 Rosie Everett
Rosie Everett Becky Cribdon
Becky Cribdon- 1Department of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, United Kingdom
- 2School of Life Sciences, University of Warwick, Coventry, United Kingdom
The use of metagenomic datasets to support ancient sedimentary DNA (sedaDNA) for paleoecological reconstruction has been demonstrated to be a powerful tool to understand multi-organism responses to climatic shifts and events. Authentication remains integral to the ancient DNA discipline, and this extends to sedaDNA analysis. Furthermore, distinguishing authentic sedaDNA from contamination or modern material also allows for a better understanding of broader questions in sedaDNA research, such as formation processes, source and catchment, and post-depositional processes. Existing tools for the detection of damage signals are designed for single-taxon input, require a priori organism specification, and require a significant number of input sequences to establish a signal. It is therefore often difficult to identify an established cytosine deamination rate consistent with ancient DNA across a sediment sample. In this study, we present MetaDamage, a tool that examines cytosine deamination on a metagenomic (all organisms) scale for multiple previously undetermined taxa and can produce a damage profile based on a few hundred reads. We outline the development and testing of the MetaDamage tool using both authentic sedaDNA sequences and simulated data to demonstrate the resolution in which MetaDamage can identify deamination levels consistent with the presence of ancient DNA. The MetaDamage tool offers a method for the initial assessment of the presence of sedaDNA and a better understanding of key questions of preservation for paleoecological reconstruction.
1. Introduction
In this study, we present the MetaDamage1 tool, which was developed to assess the levels of postmortem cytosine deamination patterns on a metagenomic scale, in which unknown multi-organism sequences can be assessed for ancient DNA damage in one process. The tool offers a novel alternative to the tools described, with key advantages including single input of metagenomic datasets, a low threshold of input sequences, and a single workflow process to produce an output summary of observed DNA damage.
The development of next-generation sequencing of ancient sedimentary DNA (sedaDNA) has allowed a wide range of metagenomic studies for paleoecological and paleoenvironmental reconstruction (Willerslev et al., 2014; Smith et al., 2015; Birks and Birks, 2016; Szczuciński et al., 2016; Slon et al., 2017; Ahmed et al., 2018; Lammers et al., 2018; Wood et al., 2018; Zobel et al., 2018; Keck et al., 2020; Seersholm et al., 2020; Murchie et al., 2021a). As a proxy for examining past vegetation, sedaDNA has demonstrated its potential as a complementary and additional tool to conventional paleoecological proxies, such as pollen, plant macrofossils, and diatoms (Parducci et al., 2015; Pedersen et al., 2016; Niemeyer et al., 2017; Zimmermann et al., 2017; Clarke et al., 2018; Epp et al., 2018; Alsos et al., 2020a; Gaffney et al., 2020; Volstad et al., 2020). The value of sedaDNA as a tool within multi-proxy research has also been supported by the development of best laboratory practices for minimizing contamination and improving data quality (Gilbert et al., 2005; Armbrecht et al., 2019; Shapiro et al., 2019). Recent discussions on the challenges of working with sedaDNA have also focused on challenges associated with conventional paleoecological research (Smith et al., 2015; Chen and Ficetola, 2020; Cribdon et al., 2020; Edwards, 2020; Dussex et al., 2021). This has included wider discussions of the issues of understanding the source area and catchment of sedaDNA, the role of taphonomic processes in the formation of the biomolecular archives, and how preservation conditions impact its contribution as a tool for paleoecological reconstruction (Alsos et al., 2018, 2020b; Parducci et al., 2018; Giguet-Covex et al., 2019; Marianne et al., 2020). This in turn has led to improvements in approaches to bioinformatic processing, such as increasing confidence in the phylogenetic assignation of taxa within ancient metagenomic sequences (Smith et al., 2015; Cribdon et al., 2020).
The number of sedaDNA studies using the shotgun sequencing approach is still limited (Smith et al., 2015; Pedersen et al., 2016; Seersholm et al., 2016; Slon et al., 2017; Ahmed et al., 2018; Parducci et al., 2019; Stahlschmidt et al., 2019; Ardelean et al., 2020; Armbrecht et al., 2020; Gaffney et al., 2020; Schulte et al., 2020; Murchie et al., 2021b; Thomas et al., 2021). The alternative, a targeted amplicon sequencing approach using organism range specific primers [e.g., chloroplast trnL (UAA) gene specific for plants; Taberlet et al., 2007] known as metabarcoding sequencing (Bell et al., 2016; Parducci et al., 2018; Edwards, 2020) has proven far more popular, with not just demonstrative capabilities of high resolution amplification of plant taxa for palaeoenvironmental reconstruction (e.g., Sønstebø et al., 2010; Jørgensen et al., 2012; Parducci et al., 2012, 2013, 2015, 2019; Pedersen et al., 2013; Giguet-Covex et al., 2014, 2019; Epp et al., 2015; Pansu et al., 2015; Alsos et al., 2016, 2020b; Sjögren et al., 2017; Clarke et al., 2018; Zale et al., 2018; Crump et al., 2019; Liu et al., 2020; Volstad et al., 2020), but for addressing key barriers in molecular research, such as financial cost (Parducci et al., 2018) and higher computational processing requirements needed for the analysis of metagenomic data. However, as outlined by Cribdon et al. (2020), one major advantage of utilizing the shotgun sequencing approach in sedaDNA research is that the sequencing approach allows for an assessment of authentication that goes beyond reliance on the absence of taxa from negative controls and replication (c.f. Clarke et al., 2018; Ficetola et al., 2018; Giguet-Covex et al., 2019; Edwards, 2020). The shotgun sequencing process amplifies whole molecules of DNA rather than targeted amplicons and, as such, captures fragment ends and allows for an assessment of any cytosine deamination damage in sequences (Briggs et al., 2007). This damage signature can then be used to discriminate between datasets containing modern sequences vs. authentic sedaDNA sequences (Sawyer et al., 2012; Key et al., 2017; Kistler et al., 2017; Parducci et al., 2019; Renaud et al., 2019; Edwards, 2020). The capacity to identify deamination has a direct impact on the understanding of wider questions in paleoecological research, such as taphonomic processes and the preservation of sedaDNA sequences in the sedimentary record (Kistler et al., 2015; Smith et al., 2015; Gaffney et al., 2020).
As a standard approach to authentication of DNA sequence, tools designed for single reference species with mathematical models describing a single coherent process of DNA modification as a property of a single sample are relied on for assessing deamination levels (mapDamage, Jónsson et al., 2013; PMD tools, Skoglund et al., 2014). In the case of sedaDNA shotgun datasets, there are rarely sufficient reads for any one taxon to apply such methods, even though the total read count may be large across all taxa. This has recently been addressed by the development of programs such as the metagenomic bacterial screening tool HOPS (Hübler et al., 2019), DamageProfiler (Neukmann et al., 2020), and PyDamage (Borry et al., 2021). Application of tools such as mapDamage to a wide range of species by concatenating reference genomes violates their mathematical framework but also becomes computationally impractical when dealing with the thousands of unknown species that may be present in a metagenomic sample.
These tools have demonstrated their capabilities in the assessment of the authenticity of sequences, in particular, the isolation of 5′-end C to T base misincorporations within bacterial metagenomic datasets. They use a sorting methodology similar to the metagenomic approach in MetaDamage described here, in which multi-organism sequences are binned either using a phylogenetic sorting tool such as MEGAN (c.f. Herbig et al., 2016; used in Hübler et al., 2019; Neukmann et al., 2020) or de novo assembly. Borry et al. (2021) has been specifically designed for ancient pathogen authentication and is limited in the formatting of the output by the incorporation of the metagenomic mapping software MALT during the mapping process (Neukmann et al., 2020). The DamageProfiler tool (Neukmann et al., 2020) offers more flexibility in input organisms with its process, but it runs in a similar way to MapDamage (Jónsson et al., 2013) in its requirement of a SAM/BAM input tool and is reliant on reference-based mapping and is therefore limited in its input potential to a computationally reasonable number of genomes. There is a similar limitation in PyDamage (Borry et al., 2021), which is reliant on the de novo assembly of bacterial genomes as input.
The MetaDamage tool follows a five-stage workflow that has been designed in Perl script and combined to support users with simple command-line use with either a FASTA or BLAST file input. In summary, MetaDamage uses a local or remote database to find a reference sequence using BLAST that corresponds directly to each input query based on the same length and direction. Each input query and its corresponding reference are then globally aligned, and the proportion of sequences where the reference and query have mismatching bases is calculated. The output is a damage profile similar in a graphical format to mapDamage (Jónsson et al., 2013), but with metagenomic data.
Initial application of the MetaDamage approach proved successful in the analysis of low-level input queries (i.e., < 100 sequences) and the identification of low-level deamination frequencies, therefore providing additional support to mapDamage analysis undertaken on an individual species level (Gaffney et al., 2020, Supplementary Figure S4.4). In this study, we demonstrate the efficacy and resolution with which a damage assessment of metagenomic datasets can help users with an early-stage analysis of the extent of ancient DNA damage on a sample-to-sample basis. The output of the MetaDamage tool can help contribute to questions in the application of sedaDNA as a paleoecological tool, such as the taphonomic processes of sedaDNA preservation and formation processes that may lead to potential modern contamination.
2. MetaDamage algorithm
2.1. Scripts
All scripts described in the methodology can be found at https://github.com/MetaDamage/MetaDamage.git.
2.2. Process
The MetaDamage tool estimates all base substitutions of sequences for the first 25 5′-end and 3′-end base positions by default, with a focus on C >T and G >A substitutions for double-stranded libraries and C>T substitutions for 5′-end and 3′-end for single-stranded libraries. The tool runs as a single pipeline and returns credible intervals on base modification estimates, which allows for a more refined understanding of the output of the substitution assessment. There are several stages to the MetaDamage pipeline, which are outlined in Figure 1.
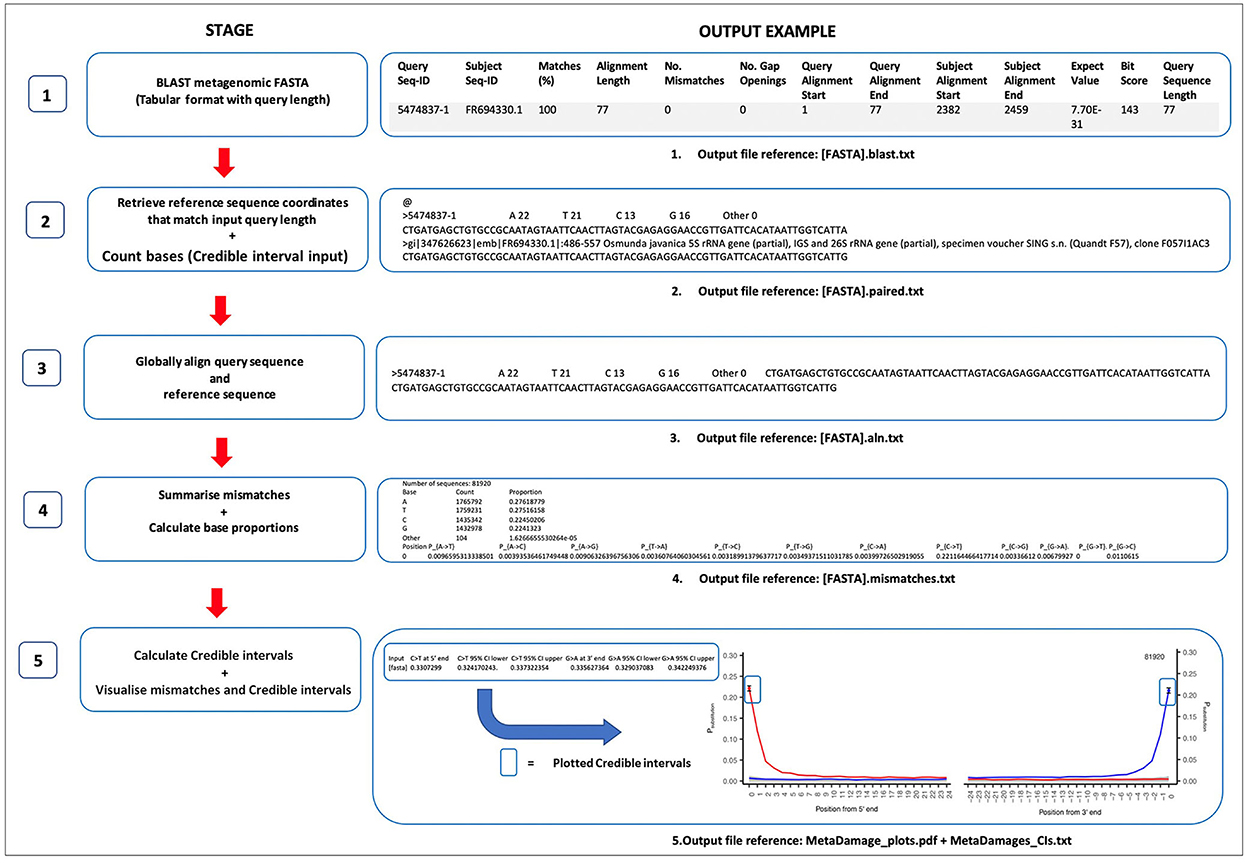
Figure 1. Stages of the MetaDamage tool, including the Credible Interval assessment.
3. MetaDamage methodology
3.1. MetaDamage pipeline stages
As outlined in Figure 1, the MetaDamage tools work in a 5-stage approach, in which each stage is detailed as follows.
The MetaDamage tool requires a FASTA or BLAST file as input and returns the subject sequence coordinates that match the input query length, given that it is likely only a portion of the query sequence will be aligned to the subject and base-modified termini are likely to be excluded. Using the BLAST output, a combined text file of the query and subject reference sequences is generated using either blastdbcmd from a local database or Efetch (Schuler et al., 1996) within the E-Utilities package, which provides access to the suite of interconnected databases of NCBI (NCBI, 2010; Harbert, 2018). The aim is to find a reference sequence that corresponds directly to the query based on the same length and same direction and is ready for alignment.
The Needleman-Wunsch algorithm is used for alignment (adapted from Needleman and Wunsch, 1970). Realignment of the query sequences to the reference sequences using a global alignment is important as it allows for alignment of the whole query sequence (end-to-end) with the reference sequence alignment so that each mismatch can be assessed in a way that is robust to the unexpected occurrence of indels.
3.1.1. Stage 1: Providing input BLAST analysis of metagenomic FASTA files
The MetaDamage tool can perform the initial BLAST search, which requires an input of a FASTA file of all query sequences, or can take a previous BLAST output with a corresponding FASTA file as an input. All sequences are subjected to BLASTn analysis (Altschul et al., 1990) with the following options, using the full NCBI nt database:
blastn -db [nucleotide database] -num_threads [x] -query [input FASTA] -out [output BLAST] -max_target_seqs 1 -max_hsps 1 -outfmt “6 std qlen.”
The applied parameters are utilized in the BLAST process for the following output:
- The “max_target_seqs” parameter is applied to limit the number of hits returned per sequence. This is set to “1” to return only the first hit (Shah et al., 2018).
- The “max_hsps” option refers to high-scoring segment pairs and will give only 1 HSP per subject for all hits in the database.
- The “6 std qlen” option determines the output format. “6” specifies a tabular format, which reduces the output footprint. “std” adds the standard output information, and “qlen” adds an additional field for the length of the query sequence.
Output: [FASTA].blast.txt.
The BLAST output in Figure 1 has been reformatted to show each header of the BLAST parameters and configured for clarity.
3.1.2. Stage 2: Retrieving reference sequence coordinates and count bases
3.1.2.1. Retrieve reference sequence
Using the input BLAST, for each query sequence:
1. Use the start and end coordinates of the reference sequence to establish whether it is reversed relative to the query.
2. Calculate new start and end coordinates that map the subject reference sequence to the whole query (matching query region, not the whole reference sequence).
3. Discard the query if the new start coordinate is before the beginning of the reference sequence because this means the 5′-end of the query is not present in the reference and therefore cannot be compared and terminal deamination signal determined.
4. Use these coordinates to extract that region of the reference sequence using either blastdbcmd on a local database or Efetch on a remote (NCBI) database and correct its orientation to the reverse complement if necessary to match the query sequence.
5. Export the reference title line, the reference sequence, and the query sequence for each query sequence into output text files.
3.1.2.2. Count bases
For every reference sequence, the number of A, T, C, and G bases is counted. This output is used later in the credible interval calculations (Stage 5).
Output: [FASTA].paired.txt.
The resulting output file contains each query sequence in the original FASTA, separated by “@” symbols (see Figure 1).
3.1.3. Stage 3: Globally align query sequence and reference sequence
Each query with the corresponding reference sequence is realigned using the Needleman-Wunsch algorithm (adapted from Needleman and Wunsch, 1970).
Output: [FASTA].aln.txt.
3.1.4. Stage 4: Summarize mismatches and calculate base proportions
3.1.4.1. Summarize mismatches
Once aligned, the first and last 25 bases of every alignment are assessed for mismatches. This approach calculates the proportion of sequences where the reference and query have mismatching bases. This process calculates proportions for all 12 possible mismatch types.
The output also includes the total number of aligned sequences.
3.1.4.2. Calculate base proportions
Using the total number of bases (for all sequences) and the total number of each base (C, T, G, and A), the Perl script calculates the empirical proportions of each base in the dataset.
Output: [FASTA].mismatches.txt.
3.1.5. Stage 5: Calculation of credible intervals and visualization of mismatches
All steps in Stage 5 are processed in R (version 4.0.3).
3.1.5.1. Calculation of credible intervals
Confidence testing is incorporated into the MetaDamage tool to allow the user to assess the estimated deamination rate given the number of input query sequences. A posterior credible interval for the observed proportion of mismatches is calculated using a beta distribution to provide a 95% range of proportions that could have given rise to the observed results at base position 0, the position most likely to demonstrate cytosine deamination. This allows the user to gauge the confidence of the deamination estimate, in particular when the number of input sequences is very low.
The parameters of the beta distribution are calculated from the output of Stage 4 ([fasta].mismatches.txt). The underlying binomial distribution is defined by a total number of trials equal to the number of instances at which a DNA fragment terminates (base position 0) in a C (defined by reference sequences), and a probability of a C > T modification equal to the proportion of C(reference) > T(query) mismatches at base position 0. The related posterior beta distribution parameters, alpha and beta, are therefore based on these and are defined as follows.
1) Generate alpha and beta values.
Alpha value: (number of trials * proportion C>T mismatch at base position 0) +1.
Beta value: [number of trials * (1 – proportion C>T mismatch at base position 0)] +1.
2) Calculate 95% credible interval boundaries.
The 95% credible interval is the upper and lower boundaries of estimated probabilities between 0.025 and 0.975. This is calculated using a cumulative beta distribution function within R using the alpha and beta values.
CI_lower_bound < qbeta (0.025, shape1 = alpha, shape2 = beta).
CI_upper_bound < qbeta (0.0975, shape1 = alpha, shape2 = beta).
Output: MetaDamage_CIs.txt.
3.1.5.2. Visualization of mismatches
The positional mismatches and credible intervals are visualized using R for the MetaDamage tool output. The mismatches are plotted as a P substitution for both the 5′ C>T (PC>T) and 3′ G>A (PG>A)-end against the ith base position for double-stranded libraries, and 5′ and 3′ C>T (PC>T) for single-stranded libraries. The total number of sequences used in the analysis is also printed in the top right of the plot.
Output: MetaDamage_plots.pdf.
An example of this output is demonstrated in Figure 2A (a reasonable number of input sequences consistent with an ancient DNA signal, n = 427 sequence reads) and Figure 2B (low number of input sequences with an ancient DNA signal, n = 26 sequence reads). This output is an example of a shotgun sedaDNA dataset taken from Everett (2021).
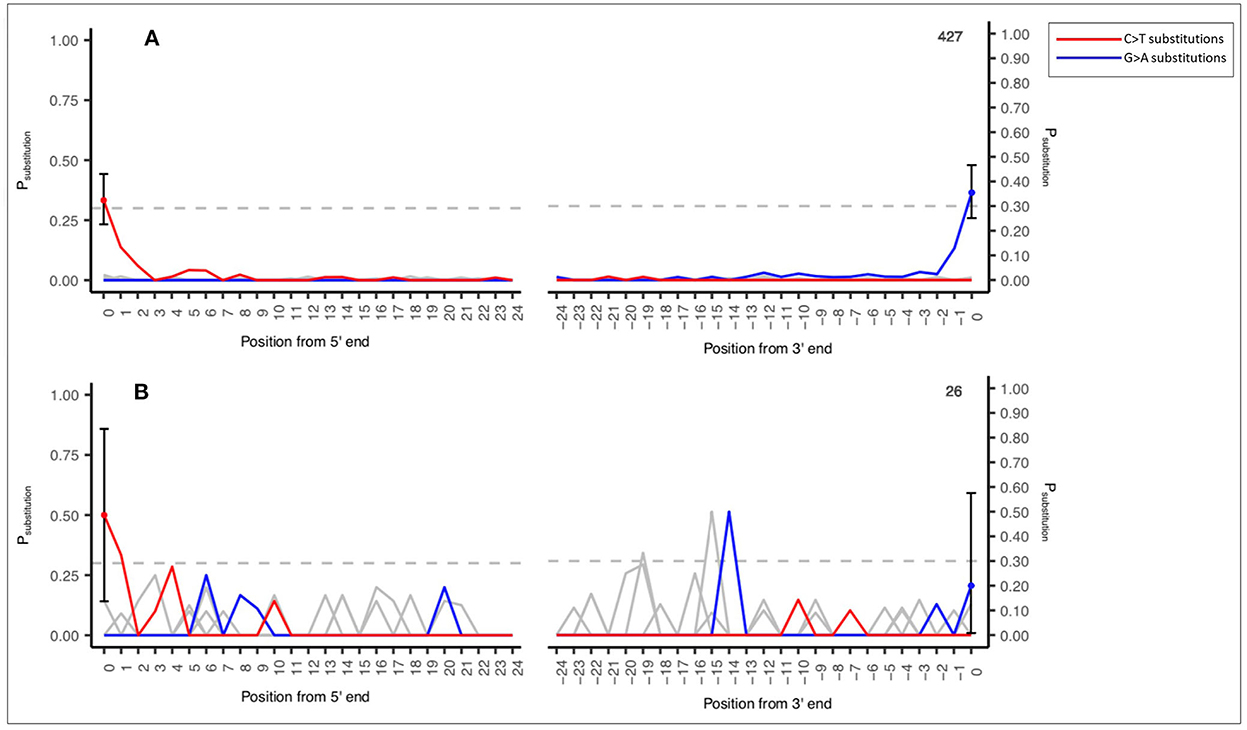
Figure 2. Example MetaDamage output—(A) High number of input sequences (427) and an observed deamination rate of 0.33333 with ancient DNA signal. (B) Low number of input sequences (26) and an observed deamination rate of 0.5 with ancient DNA signal. Shotgun datasets provided by Everett (2021).
Figures 2A, B also demonstrate the difference in output based on the observed deamination rate and the number of input sequences for a confident ancient DNA signal. Figure 2A has an observed deamination rate of 0.33333 based on 427 sequences and has produced a clearly defined ancient DNA-associated signal. Figure 2B has a higher observed deamination rate of 0.5 but is based on 26 sequences and has produced output with a signal that one cannot confidently interpret as consistent with ancient DNA. This relationship between the observed deamination rate, the number of input sequences, and the success rate of MetaDamage are discussed further in Section 4.
3.1.6. Overview of resolution testing
We tested the sensitivity and resolution of MetaDamage on simulated ancient DNA data, real sedaDNA data with authentic DNA signals, and unknown metagenomic sequences from early Holocene sediment samples to demonstrate the resolution capability of MetaDamage.
This process aimed to:
1) assess the confidence level and accuracy of the estimated deamination rates;
2) establish a baseline for the minimum number of reads from which the MetaDamage tool could identify a deamination signal;
3) compare MetaDamage outputs on known authentic sedaDNA data with simulated DNA data;
4) test MetaDamage on unpublished data with unknown damage parameters to demonstrate the capability of the tool to undertake damage assessment.
3.1.7. Testing MetaDamage on simulated datasets
The aim of using a simulated dataset was to take a known profile of double-stranded libraries, in this case, reads simulated using Gargammel (Renaud et al., 2017) from chloroplast sequences using parameters of known damage statistics from previous ancient DNA studies (Gamba et al., 2014; Schubert et al., 2014; Allentoft et al., 2015), and run through the MetaDamage tool. Chloroplast DNA was used for analytical simplicity, ease of simulation generation, and good database representation. The MetaDamage output of the observed PiC>T from the simulations would then be compared with that of the observed PiC>T value from selected ancient DNA studies.
The simulated datasets used for testing the MetaDamage tool were generated from a process that utilized the following:
1) All available chloroplast genomes (n = 4,823; Supplementary Data 1) were downloaded in the FASTA format from NCBI (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/).
2) Damage statistics from ancient DNA studies (Gamba et al., 2014; Schubert et al., 2014; Allentoft et al., 2015) were previously inferred using mapDamage as part of a paleogenomics meta-analysis (Kistler et al., 2017).
3) The chloroplast genomes and damage statistics from each DNA study were piped into the Gargammel tool (Renaud et al., 2017) to create simulated read FASTA output with the assigned damage parameters.
4) FASTAs of 100, 300, 500, and 1,000 sequences were generated by dividing the whole simulated FASTA output as subsets to test through MetaDamage.
A total of 1,600 FASTA files were generated and subjected to MetaDamage analysis (Supplementary Table S1).
3.1.8. Testing confidence in the simulated dataset
Each set of MetaDamage analysis conditions was repeated 100 times on different simulated datasets for which credible intervals were produced to test the consistency and accuracy of the deamination estimates (Figure 3), and the generated data can be found in Supplementary Data 2.
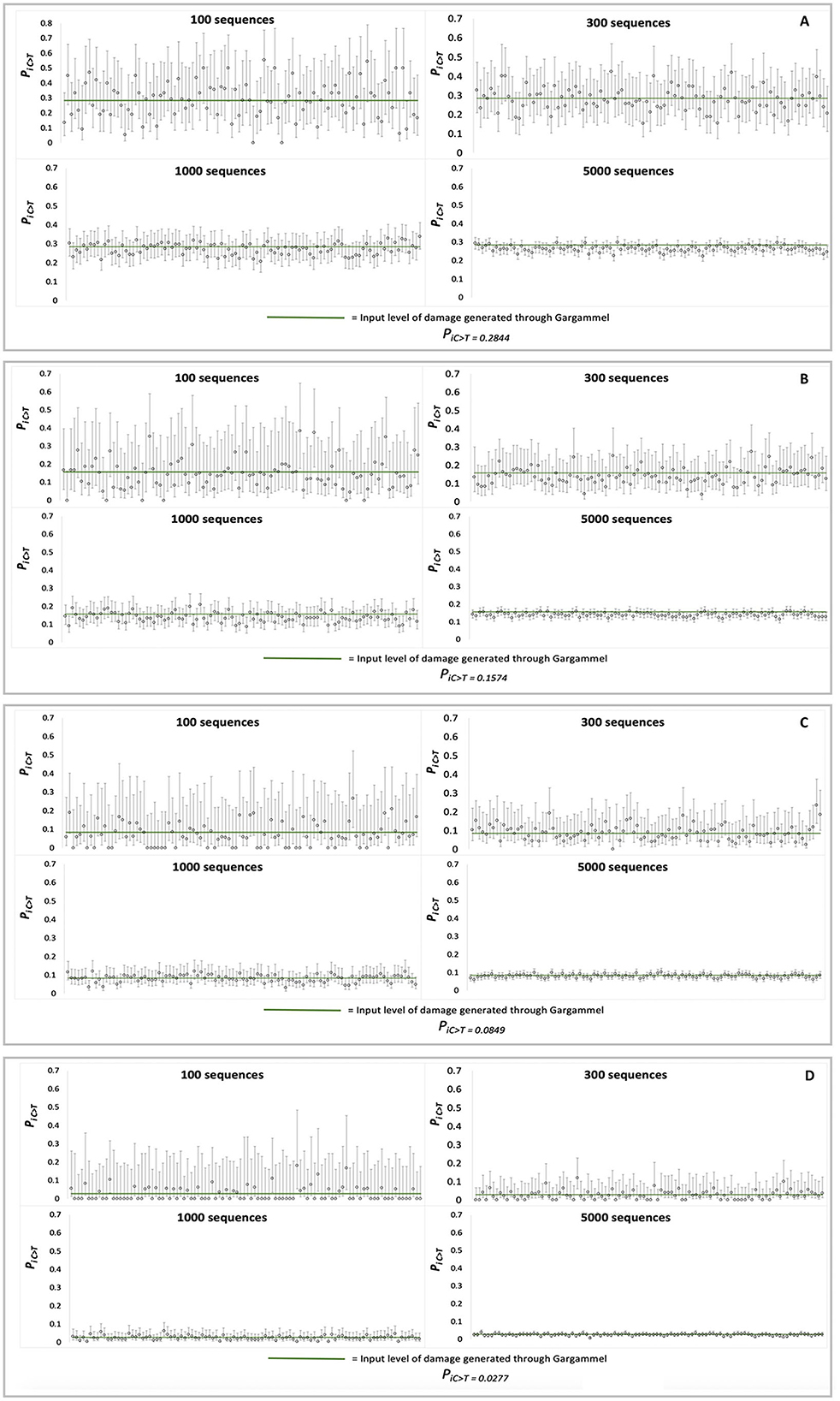
Figure 3. Observed credible intervals observed in each FASTA input of simulation data (N = 100 for each plot): (A) RISE145, (B) RISE00, (C) SRR1187907, and (D) ERR657747.
The output of the credible interval testing of the simulated data demonstrated the following observations on the efficacy of MetaDamage as a tool (see Supplementary Table S2) for examining deamination on a metagenomic scale:
1) The 95% credible interval varied as predicted with the input number of sequences, from narrowest in the 5,000-sequence trails (for example, in RISE145, the average estimated range of 95% credible interval was 0.0602) to widest in the 100-sequence trials (average estimated range of 95% credible interval was 0.3932).
2) The 95% credible interval captured the empirical deamination value in 97.25% of trials using 5,000 sequences, 96% of trials using 1,000 sequences, 95.75% of trials using 300 sequences, and 95.5% of trials using 100 sequences.
False negatives occurred in a range of 3.5% (100 sequences) to 2.75% (5,000 sequences) of the tested simulated datasets (credible intervals underestimating the empirical deamination value), in which the most common errors were driven by situations by a combination of low deamination rate and few sequences. Given the 95% range of credible intervals, these results are within expectations.
This testing has highlighted both the strength of the MetaDamage tool in its capability to assess the damage on a low number of input sequences (e.g., < 100 sequences), and under the conditions examined here, that it requires a minimum observed deamination rate (Psubstitution > 0.1) for success in identifying damage patterns where the number of input sequences is low. This is discussed further in Section 3.1.10.
3.1.9. Testing MetaDamage on published data with an authentic ancient DNA signal
The application of the MetaDamage tool to data from early Holocene sediment sequences (Gaffney et al., 2020) demonstrates sequence authenticity where the observed PiC>T was 0.1697 (Figure 4A). This input FASTA consisted of 173,727 known authentic sequences representing short reads (< 75 bp) associated with the Viridiplantae clade (processed using MEGAN; Huson et al., 2007). This clade was chosen as it had a more complex profile than the cpDNA solely tested in the simulation data and was thus used to test the capability of MetaDamage on more complex metagenomic datasets.
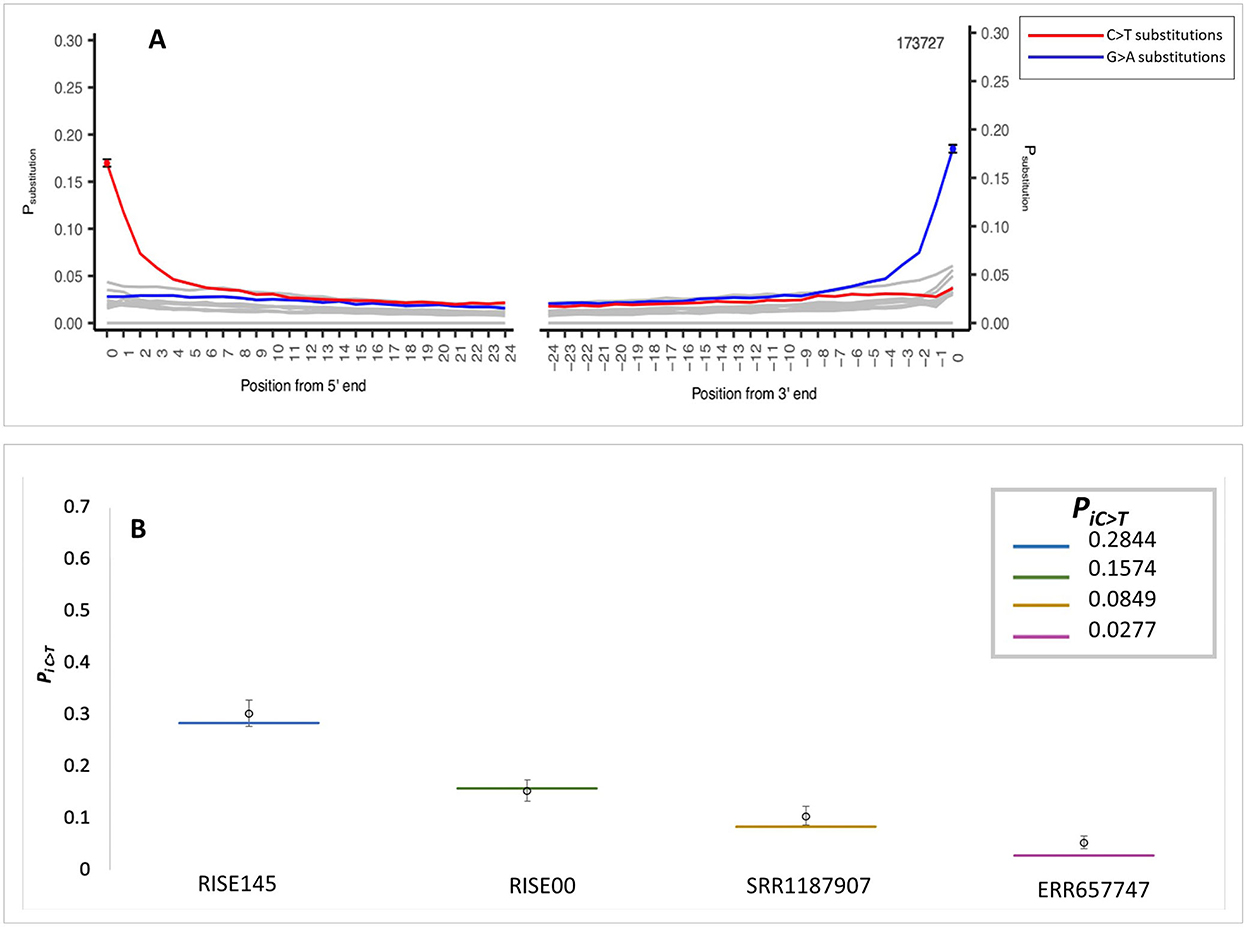
Figure 4. MetaDamage outputs—(A) Authentic Holocene Viridiplantae dataset. (B) Credible intervals observed in simulation data using authentic sequence dataset with damage parameter mapped for.
Once the MetaDamage tool demonstrated capability in identifying an authentic aDNA signal from the early Holocene sediment sequences, the input FASTA was subjected to testing in the simulation conditions, to test confidence in this initial output.
The input FASTA file was first subjected to the following processing:
- Removal of first 5 bases off 5′-end and 3′-end to remove any existing damage signal.
- Concatenation into a single FASTA format sequence and interspersal of 75 N's within FASTA to avoid the generation of chimeric simulated fragments.
Using the same damage statistics as in the initial testing (see Section 3.1.7), the modified Viridiplantae sequences (Gaffney et al., 2020) were piped into the Gargammel tool (Renaud et al., 2017) to create simulated reads with the assigned damage parameters. The simulated FASTA file was then processed with the MetaDamage tool, and credible intervals for deamination rate estimates were generated. The output of the tests is detailed (Figure 4B; see Supplementary Table S3), leading to the following observations:
1) The output of confidence testing using an authentic sedimentary DNA dataset with simulated damage characteristics supports the small range of the 95% credible interval with input sequences of over 5,000 observed in the simulated data.
2) The output demonstrates that the MetaDamage tool can process complex metagenomic datasets of mixed DNA input or unknown origin, based on the comparison of the MetaDamage PiC>T output of the authentic ancient DNA data and the damage parameters set by the simulation data.
This testing, therefore, demonstrated the capacity of the MetaDamage tool to identify damage patterns on complex metagenomic datasets. The next phase tested the resolution of the tool through the analysis of 10 datasets that had a low number of input sequences and a range of estimated deamination rates.
3.1.10. Testing MetaDamage on data where ancient DNA signal is unknown
The MetaDamage tool was tested on sedaDNA data from early Holocene sediment sequences from submerged fluvial deposits (Everett, 2021) and focused on sequences from the Viridiplantae group. The input sequences were short reads (< 150 bases) generated from double-stranded libraries. The purpose was to test the capabilities of the MetaDamage tool in dealing with a range of lower input sequences, and the relationship with observed deamination rates.
The samples analyzed included a range of input sequences from 28 to 1,103 and observed PiC>T from 0 to 0.33 (Figures 5A–J; see Supplementary Table S4). These results demonstrate the applicability of the MetaDamage tool for highly fragmented and low-read input sequences often associated with sedaDNA. The following conclusions can be drawn:
1) The MetaDamage tool can identify damage in inputs with low numbers of sequences, with the lowest success seen with 70 reads (Figure 5D).
2) Although a small dataset, the examples used demonstrate the success of the MetaDamage tool where the observed deamination rate was above Psubstitution > 0.1, except in the case of input example “D” of 70 reads where the observed PiC>T was 0.09. Although there is only one example, it demonstrates the potential for the MetaDamage tool to identify low-level damage in small input queries.
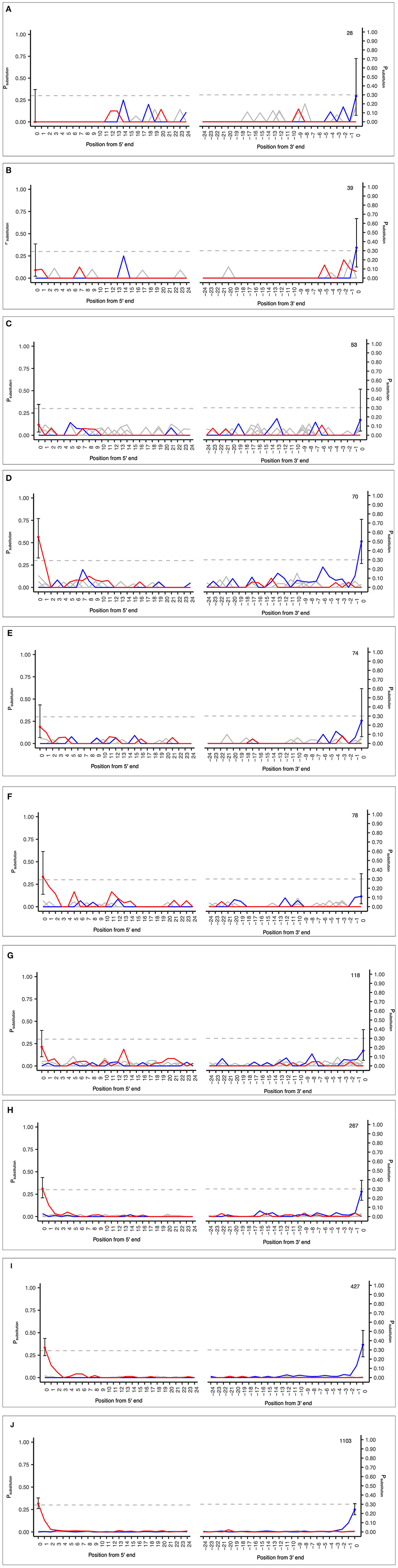
Figure 5. (A–J) Observed credible intervals in each FASTA input of empirical data from unpublished Holocene dataset (Everett, 2021).
4. Discussion
The role of authentication in ancient metagenomics and sedaDNA analysis is paramount to distinguish between authentic ancient DNA and potential contamination and for providing an understanding of wider issues relating to the accuracy of the paleoecological reconstruction, such as post-depositional preservation and the potential taxonomic bias.
The testing of MetaDamage on simulated datasets demonstrated that fewer sequences in the test sample result in less precision but similar accuracy for capturing the empirical underlying values within credible intervals that we expect to get wider with fewer input sequences. For the tested 5,000 and 1,000 input sequences, ancient DNA signals on a metagenomic scale were confidently recovered with high precision. With 300 input sequences, the credible interval range becomes wider, and with 100 input sequences, the MetaDamage tool can still establish the presence of a damage signal. However, with a varied confidence range of these observed mismatches, the results are sufficient to validate a small dataset for the initial assessment of ancient DNA signals on a metagenomic scale.
This outcome of the tests demonstrated a trade-off between the number of reads and the strength of the damage signal, with stronger damage signals (e.g., PiC>T> 0.1) being detectable with read counts of < 100, but weaker signals are not. Although more testing is required on empirical data to examine the nature of the relationship between the number of input queries and the required damage level, these tests have demonstrated the capability of MetaDamage to provide damage assessment on low-read inputs.
However, it must be noted that the MetaDamage tool has the primary aim of being used as an initial assessment of metagenomic damage analysis of multi-species input from sedaDNA samples. The low-read input that the MetaDamage testing has demonstrated the capability to assess is far below the suggested minimum input number for obtaining an accurate DNA damage profile (>1,000 mapped reads) on an individual species level (Warinner et al., 2017).
In the context of paleoecological reconstruction, the MetaDamage tool can rapidly assess the presence of authentic aDNA on a metagenomic scale without the need for reference genomes. This can contribute to the understanding of the formation and post-depositional processes associated with deposits of paleoecological value in the context of sedaDNA analyses. As a complementary tool to existing methods for ancient DNA authentication, the MetaDamage tool has demonstrated its capabilities for the initial process of authentication of metagenomic data. This initial overview allows for a more targeted approach to single-taxa authentication tools such as mapDamage (Jónsson et al., 2013) and PMDtools (Skoglund et al., 2014).
The process of authentication of sedaDNA for paleoecological reconstruction requires further research, in particular key aspects of taphonomic processes, such as determining the relationship between sediment type and preservation. Confidence in sedaDNA interpretation is not only a powerful approach for the development of the technique but overall allows users to readdress questions in key aspects, such as taphonomic processes and contextualizing the output of sedaDNA analysis with a better understanding of the potentially authentic sedaDNA.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
RE and BC wrote and designed MetaDamage. RE performed accuracy testing with some input from BC. RE was the primary author of the manuscript, with review and editing by BC. Both authors contributed to the article and approved the submitted version.
Funding
The sedaDNA dataset used for initial testing was generated under RE PhD NERC CENTA DTP funded project titled Investigating the preservation of ancient sedimentary DNA (sedaDNA) from three case study wetland environments: toward a better understanding of sedaDNA as a tool for paleoecological reconstruction.
Acknowledgments
RE would like to thank the Natural Environment Research Council (NERC) and the University of Warwick for the financial support of this research, as well as the three reviewers for comments that have improved this manuscript and the editor for support in this process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.888421/full#supplementary-material
Footnotes
References
Ahmed, E., Parducci, L., Unneberg, P., Ågren, R., Schenk, F., Rattray, J. E., et al. (2018). Archaeal community changes in Lateglacial lake sediments: evidence from ancient DNA. Quat. Sci. Rev. 181, 19–29. doi: 10.1016/j.quascirev.2017.11.037
Allentoft, M. E., Sikora, M., Sjögren, K. G., Rasmussen, S., Rasmussen, M., Stenderup, J., et al. (2015). Population genomics of Bronze Age Eurasia. Nature 522, 167–172. doi: 10.1038/nature14507
Alsos, I. G., Edwards, M. E., and Clarke, C. L. (2020b). Survival and spread of arctic plants in response to climate change: DNA-based evidence. PAGES Magazine 28, 10–11. doi: 10.22498/pages.28.1.12
Alsos, I. G., Lammers, Y., Yoccoz, N. G., Jørgensen, T., Sjögren, P., Gielly, L., et al. (2018). Plant DNA metabarcoding of lake sediments: how does it represent the contemporary vegetation. PLoS ONE 13, e0195403. doi: 10.1371/journal.pone.0195403
Alsos, I. G., Sjögren, P., Brown, A. G. L., Merkel, M. K. F., Paus, A., Lammers, Y., et al. (2020a). Last Glacial Maximum environmental conditions at Andøya, northern Norway; evidence for a northern ice-edge ecological “hotspot”. Quat. Sci. Rev. 239, 106364. doi: 10.1016/j.quascirev.2020.106364
Alsos, I. G., Sjögren, P., Edwards, M. E., Landvik, J. Y., Gielly, L., Forwick, M., et al. (2016). Sedimentary ancient DNA from Lake Skartjørna, Svalbard: assessing the resilience of arctic flora to Holocene climate change. The Holocene 26, 627–642. doi: 10.1177/0959683615612563
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ardelean, C. F., Becerra-Valdivia, L., Pedersen, M. W., Schwenninger, J. L., Oviatt, C. G., Macías-Quintero, J. I., et al. (2020). Evidence of human occupation in Mexico around the Last Glacial Maximum. Nature 584, 87–92. doi: 10.1038/s41586-020-2509-0
Armbrecht, L., Herrando-Pérez, S., Eisenhofer, R., Hallegraeff, G. M., Bolch, C. J. S., Cooper, A., et al. (2020). An optimized method for the extraction of ancient eukaryote DNA from marine sediments. Mol. Ecol. Resour. 20, 906–919. doi: 10.1111/1755-0998.13162
Armbrecht, L. H., Coolen, M. J. L., Lejzerowicz, F., George, S. C., Negandhi, K., Suzuki, Y., et al. (2019). Ancient DNA from marine sediments: precautions and considerations for seafloor coring, sample handling and data generation. Earth Sci. Rev. 196, e102887. doi: 10.1016/j.earscirev.2019.102887
Bell, K. L., de Vere Keller, N., Richardson, A., Gous, R. T., Burgess, A., Gous, A., et al. (2016). Pollen DNA barcoding: current applications and future prospects. Genome 59, 629–664. doi: 10.1139/gen-2015-0200
Birks, H. J., and Birks, H. H. (2016). How have studies of ancient DNA from sediments contributed to the reconstruction of Quaternary floras? New Phytol. 209, 499–506. doi: 10.1111/nph.13657
Borry, M., Hübner, A., Rohrlach, A. B., and Warinner, C. (2021). PyDamage: automated ancient damage identification and estimation for contigs in ancient DNA de novo assembly. PeerJ 9, e11845. doi: 10.7717/peerj.11845
Briggs, A. W., Stenzel, U., Johnson, P. L. F., Green, R. E., Kelso, J., Prüfer, K., et al. (2007). Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 104, 14616–14621. doi: 10.1073/pnas.0704665104
Chen, W., and Ficetola, G. F. (2020). Numerical methods for sedimentary-ancient-DNA-based study on past biodiversity and ecosystem functioning. Environ. DNA 2, 115–129. doi: 10.1002/edn3.79
Clarke, C. L., Edwards, M. E., Brown, A. G., Gielly, L., Lammers, Y., Heintzman, P. D., et al. (2018). Holocene floristic diversity and richness in northeast Norway revealed by sedimentary ancient DNA (sedaDNA) and pollen. Boreas 48, 12357. doi: 10.1111/bor.12357
Cribdon, B., Ware, R., Smith, O., Gaffney, V., and Allaby, R. G. (2020). PIA: more accurate taxonomic assignment of metagenomic data demonstrated on sedaDNA from the North Sea. Front. Ecol. E8, 84. doi: 10.3389/fevo.2020.00084
Crump, S. E., Miller, G. H., Power, M., Sepulveda, J., Dildar, N., Coghlan, M., et al. (2019). Arctic shrub colonization lagged peak postglacial warmth: Molecular evidence in lake sediment from Arctic Canada. Glob. Chang. Biol. 25, 4244–4256. doi: 10.1111/gcb.14836
Dussex, N., Bergfeldt, N., de Anca Prado, V., Dehasque, M., Díez-del-Molino, D., Ersmark, E., et al. (2021). Integrating multi-taxon palaeogenomes and sedimentary ancient DNA to study past ecosystem dynamics. Proc. R. Soc. B 288, 1957. doi: 10.1098/rspb.2021.1252
Edwards, M. E. (2020). The maturing relationship between Quaternary paleoecology and ancient sedimentary DNA. Quat. Res. 96, 39–47. doi: 10.1017/qua.2020.52
Epp, L. S., Gussarova, G., Boessenkool, S., Olsen, J., Haile, J., Schrøder-Nielsen, A., et al. (2015). Lake sediment multi-taxon DNA from North Greenland records early post-glacial appearance of vascular plants and accurately tracks environmental changes. Quat. Sci. Rev. 117, 152–163. doi: 10.1016/j.quascirev.2015.03.027
Epp, L. S., Kruse, S., Kath, N. J., Stoof-Leichsenring, K. R., Tiedemann, R., Pestryakova, L. A., et al. (2018). Temporal and spatial patterns of mitochondrial haplotype and species distributions in Siberian larches inferred from ancient environmental DNA and modeling. Sci. Rep. 8, 17436. doi: 10.1038/s41598-018-35550-w
Everett, R. (2021). Investigating the Preservation of Ancient Sedimentary DNA (sedaDNA) From Three Case Study Wetland Environments: Towards Better Understanding of sedaDNA as a Tool for Palaeoecological Reconstruction (Unpublished dissertation thesis). University of Warwick.
Ficetola, G. F., Poulenard, J., Sabatier, P., Messager, E., Gielly, L., Bakke, J., et al. (2018). DNA from lake sediments reveals long-term ecosystem changes after a biological invasion. Sci. Adv. 4, eaar4292. doi: 10.1126/sciadv.aar4292
Gaffney, V., Fitch, S., Bates, M., Ware, R., Kinnaird, T., Gearey, B., et al. (2020). Multi-proxy characterisation of the storegga tsunami and its impact on the early holocene landscape of the southern North Sea. Geosciences 10, e270. doi: 10.3390/geosciences10070270
Gamba, C., Jones, E. R., Teasdale, M. D., McLaughlin, R. L., Gonzalez-Fortes, G., Mattiangeli, V., et al. (2014). Genome flux and stasis in a five millennium transect of European prehistory. Nat. Commun. 5, 5257. doi: 10.1038/ncomms6257
Giguet-Covex, C., Ficetola, G. F., Walsh, K., Poulenard, J., Bajard, M., Fouinat, L., et al. (2019). New insights on lake sediment DNA from the catchment: importance of taphonomic and analytical issues on the record quality. Sci. Rep. 9, 14676. doi: 10.1038/s41598-019-50339-1
Giguet-Covex, C., Pansu, J., Arnaud, F., Rey, P. J., Griggo, C., Gielly, L., et al. (2014). Long livestock farming history and human landscape shaping revealed by lake sediment DNA. Nat. Commun. 5, 3211. doi: 10.1038/ncomms4211
Gilbert, M. T. P., Bandelt, H.-J., Hofreiter, M., and Barnes, I. (2005). Assessing ancient DNA studies. Trends Ecol. E20, 541–544. doi: 10.1016/j.tree.2005.07.005
Harbert, R. S. (2018). Algorithms and strategies in short- read shotgun metagenomic reconstruction of plant communities. Appl. Plant Sci. 6, e1034. doi: 10.1002/aps3.1034
Herbig, A., Maixner, F., Bos, K. I., Zink, A., Krause, J., and Huson, D. H. (2016). MALT: Fastaalignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv (preprint). doi: 10.1101/050559
Hübler, R., Key, F. M., Warinner, C., Bos, K. I., Krause, J., Herbig, A., et al. (2019). HOPS: automated detection and authentication of pathogen DNA in archaeological remains. Genome Biol. 20, 280. doi: 10.1186/s13059-019-1903-0
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P., and Orlando, L. (2013). mapDamage 2.0 fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Jørgensen, T., Haile, J., Möller, P. E. R., Andreev, A., Boesenkool, S., Rasmusen, M., et al. (2012). A comparative study of ancient sedimentary DNA, pollen and macrofossils from permafrost sediments of northern Siberia reveals long-term vegetational stability. Mol. Ecol. 21, 1989. doi: 10.1111/j.1365-294X.2011.05287.x
Keck, F., Millet, L., Debroas, D., Etienne, D., Galop, D., Rius, D., et al. (2020). Assessing the response of micro-eukaryotic diversity to the Great Acceleration using lake sedimentary DNA. Nat. Commun. 11, 3831. doi: 10.1038/s41467-020-17682-8
Key, F. M., Posth, C., Krause, J., Herbig, A., and Bos, K. I. (2017). Mining metagenomic data sets for ancient DNA: recommended protocols for authentication. Trends Genet. 33, 508–520. doi: 10.1016/j.tig.2017.05.005
Kistler, L., Smith, O., Ware, R., Momber, G., Bates, R., Garwood, P., et al. (2015). Thermal age, cytosine deamination and the veracity of 8,000 year old wheat DNA from sediments. bioRxiv. doi: 10.1101/032060
Kistler, L., Ware, R., Smith, O., Collins, M., and Allaby, R. G. (2017). A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res. 45, 6310–6320. doi: 10.1093/nar/gkx361
Lammers, Y., Clarke, C. L., Ers?us, C., Brown, A. G., Edwards, M. E., Gielly, L., et al. (2018). Clitellate worms (annelida) in lateglacial and holocene sedimentary dna records from the Polar Urals and Northern Norway. Boreas 48, 317–329. doi: 10.1111/bor.12363
Liu, S., Stoof-Leichsenring, K. R., Kruse, S., Pestryakova, L. A., and Herzschuh, U. (2020). Holocene vegetation and plant diversity changes in the North-Eastern Siberian treeline region from pollen and sedimentary ancient DNA. Front. Ecol. Evol. 8, 304. doi: 10.3389/fevo.2020.560243
Marianne, E., Clokie, M. R. J., Czypionka, T., Frisch, D., Godhe, A., Kremp, A., et al. (2020). Dead or alive: sediment DNA archives as tools for tracking aquatic evolution and adaptation. Commun. Biol. 3, 1–11. doi: 10.1038/s42003-020-0899-z
Murchie, T. J., Kuch, M., Duggan, A., Ledger, M., Roche, K., Klunk, J., et al. (2021a). Optimizing extraction and targeted capture of ancient environmental DNA for reconstructing past environments using PalaeoChip Arctic-1.0 bait set. Quat. Res. 99, 305–328. doi: 10.1017/qua.2020.59
Murchie, T. J., Monteath, A. J., Mahony, M. E., Long, G. S., Cocker, S., Sadoway, T., et al. (2021b). Collapse of the mammoth-steppe in central Yukon as revealed by ancient environmental DNA. Nat. Commun. 12, 7120. doi: 10.1038/s41467-021-27439-6
NCBI (2010). Entrez Programming Utilities Help. Bethesda (MD): National Center for Biotechnology Information (US). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK25501/
Needleman, S. B., and Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453. doi: 10.1016/0022-2836(70)90057-4
Neukmann, J., Peltzer, A., and Nieselt, K. (2020). DamageProfiler: fast damage pattern calculation for ancient DNA. bioRxiv (preprint). doi: 10.1101/2020.10.01.322206
Niemeyer, B., Epp, L. S., Stoof-Leichsenring, K. R., Pestryakova, L. A., and Herzschuh, U. (2017). A comparison of sedimentary DNA and pollen from lake sediments in recording vegetation composition at the Siberian treeline. Mol Ecol Res. 17, e46–e62. doi: 10.1111/1755-0998.12689
Pansu, J., Giguet-Covex, C., Ficetola, G. F., Gielly, L., Boyer, F., Zinger, L., et al. (2015). Reconstructing long-term human impacts on plant communities: an ecological approach based on lake sediment DNA. Mol. Ecol. 24, 1485–1498. doi: 10.1111/mec.13136
Parducci, L., Alsos, I. G., Unneberg, P., Pedersen, M. W., Han, L., Lammers, Y., et al. (2019). Shotgun environmental DNA, pollen, and macrofossil analysis of lateglacial lake sediments from southern Sweden. Front. Ecol. Evol. 7, 189. doi: 10.3389/fevo.2019.00189
Parducci, L., Jorgensen, T., Tollefsrud, M. M., Everland, E., Alm, T., Fontana, S. L., et al. (2012). Glacial survival of boreal trees in northern Scandinavia. Science. 335, 1083–1086. doi: 10.1126/science.1216043
Parducci, L., Matetovici, I., Fontana, S. L., Bennett, K. D., Suyama, Y., Haile, J., et al. (2013). Molecular- and pollen-based vegetation analysis in lake sediments from central Scandinavia. Mol. Ecol. 22, 3511–3524. doi: 10.1111/mec.12298
Parducci, L., Nota, K., and Wood, J. (2018). Reconstructing Past Vegetation Communities Using Ancient DNA from Lake Sediments. Cham: Springer International Publishing.
Parducci, L., Valiranta, M., Salonen, J. S., Ronkainen, T., Matetovici, I., Fontana, S. L., et al. (2015). Proxy comparison in ancient peat sediments: pollen, macrofossil and plant DNA. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 370, 1660. doi: 10.1098/rstb.2013.0382
Pedersen, M. W., Ginohlac, A., Orlando, L., Olsen, J., Andersen, K., Holm, J., et al. (2013). A comparative study of ancient environmental DNA to pollen and macrofossils from lake sediments reveals taxonomic overlap and additional plant taxa. Quat. Sci. Rev. 75, 161–168. doi: 10.1016/j.quascirev.2013.06.006
Pedersen, M. W., Ruter, A., Schweger, C., Friebe, H., Staff, R. A., Kjeldsen, K. K., et al. (2016). Postglacial viability and colonization in North America's ice-free corridor. Nature 537, 45–49. doi: 10.1038/nature19085
Renaud, G., Hanghøj, K. E., and Orlando, L. (2017). Gargammel: a sequence simulator for ancient DNA. Bioinformatics 33, 577–579. doi: 10.1093/bioinformatics/btw670
Renaud, G., Schubert, M., Sawyer, S., and Orlando, L. (2019). “Authentication and assessment of contamination in ancient DNA,” in Ancient DNA: Methods and Protocols, eds B. Shapiro, A. Barlow, P. D. Heintzman, M. Hofreiter, J. L. A. Paijmans, and A. E. R. Soares (New York, NY: Springer), 163–194. doi: 10.1007/978-1-4939-9176-1_17
Sawyer, S., Krause, J., Guschanski, K., Savolainen, V., and Pääbo, S. (2012). Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 7, e34131. doi: 10.1371/journal.pone.0034131
Schubert, M., Jónsson, H., Chang, D., Der Sarkissian, C., Ermini, L., Ginolhac, A., et al. (2014). Genetic foundation of horse domestication. Proc. Natl Acad. Sci. USA 111 E5661–E5669. doi: 10.1073/pnas.1416991111
Schuler, G. D., Epstein, J. A., Ohkawa, H., and Kans, J. A. (1996). Entrez: molecular biology database and retrieval system. Meth. Enzymol. 266, 141–162. doi: 10.1016/S0076-6879(96)66012-1
Schulte, L., Bernhardt, N., Stoof-Leichsenring, K., Zimmermann, H. H., Pestryakova, L. A., Epp, L. S., et al. (2020). Hybridization capture of larch (Larix Mill) chloroplast genes from sedimentary ancient DNA reveals past changes of Siberian forest. Mol. Ecol. Resour. 21, 801–815. doi: 10.5194/egusphere-egu2020-19733
Seersholm, F. V., Pedersen, M. W., Søe, M. J., Shokry, H., Mak, S. S. T., Ruter, A., et al. (2016). DNA evidence of bowhead whale exploitation by Greenlandic Paleo-Inuit 4,000 years ago. Nat. Commun. 7, 13389. doi: 10.1038/ncomms13389
Seersholm, F. V., Werndly, D. J., Grealy, A., Johnson, T., Keenan Early, E. M., Lundelius, E. L., et al. (2020). Rapid range shifts and megafaunal extinctions associated with late Pleistocene climate change. Nat. Commun. 11, 2770. doi: 10.1038/s41467-020-16502-3
Shah, N., Nute, M. G., Warnow, T., and Pop, M. (2018). Misunderstood parameter of NCBI BLAST impacts the correctness of bioinformatics workflows. Bioinformatics 35, 1613–1614. doi: 10.1093/bioinformatics/bty833
Shapiro, B., Barlow, A., Heintzman, P. D., Hofreiter, M., Paijmans, J. L. A., Soares, A. E. R, (eds). (2019). Ancient DNA Methods and Protocols, 2nd Edn. New York, NY: Humana Press. doi: 10.1007/978-1-4939-9176-1
Sjögren, P., Edwards, M. E., Gielly, L., Langdon, C. T., Croudace, I. W., Merkel, M. K. F., et al. (2017). Lake sedimentary DNA accurately records 20th century introductions of exotic conifers in Scotland. New Phytol. 213, 929–941. doi: 10.1111/nph.14199
Skoglund, P., Northoff, B. H., Shunkov, M. V., Derevianko, A. P., Pääbo, S., Krause, J., et al. (2014). Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. U S A. 111, 2229–2234. doi: 10.1073/pnas.1318934111
Slon, V., Hopfe, C., Weiß, C. L., Mafessoni, F., De La Rasilla, M., Lalueza-Fox, C., et al. (2017). Neandertal and Denisovan DNA from Pleistocene sediments. Science. 356, 605–608. doi: 10.1126/science.aam9695
Smith, O., Momber, G., Bates, R., Garwood, P., Fitch, S., Pallen, M., et al. (2015). Sedimentary DNA from a submerged site reveals wheat in the British Isles 8000 years ago. Science. 347, 998–1001. doi: 10.1126/science.1261278
Sønstebø, J. H., Gielly, L., Brysting, A. K., Elven, R., Edwards, M., Haile, J., et al. (2010). Using next-generation sequencing for molecular reconstruction of past Arctic vegetation and climate. Mol. Ecol. Resour. 10, 1009–1018. doi: 10.1111/j.1755-0998.2010.02855.x
Stahlschmidt, M. C., Collin, T. C., Fernandes, D. M., Bar-Oz, G., Belfer-Cohen, A., Gao, Z., et al. (2019). Ancient mammalian and plant DNA from Late Quaternary Stalagmite Layers at Solkota Cave, Georgia. Sci. Rep. 9, 6628. doi: 10.1038/s41598-019-43147-0
Szczuciński, W., Pawłowska, J., Lejzerowicz, F., Nishimura, Y., Kokociński, M., Majewski, W., et al. (2016). Ancient sedimentary DNA reveals past tsunami deposits. Mar. Geol. 381, 29–33. doi: 10.1016/j.margeo.2016.08.006
Taberlet, P., Coissac, E., Pompanon, F., Gielly, L., Miquel, C., Valentini, A., et al. (2007). Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 35, e14. doi: 10.1093/nar/gkl938
Thomas, Z. A., Mooney, S., Cadd, H., Baker, A., Turney, C., Schneider, L., et al. (2021). Late Holocene climate anomaly concurrent with fire activity and ecosystem shifts in the eastern Australian Highlands. Sci. Total Environ. 802, 149542. doi: 10.1016/j.scitotenv.2021.149542
Volstad, L. N., Alsos, I. G., Farnsworth, W. R., Heinztman, P. D., Håkansson, L., Kjellman, S. E., et al. (2020). A complete Holocene lake sediment ancient DNA record reveals long-standing high Arctic plant diversity hotspot in northern Svalbard. Quat. Sci. Rev. 234, 106207. doi: 10.1016/j.quascirev.2020.106207
Warinner, C., Herbig, A., Mann, A., Fellows Yates, J. A., Weiß, C. L., Burbano, H. A., et al. (2017). Robust Framework for Microbial Archaeology. Annu. Rev. Genom. Hum. Genet. 18, 321–356. doi: 10.1146/annurev-genom-091416-035526
Willerslev, E., Davison, J., Moora, M., Zobel, M., Coissac, E., Edwards, M. E., et al. (2014). Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51. doi: 10.1038/nature12921
Wood, J. R., Díaz, F. P., Latorre, C., Wilmshurst, J. M., Burge, O. R., et al. (2018). Plant pathogen responses to Late Pleistocene and Holocene climate change in the central Atacama Desert, Chile. Sci. Rep. 8, 1–8. doi: 10.1038/s41598-018-35299-2
Zale, R., Huang, Y. T., Bigler, C., Wood, J. R., Dalén, L., Wang, X. R., et al. (2018). Growth of plants on the Late Weichselian ice-sheet during Greenland interstadial-1? Quat. Sci. Rev. 185, 222–229. doi: 10.1016/j.quascirev.2018.02.005
Zimmermann, H., Raschke, E., Epp, L., Stoof-Leichsenring, K., Schwamborn, G., Schirrmeister, L., et al. (2017). Sedimentary ancient DNA and pollen reveal the composition of plant organic matter in Late Quaternary permafrost sediments of the Buor Khaya Peninsula (north-eastern Siberia). Biogeosciences 14, 575–596. doi: 10.5194/bg-14-575-2017
Keywords: sedaDNA, metagenomics, authentication, paleoecology, shotgun sequencing
Citation: Everett R and Cribdon B (2023) MetaDamage tool: Examining post-mortem damage in sedaDNA on a metagenomic scale. Front. Ecol. Evol. 10:888421. doi: 10.3389/fevo.2022.888421
Received: 02 March 2022; Accepted: 15 December 2022;
Published: 24 January 2023.
Edited by:
Nic Rawlence, University of Otago, New ZealandReviewed by:
Vilma Perez, University of Adelaide, AustraliaGabriel Renaud, Technical University of Denmark, Denmark
Nicola Alexandra Vogel, Technical University of Denmark Kongens Lyngby, Denmark, in collaboration with reviewer GR
Copyright © 2023 Everett and Cribdon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosie Everett,  cm9zaWUuZXZlcmV0dEBub3J0aHVtYnJpYS5hYy51aw==
cm9zaWUuZXZlcmV0dEBub3J0aHVtYnJpYS5hYy51aw==