C, 14.6% G, and 26.9% T. For the H. aff. ancistroides male, we found 31.5% A, 26.5% C, 14.7% G, and 27.1% T, while for the female, we found 31.6% A, 25,89% C, 14,51% G, and 27,87% T. In all mitogenomes, most genes are in the heavy chain, with only eight tRNAs and a PCG found in the light chain (Figure 1, Supplementary Table 1).
Protein-Coding Genes
The three mitogenomes analyzed were the same size for PCGs. Hypostomus aff. ancistroides individuals showed identical sequences, although they may have varied the starting and ending position of PCGs within the genome when compared to H. ancistroides (Supplementary Table 1). The mitochondrial genomes have thirteen PCGs: atp6 (683 bp), atp8 (168 bp), cox1 (1551 bp), cox2 (691 bp), cox3 (784 bp), cytb (1138 bp), nad1 (975 bp), nad2 (1045 bp), nad3 (349 bp), nad4 (1381 bp), nad4L (297 bp), nad5 (1827 bp), and nad6 (522 bp), representing about 70% of the mitogenomes.
There was an overlapping coding region between atp8 and atp6, nad4L and nad4, and nad5 and nad6. Except for cox1, which starts with GTG, the other PCGs use the ATG codon for initiation. The stop codons identified were TAA (for atp8, nad4L, atp6, nad1, cox1, and nad5), TAG (for nad6), and incomplete T-stop (for nad3, cox2, cox3, nad2, cytb, and nad4). Except for nad6, all the other PCGs were in the heavy chain (Figure 1, Supplementary Table 1).
Ribosomal and Transfer RNA Genes
We found two rRNA genes, 12S rRNA (954 bp) and 16S rRNA (1677 bp), located between tRNA-Phe and tRNA-Leu, separated by tRNA-Val (Figure 1, Supplementary Table 1), as commonly observed in other vertebrates (Inoue et al., 2000).
There were 22 tRNAs in both mitogenomes, with sizes ranging from 67 to 75 bp. There was a single tRNA size difference between the two populations: tRNA-Lys has 74 bp in H. ancistroides and 73 bp in H. aff. ancistroides. There are only eight tRNAs in the light chain: tRNA-Gln, tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Tyr, tRNA-Ser, tRNA-Glu, and tRNA-Pro (Supplementary Table 1). Some tRNAs had atypical codons, such as tRNA-Leu (UAA), tRNA-Ser (UGA), tRNA-Ser (GCU), and tRNA-Leu (UAG).
Control Region
As presented before, we found different sizes for the three mitochondrial genomes: H. ancistroides presented 16,826 bp, while the male of H. aff. ancistroides had 16,505 bp and the female had 17,066 bp (Figure 1, Supplementary Table 1). This difference was observed in the control region (D-loop): H. ancistroides presented 1186 bp, whereas males of H. aff. ancistroides had 865 bp and females had 1426 bp in this same region (Figure 1, Supplementary Table 1). We compared the D-loop sequences of the three individuals and found some regions that are exclusive in H. ancistroides and others that are exclusive in the female of H. aff. ancistroides (Figure 1D). This exclusivity in each individual, probably due to a 35-bp tandem repeat unit, is responsible for the significant difference in the total size of the control regions and may consequently reflect the difference in the size of their genomes. Repeated regions are a well-known problem for sequence assembly algorithms. Therefore, it was hard to assemble the D-loop region with extensive repeated units. Consequently, repetitive sequences and their repetitions resulted in different sequence length by NGS (Hahn et al., 2013; Tang et al., 2014). D-loop size variations have also been observed in other mitogenomes of different species (Wilkinson and Chapman, 1991; Miracle and Campton, 1995; Shan et al., 2014; Xue et al., 2015; Zhang et al., 2016), and differences in the size of this region between sexes were also observed in Meretrix lamarckii (Bettinazzi et al., 2016). Except for the gap in the D-loop region, all mitogenomes were assembled successfully in their entirety. Nevertheless, further studies on this variation in the D-loop are still necessary.
Phylogenetic Analysis
Almost all representatives of Hypostomus were grouped in the same clade in the tree topology obtained through Bayesian Analysis (Figure 2), except for H. plecostomus. This species position as a sister group of Pterygoplichthys may suggest an introgression of mitochondrial genes, a misidentification of the individual, or a result from the possible paraphyletic characteristic of Hypostomus (Armbruster, 2004).
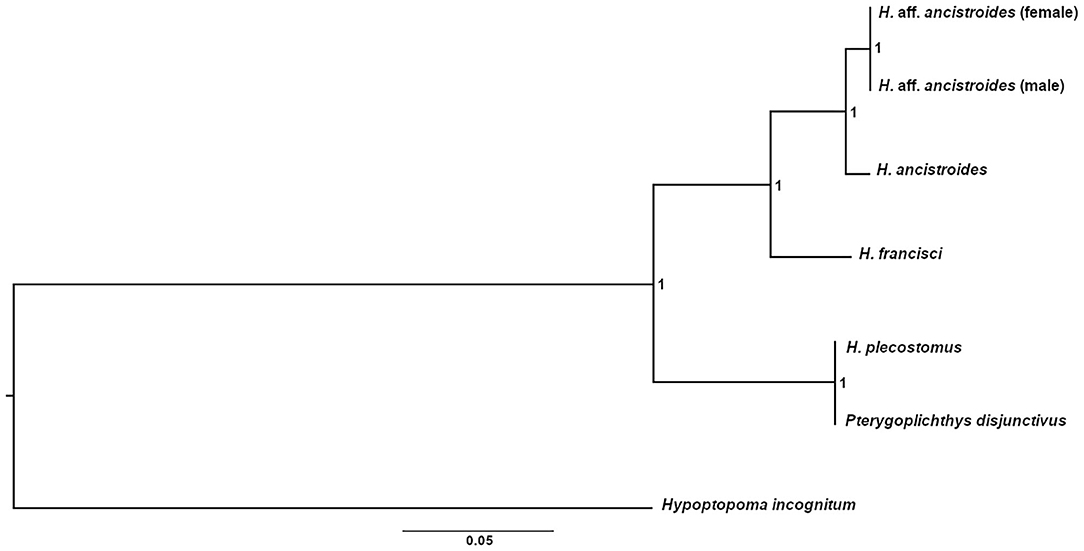
Figure 2. Bayesian phylogenetic tree for mitogenomes of Hypostomus and other Loricariidae species using Hypoptopoma incognitum as an external group. The analysis was performed with the concatenation of the 13 PCGs after choosing the best nucleotide replacement model for each gene. The analysis was carried out using four independent chains with 10,000,000 generations. The numbers on each node represent the posterior probabilities obtained.
Furthermore, females and males of H. aff. ancistroides form a well-supported clade (Figure 2). Despite being part of the same population, individuals of both sexes have sufficient differences in the PCGs that allow their separation into two distinct branches. However, they remain in a single clade, which presents itself as a sister group of H. ancistroides. Besides the proximity of these two populations, these data reinforce how different H. aff. ancistroides is when compared to H. ancistroides.
In conclusion, based on the morphological, cytogenetic, genetic, and mitochondrial differences here presented, this scenario supports H. aff. ancistroides as another species of the complex (Rocha-Reis et al., 2018), which needs to be formally described and named as a taxonomically valid species.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, MT066232; https://www.ncbi.nlm.nih.gov/genbank/, MT081402; https://www.ncbi.nlm.nih.gov/genbank/, MT396945.
Ethics Statement
The animal study was reviewed and approved by Conselho Nacional de Controle de Experimentação Animal (CONCEA).
Author Contributions
DR-R, FM, RP, and KK contributed to the conceptualization, investigation and design of the study. DR-R, RP, and FM performed the statistics analysis and were responsible for data validation. JH-H, TS, RP and KK provided resources to carry out the work. RP and KK were responsible for the administration and supervision of the research project. DR-R wrote the first draft of the manuscript. RP, KK, FM, JH-H, and TS wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The Galaxy server that we used for some calculations is partially funded by Collaborative Research Center 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and the German Federal Ministry of Education and Research [BMBF grants 031 A538A/A538C RBC, 031L0101B/031L0101C de.NBI-epi, and 031L0106 de.STAIR (de.NBI)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Henrique Peluzio for his support on the UFV computer cluster analysis. DR-R appreciates Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for the D.Sc. fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.579965/full#supplementary-material
References
Afgan, E., Baker, D., Batut, B., Van Der Beek, M., Bouvier, D., Cech, M., et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Agostinho, A. A., Gomes, L. C., and Pelicice, F. M. (2007). Ecologia e Manejo de Recursos Pesqueiros em Reservatórios do Brasil. Maringá: Eduem, 501p.
Armbruster, J. W. (2004). Phylogenetic relationships of the suckermouth armoured catfishes (Loricariidae) with emphasis on the Hypostominae and the Ancistrinae. Zool. J. Linn. Soc. 141, 1–80. doi: 10.1111/j.1096-3642.2004.00109.x
Bernt, M., Bleidorn, C., Braband, A., Dambach, J., Donath, A., Fritzsch, G., et al. (2013). A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Phylogenet. Evol. 69, 352–364. doi: 10.1016/j.ympev.2013.05.002
Bettinazzi, S., Plazzi, F., and Passamonti, M. (2016). The complete female- and male- transmitted mitochondrial genome of Meretrix lamarckii. PLoS ONE 11:e0153631. doi: 10.1371/journal.pone.0153631
Billington, N., and Hebert, P. (1991). Mitochondrial DNA diversity in fishes and its implications for introductions. C. J. Fish. Aquat. Sci. 48, 80–94. doi: 10.1139/f91-306
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Britski, H. A. (1972). “Peixes de água doce do Estado de São Paulo: sistemática”, in Poluição e Psicultura. Faculdade de Saúde Pública da Universidade de São Paulo: Instituto de Pesca da Coordenadoria da Pesquisa de Recursos Naturais da Secretaria da Agricultura, eds Comissão Interestadual da bacia Paraná-Uruguai (São Paulo), 79–108.
Caccone, A., Gentile, G., Burns, C. E., Sezzi, E., Bergman, W., Ruelle, M., et al. (2004). Extreme difference in rate of mitochondrial and nuclear DNA evolution in a large ectotherm, Galápagos tortoises. Mol. Phylogenet. Evol. 31, 794–798. doi: 10.1016/j.ympev.2004.02.004
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Fricke, R., Eschmeyer, W. N., and Van Der Laan, R. (2020). Eschmeyer's Catalog of Fishes: Genera, Species, References. Available online at: http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp. (accessed January 22, 2020).
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–abaiting and iterative mapping approach. Nucleic Acids Res. 41:e129. doi: 10.1093/nar/gkt371
Harrison, R. G. (1989). Animal mitochondrial DNA as a genetic marker in populations and evolutionary biology. Trends Ecol. Evol. 4, 6–11. doi: 10.1016/0169-5347(89)90006-2
Hirase, S., Takeshima, H., Nishida, M., and Iwasaki, W. (2016). Parallel mitogenome sequencing alleviates random rooting effect in phylogeography. Genome Biol. Evol. 8, 1267–1278. doi: 10.1093/gbe/evw063
Inoue, J. G., Miya, M., Tsukamoto, K., and Nishida, M. (2000). Complete mitochondrial DNA sequence of the Japanese sardine, Sardinops melanostictus. Fisheries Sci. 66, 924–932. doi: 10.1046/j.1444-2906.2000.00148.x
Iwasaki, W., Fukunaga, T., and Isagozawa, R. (2013). MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 30, 2531–2540. doi: 10.1093/molbev/mst141
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lane, R. (1997). “The species concept in blood-sucking vectors of human diseases”, in Species, the Units of Biodiversity, ed. M. F. Claridge, H. A. Dawah, M. R. Wilson (London: Chapman & Hall), 273–289.
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2016). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Liu, S., Zhang, J., Yao, J., and Liu, Z. (2016). The complete mitochondrial genome of the armored catfish, Hypostomus plecostomus (Siluriformes: Loricariidae). Mitochondrial DNA A 27, 1908–1909. doi: 10.3109/19401736.2014.971281
Ma, C., Yang, P., Jiang, F., Chapuis, M. P., Shali, Y., Sword, G. A., et al. (2012). Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 21, 4344–4358. doi: 10.1111/j.1365-294X.2012.05684.x
Miracle, A. L., and Campton, D. E. (1995). Tandem repeat sequence variation and length heteroplasmy in the mitochondrial DNA D-Loop of the threatened gulf of mexico sturgeon, Acipenser oxyrhynchus desotoi. J. Heredity 86, 22–27. doi: 10.1093/oxfordjournals.jhered.a111520
Moreira, D. A., Magalhães, M. G. P., Andrade, P. C. C., Furtado, C., Val, A. L., and Parente, T. E. (2015). An RNA-based approach to sequence the mitogenome of Hypoptopoma incognitum (Siluriformes: Loricariidae). Mitochondrial DNA A 27, 3784–3786. doi: 10.3109/19401736.2015.1079903
Nakatani, M., Miya, M., Mabuchi, K., Saitoh, K., and Nishida, M. (2011). Evolutionary history of otophysi (Teleostei), a major clade of the modern freshwater fishes: pangaean origin and mesozoic radiation. BMC Evol. Biol. 11:177. doi: 10.1186/1471-2148-11-177
Oyakawa, O. T., Akama, A., and Zanata, A. M. (2005). Review of the genus Hypostomus Lacépède, 1803 from Rio Ribeira de Iguape basin, with description of a new species (Pisces, Siluriformes, Loricariidae). Zootaxa 921, 1–27. doi: 10.11646/zootaxa.921.1.1
Pereira, A. H., Facchin, S., Oliveira do Carmo, A., Núñez Rodriguez, D., Cardoso Resende, L., Kalapothakis, Y., et al. (2019). Complete mitochondrial genome sequence of Hypostomus francisci (Siluriformes: Loricariidae). Mitochondrial DNA B 4, 155–157. doi: 10.1080/23802359.2018.1544860
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rocha-Reis, D. A., de Oliveira Brandão, K., de Almeida-Toledo, L. F., Pazza, R., and Kavalco, K. F. (2018). The persevering cytotaxonomy: discovery of a unique XX/XY sex chromosome system in catfishes suggests the existence of a new, endemic and rare species. Cytogenet. Genome Res. 156, 45–55. doi: 10.1159/000492959
Rocha-Reis, D. A., Pasa, R., and Kavalco, K. F. (2020). High congruence of karyotypic and molecular data on Hypostomus species from the Paraná River basin. bioRxiv [PrePrint]. doi: 10.1101/2020.09.22.308437
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MRBAYES 3.2: efficient bayesian phylogenetic inference and model selection across a large model space. Syst. Biol. 61:539–542. doi: 10.1093/sysbio/sys029
Satoh, T. P., Miya, M., Mabuchi, K., and Nishida, M. (2016). Structure and variation of the mitochondrial genome of fishes. BMC Genomics 17:719. doi: 10.1186/s12864-016-3054-y
Seemann, T., and Gladman, S. (2012). Fasta Statistics: Display Summary Statistics for a Fasta File. Available online at: https://github.com/galaxyproject/tools-iuc.
Shan, X., Xia, Y., Zheng, Y. C., Zou, F. D., and Zeng, X. M. (2014). The complete mitochondrial genome of Quasipaa boulengeri (Anura: Dicroglossidae). Mitochondrial DNA 25, 83–84. doi: 10.3109/19401736.2013.782023
Taanman, J. W. (1999). The mitochondrial genome: structure, transcription, translation and replication. BBA-Bioenergetics 1410, 103–123. doi: 10.1016/S0005-2728(98)00161-3
Tang, M., Tan, M. H., Meng, G. L., Yang, S. Z., Su, X., Liu, S. L., et al. (2014). Multiplex sequencing of pooled mitochondrial genomes–a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 42:e166. doi: 10.1093/nar/gku917
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Weber, C. (2003). “Subfamily Hypostominae”, in Check list of the freshwater fishes of South and Central America, eds R. E. Reis, S. O. Kullander, C.J. Ferraris (Porto Alegre: Edipucrs), 351–372.
Wilkinson, G. S., and Chapman, A. M. (1991). Length and sequence variation in evening bat D-loop mtDNA. Genetics 128, 607–617.
Xue, R., Liu, J. B., Yu, J. J., and Yang, J. D. (2015). The complete mitogenome of Amolops loloensis and related phylogenetic relationship among Ranidae. Mitochondrial DNA 27, 4629–4630. doi: 10.3109/19401736.2015.1101589
Keywords: Hypostomus ancistroides, phylogeny, mitogenome, Alto Paraná river basin, WGS
Citation: Rocha-Reis DA, Pasa R, Menegidio FB, Heslop-Harrison JS, Schwarzacher T and Kavalco KF (2020) The Complete Mitochondrial Genome of Two Armored Catfish Populations of the Genus Hypostomus (Siluriformes, Loricariidae, Hypostominae). Front. Ecol. Evol. 8:579965. doi: 10.3389/fevo.2020.579965
Received: 03 July 2020; Accepted: 09 November 2020;
Published: 04 December 2020.
Edited by:
Giacomo Bernardi, University of California, Santa Cruz, United StatesReviewed by:
Moises A. Bernal, Auburn University, United StatesWilliam Ludt, Natural History Museum of Los Angeles County, United States
Copyright © 2020 Rocha-Reis, Pasa, Menegidio, Heslop-Harrison, Schwarzacher and Kavalco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dinaíza Abadia Rocha-Reis, dinaizabio@gmail.com