Major Histocompatibility Complex Variation and Haplotype Associated Survival in Response to Experimental Infection of Two Bd-GPL Strains Along a Latitudinal Gradient
 Maria Cortazar-Chinarro1,2,3*†
Maria Cortazar-Chinarro1,2,3*†  Sara Meurling1†
Sara Meurling1†  Laurens Schroyens1 Mattias Siljestam1
Laurens Schroyens1 Mattias Siljestam1  Alex Richter-Boix4 Anssi Laurila1
Alex Richter-Boix4 Anssi Laurila1  Jacob Höglund1
Jacob Höglund1- 1Section of Animal Ecology, Department of Ecology and Genetics, Uppsala University, Uppsala, Sweden
- 2MEMEG/Department of Biology, Faculty of Science, Lund University, Lund, Sweden
- 3Department of Earth, Ocean and Atmospheric Sciences, Faculty of Science, University of British Columbia, Vancouver, BC, Canada
- 4Department of Political and Social Science, Pompeu Fabra University, Barcelona, Spain
While both innate and adaptive immune system mechanisms have been implicated in resistance against the chytrid fungus Batrachochytrium dendrobatidis (Bd), studies on the role of specific MHC haplotypes on Bd infection are rare. Here, we studied variation in MHC Class IIB loci in the common toad Bufo bufo along a latitudinal gradient across Sweden. In general, Swedish toad populations had few MHC Class IIB haplotypes and MHC diversity declined from south (13 haplotypes) to the north (four haplotypes). The low diversity may compromise the ability of northern populations to fight emerging disease, such as Bd. In a laboratory experiment, we infected newly metamorphosed toads with two strains of the Global Pandemic Lineage of the fungus (Bd-GPL) and compared survival with sham controls. Bd-infected toads had lower survival compared to controls. Moreover, survival was dependent on the Bd-strain and northern toads had lower Bd-mediated survival than southern individuals. MHC diversity was lower in northern toads. All northern experimental animals were monomorphic for a single MHC haplotype, whereas we found seven different haplotypes in southern experimental animals. In southern toads, survival was dependent on both Bd-strain and MHC haplotype suggesting differential infection dynamics depending on both Bd-strain and host immune system characteristics.
Introduction
Emerging infectious diseases are a severe threat to global biodiversity, causing population declines and extinctions across a range of taxa (Daszak et al., 2000). Amphibian populations world-wide are faced with emerging diseases, among them the chytrid fungus Batrachochytrium dendrobatidis (Bd) which is known to infect more than 700 amphibian species and implicated in the extinction of 90 species (Fisher et al., 2009; Lips, 2016; Scheele et al., 2019). Bd consists of numerous strains and lineages, some of which are highly pathogenic while others are more benign (Refsnider et al., 2015; Dang et al., 2017). The highly pathogenic lineage Bd-GPL has evolved within the last century in east Asia and has since spread over the world infecting naïve amphibian species and populations (O’Hanlon et al., 2018), while in other cases populations and species appear resistant to the infection (Miaud et al., 2016; Gervasi et al., 2017; Russell et al., 2019). Studies on the variation in the pathogenicity of the fungus and whether host populations vary in resistance are therefore vital, as is pinpointing the immunological and genetic basis for such differences. The ability of any amphibian species to resist the fungal infection will vary depending on a range of factors ultimately linked to differences in pathogenicity and immunogenetic variation of the species among different lineages and strains of Bd (Grogan et al., 2018).
The Major Histocompatibility Complex (MHC) is a multigene family coding for peptides involved in the adaptive immune defense of jawed vertebrates (Janeway et al., 2001). It codes for proteins that are part of the adaptive immune system and is associated with disease and parasite resistance (Hedrick, 1994). MHC class II is relevant due to its extreme polymorphism and role in combating bacterial and fungal infections (Barribeau et al., 2008). The exceptionally high diversity at MHC class II loci is believed to be maintained by various forms of balancing selection (Hughes and Nei, 1988). Genetic variation of MHC class II in wild populations can be maintained by several evolutionary mechanisms, such as various forms of frequency dependent selection and heterozygosity advantage (Sommer, 2005). Because of the immune response enhancing properties of MHC, it is suggested that heterozygous individuals are more resistant to pathogens as they have a larger available repertoire to recognize pathogens (Doherty and Zinkernagel, 1975). Previous studies of amphibians have shown a direct association between specific MHC alleles and Bd-resistance (Savage and Zamudio, 2011). However, immunogenetic variation may also be shaped by genetic drift which may vary in force within and among species (Piertney and Oliver, 2006; Cortázar-Chinarro et al., 2017).
Understanding geographic variation in immunogenetic diversity is complicated by which mechanisms shape such diversity (Potts and Slev, 1995; Hedrick, 1999). In general, historical events such as postglacial colonization processes have had a profound impact on the geographical distribution of genetic diversity. During the Last Glacial Maximum (LGM), species inhabiting northern Europe were restricted to refugia in southern Europe (Hewitt, 2004; Hampe and Petit, 2005). After the LGM, species expanded northwards, a process which involved repeated founder events (Hampe and Petit, 2005). Bottlenecks in population size caused by such founder events lead to loss of genetic variation (Nei et al., 1975). Therefore, populations residing at northern latitudes generally display less genetic diversity as compared to populations residing near former refugia (Hewitt, 2000). For example, a recent paper showed that two separate lineages of common toad Bufo bufo colonized the Scandinavian peninsula from two directions and genetic diversity was significantly higher at southern latitudes (Thörn et al., 2021). This study since it is an abundant and common species along the latitudinal gradient. This abundant species is also notably variable in Bd susceptibility (Meurling et al., in press) and MHC diversity.
Here, we experimentally infected newly metamorphosed common toads from the north and the south of Sweden with two strains of Bd-GPL, originating from Sweden and the United Kingdom, respectively. We contrasted treated toadlets with sham controls and recorded their survival over 30 days. We focused on MHC Class IIB variation of common toad populations in Sweden. The aims of the study were to quantify MHC Class II variation along the latitudinal gradient across Sweden, and to study whether Bd-mediated mortality depends on: (1) the Bd-strain, (2) the presence of certain MHC Class IIB haplotypes, or (3) the geographic origin of the experimental animals. This study should be investigated for the first time differences in pathogenicity between the Swedish and United Kingdom Bd-GPL strains.
Materials and Methods
Field Collection
For the analyses of geographical MHC-variation we collected tissue samples from 20 adult toads at each of 12 locations in four different regions along a latitudinal gradient across Sweden (Supplementary Table 1) in April–May 2015. The toads were caught in breeding aggregations and a small piece of rear leg webbing was removed and immediately stored in 96% ethanol. DNA was extracted from the tissue by using a DNeasy Blood and Tissue kit (Qiagen, Germany) according to the manufacturer’s protocol. An additional 160 individuals reared for experiments were included in tissue extractions and MHC analyses and are included in later sections (see section “Experimental infections”).
Evaluation of Major Histocompatibility Complex IIB Variation Along the Latitudinal Gradient
We amplified MHC Class II exon 2 loci (279 bp) by using the primers by Zeisset and Beebee (2013). Both forward (2F347F: GTGACCCTCTGCTCTCCATT) and reverse (2R307R: ATAATTCAGTATATACAGGGTCTCACC) primers were modified for Illumina MiSeq sequencing with an individual 8 bp barcode (custom adapters) and a sequence of three N to facilitate cluster identification, see Cortázar-Chinarro et al. (2017) for details. PCR was done in a 2720 thermal cycler with the protocol: 97°C for 30 s, followed by 35 cycles of 98°C for 10 s, 57°C for 20 s and 72°C for 15 s, and a final round of 72°C for 8 min until the samples are cooled down and stored at 4°C. PCR was conducted in a total volume of 30 μL: 0.5 μL of DNA and 22.1 μL ddH2O, 6 μL Phuside HF buffer, 0.6 μL dNTP, 0.75 μL of both forward and reverse primer and 0.3 μL Phusion Taq. Samples were amplified by using 13 different specific barcode tags, four forward primers and nine reverse primers in a single step PCR process (Cortázar-Chinarro et al., 2017). After amplification, samples were pre-pooled in five to nine pools in equimolar amounts and purified following the MiniElute Gel Extraction kit (Qiagen, Germany). Quantification of equimolar pre-pools was measured prior to purification by using Qubit (Thermo Fisher Scientific, Germany). After pre-pooling, quantifying and purifying the existing pre-pools, eight pools were prepared with a ThruPlex DNA-Seq Kit (Rubicon genomics). A final equimolar pool quality-controlled prior to sequencing using Bionalayser 2100 (Agilent). Sequencing was done in two independent Illumina Miseq V2 (250x) lanes at NGI Uppsala (Uppsala Genome Center).
In total 240 wild individuals and 160 experimental animals were included for geographical and experimental infection studies, respectively. In addition, we replicated the molecular analyses for a subset of samples, which were amplified twice and sequenced independently to check for accuracy and concordance. Replicates for the geographic analyses were selected randomly across populations, with a replication rate of ∼40% (72 samples out of 160). All experimental individuals were replicated by using a different barcoded PCR to be sure that the failing PCR was not related to the barcode used. Additionally, we used used a different Illumina tag for each replicate sample pool.
Miseq Data Analysis
We merged forward and reverse reads from pair-end sequencing data using FLASH (Magoè and Salzberg, 2011). The NextGen workbench1 was used to transform the files to ‘.fasta’ format for further analysis. We demultiplexed the sample sequences with jMHC (Stuglik et al., 2011). An Excel macro (Lighten et al., 2014) was used to estimate allele variants per individual based on Degree of Change (DoC) calculations. The DoC criterion calculates cumulative amplicon sequencing depth among the variants in the amplicon. Plotting the sequencing depth per variant shows an inflection point where the true alleles contribute to a large proportion of the cumulative depth and all the artificial variants only contribute a minor proportion. We assigned the most frequent variants as valid MHC alleles that occurred in at least 3% of the reads (Babik et al., 2009; Galan et al., 2010). Amplicons with < 300 reads were discarded from analyses for quality reasons. We thoroughly checked for chimeras by eye and if any sequences contained insertions, deletions, or stop codons these were removed. Verified MHC class IIB exon 2 alleles were named in accordance with Klein (Klein, 1975), a species abbreviation followed by gene and a following number, e.g., Bubu*01. Generalized linear mixed models (GLMM) implemented in R (lme4 package; Bates et al., 2007) were used to estimate differences in number of alleles per population.
Experimental Infections
Common toad eggs were collected in 2016 at two locations in Skåne in southernmost Sweden (PM and PH, Supplementary Table 1 and Figure 1) and two locations near Luleå in the north (LU1 and LU2). We collected ca. 10 eggs from 10 clutches per pond (∼100 eggs). Eggs were brought to the laboratory in Uppsala and reared in 20 L containers in reconstituted soft water (RSW; NaHCO3, CaSO4, MgSO4 and KCl added to deionized water; APHA, 1985). RSW was then used throughout the experiment. Each clutch was kept in a separate container under 18:6 h light/dark regime at 19°C. The tadpoles were fed ad libitum spinach and fish flakes and water was changed every third day. At metamorphosis stage 42 (Gosner, 1960), the animals were moved to another tank of the same size with access to aquatic and terrestrial (aquarium sand) habitat and a shelter. Four days after completion of tail absortion, the animals were transported to the sealed experimental facilities at the Swedish Institute for Veterinary Science, Uppsala, where they were they were experimentally infected with either Bd-GPL strain or sham controls consisting of water individually in 1.2 l plastic tanks lined with moist paper towels and a lid of a plastic bottle as a shelter. The metamorphs were kept in these tanks until the end of the experiment and fed fruit flies and crickets ad libitum under 18:6 h light/dark regime at 19°C. The condition of each animal was checked daily and the tanks were cleaned every third day.
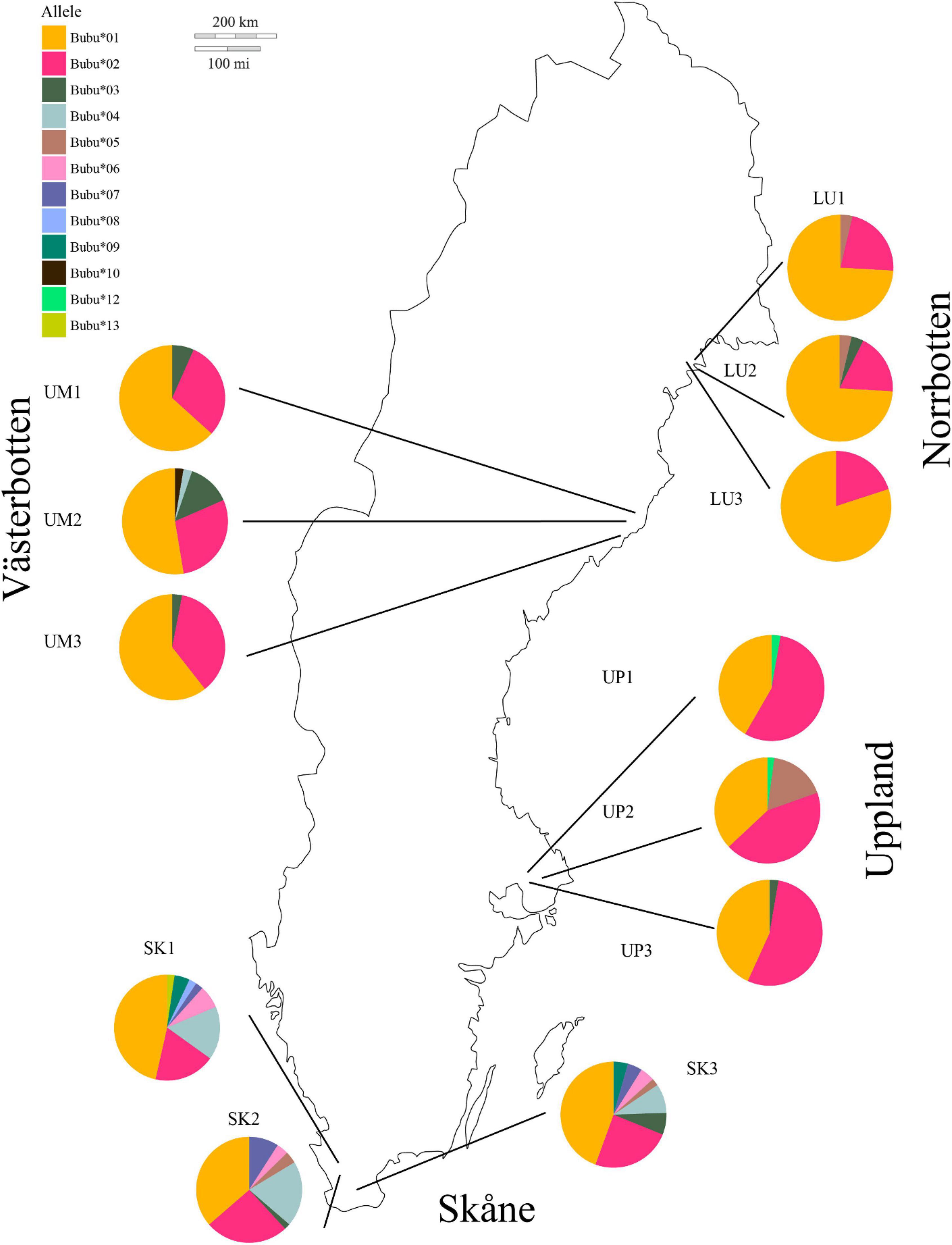
Figure 1. Haplotype frequency distribution of MHC class II sequences in Bufo bufo in 12 sites in four regions in Sweden.
The UK isolate of Bd-GPL (UK Mal 01, GPL-UK) was obtained from the Institute of Zoology, at University College of London (UCL). The Bd-GPL was isolated from an alpine newt (Ichthyosaura alpestris) in the UK in 2008. This Bd-GPL-UK strain is used by the UCL as a model system for Bd infection studies (Miaud et al., 2016). The Swedish isolate (SWED-40-5, GPL-SWED) originated from a green toad (Bufotes viridis) caught in Norra Hamnen, Malmö municipality, Sweden in 2015. For the Bd-GPL-SWED, we selected an isolate from B. viridis as a host because it was the only infected species reported showing a high Bd infection load in Sweden at the time of the experiment. Strains were passaged four times in liquid media (0.8% Tryptone, 0.2% Gelatin hydrolyzate, and 0.4% of Lactose in destilled water) prior to the experiment. Then, we carefully checked the isolates used in the experiment to be sure that they were active the day of infection. The number of zoospores were calculated with hemocytometer (standard practice) and diluted. We administered equal concentrations into tanks across all treatments.
We infected 50 (5 SK1, 14 SK2, 15 LU1, 16 LU2) post-metamorphosed toadlets with the GPL-UK strain and 52 (4, 17, 16, 15) with the GPL-SWED strain. Fifty-two (7, 17, 15, 13) toadlets were treated with a sham control. The number of individuals included in the experiment were not exactly the same between populations because not all individuals survived to the infection stage. The experimental animals were exposed individually for 5 h to 200 μl culture media containing a dosage of 60,000 zoospores from one of the Bd strains in 30 ml of reconstituted soft water (RSW). The animals in the control group were exposed for 5 h to an equivalent volume of sterile media and RSW.
Survival was recorded daily, and dead animals were preserved in 96% ethanol. Thirty days after infection, the experiment was ended and surviving toadlets were euthanized with an overdose of MS222, preserved in 96% ethanol and stored at 4°C until DNA extraction. We chose 30 days for two reasons as according to earlier studies this time is enough to detect the effects on survival in bufonids including common toad (Carey et al., 1999; Bielby et al., 2015). DNA from all animals used in the experiment were extracted from the muscle and were genotyped for MHC Class IIB sequence variation according to the protocol described above.
DNA Extraction and qPCR Analyses of Bd
To confirm infection status, we assessed the presence of Bd by using qPCR. We extracted the DNA from the hind leg using a Prepman Ultra method described in Boyle et al. (2004). The leg was excised with a sterile small blade and smashed to facilitate tissue digestion for DNA extraction later on. Presence of Bd was assessed by amplifying the internal transcribed spacer (ITS)-5.8S rRNA region (Boyle et al., 2004). Twenty-five μl reactions containing 12.5 μl 2X Taqman Master Mix (Applied Biosystem, ref. 4318157), 2.25 μl 10 μM each of forward and reverse primers, 0.625 μl 10 μM MGB probe and 5 μl of DNA (diluted x10 in water) were run. Each sample was run in triplicate. An exogenous internal positive control (IPC; Hyatt et al., 2007) was added to one well in each triplicate (1 μl 10XExo IPC master mix and 0.5 μl 50XExo IPC DNA to each sample, VICTIM dye, Applied Biosystems ref. 4304662) to avoid false negatives due to inhibitors. The qPCR assays were run on a Biorad CFX96 Real Time System machine using amplification conditions described in Boyle et al. (2004) with standards of 0.1, 1, 10, and 100 genomic equivalents “GE” and a positive control of approximately 1-10 “GE.” qPCR standards were provided by UCL (Miaud et al., 2016). For the positive control, we used samples from 2015. To generate the qPCR standard curve, we used the Global Pandemic lineage (Bd-GPL) isolated from an Alpine newt (Ichthyosaura alpestris) at the Institute of Zoology in London (see Meurling et al., in press, for more information). An individual was recorded as positive if at least one of the triplicate samples exhibited a positive signal according to Kriger et al. (2007). If the IPC showed signs of inhibition, negative samples were re-run again. Samples showed signs of inhibition or negative samples were, assigned as not scoreable (NA) and removed from the data set (see Supplementary Table 2). We note that using hind leg swab samples and this range of standards was sufficient to determine whether a sample was Bd + or Bd- but not necessarily for accurate quantification of loads. For this reason, our analyses are focused on survival but not on zoospores load values.
Experimental Infection Models
We used individual survival until the end of the experiment as a binomial response variable with Bd treatment (GPL-UK, GPL-SWED, and Control), geographic region or population (depending on the model) and presence/absence of every MHC allele in every individual as independent variables as well as their interactions in Generalized Linear Models. Due to under-dispersion, we used a quasibinomial error distribution with the logit link function where the dispersion parameter was estimated to 0.615. Geographic location was tested either as the actual population of origin or classified as region (north or south). Models followed the general structure:
Survival ∼ Bd-strain treatment + Region/Population + MHC alleles + Interactions
Model selection was based on Akaike’s Information Criterion (AIC) and all analyses were run by using the R packages “car” (Fox et al., 2012) and “nlme” (Pinheiro et al., 2017) implemented in R version 4.1.1 (R CoreTeam, 2019). We also tested individual body size at infection affected survival, and in a separate model how size in turn was affected by Region/Population and the MHC alleles (see Supplementary Table 5).
Results
Geographic Major Histocompatibility Complex Variation
Major Histocompatibility Complex Class IIB DNA amplification occurred in all but one individual. A total of 8 million reads were recovered by the two Miseq runs with on average 2 million reads per library. Thirteen unique alleles were identified (Bubu*01 – Bubu*13). We found four alleles per individual only in 11 cases, while in all other individuals we found from one to three MHC class II exon 2 alleles. If mismatches between replicates were found, we assumed that it was due to PCR problems during the DNA amplification process and were therefore consecutively excluded in further analyses. However, if one of the two replicates failed due to PCR problems, we kept the sample in consecutive analyses if the quality of the sample was optimal based on the quality score threshold.
Allelic richness (number of alleles) varied between regions and populations (Figure 1 and Supplementary Figure 1) and decreased toward the north based on the DoC analyses quality threshold. We found significant differences in number of alleles among populations along the latitudinal gradient (F3,8.010 = 10.77, p = 0.003; Supplementary Figure 1). The observed number of different alleles in each region was from south to north [e.g., “Skåne” (south): Skåne 9, Uppland 5, “Västerbotten” (north) 5 and “Norrbotten” (north) 4]. We found one allele unique to Västerbotten (Bubu*10 in one individual). In Skåne we found four alleles unique to this region: Bubu*09, Bubu*08, Bubu*07, and Bubu*06. Bubu*01 was found in all individuals in all populations (see Figure 1 and Supplementary Figure 2).
Experimental Infections
Infection was confirmed by qPCR (Supplementary Table 3). Because of failed reactions, infection could not be determined in 14.9% of the individuals. Bd was not detected in one individual that had been exposed with the UK strain. Low infection loads were found in control individuals, ranging from 0.0017 to 0.235 “GE” (an infection mean of 0.083 “GE”) in 19.2% of control animals (Supplementary Table 2). However, the infection intensity was very low in control compared to infected individuals, where we found an average infection mean of 198.283 “GE.” Control samples were amplified below the lowest standard (0.1 “GE”) and therefore considered as negative samples (Belasen et al., 2019). We recovered a total of seven unique alleles: (Bubu*01, Bubu*02, Bubu*04, Bubu*06, Bubu*07, Bubu*09, and Bubu*11) in the experimental animals. All seven were found in the southern region, whereas in the northern region all individuals were monomorphic for Bubu*01 (Supplementary Figure 1).
All animals in the control treatment survived the 30-day experimental period. Of the 102 experimentally infected animals 54 died, 45 from the northern and nine from the southern region. We observed clinical signs of chytridiomicosis like slow mobility and moribund individuals. Of the six models tested, model 3 was chosen having the lowest AIC (Table 1). The model included Region, Bd-strain and MHC alleles as main effects and the interactions between Bd-strain and MHC haplotype as well as Bd strain and Region (Table 1).
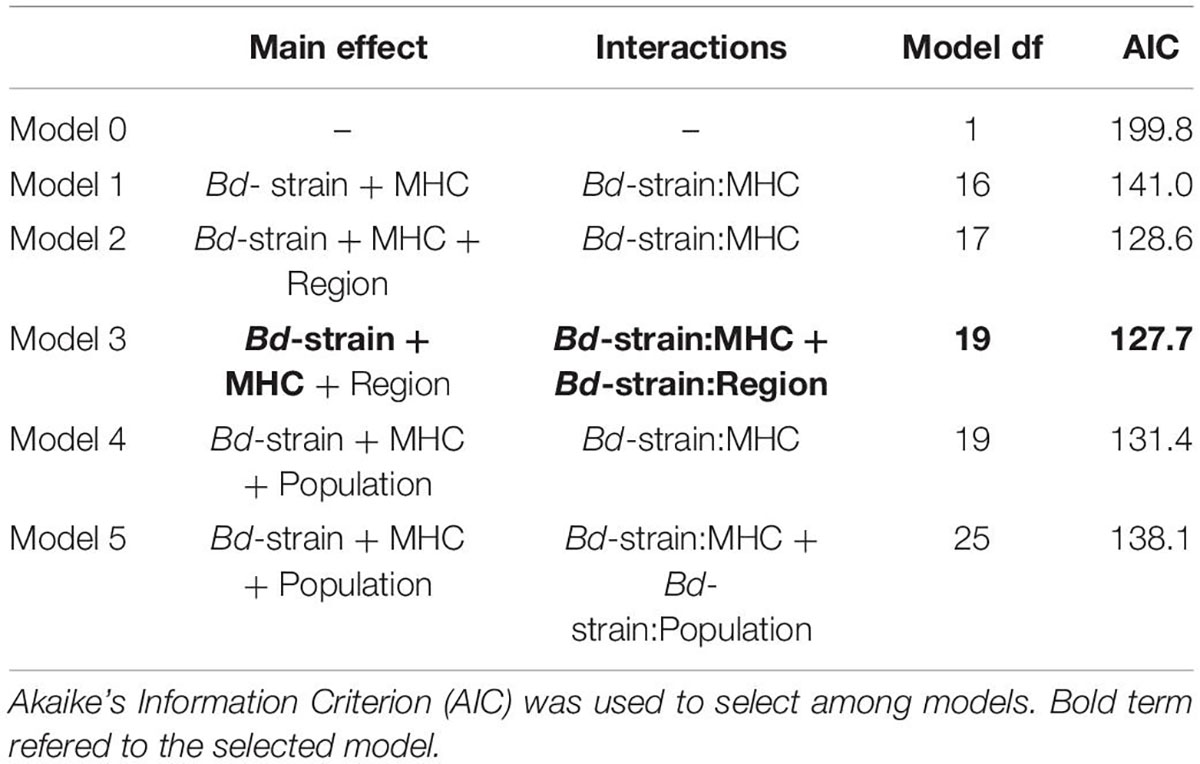
Table 1. Overview of the different models with main effects and interactions modeled.
Statistical modeling suggest that, the region from which the individuals were sampled had a strong effect on survival, with a lower survival on northern individuals (Table 2). This can for example be seen in Figure 2, looking at the survival among individuals carrying only the Bubu*01 allele (and thereby excluding any survival variation caused by the MHC-alleles), where northern individuals shows a lower survival rate in both infection treatments. Also, the interaction between the Bd-strain and the sampling region was significant (Table 2), where the UK Bd-strain caused a high mortality for individuals of the northern region, and no mortality of individuals on the southern region (Figure 2). Furthermore, in toads from the south, the haplotype Bubu*02 appeared to provide better protection against the Swedish Bd strain at the cost of less protection against the UK-strain (Table 2 and Figure 3A). The relatively rare haplotype Bubu*09 had a detrimental effect on survival with a non-significant interaction possibly suggesting that the Swedish strain was less detrimental for individuals carrying this allele (Table 2 and Figure 3B).
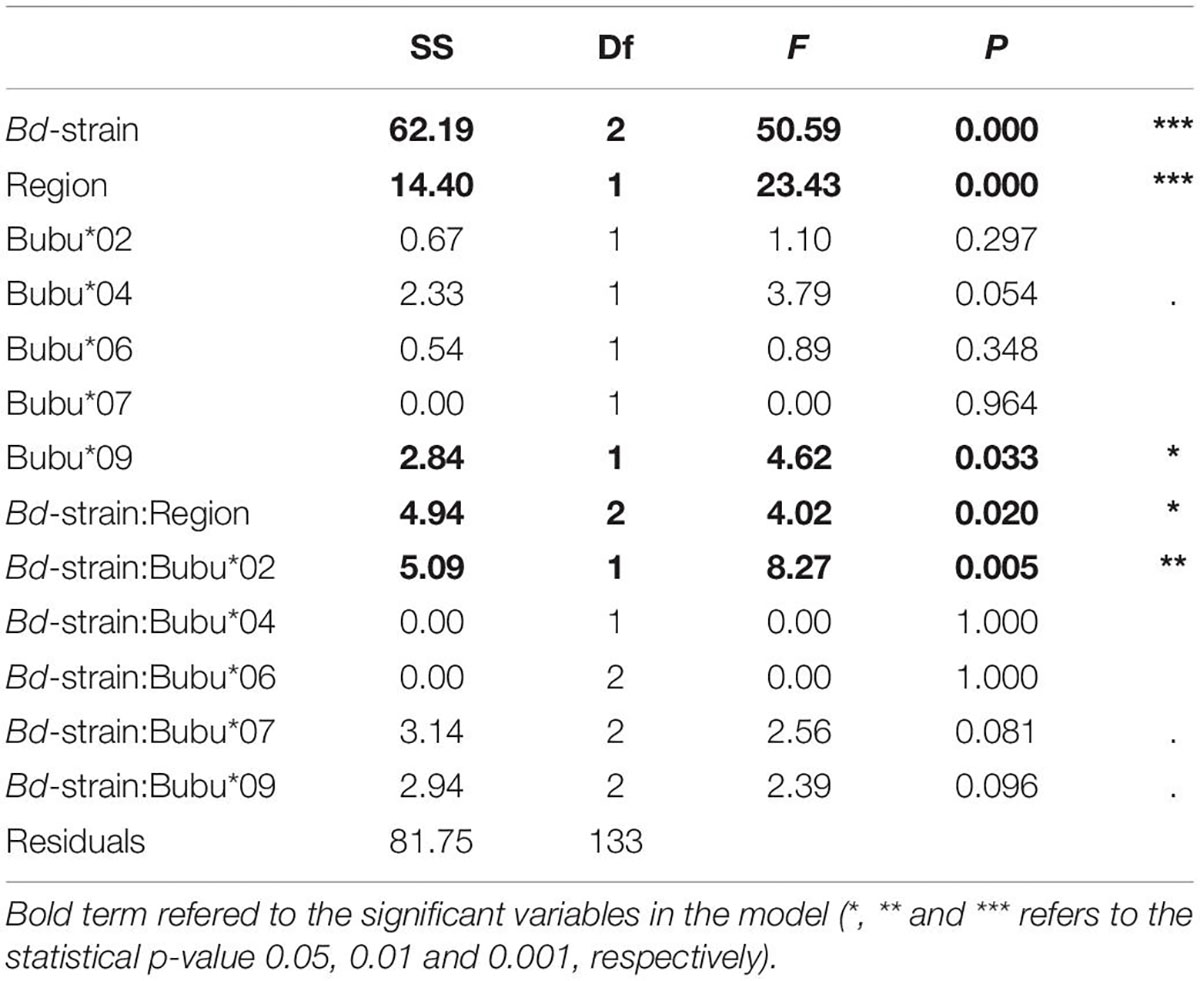
Table 2. Analysis of deviance table of the selected model (Model 3), using quasibinomial distribution and type two sums of squares.
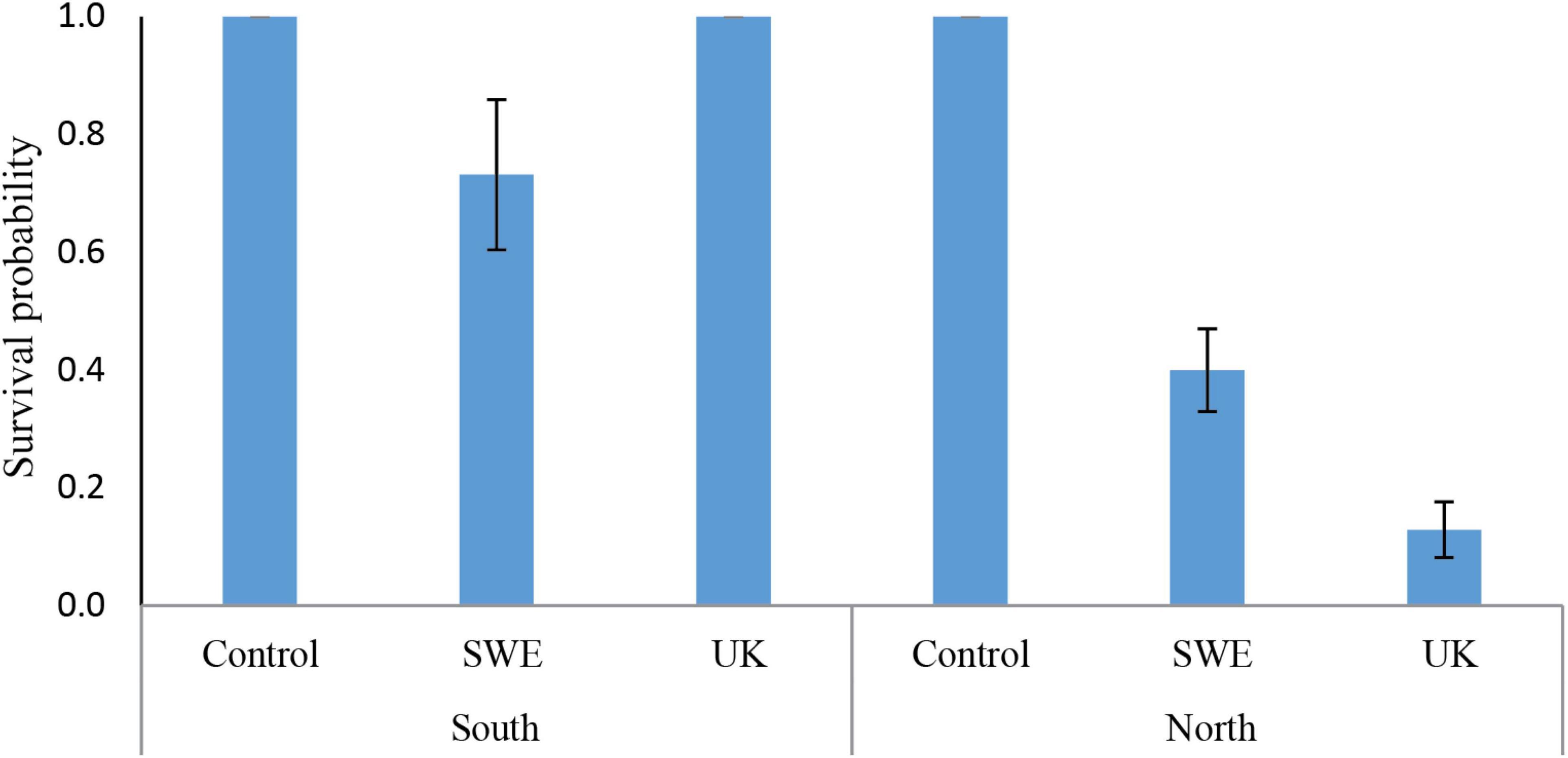
Figure 2. Predicted survival probabilities for the different Bd-infection treatments, and for individuals sample from the southern and northern region. The survival is evaluated for individuals only carrying the Bubu*01 allele, and thereby excluding any survival effects caused by the other MHC-alleles in the model. We found a significant interaction between the regions and the Bd-infection (p-value > 0.05, see Table 2), where the UK-strain cause a strong mortality for individuals of the northern region. Errors bars indicate standard errors. Absence of error bars indicate all individuals had the same value.
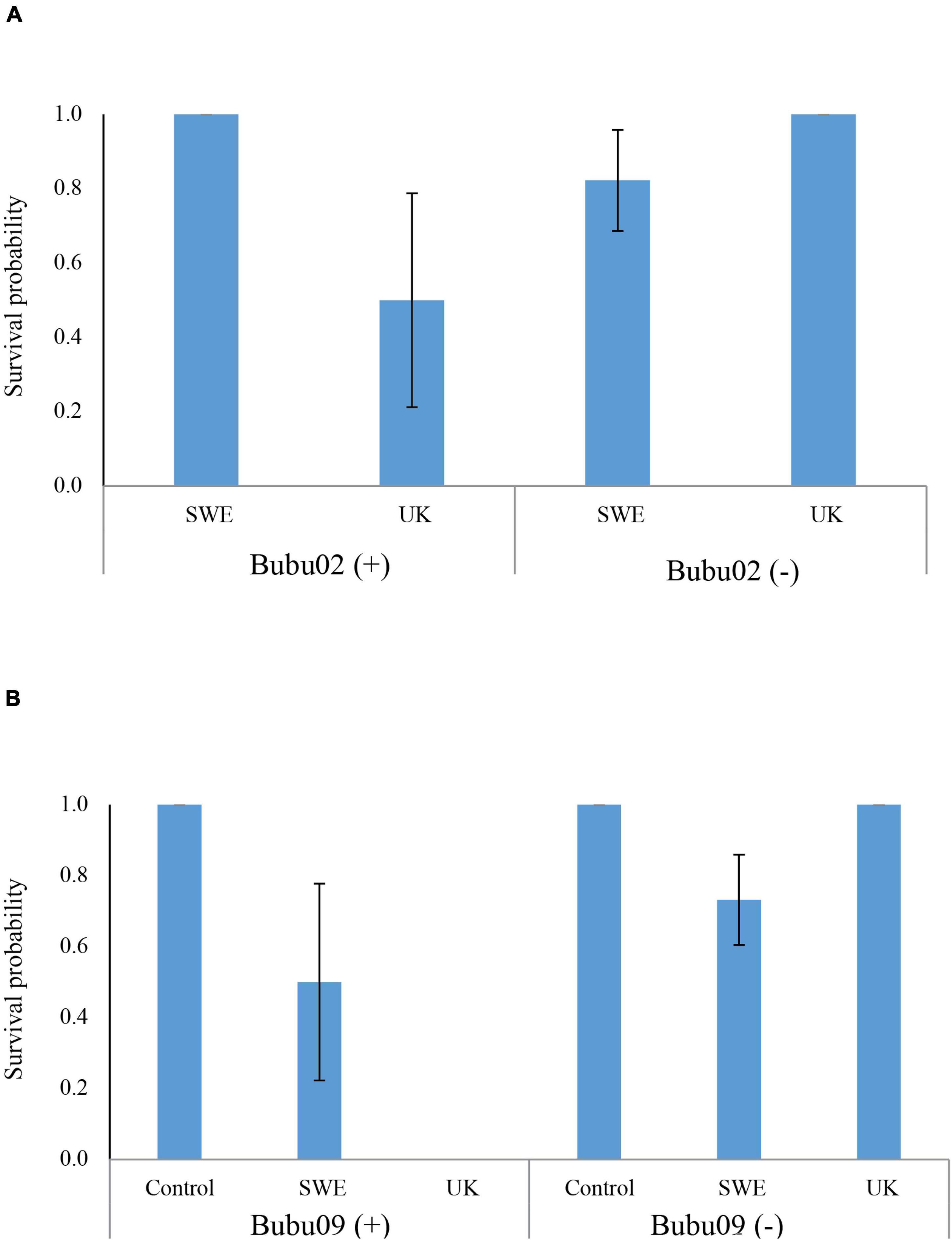
Figure 3. The predicted effects of two alleles with potential Bd-strain interactions in toads from the south. (A) The Bubu*02 haplotype provided protection against the Swedish Bd-strain but was detrimental for individuals infected with the UK Bd-strain. The predicted values are evaluated with a genetic background of carrying the Bubu*06 and Bubu*07 haplotypes as this were the case for all three individuals carrying the Bubu*02 haplotype. None of the three individuals with Bubu*02 were present in the control treatment, which was excluded. (B) Bubu*09 haplotype had a detrimental effect on survival for Bd-infected individuals, especially for the UK-strain. In both (A,B), predicted values are evaluated for individual of the southern regionals this was the case for all individuals carrying these MHC-alleles. We found a significant result for the interaction between the Bd-infection and the MHC-allele. The symbol (+) represent that the allele is present and the symbol (–) represents that the allele is absent in an individual. Errors bars indicate standard errors. Absence of error bars indicate all individuals had the same value.
Body size before infection had a strong positive effect on survival (Supplementary Table 4; see also Meurling et al., in press). We also observe that body size was in turn affected by the region, with larger individuals from the south (Supplementary Table 5). Additionally, most of the MHC alleles had an effect on body size. Whether Bubu*02, Bubu*09, and potentially Bubu*04 had a negative effect, Bubu*06 a positive effect (Supplementary Table 5).
Discussion
The main objective of this study was to study the role of specific MHC Class II haplotypes on Bd infection across a latitudinal gradient through an experimental approach using two different Bd-strains. Results obtained from laboratory experiments and field samplings suggest a differential infection dynamic depending on the Bd-strain and the host immune system, resulting in a differential mortality across the gradient: with a higher mortality in northern toads without MHC diversity, while in southern individuals’ survival was dependent on Bd-strain and MHC haplotype.
The field sampling to quantify MHC Class II variation along the latitudinal gradient across Sweden showed lower MHC diversity at higher latitudes, which is in line with previous studies performed with other species, where MHC diversity tends to decline when populations move away from putative post glacial refugia (Babik et al., 2008; Talarico et al., 2019). Such patterns are generally explained by increased genetic drift in smaller peripheral populations, and historical demographic process, such as population size fluctuations mediated via bottlenecks and founder events (Nei et al., 1975). Generally, amphibian population sizes tend to be smaller and more fragmented at northern latitudes (Johansson et al., 2006; Eckert et al., 2008) in the Northern Hemisphere, and since the force of genetic drift is inversely related to population size (Wright, 1931), northern populations are generally predicted to be more influenced by drift. Conversely, in southern populations selection is expected to be relatively more influential as populations tend to be larger and more connected. This has consequences for populations’ ability to combat disease and pathogens since immunogenetic diversity is predicted to be shaped by selection to a lesser extent at northern latitudes (Cortazar-Chinarro et al., 2018), as the experimental result obtained in this study suggest.
The infection experiment highlights a higher mortality in northern individuals, whether this result depends on the observed lack of MHC variation or some other factor differing between both regions is not possible to discern with this study. Previously we found that smaller individuals were more vulnerable to Bd, and that northern individuals were generally smaller than the southern ones (Meurling et al., in press). The role of body size in shaping host susceptibility has been observed previously in common toads (Bielby et al., 2015) as well as in other species (Garner T.W. et al., 2009; Ortiz-Santaliestra et al., 2013), and might be a contributing factor to the observed regional differences. This is the first study where we found that body size might be affected by the MHC alleles but the effect on survival is not yet conclusive. We strongly suggest that further studies are needed, where the body size is measured before and after infection, in order to investigate the associations between body size, MHC alleles it its potential effect on individual survival.
Interestingly, we also found a difference between the regions in response to the two different Bd-strains. When infected with the UK strain the southern individuals had almost complete survival, whereas the northern ones suffered high mortality. When infected with the Swedish strain, both southern and northern individuals had high mortality albeit the rate was higher in the north. Due to lack of MHC variation in the northern animals it is impossible to discern whether this was due to differences in MHC variation or some other factor, however, we have been able to find some MHC-related differences in survival in southern toads.
The presence of the haplotype Bubu*09 had a negative impact on survival when infected with the UK-strain in animals of the southern region, but the effect was limited when infected with the Swedish strain. The haplotype Bubu*02 had a negative impact on survival when infected with the UK-strain but was marginally beneficial when infected by the Swedish. To our knowledge, differences in host mortality related to different Bd-GPL strains have been only reported before in the western toad (Anaxyrus boreas) at the larval phase (Dang et al., 2017). The results obtained, where some haplotypes seem to be more susceptible to the UK-strain than to the Swedish-strain, suggest that southern populations have been coexisting with the Swedish-strain for a longer time, resulting in a reduced host susceptibility or pathogen virulence. Our result is not in concordance with what was found in the Belasen et al. (2022) meta-analysis review, where hosts presumed to have historically coexisted with endemic Bd did not show reduced mortality when exposed to Bd-GPL treatments compared with host that have not historically coexisted with endemic Bd; but like they hypothesized, endemic Bd lineages (Bd-GPL strain from Sweden) seems to pose a lower threat to amphibian species than Bd-GPL from UK. Also, Belasen et al. (2022) performed a separate analysis comparing local Bd-GPL to non-local GPL strains and did not find a significant difference. In any case, our findings highlight the importance of host- and pathogen-dependent factors in determining overall infection virulence. In addition, it shows the need to study virulence variation within the Bd-GPL lineage, which emerged only within the last century and has caused of the extinction of 90 amphibian species, while Bd-strains other than GPL are rarely associated with chytridiomycosis, population declines and species extinctions (O’Hanlon et al., 2018).
Previous studies have shown MHC Class IIB-haplotype related differences in survival in Lithobates yavapaiensis (Savage and Zamudio, 2011). In Litoria verreauxii, experimental studies showed that certain MHC Class IIB alleles conferred a survival advantage and the differences were ascribed to variation at the peptide-binding coding residues of the amino acid sequences (Bataille et al., 2015). The alleles which conferred a survival advantage in L. verreauxii were then compared to the most common alleles observed in species classified as either susceptible or resistant to Bd, and common toad was grouped among the resistant species (Bataille et al., 2015). However, common toad shows high mortality in experimental infections (Garner T. et al., 2009), and field studies on Swedish populations show that infected individuals have lower body condition than uninfected individuals (Kärvemo et al., 2019). Also, Bd has been shown to infect the sister species B. spinosus in a Spanish population (classified as common toad at the time of the study). While Bd was associated with mortality, no immediate population declines were reported (Bosch and Martínez-Solano, 2006). Yet, B. spinosus has declined in the study area since the time of the study (Bosch et al., 2018) and laboratory studies on the species found high mortality (Garner T.W. et al., 2009). All in all as these infection studies encompass different life-history stages, it is difficult to speculate on interspecific differences (Fu and Waldman, 2022).
In summary, our results show that populations suffered differently from Bd infection depending on latitude, Bd-GPL strain and we found a different immunogenetic responses associated to the specific MHC configuration of an individual. Despite the findings of an association between MHC class II alleles and amphibian susceptibility and vulnerability to infectious diseases in wild populations, further knowledge is needed to discern the effects of specific MHC alleles, Bd strain and individual sub-lethal effects when facing newly emerging diseases. The available evidence strongly suggests that susceptibility to Bd depends on a range of factors which may explain both intra- and interspecific differences. Intraspecific differences can be ascribed to immunocompetence and immunogenetic diversity. The constant co-evolutionary arms-race among hosts and pathogens is likely to lead to a range of susceptibilities and pathogenicity within and among species (Van Valen, 1973; Ejsmond and Radwan, 2015). Therefore, our study support prioritizing future studies on immunogenetic diversity and field infections to gain an understanding of wildlife vulnerability, especially at northern latitudes where amphibian populations might be more susceptible to habitat fragmentation and environmental changes.
Data Availability Statement
The data have been deposited in Figshare at the following links: https://figshare.com/s/26c6c8e25ff6cd7854e9, https://figshare.com/s/c8fdad5a5c1b6930de47. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Statement
The animal study Uppsala University was reviewed and approved by C 48/16.
Author Contributions
MC-C: conceptualization, fieldwork, data curation, formal analyses, methodology, visualization, investigation, supervision, and writing – review and editing. SM: fieldwork, methodology, and writing – review and editing. MS: formal analyses, visualization, and writing – review and editing. LS: methodology (master project) and writing – review and editing. AR-B: fieldwork and writing – review and editing. AL: conceptualization, funding acquisition, supervision, resources, and writing – review and editing. JH: conceptualization, investigation, supervision, funding acquisition, resources, and writing – original draft. All authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the Swedish Research Council Formas (grant 146400178 to JH), Stiftelsen Oscar och Lili Lamms Minne (grant DO2013-0013 to JH), Stiftelsen för Zoologisk Forskning, and Swedish Research Council (grant 621-2013- 4503 to AL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank David Åhlén, Miriam Rubin, and Cátia Chaves for invaluable help in the field.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.915271/full#supplementary-material
Footnotes
References
APHA (1985). Standard Methods for the Examination of Water and Wastewater, 16th edn. Washington, DC: American Public Health Association.
Babik, W., Pabijan, M., and Radwan, J. (2008). Contrasting patterns of variation in MHC loci in the Alpine newt. Mol. Ecol. 17, 2339–2355. doi: 10.1111/j.1365-294X.2008.03757.x
Babik, W., Pabijan, M., Arntzen, J. W., Cogalniceanu, D., Durka, W., and Radwan, J. (2009). Long-term survival of a urodele amphibian despite depleted major histocompatibility complex variation. Mol. Ecol. 18, 769–781. doi: 10.1111/j.1365-294X.2008.04057.x
Barribeau, S. M., Villinger, J., and Waldman, B. (2008). Major histocompatibility complex based resistance to a common bacterial pathogen of amphibians. PLoS One 3:e2692. doi: 10.1371/journal.pone.0002692
Bataille, A., Cashins, S. D., Grogan, L., Skerratt, L. F., Hunter, D., Mcfadden, M., et al. (2015). Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc. R. Soc. B Biol. Sci. 282:20143127. doi: 10.1098/rspb.2014.3127
Bates, D., Sarkar, D., Bates, M. D., and Matrix, L. (2007). The lme4 package. R package version 2:74.
Belasen, A. M., Bletz, M. C., Leite, D. D. S., Toledo, L. F., and James, T. Y. (2019). Long-term habitat fragmentation is associated with reduced MHC IIB diversity and increased infections in amphibian hosts. Front. Ecol. Evol. 6:236. doi: 10.3389/fevo.2018.00236
Belasen, A. M., Russell, I. D., Zamudio, K. R., and Bletz, M. (2022). Endemic lineages of Batrachochytrium dendrobatidis are associated with reduced chytridiomycosisinduced mortality in amphibians: evidence from a meta-analysis of experimentalinfection studies. Front. Vet. Sci. 9:756686. doi: 10.3389/fvets.2022.756686
Bielby, J., Fisher, M. C., Clare, F. C., Rosa, G. M., and Garner, T. W. (2015). Host species vary in infection probability, sub-lethal effects and costs of immune response when exposed to an amphibian parasite. Sci. Rep. 5, 1–8. doi: 10.1038/srep10828
Bosch, J., and Martínez-Solano, I. (2006). Chytrid fungus infection related to unusual mortalities of Salamandra salamandra and Bufo bufo in the Penalara Natural Park. Spain. Oryx 40, 84–89. doi: 10.1017/S0030605306000093
Bosch, J., Fernández-Beaskoetxea, S., Garner, T. W., and Carrascal, L. M. (2018). Long-term monitoring of an amphibian community after a climate change-and infectious disease-driven species extirpation. Glob. Chang. Biol. 24, 2622–2632. doi: 10.1111/gcb.14092
Boyle, D., Boyle, D., Olsen, V., Morgan, J., and Hyatt, A. (2004). Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Org. 60, 141–148. doi: 10.3354/dao060141
Carey, C., Cohen, N., and Rollins-Smith, L. (1999). Amphibian declines: an immunological perspective. Dev. Comp. Immunol. 23, 459–472. doi: 10.1016/S0145-305X(99)00028-2
Cortázar-Chinarro, M., Lattenkamp, E. Z., Meyer-Lucht, Y., Luquet, E., Laurila, A., and Höglund, J. (2017). Drift, selection, or migration? Processes affecting genetic differentiation and variation along a latitudinal gradient in an amphibian. BMC Evol. Biol. 17:189. doi: 10.1186/s12862-017-1022-z
Cortazar-Chinarro, M., Meyer-Lucht, Y., Laurila, A., and Höglund, J. (2018). Signatures of historical selection on MHC reveal different selection patterns in the moor frog (Rana arvalis). Immunogenetics 70, 477–484. doi: 10.1007/s00251-017-1051-1
Dang, T. D., Searle, C. L., and Blaustein, A. R. (2017). Virulence variation among strains of the emerging infectious fungus Batrachochytrium dendrobatidis (Bd) in multiple amphibian host species. Dis. Aquat. Org. 124, 233–239. doi: 10.3354/dao03125
Daszak, P., Cunningham, A. A., and Hyatt, A. D. (2000). Wildlife ecology - Emerging infectious diseases of wildlife - Threats to biodiversity and human health. Science 287, 443–449. doi: 10.1126/science.287.5452.443
Doherty, P. C., and Zinkernagel, R. M. (1975). Enhanced immunological surveillance in mice haterozygous at H-2 gene complex. Nature 256, 50–52. doi: 10.1038/256050a0
Eckert, C. G., Samis, K. E., and Lougheed, S. C. (2008). Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188. doi: 10.1111/j.1365-294X.2007.03659.x
Ejsmond, M. J., and Radwan, J. (2015). Red Queen Processes Drive Positive Selection on Major Histocompatibility Complex (MHC) Genes. PLoS Comput. Biol. 11:e1004627. doi: 10.1371/journal.pcbi.1004627
Fisher, M. C., Garner, T. W., and Walker, S. F. (2009). Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu. Rev. Microbiol. 63, 291–310. doi: 10.1146/annurev.micro.091208.073435
Fox, J., Weisberg, S., Adler, D., Bates, D., Baud-Bovy, G., Ellison, S., et al. (2012). Package ‘car’. Vienna: R Foundation for Statistical Computing.
Fu, M., and Waldman, B. (2022). Novel chytrid pathogen variants and the global amphibian pet trade. Conserv. Biol. [Epub ahead of print]. doi: 10.1111/cobi.13938
Galan, M., Guivier, E., Caraux, G., Charbonnel, N., and Cosson, J.-F. (2010). A 454 multiplex sequencing method for rapid and reliable genotyping of highly polymorphic genes in large-scale studies. BMC Genomics 11:296. doi: 10.1186/1471-2164-11-296
Garner, T. W., Walker, S., Bosch, J., Leech, S., Marcus Rowcliffe, J., Cunningham, A. A., et al. (2009). Life history tradeoffs influence mortality associated with the amphibian pathogen Batrachochytrium dendrobatidis. Oikos 118, 783–791. doi: 10.1111/j.1600-0706.2008.17202.x
Garner, T., Garcia, G., Carroll, B., and Fisher, M. (2009). Using itraconazole to clear Batrachochytrium dendrobatidis infection, and subsequent depigmentation of Alytes muletensis tadpoles. Dis. Aquat. Org. 83, 257–260. doi: 10.3354/dao02008
Gervasi, S. S., Stephens, P. R., Hua, J., Searle, C. L., Xie, G. Y., Urbina, J., et al. (2017). Linking ecology and epidemiology to understand predictors of multi-host responses to an emerging pathogen, the amphibian chytrid fungus. PLoS One 12:e0167882. doi: 10.1371/journal.pone.0167882
Gosner, K. L. (1960). A simplified table for staging anuran embryos and larvae with notes on identification. Herpetologica 16, 183–190.
Grogan, L. F., Robert, J., Berger, L., Skerratt, L. F., Scheele, B. C., Castley, J. G., et al. (2018). Review of the amphibian immune response to chytridiomycosis, and future directions. Front. Immunol. 9:2536. doi: 10.3389/fimmu.2018.02536
Hampe, A., and Petit, R. J. (2005). Conserving biodiversity under climate change: the rear edge matters. Ecol. Lett. 8, 461–467. doi: 10.1111/j.1461-0248.2005.00739.x
Hedrick, P. W. (1994). Evolutionary genetics of the Major Histocompatibility Complex. Am. Nat. 143, 945–964. doi: 10.1086/285643
Hedrick, P. W. (1999). Perspective: highly variable loci and their interpretation in evolution and conservation. Evolution 53, 313–318. doi: 10.1111/j.1558-5646.1999.tb03767.x
Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. doi: 10.1038/35016000
Hewitt, G. M. (2004). The structure of biodiversity - insights from molecular phylogeography. Front. Zool. 1:4. doi: 10.1186/1742-9994-1-4
Hughes, A. L., and Nei, M. (1988). Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335:167. doi: 10.1038/335167a0
Hyatt, A. D., Boyle, D. G., Olsen, V., Boyle, D. B., Berger, L., Obendorf, D., et al. (2007). Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis. Aquat. Organ 73, 175–192. doi: 10.3354/dao073175
Janeway, C. A., Travers, P., Walport, M., and Shlomchik, M. (2001). Immunobiology: the Immune System in Health and Disease. New York: Garland Pub.
Johansson, M., Primmer, C. R., and Merilä, J. (2006). History vs. current demography: explaining the genetic population structure of the common frog (Rana temporaria). Mol. Ecol. 15, 975–983. doi: 10.1111/j.1365-294X.2006.02866.x
Kärvemo, S., Laurila, A., and Höglund, J. (2019). Urban environment and reservoir host species are associated with Batrachochytrium dendrobatidis infection prevalence in the common toad. Dis. Aquat. Org. 134, 33–42. doi: 10.3354/dao03359
Klein, J. (1975). Biology of the Mouse Histocompatibility-2 Complex. Principles of Immunogenetics Applied to a Single System. Berlin: Springer-Verlag.
Kriger, K. M., Ashton, K. J., Hines, H. B., and Hero, J.-M. (2007). On the biological relevance of a single Batrachochytrium dendrobatidis zoospore: a reply to Smith. Dis. Aquat. Org. 73, 257–260. doi: 10.3354/dao073257
Lighten, J., Van Oosterhout, C., and Bentzen, P. (2014). Critical review of NGS analyses for de novo genotyping multigene families. Mol. Ecol. 23, 3957–3972. doi: 10.1111/mec.12843
Lips, K. R. (2016). Overview of chytrid emergence and impacts on amphibians. Philos. Trans. R. Soc. B Biol. Sci. 371:20150465. doi: 10.1098/rstb.2015.0465
Magoè, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Meurling, S., Cortazar-Chinarro, M., Siljestam, M., Åhlén, D., and Ågren, E. (in press). Body size mediates latitudinal population differences in response to Bd infection in two amphibian species. Oecologia.
Miaud, C., Dejean, T., Savard, K., Millery-Vigues, A., Valentini, A., Curt Grand Gaudin, N., et al. (2016). Invasive North American bullfrogs transmit lethal fungus Batrachochytrium dendrobatidis infections to native amphibian host species. Biol. Invasions 18, 2299–2308. doi: 10.1007/s10530-016-1161-y
Nei, M., Maruyama, T., and Chakraborty, R. (1975). Bottleneck effect and genetic-variability in populations. Evolution 29, 1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x
O’Hanlon, S. J., Rieux, A., Farrer, R. A., Rosa, G. M., Waldman, B., Bataille, A., et al. (2018). Recent Asian origin of chytrid fungi causing global amphibian declines. Science 360, 621–627. doi: 10.1126/science.aar1965
Ortiz-Santaliestra, M. E., Rittenhouse, T. A., Cary, T. L., and Karasov, W. H. (2013). Interspecific and postmetamorphic variation in susceptibility of three North American anurans to Batrachochytrium dendrobatidis. J. Herpetol. 47, 286–292. doi: 10.1670/11-134
Piertney, S. B., and Oliver, M. K. (2006). The evolutionary ecology of the major histocompatibility complex. Heredity 96, 7–21. doi: 10.1038/sj.hdy.6800724
Pinheiro, J., Bates, D., Debroy, S., Sarkar, D., Heisterkamp, S., Van Willigen, B., et al. (2017). Package ‘nlme’. Linear and Nonlinear Mixed Effects Models, version, 3-1.
Potts, W. K., and Slev, P. R. (1995). Pathogen-based models favoring MHC genetic diversity. Immunol. Rev. 143, 181–197. doi: 10.1111/j.1600-065x.1995.tb00675.x
R CoreTeam (2019). R: A Language and Environment for Statistical Computing [Computer Software Manual]. Vienna: R Foundation for Statistical Computing.
Refsnider, J. M., Poorten, T. J., Langhammer, P. F., Burrowes, P. A., and Rosenblum, E. B. (2015). Genomic correlates of virulence attenuation in the deadly amphibian chytrid fungus, Batrachochytrium dendrobatidis. G3 Genes Genomes Genet. 5, 2291–2298. doi: 10.1534/g3.115.021808
Russell, I. D., Larson, J. G., Von May, R., Holmes, I. A., James, T. Y., and Davis Rabosky, A. R. (2019). Widespread chytrid infection across frogs in the Peruvian Amazon suggests critical role for low elevation in pathogen spread and persistence. PLoS One 14:e0222718. doi: 10.1371/journal.pone.0222718
Savage, A. E., and Zamudio, K. R. (2011). MHC genotypes associate with resistance to a frog-killing fungus. Proc. Natl. Acad. Sci. U.S.A. 108, 16705–16710. doi: 10.1073/pnas.1106893108
Scheele, B. C., Pasmans, F., Skerratt, L. F., Berger, L., Martel, A., Beukema, W., et al. (2019). Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363, 1459–1463. doi: 10.1126/science.aav0379
Sommer, S. (2005). Major histocompatibility complex and mate choice in a monogamous rodent. Behav. Ecol. Sociobiol. 58, 181–189. doi: 10.1007/s00265-005-0909-7
Stuglik, M. T., Radwan, J., and Babik, W. (2011). jMHC: software assistant for multilocus genotyping of gene families using next-generation amplicon sequencing. Mol. Ecol. Resour. 11, 739–742. doi: 10.1111/j.1755-0998.2011.02997.x
Talarico, L., Babik, W., Marta, S., and Mattoccia, M. (2019). Genetic drift shaped MHC IIB diversity of an endangered anuran species within the Italian glacial refugium. J. Zool. 307, 61–70. doi: 10.1111/jzo.12617
Thörn, F., Rödin-Mörch, P., Cortazar-Chinarro, M., Richter-Boix, A., Laurila, A., and Höglund, J. (2021). The effects of drift and selection on latitudinal genetic variation in Scandinavian common toads (Bufo bufo) following postglacial recolonisation. Heredity 126, 656–667. doi: 10.1038/s41437-020-00400-x
Wright, S. (1931). Evolution in Mendelian populations. Genetics 16, 97–159. doi: 10.1093/genetics/16.2.97
Keywords: MHC, infection, disease, amphibian, chytrid fungus, Bufo bufo
Citation: Cortazar-Chinarro M, Meurling S, Schroyens L, Siljestam M, Richter-Boix A, Laurila A and Höglund J (2022) Major Histocompatibility Complex Variation and Haplotype Associated Survival in Response to Experimental Infection of Two Bd-GPL Strains Along a Latitudinal Gradient. Front. Ecol. Evol. 10:915271. doi: 10.3389/fevo.2022.915271
Received: 07 April 2022; Accepted: 21 June 2022;
Published: 04 July 2022.
Edited by:
Rosane Garcia Collevatti, Universidade Federal de Goiás, BrazilReviewed by:
Bruce Waldman, Oklahoma State University, United StatesAnat M. Belasen, University of Texas at Austin, United States
Copyright © 2022 Cortazar-Chinarro, Meurling, Schroyens, Siljestam, Richter-Boix, Laurila and Höglund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Cortazar-Chinarro, maria.cortazar@ebc.uu.se, maria.cortazar@biol.lu.se
†These authors have contributed equally to this work