 Yi Zhang
Yi Zhang Min Chen
Min Chen Xiaohui Cheng1
Xiaohui Cheng1- 1School of Information Science and Engineering, Guilin University of Technology, Guilin, China
- 2School of Computer Science and Technology, Hunan Institute of Technology, Hengyang, China
- 3School of Pharmacy, Guilin Medical University, Guilin, China
Growing evidences have indicated that microRNAs (miRNAs) play a significant role relating to many important bioprocesses; their mutations and disorders will cause the occurrence of various complex diseases. The prediction of miRNAs associated with underlying diseases via computational approaches is beneficial to identify biomarkers and discover specific medicine, which can greatly reduce the cost of diagnosis, cure, prognosis, and prevention of human diseases. However, how to further achieve a more reliable prediction of potential miRNA–disease associations with effective integration of different biological data is a challenge for researchers. In this study, we proposed a computational model by using a federated method of combined multiple-similarities fusion and space projection (MSFSP). MSFSP firstly fused the integrated disease similarity (composed of disease semantic similarity, disease functional similarity, and disease Hamming similarity) with the integrated miRNA similarity (composed of miRNA functional similarity, miRNA sequence similarity, and miRNA Hamming similarity). Secondly, it constructed the weighted network of miRNA–disease associations from the experimentally verified Boolean network of miRNA–disease associations by using similarity networks. Finally, it calculated the prediction results by weighting miRNA space projection scores and the disease space projection scores. Leave-one-out cross-validation demonstrated that MSFSP has the distinguished predictive accuracy with area under the receiver operating characteristics curve (AUC) of 0.9613 better than that of five other existing models. In case studies, the predictive ability of MSFSP was further confirmed as 96 and 98% of the top 50 predictions for prostatic neoplasms and lung neoplasms were successfully validated by experimental evidences and supporting experimental evidences were also found for 100% of the top 50 predictions for isolated diseases.
Introduction
The microRNAs (miRNAs) widely found in eukaryotes are those non-coding RNAs of about 20–25 nucleotides (Iorio et al., 2005). Life processes such as cell growth (Fernando et al., 2012; Zhu et al., 2016), differentiation (Miska, 2005), proliferation (Cheng et al., 2005), aging (Xu et al., 2004), signal transduction (Carthew and Sontheimer, 2009), etc. have been found to be associated with miRNAs. Increasing evidences continually confirm that complex diseases in humans including cancers, Alzheimer, diabetes, and lymphoma are closely related to miRNAs. In addition, some former researches proved that miRNAs can be considered as tumor genes or tumor suppressor genes. Therefore, inferring novel miRNA–disease associations have clinical significance for various human diseases due to miRNAs' potential roles in diagnosis biomarkers and treatment targets. Massive associations have been obtained via traditional biotic experiments and stored in some public databases. The traditional bio-experimental methods have high precision, but whose process is complex and time-consuming (Liang et al., 2019). Predicting and ranking potential miRNA–disease associations effectively and rapidly via computational identification methods are extremely vital to speed up the bio-experimental validation processes as well as reduce the blindness and time consumption of bio-experiments (Chen et al., 2015c, 2019c; Zeng et al., 2016b; Peng et al., 2017a, 2020).
On the basic assumption that functionally related miRNAs tend to be associated with phenotypically similar diseases and vice versa (Lu et al., 2008; Bandyopadhyay et al., 2010; Wang et al., 2010), various computational identification methods have been proposed continuously (Chen et al., 2017d, 2018c; Chen and Qu, 2018). Jiang et al. (2010a) proposed a miRNA–disease association prediction model that first used the hypergeometric distribution and constructed the functionally related miRNA network through the number of shared target genes to uncover the associations between miRNAs and diseases, but it needs to integrate other bioinformatics sources to improve model performance. Jiang et al. (2010b) proposed an approach that prioritized disease-related miRNAs based on integrating genomic data. Li et al. (2011) proposed a computational framework with which to prioritize human cancer-related miRNAs; it used the functional consistency score of miRNA-target genes and cancer-related genes to measure the associations between cancer and miRNAs. Xu et al. (2014) systematically prioritized disease-specific miRNAs by using the known disease genes and context-dependent miRNA-target interactions derived from the expression data of a matched miRNA–miRNA pair. Lack of excellent predictive performance of the above-mentioned methods may be attributed to the high false positive rate of the target genes.
Li J. et al. (2014) utilized recommendation systems to predict the associations between environmental factors, miRNAs, and diseases, but these cannot predict isolated diseases (without any known associated miRNAs) and new miRNAs (without any known associated diseases). Zhang Y. et al. (2019) used bipartite network projection (LSGSP) with known associations to reconstruct the family information, miRNA similarity network, and disease similarity network for predicting the potential miRNA–disease associations. Although LSGSP does not need negative samples, it cannot achieve good performance only with limited number of known associations. Chen et al. (2018h) proposed a bipartite recommendation algorithm to predict miRNA–disease associations (BNPMDA) that improved the prediction accuracy distinctly with the utilization of bias ratings. Chen et al. (2018b) proposed a novel information diffusion method based on network consistency (IDNC) for uncovering disease-related miRNAs. Despite not needing negative samples and simple algorithm design, too many parameters in different databases make IDNC take a long time to find the optimal values.
In recent years, some researchers have attempted to use the topological similarity of graph to predict a miRNA–disease association (Nalluri et al., 2015; Chen et al., 2016b, 2017c, 2018e; Sun et al., 2016; You et al., 2017; Zeng et al., 2018). Chen et al. (2017b) proposed the super-disease and miRNA concepts to design a novel computational model with which to infer miRNA–disease associations. Bipartite heterogeneous network method based on co-neighbor (Chen et al., 2019a), ELLPMDA of ensemble learning and link prediction (Chen et al., 2018j), and label propagation model with linear neighborhood (Li et al., 2018) were used for various types of miRNA–disease association prediction, but those did not figure out the easy way for parameter optimization. Random walk on heterogeneous network (Chen et al., 2012, 2016a, 2018a; Xuan et al., 2015; Liu et al., 2017; Luo and Xiao, 2017; Mugunga et al., 2017; Peng et al., 2018) used for inferring miRNA–disease associations has achieved excellent prediction results with global attributes, but all of their results were partial to such miRNAs that have more known associations with diseases.
Inspired by the successful application of machine learning methods in the field of bioinformatics, many researchers used supervised machine learning methods to predict a miRNA–disease association (Chen et al., 2015a,b, 2017a, 2018d,f, 2019a,b,d; Luo et al., 2017a; Xuan et al., 2018, 2019b; Wang C.-C. et al., 2019; Wang L. et al., 2019; Zhang L. et al., 2019; Zhao et al., 2019), but which need negative samples for training. Because it is hard to obtain the experimentally verified less-known miRNA–disease associations and negative samples, some semi-supervised learning approaches (such as regularized least squares) with remarkable prediction results were proposed (Chen and Huang, 2017; Chen et al., 2017c, 2018k; Peng et al., 2017b; Xu et al., 2019). Chen and Huang (2017) used Laplacian regularized sparse subspace learning for miRNA–disease association prediction (LRSSLMDA); it projected diverse statistical feature profiles into a common subspace and selected important diverse features with a L1-norm constraint. Jiang et al. (2018) proposed a novel similarity kernel fusion (SKF) method that integrated multiple-similarity kernels to construct the accurate network similarity on which to utilize Laplacian regularized least squares for potential associations inference. It can avoid to lose the initial information during the process and can eliminate some noises in integrated similarity kernels. Luo et al. (2017b) presented a semi-supervised method with Kronecker regularized least squares to predict the potential (or missing) miRNA–disease associations. However, the above semi-supervised solutions without the need for negative samples still have the limitation in initial values setting and optimal parameters of iteration selecting. Zeng et al. (2016a), Li et al. (2017), Chen et al. (2018g), Xiao et al. (2018), Xuan et al. (2019a), Xuan et al. (2019c), and Peng et al. (2017b) utilized the matrix completion to infer the potential miRNA–disease associations. Chen et al. (2018i) uncovered the potential miRNA–disease associations through integrating low-rank matrix decomposition and the sparse learning method. Qu et al. (2019) utilized matrix decomposition and label propagation to infer potential miRNA–disease associations. Tang et al. (2019) made full use of the miRNA functional similarity, the disease semantic similarity, and a dual Laplacian regularization term to work for the matrix completion of miRNA–disease associations. Chen et al. (2020) proposed a new computational model (NCMCMDA) that innovatively integrated neighborhood constraint with matrix completion to find out the absent miRNA–disease associations. Even though all of the above methods only needed experimentally validated miRNA–disease associations to make prediction with a good prediction effect, the optimal parameters selection still cannot be solved very well. Although computational prediction models have attracted a lot of interests in recent years and many distinguished research progresses have been achieved, the identification of the potential associations between miRNAs and diseases still remains to involve a large number of unclear and incomplete works that need to be further improved:
(1) The prediction accuracy still needs to be enhanced further;
(2) Isolated diseases and new miRNAs cannot be handled directly;
(3) Similarity construction processes are not accurate enough.
Around the above limitations, a global prediction method (MSFSP) that combined with multiple-similarities fusion (MSF) attribute was proposed, which could predict the associations between all diseases (including isolated diseases) and miRNAs (including new miRNAs) without needing negative samples. MSFSP mainly consisted of the following steps:
(1) Reconstructed disease similarity network (fused by disease semantic similarity, disease functional similarity, and disease Hamming similarity) and miRNA similarity network (fused by miRNA functional similarity, miRNA sequence similarity, and miRNA Hamming similarity);
(2) Reconstructed miRNA–disease network via integrated disease similarity, miRNA similarity, and verified Boolean network of miRNA–disease associations;
(3) Obtained the final prediction scores of miRNA–disease associations by using the space projections of reconstructed miRNA–disease network on similarities spaces.
Materials and Methods
Known MiRNA–Disease Associations
The experimentally verified miRNA–disease associations downloaded from HMDD v2.0 (Li Y. et al., 2014) with pre-treatment were composed of 495 processed miRNAs (formed a collection of miRNAs M = {m1, m2, …, mi, …, mnm}, nm=495), 383 diseases (formed a collection of diseases D = {d1, d2, …dj, …dnd}, nd=383), and 5,430 known miRNA–disease associations (formed a matrix ). The element value of MD(i, j) in is set to 1 if the miRNA node mi (i = 1, 2, ⋯nm) is associated with the disease node dj (j = 1, 2, …nd); otherwise, it is set to 0.
Disease Semantic Similarity and Disease Functional Similarity
According to the description in Wang et al. (2010), disease similarities based on semantic information were denoted by matrix ; it can be calculated via utilizing the arborescence attribute of disease in the MeSH database (Lowe and Barnett, 1994) where every disease node was marked in directed acyclic graph. Two diseases have more similar phenotypes when they associate with the same genes, based on which many researchers used the disease–gene associations to calculate disease functional similarity (Luo et al., 2017b; Jiang et al., 2018). As described in detail in Jiang et al. (2018), disease functional similarities were denoted by the matrix .
MiRNA Functional Similarity and MiRNA Sequence Similarity
miRNA–miRNA functional similarities were downloaded from Wang et al. (2010), and the pairwise miRNA functional similarities were denoted by the matrix . The miRNA sequence similarities obtained from the miRBase database (Kozomara and Griffiths-Jones, 2013) were denoted by the matrix .
Hamming Similarity
Hamming similarity for vectors is a function that measures the number of equal components, divided by the length of vectors (Charikar, 2002). It is known that diseases with similar phenotypes are often related to similar miRNAs. Thereby, we defined disease Hamming similarity (denoted by the matrix ), whose element value is shown as follows:
where DDhs(i, j) represents the Hamming similarity between disease node di and dj.
Similarly, we used MDT that denoted the transposed matrix of MD to define miRNA Hamming similarity (denoted by matrix ). The corresponding element value in is shown as follows:
where MMhs(i, j) represents the Hamming similarity between miRNA nodes mi and mj.
Multiple-Similarities Fusion
In this section, we used similarity kernel fusion (Wang et al., 2014; Jiang et al., 2018, 2019) to integrate three miRNA similarities (miRNA functional similarities , miRNA sequence similarities , and miRNA Hamming similarities ) into one matrix that represented integrated miRNA similarities and three disease similarities (disease functional similarities , disease semantic similarities , and disease Hamming similarities ) into one matrix that represented integrated disease similarities. Details on the integration are in the following discussion.
Firstly, using similar methods mentioned in Jiang et al. (2018, 2019), the corresponding sparse matrices for three miRNA similarities denoted by , , and , respectively, were constructed and the corresponding sparse matrices for three disease similarities were denoted by , , and , respectively.
where Nmi represents the collection of all neighbors of miRNA node mi, including mi in the corresponding three miRNA similarities matrices (MMsfs, MMsss, and MMshs), and the number of Nmi was set to 36.
Similarly, MMsss(i, j), MMshs(i, j), DDsfs(i, j), DDsss(i, j), and DDshs(i, j) were constructed by using the above representation.
Secondly, the integrated normalized matrices and sparse matrices are as follows:
where and are the normalizations for MMss and MMhs, respectively. denotes the transposed matrix of MMsfs; and are the tth iteration results of and , respectively. t was set to 10 and δ was set to 0.1, which are similar as those defined in Jiang et al. (2018, 2019). and represented the initial status of and , respectively, with the detailed calculation shown as follows:
Furthermore, similar representations of and could be obtained as that of . After t + 1 iterations, the temporarily integrated miRNA similarity denoted by matrix MMis was calculated as follows:
Thirdly, a weighted matrix for eliminating noises during the calculation process was constructed, as mentioned in Jiang et al. (2019). Then, the finally integrated miRNA similarity denoted by matrix was obtained via taking a dot product:
The finally integrated disease similarity matrix , the weighted matrix , and the temporarily integrated disease similarity matrix can be calculated by a similar calculation process as that of :
Weighted Network Construction
On account of the hypothesis that miRNAs with similar functions are often related to the diseases with similar phenotypes, many methods for miRNA–disease association prediction were proposed. Though the network MD of known experimentally verified miRNA–disease association plays a very important role in these prediction methods, network MD is only a Boolean network which can indicate if the miRNA–disease association exits or not, without any information of the extent of association. Therefore, in order to enhance the predictive validity, we used MD and similarities between miRNAs (diseases) to accurately construct a weighted network with which to uncover potential miRNA–disease associations.
Weighted Network Construction Based on MiRNA Similarities
The contribution value of the other miRNA node mk(k ≠ i) to mi (denoted by Cmk) was defined as follows:
where is the finally integrated miRNA similarity between mi and mk, and MD(k, j) represents the Boolean value of the association between mk and dj.
If there is an association between mk and dj, the more similar mk and mi are, the higher the contribution value of mk to the weight between mi and dj. Based on the discussion above, the miRNA–disease weighted network based on miRNA similarities (denoted by ) was defined as follows:
where MDm(i, j) is the weight between miRNA node mi and disease node dj, the equilibrium parameter being α ∈ [ 0, 1].
Weighted Network Construction Based on Disease Similarities
Similarly, the contribution value of the other disease nodes dk(k ≠ i) to di (denoted by Cdk) was defined as follows:
miRNA–disease weighted network based on disease similarities (denoted by ) was defined as follows:
where equilibrium parameter β ϵ [0,1].
Space Projection Scores Based on Similarities
To enhance the predictive accuracy further, we integrated and to construct miRNA space projection scores denoted by matrix , shown as follows:
where is the transposed matrix of MDd, and ||MMis*(:, j)|| is the norm of vector MMis*(:, j).
Similarly, we integrated and to construct disease space projection scores denoted by matrix , shown as follows:
where is the norm of vector .
Finally, we integrated Fpm(i, j) and Fpd(i, j) to obtain the final prediction score Fpf(i, j), shown as follows:
where is the transposed of matrix of Fpm, and the equilibrium parameter γ ∈ [0, 1] represents the importance degree of Fpm(i, j) and Fpd(i, j).
Therefore, we will integrate disease similarities, miRNA similarities, and weighted networks to obtain the final prediction scores , whose higher value means a higher probability that miRNA mi associates with disease dj. The detailed calculation steps of Fpf are shown in Figure 1 for clarity.
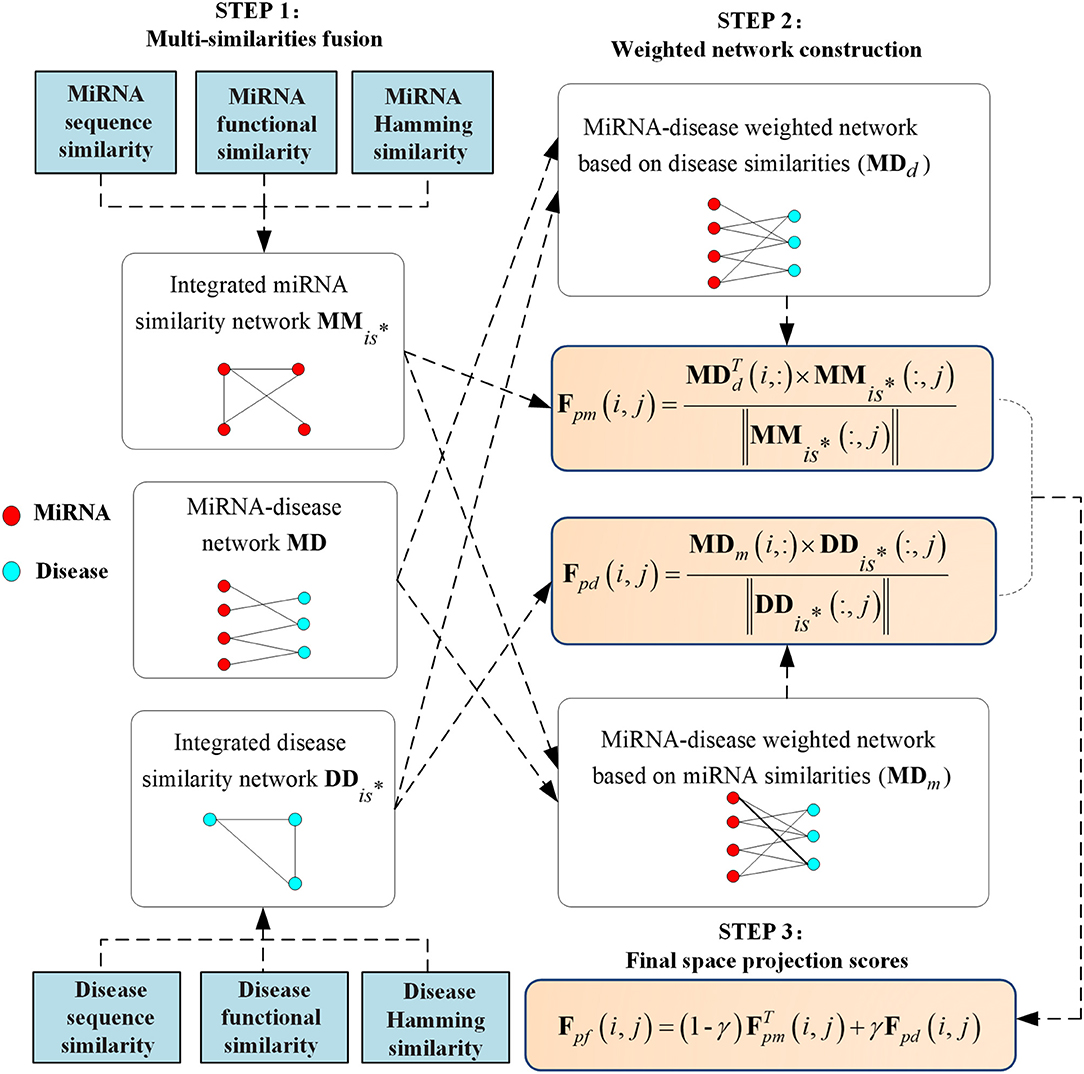
Figure 1. Flowchart of the whole modeling procedure.
Results
Influence of Parameter Selection on Performance
This section mainly discussed the influences of different types of parameters (weighting parameter α, β and equilibrium parameter γ) on the predictive performance of MSFSP. For simplicity, we set α and β to be of the same value.
Firstly, we fixed γ to 0.5 and changed α and β from 0 to 1 with a step-size of 0.1. After performing LOOCV, the results showed that AUC reached an optimal value of 0.9577 when α and β were set to 0.1. Then, the AUC values decreased gradually when α and β increased from 0.1 to 1, which caused the corresponding curve to decline linearly (shown in Figure 2). Therefore, α and β should range from 0 to 0.2 to get the optimal value.
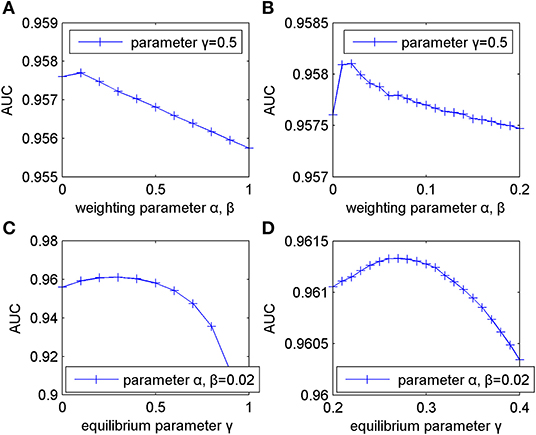
Figure 2. Influence of parameter variation on model predictive accuracy. (A) Changing weighting parameters from 0 to 1 with step-size of 0.1. (B) Changing weighting parameters from 0 to 0.2 with step-size of 0.01. (C) Changing equilibrium parameter from 0 to 1 with step-size of 0.1. (D) Changing equilibrium parameter from 0.2 to 0.4 with step-size of 0.01.
Next, in order to get more accurate weighting parameters, we fixed γ to 0.5 again and changed α and β from 0 to 0.2 with a step-size of 0.01. The corresponding changing curve is shown in Figure 2, where the optimal AUC of 0.9581 was obtained when α and β were both 0.02.
Then, based on α = β = 0.02, we evaluated the influence of γ on MSFSP in a similar way as detailed above. We increased γ from 0 to 1 with a step-size of 0.1 to obtain the corresponding results shown in Figure 2, where the optimal, suboptimal, and third-best value of AUC were obtained when γ was 0.3, 0.2, and 0.4, respectively. However, AUC decreased when γ increased from 0.4. Therefore, γ should range from 0.2 to 0.4 to get the optimal value. We increased γ from 0.2 to 0.4 with a step-size of 0.01 to get more accurate parameter values with α and β fixed to 0.02. The changing curve in Figure 2 shows the optimal value of 0.9613 when γ was 0.27.
In conclusion, our parameter selections were α = β = 0.02 and γ = 0.27.
Comparison of Predictive Performance Under Different Situations
We performed LOOCV to evaluate the predictive performance of MSFSP under the following different situations: (1) with all relevant information (MSFSP with all), (2) only with miRNA space projection (MSFSP with MSP), and (3) only with disease space projection (MSFSP with DSP). The ROC curves for the above different situations are shown in Figure 3, where the AUC value of MSFSP withr all was 0.9613, the AUC value of MSFSP with MSP was 0.9570, and the AUC value of MSFSP with DSP was 0.8489. Therefore, MSFSP showed reliable predictive performance for inferring miRNA–disease associations effectively.
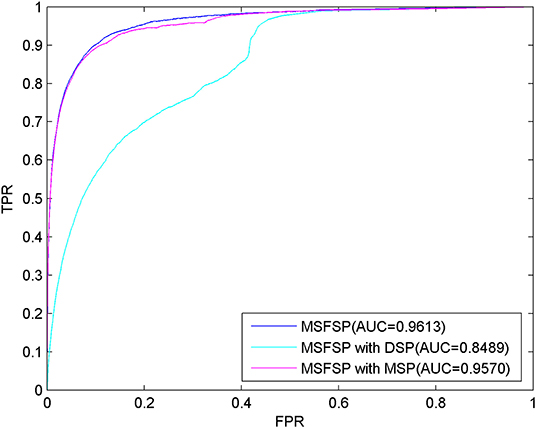
Figure 3. Receiver operating characteristic curves and area under the curve values via leave-one-out cross-validation in different situations.
Comparison of Predictive Performance With Different Integrated Similarity Constructions
Concerning the limitations of the sparsity and incompleteness existing in disease semantic similarity and miRNA functional similarity, we used MSF in MSFSP to construct the integrated disease similarity and the integrated miRNA similarity with which to solve these limitations. Some other researchers integrated disease semantic similarity (miRNA functional similarity) with Gaussian interaction profile kernel similarity to construct the integrated diseases similarity (the integrated miRNA similarity) with which to solve the same limitations (Chen et al., 2016c, 2017d; Chen and Huang, 2017; Zhao et al., 2018, 2019). In order to compare which of the two ways wherein integrated similarities were constructed has better predictive result, we compared MSF used in MSFSP with Gaussian interaction profile kernel similarity used in Chen and Huang (2017) and Zhao et al. (2019). We used the Gaussian interaction profile kernel similarity coming from Chen and Huang (2017) to replace MSF in MSFSP, and we got a new prediction model called GIPKS1SP as one object to be compared. Similarly, we used Gaussian interaction profile kernel similarity coming from Zhao et al. (2019) to replace MSF in MSFSP, and we got another new prediction model called GIPKS2SP as another object to be compared. After performing LOOCV, the AUCs of GIPKS1SP, GIPKS2SP, and MSFSP were 0.9179, 0.9212, and 0.9613, respectively (shown in Figure 4). The more reliable predictive performance obtained via MSFSP proved that MSF is better than the Gaussian interaction profile kernel similarity to construct the integrated similarities.
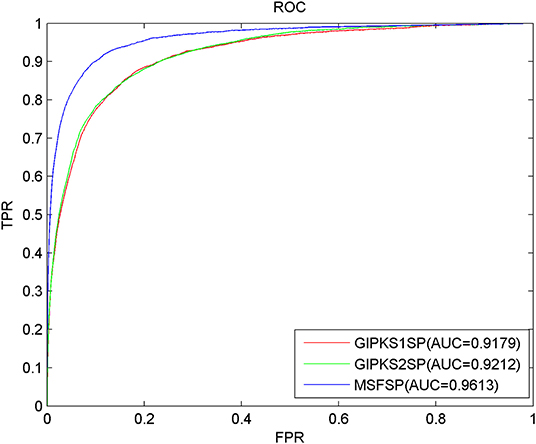
Figure 4. Receiver operating characteristic curves and area under the curve values of MSFSP against GIPKS1SP and GIPKS2SP.
Comparison to Other Methods
To our knowledge, BNPMDA (Chen et al., 2018h), MDHGI (Chen et al., 2018i), NSEMDA (Wang C.-C. et al., 2019), RFMDA (Chen et al., 2018f), and SNMFMDA (Zhao et al., 2018) are the most advanced prediction methods in inferring miRNA–disease associations so far. Due to the fact that the databases used by these five methods are similar with that of MSFSP, we compared MSFSP with these five methods on the predictive performance. The LOOCV results in Figure 5 show that the AUC values of BNPMDA, MDHGI, NSEMDA, RFMDA, SNMFMDA, and MSFSP were 0.9028, 0.8945, 0.8899,0.8891, 0.9007, and 0.9613, respectively. MSFSP achieved the superior prediction effect, at 6.09, 6.94, 7.42, 7.51, and 6.30% higher than BNPMDA, MDHGI, NSEMDA, RFMDA, and SNMFMDA, respectively.
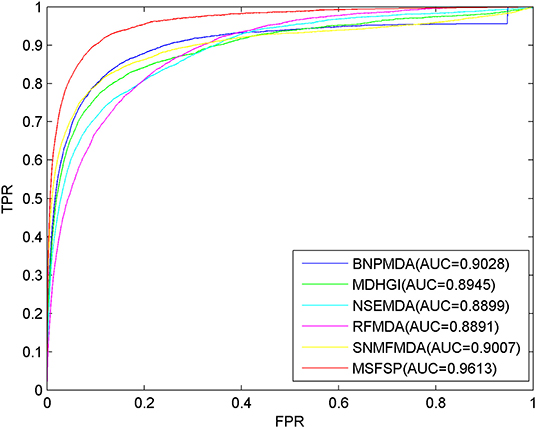
Figure 5. Receiver operating characteristic curves and area under the curve values of multiple-similarities fusion and space projection against other five methods.
Prediction of New MiRNAs and Isolated Diseases
With the continuously developing miRNA recognition technology, more and more miRNAs are being discovered, but whose associations with diseases are unknown. The prediction for isolated diseases and new miRNAs will definitely accelerate the scientists' understanding of the molecular mechanisms of diseases as well as how diseases occur. Therefore, the prediction for isolated diseases and new miRNAs has become a hot research topic in recent years.
For each miRNA, we removed all related associations with diseases to simulate the new miRNA. For each disease, we removed all related associations with miRNAs to simulate the isolated disease. Through LOOCV, the prediction results shown as AUC of 0.9493 and 0.8412, respectively, were obtained, where the ROC curve demonstrated the excellent predictive performance of MSFSP on inferring new miRNA-related diseases, as well as isolated diseases related with miRNAs (as can be seen in Figure 6).
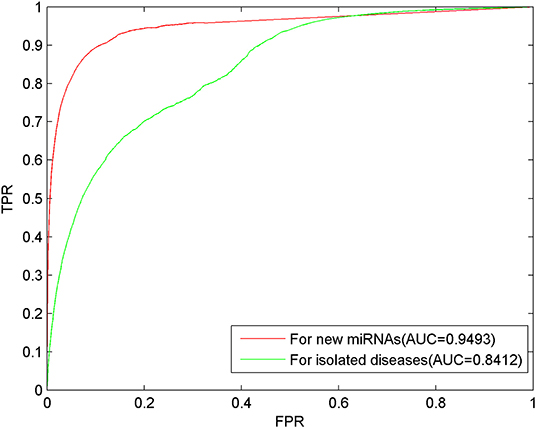
Figure 6. Predictions of new miRNAs and isolated diseases.
Case Studies
To further evaluate the predictive ability of MSFSP on inferring diseases potentially related to miRNAs, we selected prostatic neoplasms and lung neoplasms as the case studies with model training and predicting on three independent databases HMDD v3.2 (Huang et al., 2018), dbDEMC 2.0 (Yang et al., 2017), and miR2Disease (Jiang et al., 2009).
Prediction of Potential MiRNA–Disease Associations
The low detection rate of lung neoplasm, making it a common lethal disease, poses a great threat to people's lives especially in developing countries (Torre et al., 2016; Temraz et al., 2017). We used 132 known associations between lung neoplasms and miRNAs as training samples to predict the remaining unknown associations. We found the supporting evidences that 49 out of all the first 50 miRNAs related to lung neoplasms predicted by MSFSP were confirmed on the above-mentioned three databases (HMDD v3.2, dbDEMC 2.0, and miR2Disease), except hsa-mir-384 (as shown in Table 1). However, we found the association between hsa-mir-384 and lung neoplasms by searching the latest literature (Guo et al., 2019) whose publication date was after the last update of HMDD v3.2, which further confirmed the effectiveness of MSFSP for inferring diseases potentially related to miRNAs.
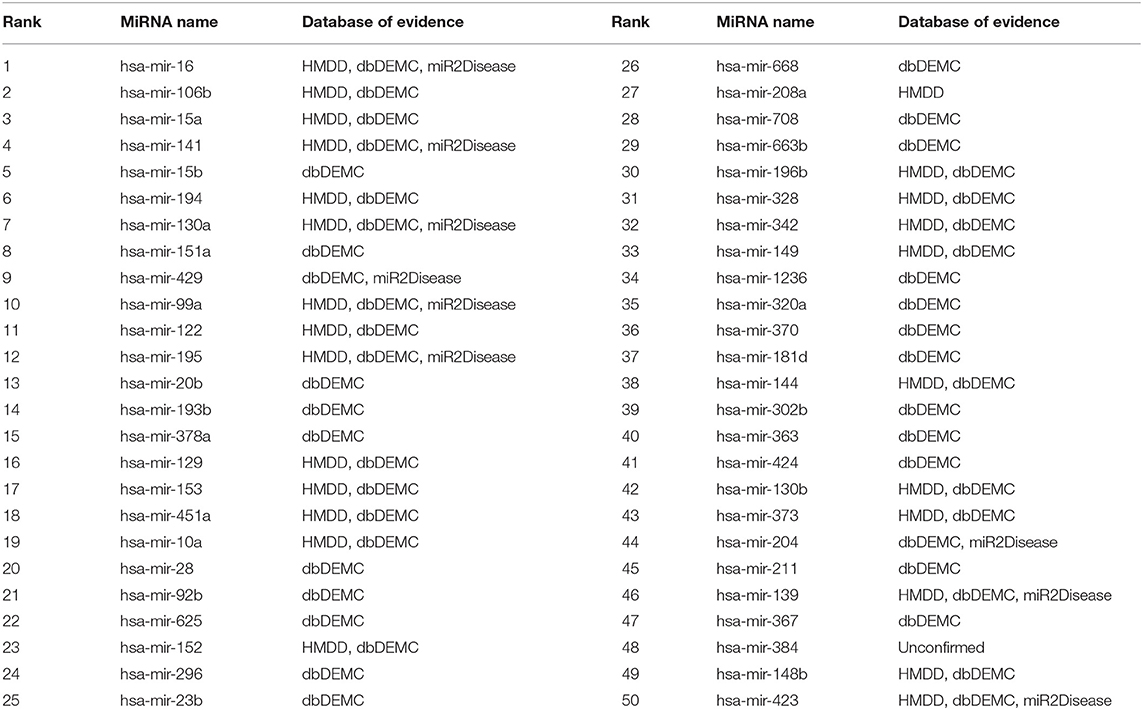
Table 1. Top 50 lung neoplasm-related miRNAs.
Prostatic neoplasm is a disease occurring in the male reproductive system, especially common in countries with severely aging population, but in recent years, more and more prostatic neoplasms occur in young people (Siegel et al., 2016). We used 118 known associations between prostatic neoplasms and miRNAs as training samples to predict the remaining unknown associations. A total of 48 out of the first 50 miRNAs related to prostatic neoplasms predicted by MSFSP were confirmed on relevant databases (HMDD v3.2, dbDEMC 2.0, and miR2Disease), except hsa-mir-633 and hsa-mir-300 (ranked 46th and 47th, respectively) (as shown in Table 2). Although there is no evidence that shows the association between these two miRNAs and prostatic neoplasms by now, we believe that some evidences will be found by scientists in the near future.
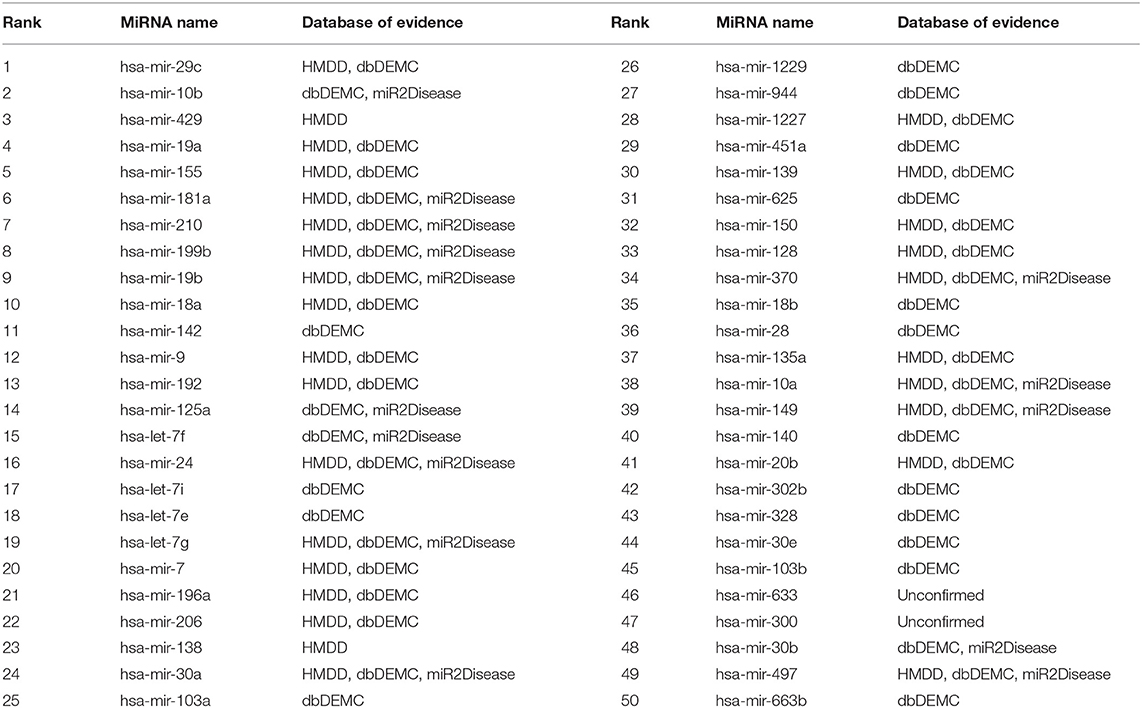
Table 2. Top 50 prostatic neoplasm-related miRNAs.
Prediction of Isolated Disease-Related MiRNAs
To further evaluate the predictive performance of MSFSP for isolated diseases which are those without any known associations, we removed all 118 known associations related to prostatic neoplasms to simulate the isolated disease condition. The supporting evidences for the top 50 prostatic neoplasm-related miRNAs predicted were all found from the relevant databases (HMDD v3.2, dbDEMC 2.0, and miR2Disease) (as shown in Table 3). Similarly, we removed all 132 known associations related to lung neoplasms to simulate the isolated disease condition. The supporting evidences on the top 50 lung neoplasm-related miRNAs predicted were all found from the above-mentioned three relevant databases (as shown in Table 4). The supporting evidences confirmed that the predictive accuracy for the above two simulated objects were both 100%, which further showed the excellent predictive performance of MSFSP on inferring diseases potentially related to miRNAs and isolated diseases related to miRNAs.
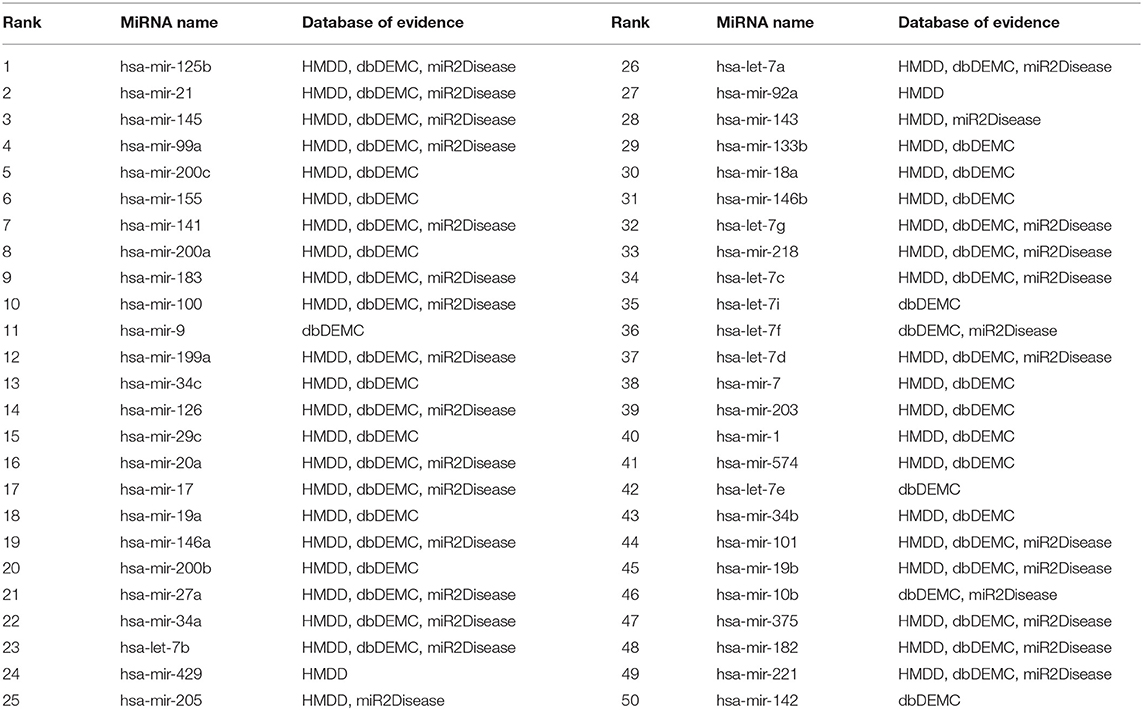
Table 3. Top 50 isolated disease-related miRNAs (prostatic neoplasm as a case).
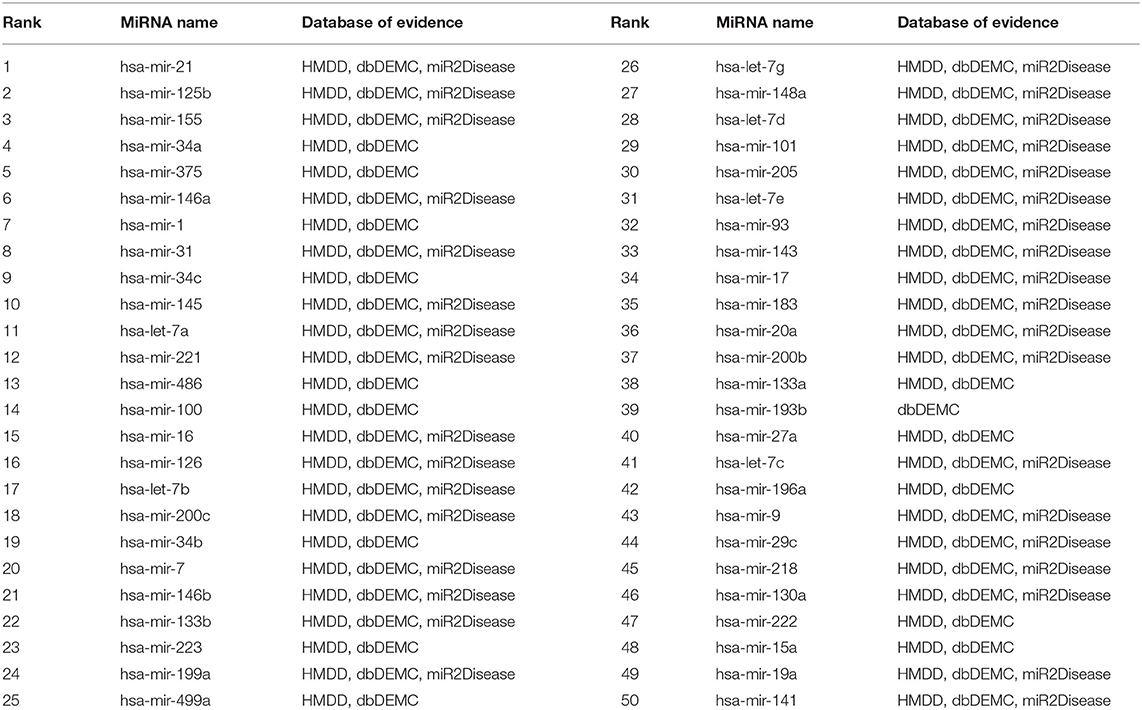
Table 4. Top 50 isolated disease-related miRNAs (lung neoplasm as a case).
Discussion and Conclusion
Considering that the identification of complex disease-related miRNAs is still a key research topic in the bio-medical field, we proposed a computational model called MSFSP that made the following contributions for the identification of miRNA–disease associations: (1) Compared to other methods, MSFSP can enhance the predictive accuracy effectively with an AUC value of 0.9613, which is higher than those of the other current classical computational models; (2) MSFSP implements prediction without needing negative samples; (3) MSFSP solved the inherent limitations of sparsity and incompleteness existing in current datasets via multiple similarities fusion; (4) MSFSP can be used to infer new miRNAs and isolated diseases, with AUC values of 0.9493 and 0.8412, respectively; (5) The predicted top 50 results for prostatic neoplasms and lung neoplasms as two cases agree well with the supporting evidences found in HMDD v3.2, dbDEMC 2.0, and miR2Disease, with the consistency of 98 and 96% respectively; (6) The predicted top 50 results for the isolated diseases simulated agree well with the supporting evidences found in HMDD v3.2, dbDEMC 2.0, and miR2Disease, with the consistency of 100% for both.
The reliable performance of MSFSP achieved can be attributed to the following factors: (1) Different biological information data were fused in MSFSP to construct the integrated miRNA similarity network and the integrated disease similarity network; (2) More accurate miRNA–disease correlations were described by weighted networks that were integrated with the disease similarity network, the miRNA similarity network, and the experimentally verified Boolean network of miRNA–disease associations; (3) MiRNA space projection scores and disease space projection scores were combined to obtain the final prediction scores, which avoided the invalid inference for new miRNAs only with disease space projection scores and the invalid inference for isolated diseases only with miRNA space projection scores.
MSFSP still has some limitations which need to be improved in the future besides its excellent prediction results. Firstly, during miRNA similarity and disease similarity calculation, the known miRNA–disease associations demand extra increase in some amount of overhead because the similarity calculation needs to be redone in LOOCV. Secondly, the construction of miRNA similarity network and disease similarity network is not accurate enough, although the accuracy has been somewhat enhanced by integrating various information. Furthermore, MSFSP can only predict if an association between miRNA and a disease exists or not, but not the specific regulatory mechanism.
Data Availability Statement
All datasets generated for this study are included in the article/supplementary material.
Author Contributions
YZ and MC conceived the concept of the work and designed the experiments and wrote the paper. MC, YZ, XC, and HW performed the literature search. MC, YZ, and XC collected and analyzed the data. All authors have approved the manuscript.
Funding
The research of this paper has been sponsored by the National Nature Science Foundation of China (Grant Nos. 61772192, 61672223, 61662017, and 61762031), the Nature Science Foundation of Hunan Province, China (Grant Nos. 2018JJ2085 and 2018JJ40064), the Scientific Research Project of the Education Department of Hunan Province, China (19A125), major cultivation projects of Hunan Institute of Technology (Grant No. 2017HGPY001), and Guangxi Key Laboratory Fund of Embedded Technology and Intelligent System (Guilin University of Technology).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bandyopadhyay, S., Mitra, R., Maulik, U., and Zhang, M. Q. (2010). Development of the human cancer microRNA network. Silence 1:6. doi: 10.1186/1758-907X-1-6
Carthew, R. W., and Sontheimer, E. J. (2009). Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655. doi: 10.1016/j.cell.2009.01.035
Charikar, M. S. (2002). “Similarity estimation techniques from rounding algorithms,” in Proceedings of the Proceedings of the thiry-fourth annual ACM symposium on Theory of computing. (Montreal, QC: ACM). doi: 10.1145/509907.509965
Chen, M., He, X., Duan, S., and Deng, Y. (2017a). A Novel Gene Selection Method Based on Sparse Representation and Max-Relevance and Min-Redundancy. Comb. Chem. High Throughput Screen 20, 158–163. doi: 10.2174/1386207320666170126114051
Chen, M., Li, Z., Zhang, Y., Chen, X., and Li, A. (2015a). A multiple platform based method for data integration. J. Comput. Theor. Nanosci. 12, 4890–4894. doi: 10.1166/jctn.2015.4457
Chen, M., Liao, B., and Li, Z. (2018a). Global similarity method based on a two-tier random walk for the prediction of microRNA–disease association. Sci Rep. 8:6481. doi: 10.1038/s41598-018-24532-7
Chen, M., Lu, X., Liao, B., Li, Z., Cai, L., and Gu, C. (2016a). Uncover miRNA-Disease Association by Exploiting Global Network Similarity. PLoS ONE 11:e0166509. doi: 10.1371/journal.pone.0166509
Chen, M., Peng, Y., Li, A., Li, Z., Deng, Y., Liu, W., et al. (2018b). A novel information diffusion method based on network consistency for identifying disease related microRNAs. RSC Adv. 8, 36675–36690. doi: 10.1039/C8RA07519K
Chen, M., Zhang, Y., Li, A., Li, Z., Liu, W., and Chen, Z. (2019a). Bipartite heterogeneous network method based on Co-neighbour for MiRNA–disease association prediction. Front. Genet. 10:385. doi: 10.3389/fgene.2019.00385
Chen, M., Zhang, Y., Li, Z., Li, A., Liu, W., Liu, L., et al. (2019b). A novel gene selection algorithm based on sparse representation and minimum-redundancy maximum-relevancy of maximum compatibility center. Curr. Proteomics. 16, 374–382. doi: 10.2174/1570164616666190123144020
Chen, M., Zhong, M., Li, Z., Li, X., and Li, A. (2015b). A novel method based on greedy algorithm for informative SNP selection. J. Comput. Theor. Nanosci. 12, 4036–4042. doi: 10.1166/jctn.2015.4316
Chen, X., Cheng, J.-Y., and Yin, J. (2018c). Predicting microRNA-disease associations using bipartite local models and hubness-aware regression. RNA Biol. 15, 1192–1205. doi: 10.1080/15476286.2018.1517010
Chen, X., Gong, Y., Zhang, D. H., You, Z. H., and Li, Z. W. (2018d). DRMDA: deep representations-based miRNA–disease association prediction. J. Cell. Mol. Med. 22, 472–485. doi: 10.1111/jcmm.13336
Chen, X., Guan, N. N., Li, J. Q., and Yan, G. Y. (2018e). GIMDA: graphlet interaction-based MiRNA-disease association prediction. J. Cell. Mol. Med. 22, 1548–1561. doi: 10.1111/jcmm.13429
Chen, X., and Huang, L. (2017). LRSSLMDA: laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput. Biol. 13:e1005912. doi: 10.1371/journal.pcbi.1005912
Chen, X., Huang, L., Xie, D., and Zhao, Q. (2018k). EGBMMDA: extreme gradient boosting machine for MiRNA-disease association prediction. Cell Death Dis. 9:3. doi: 10.1038/s41419-017-0003-x
Chen, X., Jiang, Z. C., Xie, D., Huang, D. S., Zhao, Q., Yan, G. Y., et al. (2017b). A novel computational model based on super-disease and miRNA for potential miRNA-disease association prediction. Mol. Biosyst. 13, 1202–1212. doi: 10.1039/C6MB00853D
Chen, X., Liu, M.-X., and Yan, G.-Y. (2012). RWRMDA: predicting novel human microRNA–disease associations. Mol. Biosyst. 8, 2792–2798. doi: 10.1039/c2mb25180a
Chen, X., Niu, Y. W., Wang, G. H., and Yan, G. Y. (2017c). HAMDA: hybrid approach for MiRNA-disease association prediction. J. Biomed. Inform. 76, 50–58. doi: 10.1016/j.jbi.2017.10.014
Chen, X., and Qu, J. (2018). TLHNMDA: triple layer heterogeneous network based inference for MiRNA-disease association prediction. Front. Genet. 9:234. doi: 10.3389/fgene.2018.00234
Chen, X., Sun, L.-G., and Zhao, Y. (2020). NCMCMDA: miRNA–disease association prediction through neighborhood constraint matrix completion. Brief. Bioinform. 12:bbz159. doi: 10.1093/bib/bbz159
Chen, X., Wang, C.-C., Yin, J., and You, Z.-H. (2018f). Novel human miRNA-disease association inference based on random forest. Mol Therapy-Nucleic Acids. 13, 568–579. doi: 10.1016/j.omtn.2018.10.005
Chen, X., Wang, L., Qu, J., Guan, N.-N., and Li, J.-Q. (2018g). Predicting miRNA–disease association based on inductive matrix completion. Bioinformatics 34, 4256–4265. doi: 10.1093/bioinformatics/bty503
Chen, X., Wu, Q.-F., and Yan, G.-Y. (2017d). RKNNMDA: ranking-based KNN for MiRNA-disease association prediction. RNA Biol. 14, 952–962. doi: 10.1080/15476286.2017.1312226
Chen, X., Xie, D., Wang, L., Zhao, Q., You, Z.-H., and Liu, H. (2018h). BNPMDA: bipartite network projection for MiRNA–disease association prediction. Bioinformatics 34, 3178–3186. doi: 10.1093/bioinformatics/bty333
Chen, X., Xie, D., Zhao, Q., and You, Z.-H. (2019c). MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 20, 515–539. doi: 10.1093/bib/bbx130
Chen, X., Yan, C. C., Xu, Z., You, Z. H., Yuan, H., and Yan, G. Y. (2016b). HGIMDA: heterogeneous graph inference for miRNA-disease association prediction. Oncotarget 7, 65257–65269. doi: 10.18632/oncotarget.11251
Chen, X., Yan, C. C., Zhang, X., Li, Z., Deng, L., Zhang, Y., et al. (2015c). RBMMMDA: predicting multiple types of disease-microRNA associations. Sci. Rep. 5:13877. doi: 10.1038/srep13877
Chen, X., Yan, C. C., Zhang, X., You, Z. H., Deng, L., Liu, Y., et al. (2016c). WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 6:21106. doi: 10.1038/srep21106
Chen, X., Yin, J., Qu, J., and Huang, L. (2018i). MDHGI: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 14:e1006418. doi: 10.1371/journal.pcbi.1006418
Chen, X., Zhou, Z., and Zhao, Y. (2018j). ELLPMDA: Ensemble learning and link prediction for miRNA-disease association prediction. RNA Biol. 15, 807–818. doi: 10.1080/15476286.2018.1460016
Chen, X., Zhu, C.-C., and Yin, J. (2019d). Ensemble of decision tree reveals potential miRNA-disease associations. PLoS Comput. Biol. 15:e1007209. doi: 10.1371/journal.pcbi.1007209
Cheng, A. M., Byrom, M. W., Shelton, J., and Ford, L. P. (2005). Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 33, 1290–1297. doi: 10.1093/nar/gki200
Fernando, T. R., Rodriguez-Malave, N. I., and Rao, D. S. (2012). MicroRNAs in B cell development and malignancy. J. Hematol. Oncol. 5:7. doi: 10.1186/1756-8722-5-7
Guo, Q., Zheng, M., Xu, Y., Wang, N., and Zhao, W. (2019). MiR-384 induces apoptosis and autophagy of non-small cell lung cancer cells through the negative regulation of Collagen α-1(X) chain gene. Biosci Reports. 39:BSR20181523. doi: 10.1042/BSR20181523
Huang, Z., Shi, J., Gao, Y., Cui, C., Zhang, S., Li, J., et al. (2018). HMDD v3. 0: a database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 47, D1013–D1017. doi: 10.1093/nar/gky1010
Iorio, M. V., Ferracin, M., Liu, C.-G., Veronese, A., Spizzo, R., Sabbioni, S., et al. (2005). MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 65, 7065–7070. doi: 10.1158/0008-5472.CAN-05-1783
Jiang, L., Ding, Y., Tang, J., and Guo, F. J. (2018). MDA-SKF: similarity kernel fusion for accurately discovering miRNA-disease association. Front. Genet. 9:618. doi: 10.3389/fgene.2018.00618
Jiang, L., Xiao, Y., Ding, Y., Tang, J., and Guo, F. (2019). Discovering cancer subtypes via an accurate fusion strategy on multiple profile data. Front. Genet. 10:20. doi: 10.3389/fgene.2019.00020
Jiang, Q., Hao, Y., Wang, G., Juan, L., Zhang, T., Teng, M., et al. (2010a). Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 28(4 Suppl. 1):S2. doi: 10.1186/1752-0509-4-S1-S2
Jiang, Q., Wang, G., and Wang, Y. (2010b). “An approach for prioritizing disease-related microRNAs based on genomic data integration,” in Proceedings of the International Conference on Biomedical Engineering and Informatics, (Yantai: IEEE) (2010). doi: 10.1109/BMEI.2010.5639313
Jiang, Q., Wang, Y., Hao, Y., Juan, L., Teng, M., Zhang, X., et al. (2009). miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 37, D98–D104. doi: 10.1093/nar/gkn714
Kozomara, A., and Griffiths-Jones, S. (2013). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Li, G., Luo, J., Xiao, Q., Liang, C., and Ding, P. (2018). Predicting microRNA-disease associations using label propagation based on linear neighborhood similarity. J. Biomed. Inform. 82, 169–177. doi: 10.1016/j.jbi.2018.05.005
Li, J., Wu, Z., Cheng, F., Li, W., Liu, G., and Tang, Y. (2014). Computational prediction of microRNA networks incorporating environmental toxicity and disease etiology. Sci. Rep. 4:5576. doi: 10.1038/srep05576
Li, J. Q., Rong, Z. H., Chen, X., Yan, G. Y., and You, Z. H. (2017). MCMDA: Matrix Completion for MiRNA-Disease Association prediction. Oncotarget 8, 21187–21199. doi: 10.18632/oncotarget.15061
Li, X., Wang, Q., Zheng, Y., Lv, S., Ning, S., Sun, J., et al. (2011). Prioritizing human cancer microRNAs based on genes' functional consistency between microRNA and cancer. Nucleic Acids Res. 39:e153. doi: 10.1093/nar/gkr770
Li, Y., Qiu, C., Tu, J., Geng, B., Yang, J., Jiang, T., et al. (2014). HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 42:D1070. doi: 10.1093/nar/gkt1023
Liang, C., Yu, S., and Luo, J. (2019). Adaptive multi-view multi-label learning for identifying disease-associated candidate miRNAs. PLoS Comput. Biol. 15:e1006931. doi: 10.1371/journal.pcbi.1006931
Liu, Y., Zeng, X., He, Z., and Zou, Q. (2017). “Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources,” in IEEE/ACM Transactions on Computational Biology and Bioinformatics. (IEEE), 14, 905–915. doi: 10.1109/TCBB.2016.2550432
Lowe, H. J., and Barnett, G. O. (1994). Understanding and using the medical subject headings (MeSH) vocabulary to perform literature searches. Jama 271:1103. doi: 10.1001/jama.1994.03510380059038
Lu, M., Zhang, Q., Deng, M., Miao, J., Guo, Y., Gao, W., et al. (2008). An Analysis of Human MicroRNA and Disease Associations. PLoS ONE 3:e3420. doi: 10.1371/journal.pone.0003420
Luo, J., Ding, P., Cheng, L., Cao, B., and Chen, X. (2017a). “Collective Prediction of Disease-Associated miRNAs Based on Transduction Learning,” in IEEE/ACM Transactions on Computational Biology & Bioinformatics. (IEEE), 14:7. doi: 10.1109/TCBB.2016.2599866
Luo, J., and Xiao, Q. (2017). A novel approach for predicting microRNA-disease associations by unbalanced bi-random walk on heterogeneous network. J. Biomed. Inform. 66, 194–203. doi: 10.1016/j.jbi.2017.01.008
Luo, J., Xiao, Q., Liang, C., and Ding, P. (2017b). Predicting MicroRNA-Disease associations using kronecker regularized least squares based on heterogeneous omics data. IEEE Access. 5, 2503–2513. doi: 10.1109/ACCESS.2017.2672600
Miska, E. A. (2005). How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev.15:563–568. doi: 10.1016/j.gde.2005.08.005
Mugunga, I., Ju, Y., Liu, X., and Huang, X. (2017). Computational prediction of human disease-related microRNAs by path-based random walk. Oncotarget 8, 58526–58535. doi: 10.18632/oncotarget.17226
Nalluri, J. J., Kamapantula, B. K., Barh, D., Jain, N., Bhattacharya, A., de Almeida, S. S., et al. (2015). DISMIRA: Prioritization of disease candidates in miRNA-disease associations based on maximum weighted matching inference model and motif-based analysis. BMC Genomics 16:S12. doi: 10.1186/1471-2164-16-S5-S12
Peng, L., Chen, Y., Ma, N., and Chen, X. (2017a). NARRMDA: negative-aware and rating-based recommendation algorithm for miRNA-disease association prediction. Mol. Biosyst. 2650–2659. doi: 10.1039/C7MB00499K
Peng, L., Peng, M., Liao, B., Huang, G., Liang, W., and Li, K. (2017b). Improved low-rank matrix recovery method for predicting miRNA-disease association. Sci. Rep. 7:6007. doi: 10.1038/s41598-017-06201-3
Peng, L.-H., Sun, C.-N., Guan, N.-N., Li, J.-Q., and Chen, X. (2018). HNMDA: heterogeneous network-based miRNA–disease association prediction. Mol Genet. Genomics. 293, 983–995. doi: 10.1007/s00438-018-1438-1
Peng, L-H., Zhou, L-Q., Chen, X., and Piao, X. (2020). A computational study of potential miRNA-disease association inference based on ensemble learning and kernel ridge regression. Front. Bioeng. Biotechnol. 8:40. doi: 10.3389/fbioe.2020.00040
Qu, J., Chen, X., Yin, J., Zhao, Y., and Li, Z.-W. (2019). Prediction of potential miRNA-disease associations using matrix decomposition and label propagation. Knowl. Based Syst. 186:104963. doi: 10.1016/j.knosys.2019.104963
Siegel, R. L., Miller, K. D., and Jemal, A. (2016). Cancer statistics, (2016). CA Cancer J. Clin. 66:7–30. doi: 10.3322/caac.21332
Sun, D., Li, A., Feng, H., and Wang, M. (2016). NTSMDA: prediction of miRNA-disease associations by integrating network topological similarity. Mol. Biosyst. 12:2224. doi: 10.1039/C6MB00049E
Tang, C., Zhou, H., Zheng, X., Zhang, Y., and Sha, X. (2019). Dual Laplacian regularized matrix completion for microRNA-disease associations prediction. RNA Biol. 16:601–611. doi: 10.1080/15476286.2019.1570811
Temraz, S., Charafeddine, M., Mukherji, D., and Shamseddine, A. (2017). Trends in lung cancer incidence in Lebanon by gender and histological type over the period 2005–2008. J. Epidemiol. Glob. Health.7:161–167. doi: 10.1016/j.jegh.2017.04.003
Torre, L. A., Siegel, R. L., and Jemal, A. (2016). “Lung cancer statistics,” in Lung Cancer and Personalized Medicine. (Springer). 1–19. doi: 10.1007/978-3-319-24223-1_1
Wang, B., Mezlini, A. M., Demir, F., Fiume, M., Tu, Z., Brudno, M., et al. (2014). Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 11:333. doi: 10.1038/nmeth.2810
Wang, C.-C., Chen, X., Yin, J., and Qu, J. (2019). An integrated framework for the identification of potential miRNA-disease association based on novel negative samples extraction strategy. RNA Biol. 16, 257–269. doi: 10.1080/15476286.2019.1568820
Wang, D., Wang, J., Lu, M., Song, F., and Cui, Q. (2010). Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics26, 1644–1650. doi: 10.1093/bioinformatics/btq241
Wang, L., You, Z.-H., Chen, X., Li, Y.-M., Dong, Y.-N., Li, L.-P., et al. (2019). LMTRDA: using logistic model tree to predict MiRNA-disease associations by fusing multi-source information of sequences and similarities. PLoS Comput. Biol. 15:e1006865. doi: 10.1371/journal.pcbi.1006865
Xiao, Q., Luo, J., Liang, C., Cai, J., and Ding, P. (2018). A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 34, 239–248. doi: 10.1093/bioinformatics/btx545
Xu, C., Ping, Y., Li, X., Zhao, H., Wang, L., Fan, H., et al. (2014). Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol. Biosyst. 10, 2800–2809. doi: 10.1039/C4MB00353E
Xu, J., Cai, L., Liao, B., Zhu, W., Wang, P., Meng, Y., et al. (2019). Identifying Potential miRNAs -Disease Associations With Probability Matrix Factorization. Front. Genet. 10.
Xu, P., Guo, M., and Hay, B. A. (2004). MicroRNAs and the regulation of cell death. Trends Genet. 20, 617–624. doi: 10.1016/j.tig.2004.09.010
Xuan, P., Dong, Y., Guo, Y., Zhang, T., and Liu, Y. (2018). Dual convolutional neural network based method for predicting disease-related miRNAs. Int. J. Mol. Sci. 19:3732. doi: 10.3390/ijms19123732
Xuan, P., Han, K., Guo, Y., Li, J., Li, X., Zhong, Y., et al. (2015). Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 31, 1805–1815. doi: 10.1093/bioinformatics/btv039
Xuan, P., Li, L., Zhang, T., Zhang, Y., and Song, Y. (2019a). Prediction of Disease-related microRNAs through integrating attributes of microRNA Nodes and multiple kinds of connecting edges. Molecules 24:3099. doi: 10.3390/molecules24173099
Xuan, P., Sun, H., Wang, X., Zhang, T., and Pan, S. (2019b). Inferring the Disease-Associated miRNAs Based on Network Representation Learning and Convolutional Neural Networks. Int. J. Mol. Sci. 20:3648. doi: 10.3390/ijms20153648
Xuan, P., Zhang, Y., Zhang, T., Li, L., and Zhao, L. (2019c). Predicting miRNA-disease associations by incorporating projections in low-dimensional space and local topological information. Genes 10:685. doi: 10.3390/genes10090685
Yang, Z., Wu, L., Wang, A., Tang, W., Zhao, Y., Zhao, H., et al. (2017). dbDEMC 2.0: updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res 45, D812–D818. doi: 10.1093/nar/gkw1079
You, Z. H., Huang, Z. A., Zhu, Z., Yan, G. Y., Li, Z. W., Wen, Z., et al. (2017). PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 13:e1005455. doi: 10.1371/journal.pcbi.1005455
Zeng, X., Ding, N., Rodríguez-Patón, A., Lin, Z., and Ju, Y. (2016a). Prediction of MicroRNA–disease associations by matrix completion. Curr. Proteomics 13, 151–157. doi: 10.2174/157016461302160514005711
Zeng, X., Liu, L., Lü, L, and Zou, Q. (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34, 2425–2432. doi: 10.1093/bioinformatics/bty112
Zeng, X., Zhang, X., and Zou, Q. (2016b). Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 17, 193–203. doi: 10.1093/bib/bbv033
Zhang, L., Chen, X., and Yin, J. (2019). Prediction of Potential miRNA–disease associations through a novel unsupervised deep learning framework with variational autoencoder. Cells 8:1040. doi: 10.3390/cells8091040
Zhang, Y., Chen, M., Cheng, X., and Chen, Z. (2019). LSGSP: a novel miRNA–disease association prediction model using a Laplacian score of the graphs and space projection federated method. RSC Adv. 9, 29747–29759. doi: 10.1039/C9RA05554A
Zhao, Y., Chen, X., and Yin, J. (2018). A novel computational method for the identification of potential miRNA-disease association based on symmetric non-negative matrix factorization and Kronecker regularized least square. Front. Genet. 9:324. doi: 10.3389/fgene.2018.00324
Zhao, Y., Chen, X., and Yin, J. (2019). Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics 35, 4730–4738. doi: 10.1093/bioinformatics/btz297
Keywords: disease similarity, miRNA similarity, multiple-similarities fusion, space projection, computational prediction model
Citation: Zhang Y, Chen M, Cheng X and Wei H (2020) MSFSP: A Novel miRNA–Disease Association Prediction Model by Federating Multiple-Similarities Fusion and Space Projection. Front. Genet. 11:389. doi: 10.3389/fgene.2020.00389
Received: 28 January 2020; Accepted: 27 March 2020;
Published: 30 April 2020.
Edited by:
Xianwen Ren, Peking University, ChinaReviewed by:
Xing Chen, China University of Mining and Technology, ChinaNana Guan, Guizhou University of Finance and Economics, China
Copyright © 2020 Zhang, Chen, Cheng and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Chen, chenmin@hnit.edu.cn; Hanyan Wei, why@glmc.edu.cn
†These authors share first authorship