 Marek Wieczorek1†
Marek Wieczorek1† Esam T. Abualrous2†
Esam T. Abualrous2† Jana Sticht1
Jana Sticht1 Miguel Álvaro-Benito1
Miguel Álvaro-Benito1 Sebastian Stolzenberg2
Sebastian Stolzenberg2 Frank Noé2
Frank Noé2 Christian Freund1*
Christian Freund1*
- 1Protein Biochemistry, Institute for Biochemistry, Freie Universität Berlin, Berlin, Germany
- 2Computational Molecular Biology Group, Institute for Mathematics, Berlin, Germany
Antigen presentation by major histocompatibility complex (MHC) proteins is essential for adaptive immunity. Prior to presentation, peptides need to be generated from proteins that are either produced by the cell’s own translational machinery or that are funneled into the endo-lysosomal vesicular system. The prolonged interaction between a T cell receptor and specific pMHC complexes, after an extensive search process in secondary lymphatic organs, eventually triggers T cells to proliferate and to mount a specific cellular immune response. Once processed, the peptide repertoire presented by MHC proteins largely depends on structural features of the binding groove of each particular MHC allelic variant. Additionally, two peptide editors—tapasin for class I and HLA-DM for class II—contribute to the shaping of the presented peptidome by favoring the binding of high-affinity antigens. Although there is a vast amount of biochemical and structural information, the mechanism of the catalyzed peptide exchange for MHC class I and class II proteins still remains controversial, and it is not well understood why certain MHC allelic variants are more susceptible to peptide editing than others. Recent studies predict a high impact of protein intermediate states on MHC allele-specific peptide presentation, which implies a profound influence of MHC dynamics on the phenomenon of immunodominance and the development of autoimmune diseases. Here, we review the recent literature that describe MHC class I and II dynamics from a theoretical and experimental point of view and we highlight the similarities between MHC class I and class II dynamics despite the distinct functions they fulfill in adaptive immunity.
Introduction
Major histocompatibility complex (MHC) class I and class II proteins play a pivotal role in the adaptive branch of the immune system. Both classes of proteins share the task of presenting peptides on the cell surface for recognition by T cells. Immunogenic peptide–MHC class I (pMHCI) complexes are presented on nucleated cells and are recognized by cytotoxic CD8+ T cells. The presentation of pMHCII by antigen-presenting cells [e.g., dendritic cells (DCs), macrophages, or B cells], on the other hand, can activate CD4+ T cells, leading to the coordination and regulation of effector cells. In all cases, it is a clonotypic T cell receptor that interacts with a given pMHC complex, potentially leading to sustained cell:cell contact formation and T cell activation.
Major histocompatibility complex class I and class II share an overall similar fold. The binding platform is composed of two domains, originating from a single heavy α-chain (HC) in the case of MHC class I and from two chains in the case of MHC class II (α-chain and β-chain) (Figure 1A). The two domains evolved to form a slightly curved β-sheet as a base and two α-helices on top, which are far enough apart to accommodate a peptide chain in-between. Two membrane-proximal immunoglobulin (Ig) domains support the peptide-binding unit. One Ig domain is present in each chain of MHC class II, while the second Ig-type domain of MHC class I is provided by non-covalent association of the invariant light chain beta-2 microglobulin (β2m) with the HC. Transmembrane helices anchor the HC of MHC class I and both chains of MHC class II in the membrane (Figure 1A).
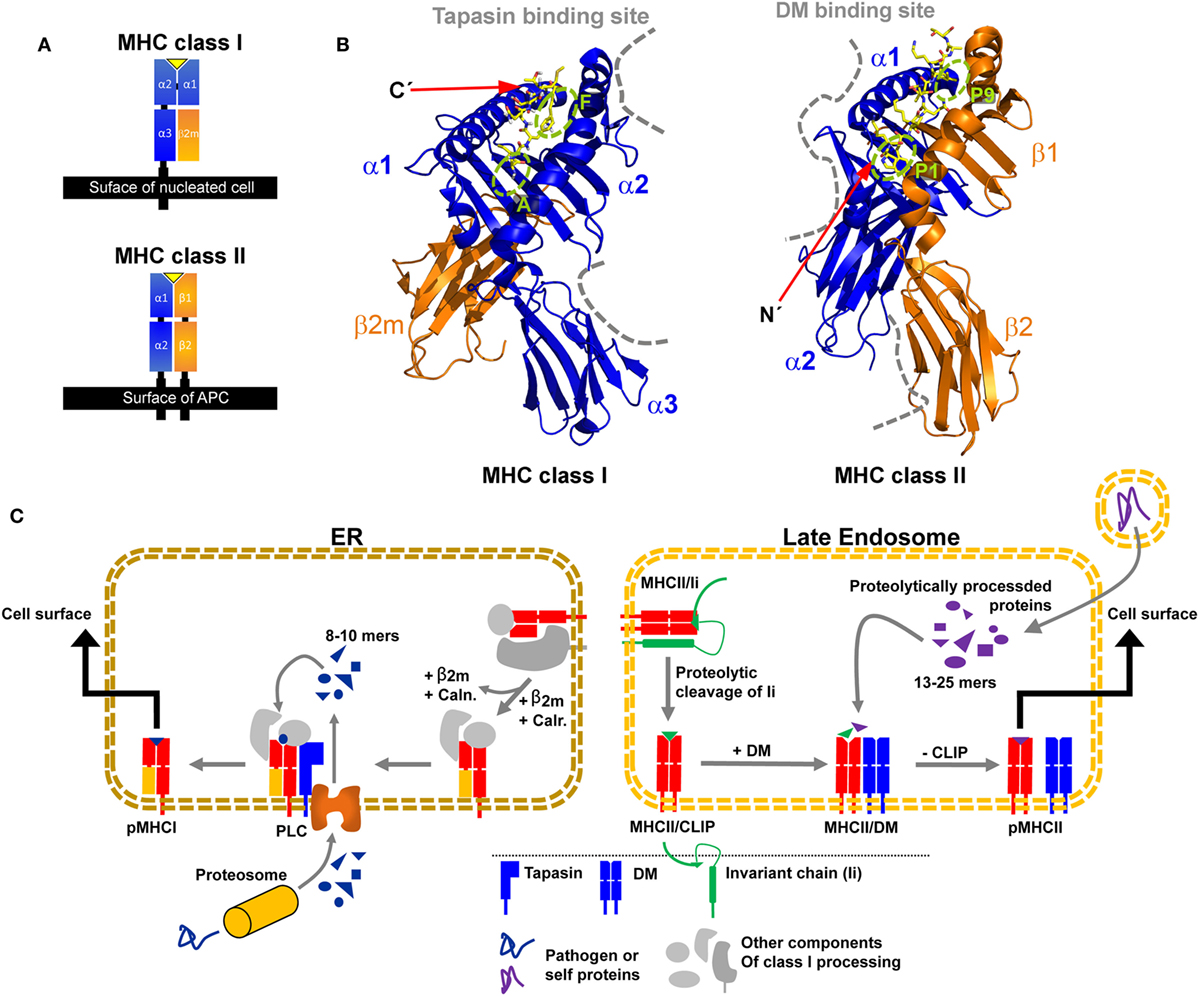
Figure 1. Structural characteristics of major histocompatibility complex (MHC) class I and MHC class II proteins and their compartment-dependent loading with processed peptides. (A) Domain topology of a pMHC class I and pMHC class II complex. (B) Structure of HLA-A68 in complex with an HIV-derived peptide (PDB: 4HWZ, left) and HLA-DR1 in complex with a hemagglutinin-derived peptide (1DLH, right). Indicated are the supposed interaction sites of MHC class I with tapasin and of MHC class II with DM as dashed gray lines. The peptide is shown in yellow with its N and C-terminus marked and relevant pockets are labeled green (C) Simplified illustration of MHC class I (left) and II (right) processing and peptide-editing pathways. CLIP, class II-associated invariant chain peptide; Caln., calnexin; Calr., calreticulin; ER, endoplasmic reticulum; PLC, peptide loading complex.
The groove in-between the two helices accommodates peptides based on (i) the formation of a set of conserved hydrogen bonds between the side-chains of the MHC molecule and the backbone of the peptide and (ii) the occupation of defined pockets by peptide side chains (anchor residues P2 or P5/6 and PΩ in MHC class I and P1, P4, P6, and P9 in MHC class II) (1–4). The type of interactions of individual peptide side-chains with the MHC depend on the geometry, charge distribution, and hydrophobicity of the binding groove. Predicting the affinity of these distinct MHC–antigen interactions for individual allotypes has been a long-standing goal in the community. While good progress has been made in developing and optimizing bioinformatic algorithms to estimate peptide binding to MHC proteins, these in silico predictions, however, still yield false positives (5, 6), and often fail in predicting immunodominance. We argue that understanding the relevance of transient or energetically excited protein conformations that are visited during the equilibrium fluctuations of the molecular structure is important for making good predictions.
In MHC class I, the binding groove is closed at both ends by conserved tyrosine residues leading to a size restriction of the bound peptides to usually 8–10 residues with its C-terminal end docking into the F-pocket (7–9). In contrast, MHC class II proteins usually accommodate peptides of 13–25 residues in length in their open binding groove, with the peptide N-terminus usually extruding from the P1 pocket (10). It has been reported that the interactions at the F pocket region in MHC class I and the P1 region (including the P2 site) in MHC class II appear to have a dominant effect on the presentation of stable pMHC complexes and on the immunodominance of certain peptidic epitopes (11–16). Interestingly, these pockets are located at opposite ends of the binding groove of the respective MHC class I and MHC class II structures (Figure 1B).
The most polymorphic human MHC class I and class II proteins (human leukocyte antigens, HLAs) are each expressed from three gene regions (MHC class I: HLA-A, -B, -C; MHC class II: HLA-DR, -DP, -DQ), which are all highly polymorphic. This allelic variation mainly affects the nature and composition of the peptide-binding groove and thus modulates the peptide repertoire that is presented on the surface by MHC class I or MHC class II proteins for CD8+ or CD4+ T cell recognition, respectively. A good match of the peptide and the MHC binding groove is an important, but certainly not the sole determinant of its presentation. In fact, the formation of a pMHC complex depends on its peptide-loading pathway, in which the selection of peptides is influenced by several factors, such as antigen availability, protease activity, or the availability of chaperones. In addition, for each MHC class, a “catalyst” is available to enhance peptide exchange for certain peptides: tapasin for MHC class I and HLA-DM for MHC class II. These molecules edit the presented peptide repertoire and bias the exchange reaction toward the presentation of thermodynamically stable complexes. Tapasin and HLA-DM thus act similar to typical enzymes by reducing the energy barrier for peptide exchange. However, in the case of HLA-DM and tapasin, no covalent bonds are formed or cleaved during the exchange reaction.
The MHC class I HC folds and assembles with β2m in the lumen of the endoplasmic reticulum (ER). The partially folded heterodimer is then incorporated into the peptide-loading complex (PLC) for peptide binding and exchange. In the PLC, tapasin is a protein that catalyzes, together with other chaperones, the loading of high-affinity peptides derived from proteolysis of endogenously expressed proteins (Figure 1C, left panel) (17, 18). In the absence of tapasin, some class I allotypes (such as HLA-B*44:02) are retained in the ER (tapasin-dependent), whereas other class I proteins (tapasin-independent, such as HLA-B*44:05 and HLA-B*27:09) can bind peptides and travel to the cell surface (19–22). There is no crystal structure of the MHC class I/tapasin complex, but several structural models and mutational studies suggested that tapasin binds two regions in the HC of MHC class I, a loop in the α3 domain (residues 222–229), and a region of the α2 domain (residues 128–137) adjacent to the F-pocket (Figure 1B) (18, 21, 23–29).
Major histocompatibility complex class II proteins fold in the ER in complex with a protein called invariant chain (Ii) (30) and are then transported to late endosomal compartments (also coined MHC class II compartment, MIIC). There, Ii is cleaved by cathepsin proteases and a short fragment remains bound to the peptide-binding groove of MHC class II proteins, termed class II-associated invariant chain peptide (CLIP). This placeholder peptide is then normally exchanged against higher affinity peptides, which are derived from proteolytically degraded proteins available in endocytic compartments (Figure 1C, right panel). HLA-DM accelerates peptide exchange, with different allelic variants being more or less susceptible to catalysis. HLA-DM has a highly similar structural fold compared to classical MHC class II proteins, but its closed-up binding groove prevents peptide binding. Crystal structures of HLA-DM in complex with the MHC class II protein HLA-DR1 (31) and in complex with the competitive inhibitor HLA-DO (32) revealed that HLA-DM mainly contacts the α1-domain of MHC class II proteins close to the P1 pocket and additionally the membrane-proximal β2-domain, in line with previous mutational analyses (Figure 1B) (13, 33–35).
Despite the structural differences between tapasin and HLA-DM as well as their presumably opposite sites of interaction with regard to the orientation of the binding groove, a similar mode of action has been suggested, hinting at a possible convergent evolution of the two exchange catalysts (Figure 1B) (36). A common feature seems to be that both catalysts target regions in the vicinity of those pockets in the peptide-binding groove that are of great relevance for the stability of the respective pMHC complex (11, 13, 15, 31, 37). Furthermore, in both cases, the binding of a high-affinity peptide is able to release the interaction with tapasin/DM (13, 26, 38, 39) and allows for transport of stable pMHC complexes to the cell surface.
While the general hallmarks of antigen processing and editing have been established, the discussion is now moving toward the dynamics of the system, both at the cellular and molecular level. The mechanistic questions relate to a description of how exactly peptides are selected for presentation and how tapasin and HLA-DM catalyze this reaction in an allele-specific manner.
Structural Variations in MHC Complexes
Many allelic variants of MHC class I and MHC class II bound to individual peptide antigens display different biochemical features, but surprisingly, their “ground-state,” i.e., thermodynamically most stable conformations reported by the many available pMHC X-ray structures are very similar. In contrast, increasing experimental and computational evidence of wild type (WT) and mutant MHC complexes over the past years incontestably revealed that changes in conformational dynamics in MHC proteins have to accompany peptide loading and exchange (22, 40–46).
To highlight possible dynamic regions within ground-state crystal structures of human MHC class I and class II proteins bound to a peptide, we performed a global B-factor analysis of all available X-ray crystal structures of human MHC complexes in the absence of any other binding partner. In each structure, we normalized the B-factor values of each alpha carbon (CA) atom to the global mean. Then the variance of all the normalized B-factor values for each CA atom, in 297 human pMHC class I and 41 human pMHC class II structures, was calculated and depicted with a blue to red color spectrum, respectively (heat map on structures of HLA-A*0201 and HLA-DR1, Figures 2A,B). Overall in the binding groove of class I (class II), the α-helix in the α2 (β1) domain displays higher B-factor variation values than the α-helix in the α1 (α1) domain and the β-strands from both domains. Among the pMHCI structures, although B-factor variation values in the N-terminus-proximal helical segments indicate the existence of a certain degree of dynamics, the α2-helical region around the peptide C-terminus displays the highest variation in B-factors (Figure 2A). Among the class II structures, high B-factor variation values are found especially in β-strands 2 and 4 of the α-chain, the 310 helical region and almost the entire β-chain α-helix (Figure 2B). Our analysis is corroborated by a previous comparison of 91 different pMHC class II crystal structures (47). In this analysis, conformational heterogeneities were observed in three regions: the 310-helical region (α45–54), the kink region in the β1-helix (β62–71), and the β2-domain (β105–112).
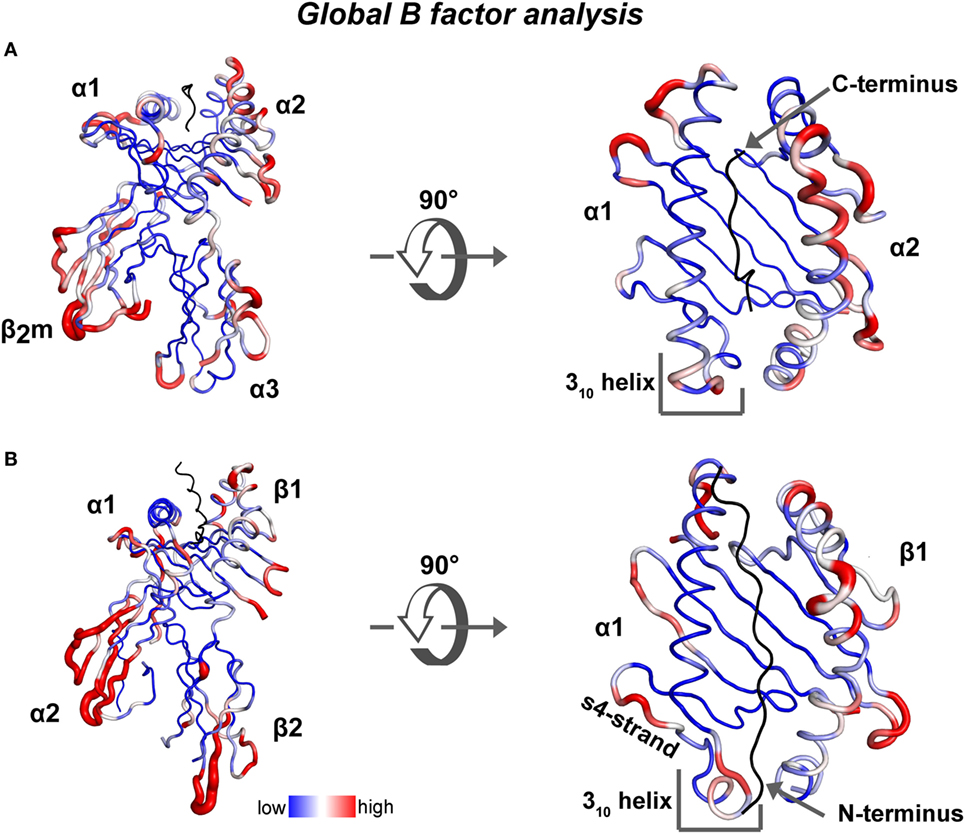
Figure 2. Global B-factor analysis of X-ray crystal structures of MHC class I and MHC class II. Shown is the variance of the normalized residual B factor values of CA atoms (A) derived from 297 human pMHC class I structures is plotted as blue to red spectrum on a HLA-A*0201/peptide complex (PDB: 5HHN) and (B) from 41 human pMHC class II structures is plotted on a DR1/peptide complex (PDB: 4X5W).
It is known that, to some extent, structural variations can be introduced by variable peptide-binding modes. In this context, peptides longer than 8–10 residues have been reported to bind to the MHC class I binding groove (48–52). To accommodate the increase in length, the peptides have to bulge out, leaving the central residues (between p2 and pΩ) exposed to solvent. This is usually achieved by a kink in the backbone in the middle part of the peptide. Recently, two crystal structures of HLA-A2 bound to15-mer peptides have been solved (53). The two peptides follow a binding mode similar to that of the canonical peptides with two anchor residues in the B and F pockets. While one of the peptides showed a mobile central conformation similar to another reported long peptide (48, 54), the other peptide adopted an unusual rigid β-hairpin secondary structure. Furthermore, although the binding of the N- and C-termini at both ends of the binding groove is conserved in almost all the MHC class I complexes, some exceptions have been reported. For example, in the F pocket of HLA-A2, the C terminal residue of the peptide extends by ~1 Å leading to a significant rearrangement of the pocket with only one of the standard hydrogen bonds (at Thr143) preserved (55). Another example is seen in the HLA-B35 protein, the short N-terminus of the 8-mer peptide does not reach the A pocket. Instead, the hydrogen bonds between the amino group of P1 residue and residue 45 of MHC class I are mediated by a water molecule (56).
Since, in the case of MHC class II proteins, the peptide ligand within the binding groove usually adopts a pseudosymmetrical PPII helix-like conformation, bidirectional binding is theoretically possible. An interesting case represents a crystal structure of a DR1/CLIP complex, in which the peptide binds in a very unusual, inverted orientation (C-terminus close to the P1 pocket). The driving force for this peptide inversion is the formation of three additional H-bonds of P1-close residues and the backbone of the peptide’s C-terminus (57, 58). Biological and biochemical evidence for the existence of other pMHC class II isomers have been described in the context of autoimmunity (59). Since MHC class II proteins have open binding grooves, peptides can protrude outwards and even bind in different registers. In this regard, an insulin B chain-derived peptide (InsB9–23) was suggested to induce type 1 diabetes (T1D) in a thermodynamically less favored, low-affinity binding register (60, 61).
Apart from variable peptide binding, catalyst binding can induce more significant conformational variation, as seen in DM-bound DR. By designing a P1-anchor-free pMHC class II complex, Pos et al. could increase the affinity for the pMHC class II to DM and solve the crystal structure for the covalently tethered DR1/HA/DM complex (31). In this MHC class II/DM complex, the interaction interface is primarily composed by the α-subunits of DM and DR1 (~65% of the entire interaction surface, Figure 1B). DM binding to DR stabilizes a rearranged conformation in the vicinity of the P1 pocket of DR1 resulting from the absence of critical peptide-MHC class II interactions in this region. The extended region in the DR1α-chain (α52–55) and the 310-helix adopt an α-helical fold (Figure 3A). Compared to other parts of the pMHC class II structure, it was shown that this site indeed represents a conformationally labile region (35, 46). In addition, the C-terminal part of the β1-α-helix (β86–91) becomes slightly less structured (Figure 3B).
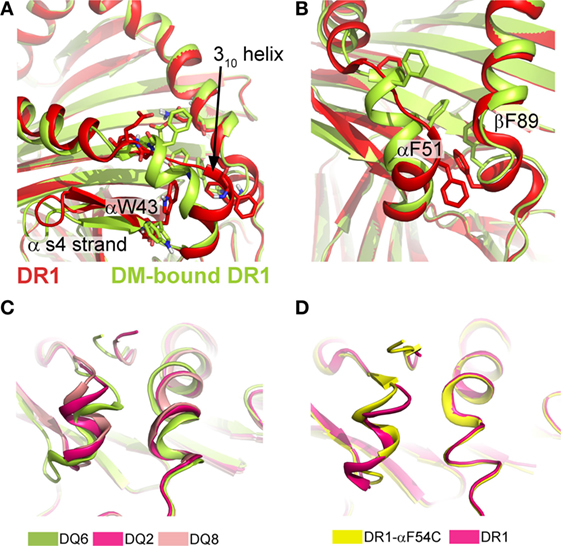
Figure 3. Conformational rearrangements upon DM binding and structural variations in type 1 diabetes-susceptible DQ complexes. (A) Structural rearrangement in the α1-S4 strand and 310-helical region seen in DR1 when bound to DM (limon cartoon) compared to DR1 unbound DM (red). (B) DM-induced rearrangements in the P1-pocket and the surrounding helical segments. PDBs used in (A,B) 1DLH and 4FQX. (C) Overlay of DQ2/ag (PDB: 1S9V), DQ6/hyp1–13 (PDB:1UVQ) and DQ8/InsB9–23 (PDB: 1JK8) showing the structural variations of the 310 helix and the P1-proximal β1-helix. Interdomain communication as exemplarily indicated by the hydrogen bond between αR52 and βE86/βT89 in the DQ8 allele variant is thought to increase the stability of these regions and was previously discussed to be linked to a lowered DM-susceptibility (62, 63). ag, αI-gliadin; hyp, hypocretin peptide 1–13; InsB, insulin B chain 9–23. (D) Structural alignment of DR1/CLIP (PDB: 3QXA) and DR1-αF54C/CLIP (PDB: 3QXD), a mutant that shows an altered conformation in the 310 helix and an increased DM susceptibility.
A feature, which is also present in DM-bound DO, is the intermolecular H-bonds between two conserved residues (DRα W43 and DMα N125). The formation of these critical bonds (as well as other interactions) likely depends on the “flipping out” movement of αW43 from the P1 pocket of DR1 toward DMα (Figure 3A). This movement of DR1 αW43 was suggested to be triggered by partial dissociation of the peptide’s N-terminus or by transient destabilization of contacts of the peptide N-terminus when bound to MHC class II (13, 64). As a consequence, the P1 site is stabilized by a repositioning of two phenylalanine sidechains (DRα F51 and DRβ F89), thereby compensating for the loss of peptide anchors and αW43 from this region (Figure 3B). Incoming peptides have to compete with these repositioned Phe residues for the P1/P2 site in order to be selected for display. Interestingly, structural characteristics of T1D-conferring HLA-DQ alleles were indeed discussed to be linked to decreased DM-sensitivity (62). The analysis of several DQ variants indicated structural differences of the T1D-risk variants DQ2 and DQ8 when compared to DQ1, DQ 6, or DR variants. The decrease in DM-susceptibility of these two proteins was explained by a stabilization of the 310 helical region (63) (Figure 3C). However, the exact relationship between structural variations in the 310 helix and DM-susceptibility are not clear, as highly DM-susceptible DR complexes can display a different conformational mode, compared to DM-susceptible DQ alleles (Figure 3D).
The observed changes lead to the question as to how much structural plasticity in these regions preexists in the peptide-loaded form and provide a prerequisite for catalyzed as well as for spontaneous peptide exchange. For example, do pMHC complexes sample conformations observed in simulations of empty proteins or in complex with the catalyst? How would allelic variation affect the distribution of MHC proteins within the conformational space and thereby influence the presented peptide repertoire? Could variation in protein plasticity also account for the association of specific MHC alleles with immune diseases? Since the polymorphic peptide-binding groove of MHC proteins defines its affinity for a certain peptide, substitutions of even a single amino acid may lead to significantly different affinities for individual peptides. In general, the critical factors defining whether a peptide is presented or not are determined at the different levels of antigen processing and presentation such as uptake route, amount, and folding state of the antigenic protein, amenability to proteolytic degradation, and catalysis of the complex, etc. However, at the molecular level, it has been shown that certain polymorphisms shape individual pockets in the peptide-binding groove to optimally present an autoimmunogenic self-peptide (65–67). In other cases, the functional impact of disease-associated polymorphisms remained enigmatic and suggests that dynamics might account for the observed differences.
Dynamics of Peptide-Free MHC Proteins
While simulations and experimental studies vary in the features ascribed to peptide-free MHC proteins, they certainly agree in attributing a substantial degree of dynamics to the peptide-binding groove. Thus, binding of peptides to MHC proteins is of utmost importance for the stabilization of the known MHC fold (40, 68). The lack of a crystal or an NMR structure of peptide-free MHC protein hinders an accurate description of the structural changes upon peptide binding and this is probably due to the ensemble character of the peptide-free conformers. This ensemble character, however, has been probed by computational techniques, as discussed in the following paragraphs.
MHC Class I
Most of the conformational dynamics information on the peptide-free class I have been revealed by molecular dynamics (MD) simulations. In such simulations, peptide-free class I protein is modeled from the crystal structure by deleting the atoms of the bound peptide. In the absence of peptide, an increased conformational flexibility of the F pocket region was observed for several allelic variants (HLA-A*02:01, HLA-B*44:02, HLA-B*44:05, HLA-B*27:05, HLA-B*27:09, H-2Db, and H-2Kb) (9, 15, 21, 22, 69–71). Longer simulations of chicken and human class I allotypes showed increased global motion in the peptide-free form when compared to the peptide-bound proteins (72, 73). By combining molecular docking and MD simulations, a conformational transition of the 310 helical segment of H-2Ld between the peptide-bound and peptide-free class I was observed. Thus, a conformational reorganization close to the A and B pockets upon peptide binding was proposed (74).
Experimentally, circular dichorism (CD) was used to measure the thermal denaturation temperature (Tm) of the peptide-free HLA-B*07:02 (B7/β2m). By increasing the temperature, a gradual loss of the structure-specific signal of B7/β2m in the CD spectrum of peptide-free class I was detected, indicating a more heterogeneous conformational population. Furthermore, in the absence of peptide, the binding groove of B7/β2m was more sensitive to enzymatic proteolysis when compared to the peptide-bound form (40). Saini et al. studied the unfolding of H-2Kb by measuring the intrinsic tryptophan florescence. The results pointed to a folding intermediate of peptide-free class I proteins that are more structured than a molten globule (75). This was in line with a previous study arguing for a native-like conformation of in vitro refolded empty murine class I proteins (76).
Structural studies using NMR indicated a loss of the binding-groove fold in the peptide-free form of a HLA-C allotype. In particular, NMR spectra of these peptide-free MHC class I protein show the loss of selected methionine NH resonances of the β-sheet floor of the peptide-binding groove, which indicated unfolding or conformational exchange of this part of the protein (77). This is consistent with previous reports indicating that especially the binding groove is undergoing conformational exchange in the absence of the bound peptide (40, 78).
A work that focused on complexes with a partly filled MHC class I binding groove pointed to a requirement of the stabilization of the F pocket region. By using a refolded H-2Db in the presence of a pentamer peptide (NYPAL), which binds to the C to F pockets in the binding groove, a X-ray crystal structure of the pMHCI complex could be solved (79). Thus, peptide/class I interactions at the F pocket region seem to be sufficient to keep class I in a folded state. In agreement, short dipeptides mimicking the peptide C-terminus of high-affinity ligands support the folding of HLA-A*0201, displaying high peptide-receptivity (14). To stabilize peptide-free class I in a folded form independently from the peptide, the Springer group created a novel variant by introducing a disulfide bond to restrain the high flexibility of the F pocket region. The disulfide mutant showed an increased peptide and β2m affinity and bypassed the cellular quality control (80).
MHC Class II
Physiologically, the question if peptide-free MHC class II proteins play a role in adaptive immunity is posed by studies indicating that unloaded MHC class II proteins are abundantly present on the surface of immature DCs. There, they are able to bind ligands from the extracellular milieu and activate T cells (81, 82). Two isomers of peptide-unloaded MHC proteins seem to exist, each displaying different kinetic properties (83). While the peptide-receptive empty isomer of DR1 binds peptide rapidly, the conversion to the non-receptive isomer within less than 5 min dramatically reduces the peptide-binding capacity of human DR (41, 84, 85). Studies using circular dichroism and size exclusion chromatography predicted a conformational change of peptide-free MHC class II upon peptide binding, and an increase in the overall stability (44, 68, 86). Peptide-free MHC class II proteins thus show a lower degree of helicity and an increased hydrodynamic radius compared to peptide-loaded MHC class II.
Carven and Stern studied ligand-induced conformational changes by selective chemical side-chain modification of peptide-free DR1 followed by mass spectroscopy analysis (87). The results of this study were inconsistent with a partly unfolded state of DR1 in the absence of ligand, but rather indicated a more localized conformational change induced upon peptide binding. However, empty MHC class II proteins harbor the hallmarks of partially unstructured peptide-binding domains when studied spectroscopically. NMR-spectra of the α-chain of such peptide-free MHC class II proteins barely display any signals for residues corresponding to the folded peptide-binding groove, indicating that the binding groove undergoes conformational exchange (46). Taken together, it appears likely that the empty binding groove dynamically samples different native-like conformations. Interestingly, similar to MHC class I, certain small molecules and dipeptides increase peptide-receptivity of peptide-free MHC class II proteins, presumably by preventing a “closure” of the binding groove (88, 89). In similar to the F pocket of MHCI, the predicted site of interaction is the most dominant pocket of the binding groove (P1).
In a MD simulation study combined with peptide-binding assays, it was shown that a well-conserved residue, βN82, which also contributes disproportionally to pMHCII stability (12, 90), is participating in the control of peptide receptivity (91). The authors suggested that the “non-receptive” state of peptide-free DR1 is induced by a molecular lock through the formation of a hydrogen-bond between DRβ N82A and DRα Q9. The observed narrowing of parts of the binding groove-flanking α-helices likely represents the trigger for such a clamped conformation. Using MD simulations, another investigation suggested a movement of the α51–59 region into the P1–P4 site of the binding groove in the empty state. In this state, the α51–59 region adopted a ligand-like conformation. In addition, an increased flexibility of the β2 domain as well as the β50–70 helical region were observed (92). A higher flexibility of the β58–69 helical region was also seen in MD simulations of the HLA-DR3 protein upon in silico peptide removal (93) (Table 1). Interestingly, this helical segment is also recognized by monoclonal antibodies designed to bind to the peptide-free conformation (81, 94).
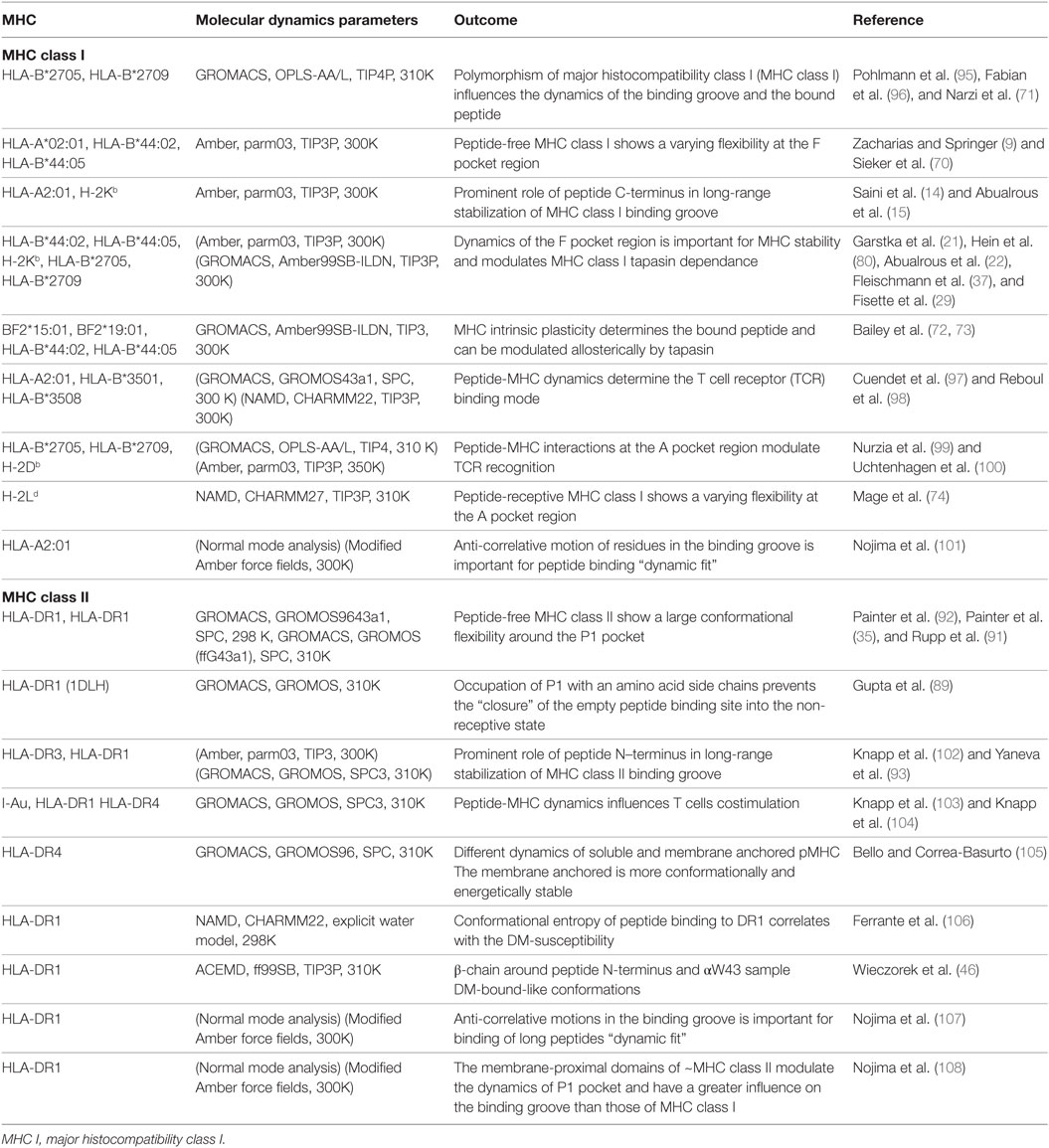
Table 1. Computational studies on MHC class I and II dynamics.
Finally, it has to be noted that the timescale of the experimental descriptions of empty MHC molecules differs vastly from the theoretical studies. While the latter describe the initial events of conformational changes accompanying peptide removal, the experimental investigations observe the properties of the empty MHC species at or near equilibrium.
Dynamic Features of Peptide-Bound MHC Complexes
While the study of empty MHC proteins is of theoretical and conceptual interest, nature has engineered the antigen-presenting system in a way that prevents the accumulation of isolated, non-peptide-bound MHC molecules. Endogenous peptides derived from the proteasome in case of class I or from the invariant chain in the case of class II first bind and eventually are replaced by antigenic peptide. This inherently dynamic process is enabled by intrinsic features of the MHC molecules and several studies suggest that pMHC complexes sample different and transient conformations dependent on the bound peptide and the allelic variant under investigation (46, 64, 71, 72, 95, 100, 106, 113, 114).
MHC Class I
Changes in conformational dynamics in MHC class I are heterogeneously distributed along its peptide-binding groove, as suggested by both computational and experimental studies. For example, MD simulations showed a subtype-dependent conformational flexibility of the F pocket region. Residues 114 and 116 of the HC, at the bottom of the F pocket, and residues 74 and 77 from the α1-helix, engaging the peptide’s C-terminus, show an altered mobility in different MHC class I allotypes (9, 22, 70, 71, 96, 115, 116). Consistently, it was shown that the dynamics of the MHC class I binding groove was most profoundly affected by C-terminal residues of the peptide (15). In longer MD simulations, in addition to varying protein plasticity in the F pocket region, an enhanced sampling of conformations in the α3-domain upon peptide binding was observed (72, 73) (Table 1).
Experimental observations at the atomistic level, derived from NMR-based relaxation-dispersion experiments, have elucidated the peptide dependency of minor states on the stability of pMHCI complexes. Conformational fluctuations of different HLA-B*35:01 complexes were localized to the peptide-binding groove, including residues of the B, E, and F-pocket, but not in the IgG-like domains (113). Interestingly, the presence of minor conformations in pMHCI complexes (ranging from approximately >1 to 4.5%) could be positively correlated to the thermostability and surface presentation of the pMHCI complex under investigation, implying that a minor conformation considerably contributes to pMHCI stability. Similar, investigations of HLA-A*02:01 loaded with different peptides by HD exchange/MS and fluorescence anisotropy revealed that fluctuations within the binding groove depend on the ligand bound to MHC class I (112). Despite these ligand-sensitive changes in dynamics, the α2-helix showed a general higher flexibility than the α1-helix. The authors concluded that the observed variations in dynamics throughout the peptide-binding site could influence receptor engagement, entropic penalties during receptor binding, and the population of binding-competent states (see also Table 2).
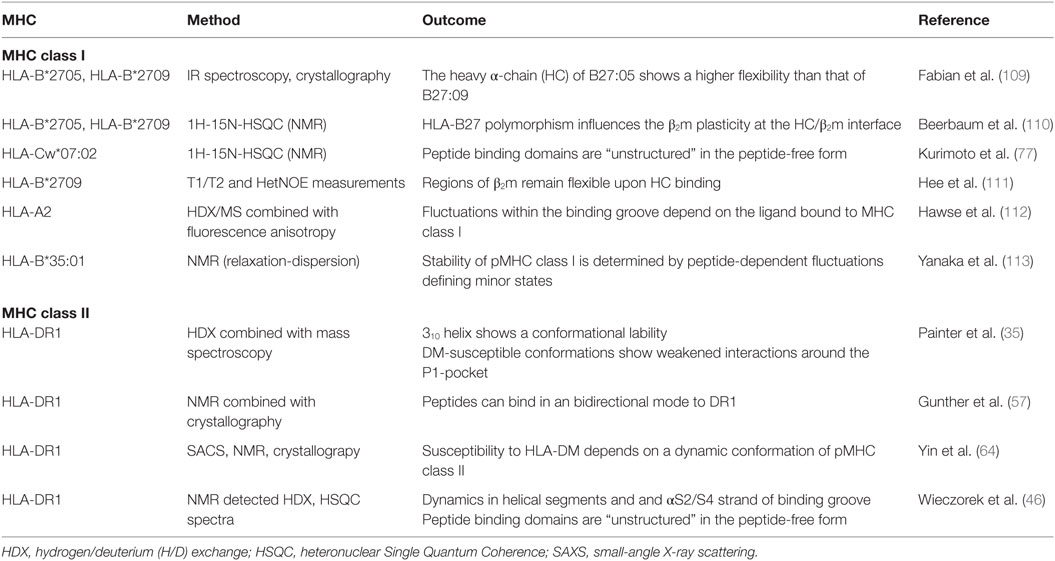
Table 2. Experimental studies on major histocompatibility complex (MHC) class I and II dynamics.
Another study also showed that β2m seems to sense allelic as well as peptide-induced conformational variations and accommodates to them, showing a high degree of plasticity within the inter-domain interface with the HC domains (110). NMR-chemical-shift changes of complexes were most pronounced in the region close to the F-pocket. By comparing the dynamics in the ns–ms timescale of HC-bound and free β2m, Hee et al. demonstrated that most residues gain rigidity upon HC binding (111). Nevertheless, three sites (region around His31, site around Asp53 and Lys58, and region around Ser88) remained flexible in the mature complex. Interestingly, His31 and Ser88 are located underneath the F-pocket, which stability is known to be important for tapasin function. Moreover, Lys58 and Ser88 are also known to interact with several other proteins, including the natural killer cell receptors Ly49A CD8 and LIR1 (117–119). Conformational sampling of these regions could thus be critical for the interaction with these receptors.
MHC Class II
A similarly heterogeneous picture in regards to altered binding groove dynamics can be observed for MHCII: by dissecting the thermodynamics of peptide MHC class II interactions, Ferrante et al. conclusively demonstrated how dynamics of pMHCII complexes are linked to peptide affinity and DM-susceptibility (106). Using different biophysical techniques and MD simulations, the authors showed that peptide binding events can be driven by a considerable proportion of conformational entropy (if enthalpic interactions are less favored). MD simulations suggest that peptide-dependent conformational fluctuations involve alterations of α-chain residues DR1α-43–54 and β-chain residues 63–68 and 79–90. Wieczorek et al. performed H/D-exchange measurements in combination with NMR spectroscopy to obtain residue-specific experimental information about the stability of individual secondary structure elements. Several regions undergoing conformational fluctuation even in highly stable pMHCII complexes were thus revealed. These fluctuations are confirmed by extensive (~100 μs) MD simulations and Markov model analyses that reveal transient conformations with obvious relevance for the peptide-exchange pathway (see below for details) (46). In particular, the highest lability was seen in DRα 46–62, in parts of β-strands s2–s4 sitting underneath the N-terminal part of the α1 helix, β65–93 and several loops connecting the β-strands of the peptide-binding site. Interestingly, this experimental piece of evidence is also in line with the global B-factor analysis presented here (Figure 2B); a certain degree of dynamics thus seems to already be encoded even in the context of a high affinity pMHC complex. Earlier on, it was shown by HD-exchange/MS measurements that conserved peptide–MHC class II contacts (H–bonds) are strong at the P1 pocket-proximal site of the peptide (especially position βN82) in highly stable immunodominant complexes, which is in agreement with previous biochemical studies (12, 35, 90). However, local destabilization induced by a point mutation in the DR1 complex and the use of a different ligand (DR1-αF54C/CLIP) strongly enhanced fluctuations of the peptide and especially weakened contacts around the P1 site (35). This implies that weakening interactions by substitutions in the peptide or MHC (allelic variation) would have an influence on conformational fluctuations that correlate with DM-susceptibility.
MHC Dynamics During Peptide Exchange
While the studies described in the previous section unambiguously demonstrate the dynamic features of pMHC complexes, the question arises naturally in how far these properties translate into peptide exchange. For pMHCII complexes experimental progress has been made in identifying intermediate or transient conformations of pMHCII with regard to catalyzed or intrinsic peptide exchange (13, 31, 32, 35, 46). For pMHCI molecules the atomistic description of structural changes during peptide exchange mostly relies on MD simulation studies, supported by mutational analysis and circumstantial biophysical evidence (14, 22, 37, 73).
MHC Class I
Within cells, tapasin is a key protein that mediates the binding of high-affinity peptides to most class I proteins. To date, there is no crystal structure of the tapasin/MHC class I complex. Based on mutational studies, however, two regions have been shown to be essential for tapasin interaction with the HC of MHC class I: a loop in the α3 domain (residues 222–229) and a part of the α2 domain (residues 128–137) (17, 23, 25, 120, 121). Two major functions have been proposed for tapasin: (i) a chaperone-like stabilization of empty class I proteins (20–22, 24, 122) and (ii) a peptide-editing function through peptide-exchange catalysis (26, 123, 124). Several computational models have been published to describe the mechanism of action of tapasin on MHC class I (28, 29, 121). Most researchers agree on the importance of the F-pocket region for peptide exchange (14, 21, 37, 80, 125). However, the association of F-pocket dynamics and the peptide-exchange mechanism remain a matter of debate. So far, dynamics in the F-pocket region in the presence of peptide have not revealed any significant conformational exchange phenomena in most MD simulations (Table 1). Recently, longer MD simulations (microsecond timescale) of tapasin/pMHCI complexes indicated that binding of tapasin to HLA-B*44:02 accelerates the dissociation of low-affinity peptides (29, 37). Using a computational systems model (73, 126), it was shown that peptide exchange seems to depend on the opening and closing rate of the binding groove in the presence of peptide. According to this study, the pMHCI opening rate is peptide-dependent, but pMHCI closing is allele-dependent. Consequently, a low-affinity peptide complex would display fast opening rates, but only if the MHC allele variant has an F-pocket signature (more plasticity) that allows for fast closing in the presence of a high-affinity peptide (as B44:05), it would lead to efficient peptide exchange in the absence of catalyst. Allele variants with a rigid F-pocket conformation (as B44:02) in contrast depend on tapasin to sample the necessary conformational states to close the binding groove quickly.
It has to be considered that as a default, tapasin is present in the cell and that it may also provide the necessary function as a chaperone to prevent the collapse of empty MHC class I molecules into a non-receptive state (29, 127) as it is experimentally measured in tapasin-deficient cells (21, 22). However, in the absence of a crystal structure of the tapasin/MHC class I complex, it is difficult to rationalize the dynamics with regard to tapasin binding and exchange.
MHC Class II
Two seminal studies made it unambiguously clear that HLA-DM recognizes complexes showing a P1-destabilized conformation (13, 31). However, since DM-susceptible structures rarely show any of the changes present in the DM-bound structure (e.g., folding of α-46–55 into an α-helix and unfolding of C-terminal β-helix), the question of the conformational prerequisites for DM binding arises. As mentioned previously, an important study indeed demonstrated that HLA-DM attacks pMHC class II complexes at a site of conformational lability, the 310-helical region (35). This segment, together with a neighboring unstructured segment (α52–55) folds into an α-helix when bound to DM (31). Interestingly, increased fluctuations of this region could be observed by other computational and experimental studies, implying the existence of higher conformational entropy within this region (46, 106). Noteworthy, the α-helix in the region β86–β91 opens up to a certain degree in the DM-bound structure of DR1. However, a considerable influence of P1-remote sites on conformational dynamics and DM-dependence was recently demonstrated (64). In this study, alteration of P9-pocket/peptide interactions influenced dynamics of the pMHCII, likely in regions relevant for DM binding. The authors concluded that the key determinants for HLA-DM recognition are conformational dynamics present in HLA-DR1. Similarly, and as already mentioned above, Ferrante et al. explained the relationship between entropic penalties and DM binding in a thermodynamic context (106). According to their experimental and computational results, higher conformational entropy of pMHCII complexes correlates with DM susceptibility.
A recent study by our group in the MHC class II field explored internal motions of pMHC class II molecules along the conformational peptide-exchange pathway in a more conceptual model (46). Using NMR/HDX (hydrogen deuterium exchange) and MD simulations of over 200 μs in total, followed by Markov State model (MSM) analysis (128–132), we have identified transient conformations relevant for the DM-catalyzed and non-catalyzed (spontaneous) peptide-editing process (46). In agreement with the general view, the catalyzed pathway depends on the particular destabilization of the region surrounding the P1 pocket, sharing in part features of MHC class II bound to DM. More specifically, it has been suggested for MHC class II that pMHC complexes have to sample P1-pocket-destabilized conformations to allow for HLA-DM binding (13).
The non-catalyzed pathway, however, was correlated to the ground state of the pMHCII complex and, therefore, is directly correlated with thermodynamic stability. Indeed, it was shown that, removal of two hydrogen bonds between β82N of the MHC class II and the backbone of the peptide in the mutant DR1βN82A drastically reduces stability and, at the same time, dramatically enhances non-catalyzed peptide exchange. Nevertheless, binding to HLA-DM is also enhanced for the βN82A mutant, leading to the somewhat paradoxical finding that an MHCII molecule might bind tightly to a catalyst that it does not need for exchanging peptide. This can be best conceptualized when assuming that the βN82A mutant of HLA-DR1 in addition to increased spontaneous exchange more frequently samples a rare conformation along the pathway of catalyzed exchange. MD simulations in conjunction with MSM analysis indeed show that an excited state structurally correlated with features of the HLA-DM bound conformation. This excited state was seen to be significantly more frequently sampled in the mutant compared to WT HLA-DR1.
However, a similar intermediate state can be defined for the very stable WT protein, where peptide release from the pockets was not mandatory for the observation of the early intermediates. Thus, if the pMHCII forms a stable complex, the peptide editing depends on the population of rare conformations that can be selected by the catalyst DM for binding. This study demonstrated the critical importance of residues 80–93 of the β1 helix for catalyzed exchange, suggesting that β1 helical unfolding is critical for the rearrangement of this segment as it is observed in the DR1-DM structure (31). As the study shows, mutations in the β1 helix (e.g., βE87P) designed to specifically destabilize the C-terminal part of the β1 helix without disrupting H-bonds to the peptide, are able to over-proportionally shift the dynamics toward the HLA-DM-dependent pathway (46).
In conclusion, this model helps to reconcile discrepancies in the hypothesized correlations of peptide affinity, pMHC stability, DM susceptibility, and catalytic effect (133).
Conclusion and Outstanding Questions
Major histocompatibility complex proteins are encoded by oligogenic and highly polymorphic genes and most polymorphisms map to the regions important for peptide binding. The pMHC complexes display various degrees of flexibility along the binding groove, and these dynamic features seem to correlate with the propensity for peptide exchange. Of interest is the fact that tapasin and DM both bind their MHC targets in regions of enhanced dynamics. Short, destabilized helical segments together with their adjacent structural elements seem to represent the requirements for transient binding of the respective catalyst. The degree of these local flexibilities can be correlated to a higher dependence of a particular pMHC complex on tapasin or DM. Polymorphic substitutions might not only change the binding preference for certain ligands but also the overall stability and dynamics of the corresponding allelic variants. In turn, this will affect the conformational ensemble recognized by the peptide editors and in principle should be able to explain why certain alleles seem to possess a generally lower taspasin or DM dependence. In this way, MHC molecules may become a paradigmatic example of how differences in the dynamic landscape of protein complexes translate into distinct functional outcomes of physiological relevance.
How far have we come and what has to be done to achieve this goal? Figures 4 and 5 summarize the findings described in this review and also emphasize the most daunting questions in the field that need to be answered in order to formulate a unifying concept of antigen exchange. What seems to be clear is that both type of MHC molecules can exchange peptide along two distinct pathways, with the ratio of spontaneous versus catalyzed exchange certainly being different for the allelic protein variants and pMHC complex. While dynamics often correlates with thermodynamic stability, it has yet to be seen which type of motions are critical for catalysis and which structural elements are indispensable for these transitions to occur. For MHC class II molecules, the structure of the MHCII–DM complexes provides a cornerstone (31), and the early intermediates (μs-ms timescale) toward the DM-bound form could be defined (35, 46) (Figure 5). However, there are no structural insights about the replacement of DM by incoming peptide, thus requiring experimental and simulation strategies to follow the fate of DM-prebound MHCII molecules. In the case of MHCI, a tapasin-class I complex structure is required in order to provide a reliable framework for further experimental and theoretical studies. Similarly, characterization of empty MHC molecules will certainly aid in defining the dynamic modes that are explored by the peptide-binding domains. Since it has been shown that empty MHC molecules can be rescued by the chaperoning function of the exchange catalysts (134, 135) and thus the dynamics that occur upon peptide exchange are likely to show features of the empty state. It seems, therefore, highly desirable to compare the two systems on time scales down to a few milliseconds.
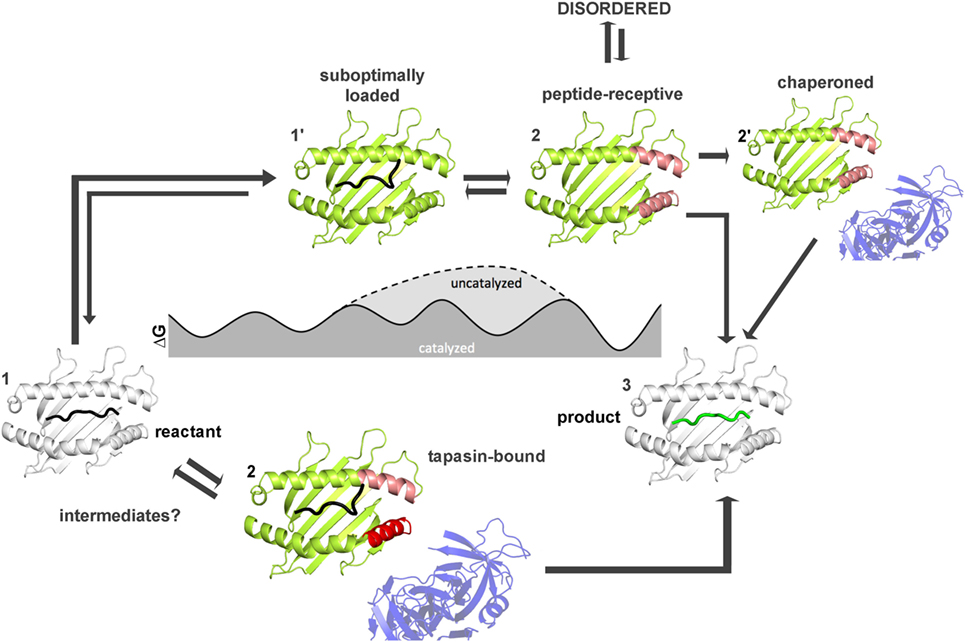
Figure 4. Thermodynamic model for peptide exchange of major histocompatibility complex (MHC) class I. Peptide–MHC class I (pMHCI) complexes can follow two mechanistic pathways for peptide exchange starting from pMHCI ground state (state 1). In the tapasin-catalyzed pathway, tapasin modulates conformational changes in the α2-1 helix (red) of the F pocket region (pink) and the α3 domain (not shown) that accelerate the kinetics of peptide dissociation (state 2) and the loading of a high-affinity peptide (3). More intermediates states (between state 1 and state 3) need to be identified by computational studies and/or NMR and X-ray crystallography. In the non-catalyzed pathway, the peptide dissociates from the sub-optimally-loaded intermediate state (state 1′). The resulting empty MHC molecule shows subtype-dependent dynamics (especially at the F pocket region, pink) and thus can exist in a stable peptide-receptive form (state 2′) or in an unstable form (state 2″) that is chaperoned by tapasin for peptide binding. The structures used in states 1 and state 3 were modified from PDB: 1UXS (shown in white). The models used in states 1′, 2, 2′, and 2″ represent suggested states by computational and experimental studies (shown in limon).
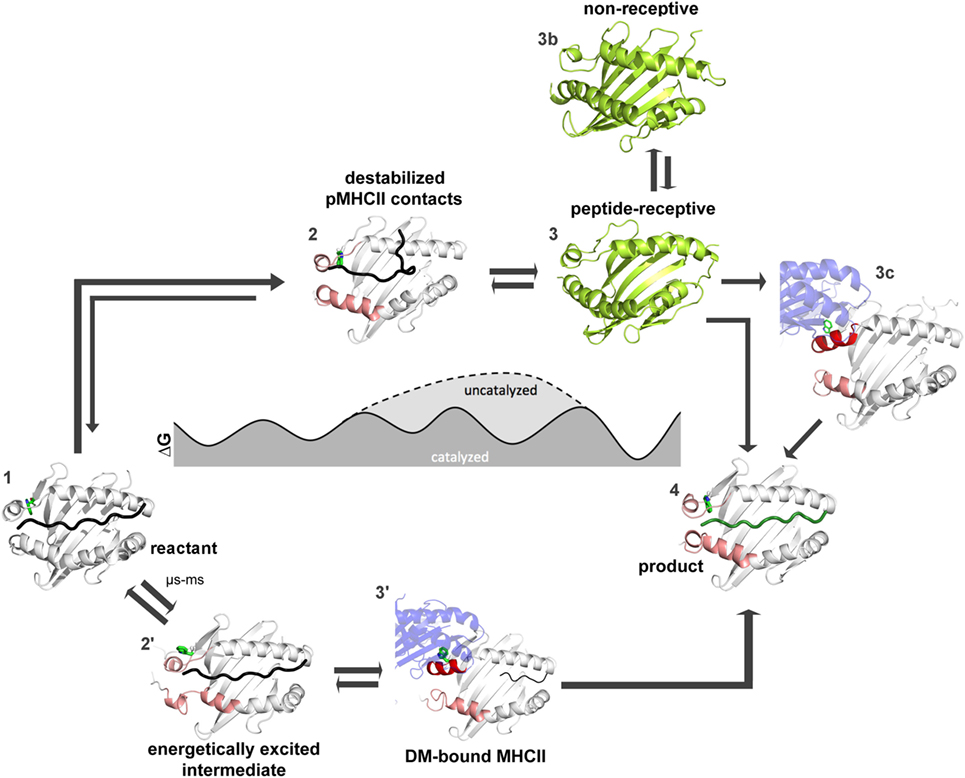
Figure 5. Thermodynamic model for peptide editing of major histocompatibility complex class II. pMHCII complexes can follow two mechanistic pathways for peptide exchange. The DM-catalyzed route requires multi-step transitions starting from the pMHCII ground state (1). This includes initially an out-flip movement of αW43 or the destabilization of β80–93 region via spontaneous conformational sampling of rare conformations (state 2′). DM would preferably select for conformations that are sampled on longer timescales and which show both, an out-flip movement of αW43 and a destabilized β80–93 region. Binding of DM to the energetically excited intermediate (which shows in part features of the DM-bound state) would then induce further rearrangements in the 310-helical region (state 3′) and thereby accelerate peptide-release. Binding of peptides which can displace the stabilizing interactions complete the peptide exchange process (state 4). Spontaneous (non-catalyzed) peptide exchange depends on the intrinsic stability of the pMHCII complex and does not rely on the sampling of rare conformations (state 2). Binding of a new peptide would likely require dissociation of the bound peptide, leading to the empty state (state 3) which rapidly converts into the non-receptive state (state 3b) but can also be chaperone by DM (state 3c) in order to allow for high-affinity peptide binding (state 4). Structures used in state 1, 3′, and 4 were derived from PDB: 4QXA, 4FQX, and 1DLH, respectively. Cartoons shown in 2, 2′, 3, and 3b were derived from molecular dynamic simulations (46, 91).
For both MHC classes, more sophisticated NMR experiments capitalizing on selective amino acid side-chain labeling protocols are probably required and methods relying on CEST or relaxation dispersion should be able to yield more direct information on the anticipated intermediate states (136, 137). So far, in-depth NMR experiments are restricted to certain stable pMHC complexes and the investigation of other alleles have been hampered by the in-availability of other variants such as the disease-relevant DQ alleles. There is a need to expand the experimental basis of dynamically investigated pMHC complexes in order to test the predictions made on the basis of the dynamic features of just a few alleles. Solutions are most likely to come from protein engineering approaches in combination with the use of different expression systems. The increasing importance of MD simulations arises from the fact that micro-to-milli-second simulations in combination with Markov State Modeling will become more of a standard in the field. This is essential, because the critical intermediates of antigen exchange seem to be populated at this time scale. Once we are able to conceptualize conformational peptide exchange, we will be in the position to better predict MHC peptide occupancies in the context of cellular editing mechanisms and we will understand and be able to manipulate the action of small molecules or biological macromolecules that modulate peptide exchange.
Author Contributions
MW, EA, and CF designed and wrote the manuscript. JS, MÁ-B, SS, and FN critically read, discussed, and edited the content prior to submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
CF is thankful for funding by the DFG (SFB958, SFB854, TR186 and SPP1623).
References
1. Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature (1991) 351:290–6. doi:10.1038/351290a0
2. Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, et al. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science (1992) 255:1261–3. doi:10.1126/science.1546328
3. Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature (1993) 364:33–9. doi:10.1038/364033a0
4. Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature (1994) 368:215–21. doi:10.1038/368215a0
5. Paul S, Kolla RV, Sidney J, Weiskopf D, Fleri W, Kim Y, et al. Evaluating the immunogenicity of protein drugs by applying in vitro MHC binding data and the immune epitope database and analysis resource. Clin Dev Immunol (2013) 2013:467852. doi:10.1155/2013/467852
6. Lyngaa R, Pedersen NW, Schrama D, Thrue CA, Ibrani D, Met O, et al. T-cell responses to oncogenic Merkel cell polyomavirus proteins distinguish patients with Merkel cell carcinoma from healthy donors. Clin Cancer Res (2014) 20:1768–78. doi:10.1158/1078-0432.CCR-13-2697
7. Matsumura M, Fremont DH, Peterson PA, Wilson IA. Emerging principles for the recognition of peptide antigens by MHC class I molecules. Science (1992) 257:927–34. doi:10.1126/science.1323878
8. Bouvier M, Wiley DC. Importance of peptide amino and carboxyl termini to the stability of MHC class I molecules. Science (1994) 265:398–402. doi:10.1126/science.8023162
9. Zacharias M, Springer S. Conformational flexibility of the MHC class I alpha1-alpha2 domain in peptide bound and free states: a molecular dynamics simulation study. Biophys J (2004) 87:2203–14. doi:10.1529/biophysj.104.044743
10. Chicz RM, Urban RG, Lane WS, Gorga JC, Stern LJ, Vignali DA, et al. Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature (1992) 358:764–8. doi:10.1038/358764a0
11. Natarajan SK, Stern LJ, Sadegh-Nasseri S. Sodium dodecyl sulfate stability of HLA-DR1 complexes correlates with burial of hydrophobic residues in pocket 1. J Immunol (1999) 162:3463–70.
12. McFarland BJ, Katz JF, Beeson C, Sant AJ. Energetic asymmetry among hydrogen bonds in MHC class II*peptide complexes. Proc Natl Acad Sci U S A (2001) 98:9231–6. doi:10.1073/pnas.151131498
13. Anders AK, Call MJ, Schulze MS, Fowler KD, Schubert DA, Seth NP, et al. HLA-DM captures partially empty HLA-DR molecules for catalyzed removal of peptide. Nat Immunol (2011) 12:54–61. doi:10.1038/ni.1967
14. Saini SK, Ostermeir K, Ramnarayan VR, Schuster H, Zacharias M, Springer S. Dipeptides promote folding and peptide binding of MHC class I molecules. Proc Natl Acad Sci U S A (2013) 110:15383–8. doi:10.1073/pnas.1308672110
15. Abualrous ET, Saini SK, Ramnarayan VR, Ilca FT, Zacharias M, Springer S. The carboxy terminus of the ligand peptide determines the stability of the MHC class I molecule H-2Kb: a combined molecular dynamics and experimental study. PLoS One (2015) 10:e0135421. doi:10.1371/journal.pone.0135421
16. Kim A, Sadegh-Nasseri S. Determinants of immunodominance for CD4 T cells. Curr Opin Immunol (2015) 34:9–15. doi:10.1016/j.coi.2014.12.005
17. Wright CA, Kozik P, Zacharias M, Springer S. Tapasin and other chaperones: models of the MHC class I loading complex. Biol Chem (2004) 385:763–78. doi:10.1515/BC.2004.100
18. Dong G, Wearsch PA, Peaper DR, Cresswell P, Reinisch KM. Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity (2009) 30:21–32. doi:10.1016/j.immuni.2008.10.018
19. Park B, Lee S, Kim E, Ahn K. A single polymorphic residue within the peptide-binding cleft of MHC class I molecules determines spectrum of tapasin dependence. J Immunol (2003) 170:961–8. doi:10.4049/jimmunol.170.9.4869-a
20. Zarling AL, Luckey CJ, Marto JA, White FM, Brame CJ, Evans AM, et al. Tapasin is a facilitator, not an editor, of class I MHC peptide binding. J Immunol (2003) 171:5287–95. doi:10.4049/jimmunol.171.10.5287
21. Garstka MA, Fritzsche S, Lenart I, Hein Z, Jankevicius G, Boyle LH, et al. Tapasin dependence of major histocompatibility complex class I molecules correlates with their conformational flexibility. FASEB J (2011) 25:3989–98. doi:10.1096/fj.11-190249
22. Abualrous ET, Fritzsche S, Hein Z, Al-Balushi MS, Reinink P, Boyle LH, et al. F pocket flexibility influences the tapasin dependence of two differentially disease-associated MHC class I proteins. Eur J Immunol (2015) 45:1248–57. doi:10.1002/eji.201445307
23. Lewis JW, Elliott T. Evidence for successive peptide binding and quality control stages during MHC class I assembly. Curr Biol (1998) 8:717–20. doi:10.1016/S0960-9822(98)70280-5
24. Suh WK, Derby MA, Cohen-Doyle MF, Schoenhals GJ, Fruh K, Berzofsky JA, et al. Interaction of murine MHC class I molecules with tapasin and TAP enhances peptide loading and involves the heavy chain alpha3 domain. J Immunol (1999) 162:1530–40.
25. Turnquist HR, Kohlgraf KG, Mcilhaney MM, Mosley RL, Hollingsworth MA, Solheim JC. Tapasin decreases immune responsiveness to a model tumor antigen. J Clin Immunol (2004) 24:462–70. doi:10.1023/B:JOCI.0000029118.51587.d9
26. Chen M, Bouvier M. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. EMBO J (2007) 26:1681–90. doi:10.1038/sj.emboj.7601624
27. Van Hateren A, James E, Bailey A, Phillips A, Dalchau N, Elliott T. The cell biology of major histocompatibility complex class I assembly: towards a molecular understanding. Tissue Antigens (2010) 76:259–75. doi:10.1111/j.1399-0039.2010.01550.x
28. Simone LC, Georgesen CJ, Simone PD, Wang X, Solheim JC. Productive association between MHC class I and tapasin requires the tapasin transmembrane/cytosolic region and the tapasin C-terminal Ig-like domain. Mol Immunol (2012) 49:628–39. doi:10.1016/j.molimm.2011.11.002
29. Fisette O, Wingbermuhle S, Tampe R, Schafer LV. Molecular mechanism of peptide editing in the tapasin-MHC I complex. Sci Rep (2016) 6:19085. doi:10.1038/srep19085
30. Roche PA, Cresswell P. Invariant chain association with HLA-DR molecules inhibits immunogenic peptide binding. Nature (1990) 345:615–8. doi:10.1038/345615a0
31. Pos W, Sethi DK, Call MJ, Schulze MS, Anders AK, Pyrdol J, et al. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell (2012) 151:1557–68. doi:10.1016/j.cell.2012.11.025
32. Guce AI, Mortimer SE, Yoon T, Painter CA, Jiang W, Mellins ED, et al. HLA-DO acts as a substrate mimic to inhibit HLA-DM by a competitive mechanism. Nat Struct Mol Biol (2013) 20:90–8. doi:10.1038/nsmb.2460
33. Doebele RC, Busch R, Scott HM, Pashine A, Mellins ED. Determination of the HLA-DM interaction site on HLA-DR molecules. Immunity (2000) 13:517–27. doi:10.1016/S1074-7613(00)00051-0
34. Pashine A, Busch R, Belmares MP, Munning JN, Doebele RC, Buckingham M, et al. Interaction of HLA-DR with an acidic face of HLA-DM disrupts sequence-dependent interactions with peptides. Immunity (2003) 19:183–92. doi:10.1016/S1074-7613(03)00200-0
35. Painter CA, Negroni MP, Kellersberger KA, Zavala-Ruiz Z, Evans JE, Stern LJ. Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc Natl Acad Sci U S A (2011) 108:19329–34. doi:10.1073/pnas.1108074108
36. Sadegh-Nasseri S, Chen M, Narayan K, Bouvier M. The convergent roles of tapasin and HLA-DM in antigen presentation. Trends Immunol (2008) 29:141–7. doi:10.1016/j.it.2008.01.001
37. Fleischmann G, Fisette O, Thomas C, Wieneke R, Tumulka F, Schneeweiss C, et al. Mechanistic basis for epitope proofreading in the peptide-loading complex. J Immunol (2015) 195:4503–13. doi:10.4049/jimmunol.1501515
38. Praveen PV, Yaneva R, Kalbacher H, Springer S. Tapasin edits peptides on MHC class I molecules by accelerating peptide exchange. Eur J Immunol (2010) 40:214–24. doi:10.1002/eji.200939342
39. Van Hateren A, Carter R, Bailey A, Kontouli N, Williams AP, Kaufman J, et al. A mechanistic basis for the co-evolution of chicken tapasin and major histocompatibility complex class I (MHC I) proteins. J Biol Chem (2013) 288:32797–808. doi:10.1074/jbc.M113.474031
40. Bouvier M, Wiley DC. Structural characterization of a soluble and partially folded class I major histocompatibility heavy chain/beta 2m heterodimer. Nat Struct Biol (1998) 5:377–84. doi:10.1038/nsb0598-377
41. Schmitt L, Boniface JJ, Davis MM, Mcconnell HM. Kinetic isomers of a class II MHC-peptide complex. Biochemistry (1998) 37:17371–80. doi:10.1021/bi9815593
42. Springer S, Doring K, Skipper JC, Townsend AR, Cerundolo V. Fast association rates suggest a conformational change in the MHC class I molecule H-2Db upon peptide binding. Biochemistry (1998) 37:3001–12. doi:10.1021/bi9717441
43. Schmitt L, Boniface JJ, Davis MM, Mcconnell HM. Conformational isomers of a class II MHC-peptide complex in solution. J Mol Biol (1999) 286:207–18. doi:10.1006/jmbi.1998.2463
44. Zarutskie JA, Sato AK, Rushe MM, Chan IC, Lomakin A, Benedek GB, et al. A conformational change in the human major histocompatibility complex protein HLA-DR1 induced by peptide binding. Biochemistry (1999) 38:5878–87. doi:10.1021/bi983048m
45. Hansen TH, Lybarger L, Yu L, Mitaksov V, Fremont DH. Recognition of open conformers of classical MHC by chaperones and monoclonal antibodies. Immunol Rev (2005) 207:100–11. doi:10.1111/j.0105-2896.2005.00315.x
46. Wieczorek M, Sticht J, Stolzenberg S, Gunther S, Wehmeyer C, El Habre Z, et al. MHC class II complexes sample intermediate states along the peptide exchange pathway. Nat Commun (2016) 7:13224. doi:10.1038/ncomms13224
47. Painter CA, Stern LJ. Conformational variation in structures of classical and non-classical MHCII proteins and functional implications. Immunol Rev (2012) 250:144–57. doi:10.1111/imr.12003
48. Guo HC, Jardetzky TS, Garrett TP, Lane WS, Strominger JL, Wiley DC. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature (1992) 360:364–6. doi:10.1038/360364a0
49. Wilson IA, Fremont DH. Structural analysis of MHC class I molecules with bound peptide antigens. Semin Immunol (1993) 5:75–80. doi:10.1006/smim.1993.1011
50. Chen Y, Sidney J, Southwood S, Cox AL, Sakaguchi K, Henderson RA, et al. Naturally processed peptides longer than nine amino acid residues bind to the class I MHC molecule HLA-A2.1 with high affinity and in different conformations. J Immunol (1994) 152:2874–81.
51. Urban RG, Chicz RM, Lane WS, Strominger JL, Rehm A, Kenter MJ, et al. A subset of HLA-B27 molecules contains peptides much longer than nonamers. Proc Natl Acad Sci U S A (1994) 91:1534–8. doi:10.1073/pnas.91.4.1534
52. Tynan FE, Borg NA, Miles JJ, Beddoe T, El-Hassen D, Silins SL, et al. High resolution structures of highly bulged viral epitopes bound to major histocompatibility complex class I. Implications for T-cell receptor engagement and T-cell immunodominance. J Biol Chem (2005) 280:23900–9. doi:10.1074/jbc.M503060200
53. Hassan C, Chabrol E, Jahn L, Kester MG, De Ru AH, Drijfhout JW, et al. Naturally processed non-canonical HLA-A*02:01 presented peptides. J Biol Chem (2015) 290:2593–603. doi:10.1074/jbc.M114.607028
54. Bade-Doding C, Theodossis A, Gras S, Kjer-Nielsen L, Eiz-Vesper B, Seltsam A, et al. The impact of human leukocyte antigen (HLA) micropolymorphism on ligand specificity within the HLA-B*41 allotypic family. Haematologica (2011) 96:110–8. doi:10.3324/haematol.2010.030924
55. Madden DR, Garboczi DN, Wiley DC. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell (1993) 75:693–708. doi:10.1016/0092-8674(93)90490-H
56. Smith KJ, Reid SW, Stuart DI, Mcmichael AJ, Jones EY, Bell JI. An altered position of the alpha 2 helix of MHC class I is revealed by the crystal structure of HLA-B*3501. Immunity (1996) 4:203–13. doi:10.1016/S1074-7613(00)80429-X
57. Gunther S, Schlundt A, Sticht J, Roske Y, Heinemann U, Wiesmuller KH, et al. Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proc Natl Acad Sci U S A (2010) 107:22219–24. doi:10.1073/pnas.1014708107
58. Schlundt A, Gunther S, Sticht J, Wieczorek M, Roske Y, Heinemann U, et al. Peptide linkage to the alpha-subunit of MHCII creates a stably inverted antigen presentation complex. J Mol Biol (2012) 423:294–302. doi:10.1016/j.jmb.2012.07.008
59. Mohan JF, Unanue ER. A novel pathway of presentation by class II-MHC molecules involving peptides or denatured proteins important in autoimmunity. Mol Immunol (2013) 55:166–8. doi:10.1016/j.molimm.2012.10.024
60. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A (2010) 107:10978–83. doi:10.1073/pnas.1006545107
61. Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med (2011) 208:2375–83. doi:10.1084/jem.20111502
62. Zhou Z, Jensen PE. Structural characteristics of HLA-DQ that may impact DM editing and susceptibility to type-1 diabetes. Front Immunol (2013) 4:262. doi:10.3389/fimmu.2013.00262
63. Zhou Z, Reyes-Vargas E, Escobar H, Rudd B, Rockwood AL, Delgado JC, et al. Type 1 diabetes associated HLA-DQ2 and DQ8 molecules are relatively resistant to HLA-DM mediated release of invariant chain-derived CLIP peptides. Eur J Immunol (2016) 46:834–45. doi:10.1002/eji.201545942
64. Yin L, Trenh P, Guce A, Wieczorek M, Lange S, Sticht J, et al. Susceptibility to HLA-DM protein is determined by a dynamic conformation of major histocompatibility complex class II molecule bound with peptide. J Biol Chem (2014) 289:23449–64. doi:10.1074/jbc.M114.585539
65. Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol (2009) 10:1096–101. doi:10.1038/ni.1780
66. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet (2012) 44:291–6. doi:10.1038/ng.1076
67. Sollid LM, Pos W, Wucherpfennig KW. Molecular mechanisms for contribution of MHC molecules to autoimmune diseases. Curr Opin Immunol (2014) 31:24–30. doi:10.1016/j.coi.2014.08.005
68. Reich Z, Altman JD, Boniface JJ, Lyons DS, Kozono H, Ogg G, et al. Stability of empty and peptide-loaded class II major histocompatibility complex molecules at neutral and endosomal pH: comparison to class I proteins. Proc Natl Acad Sci U S A (1997) 94:2495–500. doi:10.1073/pnas.94.6.2495
69. Sieker F, Springer S, Zacharias M. Comparative molecular dynamics analysis of tapasin-dependent and -independent MHC class I alleles. Protein Sci (2007) 16:299–308. doi:10.1110/ps.062568407
70. Sieker F, Straatsma TP, Springer S, Zacharias M. Differential tapasin dependence of MHC class I molecules correlates with conformational changes upon peptide dissociation: a molecular dynamics simulation study. Mol Immunol (2008) 45:3714–22. doi:10.1016/j.molimm.2008.06.009
71. Narzi D, Becker CM, Fiorillo MT, Uchanska-Ziegler B, Ziegler A, Bockmann RA. Dynamical characterization of two differentially disease associated MHC class I proteins in complex with viral and self-peptides. J Mol Biol (2012) 415:429–42. doi:10.1016/j.jmb.2011.11.021
72. Bailey A, Van Hateren A, Elliott T, Werner JM. Two polymorphisms facilitate differences in plasticity between two chicken major histocompatibility complex class I proteins. PLoS One (2014) 9:e89657. doi:10.1371/journal.pone.0089657
73. Bailey A, Dalchau N, Carter R, Emmott S, Phillips A, Werner JM, et al. Selector function of MHC I molecules is determined by protein plasticity. Sci Rep (2015) 5:14928. doi:10.1038/srep14928
74. Mage MG, Dolan MA, Wang R, Boyd LF, Revilleza MJ, Robinson H, et al. The peptide-receptive transition state of MHC class I molecules: insight from structure and molecular dynamics. J Immunol (2012) 189:1391–9. doi:10.4049/jimmunol.1200831
75. Saini SK, Abualrous ET, Tigan AS, Covella K, Wellbrock U, Springer S. Not all empty MHC class I molecules are molten globules: tryptophan fluorescence reveals a two-step mechanism of thermal denaturation. Mol Immunol (2013) 54:386–96. doi:10.1016/j.molimm.2013.01.004
76. Fahnestock ML, Tamir I, Narhi L, Bjorkman PJ. Thermal stability comparison of purified empty and peptide-filled forms of a class I MHC molecule. Science (1992) 258:1658–62. doi:10.1126/science.1360705
77. Kurimoto E, Kuroki K, Yamaguchi Y, Yagi-Utsumi M, Igaki T, Iguchi T, et al. Structural and functional mosaic nature of MHC class I molecules in their peptide-free form. Mol Immunol (2013) 55:393–9. doi:10.1016/j.molimm.2013.03.014
78. Schumacher TN, Heemels MT, Neefjes JJ, Kast WM, Melief CJ, Ploegh HL. Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell (1990) 62:563–7. doi:10.1016/0092-8674(90)90020-F
79. Glithero A, Tormo J, Doering K, Kojima M, Jones EY, Elliott T. The crystal structure of H-2D(b) complexed with a partial peptide epitope suggests a major histocompatibility complex class I assembly intermediate. J Biol Chem (2006) 281:12699–704. doi:10.1074/jbc.M511683200
80. Hein Z, Uchtenhagen H, Abualrous ET, Saini SK, Janssen L, Van Hateren A, et al. Peptide-independent stabilization of MHC class I molecules breaches cellular quality control. J Cell Sci (2014) 127:2885–97. doi:10.1242/jcs.145334
81. Santambrogio L, Sato AK, Fischer FR, Dorf ME, Stern LJ. Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc Natl Acad Sci U S A (1999) 96:15050–5. doi:10.1073/pnas.96.26.15050
82. Potolicchio I, Chitta S, Xu X, Fonseca D, Crisi G, Horejsi V, et al. Conformational variation of surface class II MHC proteins during myeloid dendritic cell differentiation accompanies structural changes in lysosomal MIIC. J Immunol (2005) 175:4935–47. doi:10.4049/jimmunol.175.8.4935
83. Rabinowitz JD, Vrljic M, Kasson PM, Liang MN, Busch R, Boniface JJ, et al. Formation of a highly peptide-receptive state of class II MHC. Immunity (1998) 9:699–709. doi:10.1016/S1074-7613(00)80667-6
84. Natarajan SK, Assadi M, Sadegh-Nasseri S. Stable peptide binding to MHC class II molecule is rapid and is determined by a receptive conformation shaped by prior association with low affinity peptides. J Immunol (1999) 162:4030–6.
85. Grotenbreg GM, Nicholson MJ, Fowler KD, Wilbuer K, Octavio L, Yang M, et al. Empty class II major histocompatibility complex created by peptide photolysis establishes the role of DM in peptide association. J Biol Chem (2007) 282:21425–36. doi:10.1074/jbc.M702844200
86. Frayser M, Sato AK, Xu L, Stern LJ. Empty and peptide-loaded class II major histocompatibility complex proteins produced by expression in Escherichia coli and folding in vitro. Protein Expr Purif (1999) 15:105–14. doi:10.1006/prep.1998.0987
87. Carven GJ, Stern LJ. Probing the ligand-induced conformational change in HLA-DR1 by selective chemical modification and mass spectrometric mapping. Biochemistry (2005) 44:13625–37. doi:10.1021/bi050972p
88. Hopner S, Dickhaut K, Hofstatter M, Kramer H, Ruckerl D, Soderhall JA, et al. Small organic compounds enhance antigen loading of class II major histocompatibility complex proteins by targeting the polymorphic P1 pocket. J Biol Chem (2006) 281:38535–42. doi:10.1074/jbc.M606437200
89. Gupta S, Hopner S, Rupp B, Gunther S, Dickhaut K, Agarwal N, et al. Anchor side chains of short peptide fragments trigger ligand-exchange of class II MHC molecules. PLoS One (2008) 3:e1814. doi:10.1371/journal.pone.0001814
90. Zhou Z, Callaway KA, Weber DA, Jensen PE. Cutting edge: HLA-DM functions through a mechanism that does not require specific conserved hydrogen bonds in class II MHC-peptide complexes. J Immunol (2009) 183:4187–91. doi:10.4049/jimmunol.0901663
91. Rupp B, Gunther S, Makhmoor T, Schlundt A, Dickhaut K, Gupta S, et al. Characterization of structural features controlling the receptiveness of empty class II MHC molecules. PLoS One (2011) 6:e18662. doi:10.1371/journal.pone.0018662
92. Painter CA, Cruz A, Lopez GE, Stern LJ, Zavala-Ruiz Z. Model for the peptide-free conformation of class II MHC proteins. PLoS One (2008) 3:e2403. doi:10.1371/journal.pone.0002403
93. Yaneva R, Springer S, Zacharias M. Flexibility of the MHC class II peptide binding cleft in the bound, partially filled, and empty states: a molecular dynamics simulation study. Biopolymers (2009) 91:14–27. doi:10.1002/bip.21078
94. Carven GJ, Chitta S, Hilgert I, Rushe MM, Baggio RF, Palmer M, et al. Monoclonal antibodies specific for the empty conformation of HLA-DR1 reveal aspects of the conformational change associated with peptide binding. J Biol Chem (2004) 279:16561–70. doi:10.1074/jbc.M314315200
95. Pohlmann T, Bockmann RA, Grubmuller H, Uchanska-Ziegler B, Ziegler A, Alexiev U. Differential peptide dynamics is linked to major histocompatibility complex polymorphism. J Biol Chem (2004) 279:28197–201. doi:10.1074/jbc.C400128200
96. Fabian H, Huser H, Narzi D, Misselwitz R, Loll B, Ziegler A, et al. HLA-B27 subtypes differentially associated with disease exhibit conformational differences in solution. J Mol Biol (2008) 376:798–810. doi:10.1016/j.jmb.2007.12.009
97. Cuendet MA, Zoete V, Michielin O. How T cell receptors interact with peptide-MHCs: a multiple steered molecular dynamics study. Proteins (2011) 79:3007–24. doi:10.1002/prot.23104
98. Reboul CF, Meyer GR, Porebski BT, Borg NA, Buckle AM. Epitope flexibility and dynamic footprint revealed by molecular dynamics of a pMHC-TCR complex. PLoS Comput Biol (2012) 8:e1002404. doi:10.1371/journal.pcbi.1002404
99. Nurzia E, Narzi D, Cauli A, Mathieu A, Tedeschi V, Caristi S, et al. Interaction pattern of Arg 62 in the A-pocket of differentially disease-associated HLA-B27 subtypes suggests distinct TCR binding modes. PLoS One (2012) 7:e32865. doi:10.1371/journal.pone.0032865
100. Uchtenhagen H, Abualrous ET, Stahl E, Allerbring EB, Sluijter M, Zacharias M, et al. Proline substitution independently enhances H-2D(b) complex stabilization and TCR recognition of melanoma-associated peptides. Eur J Immunol (2013) 43:3051–60. doi:10.1002/eji.201343456
101. Nojima H, Takeda-Shitaka M, Kurihara Y, Adachi M, Yoneda S, Kamiya K, et al. Dynamic characteristics of a peptide-binding groove of human HLA-A2 class I MHC molecules: normal mode analysis of the antigen peptide-class I MHC complex. Chem Pharm Bull (Tokyo) (2002) 50:1209–14. doi:10.1248/cpb.50.1209
102. Knapp B, Omasits U, Bohle B, Maillere B, Ebner C, Schreiner W, et al. 3-Layer-based analysis of peptide-MHC interaction: in silico prediction, peptide binding affinity and T cell activation in a relevant allergen-specific model. Mol Immunol (2009) 46:1839–44. doi:10.1016/j.molimm.2009.01.009
103. Knapp B, Omasits U, Schreiner W, Epstein MM. A comparative approach linking molecular dynamics of altered peptide ligands and MHC with in vivo immune responses. PLoS One (2010) 5:e11653. doi:10.1371/journal.pone.0011653
104. Knapp B, Fischer G, Van Hemelen D, Fae I, Maillere B, Ebner C, et al. Association of HLA-DR1 with the allergic response to the major mugwort pollen allergen: molecular background. BMC Immunol (2012) 13:43. doi:10.1186/1471-2172-13-43
105. Bello M, Correa-Basurto J. Molecular dynamics simulations to provide insights into epitopes coupled to the soluble and membrane-bound MHC-II complexes. PLoS One (2013) 8:e72575. doi:10.1371/journal.pone.0072575
106. Ferrante A, Templeton M, Hoffman M, Castellini MJ. The thermodynamic mechanism of peptide-MHC class II complex formation is a determinant of susceptibility to HLA-DM. J Immunol (2015) 195:1251–61. doi:10.4049/jimmunol.1402367
107. Nojima H, Takeda-Shitaka M, Kurihara Y, Kamiya K, Umeyama H. Dynamic flexibility of a peptide-binding groove of human HLA-DR1 class II MHC molecules: normal mode analysis of the antigen peptide-class II MHC complex. Chem Pharm Bull (Tokyo) (2003) 51:923–8. doi:10.1248/cpb.51.923
108. Nojima H, Kanou K, Kamiya K, Atsuda K, Umeyama H, Takeda-Shitaka M. Dynamic influence of the two membrane-proximal immunoglobulin-like domains upon the peptide-binding platform domain in class I and class II major histocompatibility complexes: normal mode analysis. Chem Pharm Bull (Tokyo) (2009) 57:1193–9. doi:10.1248/cpb.57.1193
109. Fabian H, Loll B, Huser H, Naumann D, Uchanska-Ziegler B, Ziegler A. Influence of inflammation-related changes on conformational characteristics of HLA-B27 subtypes as detected by IR spectroscopy. FEBS J (2011) 278:1713–27. doi:10.1111/j.1742-4658.2011.08097.x
110. Beerbaum M, Ballaschk M, Erdmann N, Schnick C, Diehl A, Uchanska-Ziegler B, et al. NMR spectroscopy reveals unexpected structural variation at the protein-protein interface in MHC class I molecules. J Biomol NMR (2013) 57:167–78. doi:10.1007/s10858-013-9777-z
111. Hee CS, Beerbaum M, Loll B, Ballaschk M, Schmieder P, Uchanska-Ziegler B, et al. Dynamics of free versus complexed beta2-microglobulin and the evolution of interfaces in MHC class I molecules. Immunogenetics (2013) 65:157–72. doi:10.1007/s00251-012-0667-4
112. Hawse WF, Gloor BE, Ayres CM, Kho K, Nuter E, Baker BM. Peptide modulation of class I major histocompatibility complex protein molecular flexibility and the implications for immune recognition. J Biol Chem (2013) 288:24372–81. doi:10.1074/jbc.M113.490664
113. Yanaka S, Ueno T, Shi Y, Qi J, Gao GF, Tsumoto K, et al. Peptide-dependent conformational fluctuation determines the stability of the human leukocyte antigen class I complex. J Biol Chem (2014) 289:24680–90. doi:10.1074/jbc.M114.566174
114. Chou CL, Sadegh-Nasseri S. HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J Exp Med (2000) 192:1697–706. doi:10.1084/jem.192.12.1697
115. Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol (1995) 13:587–622. doi:10.1146/annurev.iy.13.040195.003103
116. Blanco-Gelaz MA, Suarez-Alvarez B, Diaz-Pena R, Lopez-Larrea C. HLA-B27 polymorphism at position 116 critically influences the association with TAP/tapasin, intracellular trafficking and conformational homodimers formation. Mol Immunol (2009) 46:1304–11. doi:10.1016/j.molimm.2008.11.023
117. Gao GF, Tormo J, Gerth UC, Wyer JR, Mcmichael AJ, Stuart DI, et al. Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature (1997) 387:630–4. doi:10.1038/42523
118. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol (2003) 4:913–9. doi:10.1038/ni961
119. Deng L, Cho S, Malchiodi EL, Kerzic MC, Dam J, Mariuzza RA. Molecular architecture of the major histocompatibility complex class I-binding site of Ly49 natural killer cell receptors. J Biol Chem (2008) 283:16840–9. doi:10.1074/jbc.M801526200
120. Harris MR, Lybarger L, Myers NB, Hilbert C, Solheim JC, Hansen TH, et al. Interactions of HLA-B27 with the peptide loading complex as revealed by heavy chain mutations. Int Immunol (2001) 13:1275–82. doi:10.1093/intimm/13.10.1275
121. Hulpke S, Baldauf C, Tampe R. Molecular architecture of the MHC I peptide-loading complex: one tapasin molecule is essential and sufficient for antigen processing. FASEB J (2012) 26:5071–80. doi:10.1096/fj.12-217489
122. Grandea AG III, Golovina TN, Hamilton SE, Sriram V, Spies T, Brutkiewicz RR, et al. Impaired assembly yet normal trafficking of MHC class I molecules in Tapasin mutant mice. Immunity (2000) 13:213–22. doi:10.1016/S1074-7613(00)00021-2
123. Gao B, Adhikari R, Howarth M, Nakamura K, Gold MC, Hill AB, et al. Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity (2002) 16:99–109. doi:10.1016/S1074-7613(01)00260-6
124. Schneeweiss C, Garstka M, Smith J, Hutt MT, Springer S. The mechanism of action of tapasin in the peptide exchange on MHC class I molecules determined from kinetics simulation studies. Mol Immunol (2009) 46:2054–63. doi:10.1016/j.molimm.2009.02.032
125. Saini SK, Schuster H, Ramnarayan VR, Rammensee HG, Stevanovic S, Springer S. Dipeptides catalyze rapid peptide exchange on MHC class I molecules. Proc Natl Acad Sci U S A (2015) 112:202–7. doi:10.1073/pnas.1418690112
126. Dalchau N, Phillips A, Goldstein LD, Howarth M, Cardelli L, Emmott S, et al. A peptide filtering relation quantifies MHC class I peptide optimization. PLoS Comput Biol (2011) 7:e1002144. doi:10.1371/journal.pcbi.1002144
127. Kienast A, Preuss M, Winkler M, Dick TP. Redox regulation of peptide receptivity of major histocompatibility complex class I molecules by ERp57 and tapasin. Nat Immunol (2007) 8:864–72. doi:10.1038/ni1483
128. Noe F, Horenko I, Schutte C, Smith JC. Hierarchical analysis of conformational dynamics in biomolecules: transition networks of metastable states. J Chem Phys (2007) 126:155102. doi:10.1063/1.2714539
129. Buchete NV, Hummer G. Coarse master equations for peptide folding dynamics. J Phys Chem B (2008) 112:6057–69. doi:10.1021/jp0761665
130. Prinz JH, Wu H, Sarich M, Keller B, Senne M, Held M, et al. Markov models of molecular kinetics: generation and validation. J Chem Phys (2011) 134:174105. doi:10.1063/1.3565032
131. Bowman GR, Pande VS, Noé F. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation. Heidelberg: Springer Science & Business Media (2014).
132. Scherer MK, Trendelkamp-Schroer B, Paul F, Perez-Hernandez G, Hoffmann M, Plattner N, et al. PyEMMA 2: a software package for estimation, validation, and analysis of Markov models. J Chem Theory Comput (2015) 11:5525–42. doi:10.1021/acs.jctc.5b00743
133. Weber DA, Evavold BD, Jensen PE. Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science (1996) 274:618–20. doi:10.1126/science.274.5287.618
134. Kropshofer H, Arndt SO, Moldenhauer G, Hammerling GJ, Vogt AB. HLA-DM acts as a molecular chaperone and rescues empty HLA-DR molecules at lysosomal pH. Immunity (1997) 6:293–302. doi:10.1016/S1074-7613(00)80332-5
135. Grandea AG III, Van Kaer L. Tapasin: an ER chaperone that controls MHC class I assembly with peptide. Trends Immunol (2001) 22:194–9. doi:10.1016/S1471-4906(01)01861-0
136. Rosen MK, Gardner KH, Willis RC, Parris WE, Pawson T, Kay LE. Selective methyl group protonation of perdeuterated proteins. J Mol Biol (1996) 263:627–36. doi:10.1006/jmbi.1996.0603
Keywords: antigen presentation, major histocompatibility complex, HLA, protein dynamics, peptide exchange, tapasin, HLA-DM, adaptive immunity
Citation: Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F and Freund C (2017) Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 8:292. doi: 10.3389/fimmu.2017.00292
Received: 13 December 2016; Accepted: 28 February 2017;
Published: 17 March 2017
Edited by:
Joern Martin Werner, University of Southampton, UKCopyright: © 2017 Wieczorek, Abualrous, Sticht, Álvaro-Benito, Stolzenberg, Noé and Freund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Freund, christian.freund@fu-berlin.de
†These authors have contributed equally to this work.