
- Department of Cellular and Molecular Physiology, Institute of Translational Medicine, University of Liverpool, Liverpool, UK
Non-coding RNAs (ncRNAs) have long been recognized at imprinted gene loci and provided early paradigms to investigate their functions and molecular mechanisms of action. The characteristic feature of imprinted genes, their monoallelic, parental-origin-dependent expression, is achieved through complex epigenetic regulation, which is modulated by ncRNAs. This minireview focuses on two imprinted gene clusters, in which changes in ncRNA expression contribute to human disorders. At the GNAS locus loss of NESP RNA can cause autosomal dominant Pseudohypoparathyroidism type 1b (AD-PHP-Ib), while at the SNRPN-UBE3A locus a long ncRNA and processed snoRNAs play a role in Angelman-Syndrome (AS) and Prader–Willi-Syndrome (PWS). The ncRNAs silence overlapping protein-coding transcripts in sense or anti-sense orientation through changes in histone modifications as well as DNA methylation at CpG-rich sequence motifs. Their epigenetic modulatory functions are required in early development in the pre-implantation embryo or already in the parental germ cells. However, it remains unclear whether the sequence homology-carrying ncRNA itself is required, or whether the process of its transcription through other promoters causes the silencing effect.
Imprinted gene loci provided early model systems, in which non-coding RNAs (ncRNAs) have been investigated (Barlow, 2011; Ferguson-Smith, 2011). Imprinted genes are defined as being monoallelically expressed dependent on their parental origin. During the mammalian imprinting process, epigenetic marks are established in the female or male germlines at imprinting control regions (ICRs), which results in the silencing of one parental allele in somatic cells of the offspring. A common feature of imprinted genes is their occurrence in clusters, whereby one ICR regulates the monoallelic expression of several neighboring genes, although single units of an imprinted gene and associated retrogene have also be identified (Cowley and Oakey, 2010). Imprinted gene clusters often contain ncRNAs, which are now increasingly recognized for their regulatory effects on nearby imprinted protein-coding genes. This minireview will focus on the GNAS and SNRPN-UBE3A imprinting clusters, in which disturbances of the ncRNAs are associated with human hereditary disorders. Functions of ncRNAs at other clusters have been covered in recent excellent reviews (Barlow, 2011; Ferguson-Smith, 2011; Pauler et al., 2012).
Roles of Non-Coding RNAs at the GNAS Locus and Their Involvement in AD-PHP-Ib
The roles of ncRNAs at the complex imprinted Gnas locus (Figure 1A) have become more evident through recent studies in human and mouse (Williamson et al., 2004, 2006, 2011; Bastepe et al., 2005; Liu et al., 2005a; Chotalia et al., 2009; Chillambhi et al., 2010). The locus, which is largely conserved between both species, consists of two main protein-coding transcripts (Gnas and Gnasxl) and two regulatory non-coding transcripts termed Nespas and Exon 1A (EXON A/B in human). A fifth transcription unit, Nesp, exerts a dual function within the locus through epigenetically regulating other transcripts and by encoding a protein (see below; Ischia et al., 1997; Plagge et al., 2005; Chotalia et al., 2009; Fröhlich et al., 2010). Gnas encodes Gαs, the α-stimulatory subunit of trimeric G-proteins, which mediates signal transduction from seven-transmembrane receptors to adenylate cyclase (Weinstein et al., 2001; Plagge et al., 2008). In some cell types, e.g., renal proximal tubules, brain subregions, thyroid, pituitary somatotroph cells among others, Gnas is preferentially or exclusively expressed from the maternally inherited allele (Plagge et al., 2008; Chen et al., 2009; Zazo et al., 2011). Defects in Gnas expression from the maternal allele can, therefore, disrupt various hormone signaling pathways, which leads to a range of disease symptoms termed “Pseudohypoparathyroidisms (PHP)” with or without “Albright’s Hereditary Osteodystrophy (AHO).” Typically, these comprise resistance to parathyroid hormone, thyroid stimulating hormone, growth hormone releasing hormone, gonadotrophins, and α-melanocyte stimulating hormone (Plagge et al., 2008; Chen et al., 2011; Mantovani, 2011). The molecular defects causing PHP/AHO can be categorized into two types: (a) mutations in the coding exons of GNAS or (b) epigenetic changes at the differentially methylated regions (DMRs) of the GNAS locus (Figure 1A). Maternally inherited coding exon mutations invariably lead to a severe combination of many PHP/AHO features, while epigenetic changes often only result in a limited spectrum of hormone resistance symptoms, mainly parathyroid hormone resistance (then also termed PHP-Ib; de Nanclares et al., 2007; Mariot et al., 2008; Kelsey, 2010; Mantovani et al., 2010; Mantovani, 2011).
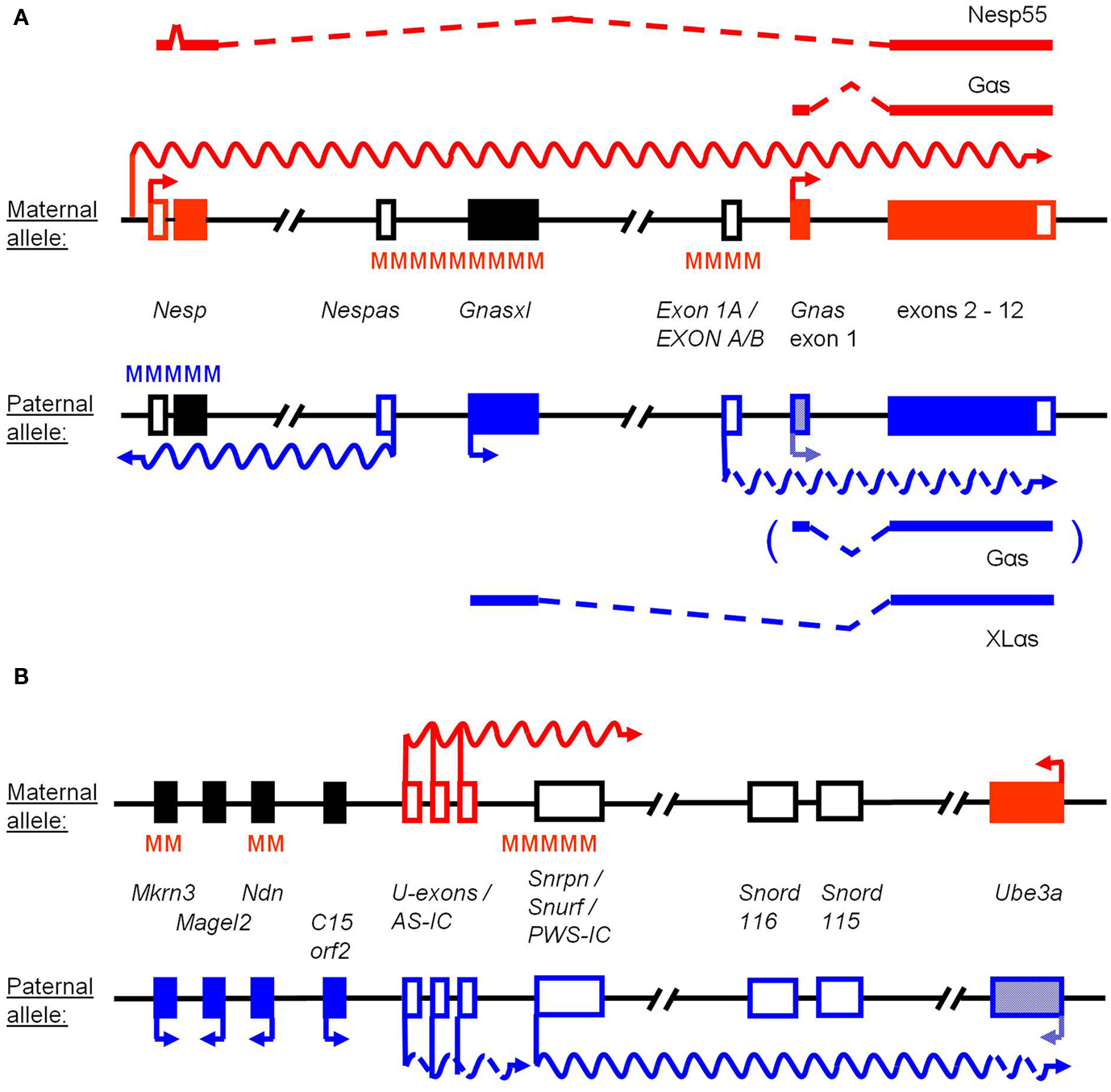
Figure 1. Simplified schemes of the Gnas and Snrpn-Ube3a imprinted loci. The features of the maternal and paternal alleles are indicated in red and blue, respectively. Genes or transcripts are named in the central part. Arrows mark transcription start sites and undulating lines ncRNAs. Open and filled boxes represent non-coding and coding exons, respectively, while black boxes indicate silenced genes. Differentially methylated regions (DMRs) of DNA are marked by MMM. (A) For the Gnas locus the alternatively spliced coding transcripts and proteins are named above and below the alleles. DMRs at Nespas and Exon 1A (EXON A/B in human) are established in the maternal germline, while methylation at Nesp occurs during early embryonic development. Gnas is expressed biallelically, but is silenced on the paternal allele in some tissues (hatched box), in which Exon 1A shows comparatively high expression levels (interrupted undulating line). Nesp represents a coding transcript (ORF limited to Nesp exon 2), but a regulatory RNA is initiated from a separate promoter in oocytes (red undulating line). Nespas is expressed from the unmethylated imprinting control region (ICR) of the locus. (B) The Snrpn-Ube3a imprinting cluster contains several genes (represented by single-exon boxes) that show monoallelic expression in brain. Ube3a is biallelically expressed in most tissues, but silenced on the paternal allele in neurons (hatched box). The Snrpn long ncRNA occurs in multiple, variably processed forms, including brain-specific variants that contain upstream promoters/exons (U-exons) and sequences overlapping with Ube3a (interrupted undulating lines). Snord115 and Snord116 represent clusters of C/D box snoRNAs, which are generated from the Snrpn ncRNA. The bipartite ICR of the human locus is indicated as AS-IC and PWS-IC, the latter being conserved in mouse around the main Snrpn start site. C15orf2 is not present in the murine locus. Somatic DMRs at Ndn and Mkrn3 are established during development. Transcription from U-exons occurs in the female germline in growing oocytes (red undulating line) and is involved in the establishment of methylation at the PWS-IC.
The epigenetic abnormalities associated with PHP consist of changes in DNA methylation at DMRs as well as changes in ncRNA expression. Furthermore, these can occur in a familial pattern upon maternal inheritance of a mutation, i.e., autosomal dominant pseudohypoparathyroidism type 1b (AD-PHP-Ib; Bastepe et al., 2003, 2005), or sporadically, in which case the molecular cause is unknown, but most likely due to abnormalities in epigenetic regulators that act at several imprinted loci (Liu et al., 2005b; Linglart et al., 2007; Fernandez-Rebollo et al., 2011; Mantovani, 2011; Maupetit-Mehouas et al., 2011). A common epigenetic change in AD-PHP-Ib is the loss of maternal allele-specific methylation at the EXON A/B DMR, located a few kb upstream of GNAS exon 1 (Figure 1A). Interestingly, on the unmethylated paternal allele this exon acts as the start site of a ncRNA, which is transcribed across the GNAS promoter in the same direction (Liu et al., 2000a,b). Evidence from AD-PHP-Ib patients as well as mouse models indicates that the expression levels of the two transcripts, EXON A/B and GNAS, are oppositely regulated in cis. On the maternal allele methylation across EXON A/B inhibits expression of the ncRNA, while the downstream GNAS promoter drives the expression of the coding RNA (Liu et al., 2000a,b). On the paternal allele lack of methylation allows EXON A/B expression, while the GNAS promoter is suppressed in at least some cell types, i.e., GNAS expression becomes imprinted, for example in renal proximal tubules. In AD-PHP-Ib patients two types of deletion mutations upstream of GNAS result in loss of methylation at the EXON A/B DMR. One of these comprises a 1.3 kb region in the neighboring gene STX16 ∼220 kb upstream of GNAS (Bastepe et al., 2003; Linglart et al., 2005), and the other deletion affects the most 5′ region within the complex GNAS locus (i.e., NESP and NESPAS exons; Bastepe et al., 2005; Chillambhi et al., 2010; Richard et al., 2012). For the latter type of deletion it has been shown that the loss of methylation at EXON A/B is not only associated with a loss of GNAS expression, but also with an increase in the levels of the non-coding EXON A/B RNA (Bastepe et al., 2005; Fröhlich et al., 2010). This raises the question of whether the EXON A/B RNA, or the process of its transcription, regulates GNAS expression. Further insights have been provided recently through a mouse model, in which the Exon 1A RNA has been truncated by insertion of a polyadenylation cassette (Eaton et al., 2012). Paternal inheritance of this allele results in early termination of Exon 1A transcription, thus avoiding extension across the downstream Gnas promoter region. DNA methylation across the Exon 1A DMR is not changed in this model. However, the expression of Gnas becomes up-regulated in tissues where it is normally imprinted and repressed (and in which normally comparatively high levels of Exon 1A RNA are found; Eaton et al., 2012). These observations favor a mechanism, whereby the non-coding Exon 1A RNA, or the process of its transcription through the Gnas promoter, inhibits the expression of the latter (transcriptional interference), although alternative mechanisms (e.g., impairment of a silencer element for Gnas in the Exon 1A region) cannot be excluded at this stage (Liu et al., 2000a; Eaton et al., 2012).
In contrast to the Exon 1A transcript, the Nespas ncRNA functions in an anti-sense orientation, to counter-regulate, and silence the transcription of the Nesp55 coding RNA (Hayward and Bonthron, 2000; Wroe et al., 2000; Williamson et al., 2011). Nespas is expressed from a separate promoter, which is located upstream of Exon 1A and Gnas in the imprinting control center (ICR) of the locus (Figure 1A; Williamson et al., 2006). It is transcribed in the opposite direction of Exon 1A and Gnas and from the paternal allele only. Its promoter is methylated on the maternal allele (Hayward and Bonthron, 2000; Wroe et al., 2000; Williamson et al., 2002). The role of the Nespas ncRNA was investigated using a polyadenylation cassette again, to terminate the transcript a short distance downstream of its initiation site (Williamson et al., 2011). The authors showed that the truncated RNA had lost its anti-sense silencing function for Nesp on the paternal allele. The DNA methylation at the Nesp promoter on the paternal allele, which is normally established during early embryogenesis, was lost and Nesp became biallelically expressed. A second, hypomorphic Nespas mutant (>90% loss of RNA levels, but no truncation) revealed further insights into the mechanisms, by which the ncRNA silences Nesp (Williamson et al., 2011). In this model DNA methylation on the paternal allele of the Nesp DMR was also lost, but, in contrast to the truncation model, expression of Nesp RNA was only partially up-regulated. The reason for the different levels of induction of Nesp transcription in the Nespas truncation vs. hypomorph model was found to be related to histone modifications. While the activating histone mark H3K4me3 is usually depleted at the Nesp promoter on the paternal allele, increased levels were found in both Nespas mutants, in line with Nesp transcriptional activation. Moreover, the H3K4me3 levels correlated with the degree of transcriptional activation of Nesp as these were higher in the Nespas truncation model (Williamson et al., 2011). Thus, under normal conditions Nespas anti-sense transcription through the Nesp promoter results in low H3K4me3 levels and suppression of Nesp. Furthermore, the increased H3K4me3 levels in both Nespas mutants are a likely cause for the lack of DNA methylation at the Nesp DMR, since this histone modification is incompatible with the action of the DNA methyltransferases Dnmt3a and 3L (Ooi et al., 2007; Zhang et al., 2010). Other changes in histone marks of both Nespas mouse models included a depletion of repressive H3K9me3 downstream of the Nesp start site and a depletion of the transcription elongation mark H3K36me3 upstream of Nesp, which is consistent with up-regulation of Nesp and loss of Nespas transcription, respectively, on the paternal allele (Williamson et al., 2011). Overall, these findings indicate a major role for the non-coding anti-sense RNA Nespas (or for its transcription process) in mediating the silencing and stable suppression of Nesp in cis through low levels of H3K4me3 and, consequently, DNA methylation via Dnmt3a/L. The attraction of histone modifying enzymes, e.g., a histone demethylase, through the ncRNA appears to be a likely molecular mechanism. Whether the changes of Nespas and Nesp expression on the paternal allele of these mouse models further affect the downstream Exon 1A and Gnas transcription units, potentially leading to loss of imprinting of Gnas as described in the Nespas ICR deletion mice, remains to be analyzed (Williamson et al., 2006, 2011).
The theme of epigenetic regulation through RNAs (or through the process of their transcription across other promoters) extends to a third RNA of the Gnas locus, namely the protein-coding Nesp (Ischia et al., 1997; Plagge et al., 2005; Chotalia et al., 2009). Dual functions as a coding and ncRNA are unusual, but have recently been described in several cases (Cooper et al., 2011; Kageyama et al., 2011). The regulatory role of Nesp occurs in the female germline in growing oocytes at postnatal stages when maternal DNA methylation imprints are established at the Nespas ICR and Exon 1A germline DMR (Chotalia et al., 2009). At this stage, an oocyte-specific promoter drives the transcription of Nesp, which extends through all other promoters and exons of the Gnas locus (Figure 1A). When a polyadenylation/transcriptional termination cassette was used to truncate the Nesp RNA shortly after its single-exon open reading frame on the maternal allele, the DNA methylation marks at the Nespas ICR and Exon 1A germline DMR failed to be established (Chotalia et al., 2009). Offspring that inherited this truncation maternally showed epigenetic and transcriptional patterns on the maternal chromosome in somatic tissues that resembled those normally found on the paternal chromosome, i.e., loss of DNA methylation was associated with expression of the Nespas, Gnasxl, and Exon 1A transcripts. Consequently, transcription of Nesp and Gnas (in imprinted tissues) became silenced and the somatic Nesp DMR methylated on the maternal chromosome (Chotalia et al., 2009). The consequences of the Nesp truncation are, therefore, very similar to the NESP deletion mutations found in patients with AD-PHP-Ib described above (Bastepe et al., 2005; Chillambhi et al., 2010; Richard et al., 2012) and to the mouse Nesp deletion model (Fröhlich et al., 2010). The comparison of these various mutants emphasizes the importance of the process of transcription (or generation of overlapping RNA) for establishing epigenetic modifications, which then last throughout development. Regarding details of the molecular mechanisms involved, several possibilities have been discussed and a crucial role of histone modifications/modifying enzymes and their interaction with DNA methyltransferase and/or RNA Polymerase complexes appear most likely (Smallwood and Kelsey, 2012).
Roles of the Long ncRNA SNRPN at the PWS/AS Locus
Another cluster of imprinted genes that is regulated by a large ∼0.5–1.0 Mb ncRNA, termed SNRPN, is located on human chromosome 15q11-13 and is largely conserved on mouse chromosome 7 (Figure 1B; Landers et al., 2004; Buiting, 2010). In humans, the locus is associated with the neurogenetic imprinting disorders Prader–Willi-Syndrome (PWS) and Angelman-Syndrome (AS; Buiting, 2010; Mabb et al., 2011). AS is caused by loss of expression of UBE3A from the maternal allele. UBE3A encodes an E3 ubiquitin ligase, which is only imprinted in brain and biallelically expressed in other tissues. Among other symptoms, the disorder is characterized by developmental delays, intellectual disability, and behavioral abnormalities (happy demeanor; Mabb et al., 2011). By contrast, the symptoms of PWS, which include neonatal hypotonia, feeding difficulties, and growth retardation followed from early childhood onward by hyperphagia, severe obesity, hypogonadism, and behavioral abnormalities, is caused by loss of expression of several paternally expressed genes of the locus (Buiting, 2010). Mouse models and human genetic studies indicate that the genes NECDIN (NDN), MAGEL2, and parts of the long ncRNA (lncRNA) SNRPN contribute to this complex disorder (Figure 1B; Gerard et al., 1999; Muscatelli et al., 2000; Kozlov et al., 2007; Sahoo et al., 2008; de Smith et al., 2009; Duker et al., 2010). The lncRNA SNRPN can be processed and spliced in many ways and is expressed in most tissues. Some of its transcript variants are neuron-specific, initiated at separate upstream exons (U-exons), and overlap anti-sense with Ube3a (then also termed UBE3A-ATS; Bressler et al., 2001; Runte et al., 2001; Yamasaki et al., 2003; Landers et al., 2004; Le Meur et al., 2005). The U-exon/SNRPN/UBE3A-ATS lncRNA has a crucial role in silencing UBE3A on the paternal allele in neural tissue. The SNRPN main transcriptional start site is located within the PWS-IC, a 4.3 kb (or 6.0 kb in mouse) region, which remains unmethylated on the paternal allele and is thought to contain an activator function for all paternally expressed genes, including those located upstream of the lncRNA (Figure 1B; Buiting, 2010; DuBose et al., 2011). This function of the PWS-IC, to guarantee paternal gene expression, appears to be required at a critical time point during pre-implantation development, but not anymore at later stages, e.g., in neuronal precursor cells (DuBose et al., 2012; Rabinovitz et al., 2012). It remains to be clarified whether transcription of the Snrpn lncRNA in early embryos might attract histone modifications, which consequently lead to the permanent setting of all epigenetic marks of the paternal allele (Makedonski et al., 2005; Mabb et al., 2011), resulting not only in activation of paternally expressed genes, but also silencing of Ube3a in brain. Alternatively, Ube3a might be silenced directly on the level of transcriptional processes, since no differential DNA methylation occurs at this gene (Mabb et al., 2011). A truncated Ube3a-ats RNA that still contains the Snord RNA clusters (Figure 1B; see below), but does not overlap with Ube3a anymore, lost its silencing function on the latter in a cell culture model, indicating that the lncRNA needs to contain anti-sense sequences to Ube3a or that the process of transcription through the coding gene is required (Meng et al., 2012).
The SNRPN lncRNA has a unique additional feature as it contains two large clusters of small nucleolar RNAs (C/D box snoRNAs), termed SNORD115 and SNORD116, which are predominantly expressed in the brain and, in the case of SNORD116, contribute to many of the PWS symptoms (Sahoo et al., 2008; de Smith et al., 2009; Duker et al., 2010; Bortolin-Cavaille and Cavaille, 2012). The evidence from human PWS patients carrying microdeletions of the SNORD116 cluster has been confirmed in mice with targeted deletion of the corresponding elements, although the murine phenotype is largely restricted to the neonatal hypotonia and growth retardation of PWS and does not reproduce the obesity and infertility found in adult PWS patients (Skryabin et al., 2007; Ding et al., 2008). The Snord115 and 116 clusters are processed from Snrpn and show the typical features of C/D box snoRNAs. However, their precise molecular functions remain elusive as they do not show the usual anti-sense sequence characteristics to ribosomal or spliceosomal RNAs (Bortolin-Cavaille and Cavaille, 2012).
In humans, the expression of the SNRPN lncRNA from the PWS-IC is itself regulated by a second element, which is located ∼30 kb upstream and termed AS-IC (Figure 1B; Buiting, 2010; Mabb et al., 2011). The AS-IC mediates methylation of the PWS-IC on the maternal chromosome and silencing of SNRPN, which in turn allows UBE3A expression. The AS-IC element was shown to bind a number of proteins, including transcription factors, but is not conserved as an orthologous element in mice (Kaufman et al., 2009). However, details of the PWS-IC imprinting mechanism in mice have recently emerged, showing that transcription through the Snrpn promoter from upstream exons (U-exons) in oocytes brings about the DNA methylation and silencing of Snrpn exon 1 on the maternal allele (Mapendano et al., 2006; Smith et al., 2011). In a BAC transgenic model a subset of U-exons (contained within a ∼100 kb region 5′ of Snrpn exon 1) caused transcription through the PWS-IC/Snrpn promoter in growing oocytes. This transcriptional activity led to DNA methylation at the PWS-IC in cis and silencing of the Snrpn lncRNA expression, i.e., establishment of the maternal imprint mark. There is also evidence for the presence of a corresponding U-exon in the human AS-IC region, which might allow for a similar mechanism of transcription/RNA mediated epigenetic modification (Farber et al., 1999). As with the Gnas locus, it remains unclear whether the RNA is required or whether the process of transcription through the Snrpn promoter suffices, to establish the epigenetic/imprinting mark at the PWS-IC (Chotalia et al., 2009; Smith et al., 2011). However, the functional similarities in oocytes between the U-exons of Snrpn and Nesp of the Gnas locus are evident, and it has been shown recently that transcription is a common feature associated with the establishment of DNA methylation in oocytes (Smallwood et al., 2011).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in the laboratory has been supported by the UK Medical Research Council (MRC), the Royal Society, and the Center for Integrative Mammalian Biology of the Universities of Liverpool and Manchester.
References
Barlow, D. P. (2011). Genomic imprinting: a mammalian epigenetic discovery model. Annu. Rev. Genet. 45, 379–403.
Bastepe, M., Fröhlich, L. F., Hendy, G. N., Indridason, O. S., Josse, R. G., Koshiyama, H., et al. (2003). Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J. Clin. Invest. 112, 1255–1263.
Bastepe, M., Fröhlich, L. F., Linglart, A., Abu-Zahra, H. S., Tojo, K., Ward, L. M., et al. (2005). Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat. Genet. 37, 25–27.
Bortolin-Cavaille, M. L., and Cavaille, J. (2012). The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader-Willi locus generate canonical box C/D snoRNAs. Nucleic Acids Res. 40, 6800–6807.
Bressler, J., Tsai, T. F., Wu, M. Y., Tsai, S. F., Ramirez, M. A., Armstrong, D., et al. (2001). The SNRPN promoter is not required for genomic imprinting of the Prader-Willi/Angelman domain in mice. Nat. Genet. 28, 232–240.
Buiting, K. (2010). Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 365–376.
Chen, M., Nemechek, N. M., Mema, E., Wang, J., and Weinstein, L. S. (2011). Effects of deficiency of the G protein Gsα on energy and glucose homeostasis. Eur. J. Pharmacol. 660, 119–124.
Chen, M., Wang, J., Dickerson, K. E., Kelleher, J., Xie, T., Gupta, D., et al. (2009). Central nervous system imprinting of the G protein G(s)α and its role in metabolic regulation. Cell Metab. 9, 548–555.
Chillambhi, S., Turan, S., Hwang, D. Y., Chen, H. C., Jüppner, H., and Bastepe, M. (2010). Deletion of the noncoding GNAS antisense transcript causes pseudohypoparathyroidism type Ib and biparental defects of GNAS methylation in cis. J. Clin. Endocrinol. Metab. 95, 3993–4002.
Chotalia, M., Smallwood, S. A., Ruf, N., Dawson, C., Lucifero, D., Frontera, M., et al. (2009). Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 23, 105–117.
Cooper, C., Vincett, D., Yan, Y., Hamedani, M. K., Myal, Y., and Leygue, E. (2011). Steroid receptor RNA activator bi-faceted genetic system: heads or tails? Biochimie 93, 1973–1980.
Cowley, M., and Oakey, R. J. (2010). Retrotransposition and genomic imprinting. Brief. Funct. Genomics 9, 340–346.
de Nanclares, G. P., Fernandez-Rebollo, E., Santin, I., Garcia-Cuartero, B., Gaztambide, S., Menendez, E., et al. (2007). Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J. Clin. Endocrinol. Metab. 92, 2370–2373.
de Smith, A. J., Purmann, C., Walters, R. G., Ellis, R. J., Holder, S. E., Van Haelst, M. M., et al. (2009). A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity, and hypogonadism. Hum. Mol. Genet. 18, 3257–3265.
Ding, F., Li, H. H., Zhang, S., Solomon, N. M., Camper, S. A., Cohen, P., et al. (2008). SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 3, e1709. doi:10.1371/journal.pone.0001709
DuBose, A. J., Smith, E. Y., Johnstone, K. A., and Resnick, J. L. (2012). Temporal and developmental requirements for the Prader-Willi imprinting center. Proc. Natl. Acad. Sci. U.S.A. 109, 3446–3450.
DuBose, A. J., Smith, E. Y., Yang, T. P., Johnstone, K. A., and Resnick, J. L. (2011). A new deletion refines the boundaries of the murine Prader-Willi syndrome imprinting center. Hum. Mol. Genet. 20, 3461–3466.
Duker, A. L., Ballif, B. C., Bawle, E. V., Person, R. E., Mahadevan, S., Alliman, S., et al. (2010). Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 18, 1196–1201.
Eaton, S. A., Williamson, C. M., Ball, S. T., Beechey, C. V., Moir, L., Edwards, J., et al. (2012). New mutations at the imprinted Gnas cluster show gene dosage effects of Gsα in postnatal growth and implicate XLαs in bone and fat metabolism but not in suckling. Mol. Cell. Biol. 32, 1017–1029.
Farber, C., Dittrich, B., Buiting, K., and Horsthemke, B. (1999). The chromosome 15 imprinting centre (IC) region has undergone multiple duplication events and contains an upstream exon of SNRPN that is deleted in all Angelman syndrome patients with an IC microdeletion. Hum. Mol. Genet. 8, 337–343.
Ferguson-Smith, A. C. (2011). Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 12, 565–575.
Fernandez-Rebollo, E., Perez de Nanclares, G., Lecumberri, B., Turan, S., Anda, E., Perez-Nanclares, G., et al. (2011). Exclusion of the GNAS locus in PHP-Ib patients with broad GNAS methylation changes: evidence for an autosomal recessive form of PHP-Ib? J. Bone Miner. Res. 26, 1854–1863.
Fröhlich, L. F., Mrakovcic, M., Steinborn, R., Chung, U. I., Bastepe, M., and Jüppner, H. (2010). Targeted deletion of the Nesp55 DMR defines another Gnas imprinting control region and provides a mouse model of autosomal dominant PHP-Ib. Proc. Natl. Acad. Sci. U.S.A. 107, 9275–9280.
Gerard, M., Hernandez, L., Wevrick, R., and Stewart, C. L. (1999). Disruption of the mouse necdin gene results in early post-natal lethality. Nat. Genet. 23, 199–202.
Hayward, B. E., and Bonthron, D. T. (2000). An imprinted antisense transcript at the human GNAS1 locus. Hum. Mol. Genet. 9, 835–841.
Ischia, R., Lovisetti-Scamihorn, P., Hogue-Angeletti, R., Wolkersdorfer, M., Winkler, H., and Fischer-Colbrie, R. (1997). Molecular cloning and characterization of NESP55, a novel chromogranin- like precursor of a peptide with 5-HT1B receptor antagonist activity. J. Biol. Chem. 272, 11657–11662.
Kageyama, Y., Kondo, T., and Hashimoto, Y. (2011). Coding vs non-coding: translatability of short ORFs found in putative non-coding transcripts. Biochimie 93, 1981–1986.
Kaufman, Y., Heled, M., Perk, J., Razin, A., and Shemer, R. (2009). Protein-binding elements establish in the oocyte the primary imprint of the Prader-Willi/Angelman syndromes domain. Proc. Natl. Acad. Sci. U.S.A. 106, 10242–10247.
Kelsey, G. (2010). Imprinting on chromosome 20: tissue-specific imprinting and imprinting mutations in the GNAS locus. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 377–386.
Kozlov, S. V., Bogenpohl, J. W., Howell, M. P., Wevrick, R., Panda, S., Hogenesch, J. B., et al. (2007). The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 39, 1266–1272.
Landers, M., Bancescu, D. L., Le Meur, E., Rougeulle, C., Glatt-Deeley, H., Brannan, C., et al. (2004). Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res. 32, 3480–3492.
Le Meur, E., Watrin, F., Landers, M., Sturny, R., Lalande, M., and Muscatelli, F. (2005). Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region. Dev. Biol. 286, 587–600.
Linglart, A., Bastepe, M., and Jüppner, H. (2007). Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus. Clin. Endocrinol. (Oxf) 67, 822–831.
Linglart, A., Gensure, R. C., Olney, R. C., Jüppner, H., and Bastepe, M. (2005). A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am. J. Hum. Genet. 76, 804–814.
Liu, J., Chen, M., Deng, C., Bourc’his, D., Nealon, J. G., Erlichman, B., et al. (2005a). Identification of the control region for tissue-specific imprinting of the stimulatory G protein α-subunit. Proc. Natl. Acad. Sci. U.S.A. 102, 5513–5518.
Liu, J., Nealon, J. G., and Weinstein, L. S. (2005b). Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum. Mol. Genet. 14, 95–102.
Liu, J., Litman, D., Rosenberg, M. J., Yu, S., Biesecker, L. G., and Weinstein, L. S. (2000a). A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J. Clin. Invest. 106, 1167–1174.
Liu, J., Yu, S., Litman, D., Chen, W., and Weinstein, L. S. (2000b). Identification of a methylation imprint mark within the mouse Gnas locus. Mol. Cell. Biol. 20, 5808–5817.
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303.
Makedonski, K., Abuhatzira, L., Kaufman, Y., Razin, A., and Shemer, R. (2005). MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum. Mol. Genet. 14, 1049–1058.
Mantovani, G. (2011). Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J. Clin. Endocrinol. Metab. 96, 3020–3030.
Mantovani, G., de Sanctis, L., Barbieri, A. M., Elli, F. M., Bollati, V., Vaira, V., et al. (2010). Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of albright hereditary osteodystrophy and molecular analysis in 40 patients. J. Clin. Endocrinol. Metab. 95, 651–658.
Mapendano, C. K., Kishino, T., Miyazaki, K., Kondo, S., Yoshiura, K., Hishikawa, Y., et al. (2006). Expression of the Snurf-Snrpn IC transcript in the oocyte and its putative role in the imprinting establishment of the mouse 7C imprinting domain. J. Hum. Genet. 51, 236–243.
Mariot, V., Maupetit-Mehouas, S., Sinding, C., Kottler, M. L., and Linglart, A. (2008). A maternal epimutation of GNAS leads to Albright osteodystrophy and parathyroid hormone resistance. J. Clin. Endocrinol. Metab. 93, 661–665.
Maupetit-Mehouas, S., Mariot, V., Reynes, C., Bertrand, G., Feillet, F., Carel, J. C., et al. (2011). Quantification of the methylation at the GNAS locus identifies subtypes of sporadic pseudohypoparathyroidism type Ib. J. Med. Genet. 48, 55–63.
Meng, L., Person, R. E., and Beaudet, A. L. (2012). Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 21, 3001–3012.
Muscatelli, F., Abrous, D. N., Massacrier, A., Boccaccio, I., Le Moal, M., Cau, P., et al. (2000). Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 9, 3101–3110.
Ooi, S. K., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717.
Pauler, F. M., Barlow, D. P., and Hudson, Q. J. (2012). Mechanisms of long range silencing by imprinted macro non-coding RNAs. Curr. Opin. Genet. Dev. 22, 283–289.
Plagge, A., Isles, A. R., Gordon, E., Humby, T., Dean, W., Gritsch, S., et al. (2005). Imprinted Nesp55 influences behavioral reactivity to novel environments. Mol. Cell. Biol. 25, 3019–3026.
Plagge, A., Kelsey, G., and Germain-Lee, E. L. (2008). Physiological functions of the imprinted Gnas locus and its protein variants Gαs and XLαs in human and mouse. J. Endocrinol. 196, 193–214.
Rabinovitz, S., Kaufman, Y., Ludwig, G., Razin, A., and Shemer, R. (2012). Mechanisms of activation of the paternally expressed genes by the Prader-Willi imprinting center in the Prader-Willi/Angelman syndromes domains. Proc. Natl. Acad. Sci. U.S.A. 109, 7403–7408.
Richard, N., Abeguile, G., Coudray, N., Mittre, H., Gruchy, N., Andrieux, J., et al. (2012). A new deletion ablating NESP55 causes loss of maternal imprint of A/B GNAS and autosomal dominant pseudohypoparathyroidism type Ib. J. Clin. Endocrinol. Metab. 97, E863–E867.
Runte, M., Huttenhofer, A., Gross, S., Kiefmann, M., Horsthemke, B., and Buiting, K. (2001). The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 10, 2687–2700.
Sahoo, T., del Gaudio, D., German, J. R., Shinawi, M., Peters, S. U., Person, R. E., et al. (2008). Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 40, 719–721.
Skryabin, B. V., Gubar, L. V., Seeger, B., Pfeiffer, J., Handel, S., Robeck, T., et al. (2007). Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet. 3, e235. doi:10.1371/journal.pgen.0030235
Smallwood, S. A., and Kelsey, G. (2012). De novo DNA methylation: a germ cell perspective. Trends Genet. 28, 33–42.
Smallwood, S. A., Tomizawa, S., Krueger, F., Ruf, N., Carli, N., Segonds-Pichon, A., et al. (2011). Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 43, 811–814.
Smith, E. Y., Futtner, C. R., Chamberlain, S. J., Johnstone, K. A., and Resnick, J. L. (2011). Transcription is required to establish maternal imprinting at the Prader-Willi syndrome and Angelman syndrome locus. PLoS Genet. 7, e1002422. doi:10.1371/journal.pgen.1002422
Weinstein, L. S., Yu, S., Warner, D. R., and Liu, J. (2001). Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr. Rev. 22, 675–705.
Williamson, C. M., Ball, S. T., Dawson, C., Mehta, S., Beechey, C. V., Fray, M., et al. (2011). Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet. 7, e1001347. doi:10.1371/journal.pgen.1001347
Williamson, C. M., Ball, S. T., Nottingham, W. T., Skinner, J. A., Plagge, A., Turner, M. D., et al. (2004). A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat. Genet. 36, 894–899.
Williamson, C. M., Skinner, J. A., Kelsey, G., and Peters, J. (2002). Alternative non-coding splice variants of Nespas, an imprinted gene antisense to Nesp in the Gnas imprinting cluster. Mamm. Genome 13, 74–79.
Williamson, C. M., Turner, M. D., Ball, S. T., Nottingham, W. T., Glenister, P., Fray, M., et al. (2006). Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat. Genet. 38, 350–355.
Wroe, S. F., Kelsey, G., Skinner, J. A., Bodle, D., Ball, S. T., Beechey, C. V., et al. (2000). An imprinted transcript, antisense to Nesp, adds complexity to the cluster of imprinted genes at the mouse Gnas locus. Proc. Natl. Acad. Sci. U.S.A. 97, 3342–3346.
Yamasaki, K., Joh, K., Ohta, T., Masuzaki, H., Ishimaru, T., Mukai, T., et al. (2003). Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 12, 837–847.
Zazo, C., Thiele, S., Martin, C., Fernandez-Rebollo, E., Martinez-Indart, L., Werner, R., et al. (2011). Gsα activity is reduced in erythrocyte membranes of patients with psedohypoparathyroidism due to epigenetic alterations at the GNAS locus. J. Bone Miner. Res. 26, 1864–1870.
Keywords: genomic imprinting, non-coding RNA, Gnas, pseudohypoparathyroidism, Snrpn, Ube3a, Prader–Willi-syndrome, Angelman-syndrome
Citation: Plagge A (2012) Non-coding RNAs at the Gnas and Snrpn-Ube3a imprinted gene loci and their involvement in hereditary disorders. Front. Gene. 3:264. doi: 10.3389/fgene.2012.00264
Received: 30 August 2012; Paper pending published: 25 September 2012;
Accepted: 05 November 2012; Published online: 26 November 2012.
Edited by:
Peng Jin, Emory University School of Medicine, USAReviewed by:
Yi-Tao Yu, University of Rochester Medical Center, USAFei Li, Nanjing Agricultural University, China
Copyright: © 2012 Plagge. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Antonius Plagge, Department of Cellular and Molecular Physiology, Institute of Translational Medicine, University of Liverpool, Crown Street, Liverpool L69 3BX, UK. e-mail: a.plagge@liv.ac.uk