CRISPR/Cas-Based Gene Editing Strategies for DOCK8 Immunodeficiency Syndrome
 Sujan Ravendran
Sujan Ravendran Sabina Sánchez Hernández
Sabina Sánchez Hernández - Department of Biomedicine, Aarhus University, Aarhus, Denmark
Defects in the DOCK8 gene causes combined immunodeficiency termed DOCK8 immunodeficiency syndrome (DIDS). DIDS previously belonged to the disease category of autosomal recessive hyper IgE syndrome (AR-HIES) but is now classified as a combined immunodeficiency (CID). This genetic disorder induces early onset of susceptibility to severe recurrent viral and bacterial infections, atopic diseases and malignancy resulting in high morbidity and mortality. This pathological state arises from impairment of actin polymerization and cytoskeletal rearrangement, which induces improper immune cell migration-, survival-, and effector functions. Owing to the severity of the disease, early allogenic hematopoietic stem cell transplantation is recommended even though it is associated with risk of unintended adverse effects, the need for compatible donors, and high expenses. So far, no alternative therapies have been developed, but the monogenic recessive nature of the disease suggests that gene therapy may be applied. The advent of the CRISPR/Cas gene editing system heralds a new era of possibilities in precision gene therapy, and positive results from clinical trials have already suggested that the tool may provide definitive cures for several genetic disorders. Here, we discuss the potential application of different CRISPR/Cas-mediated genetic therapies to correct the DOCK8 gene. Our findings encourage the pursuit of CRISPR/Cas-based gene editing approaches, which may constitute more precise, affordable, and low-risk definitive treatment options for DOCK8 deficiency.
Introduction
Primary immunodeficiencies (PIDs) include more than 400 rare congenital monogenic disorders characterized by impairment of immunity, susceptibility to infectious diseases, autoimmunity, autoinflammatory diseases, allergy and/or malignancy (Tangye et al., 2020). In recent years, there has been an increase in the recognition and diagnosis of previously undefined genetically caused abnormalities in the immune system (Tangye et al., 2020). This has been made possible through the completion of the Human Genome project in the early 2000s, improved definition of clinical phenotypes, and advancement of cost-effective and time-efficient sequencing through implementation of next generation DNA sequencing technologies (Meyts et al., 2016; Bousfiha et al., 2020; Tangye et al., 2020; Gates et al., 2021).
Among these disorders is DOCK8 immunodeficiency syndrome (DIDS) also known as DOCK8 deficiency. Until recently DOCK8 deficiency was termed DOCK8-related Hyper Immunoglobulin E (IgE) Syndrome (HIES), as it is characterized by elevated IgE levels, eosinophilia, and recurrent infections. The majority of HIES is caused by either autosomal dominant inheritance (AD-HIES) of mutations in the signal transducer and activator of transcriptase 3 (STAT3) gene (Grimbacher et al., 1999), or autosomal recessive inheritance (AR-HIES) primarily of mutations in the guanine-nucleotide exchange factor dedicator of cytokinesis 8 (DOCK8) gene (Su et al., 2011). However, an increased insight into the functionality of DOCK8 illuminates its impact on both the T- and B-cell compartment of the immune system, which has promoted the reclassification as a DOCK8-related combined immunodeficiency (CID). DOCK8 deficiency is a severe disorder with early onset of morbidity and high mortality rates exceeding those associated with STAT3 HIES (Aydin et al., 2015; Tsilifis et al., 2021). Since the majority of the clinical manifestations of DOCK8 deficiency pertain to the immune system, hematopoietic stem cell transplantation (HSCT) in early childhood is encouraged. However, this is challenged by the need for a HLA-matched donor and associated with adverse events such as immune rejection and graft-versus-host disease (Copelan, 2006; Aydin et al., 2019).
The ideal treatment of DOCK8 deficiency would be correcting the disease-causing mutation in the patient’s own cells, thereby restoring the DOCK8 functionality. This would circumvent the obstacle of identifying HLA compatible donors for allogeneic transplantation and eliminate the associated risks. During the past two decades, genetic therapies have shown promising results for an expanding numbers of genetic disorders (Booth et al., 2019; Porteus, 2019). Meanwhile, precise genome editing tools were developed and applied in a range of pre-clinical and even a few clinical gene therapy studies. In particular, the discovery of the CRISPR/Cas system as a highly versatile genome editing platform accelerated the development of genome editing methods (Bak et al., 2018a). This ultimately led to the 2020 Nobel Prize in Chemistry awarded to Emmanuelle Charpentier and Jennifer Doudna for ‘the development of a method for genome editing’. CRISPR/Cas offers unprecedented simplicity in facilitating genome editing, and has proven highly precise and efficient (Jensen et al., 2019; Porteus, 2019). Since the first injections of CRISPR gene edited cells into patients in 2016 (Cyranoski, 2016), there have been published reports on only a few clinical trials. These trials have marked important milestones by providing evidence on safety, whereas recent clinical trials on sickle cell disease, β-thalassemia, and transthyretin amyloidosis have been the first to demonstrate therapeutic and potentially curative potential (Frangoul et al., 2021; Gillmore et al., 2021).
In preclinical studies, CRISPR/Cas gene editing has shown tremendous potential in a wide range of diseases, but has so far not been applied to DOCK8. Here, we will elaborate on why gene editing is within the realm of possibility for treating DOCK8 deficiency. First, we briefly present our current understanding of the genetic, molecular, and cellular mechanisms involved in DOCK8 deficiency. Second, we portray the common disease manifestations and discuss current diagnostic and treatment approaches. Third, after describing recent advancements in the field of genome editing and discussing advantages and disadvantages of the different precise gene editing platforms, we define suitable CRISPR/Cas strategies for treating, which may constitute a definitive cure for DOCK8 deficiency. Finally, we give a concise summary of hurdles and challenges for using gene editing in the clinical setting.
The Genetics of DOCK8 Deficiency
The large DOCK8 gene is located on the short arm of chromosome 9, includes 48 exons, spans over 250 kilobases, and encodes a protein of approximately 190 kDa. Bi-allelic loss-of-function mutations in the DOCK8 gene is associated with DOCK8 deficiency (Zhang et al., 2010; Database resources of the, 2018). DOCK8 deficiency is estimated to affect less than one person per million, but the exact prevalence is unknown (Biggs et al., 2017). The disease was not recognized until 2009, and only about 200 cases have been described world-wide so far, which have been identified predominantly in populations with consanguineous marriage (Zhang et al., 2009; Biggs et al., 2017).
To get a collected overview on the different patient mutations, we performed a comprehensive data collection of 60 disease-causing DOCK8 variants described in the literature and registered in the ClinVar database. These variants are represented in Figure 1 and listed in Supplementary Table S1. Even though no specific mutation hotspot regions were identified, the majority of disease-causing mutations in DOCK8 were deletions which cover 61.5% of the variants and range from a few base pairs to deletions spanning several hundred base pairs. The high propensity for deletions has been hypothesized to be partly caused by the occurrence of repetitive genomic sequences leading to abnormal recombinations in this region (Engelhardt et al., 2009). The pathogenic variants in DOCK8 are predominantly loss of function, thus abolishing the expression of DOCK8, but occurrences of DOCK8 duplication has been shown to associate with neurodevelopmental conditions (Jing et al., 2014).
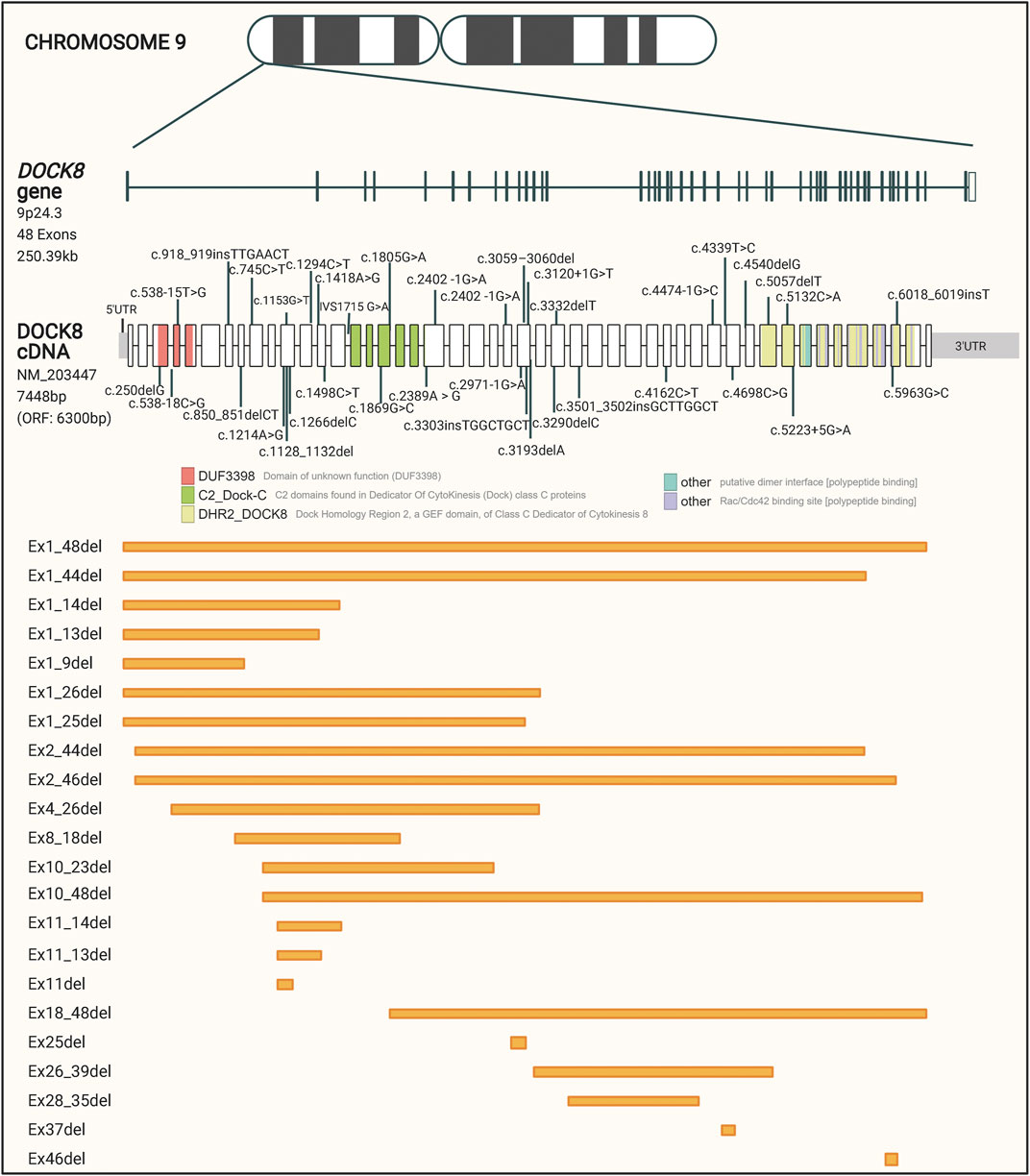
FIGURE 1. Schematic representation of reported patient mutations in the DOCK8 gene. The DOCK8 gene, composed of 48 exons, is located on the short arm of chromosome nine and spans over 250 kb. The distribution of mutations associated with DOCK8 deficiency collected from the ClinVar database is represented along the DOCK8 cDNA. Boxes represent the 48 exons of the gene and different colors indicate major domain-encoding regions. Out of a total of 1,139 DOCK8 variants reported to date, 60 have been found in patients where DOCK8 deficiency has been diagnosed. Orange boxes represent deletions of one or more exons.
As with a few other primary immunodeficiencies, there have been reported cases of somatic reversion leading to partial re-expression of DOCK8 protein in some cell linages. These occurrences of “natural gene-therapy” may reflect the location of DOCK8 within a recombination hotspot, promoting either a somatic repair of a point-mutation, recombination-mediated gene conversion, or recombination-mediated intragenic single crossover (Pillay et al., 2021). Jing et al. observed some clinical improvement in seventeen patients with somatic reversion, with significant improvement in overall survival and age-stratified morbidity. However, these improvements were insufficient for disease elimination presumed to be due to inadequate DOCK8 re-establishment, particularly within the T cells (Pillay et al., 2021). In contrast, Pillay et al. identified three patients with biallelic compound heterozygous DOCK8 germline variants, who displayed significant DOCK8 expression in their lymphocyte subsets ranging from 10% of all B cells to 75% of all CD8+ T cells, while myeloid cells did not express DOCK8. DNA sequencing analyses revealed that one pathogenic allele had been genetically repaired, which was hypothesized to have occurred in either a single common lymphoid progenitor cell or a single hematopoietic stem cell. In all three patients, the somatic reversion improved survival, differentiation, and function of lymphocytes and provided great clinical improvement to the patients (Namekata et al., 2014). Such single progenitor/stem cell reversions signify that only modest DOCK8 correction frequencies by gene therapy in autologous hematopoietic stem cells or lymphoid progenitor cells could provide significant clinical benefit to the patients.
The Role of DOCK8
Until recently, the molecular mechanism of DOCK8 and its influence in cell homeostasis was unknown and unexplored. However, recent discoveries have shed light on these, and we present here these recent discoveries with a focus on the immunoregulatory influence of DOCK8.
Molecular Homeostasis of DOCK8
DOCK8 belongs to the subfamily of DOCK proteins, which are atypical guanine nucleotide exchange factors (GEFs), which to date consists of 11 proteins numerically named from DOCK1 to 11 (Côté and Vuori, 2007). DOCK proteins activate small G proteins (guanine nucleotide-binding proteins), which are GTPases involved in signal transduction. G proteins bind GTP in their on-state but hydrolyse GTP to GDP and then transition into an off-state. Re-activation requires dissociation of GDP and binding of GTP—an exchange that is facilitated by DOCK proteins. DOCK proteins consist of two conserved protein domains known as Dock Homology Region 1 and 2, (DHR1 and -2) (Côté et al., 2005). The DHR1 domain is located upstream of the DHR2 domain and mediates the binding to phosphoinositide (PI), which leads to localized GEF activity near the cell membrane (Sakurai et al., 2021; Côté and Vuori, 2002). The DHR2 domain interacts with the nucleotide-free form of Rho GTPases such as RhoA, Rac, or Cdc42, depending on the DOCK protein. This interaction induces the catalytic activation of the GTPases mediated by GDP-GTP exchange (Harada et al., 2012).
The DHR2 domain of DOCK8 acts as a Cdc42-specific GEF (Kunimura et al., 2020) and therefore regulates Cdc42-specific activities such as cytoskeleton remodelling and actin polymerization, which in turn influence diverse signalling pathways and controls cellular morphology, migration, and protein trafficking (Begum et al., 2004; Li and Gundersen, 2008; Kumari et al., 2014) (Figure 2). This has particularly been reported in T and B cells where Cdc42 have been shown to be implicated in cytoskeletal remodelling necessary for functional T cell activation and cytokine secretion as well as defects in B cell receptor signalling and differentiation into plasma cells (Chemin et al., 2012; Burbage et al., 2015; Su and Orange, 2020) Mutations in Cdc42 are associated with an unusual broad spectrum of diverse abnormal phenotypical characteristics which alters morphological appearance and somatic/non-somatic functions. In some patients, cases of immunodeficiency have been reported although the phenotypic spectrum associated with Cdc42 mutations seems wider than that of DOCK8 deficiency (Al-Herz et al., 2016). This would be explained by the ubiquitous expression of Cdc42 whereas DOCK8 expression is largely confined to cells of the immune system, leading to the immune-specific phenotypical characteristics of DOCK8 deficiency1.
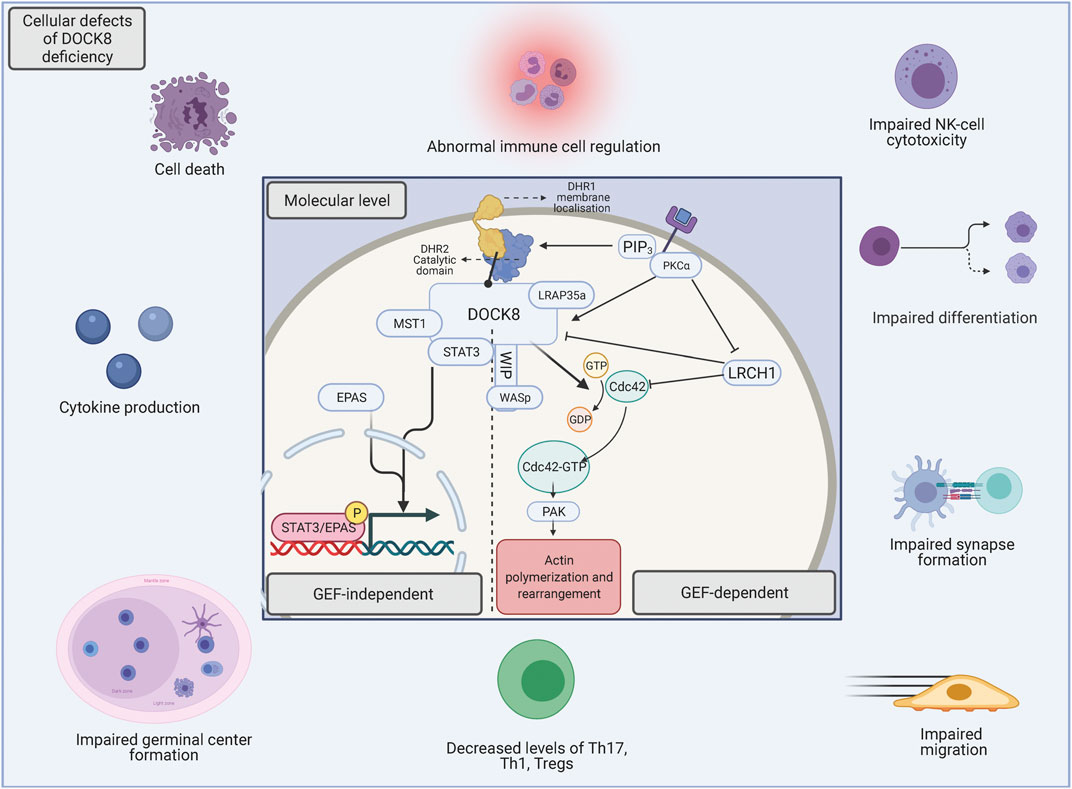
FIGURE 2. The underlying molecular foundation for DOCK8 deficiency. The perturbation of DOCK8 expression disturbs a broad spectrum of immune cell functions such as differentiation, survival, migration, activation, immunotolerance and -function (McGhee and Chatila, 2010). The basis of the various functions of DOCK8 can be divided into either GEF-dependent actin regulation or functions within GEF-independent pathways. When chemokines bind to extracellular receptors, phosphoinositide 3 kinase (PI3K) is activated and initiates the production of phosphatidylinositol (3,4,5)-triphosphate (PIP3), which recruits DOCK8 via the DHR-1 subunit, which consequently leads to membrane-adjacent GEF activity (Xu et al., 2017; Sakurai et al., 2021). In addition, when chemokines bind extracellular receptors, PKCα is activated, which phosphorylate DOCK8 for dissociation from Leuchine Rich Repeats And Calponin Homology Domain Containing (LRCH1) (2), thus diminishing its inhibitory impact (Xu et al., 2017). The catalytic DHR2 domain of DOCK8 interacts with the nucleotide-free form of the Rho GTPase Cdc42, and mediates activation through GDP-GTP exchange (Harada et al., 2012) and leads to down-stream regulation of several biological activities such as cell morphology, -survival, -signaling and -cytoskeletal dynamics, all mediated through p21-activated kinases (PAK) (Bokoch, 2003). In addition to the aforementioned functions, DOCK8 loss in the GEF-independent pathways leads to nuclear translocation of EPAS1 promoting IL-31 production (Yamamura et al., 2017). Furthermore, DOCK8 associates with the transcription factor STAT3 and facilitates activation-induced STAT3 translocation to the nucleus. Here, the guanine nucleotide exchange function of DOCK8 is also necessary for optimal STAT3 phosphorylation and Th17 differentiation (Su et al., 2019).
DOCK8 specifically associates with the transcription factor STAT3, which is mutated in AD-HIES. This interaction facilitates activation-induced STAT3 translocation to the nucleus, and the guanine nucleotide exchange function of DOCK8 is also necessary for optimal STAT3 phosphorylation and Th17 differentiation (Su et al., 2019). This functional relationship between DOCK8 and STAT3 explains the phenotypic overlap between DOCK8 deficiency and AD-HIES.
Immunological Impairment
There is a large diversity in immunophenotypical appearance of DOCK8 deficiency patients, which reflects the prominent role of DOCK8 in several key immunological processes (McGhee and Chatila, 2010) either in a cytoskeleton-dependent or -independent immune response in both innate and adaptive immunity (Figure 2). DOCK8 therefore serves critical roles in several immune cell types to preserve a broad immune response against bacterial, viral, and fungal agents as well as to sustain self-tolerance.
DOCK8 regulates actin cytoskeletal rearrangement (Dustin, 2002), which has been deemed crucial for facilitating adhesion and formation and functionality of the immunological synapses. This interaction between an immune cell and an antigen presenting cell is mediated by surface components such as the lymphocyte function associated-1 (LFA-1) and the counter receptor Intercellular Adhesion Molecule (ICAM-1), which plays an essential role in the complex cascade of molecular events inducing optimal function and homeostasis of immune cells (Janssen et al., 2016). In the absence of DOCK8, a significant impairment of LFA-1/ICAM-1 binding capacity is observed in CD8+ T cells, Regulatory T cells (Tregs), B cells, T follicular helper cells (Tfh), and T helper (Th) cells explaining some of the broad implications of DOCK8 deficiciency (Randall et al., 2011; Randall et al., 2012; Zhang et al., 2016; Janssen et al., 2017; Janssen et al., 2020).
DOCK8 deficiency causes reduced humoral immunity and self-tolerance. The germinal centers, located in secondary lymphoid organs, facilitate the selection and maturation of antigen-activated B-cell clones and provide an optimal immunological response to infections or immunization (Meshaal et al., 2018). However, in absence of DOCK8, the migration of Tfh cells into the germinal center is impaired (Zhang et al., 2016). This may play a significant part in the impaired maturation of B cells into memory cells (Randall et al., 2009; Caracciolo et al., 2014) and reduced persistence of the germinal centers (Biram et al., 2019). Furthermore, the compromised immunological synapse formation (Zhang et al., 2016) and deficient LFA-1 polarization consequently results in reduced production of high affinity IgG antibodies (Jabara et al., 2012; Zhang et al., 2016; Tang et al., 2019), reduced receptor repertoire, and antibody avidity (Janssen et al., 2014).
A heightened immune response, caused by abnormal regulation of T helper and Tregs and increased IgE production, consequently leading to atopic diseases, is common in DOCK8 deficient patients (Aydin et al., 2015). This may partly be due to the significant numerical reduction of Tregs (Caracciolo et al., 2014; Du et al., 2021). Tregs, a subset of CD4+ T cells, provide an essential negative immunomodulatory function in immune homeostasis and maintaining immune tolerance towards self-antigens (Singh et al., 2017). In the absence of DOCK8, the capacity of Tregs to suppress the proliferation of T cells is absent causing autoimmunity (Shi et al., 2018; Du et al., 2021). This may be attributed to an impaired function of the role of DOCK8 in IL-2 signalling (Randall et al., 2011; Shi et al., 2018), impaired Treg migration (Randall et al., 2021), and defective thymocyte differentiation to Treg (Janssen et al., 2021). Susceptibility towards atopic diseases may in addition be caused by the bias towards Th2 (Engelhardt et al., 2015), lack of peripheral B cell tolerance, and increases in autoantibody production (Du et al., 2021).
One of the key features of DOCK8 deficiency patients is their predisposition for cutaneous infections (Zhang et al., 2014; Aydin et al., 2015). This may stem from abnormal trafficking of immune cells to the skin as DOCK8-mutated T-cells and NK-cells show impaired morphological integrity leading to cytothripsis during prolonged migration through confined spaces (Lambe et al., 2011). The increased susceptibility to non-skin centred viruses may reflect the progressive lymphopenia, particular of the T cell population, and atypical functionality of T cells due to impaired persistence and recall of antigen-stimulated CD8 T cells, irregular synapse formation with the antigen presenting cells, altered differentiation and impaired proliferation of T cells and DCs (Zhang et al., 2010; Keles et al., 2014; Janssen et al., 2020). In addition, the decreased circulating plasmacytoid dendritic cells and impaired migration of dendritic inhibits normal trafficking to lymph nodes leading to insufficient dendritic cell accumulation in parenchyma for optimal conditions for T-cell priming (Mizesko et al., 2013; Krishnaswamy et al., 2015; Krishnaswamy et al., 2017; Kunimura et al., 2020).
The Natural Killer (NK) cell population exerts an essential antiviral effect by enforcing cellular death of virus-infected cells and is essential for tumour surveillance. The cytotoxic effector function of NK cells in DOCK8 deficient patients is also defective due to impaired lytic synapse formation, abnormal actin accumulation and granule polymerisation (Crawford et al., 2013). Furthermore, DOCK8 is involved in the development of Natural Killer T (NKT) cells and their cytokine production meaning that DOCK8 deficient patients have immature NKT cells and/or NKT cells that display compromised survival (Tangye et al., 2017).
The quantity and function of Th17 T cells is also diminished in DOCK8 deficiency patients (Milner et al., 2008; Caracciolo et al., 2014; Su et al., 2019). Decreased differentiation of Th17 T cells has been accentuated as one of the primary characteristics of HIES (Sandquist and Kolls, 2018). Reduced Th17 differentiation leads to suboptimal activity of anti-fungal and anti-bacterial immunity mainly due to impaired recruitment of neutrophils (Keles et al., 2016). This is partially due to memory CD4+ T cells favouring the production of Th2 cytokines at the expense of Th1 and Th17 promoting cytokines (Milner et al., 2008). In addition, intrinsic factors inhibiting Th17 differentiation due to impaired STAT3 phosphorylation, translocation, and transcriptional activity is also implicated (Al-Herz et al., 2016).
Clinical Manifestations of DOCK8 Deficiency
The initial diagnosis of DOCK8 deficiency is based on the clinical characteristics in combination with the laboratory immunological findings, with final verification through genetic analysis. DOCK8 deficiency was described clinically for the first time in 2009 and is characterized by early-onset and severe morbidity. Cohort studies have reported around 50% probability to survive beyond 20 years of age with a mean age at death of 9–12 years (Aydin et al., 2015; Zhang et al., 2014). The disease primarily presents with atopic disease, upper and/or lower respiratory infection, frequent viral cutaneous infections, and malignancy (Figure 3) (Zhang et al., 2009; Zhang et al., 2014; Aydin et al., 2015; Haskologlu et al., 2020). Mortality occurs primarily due to infectious agents particularly affecting the skin and respiratory tracts, followed by malignancy, and less commonly CNS vasculitis (Aydin et al., 2015). Almost obligatory findings in these patients are eczema and markedly elevated IgE levels, and there is a high frequency of atopic diseases like food allergies and asthma (Chu et al., 2012; Broides et al., 2017).
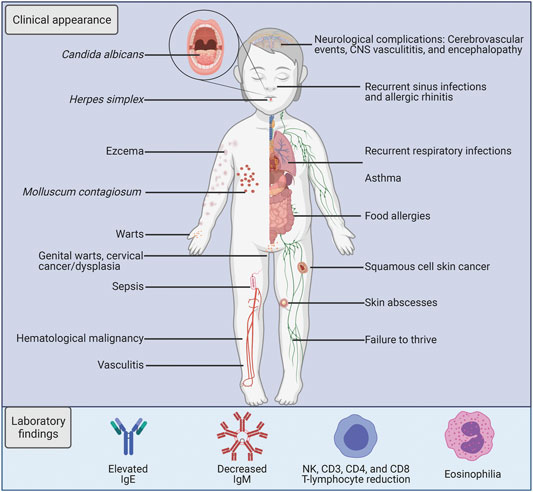
FIGURE 3. Characteristics of DOCK8 deficiency. A schematic illustration listing the key clinical and laboratory findings in DOCK8 deficiency patients.
Flow cytometric analysis is used to analyse intracellular expression levels of DOCK8, but it is also used to identify B cell maturation arrest and altered frequencies of CD4+ and CD8+ T cells (Caracciolo et al., 2014). The circulating peripheral blood of DOCK8-deficient patients is characterised by a decreased CD4+ T cell count and a shift in the CD8+ T cell compartment towards a more exhausted phenotypic subset (Janssen et al., 2020). The B cell compartment displays an increase in naive B cells and a reduction in memory B cells (Tang et al., 2019). However, differential counts of lymphoid cells can show a significant heterogeneity in abnormal findings, thus warranting additional clinical parameters for diagnosis (Alsum et al., 2013).
DOCK8-deficiency leads to a predisposition of cancer, particularly subtypes of haematological or epithelial origin which are often virally-induced either by Epstein Barr Virus (EBV)-driven leiomyosarcomas and lymphomas, and/or Human Papilloma Virus (HPV)-associated squamous cell carcinomas (Papan et al., 2014; Zhang et al., 2014; Aydin et al., 2015; Haskologlu et al., 2020) In the aforementioned cohort studies, 8–17% of patients had developed malignancies during the follow-up period, which included cases of lymphoma (Burkitt and non-Hodgkin lymphoma), squamous cell carcinoma, and sarcoma (Zhang et al., 2014; Aydin et al., 2015).
To summarize, physicians are encouraged to be vigilant about the clinical manifestations of DOCK8-related primary immunodeficiency. It is mainly characterized as HIES and enhances the susceptibility of recurrent viral and bacterial infection, atopic disease, and higher probability of malignancy.
Current Treatment Strategies
Present management of DOCK8 deficiency includes frequent screening for disease progression and treatment of complications through administration of immunoprophylaxis, antiviral and antibacterial treatments prior to a definitive cure through HSCT, if a compatible donor can be identified.
The evidence supporting allogeneic HSCT for treatment of DOCK8 has been described through multiple reports (Chu et al., 2012; Aydin et al., 2019). HSCT is performed after an initial myeloablative or reduced intensity conditioning regimen consisting primarily of chemotherapy and occasionally with additional radiotherapy (Aydin et al., 2019). The purpose of conditioning is to induce adequate immunosuppression and ablation of the recipient’s hematopoietic stem cells. Administration of interferon alpha has shown efficacious as a rescue therapy for viral infections such as Herpex Simplex virus (HSV) and Human Papilloma virus (HPV) (Al-Zahrani et al., 2014; Gernez et al., 2018). In addition, patients awaiting stem cell transplantation benefit from immunoglobulin replacement therapy (IVIG) and prophylactic treatment (Bazinet and Popradi, 2019). Antibodies in DOCK8 patients have been shown to display reduced avidity, which is why IVIG is recommended despite normo- or hyperphysiological antibody levels (Janssen et al., 2014). Furthermore, prophylactic treatment is given post-transplantation to prevent infection and non-infectious complications in the period until immune reconstitution (Devetten and Armitage, 2007).
The detection and aggressive treatment of infectious disease is paramount to avert fatal progression leading to death. However, a large retrospective report consisting of 136 patients with DOCK8 immunodeficiency accentuates the severe disease progression. Unfortunately, even with early intervention with aggressive therapies or prophylaxis such as anti-bacterial, fungal, viral, immunomodulatory, and immunoglobulin replacement treatment, 63% of the patients succumb by their fourth decade of life (Aydin et al., 2015). Therefore, early allogeneic HSCT is clearly indicated, which is also the sole possibility for a curative treatment. Advancements in HSCT have vastly increased the post-HSCT survival for DOCK8 deficiency patients. Hence, patients undergoing HSCT between 1995 and 2010 had a 2-years overall survival of 57 versus 92% for patients transplanted between 2011 and 2015 (Aydin et al., 2019). However, HSCT still entails several risks of severe adverse events, particularly from haploidentical relatives or unrelated donors which pose risks of graft-versus host disease and graft failure (Fox et al., 2018). Reports show that among DOCK8 deficient patients undergoing HSCT, 33% develop acute graft versus host disease (Aydin et al., 2019). Furthermore, HSCT is not always available due to disease progression because of late or misguided diagnosis, and the lack of HLA-compatible donors (Broder et al., 2017; Slatter and Gennery, 2018; Gavrilova, 2019).
Finally, HSCT is associated with high expenses and its use is gradually increasing thus indicating the need for novel therapies that reduce the overall medical cost associated with HSCT and its side effects (Morgan et al., 2017; Mayerhoff et al., 2019). Autologous HSCT with genetically modified stem cells may constitute a promising therapy with fewer adverse outcomes and higher availability to patients with DOCK8 deficiency (Kim et al., 1996; Talib and Shepard, 2020).
The Therapeutic Promises of Genome Editing
Our ability to precisely rewrite and manipulate the instructions encoded in the genome has greatly expanded over the last few decades. By using programmable nucleases, such as Zinc Finger Nucleases (ZFNs) (Miller et al., 2007; Szczepek et al., 2007; Christian et al., 2010), TALE nucleases (TALENs) (Miller et al., 2011; Mussolino et al., 2011; Jinek et al., 2012) and RNA-guided Cas nucleases (Cong et al., 2013; Tebas et al., 2014), researchers can direct the creation of double-strand breaks (DSBs) to specific sites in the DNA and hereby exploit the cellular DNA repair machinery to introduce desired genetic modifications. Induced DSBs are repaired through either Non-Homologous End-joining (NHEJ), an error-prone repair mechanism that induces insertion or deletions (INDELs) at the DNA breakpoint, or Homology-Directed Repair (HDR), a precise repair pathway that uses homologous repair templates to copy from during repair of the DSB, allowing the inclusion of foreign DNA sequences into a specific locus (Figure 4A).
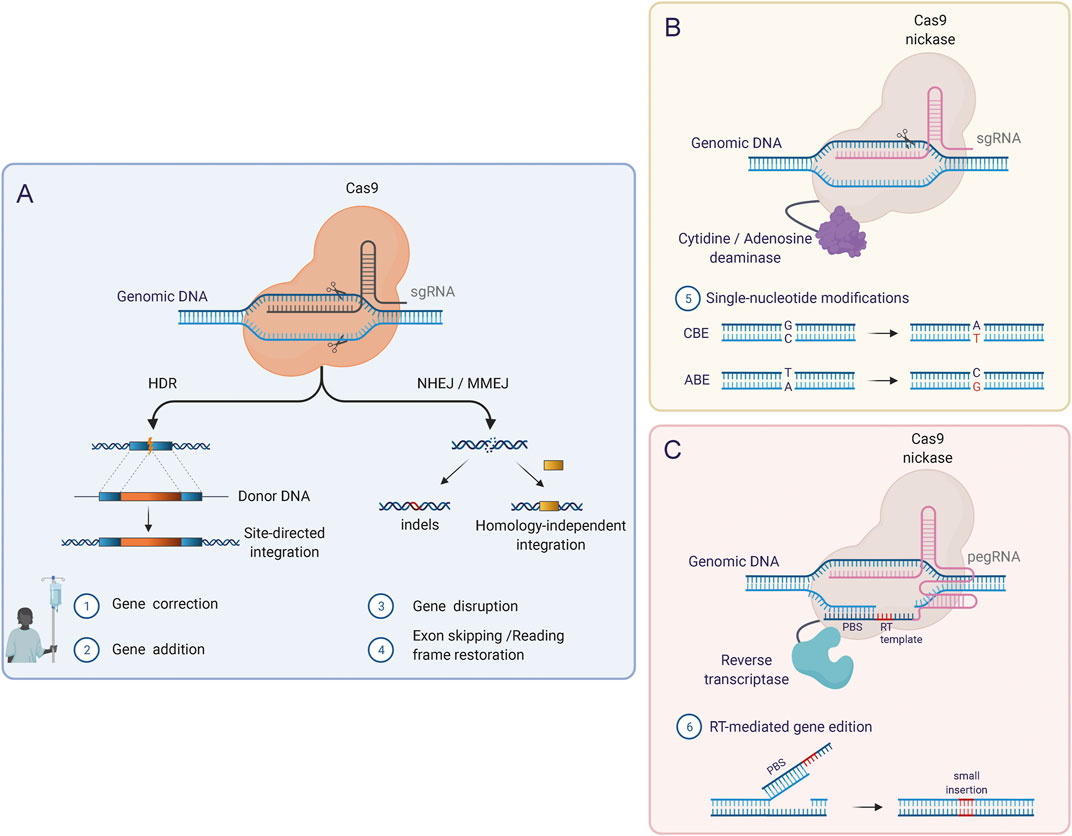
FIGURE 4. Genome editing tools based on CRISPR/Cas. (A) The original CRISPR/Cas RNA-guided nuclease system induces DSBs in the genome in a targeted manner and requires binding of the single guide RNA molecule (sgRNA) to the target DNA as well as the recognition of a specific PAM sequence adjacent to the target sequence. Using this system, the two major DNA repair pathways (HDR and NHEJ) can be exploited to introduce modifications at the target locus. (B) Base Editors (BEs) combine Cas nickases lacking one nuclease domain with DNA deaminases. BEs mediate single-nucleotide conversion, which enables the correction of point mutations. Two types of BEs can be distinguished: Cytosine base editors (CBEs), which mediate C-G to T-A conversions, and adenine base editors (ABEs) which induce A-T to G-C conversions. (C) Prime Editors (PEs) can induce point mutations, small insertions, and small deletions and consist of a Cas nickase fused to a reverse transcriptase (RT) domain. In this system, a reformulated prime editing gRNA (pegRNA) confers the specificity (like the sgRNAs) but additionally contains the template for the desired DNA modifications in the 3′ end. After the induction of a single-strand break (SSB) by the Cas9 nickase, the 3′ end of the pegRNA (primer binding site; PBS) hybridizes with the free 3′ DNA end and acts as a reverse transcription template.
Using programmable nucleases, gene disruption or remodeling of regulatory sequences can be easily achieved by NHEJ. These are strategies that have already been used in clinical trials with promising results (Bushman, 2007; Xu et al., 2019; Stadtmauer et al., 2020; Frangoul et al., 2021; Gillmore et al., 2021). However, for most autosomal recessive PIDs, the addition, replacement, or correction of the affected gene is required thereby necessitating the use of the HDR pathway. This can be achieved by direct correction of the genetic mutation or through the integration of the complete or partial open reading frame cDNA sequence either directly after the endogenous promoter or into a safe harbor site in the genome with a heterologous promoter.
The ability of hematopoietic stem cells (HSCs) to perpetually self-renew and differentiate into all hematopoietic lineages makes them an ideal therapeutic target for gene therapy for treating blood and immune system diseases, including PIDs. Several gene therapy clinical trials have been performed in HSCs since the first one in 1990, and they have mainly been performed using retroviral vectors that integrate the transgene into the chromosomes of HSCs in a semi-random fashion (Booth et al., 2016). Despite clinical trials for several PIDs showing curative potential of these gene therapies, there are also multiple reports of patients developing leukemia due to insertional mutagenesis caused by the retroviral vector (Dever et al., 2016) These events were caused by vector integration close to cancer-related genes such as LMO2 and transactivation of these genes by the strong viral promoter/enhancer elements present in the retroviral vectors. Although lentiviral vectors with self-inactivating mechanisms have proven a safer alternative, they have not entirely eliminated the risks associated with random vector integration, thereby rationalizing the pursuit for safer alternatives such as precise genome editing. Undoubtedly, the advent of the CRISPR/Cas gene editing system supports the onward march towards precision gene therapy.
Initially the CRISPR/Cas system was comprised of the Cas9 endonuclease and two small RNAs: CRISPR RNA (crRNA) and trans-activating RNA (tracrRNA). While crRNA confers specificity to a complementary region in the genome and thereby serves to guide Cas9 to its target, the tracrRNA acts as a Cas9 binding handle to enable the formation of a ribonucleoprotein (RNP) complex. In seminal work from the 2020 Nobel prize winners Jennifer Doudna and Emmanuelle Charpentier they merged the crRNA and tracrRNA into a single guide molecule (sgRNA) thereby reducing the system from three to two components (Cong et al., 2013).
The therapeutic potential of targeted gene editing in long-term repopulating HSCs (LT-HSCs) was first demonstrated in a preclinical study for X-linked Severe Combined Immunodeficiency (SCID-X1) using ZFNs delivered by mRNA electroporation and repair template delivery by an integration-defective lentiviral vector (IDLVs). In this study, Genovese et al. achieved targeted integration of a partial IL2RG cDNA comprising a super-exon of exons 5–8 of IL2RG into exon 5 of the endogenous IL2RG gene. Thus, transcription occurs from the endogenous IL2RG promoter and exon 4 splices with the newly inserted super-exon to generate a correct and full-lenght IL2RG reading frame, thereby providing a platform with the potential to correct all SCID-X1 IL2RG mutations downstream of exon 4. Long-term engraftment of the targeted HSCs in transplanted NSG mice was confirmed and they were also able to correct the defective IL2RG gene in HSCs from a patient with SCID-X1. The CRISPR/Cas system was similarly applied in HSCs for the first time to correct the mutation in the β-globin gene responsible for Sickle Cell Disease (SCD) (Lattanzi et al., 2021). Here, precise correction of the disease-causing mutation was performed with similar evidence of long-term engraftment in mice and reconstitution of functional β-globin (Mohrin et al., 2010). However, both studies also revealed what is now considered the largest challenge for applying precise HDR-mediated gene editing in HSCs, which is the low efficiencies of HDR-mediated editing in the long-term (LT)-HSC compartment compared to the progenitor cell population of the total CD34+ cells. This is mainly believed to be caused by HDR only being active in the late S and G2 phases of the cell cycle whereas NHEJ is the prevailing repair mechanism in quiescent cells (Wilson et al., 2008). This poses a paradoxical challenge in HSC-based gene editing since otherwise quiescent HSCs must be forced into cycling, but cycling is known to be associated with loss of stem cell properties (Song et al., 2016). Hence, NHEJ-focused HSC therapies have shown higher efficiencies in HSCs, confirmed in a recent clinical trial (Frangoul et al., 2021), but is generally more difficult to apply for recessive disorders where expression of a functional gene must be restored.
To enhance HDR frequencies, researchers have been working on different strategies that include the use of repair pathway-modulating small molecules that promote HDR or inhibit NHEJ (Lin et al., 2014; Chu et al., 2015; Robert et al., 2015; Nambiar et al., 2019; De Ravin et al., 2021; Fu et al., 2021), cell cycle synchronization to ensure S/G2 status upon editing (Charpentier et al., 2018; Shin et al., 2020), and the development of novel engineered Cas9 variants for example fusing HDR-promoting or NHEJ-inhibiting proteins to Cas9 (Jayavaradhan et al., 2019; Ferrari et al., 2020). New protocols also transiently inhibit the p53 pathway to achieve high percentages of HDR editing in LT-HSCs (Vavassori et al., 2021). The most advanced example of this is a recent study correcting the CD40 ligand gene (CD40LG), which has deactivating mutations in X‐linked hyper‐IgM syndrome type I (HIGM1) (Schiroli et al., 2019). Here, the authors introduce mRNA encoding a dominant negative p53 variant (GSE53) along the CRISPR/Cas9 gene editing components. Prior studies have shown that during gene editing in HSCs, DSBs and the presence of adeno-associated virus (AAV) vector genomes, used to deliver the HDR repair template, activate p53, which constrains HSC yield, proliferation, and engraftment of gene-edited HSCs (Cromer et al., 2021). With the addition of GSE53, the authors showed up to 30% CD40LG correction frequencies (cDNA integration) in LT-HSCs.
The HDR pathway also provides the possibility of replacing entire gene sequences or large genomic regions. This was recently showcased in LT-HSCs where a DNA repair template was designed in a way that the copy-paste mechanism replaced the one pf the α-globin genes (HBA1) with that of β-globin. This approach could prove therapeutic in patients with β-thalassemia to normalize the balance between α and β chains, thus restoring adult hemoglobin functionality (Maresca et al., 2013). Moreover, this study showed that whole gene replacement is possible in LT-HSCs, thereby providing an additional genome editing strategy for genetic diseases.
As alternatives to HDR, novel gene correction approaches based on HDR-independent targeted gene integration (Sakuma et al., 2016; Yao et al., 2017; Porto et al., 2020), base editing (Anzalone et al., 2019), and more recently prime editing (Newby et al., 2021) try to overcome the inherent limitations of HDR-mediated genome editing. In base and prime editing, the functional properties of Cas9 can be extended by fusing new effector domains to catalytically inactive Cas9 protein or Cas9 nickase. In this way, without requiring DSBs or donor DNA templates, Base Editors (BEs) mediate single-nucleotide conversions in a targeted manner, while Prime Editors (PEs) write new genetic information into a specific nicked locus directed by a small template present on the sgRNA (Figures 4B,C) (Newby et al., 2021). These are promising alternatives for genome editing, and BEs have already shown as high as 68% base editing of the β-globin gene in human LT-HSCs evaluated 16 weeks after transplantation into mice (Vaidyanathan et al., 2018) Similar evidence for PEs must be provided to reinforce their applicability in HSCs.
CRISPR/Cas9 Delivery Strategies in HSCs
Delivery has for a long time been the main hurdle for advancing gene therapy. Since HSCs were the first stem cells to be discovered, purified, and used for therapy (bone marrow transplants), HSCs were also obvious first choice for gene therapy since ex vivo gene therapy is much simpler than in vivo gene therapy.
In general, three different approaches exist to introduce the two components of the CRISPR/Cas-system into cells. In the first approach, DNA such as plasmid DNA is used, encoding the Cas9 and sgRNA. Plasmid delivery is associated with rather slow onset of editing, when compared to the other modalities (Vaidyanathan et al., 2018). The second “all-RNA” approach delivers mRNA encoding Cas9 along in vitro-transcribed or chemically synthesized sgRNAs (Laustsen et al., 2019). Lastly, a recombinant Cas9 protein precomplexed to the sgRNA as an RNP complex can be delivered into cells (Genovese et al., 2014; Lino et al., 2018; Vakulskas et al., 2018). However, in most primary cells, the cost-effective plasmid delivery approach leads to high undesirable cytotoxicity (Cromer et al., 2018; Lino et al., 2018). All-RNA delivery using Cas9 mRNA and sgRNAs is better tolerated by primary cells, even though they still induce a higher innate immune response than RNP delivery (Hendel et al., 2015). Hence, the preferred delivery format for HSC gene editing is using RNP complexes with sgRNAs that are chemically synthetized with modified nucleotides at both ends to protects them from degradation by exonucleases (Laustsen et al., 2019). The delivery mode of choice in HSCs is using electroporation, which relies on short pulses of electrical current to induce small pores in the cell membrane that allows diffusion of macromolecules (Gundry et al., 2017). Combined with Cas9 RNP, this mode has shown exceptionally high on-target efficiency. At the same time, the short half-life of the RNP complex provides a hit-and-run modality that reduces the risk of off-target activity at sites that resemble the intended target in the genome (Genovese et al., 2014; De Ravin et al., 2017; Lino et al., 2018; Vaidyanathan et al., 2018).
In addition to the Cas9 and sgRNA, HDR requires the introduction of a repair template. Here, non-viral repair templates like chemically synthesized single-stranded oligodeoxynucleotide (ssODN) of up to 200 nt have shown effective in HSC gene editing (DeWitt et al., 2016; Romero et al., 2019). However, ssODNs have been reported to be less efficient and induce higher toxicity compared to repair template delivery approaches that rely on viral vectors. At the same time, ssODNs suffer from size constraints associated with chemical DNA synthesis (Roth et al., 2018). However, recent work establishes evidence for surpassing the size constrains of ssODNs enabling delivery of >1 kb long dsDNA co-electroporated with Cas9 RNP complexes in primary human T cells with a tolerable toxicity profile (Wang et al., 2015). The applicability of this platform in HSC is intriguing, but needs further investigation. Lentiviral vectors, which have been employed in numerous clinical trials for ex vivo HSC gene therapy, have also been employed as repair template for HDR. Here, the natural integrating mechanism of lentiviral vectors is removed by introducing an inactivating mutation in the viral Integrase enzyme to generate integration-defective lentiviral vectors (IDLVs) suitable for donor DNA delivery. IDLVs do have higher carrying capacity than AAV vectors, but have been shown to be inferior to AAVs when used for repair template delivery, where specifically AAV serotype 6 has proven effective in HSCs with tolerable cytotoxicy (Grieger and Samulski, 2005). Despite the relatively low packaging capacity of AAV vectors at around 4.5 kb (Bak and Porteus, 2017), this is sufficient for most genome editing purposes, and an approach splitting a large transgene between two AAV donors that undergo sequential HDR at the target locus has been devised to overcome this limit (Bak et al., 2018b). Hence, the combination of RNP complexes with simultaneous delivery of DNA donor template using AAV6 currently represents the most promising technology for versatile gene editing in HSCs (Hacein-Bey-Abina et al., 2002; Bak et al., 2017; Wilkinson et al., 2021).
Towards a Curative CRISPR/Cas9-Based Gene Editing Approach for DOCK8-Related Primary Immunodeficiency
Retroviral gene therapy in autologous HSCs has provided clinical benefit in several PIDs including SCID-X1 (Aiuti et al., 2002; Gaspar et al., 2004; Hacein-Bey-Abina et al., 2010; Gaspar et al., 2011), Adenosine deaminase deficiency (ADA-SCID) (Aiuti et al., 2009; Boztug et al., 2010; Cicalese et al., 2016; Scott et al., 2017), Wiskott Aldrich Syndrome (WAS) (Ott et al., 2006; Aiuti et al., 2013; Hacein-Bey Abina et al., 2015), and X-linked chronic granulomatous disease (X-CGD) (Cavazzana-Calvo et al., 2010; Kang et al., 2010). However, integration of the transgene in these approaches occurs semi-randomly into the patient’s genome, and as mentioned earlier this can lead to oncogenic transformation due to insertional mutagenesis. Even vectors with lower genotoxic potential, such as lentiviral vectors (LVs), can give rise to insertional mutagenesis, which is a risk that needs to be considered in clinical applications (Brown et al., 1998; Modlich et al., 2009). Furthermore, transgene expression levels often differ from the physiological levels of the affected gene due to the use of a constitutive promoter that does not allow tissue-specific or temporal regulation of expression. Unregulated gene expression can for some diseases be detrimental exemplified by the CD40LG gene, which must be expressed at controlled levels as evidenced by preclinical mouse studies where ex vivo gene therapy in a mouse model of X-linked hyper-IgM syndrome with CD40LG-encoding murine gamma-retroviral vectors in HSCs led to lymphoproliferative disorder assumingly as a consequence of unregulated expression of the CD40L transgene (Glessner et al., 2017). DOCK8 deficiency has not been approached with retro- or lentiviral gene delivery, but copy number variation analyses have identified DOCK8 duplications to be significantly associated with a spectrum of neuropsychiatric disorders (Jing et al., 2014). Even though a direct link from DOCK8 CNV to immunological defects has not been established, this might suggest that elevated levels of DOCK8 gene expression can impede normal cellular function. Tight DOCK8 expression in different mature immune cell subsets and regulated DOCK8 expression during hematopoiesis would be impossible to establish with LVs carrying constitutive heterologous promoters and would require full delineation of the regulatory mechanisms that govern DOCK8 expression and reconstruction of a DOCK8 promoter suitable for LV use. Hence, LV-mediated gene delivery may not be a therapeutic option in DOCK8 deficiency, whereas precise gene editing approaches may be optimally suited for such diseases. In the following section, we summarize essential considerations for gene editing and describe different gene editing strategies and their potential use for correcting DOCK8-mediated immunodeficiency.
Potential Gene Correction Strategies for DOCK8 Deficiency
Utilizing the Non-Homologous End-Joining Pathway
An NHEJ-based treatment strategy for DOCK8 deficiency would be highly desirable since the NHEJ pathway is much more active in HSCs compared to the HDR pathway. However, due to the autosomal recessive nature of DOCK8 deficiency, an NHEJ-based strategy that introduces INDELs in the genome is challenging to apply to DOCK8. For disease-causing variants where the reading frame is disrupted, one option for the NHEJ pathway is to use the “reframing” approach which relies on introduced INDELs to restore the correct reading frame (Figure 4A). However, CRISPR/Cas-generated INDELs occur in a semi-stochastic fashion, which means that a population of edited cells will contain a mix of different INDELs, which are specific to the sgRNA used. Hence, this approach depends on the availability of a sgRNA in the vicinity of the mutation, which creates reframing INDELs. Depending on the type and location of these reframing INDELs there may be a loss or addition of amino acids to the reading frame, which may in some rare instances establish a dominant gain of function variant. However, the frequency of reframing INDELs may only need to be low, as evidenced by the aforementioned cases of somatic reversion establishing the possibility that correction of a single lymphoid progenitor or stem cell may be sufficient to provide therapeutic benefit. Depending on the type and location of these reframing INDELs there may be a loss or addition of amino acids to the reading frame, which may perturb protein function. However, for some patient mutations, this approach may be applied. Recent preclinical studies have in fact used reframing to correct mutations in HSCs from patients with Fanconi Anemia where there is a great survival adantage (Román-Rodríguez et al., 2019). The downside of this approach is that it cannot be generalized but would need a different sgRNA for each patient mutation. Also, for longer gene deletions, this approach would not be feasible and 61.5% of patients harbour deletions. This individualized approach is costly to develop and therefore difficult to bring to clinical trials.
Base and Prime Editing
BEs are promising new tools for gene editing, but they can only address a subset of mutations (Figure 4B). In theory, Cytosine BEs enable correction of 26% of all known pathogenic SNP variants, while the Adenine BEs could potentially correct 28%. However, in DOCK8 deficient patients, only a subset of approximately 26% of patients carry pathogenic SNPs in DOCK8. Therefore, even the pursuit to develop individualized base editors for specific patient mutations would only be possible for a subset of the patients. Similar challenges exist for the recently developed PE (Newby et al., 2021) (Figure 4C). The most distinctive attribute of this technology and advantage over BEs is its ability to make any small sequence changes. Like the BEs, this occurs without inducing a DSB - thus mitigating the error-prone NHEJ pathway and the low rates of HDR in post-miotic cells (Scholefield and Harrison, 2021). However, PE is limited to make insertions of less than 80 bp and deletions smaller than 50 bp (Kim et al., 2014). This only enables correction of around 15% of the current DOCK8 patient mutations. Furthermore, there have not been any reports of efficient PE in HSCs.
Homology-Independent Targeted Insertion
Homology-independent targeted insertion (HITI) is a gene editing approach that enables integration of DNA at a specific target site without relying on homologous sequencies in the DNA donor (Figure 5F). Instead, it uses the NHEJ pathway to integrate the linear DNA donor at the break site through an end-joining mechanism, but its efficiency has been reported to be less than 5% in most cases (Suzuki and Izpisua Belmonte, 2018). The applicability of HITI in CD34+ HSPC might be high as these quiescent cells are often in the G0/G1 phases of the cell cycle where NHEJ is the primary DNA repair pathway (Suzuki and Izpisua Belmonte, 2018). Recently, Hanan Bloomer et. al. utilized the HITI system reaching an average of 21% integration in long-term repopulating HSC in mouse xenotransplantation studies (Bloomer et al., 2021). However, the integration mechanism allows transgene cassette insertion in both orientation and the NHEJ mechanism can also lead to end-trimming of the DNA donor template and/or the genomic target site. Hence, this pathway does not lead to exact genome editing outcomes, which might impede its application for DOCK8 deficiency. At present, the technology would need to mature and further studies would need to be conducted to proof its applicability in HSCs.
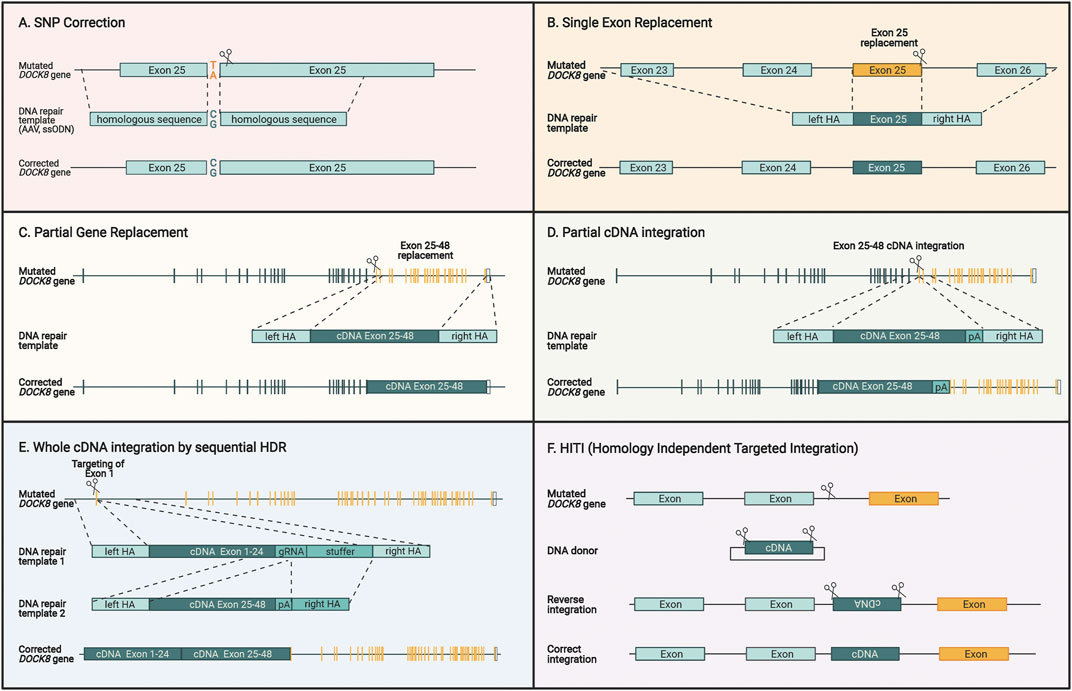
FIGURE 5. Different genome editing strategies for restoring DOCK8 gene expression. (A) Specific correction of SNPs by providing DNA repair templates with homologous sequences surrounding the mutation. (B) Single exons can be replaced by introducing a double strand break close to the end of the exon in addition to supplying a DNA repair template encoding homologous sequences surrounding the exon. (C) Multiple exons can be replaced by cutting close to the region which is intended to be replaced and providing a DNA repair template with homology arms that are homologous to the adjacent regions. (D) A cDNA sequence covering several exons can be introduced upstream to the mutated sequence and the homology arms contain the sequences surrounding the cut site. This way the cDNA will be fused directly to the previous exons, and the downstream exon will be inactivated. (E) To replace the entire DOCK reading frame, a two-step HDR approach can be applied due. For utilizing AAV vectors for repair template delivery, this two-step approach is necessary due to the large size of the gene and the limited capacity of AAV vectors. (F) HITI is different to the previous HDR-based strategies since it uses the NHEJ pathway to insert a DNA sequence without homology arms directly at the cut site. Since this insertion is not exact, intronic regions are targeted to insert a desired sequence that maintains the normal splicing mechanisms and keeps endogenous expression intact. For all figures, the yellow sequences designate exons where patient mutations will be corrected by the specific approach.
Homology-Directed Repair
The most versatile method in terms of possibilities of gene editing outcome is utilizing the HDR pathway, which requires co-delivery of a homologous DNA repair template. In this template, the new DNA sequence is flanked by DNA sequences that are homologous to the target gene sequence around the cut site (Figure 4A). HDR enables various kinds of genetic alterations ranging from single base pair changes to whole cDNA insertion strategies. In contrast to gene delivery by retroviral vectors, HDR preserves endogenous DOCK8 gene regulatory elements with the possibility to re-establish physiological gene expression levels.
Single base pair correction has previously been shown in CD34+ HSCs and is particularly advanced for Sickle Cell Disease (Magis et al., 2019; Lattanzi et al., 2021) This disease is one of the most prevalent genetic disorders and is caused by a single nucleotide substitution that changes a glutamic acid into valine. A direct base pair correction approach is highly desirable since no major perturbations are made to the gene and all regulatory elements of the promoter, untranslated regions, splice elements, and introns are maintained (Figure 5A). This approach could also be applied to DOCK8 mutations but suffers from the same challenge as base and prime editors as mutation-specific CRISPR/Cas reagents must be developed, constituting a major financial burden.
A more universal approach would be to insert part of the DOCK8 reading frame into the endogenous DOCK8 locus thereby spanning larger segments of the gene and potentially covering several patient mutations. This could be used to replace single exons, multiple exons, and potentially all exons (Figures 5B–D). Such strategies have been used preclinically before for multiple hematopoietic diseases including β-thalassemia (Lattanzi et al., 2021), X-SCID (Cromer et al., 2018; Pavel-Dinu et al., 2019), hyper-IgM syndrome (Hubbard et al., 2016; Kuo et al., 2018; Schiroli et al., 2019), and X-CGD (De Ravin et al., 2016; DeWitt et al., 2016). Since AAV is the preferred vector for delivery of the repair template, DOCK8 represents a particular challenge due to its large ORF size of 6.3 kb. The maximum size of AAV packaging is around 4.7kb, which must include the homology arms, which are normally 2 x 400bp leaving around 3.9 kb for the cDNA to be inserted. For DOCK8, this would allow inserting approximately 60% of the DOCK8 ORF. To include as many patient mutations as possible, the optimal region to target would be exons 25–48 encompassing 39% of the known patient mutations (Figure 5D).
We have previously devised an HDR strategy for integrating large gene segments exceeding the capacity of AAV6 vectors. Here, the large transgene is delivered using two separate AAV repair template vectors (Bak et al., 2018b). This makes use of two consecutive HDR steps that first integrate one half of the gene and then the next half, hence overcoming the capacity limit for a single AAV, which potentially would enable a universal HDR approach to target all known DOCK8 mutations present in the reading frame (Figure 5E) (Balakrishnan and Jayandharan, 2014). This approach has recently shown promise as a curative correction strategy for cystic fibrosis, which involves the large 4.4 kb CFTR ORF (Vaidyanathan et al., 2021).
One key aspect is tailoring the donor design and targeting strategies for optimal expression of the target gene. Studies show that for some genes, mere integration of the full reading frame cDNA into the start codon of the endogenous locus leads to suboptimal gene expression levels. Physiological transgene expression can be reached by improving several steps including biallelic integration rates, cDNA codon usage, and inclusion of transcriptional and posttranscriptional regulatory elements. Some studies have used the cDNA of the reading frame only, thereby excluding the 3′UTR from the constructs. This exclusion has advantages during HDR since the 3′UTR sequence in the DNA donor template cannot be diverged from the endogenous 3′UTR like is possible for the reading frame using synonymous codons. This “internal homology” creates two sites of homology between the DNA donor template and the genome: (1) the site of the DSB to which the homology arms have homology and (2) the 3′UTR which is distant from the DSB when designed for the region around the endogenous start codon. This double homology creates the possibility of unpredictable HDR events, which is the reason why the exclusion of the 3′ UTR is preferred. However, 3′ UTRs are known to include several regulatory elements like miRNA binding sites and AU-rich elements that can be stabilizing or destabilising to the mRNA. An example of this is gene editing with CD40LG replacement by HDR where the inclusion of the 3′ UTR is essential to ensure high gene expression levels (Hubbard et al., 2016; Kuo et al., 2018). Studies have also shown that retaining intron one or the terminal intron in the cDNA can contribute positively to efficient expression levels (Gray et al., 2021; Sweeney et al., 2021).
The Societal Challenges of Bringing CRISPR/Cas9 Gene Therapies for DOCK8 Deficiency to Patients
The European Commission defines rare diseases as those with a prevalence below five of every 2,000 people (<0.25%) (European Commission (2021, 2021). Although no official definition of “ultra-rare” disease has been established, the European Union defines orphan medicinal products as those that address individuals affected by severe or life-threatening diseases which affect no more than 5 persons in 10,000 (<0.05%) withing the European union. Therefore, based on the current literature entailing DOCK8 deficiency diagnoses, this disease may be defined as an ultra-rare disease.
Several financial, logistical, and ethical questions arise when considering orphan drugs for rare diseases. There are few to no financial incentives for biopharmaceutical companies to venture into ultra-rare disorders due to high development costs and the prospect of low revenues, which is only circumvented with soaring treatment prices. This was demonstrated, with the recently approved one time curative gene replacement therapy for Spinal Muscular Atrophy, which cost more than two million US dollars (Dean et al., 2021). Furthermore, non-economic aspects such as the need for accelerated approval of orphan drugs and how to encourage cooperation between countries and stakeholders in the pursuit of bringing orphan drugs to the marked is worth considering (Kacetl et al., 2020). There has been rapid growth in orphan drug policy establishment, and it is important that governments establish incentives that promote research and development for these indications (Chan et al., 2020). For ultra-rare disease gene therapy development, it is pivotal that translational research and clinical trials are performed in international collaborations to promote access to patient samples and ultimately to centralize clinical trials and coordinate logistical challenges. While this might be possible in high-income countries, several stakeholder are now recognizing that the single largest challenge will be providing access to novel and expensive treatment modalities for patients from low- and middle-income countries and patients from disadvantaged communities and ethnic groups (Gene, 2021). Because gene therapies involve complex procedures during the GMP-compliant manufacturing of a living cell product (Figure 6), there is a need for advanced infrastructure and highly educated personnel. This demand constitutes a major bottleneck for many institutions with the desire to treat patients with novel gene therapies, and they often fall short in meeting the demands from patients who are in critical need to gain access to potential life-saving therapies. In the future, semi-automated closed cell manufacturing systems might make it easier to implement gene therapies locally (Adair et al., 2016).

FIGURE 6. Principles of ex vivo gene editing. For a patient specific treatment, CD34+ hematopoietic stem cells are isolated from a patient’s blood through apheresis followed by an automated cell processing system that enrich CD34+ cells. Afterwards, the genome editing tools (Cas9 and sgRNA) are delivered to the cells by electroporation and the DNA repair templates are delivered by addition of AAV6 vectors to the cell culture. The genetically modified cells can be cryopreserved before infusion into the patient, which has typically undergone myeloablative conditioning prior to infusion.
As the possible applications of gene therapy exceed beyond the field of research, ethical concerns arise and are discussed to establish a framework for appropriate application of genetic therapies. One important ethical consideration in gene therapy is the distinction between somatic and germline gene editing. The 2018 reports of gene edited Chinese twin babies sparked multiple calls for a global moratorium on clinical uses of human germline editing (Lander et al., 2019). Important discussions arose from this event concerning ethical issues such as patient safety, missing consent from the unborn child, uncertainty of the monitoring period of adverse events, how to justify the defying of natural order, and the potential future implementation of gene therapy as a preventive treatment (Araki and Ishii, 2014). While these discussions on germline gene therapy are ongoing, it is important to remember that somatic gene therapy is bringing about an increasing number of success stories, thereby challenging the need for germ line therapies.
Current Technological Challenges of Gene Editing Reaching the Clinical Setting
Human cells have acquired several mechanisms to detect and correct genomic lesions as each cell experiences several thousand DNA lesions daily (Carusillo and Mussolino, 2020). Most gene editing technologies piggyback on these mechanisms, but also suffer from the adverse events of activating DNA damage responses. This is not only caused by DNA double strand breaks but are also invoked by exposure to AAV vectors. This can lead to cumulative p53 pathway activation which can negatively impact engraftment of edited HSCs (Cromer et al., 2021).
Off-target INDEL induction, translocations, chromothripsis, large on-target INDELs, and off-target integration of DNA donor template are other non-intended consequences that can potentially lead to adverse events and must therefore be evaluated (Urnov, 2021). Off-target activity is based on following three elements: the uniqueness of the target site, the chromatin state of the genome, and nuclease exposure duration and efficiency. Cas9 specificity is dependent on target homology with the 20-nt spacer region of the gRNA, but this sequence can tolerate a mismatch of several bases, consequently enabling possible binding to secondary unintended regions (Mali et al., 2013). In addition, off-target activity is more common to occur at open chromatin regions rather than at closed chromatin region (Singh et al., 2015; Kim and Kim, 2018). Minimizing the off-target activity can theoretically be reached through either increasing the nuclease dissociation from Watson-Crick base paired genomic regions or reducing its cleavage rate (Bisaria et al., 2017). Several Cas9 variants have been engineered with such overall reduction in DNA affinity, thereby maintaining on-target affinity within a window of maximal cleavage while reducing off-target affinity with concomitant reduction in cleavage (Genovese et al., 2014). Several unbiased detection methods have been developed to identify off-target site, like GUIDE-seq and DISCOVER-Seq (Tsai et al., 2015; Wienert et al., 2019) while other methods like CAST-seq evaluates translocations and other gross rearrangements (Turchiano et al., 2021). Overall, with careful sgRNA design and implementation of novel technologies like high-fidelity Cas9 proteins, these adverse genomic events can be reduced and are often benchmarked against existing lentiviral vectors that integrate their cargo semi-randomly in the genome. Off-target activity and insertional mutagenesis remain important concerns in gene therapy and more clinical trials and long-term follow-up is the only means to truly gauge the proportions and clinical relevance of these events.
Conclusion and Future Direction
In the near future, we expect several genetic diseases such as DOCK8 deficiency, which lack efficient and safe treatment regimens, to be treatable with appropriate gene therapy strategies. The great advance compared to conventional pharmaceutical approaches is that gene therapy directly corrects the underlying genomic abnormality of the disease. This provides the possibility to cure these diseases rather than treat symptoms. Given the vast diversity in gene-editing tools, we emphasize the requirement of selecting proper therapeutic strategies that match the underlying genetics of the disease. These strategies have distinct advantages, limitations, and potential adverse effects and for many indications the choices might not be straight-forward. Therefore, when pursuing a curative treatment for DOCK8 deficiency it is imperative that several modalities are explored at the developmental stage to maximize the therapeutic effects while considering disease mechanism, mutation locations, and strategies for delivery and correction. We suggest that efficient and patient-universal correction may be achieved by exploiting the HDR pathway. Additionally, ex vivo introduction of repair templates using AAV donor templates currently seems to be the most proper delivery mechanism and may be designed to replace single or multiple exons with hot-spot mutations or perhaps even include a full cDNA through a two-step HDR strategy. However, further experimental work is warranted to evaluate the efficiency of the different strategies in pursuit of a definitive cure for DOCK8 deficiency. Meanwhile, intensive research in genome editing leads to a continuous emergence of technologies that are bound to enable future breakthroughs in clinical gene therapy. Already, CRISPR/Cas9-based clinical trials have proven successful for sickle cell disease, beta-thalassemia, and transthyretin amyloidosis (Frangoul et al., 2021; Gillmore et al., 2021), and many efforts are focused on optimizing the conditioning regimen, such as the development of anti-CD117 antibodies that deplete HSCs in a targeted and safe manner (Kwon et al., 2019). With these advances and accumulation of experience from clinical trials, the future looks bright for bringing more CRISPR/Cas9-based gene therapies to patients that are safer and more efficient.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
SR took lead in writing the manuscript with input from all co-authors. SK was lead in generating figures with input from all authors. ROB supervised the writing process and edited the final manscript for submission.
Conflict of Interest
ROB holds equity in Graphite Bio and UNIKUM Therapeutics and is a part-time employee of UNIKUM Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
ROB gratefully acknowledges the support from a Lundbeck Foundation Fellowship (R238-2016-534 3349), the Independent Research Fund Denmark (0134-00113B, 0242-00009B, and 9144-00001B), an AIAS-COFUND (Marie Curie) fellowship from Aarhus Institute of Advanced Studies (AIAS) co-funded by Aarhus University’s Research Foundation and the European Union’s seventh Framework Program under grant agreement no 609033, the Novo Nordisk Foundation (NNF19OC0058238 and NNF17OC0028894), Innovation Fund Denmark (8056-00010B), the Carlsberg Foundation (CF20-0424 and CF17-0129), Slagtermester Max Wørzner og Hustru Inger Wørzners Mindelegat, the AP Møller Foundation, the Riisfort Foundation, and a Genome Engineer Innovation Grant from Synthego. All figures were created with BioRender.com.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgeed.2022.793010/full#supplementary-material
Footnotes
References
Adair, J. E., Waters, T., Haworth, K. G., Kubek, S. P., Trobridge, G. D., Hocum, J. D., et al. (2016). Semi-Automated Closed System Manufacturing of Lentivirus Gene-Modified Haematopoietic Stem Cells for Gene Therapy. Nat. Commun. 7, 1–10. doi:10.1038/ncomms13173
Aiuti, A., Biasco, L., Scaramuzza, S., Ferrua, F., Cicalese, M. P., Baricordi, C., et al. (2013). Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 341 (6148), 1233151. doi:10.1126/science.1233151
Aiuti, A., Cattaneo, F., Galimberti, S., Benninghoff, U., Cassani, B., Callegaro, L., et al. (2009). Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. N. Engl. J. Med. 360 (5), 447–458. doi:10.1056/NEJMoa0805817
Aiuti, A., Slavin, S., Aker, M., Ficara, F., Deola, S., Mortellaro, A., et al. (2002). Correction of ADA-SCID by Stem Cell Gene Therapy Combined with Nonmyeloablative Conditioning. Science 296 (5577), 2410–2413. doi:10.1126/science.1070104
Al-Herz, W., Chu, J. I., van der Spek, J., Raghupathy, R., Massaad, M. J., Keles, S., et al. (2016). Hematopoietic Stem Cell Transplantation Outcomes for 11 Patients with Dedicator of Cytokinesis 8 Deficiency. J. Allergy Clin. Immunol. 138 (3), 852–859. doi:10.1016/j.jaci.2016.02.022
Al-Zahrani, D., Raddadi, A., Massaad, M., Keles, S., Jabara, H. H., Chatila, T. A., et al. (2014). Successful Interferon-Alpha 2b Therapy for Unremitting Warts in a Patient with DOCK8 Deficiency. Clin. Immunol. 153 (1), 104–108. doi:10.1016/j.clim.2014.04.005
Alsum, Z., Hawwari, A., Alsmadi, O., Al-Hissi, S., Borrero, E., Abu-staiteh, A., et al. (2013). Clinical, Immunological and Molecular Characterization of DOCK8 and DOCK8-like Deficient Patients: Single center Experience of Twenty Five Patients. J. Clin. Immunol. 33 (1), 55–67. doi:10.1007/s10875-012-9769-x
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 576 (7785), 149–157. doi:10.1038/s41586-019-1711-4
Araki, M., and Ishii, T. (2014). International Regulatory Landscape and Integration of Corrective Genome Editing into In Vitro Fertilization. Reprod. Biol. Endocrinol. 12 (1), 108–112. doi:10.1186/1477-7827-12-108
Aydin, S. E., Freeman, A. F., Al-Herz, W., Al-Mousa, H. A., Arnaout, R. K., Aydin, R. C., et al. (2019). Hematopoietic Stem Cell Transplantation as Treatment for Patients with DOCK8 Deficiency. J. Allergy Clin. Immunol. Pract. 7 (3), 848–855. doi:10.1016/j.jaip.2018.10.035
Aydin, S. E., Kilic, S. S., Kilic, S. S., Aytekin, C., Kumar, A., Porras, O., et al. (2015). DOCK8 Deficiency: Clinical and Immunological Phenotype and Treatment Options - a Review of 136 Patients. J. Clin. Immunol. 35 (2), 189–198. doi:10.1007/s10875-014-0126-0
Bak, R. O., Dever, D. P., and Porteus, M. H. (2018). CRISPR/Cas9 Genome Editing in Human Hematopoietic Stem Cells. Nat. Protoc. 13 (2), 358–376. doi:10.1038/nprot.2017.143
Bak, R. O., Dever, D. P., Reinisch, A., Cruz Hernandez, D., Majeti, R., and Porteus, M. H. (2017). Multiplexed Genetic Engineering of Human Hematopoietic Stem and Progenitor Cells Using CRISPR/Cas9 and AAV6. Elife 6, 1–19. doi:10.7554/eLife.27873
Bak, R. O., Gomez-Ospina, N., and Porteus, M. H. (2018). Gene Editing on Center Stage. Trends Genet. 34 (8), 600–611. doi:10.1016/j.tig.2018.05.004
Bak, R. O., and Porteus, M. H. (2017). CRISPR-mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cel Rep. 20 (3), 750–756. doi:10.1016/j.celrep.2017.06.064
Balakrishnan, B., and Jayandharan, G. (2014). Basic Biology of Adeno-Associated Virus (AAV) Vectors Used in Gene Therapy. Cgt 14 (2), 86–100. doi:10.2174/1566523214666140302193709
Bazinet, A., and Popradi, G. (2019). A General Practitioner's Guide to Hematopoietic Stem-Cell Transplantation. Curr. Oncol. 26 (3), 187–191. doi:10.3747/co.26.5033
Begum, R., Nur-E-Kamal, M. S. A., and Zaman, M. (2004). The Role of Rho GTPases in the Regulation of the Rearrangement of Actin Cytoskeleton and Cell Movement. Exp. Mol. Med. 36 (4), 358–366. doi:10.1038/emm.2004.47
Biggs, C. M., Keles, S., and Chatila, T. A. (2017). DOCK8 Deficiency: Insights into Pathophysiology, Clinical Features and Management. Clin. Immunol. 181 (181), 75–82. doi:10.1016/j.clim.2017.06.003
Biram, A., Davidzohn, N., and Shulman, Z. (2019). T Cell Interactions with B Cells during Germinal center Formation, a Three‐step Model. Immunol. Rev. 288 (1), 37–48. doi:10.1111/imr.12737
Bisaria, N., Jarmoskaite, I., and Herschlag, D. (2017). Lessons from Enzyme Kinetics Reveal Specificity Principles for RNA-Guided Nucleases in RNA Interference and CRISPR-Based Genome Editing. Cel Syst. 4 (1), 21–29. doi:10.1016/j.cels.2016.12.010
Bloomer, H., Smith, R. H., Hakami, W., and Larochelle, A. (2021). Genome Editing in Human Hematopoietic Stem and Progenitor Cells via CRISPR-Cas9-Mediated Homology-independent Targeted Integration. Mol. Ther. 29 (4), 1611–1624. doi:10.1016/j.ymthe.2020.12.010
Bokoch, G. M. (2003). Biology of the P21-Activated Kinases. Annu. Rev. Biochem. 72, 743–781. doi:10.1146/annurev.biochem.72.121801.161742
Booth, C., Gaspar, H. B., and Thrasher, A. J. (2016). Treating Immunodeficiency through HSC Gene Therapy. Trends Mol. Med. 22 (4), 317–327. doi:10.1016/j.molmed.2016.02.002
Booth, C., Romano, R., Roncarolo, M. G., and Thrasher, A. J. (2019). Gene Therapy for Primary Immunodeficiency. Hum. Mol. Genet. 28 (R1), R15–R23. doi:10.1093/hmg/ddz170
Bousfiha, A., Jeddane, L., Picard, C., Al-Herz, W., Ailal, F., Chatila, T., et al. (2020). Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 40 (1), 66–81. doi:10.1007/s10875-020-00758-x
Boztug, K., Schmidt, M., Schwarzer, A., Banerjee, P. P., Díez, I. A., Dewey, R. A., et al. (2010). Stem-Cell Gene Therapy for the Wiskott-Aldrich Syndrome. N. Engl. J. Med. 363 (20), 1918–1927. doi:10.1056/NEJMoa1003548
Broder, M. S., Quock, T. P., Chang, E., Reddy, S. R., Agarwal-Hashmi, R., Arai, S., et al. (2017). The Cost of Hematopoietic Stem-Cell Transplantation in the United States. Am. Health Drug Benefits 10 (7), 366–374.
Broides, A., Mandola, A. B., Levy, J., Yerushalmi, B., Pinsk, V., Eldan, M., et al. (2017). The Clinical and Laboratory Spectrum of Dedicator of Cytokinesis 8 Immunodeficiency Syndrome in Patients with a Unique Mutation. Immunol. Res. 65 (3), 651–657. doi:10.1007/s12026-016-8883-x
Brown, M. P., Topham, D. J., Sangster, M. Y., Zhao, J., Flynn, K. J., Surman, S. L., et al. (1998). Thymic Lymphoproliferative Disease after Successful Correction of CD40 Ligand Deficiency by Gene Transfer in Mice. Nat. Med. 4 (11), 1253–1260. doi:10.1038/3233
Burbage, M., Keppler, S. J., Gasparrini, F., Martínez-Martín, N., Gaya, M., Feest, C., et al. (2015). Cdc42 Is a Key Regulator of B Cell Differentiation and Is Required for Antiviral Humoral Immunity. J. Exp. Med. 212 (1), 53–72. doi:10.1084/jem.20141143
Bushman, F. D. (2007). Retroviral Integration and Human Gene Therapy. J. Clin. Invest. 117 (8), 2083–2086. doi:10.1172/JCI32949
Caracciolo, S., Moratto, D., Giacomelli, M., Negri, S., Lougaris, V., Porta, F., et al. (2014). Expansion of CCR4+ Activated T Cells Is Associated with Memory B Cell Reduction in DOCK8-Deficient Patients. Clin. Immunol. 152 (1-2), 164–170. doi:10.1016/j.clim.2014.03.008
Carusillo, A., and Mussolino, C. (2020). DNA Damage: From Threat to Treatment. Cells 9 (7), 1665. doi:10.3390/cells9071665
Cavazzana-Calvo, M., Payen, E., Negre, O., Wang, G., Hehir, K., Fusil, F., et al. (2010). Transfusion independence and HMGA2 Activation after Gene Therapy of Human β-thalassaemia. Nature 467 (7313), 318–322. doi:10.1038/nature09328
Chan, A. Y. L., Chan, V. K. Y., Olsson, S., Fan, M., Jit, M., Gong, M., et al. (2020). Access and Unmet Needs of Orphan Drugs in 194 Countries and 6 Areas: A Global Policy Review with Content Analysis. Value in Health 23 (12), 1580–1591. doi:10.1016/j.jval.2020.06.020
Charpentier, M., Khedher, A. H. Y., Menoret, S., Brion, A., Lamribet, K., Dardillac, E., et al. (2018). CtIP Fusion to Cas9 Enhances Transgene Integration by Homology-dependent Repair. Nat. Commun. 9 (1), 1133. doi:10.1038/s41467-018-03475-7
Chemin, K., Bohineust, A., Dogniaux, S., Tourret, M., Guégan, S., Miro, F., et al. (2012). Cytokine Secretion by CD4+ T Cells at the Immunological Synapse Requires Cdc42-dependent Local Actin Remodeling but Not Microtubule Organizing center Polarity. J.I. 189 (5), 2159–2168. doi:10.4049/jimmunol.1200156
Christian, M., Cermak, T., Doyle, E. L., Schmidt, C., Zhang, F., Hummel, A., et al. (2010). Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 186 (2), 757–761. doi:10.1534/genetics.110.120717
Chu, E. Y., Freeman, A. F., and Jing, H. (2012). Cutaneous Manifestations of DOCK8 Deficiency Syndrome. Arch. Dermatol. 148 (1), 79–84. doi:10.1001/archdermatol.2011.262
Chu, V. T., Weber, T., Wefers, B., Wurst, W., Sander, S., Rajewsky, K., et al. (2015). Increasing the Efficiency of Homology-Directed Repair for CRISPR-Cas9-Induced Precise Gene Editing in Mammalian Cells. Nat. Biotechnol. 33 (5), 543–548. doi:10.1038/nbt.3198
Cicalese, M. P., Ferrua, F., Castagnaro, L., Pajno, R., Barzaghi, F., Giannelli, S., et al. (2016). Update on the Safety and Efficacy of Retroviral Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. Blood 128 (1), 45–54. doi:10.1182/blood-2016-01-688226
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339 (6121), 819–823. doi:10.1126/science.1231143
Copelan, E. A. (2006). Hematopoietic Stem-Cell Transplantation. N. Engl. J. Med. 354, 1813–1826. doi:10.1056/nejmra052638
Côté, J.-F., Motoyama, A. B., Bush, J. A., and Vuori, K. (2005). A Novel and Evolutionarily Conserved PtdIns(3,4,5)P3-Binding Domain Is Necessary for DOCK180 Signalling. Nat. Cel Biol. 7 (8), 797–807. doi:10.1038/ncb1280
Côté, J.-F., and Vuori, K. (2007). GEF what? Dock180 and Related Proteins Help Rac to Polarize Cells in New Ways. Trends Cel Biol. 17 (8), 383–393. doi:10.1016/j.tcb.2007.05.001
Côté, J.-F., and Vuori, K. (2002). Identification of an Evolutionarily Conserved Superfamily of DOCK180-Related Proteins with Guanine Nucleotide Exchange Activity. J. Cel Sci. 115 (24), 4901–4913. doi:10.1242/jcs.00219
Crawford, G., Enders, A., Gileadi, U., Stankovic, S., Zhang, Q., Lambe, T., et al. (2013). DOCK8 Is Critical for the Survival and Function of NKT Cells. Blood 122 (12), 2052–2061. doi:10.1182/blood-2013-02-482331
Cromer, M. K., Camarena, J., Martin, R. M., Lesch, B. J., Vakulskas, C. A., Bode, N. M., et al. (2021). Gene Replacement of α-globin with β-globin Restores Hemoglobin Balance in β-thalassemia-derived Hematopoietic Stem and Progenitor Cells. Nat. Med. 27, 677–687. doi:10.1038/s41591-021-01284-y
Cromer, M. K., Vaidyanathan, S., Ryan, D. E., Curry, B., Lucas, A. B., Camarena, J., et al. (2018). Global Transcriptional Response to CRISPR/Cas9-AAV6-Based Genome Editing in CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther. 26 (10), 2431–2442. doi:10.1016/j.ymthe.2018.06.002
Cyranoski, D. (2016). CRISPR Gene-Editing Tested in a Person for the First Time. Nature 539, 479. doi:10.1038/nature.2016.20988
Database resources of the National Center for Biotechnology Information (2018). Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46 (D1), D8–D13. doi:10.1093/nar/gkx1095
De Ravin, S. S., Brault, J., Meis, R. J., Liu, S., Li, L., Pavel-Dinu, M., et al. (2021). Enhanced Homology-Directed Repair for Highly Efficient Gene Editing in Hematopoietic Stem/Progenitor Cells. Blood 137, 2598–2608. doi:10.1182/blood.2020008503
De Ravin, S. S., Li, L., Wu, X., Choi, U., Allen, C., Koontz, S., et al. (2017). CRISPR-Cas9 Gene Repair of Hematopoietic Stem Cells from Patients with X-Linked Chronic Granulomatous Disease. Sci. Transl. Med. 9, 9. doi:10.1126/scitranslmed.aah3480
De Ravin, S. S., Reik, A., Liu, P.-Q., Li, L., Wu, X., Su, L., et al. (2016). Targeted Gene Addition in Human CD34+ Hematopoietic Cells for Correction of X-Linked Chronic Granulomatous Disease. Nat. Biotechnol. 34 (4), 424–429. doi:10.1038/nbt.3513
Dean, R., Jensen, I., Cyr, P., Miller, B., Maru, B., Sproule, D. M., et al. (2021). An Updated Cost-Utility Model for Onasemnogene Abeparvovec (Zolgensma) in Spinal Muscular Atrophy Type 1 Patients and Comparison with Evaluation by the Institute for Clinical and Effectiveness Review (ICER). J. Market Access Health Pol. 9 (1), 1889841. doi:10.1080/20016689.2021.1889841
Dever, D. P., Bak, R. O., Reinisch, A., Camarena, J., Washington, G., Nicolas, C. E., et al. (2016). CRISPR/Cas9 β-globin Gene Targeting in Human Haematopoietic Stem Cells. Nature 539 (7629), 384–389. doi:10.1038/nature20134
Devetten, M., and Armitage, J. O. (2007). Hematopoietic Cell Transplantation: Progress and Obstacles. Ann. Oncol. 18 (9), 1450–1456. doi:10.1093/annonc/mdm064
DeWitt, M. A., Magis, W., Bray, N. L., Wang, T., Berman, J. R., Urbinati, F., et al. (2016). Selection-free Genome Editing of the Sickle Mutation in Human Adult Hematopoietic Stem/progenitor Cells. Sci. Transl. Med. 8 (360), 360ra134. doi:10.1126/scitranslmed.aaf9336
Du, Y., Fang, Q., and Zheng, S.-G. (2021). “Regulatory T Cells: Concept, Classification, Phenotype, and Biological Characteristics,” in T Regulatory Cells in Human Health and Diseases. Editor S-G. Zheng (Singapore: Springer), 1–31. doi:10.1007/978-981-15-6407-9_1
Dustin, M. L. (2002). Membrane Domains and the Immunological Synapse: Keeping T Cells Resting and Ready. J. Clin. Invest. 109 (2), 155–160. doi:10.1172/JCI0214842
Engelhardt, K. R., Gertz, M. E., Keles, S., Schäffer, A. A., Sigmund, E. C., Glocker, C., et al. (2015). The Extended Clinical Phenotype of 64 Patients with Dedicator of Cytokinesis 8 Deficiency. J. Allergy Clin. Immunol. 136 (2), 402–412. doi:10.1016/j.jaci.2014.12.1945
Engelhardt, K. R., McGhee, S., Winkler, S., Sassi, A., Woellner, C., Lopez-Herrera, G., et al. (2009). Large Deletions and point Mutations Involving the Dedicator of Cytokinesis 8 (DOCK8) in the Autosomal-Recessive Form of Hyper-IgE Syndrome. J. Allergy Clin. Immunol. 124, 1289–1302. doi:10.1016/j.jaci.2009.10.038
European Commission (2021). Rare Diseases. Availableat: https://ec.europa.eu/info/research-and-innovation/research-area/health-research-and-innovation/rare-diseases_en (Accessed February 11, 2021).
Ferrari, S., Jacob, A., Beretta, S., Unali, G., Albano, L., Vavassori, V., et al. (2020). Efficient Gene Editing of Human Long-Term Hematopoietic Stem Cells Validated by Clonal Tracking. Nat. Biotechnol. 38 (11), 1298–1308. doi:10.1038/s41587-020-0551-y
Fox, T. A., Chakraverty, R., Burns, S., Carpenter, B., Thomson, K., Lowe, D., et al. (2018). Successful Outcome Following Allogeneic Hematopoietic Stem Cell Transplantation in Adults with Primary Immunodeficiency. Blood 131 (8), 917–931. doi:10.1182/blood-2017-09-807487
Frangoul, H., Altshuler, D., Cappellini, M. D., Chen, Y.-S., Domm, J., Eustace, B. K., et al. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 384 (3), 252–260. doi:10.1056/NEJMoa2031054
Fu, Y.-W., Dai, X.-Y., Wang, W.-T., Yang, Z.-X., Zhao, J.-J., Zhang, J.-P., et al. (2021). Dynamics and Competition of CRISPR-Cas9 Ribonucleoproteins and AAV Donor-Mediated NHEJ, MMEJ and HDR Editing. Nucleic Acids Res. 49 (2), 969–985. doi:10.1093/nar/gkaa1251
Gaspar, H. B., Cooray, S., Gilmour, K. C., Parsley, K. L., Adams, S., Howe, S. J., et al. (2011). Long-term Persistence of a Polyclonal T Cell Repertoire after Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 3 (97), 97ra79. doi:10.1126/scitranslmed.3002715
Gaspar, H. B., Parsley, K. L., Howe, S., King, D., Gilmour, K. C., Sinclair, J., et al. (2004). Gene Therapy of X-Linked Severe Combined Immunodeficiency by Use of a Pseudotyped Gammaretroviral Vector. The Lancet 364 (9452), 2181–2187. doi:10.1016/S0140-6736(04)17590-9
Gates, A. J., Gysi, D. M., Kellis, M., and Barabási, A.-L. (2021). A Wealth of Discovery Built on the Human Genome Project - by the Numbers. Nature 590 (7845), 212–215. doi:10.1038/d41586-021-00314-6
Gavrilova, T. (2019). Considerations for Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Disorders. Wjt 9 (3), 48–57. doi:10.5500/wjt.v9.i3.48
Gene, (2021). Gene Therapies Should Be for All. Nat. Med. 27 (8), 1311. doi:10.1038/s41591-021-01481-9
Genovese, P., Schiroli, G., Escobar, G., Di Tomaso, T., Firrito, C., Calabria, A., et al. (2014). Targeted Genome Editing in Human Repopulating Haematopoietic Stem Cells. Nature 510 (7504), 235–240. doi:10.1038/nature13420
Gernez, Y., Baker, M. G., and Maglione, P. J. (2018). Humoral Immunodeficiencies: Conferred Risk of Infections and Benefits of Immunoglobulin Replacement Therapy. Transfusion 58 (December), 3056–3064. doi:10.1111/trf.15020
Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M. L., et al. (2021). CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 385, 493–502. doi:10.1056/nejmoa2107454
Glessner, J. T., Li, J., Li, J., Wang, D., March, M., Lima, L., et al. (2017). Copy Number Variation Meta-Analysis Reveals a Novel Duplication at 9p24 Associated with Multiple Neurodevelopmental Disorders. Genome Med. 9 (1), 1–11. doi:10.1186/s13073-017-0494-1
Gray, D. H., Villegas, I., Long, J., Santos, J., Keir, A., Abele, A., et al. (2021). Optimizing Integration and Expression of Transgenic Bruton's Tyrosine Kinase for CRISPR-Cas9-Mediated Gene Editing of X-Linked Agammaglobulinemia. CRISPR J. 4 (2), 191–206. doi:10.1089/crispr.2020.0080
Grieger, J. C., and Samulski, R. J. (2005). Packaging Capacity of Adeno-Associated Virus Serotypes: Impact of Larger Genomes on Infectivity and Postentry Steps. J. Virol. 79 (15), 9933–9944. doi:10.1128/jvi.79.15.9933-9944.2005
Grimbacher, B., Holland, S. M., Gallin, J. I., Greenberg, F., Hill, S. C., Malech, H. L., et al. (1999). Hyper-IgE Syndrome with Recurrent Infections - an Autosomal Dominant Multisystem Disorder. N. Engl. J. Med. 340 (9), 692–702. doi:10.1056/NEJM199903043400904
Gundry, M. C., Dever, D. P., Yudovich, D., Bauer, D. E., Haas, S., Wilkinson, A. C., et al. (2017). Technical Considerations for the Use of CRISPR/Cas9 in Hematology Research. Exp. Hematol. 54, 4–11. doi:10.1016/j.exphem.2017.07.006
Hacein-Bey Abina, S., Gaspar, H. B., Blondeau, J., Caccavelli, L., Charrier, S., Buckland, K., et al. (2015). Outcomes Following Gene Therapy in Patients with Severe Wiskott-Aldrich Syndrome. JAMA 313 (15), 1550–1563. doi:10.1001/jama.2015.3253
Hacein-Bey-Abina, S., Hauer, J., Lim, A., Picard, C., Wang, G. P., Berry, C. C., et al. (2010). Efficacy of Gene Therapy for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 363 (4), 355–364. doi:10.1056/NEJMoa1000164
Hacein-Bey-Abina, S., Le Deist, F., Carlier, F., Bouneaud, C., Hue, C., De Villartay, J.-P., et al. (2002). Sustained Correction of X-Linked Severe Combined Immunodeficiency by Ex Vivo Gene Therapy. N. Engl. J. Med. 346 (16), 1185–1193. doi:10.1056/NEJMoa012616
Harada, Y., Tanaka, Y., Terasawa, M., Pieczyk, M., Habiro, K., Katakai, T., et al. (2012). DOCK8 Is a Cdc42 Activator Critical for Interstitial Dendritic Cell Migration during Immune Responses. Blood 119 (19), 4451–4461. doi:10.1182/blood-2012-01-407098
Haskologlu, S., Kostel Bal, S., Islamoglu, C., Aytekin, C., Guner, S., Sevinc, S., et al. (2020). Clinical, Immunological Features and Follow up of 20 Patients with Dedicator of Cytokinesis 8 (DOCK8) Deficiency. Pediatr. Allergy Immunol. 31 (5), 515–527. doi:10.1111/pai.13236
Hendel, A., Bak, R. O., Clark, J. T., Kennedy, A. B., Ryan, D. E., Roy, S., et al. (2015). Chemically Modified Guide RNAs Enhance CRISPR-Cas Genome Editing in Human Primary Cells. Nat. Biotechnol. 33 (9), 985–989. doi:10.1038/nbt.3290
Hubbard, N., Hagin, D., Sommer, K., Song, Y., Khan, I., Clough, C., et al. (2016). Targeted Gene Editing Restores Regulated CD40L Function in X-Linked Hyper-IgM Syndrome. Blood 127 (21), 2513–2522. doi:10.1182/blood-2015-11-683235
Jabara, H. H., McDonald, D. R., Janssen, E., Massaad, M. J., Ramesh, N., Borzutzky, A., et al. (2012). DOCK8 Functions as an Adaptor that Links TLR-MyD88 Signaling to B Cell Activation. Nat. Immunol. 13 (6), 612–620. doi:10.1038/ni.2305
Janssen, E., Kumari, S., Tohme, M., Ullas, S., Barrera, V., Tas, J. M. J., et al. (2017). DOCK8 Enforces Immunological Tolerance by Promoting IL-2 Signaling and Immune Synapse Formation in Tregs. JCI Insight 2 (19), 1–18. doi:10.1172/jci.insight.94298
Janssen, E., Morbach, H., Ullas, S., Bannock, J. M., Massad, C., Menard, L., et al. (2014). Dedicator of Cytokinesis 8-deficient Patients Have a Breakdown in Peripheral B-Cell Tolerance and Defective Regulatory T Cells. J. Allergy Clin. Immunol. 134 (6), 1365–1374. doi:10.1016/j.jaci.2014.07.042
Janssen, E., Tohme, M., Butts, J., Giguere, S., Sage, P. T., Velázquez, F. E., et al. (2020). DOCK8 Is Essential for LFA-1-dependent Positioning of T Follicular Helper Cells in Germinal Centers. JCI Insight 5, 5. doi:10.1172/jci.insight.134508
Janssen, E., Tohme, M., Hedayat, M., Leick, M., Kumari, S., Ramesh, N., et al. (2016). A DOCK8-WIP-WASp Complex Links T Cell Receptors to the Actin Cytoskeleton. J. Clin. Invest. 126 (10), 3837–3851. doi:10.1172/JCI85774
Janssen, E., Wilkie, H., and Geha, R. S. (2021). Macabre TH2 Skewing in DOCK8 Deficiency. J. Allergy Clin. Immunol. 148 (1), 73–75. doi:10.1016/j.jaci.2021.02.025
Jayavaradhan, R., Pillis, D. M., Goodman, M., Zhang, F., Zhang, Y., Andreassen, P. R., et al. (2019). CRISPR-Cas9 Fusion to Dominant-Negative 53BP1 Enhances HDR and Inhibits NHEJ Specifically at Cas9 Target Sites. Nat. Commun. 10 (1), 2866. doi:10.1038/s41467-019-10735-7
Jensen, T. I., Axelgaard, E., and Bak, R. O. (2019). Therapeutic Gene Editing in Haematological Disorders withCRISPR/Cas9. Br. J. Haematol. 185 (5), 821–835. doi:10.1111/bjh.15851
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337 (6096), 816–821. doi:10.1126/science.1225829
Jing, H., Zhang, Q., Zhang, Y., Hill, B. J., Dove, C. G., Gelfand, E. W., et al. (2014). Somatic Reversion in Dedicator of Cytokinesis 8 Immunodeficiency Modulates Disease Phenotype. J. Allergy Clin. Immunol. 133 (6), 1667–1675. doi:10.1016/j.jaci.2014.03.025
Kacetl, J., Marešová, P., Maskuriy, R., and Selamat, A. (2020). Ethical Questions Linked to Rare Diseases and Orphan Drugs - A Systematic Review. Rmhp 13, 2125–2148. doi:10.2147/RMHP.S260641
Kang, E. M., Choi, U., Theobald, N., Linton, G., Long Priel, D. A., Kuhns, D., et al. (2010). Retrovirus Gene Therapy for X-Linked Chronic Granulomatous Disease Can Achieve Stable Long-Term Correction of Oxidase Activity in Peripheral Blood Neutrophils. Blood 115 (4), 783–791. doi:10.1182/blood-2009-05-222760
Keles, S., Charbonnier, L. M., Kabaleeswaran, V., Reisli, I., Genel, F., Gulez, N., et al. (2016). Dedicator of Cytokinesis 8 Regulates Signal Transducer and Activator of Transcription 3 Activation and Promotes TH17 Cell Differentiation. J. Allergy Clin. Immunol. 138 (5), 1384–1394. doi:10.1016/j.jaci.2016.04.023.DOCK8
Keles, S., Jabara, H. H., Reisli, I., McDonald, D. R., Barlan, I., Hanna-Wakim, R., et al. (2014). Plasmacytoid Dendritic Cell Depletion in DOCK8 Deficiency: Rescue of Severe Herpetic Infections with IFN-α 2b Therapy. J. Allergy Clin. Immunol. 133 (6), 1753–1755. doi:10.1016/j.jaci.2014.03.032
Kim, D., and Kim, J.-S. (2018). DIG-seq: A Genome-wide CRISPR Off-Target Profiling Method Using Chromatin DNA. Genome Res. 28 (12), 1894–1900. doi:10.1101/gr.236620.118
Kim, S., Kim, D., Cho, S. W., Kim, J., and Kim, J.-S. (2014). Highly Efficient RNA-Guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 24 (6), 1012–1019. doi:10.1101/gr.171322.113
Kim, Y. G., Cha, J., and Chandrasegaran, S. (1996). Hybrid Restriction Enzymes: Zinc finger Fusions to Fok I Cleavage Domain. Proc. Natl. Acad. Sci. 93 (3), 1156–1160. doi:10.1073/pnas.93.3.1156
Krishnaswamy, J. K., Gowthaman, U., Zhang, B., Mattsson, J., Szeponik, L., Liu, D., et al. (2017). Migratory CD11b + Conventional Dendritic Cells Induce T Follicular Helper Cell-dependent Antibody Responses. Sci. Immunol. 2 (18), 1–14. doi:10.1126/sciimmunol.aam9169
Krishnaswamy, J. K., Singh, A., Gowthaman, U., Wu, R., Gorrepati, P., Sales Nascimento, M., et al. (2015). Coincidental Loss of DOCK8 Function in NLRP10-Deficient and C3H/HeJ Mice Results in Defective Dendritic Cell Migration. Proc. Natl. Acad. Sci. USA 112 (10), 3056–3061. doi:10.1073/pnas.1501554112
Kumari, S., Curado, S., Mayya, V., and Dustin, M. L. (2014). T Cell Antigen Receptor Activation and Actin Cytoskeleton Remodeling. Biochim. Biophys. Acta (Bba) - Biomembranes 1838 (2), 546–556. doi:10.1016/j.bbamem.2013.05.004
Kunimura, K., Uruno, T., and Fukui, Y. (2020). DOCK Family Proteins: Key Players in Immune Surveillance Mechanisms. Int. Immunol. 32 (1), 5–15. doi:10.1093/intimm/dxz067
Kuo, C. Y., Long, J. D., Campo-Fernandez, B., de Oliveira, S., Cooper, A. R., Romero, Z., et al. (2018). Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cel Rep. 23 (9), 2606–2616. doi:10.1016/j.celrep.2018.04.103
Kwon, H.-S., Logan, A. C., Chhabra, A., Pang, W. W., Czechowicz, A., Tate, K., et al. (2019). Anti-human CD117 Antibody-Mediated Bone Marrow Niche Clearance in Nonhuman Primates and Humanized NSG Mice. Blood 133 (19), 2104–2108. doi:10.1182/blood-2018-06-853879
Lambe, T., Crawford, G., Johnson, A. L., Crockford, T. L., Bouriez-Jones, T., Smyth, A. M., et al. (2011). DOCK8 Is Essential for T-Cell Survival and the Maintenance of CD8+ T-Cell Memory. Eur. J. Immunol. 41 (12), 3423–3435. doi:10.1002/eji.201141759
Lander, E. S., Baylis, F., Zhang, F., Charpentier, E., Berg, P., Bourgain, C., et al. (2019). Adopt a Moratorium on Heritable Genome Editing. Nature 567 (7747), 165–168. doi:10.1038/d41586-019-00726-5
Lattanzi, A., Camarena, J., Lahiri, P., Segal, H., Srifa, W., Vakulskas, C. A., et al. (2021). Development of β-globin Gene Correction in Human Hematopoietic Stem Cells as a Potential Durable Treatment for Sickle Cell Disease. Sci. Transl. Med. 13, 13. doi:10.1126/scitranslmed.abf2444
Laustsen, A., and Bak, R. O. (2019). “Electroporation-Based CRISPR/Cas9 Gene Editing Using Cas9 Protein and Chemically Modified sgRNAs: Methods and Protocols,” in Methods in Molecular Biology (Clifton, N.J.). Editor Y. Luo (New York, NY: United States: Springer New York), 1961, 127–134. doi:10.1007/978-1-4939-9170-9_9
Li, R., and Gundersen, G. G. (2008). Beyond Polymer Polarity: How the Cytoskeleton Builds a Polarized Cell. Nat. Rev. Mol. Cel Biol. 9 (11), 860–873. doi:10.1038/nrm2522
Lin, S., Staahl, B. T., Alla, R. K., and Doudna, J. A. (2014). “Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery,”Elife. Editor D. Weigel, 3, e04766. doi:10.7554/eLife.04766
Lino, C. A., Harper, J. C., Carney, J. P., and Timlin, J. A. (2018). Delivering Crispr: A Review of the Challenges and Approaches. Drug Deliv. 25 (1), 1234–1257. doi:10.1080/10717544.2018.1474964
Magis, W., DeWitt, M. A., Wyman, S. K., Vu, J. T., Heo, S.-J., Shao, S. J., et al. (2019). High-level Correction of the Sickle Mutation Amplified In Vivo during Erythroid Differentiation. bioRxiv, 432716. doi:10.1101/432716
Mali, P., Aach, J., Stranges, P. B., Esvelt, K. M., Moosburner, M., Kosuri, S., et al. (2013). CAS9 Transcriptional Activators for Target Specificity Screening and Paired Nickases for Cooperative Genome Engineering. Nat. Biotechnol. 31 (9), 833–838. doi:10.1038/nbt.2675
Maresca, M., Lin, V. G., Guo, N., and Yang, Y. (2013). Obligate Ligation-Gated Recombination (ObLiGaRe): Custom-Designed Nuclease-Mediated Targeted Integration through Nonhomologous End Joining. Genome Res. 23 (3), 539–546. doi:10.1101/gr.145441.112
Mayerhoff, L., Lehne, M., Hickstein, L., Salimullah, T., Prieur, S., Thomas, S. K., et al. (2019). Cost Associated with Hematopoietic Stem Cell Transplantation: A Retrospective Claims Data Analysis in Germany. J. Comp. Effectiveness Res. 8 (2), 121–131. doi:10.2217/cer-2018-0100
McGhee, S. A., and Chatila, T. A. (2010). DOCK8 Immune Deficiency as a Model for Primary Cytoskeletal Dysfunction. Dis. Markers 29 (3-4), 151–156. doi:10.3233/DMA-2010-0740
Meshaal, S. S., El Hawary, R. E., Eldash, A., Grimbacher, B., Camacho-Ordonez, N., Abd Elaziz, D. S., et al. (2018). Diagnosis of DOCK8 Deficiency Using Flow Cytometry Biomarkers: an Egyptian Center Experience. Clin. Immunol. 195 (195), 36–44. doi:10.1016/j.clim.2018.07.011
Meyts, I., Bosch, B., Bolze, A., Boisson, B., Itan, Y., Belkadi, A., et al. (2016). Exome and Genome Sequencing for Inborn Errors of Immunity. J. Allergy Clin. Immunol. 138 (4), 957–969. doi:10.1016/j.jaci.2016.08.003
Miller, J. C., Holmes, M. C., Wang, J., Guschin, D. Y., Lee, Y.-L., Rupniewski, I., et al. (2007). An Improved Zinc-finger Nuclease Architecture for Highly Specific Genome Editing. Nat. Biotechnol. 25 (7), 778–785. doi:10.1038/nbt1319
Miller, J. C., Tan, S., Qiao, G., Barlow, K. A., Wang, J., Xia, D. F., et al. (2011). A TALE Nuclease Architecture for Efficient Genome Editing. Nat. Biotechnol. 29 (2), 143–148. doi:10.1038/nbt.1755
Milner, J. D., Brenchley, J. M., Laurence, A., Freeman, A. F., Hill, B. J., Elias, K. M., et al. (2008). Impaired TH17 Cell Differentiation in Subjects with Autosomal Dominant Hyper-IgE Syndrome. Nature 452 (7188), 773–776. doi:10.1038/nature06764
Mizesko, M. C., Banerjee, P. P., Monaco-Shawver, L., Mace, E. M., Bernal, W. E., Sawalle-Belohradsky, J., et al. (2013). Defective Actin Accumulation Impairs Human Natural Killer Cell Function in Patients with Dedicator of Cytokinesis 8 Deficiency. J. Allergy Clin. Immunol. 131 (3), 840–848. doi:10.1016/j.jaci.2012.12.1568
Modlich, U., Navarro, S., Zychlinski, D., Maetzig, T., Knoess, S., Brugman, M. H., et al. (2009). Insertional Transformation of Hematopoietic Cells by Self-Inactivating Lentiviral and Gammaretroviral Vectors. Mol. Ther. 17 (11), 1919–1928. doi:10.1038/mt.2009.179
Mohrin, M., Bourke, E., Alexander, D., Warr, M. R., Barry-Holson, K., Le Beau, M. M., et al. (2010). Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell. 7 (2), 174–185. doi:10.1016/j.stem.2010.06.014
Morgan, R. A., Gray, D., Lomova, A., and Kohn, D. B. (2017). Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell Stem Cell. 21 (5), 574–590. doi:10.1016/j.stem.2017.10.010
Mussolino, C., Morbitzer, R., Lütge, F., Dannemann, N., Lahaye, T., and Cathomen, T. (2011). A Novel TALE Nuclease Scaffold Enables High Genome Editing Activity in Combination with Low Toxicity. Nucleic Acids Res. 39 (21), 9283–9293. doi:10.1093/nar/gkr597
Nambiar, T. S., Billon, P., Diedenhofen, G., Hayward, S. B., Taglialatela, A., Cai, K., et al. (2019). Stimulation of CRISPR-Mediated Homology-Directed Repair by an Engineered RAD18 Variant. Nat. Commun. 10 (1), 3395. doi:10.1038/s41467-019-11105-z
Namekata, K., Kimura, A., Kawamura, K., Harada, C., and Harada, T. (2014). Dock GEFs and Their Therapeutic Potential: Neuroprotection and Axon Regeneration. Prog. Retin. Eye Res. 43, 1–16. doi:10.1016/j.preteyeres.2014.06.005
Newby, G. A., Yen, J. S., Woodard, K. J., Mayuranathan, T., Lazzarotto, C. R., Li, Y., et al. (2021). Base Editing of Haematopoietic Stem Cells Rescues Sickle Cell Disease in Mice. Nature 595 (7866), 295–302. doi:10.1038/s41586-021-03609-w
Ott, M. G., Schmidt, M., Schwarzwaelder, K., Stein, S., Siler, U., Koehl, U., et al. (2006). Correction of X-Linked Chronic Granulomatous Disease by Gene Therapy, Augmented by Insertional Activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 12 (4), 401–409. doi:10.1038/nm1393
Papan, C., Hagl, B., Heinz, V., Albert, M. H., Ehrt, O., Sawalle-Belohradsky, J., et al. (2014). Beneficial IFN-α Treatment of Tumorous Herpes Simplex Blepharoconjunctivitis in Dedicator of Cytokinesis 8 Deficiency. J. Allergy Clin. Immunol. 133 (5), 1456–1458. doi:10.1016/j.jaci.2014.02.008
Pavel-Dinu, M., Wiebking, V., Dejene, B. T., Srifa, W., Mantri, S., Nicolas, C. E., et al. (2019). Gene Correction for SCID-X1 in Long-Term Hematopoietic Stem Cells. Nat. Commun. 10 (1), 1634. doi:10.1038/s41467-019-09614-y
Pillay, B. A., Fusaro, M., Gray, P. E., Statham, A. L., Burnett, L., Bezrodnik, L., et al. (2021). Somatic Reversion of Pathogenic DOCK8 Variants Alters Lymphocyte Differentiation and Function to Effectively Cure DOCK8 Deficiency. J. Clin. Invest. 131, 131. doi:10.1172/JCI142434
Porteus, M. H. (2019). A New Class of Medicines through DNA Editing. N. Engl. J. Med. 380 (10), 947–959. doi:10.1056/nejmra1800729
Porto, E. M., Komor, A. C., Slaymaker, I. M., and Yeo, G. W. (2020). Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discov. 19 (12), 839–859. doi:10.1038/s41573-020-0084-6
Randall, K. L., Lambe, T., Johnson, A. L., Treanor, B., Kucharska, E., Domaschenz, H., et al. (2012). Europe PMC Funders Group DOCK8 Mutations Cripple B Cell Immune Synapse, Germinal Centers and Long-Lived Antibody Production Nat. Immunol. 10 (12), 1283–1291. doi:10.1038/ni.1820.DOCK8
Randall, K. L., Chan, S. S.-Y., Ma, C. S., Fung, I., Mei, Y., Yabas, M., et al. (2011). DOCK8 Deficiency Impairs CD8 T Cell Survival and Function in Humans and Mice. J. Exp. Med. 208 (11), 2305–2320. doi:10.1084/jem.20110345
Randall, K. L., Lambe, T., Johnson, A. L., Treanor, B., Kucharska, E., Domaschenz, H., et al. (2009). Dock8 Mutations Cripple B Cell Immunological Synapses, Germinal Centers and Long-Lived Antibody Production. Nat. Immunol. 10 (12), 1283–1291. doi:10.1038/ni.1820
Randall, K. L., Law, H. D., Ziolkowski, A. F., Wirasinha, R. C., Goodnow, C. C., and Daley, S. R. (2021). DOCK8 Deficiency Diminishes Thymic T‐regulatory Cell Development but Not Thymic Deletion. Clin. Transl Immunol. 10 (1), 1–8. doi:10.1002/cti2.1236
Robert, F., Barbeau, M., Éthier, S., Dostie, J., and Pelletier, J. (2015). Pharmacological Inhibition of DNA-PK Stimulates Cas9-Mediated Genome Editing. Genome Med. 7 (1), 93. doi:10.1186/s13073-015-0215-6
Román-Rodríguez, F. J., Ugalde, L., Álvarez, L., Díez, B., Ramírez, M. J., Risueño, C., et al. (2019). NHEJ-mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell. 25 (5), 607–621. doi:10.1016/j.stem.2019.08.016
Romero, Z., Lomova, A., Said, S., Miggelbrink, A., Kuo, C. Y., Campo-Fernandez, B., et al. (2019). Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 27 (8), 1389–1406. doi:10.1016/j.ymthe.2019.05.014
Roth, T. L., Puig-Saus, C., Yu, R., Shifrut, E., Carnevale, J., Li, P. J., et al. (2018). Reprogramming Human T Cell Function and Specificity with Non-viral Genome Targeting. Nature 559 (7714), 405–409. doi:10.1038/s41586-018-0326-5
Sakuma, T., Nakade, S., Sakane, Y., Suzuki, K.-I. T., and Yamamoto, T. (2016). MMEJ-assisted Gene Knock-In Using TALENs and CRISPR-Cas9 with the PITCh Systems. Nat. Protoc. 11 (1), 118–133. doi:10.1038/nprot.2015.140
Sakurai, T., Kukimoto-Niino, M., Kunimura, K., Yamane, N., Sakata, D., Aihara, R., et al. (2021). A Conserved PI(4,5)P2-binding Domain Is Critical for Immune Regulatory Function of DOCK8. Life Sci. Alliance 4 (4), e202000873. doi:10.26508/lsa.202000873
Sandquist, I., and Kolls, J. (2018). Update on Regulation and Effector Functions of Th17 Cells. F1000Res 7 (0), 205–208. doi:10.12688/f1000research.13020.1
Schiroli, G., Conti, A., Ferrari, S., della Volpe, L., Jacob, A., Albano, L., et al. (2019). Precise Gene Editing Preserves Hematopoietic Stem Cell Function Following Transient P53-Mediated DNA Damage Response. Cell Stem Cell. 24 (4), 551–565. doi:10.1016/j.stem.2019.02.019
Scholefield, J., and Harrison, P. T. (2021). Prime Editing - an Update on the Field. Gene Ther. 28, 396–401. doi:10.1038/s41434-021-00263-9
Scott, O., Kim, V. H.-D., Reid, B., Pham-Huy, A., Atkinson, A. R., Aiuti, A., et al. (2017). Long-Term Outcome of Adenosine Deaminase-Deficient Patients-A Single-Center Experience. J. Clin. Immunol. 37 (6), 582–591. doi:10.1007/s10875-017-0421-7
Shi, H., Liu, C., Tan, H., Li, Y., Nguyen, T.-L. M., Dhungana, Y., et al. (2018). Hippo Kinases Mst1 and Mst2 Sense and Amplify IL-2R-STAT5 Signaling in Regulatory T Cells to Establish Stable Regulatory Activity. Immunity 49 (5), 899–914. doi:10.1016/j.immuni.2018.10.010
Shin, J. J., Schröder, M. S., Caiado, F., Wyman, S. K., Bray, N. L., Bordi, M., et al. (2020). Controlled Cycling and Quiescence Enables Efficient HDR in Engraftment-Enriched Adult Hematopoietic Stem and Progenitor Cells. Cel Rep. 32 (9), 108093. doi:10.1016/j.celrep.2020.108093
Singh, A. K., Eken, A., Hagin, D., Komal, K., Bhise, G., Shaji, A., et al. (2017). DOCK8 Regulates Fitness and Function of Regulatory T Cells through Modulation of IL-2 Signaling. JCI Insight 2 (19), 1–15. doi:10.1172/jci.insight.94275
Singh, R., Kuscu, C., Quinlan, A., Qi, Y., and Adli, M. (2015). Cas9-chromatin Binding Information Enables More Accurate CRISPR Off-Target Prediction. Nucleic Acids Res. 43 (18), e118. doi:10.1093/nar/gkv575
Slatter, M. A., and Gennery, A. R. (2018). Hematopoietic Cell Transplantation in Primary Immunodeficiency - Conventional and Emerging Indications. Expert Rev. Clin. Immunol. 14 (2), 103–114. doi:10.1080/1744666X.2018.1424627
Song, J., Yang, D., Xu, J., Zhu, T., Chen, Y. E., and Zhang, J. (2016). RS-1 Enhances CRISPR/Cas9- and TALEN-Mediated Knock-In Efficiency. Nat. Commun. 7 (1), 10548. doi:10.1038/ncomms10548
Stadtmauer, E. A., Fraietta, J. A., Davis, M. M., Cohen, A. D., Weber, K. L., Lancaster, E., et al. (2020). CRISPR-engineered T Cells in Patients with Refractory Cancer. Science 367, 367. doi:10.1126/science.aba7365
Su, H. C., Jing, H., Angelus, P., and Freeman, A. F. (2019). Insights into Immunity from Clinical and Basic Science Studies of DOCK8 Immunodeficiency Syndrome. Immunol. Rev. 287 (1), 9–19. doi:10.1111/imr.12723
Su, H. C., Jing, H., and Zhang, Q. (2011). DOCK8 Deficiency. Ann. N. Y Acad. Sci. 1246 (1), 26–33. doi:10.1111/j.1749-6632.2011.06295.x
Su, H. C., and Orange, J. S. (2020). The Growing Spectrum of Human Diseases Caused by InheritedCDC42 Mutations. J. Clin. Immunol. 40 (4), 551–553. doi:10.1007/s10875-020-00785-8
Suzuki, K., and Izpisua Belmonte, J. C. (2018). In Vivo genome Editing via the HITI Method as a Tool for Gene Therapy. J. Hum. Genet. 63 (2), 157–164. doi:10.1038/s10038-017-0352-4
Sweeney, C. L., Pavel-Dinu, M., Choi, U., Brault, J., Liu, T., Koontz, S., et al. (2021). Correction of X-CGD Patient HSPCs by Targeted CYBB cDNA Insertion Using CRISPR/Cas9 with 53BP1 Inhibition for Enhanced Homology-Directed Repair. Gene Ther. 28, 373–390. doi:10.1038/s41434-021-00251-z
Szczepek, M., Brondani, V., Büchel, J., Serrano, L., Segal, D. J., and Cathomen, T. (2007). Structure-based Redesign of the Dimerization Interface Reduces the Toxicity of Zinc-finger Nucleases. Nat. Biotechnol. 25 (7), 786–793. doi:10.1038/nbt1317
Talib, S., and Shepard, K. A. (2020). Unleashing the Cure: Overcoming Persistent Obstacles in the Translation and Expanded Use of Hematopoietic Stem Cell-Based Therapies. Stem Cell Transl Med 9 (4), 420–426. doi:10.1002/sctm.19-0375
Tang, W., Dou, Y., Qin, T., Ding, Y., Tang, X., Zhao, X., et al. (2019). Skewed B Cell Receptor Repertoire and Reduced Antibody Avidity in Patients with DOCK8 Deficiency. Scand. J. Immunol. 89 (6), e12759–9. doi:10.1111/sji.12759
Tangye, S. G., Al-Herz, W., Bousfiha, A., Chatila, T., Cunningham-Rundles, C., Etzioni, A., et al. (2020). Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 40 (1), 24–64. doi:10.1007/s10875-019-00737-x
Tangye, S. G., Pillay, B., Randall, K. L., Avery, D. T., Phan, T. G., Gray, P., et al. (2017). Dedicator of Cytokinesis 8-deficient CD4 + T Cells Are Biased to a T H 2 Effector Fate at the Expense of T H 1 and T H 17 Cells. J. Allergy Clin. Immunol. 139 (3), 933–949. doi:10.1016/j.jaci.2016.07.016
Tebas, P., Stein, D., Tang, W. W., Frank, I., Wang, S. Q., Lee, G., et al. (2014). Gene Editing ofCCR5in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 370 (10), 901–910. doi:10.1056/NEJMoa1300662
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2015). GUIDE-seq Enables Genome-wide Profiling of Off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 33 (2), 187–197. doi:10.1038/nbt.3117
Tsilifis, C., Freeman, A. F., and Gennery, A. R. (2021). STAT3 Hyper-IgE Syndrome-An Update and Unanswered Questions. J. Clin. Immunol. 41 (5), 864–880. doi:10.1007/s10875-021-01051-1
Turchiano, G., Andrieux, G., Klermund, J., Blattner, G., Pennucci, V., el Gaz, M., et al. (2021). Quantitative Evaluation of Chromosomal Rearrangements in Gene-Edited Human Stem Cells by CAST-Seq. Cell Stem Cell. 28 (6), 1136–1147. doi:10.1016/j.stem.2021.02.002
Urnov, F. D. (2021). CRISPR-Cas9 Can Cause Chromothripsis. Nat. Genet. 53 (6), 768–769. doi:10.1038/s41588-021-00881-4
Vaidyanathan, S., Azizian, K. T., Haque, A. K. M. A., Henderson, J. M., Hendel, A., Shore, S., et al. (2018). Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. - Nucleic Acids 12, 530–542. doi:10.1016/j.omtn.2018.06.010
Vaidyanathan, S., Baik, R., Chen, L., Bravo, D. T., Suarez, C. J., Abazari, S. M., et al. (2021). Targeted Replacement of Full-Length CFTR in Human Airway Stem Cells by CRISPR/Cas9 for Pan-Mutation Correction in the Endogenous Locus. bioRxiv. doi:10.1101/2021.02.26.432961
Vakulskas, C. A., Dever, D. P., Rettig, G. R., Turk, R., Jacobi, A. M., Collingwood, M. A., et al. (2018). A High-Fidelity Cas9 Mutant Delivered as a Ribonucleoprotein Complex Enables Efficient Gene Editing in Human Hematopoietic Stem and Progenitor Cells. Nat. Med. 24 (8), 1216–1224. doi:10.1038/s41591-018-0137-0
Vavassori, V., Mercuri, E., Marcovecchio, G. E., Castiello, M. C., Schiroli, G., Albano, L., et al. (2021). Modeling, Optimization, and Comparable Efficacy of T Cell and Hematopoietic Stem Cell Gene Editing for Treating hyper‐IgM Syndrome. EMBO Mol. Med. 13 (3), 1–25. doi:10.15252/emmm.202013545
Wang, J., Exline, C. M., Declercq, J. J., Llewellyn, G. N., Hayward, S. B., Li, P. W.-L., et al. (2015). Homology-driven Genome Editing in Hematopoietic Stem and Progenitor Cells Using ZFN mRNA and AAV6 Donors. Nat. Biotechnol. 33 (12), 1256–1263. doi:10.1038/nbt.3408
Wienert, B., Wyman, S. K., Richardson, C. D., Yeh, C. D., Akcakaya, P., Porritt, M. J., et al. (2019). Unbiased Detection of CRISPR Off-Targets In Vivo Using DISCOVER-Seq. Science 364 (6437), 286–289. doi:10.1126/science.aav9023
Wilkinson, A. C., Dever, D. P., Baik, R., Camarena, J., Hsu, I., Charlesworth, C. T., et al. (2021). Cas9-AAV6 Gene Correction of Beta-Globin in Autologous HSCs Improves Sickle Cell Disease Erythropoiesis in Mice. Nat. Commun. 12 (1), 1–9. doi:10.1038/s41467-021-20909-x
Wilson, A., Laurenti, E., Oser, G., van der Wath, R. C., Blanco-Bose, W., Jaworski, M., et al. (2008). Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell. 135 (6), 1118–1129. doi:10.1016/j.cell.2008.10.048
Xu, L., Wang, J., Liu, Y., Xie, L., Su, B., Mou, D., et al. (2019). CRISPR-edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 381 (13), 1240–1247. doi:10.1056/NEJMoa1817426
Xu, X., Han, L., Zhao, G., Xue, S., Gao, Y., Xiao, J., et al. (2017). LRCH1 Interferes with DOCK8-Cdc42-Induced T Cell Migration and Ameliorates Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 214 (1), 209–226. doi:10.1084/jem.20160068
Yamamura, K., Uruno, T., Shiraishi, A., Tanaka, Y., Ushijima, M., Nakahara, T., et al. (2017). The Transcription Factor EPAS1 Links DOCK8 Deficiency to Atopic Skin Inflammation via IL-31 Induction. Nat. Commun. 8, 8. doi:10.1038/ncomms13946
Yao, X., Wang, X., Hu, X., Liu, Z., Liu, J., Zhou, H., et al. (2017). Homology-mediated End Joining-Based Targeted Integration Using CRISPR/Cas9. Cell Res. 27 (6), 801–814. doi:10.1038/cr.2017.76
Zhang, Q., Davis, J. C., Dove, C. G., and Su, H. C. (2010). Genetic, Clinical, and Laboratory Markers for DOCK8 Immunodeficiency Syndrome. Dis. Markers 29 (3-4), 131–139. doi:10.3233/DMA-2010-0737
Zhang, Q., Davis, J. C., Lamborn, I. T., Freeman, A. F., Jing, H., Favreau, A. J., et al. (2009). Combined Immunodeficiency Associated withDOCK8Mutations. N. Engl. J. Med. 361 (21), 2046–2055. doi:10.1056/nejmoa0905506
Zhang, Q., Dove, C. G., Hor, J. L., Murdock, H. M., Strauss-Albee, D. M., Garcia, J. A., et al. (2014). DOCK8 Regulates Lymphocyte Shape Integrity for Skin Antiviral Immunity. J. Exp. Med. 211 (13), 2549–2566. doi:10.1084/jem.20141307
Keywords: gene editing (CRISPR-Cas9), CRISPR/Cas 9, hematopoietic stem cell, DOCK8 immunodeficiency syndrome, DOCK8 deficiency, DOCK8, gene therapy, Primary immunodeficiency
Citation: Ravendran S, Hernández SS, König S and Bak RO (2022) CRISPR/Cas-Based Gene Editing Strategies for DOCK8 Immunodeficiency Syndrome. Front. Genome Ed. 4:793010. doi: 10.3389/fgeed.2022.793010
Received: 11 October 2021; Accepted: 14 February 2022;
Published: 17 March 2022.
Edited by:
David Hagin, Tel Aviv Sourasky Medical Center, IsraelReviewed by:
Alexandra Freeman, National Institutes of Health (NIH), United StatesCaroline Y. Kuo, David Geffen School of Medicine at UCLA, University of California, Los Angeles, United States
Copyright © 2022 Ravendran, Hernández, König and Bak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rasmus O. Bak, bak@biomed.au.dk