 Rob J. W. Arts
Rob J. W. Arts Leo A. B. Joosten
Leo A. B. Joosten Mihai G. Netea
Mihai G. Netea- 1Department of Internal Medicine, Radboud Center for Infectious Diseases, Radboud University Medical Center, Nijmegen, Netherlands
- 2Department of Medical Genetics, Iuliu Haţieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 3Department for Genomics and Immunoregulation, Life and Medical Sciences Institute (LIMES), University of Bonn, Bonn, Germany
During induction of trained immunity, monocytes and macrophages undergo a functional and transcriptional reprogramming toward increased activation. Important rewiring of cellular metabolism of the myeloid cells takes place during induction of trained immunity, including a shift toward glycolysis induced through the mTOR pathway, as well as glutaminolysis and cholesterol synthesis. Subsequently, this leads to modulation of the function of epigenetic enzymes, resulting in important changes in chromatin architecture that enables increased gene transcription. However, in addition to the beneficial effects of trained immunity as a host defense mechanism, we hypothesize that trained immunity also plays a deleterious role in the induction and/or maintenance of autoimmune and autoinflammatory diseases if inappropriately activated.
Introduction
Since the introduction of the term trained immunity for the non-specific memory of the innate immune system in 2011 (1), an increasing number of studies have investigated its role in homeostasis and disease. Innate immune memory has been described in plants and non-vertebrates for a relatively long time, representing the immune adaptation in these species that lack an adaptive immune system (2). In vertebrates, it was firstly shown that NK cells possess a non-specific memory that contributes in the innate host defense (3, 4). Later also monocytes and macrophages were shown to have a non-specific memory (5, 6). When human monocytes were exposed in vitro with microbial components such as β-glucan of the Candida albicans cell wall or the Bacillus Calmette-Guérin (BCG) vaccine, and a week later restimulated with non-related stimuli, the capacity of cells to produce cytokines was increased compared with non-trained (naive) cells (3, 5). Moreover, when mice were challenged (or “trained”) with β-glucan or BCG in vivo, they showed lower mortality after lethal C. albicans or Staphyococcus aureus infections, a process that was largely dependent on the innate immune system (3, 5, 6). Moreover, in humans, BCG vaccination results in trained monocytes with increased responsiveness against microorganisms, which probably explains at least partly the lower mortality from a variety of infections in vaccinated children (3, 6–9).
There are a set of important characteristics that distinguish innate and adaptive immune memory processes. In case of the classical adaptive immune memory, a specific antigen is recognized and specific T- and/or B-lymphocytes expand that specifically respond to that antigen. The breadth of needed responses is insured by gene recombination of V-D-J system. Upon reinfection, long-term memory cells will respond very specifically to the same antigen: thus, adaptive memory is both specific and enhanced, compared to the primary response. In contrast, the molecular substrate of trained immunity is epigenetically mediated, with genome-wide changes in histone marks and thus chromatin architecture playing a major role in the change of the phenotype of monocytes and macrophages (6, 10). By stimulation of an innate immune cell (or its precursors) an enhanced non-specific immunological reaction will be evoked due to differences in which gene transcription takes place due to changes in chromatin configuration (11). Innate immune memory (trained immunity) will thus evoke an increased response, but which is non-specific.
Also changes in cellular metabolism of trained monocytes and macrophages were shown to be a major important component of the trained immunity phenotype. Induction of glycolysis in an mTOR/HIF-1α-dependent manner is indispensible for the induction of trained immunity (12, 13), just as induction of glutamine metabolism that results in fumarate accumulation (14, 15). Interestingly, there is a tight link between metabolic and epigenetic changes that occur in cells (16). We have for example recently shown, that induction of glycolysis is essential for the induction of epigenetic changes seen in trained immunity. When glycolysis was inhibited also the induction of trained immunity by its epigenetic changes was inhibited (12, 13). How induction of glycolysis exactly leads to epigenetic changes is still unclear, but accumulation of acetyl-CoA has been suggested as a mechanism, inducing histone acetylation (11). Moreover, also accumulation of fumarate (one of the intermediates of the TCA cycle) was shown to induce trained immunity by inhibiting histone demethylases and therefore inducing epigenetic changes in human monocytes (14). Recently, we have shown that induction of the cholesterol synthesis pathway, which results in mevalonate accumulation, is also one of the contributors to the induction of epigenetic changes in trained immunity (17). However, this remains a rather new topic of research and more research has to be performed to better understand the exact link between metabolism en epigenetics and its role in trained immunity.
Induction of trained immunity may be important for diseases characterized by defective function of innate immune responses. We have recently shown that the induction of trained immunity by β-glucan is able to counteract the epigenetic changes induced in monocytes in postsepsis immunoparalysis (18): this may represent a potential new therapy. However, trained immunity could also be the cause or play a role in maintaining disease activity in diseases characterized by excessive inflammation, although this needs to be investigated in future studies. In this review, we give an overview of literature that provides indications for the potential role of trained immunity in autoimmune and autoinflammatory diseases. We focus on the possibility that increased function of the myeloid cells in these conditions may be mediated by epigenetic rewiring, and thus possibly a trained immunity phenotype. Another possible mechanism how monocytes of patients with autoimmune and autoinflammatory diseases are more responsive is by specific genetic variations: this hypothesis has been extensively discussed in other reviews, and it will therefore be not presented here. Importantly, adaptive memory responses are also very well known to play a crucial part in autoimmune diseases. However, the role of the adaptive immune system in these diseases has been discussed elsewhere in very good recent review articles (19–21) and was therefore not included in this review.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is one of the most common inflammatory arthritis. The pathogenesis of this autoimmune disease is complex and remains partially elusive; it is partly genetically regulated, but environmental factors also play a role, finally leading to synovial inflammation and destruction of bone and cartilage (22). Initially, the role of the adaptive immune system was mainly studied, as the loss of self tolerance of CD4+ T lymphocytes was considered the main defect in RA (23). However, this paradigm is changing and nowadays the focus has also shifted to the role of the innate immune system in RA (24–26). Innate immune cells have been defined as being the cause of the tissue-damaging inflammatory lesions, with macrophages as main producers of proinflammatory cytokines. Macrophages are considered the cause of cartilage and bone destruction by inducing inflammation and regulating osteoclast activity, e.g., by inducing RANKL production (27–30). The number of infiltrated macrophages seems to negatively correlate with response to therapy and the degree of joint erosion (31, 32) and patients with highly active RA show a greater amount of M1-type macrophages in the synovial fluid compared to those with lower disease scores (33). Targeting the cytokines produced by macrophages [e.g., tumor necrosis factor α (TNFα)] has been proven to be a very successful treatment (34).
When circulating monocytes from RA patients were analyzed, gene expression of several proinflammatory cytokines was increased (35, 36). CD14+ monocytes showed an increased expression of CD11b, and when ex vivo stimulated, increased production of IL-1β and IL-6 is observed (37). Interestingly, PI3K/mTOR and MAPK signaling pathways are also activated in RA monocytes (36, 38), and inhibiting mTOR reduced synovial osteoclast formation and protected against local bone erosions and cartilage loss (39, 40). In addition, epigenetic changes have been suggested to play a role, although no genome wide epigenetic analyses in monocytes/macrophages have been performed. In RA synovial tissue the HAT/HDAC balance is moved into the direction of histone acetylation (41). HDAC1 and HDAC2 activity has been specifically analyzed in synovial fibroblasts (not in synovial macrophages), but no difference was found (42). Interestingly, etanercept and adalimumab downregulate trimetylation of H3K4, H3K27, H3K36, and H3K79, as well as acetylation of histone 3 and 4 at the promoter site of CCL2 (MCP-1) in monocytes which correlated with RA disease activity (43). In contrast, a recent study did not identify an increase in H3K4me3 at TNFA and IL6 genes of circulating monocytes in RA patients (44). In addition, the metabolic changes that occur in trained immunity also occur in macrophages from RA patients: upon stimulation with LPS higher levels of ATP are present in the patient-derived macrophages (45). Glycolysis is also upregulated, as are rate-limiting enzymes (e.g., PKM2, PFKFB3, and HK2) and the glucose transporters GLUT1 and GLUT3 (46). Functionally, glucose uptake and oxygen consumption are increased (46), whereas accumulation of glutamate, succinate, and fumarate are present as signs of a highly metabolic active state of an RA proinflammatory macrophage (47, 48), similar to a trained immunity phenotype (12, 14).
Hence, multiple similarities exist between tissue macrophages or circulating monocytes in RA and trained monocytes or macrophages. Interestingly, in RA patients an increased risk for atherosclerotic disease is seen (49), which corresponds with the hypothesis that trained macrophages contribute to the development of atherosclerotic lesions (50). How the training in RA is induced remains unknown, although several danger-associated molecular patterns (DAMPs), that could be released by local sterile inflammation and tissue damage in the joint have been proposed to induce trained immunity (51), just as the increased production of IL-32 (52). Future studies to investigate these mechanisms are warranted.
Systemic Lupus Erythematosus (SLE)
In SLE several immunological abnormalities are present. The production of a number of antinuclear antibodies is the most prominent and well-known process, and they are used for diagnostic purposes. However, monocytes and macrophages also play a prominent role in the disease activity, which increasingly becomes a topic of research (53–55). In SLE patients, macrophages are characterized by a proinflammatory status and show an increased production of proinflammatory cytokines, such as IFNα, TNFα, and IL-6 (56–58). CD16+ monocytes express more CD80, CD86, HLA-DR, and CX3CR1 (59) and also expression data of CD14+ and non-classical monocytes demonstrate a proinflammatory phenotype (60–64). Inflammasome and interferon-regulated genes that were regulated by IRF1 (65), with IFNα as one of the main mediators (66–70), are induced in SLE monocytes. Moreover, monocytes show an improved antigen presentation capacity and are easily activated (71). It is therefore suggested that monocytes and macrophages are better capable of presenting self-antigens to autoreactive T cells and therefore inducing or maintaining disease activity (56). Interestingly, a SNP in the IL1B gene that was associated with lower IL-1β production upon LPS stimulation was protective for SLE (72).
Epigenetic modulation in monocytes clearly plays a role in SLE. Histones around the TNFA genomic region are highly acetylated in SLE monoyctes resembling high accessibility for transcription (73). Also whole-genome assessment of histone H4 of monocytes reveals hyperacetylation (74–76) and also at enhancers of monocytes SLE-specific alterations for H3K4me3 and H3K27me3 could be determined (77). Whole-genome epigenetic analysis of H3K4me3 of primary monocytes of SLE patients reveals a strong association with inflammation and immune responses related genes (78, 79). When H3K4me3 modulations were related to gene promoter site, and these were compared with the major epigenetically modulated promoter sites in in vitro β-glucan-trained monocytes (10), the M2 and M3 clusters (defined in β-glucan training) were also significantly modulated in SLE monocytes (Table 1). In another article in which monocytes from six SLE patients were compared with six controls, 136,573 DNase hypersensitivity sites (DHS) were defined. Of these DHS, 4,583 showed SLE-specific changes for H3K4me3 and 1,714 for H3K27me3 at promoter sites. At enhancer site theses numbers were 12,109 and 3,046, respectively (55). Transcription factor binding motifs analysis revealed PU.1 and CEBPB as main transcription factors related to the H3K4me3 induced genomic areas, just as seen for β-glucan-induced training (10). Also BLIMP1 and interferon-related transcription factors as STAT1/6, and IRF1/4/8 were defined (55).
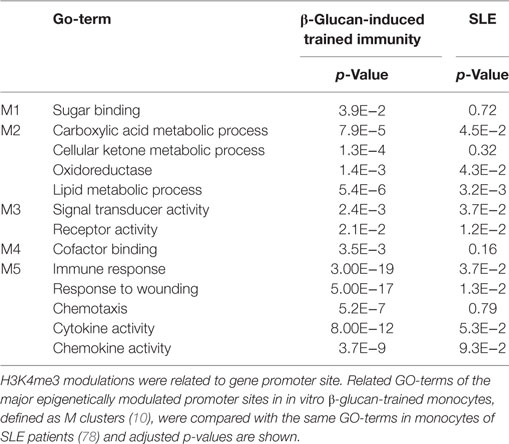
Table 1. Comparison of H3K4me3-related GO-terms in β-trained monocytes and monocytes of SLE patients.
In trained immunity induced by β-glucan and also by BCG, it has been shown that important changes in cellular metabolism are induced, and that changing concentrations of metabolites play a role in the modulation of epigenetic rewiring (10, 12–14).
In immune cells from SLE patients, metabolic changes are present and might influence the epigenetic landscape as well (80). SAMs are cofactors in DNA and histone methyltranferase reactions and in T cells in SLE SAMs have been shown to be modulated and influence the epigenetic landscape, both at DNA (81, 82) and histone methylation level (83, 84). Histone demethylases need α-ketogluterate and Fe2+ as cofactors, which were shown to be modulated in T cells of SLE patients (84). Acetyl CoA functions as an acetyl donor for histone acetyl transferases. Histone acetylation is defective in SLE, and when in mice in an SLE model HDACs were inhibited, this showed a positive effect on disease activity and nephritis (85–87).
Finally, also activation of the mTOR pathway is a marker of disease and of onset of disease in SLE. The mTOR pathway is activated in many immune cells, among others also in monocytes (88–90). Inhibition of mTOR with rapamycin has shown beneficial effects on several outcome measures, but monocytes/macrophages were unfortunately not part of the analysis [reviewed in Ref. (88)].
The modulation of monocytes from the peripheral blood could be due to changes induced in the bone marrow. Gene expression analysis of mononuclear bone marrow cells from SLE patients reveals that ERK, JNK, and p38 MAP kinases and STAT3 are significantly upregulated (91). As anti-dsDNA antibodies can bind to TLR4 and activate the NLRP3 inflammasome, it is tempting to speculate whether they might be the factor inducing trained immunity in SLE patients (92, 93).
Sjögren’s Syndrome (SS)
Sjögren’s syndrome is a chronic autoimmune disease characterized by salivary and lacrimal gland dysfunction (94). Also in the case of SS, clues can be found in literature to suggest that a trained immunity profile may characterize its myeloid cells. Immune cell infiltrates of the salivary glands in SS mainly contain macrophages and DCs with increased IL-12 and IL-18 expression and depletion of macrophages in the gland tissue improved disease symptoms in a mouse model (95–97). When peripheral CD14+ monocytes were stimulated with apoptotic cells, they showed an increased production of TNFα or IL-1β and a decreased production of IL-10, thus displaying a proinflammatory profile (98, 99). Also when monocyte-derived dendritic cells were stimulated with LPS a clear proinflammatory profile was seen, with among others higher levels of TNFα, MIG, IFNα, IL-6, IL-12, MIP1α/β, MCP1 compared to healthy volunteers (100). In non-stimulated circulating monocytes, type I IFN-related genes were higher expressed compared to control monocytes, which correlated with the induction of B-cell activating factor (BAFF) (101–103). When monocytes were stimulated with IFNγ, increased IL-6 and BAFF production was seen as well (104). However, others have reported different effects, with decreased cytokine production upon PPD stimulation of PBMCs isolated from SS patients (105). Furthermore, NFκB activation is promoted by reduced IκBα expression in Sjögren monocytes (106). Increased STAT1 is seen in SS monocytes (107), similar to constitutive STAT5 activation (108).
No epigenetic assessment of monocytes from SS patients has been performed, apart from increased expression of miRNA 146a and 181a in PBMCs (109–113), and miR-34b-3p, miR-4701-5p, miR-609, miR-300, miR-3162-3p, and miR-877-3p upregulation in monocytes. These miRNAs collectively suppress TGFβ signaling, as opposed to proinflammatory interleukin-12 and Toll-like receptor/NFκB pathways (114). Analysis of cellular metabolic processes and induction of mTOR activation in monocytes/macrophages in SS has not been performed, but treatment with a rapamycin nanoparticle reduced destruction of lacrimal glands in a mouse model (115).
Behçet’s Disease (BD)
Behçet’s disease is an inflammatory disorder that is characterized by oral en genital lesions, arthritis, and uveitis. The exact cause of BD is unknown (116). Myeloid cells play an important role in Behçet’s disease activity, as granulocyte and monocyte adsorption apheresis reduced disease symptoms in two patients (117). Furthermore, peripheral monocytes of BD patients are activated and produce more proinflammatory cytokines, which might be a first clue for the presence of a trained immunity phenotype in monocytes of BD patients (118–122). The P2X7 receptor, involved in the activation of IL-1β production, has an increased expression on monocytes in BD, which is regulated by TNFα (123), while the expression of TLR2 and TLR4 is also upregulated (124, 125). The response of CD14+ monocytes from BD patients to IFNγ showed increased production of CXCL9 and CXCL10; however, the CXCL10 production might be increased due to dysregulated posttranslational regulation (126). A whole-genome DNA methylation profiling of monocytes revealed 383 CpG sites to be differently regulated compared to healthy controls (and only 123 were found in CD4+ T cells), with cytoskeleton modulation as one of the main regulated pathways (127).
Systemic Sclerosis (SSc)
Systemic sclerosis is a complex autoimmune disease with extensive fibrosis, vascular alterations and immune activation among its principal features (128). Also in SSc the monocyte compartment appears to play an important role. CD14+ monocytes levels are increased in peripheral blood and in skin infiltrates (129, 130). An increased type I IFN signature was observed in early and definite SSc patient monocytes, which correlated with increased BAFF mRNA expression. Production of other cytokines, chemokines and their receptors (among others IL6, TNFα, and TGFβ) is upregulated as well (131–145). Monocytes of SSc patients produce more reactive oxygen species (146, 147), whereas nitric oxide production was decreased (148). SSc PBMCs produce more IL-8 and CCL18 when stimulated with IgG (149). In the vascular alterations present in SSc patients, monocytes appear to play a role as they can differentiate into fibroblast-like cells and produce extracellular matrix (modulating) proteins, resulting in a proangiogenic but impaired vasculogenic environment (132, 150–156). The macrophage activation markers CD163 and CD204 are more expressed in SSc (129, 132), which by some authors has been linked to a M2 macrophage phenotype with anti-inflammatory and profibrotic features (157). The alveolar macrophages in SSc patients with pulmonary fibrosis have a strong M2 phenotype with expression of CCL17, CCL18, and CCL22 and increased activation of STAT 3 (158). Increased TNFα production by these cells was also reported (159).
On the level of epigenetic profiles, hardly any data are available in monocytes or macrophages from SSc patients. The only data available in monocytes show that histone demethylation plays a role in the production of tissue inhibitor of metalloproteinases 1 in the presence of TLR8 stimulation (160). However, given the broad amount of data present in the literature, an epigenetic analysis on circulating monocytes in SSc would be a logical next step.
Wegener’s Granulomatosis (WG)
Wegener’s granulomatosis is a systemic inflammatory disorder characterized by vasculitis of the small- and medium-size vessels in many organs. The exact cause remains unknown, but monocytes have been suggested to play a role in disease activity (161).
Monocytes of WG patients are activated, as shown by increased CD11b and CD64 expression, and increased concentrations of neopterin and IL-6 are found in the serum (162). The expression of adhesion molecules is increased on monocytes from WG patients (163). Interestingly, anti-PR3 induces cytokine production by monocytes and it was shown that when monocytes are primed with ANCA or PR3 antibodies, their responses to LPS and LTA stimulation increase, with more TNFα, IL-6 and IL-8 production (164) and increased expression of CD14, CD18, and several PRRs (165, 166). Monocytes of WG patients that did not respond to methotrexate showed increased intracellular levels of IL-12 and TNFα (but no IL-8) that normalized after cyclophosphamide treatment (167). Also IL-8, IL-12, and MCP-1 production was increased in WG monocytes (166, 168–170). In contrast, other studies have shown that monocytes from WG patients are shown to produce less reactive oxygen species and have impaired phagocytosis (171) and in one report, monocytes of WG patients were stimulated with LPS, they produced less TNFα compared to healthy controls (172). No studies on epigenetic modulation of cellular metabolism of monocytes in WG are available. In order to assess whether a trained immunity phenotype is indeed responsible for the functional changes observed, such studies should be performed.
Sarcoidosis
Sarcoidosis is a complex systemic granulomatous disorder of unknown etiology. Circulating monocytes op sarcoidosis patients produce less IL-10 (173), express more CD16+ (174, 175), BAFF (166), TLR2 and TLR4 (176), IL-2R (177), adhesion molecules (178), produce more proinflammatory cytokines (176, 179–181) and oxygen radicals (182), and have increased phagocytic activity (183), thus showing an increased activated phenotype (184). Moreover, monocytes of sarcoidosis patients are more likely to form giant cells (185). Fibrin, which is newly formed in granulomas, is able to induce IL-1β production and might therefore serve as an inductor of trained immunity (186).
RNA-sequencing analysis of monocytes of sarcoidosis patients revealed several differentially expressed genes, with enrichment of ribosome, phagocytosis, lysosome, proteasome, oxidative phosphorylation, and metabolic pathways are the main pathways (187). No epigenetic analysis of monocytes in sarcoidosis has been done so far (188).
Type 1 Diabetes Mellitus (T1DM)
Type 1 diabetes mellitus is an important autoimmune disease resulting in profound defects in glucose metabolism. The exact underlying mechanism is unknown, but autoimmune destruction of β-cells of the pancreas is the cause of insulin deficiency (189). Monocytes and macrophages are thought to play a key role in the development of T1DM (190, 191). When peripheral blood monocytes from recent-onset T1DM patients were assessed, more CD14+CD16+ monocytes were found, which was negatively correlated with insulin and C-peptide serum levels. Furthermore, these monocytes showed higher expression of HLA-DR and CD86, and showed an activated proinflammatory phenotype (192–196). In recent-onset T1DM patients, plasma TNFα levels were higher which correlated with CD14 expression (197). However, another study showed decreased total number of monocytes in T1DM (198). Serum levels of MIF and MCP-1 are also increased (199). Prior to development of T1DM, children show an IFN transcriptional signature in PBMCs, which might be a result of a recent upper respiratory infection. Increased expression of SIGLEC-1 on CD14+ monocytes was also found (200). Furthermore, it was shown that IL-1β plays a central role in the inflammation seen in T1DM (201) and that during the development of T1DM TLR-induced IL-1β and IL-6 production from monocytes is enhanced (202). Importantly, inhibiting IL-1β production or IL-1β signaling can improve T1DM outcome (203–206). In contrast, monocyte-derived DCs did not appear to be affected in recent-onset T1DM (191).
Epigenetics has been proposed to play a role in T1DM as well (207). Assessment of DNA methylation profiles of 32 T1DM patients versus 31 healthy individuals revealed 153 hypomethylated and 225 hypermethylated loci in whole blood, and, respectively, 155 and 247 in monocytes. However, whether these are causative to the disease or a consequence of the pathological process remains unclear (208). In another study methylation variable positions were assessed in monocytes prior to development of T1DM: 132 positions were identified and associated with a gene, with important immunology-related genes as HLA-DQB1, HLA-DRB1, NFKB1A, and TNF as major examples (209). Also important histone modifications were seen in H3K9ac in monocytes, but also these were more likely to be induced after development of the disease (210). However, in one study, H3K9ac at HLA-DRB1 and HLA-DQB1 of monocytes was shown to correlate with T1DM susceptibility prior to disease (211). Differences in H3K9me3 in inflammatory and autoimmune-related pathways were found in lymphocytes of T1DM patients, but no differences were found in monocytes (212).
Cellular metabolism of monocytes in T1DM prior to disease onset is still poorly known. Only one study revealed changes in the transcriptional signature of cell metabolism, cell survival, and oxidative stress in monocytes of recent-onset DM1. Interestingly, one of the main induced genes was HIF1A (213), although mTOR does not appear among the significantly differently regulated genes.
Lastly, as BCG is able to induce trained immunity, it could be expected that BCG-vaccinated children are at higher risk to develop T1DM. However, in an observational trial no such correlation has been found (214, 215). Interestingly, in a randomized controlled trial, BCG was suggested to have a beneficial effect on insulin production, as induction of TNF production resulted in reduced autoimmune phenotype of innate immune cells and induction of Tregs (216, 217).
Autoinflammatory Disorders
Autoinflammatory disorders or periodic fever syndromes consist of a set of diseases that are characterized by periodic episodes of fever and inflammation. Common symptoms are joint pain, rash, abdominal pain, and long-term disease can result in amyloidosis. These diseases are not induced by autoantibodies or autoreactive T-cells but are the result of a hyperfunctional innate immune system, which is due to genetic defects. The familial autoinflammatory syndromes are generally rare, and the pathogenesis is not well understood for some of them (218). Here, we argue that trained immunity could be a likely contributor in these diseases.
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) is a multisystemic autoinflammatory condition associated with heterozygous TNFRSF1A mutations, presenting with a variety of clinical symptoms, many of which still unexplained. TRAPS monocytes shown an inflammatory baseline state, with enhanced IL1B and IL1R1 gene expression, also in non-active disease, whereas IFN and TGFβ are downregulated (219, 220). Also CD16 expression is upregulated (221) and MAPK can be spontaneously activated (220). Interestingly, a TRAPS patient with a monocytic fasciitis has been successfully treated with tacrolimus (an mTOR inhibitor) (222).
Cryopyrin-associated periodic syndromes (CAPS) are caused by a mutation in NLRP3 inflammasome, resulting in increased IL-1β and IL-18 production, but impaired production of the anti-inflammatory IL-6 and IL-1RA cytokines (223, 224). Interestingly, although the genetic defect is the main cause of these abnormalities, epigenetic analysis of CAPS monocytes revealed that DNA methylation was also affected, resulting at increased expression of inflammasome-related genes. When CAPS patients were treated with IL-1 neutralizing therapies, their methylation profile reversed toward that of healthy controls (225).
In familial Mediterranean fever (FMF), ex vivo LPS-stimulated PBMCs and monocytes produce more IL-1α and β, IL-6, IL-8, IL-12, IL-18, and TNFα, and in non-stimulated PBMCs higher production of IL-6 and TNFα was found (226–230), and also expression of CD11b was increased (231).
Hyper-IgD syndrome (HIDS) is an autoinflammatory disorder caused by a mutation in mevalonate kinase (232, 233), in which patients experience periodic attacks of sterile inflammation with symptoms such as fever, skin lesions, lymphadenopathy, and arthralgia (234). Interestingly, PBMCs of HIDS patients produce more cytokines in unstimulated as well as stimulated state and the IgD itself was able to induce cytokine production (235–238). In a whole-blood transcriptome analysis several glycolysis-related genes were higher expressed in HIDS patients, which decreased after canakinumab treatment (239). In addition, we recently have shown broad genome-wide changes in the H3K27Ac marker in monocytes of HIDS patients, which are due to mevalonate accumulation and are believed to express a trained immunity profile (Bekkering et al., 2018 Cell in press).
The common factor in these autoinflammatory syndromes is overproduction of IL-1β, which is one of the reasons why anti-IL-1 therapeutic approaches are successful (240). As IL-1β is considered as a causal factor and plausibly plays a role in maintaining disease, accumulating evidence suggests that IL-1β is an important inducer of trained immunity. Interestingly, old studies have shown that injection of IL-1β in mice prevented death from subsequent bacterial and fungal infections (241, 242). It is tempting to hypothesize that IL-1β induces trained immunity and prevent from the subsequent infections, by epigenetically modifying monocytes and macrophages resulting in more proinflammatory immune cells. To further substantiate this hypothesis, we have recently performed experiments in which human monocytes are ex vivo trained with IL-1β. This indeed resulted in trained monocytes that produced more IL-6 and TNFα upon restimulation with LPS, and also increased H3K4me3 occupancy at the promoters of IL6 and TNFA was observed (Arts et al., Cell Host Microbe 2018, in press). While IL-1 can induce beneficial effects in infections through induction of trained immunity, a negative consequence could be induction of overinflammation, which might result in an autoinflammatory disease. This thus opens a potential new field of research, where trained immunity in these autoinflammatory diseases should be further evaluated.
Chronic Granulomatous Disease (CGD)
Chronic granulomatous disease is an inherited immunological disorder, in which intracellular superoxide radical production is deficient. Although CGD is an immunodeficiency, it also has autoinflammatory characteristics, which is why it is discussed here as well. Normally CGD presents in the first years of life with severe recurrent bacterial and fungal infections, but it can also present later in life (243). CGD phagocytes are impaired in destroying phagocytozed microorganisms, rendering the patients susceptible to bacterial and fungal infections. Besides this immunodeficiency, CGD patients suffer from various auto inflammatory symptoms, such as granuloma formation and Crohn-like colitis (244). Also monocytes from CGD patients display a proinflammatory phenotype with increased secretion of inflammasome-mediated cytokines (IL-1β, IL-18) possibly due the inflammasome triggering effect of ROS, but also increases of other cytokines and chemokines, and NKκB and ERK expression upon stimulation (245–247). Circulating monocytes display an inflammatory phenotype with more CD16+ expression and more intracellular IL-1β and TNFα (247). However, others have shown lower TNFα production by CGD monocytes (248). Incubation of CGD monocytes with rapamycin (an mTOR inhibitor) counterbalanced the preactivation state of monocytes ex vivo (247), hence implicating a role for mTOR. IL-1 inhibition reduced inflammation in humans and reduced disease activity of, e.g., CGD-associated colitis, possibly also by restoring autophagy (249, 250). Interestingly, injection of fungal β-glucan results in hyperinflammation and necrosis in CGD mice associated with increased IL-1β, IL-6, and TNFα production (251–253).
Metabolically also clear differences were found in CGD monocytes. Several metabolites of the tryptophan pathway accumulate and indoleamine 2,3-dioxygenase is activated (254), just as seen in monocytes stimulated with LPS or IFNγ (255). CGD monocytes have been shown to higher acidification (256), which might be the result of increased lactate production.
Conclusion
In this review, we present an overview of the data supporting the concept that monocytes from patients with several autoimmune and autoinflammatory diseases display features consistent with a trained immunity phenotype. The phenotype of a trained monocyte has been defined with characteristics as (1) increased cytokine production, (2) changes in cellular metabolism (mainly increased glycolysis and lactate production), and (3) epigenetic rewiring (Table 2). Trained immunity could serve a role in the initiation of the disease and in the maintenance or aggravation of the symptoms. In the case of disease initiation, a genetic or environmental factor (or combinations of both) would induce trained monocytes/macrophages that initiate the disease. In the case of disease progression, monocytes/macrophages become trained and are therefore easier activated, which would result in the maintenance or deterioration of disease symptoms. This is an important distinction to take into account, as different experimental approaches would apply.
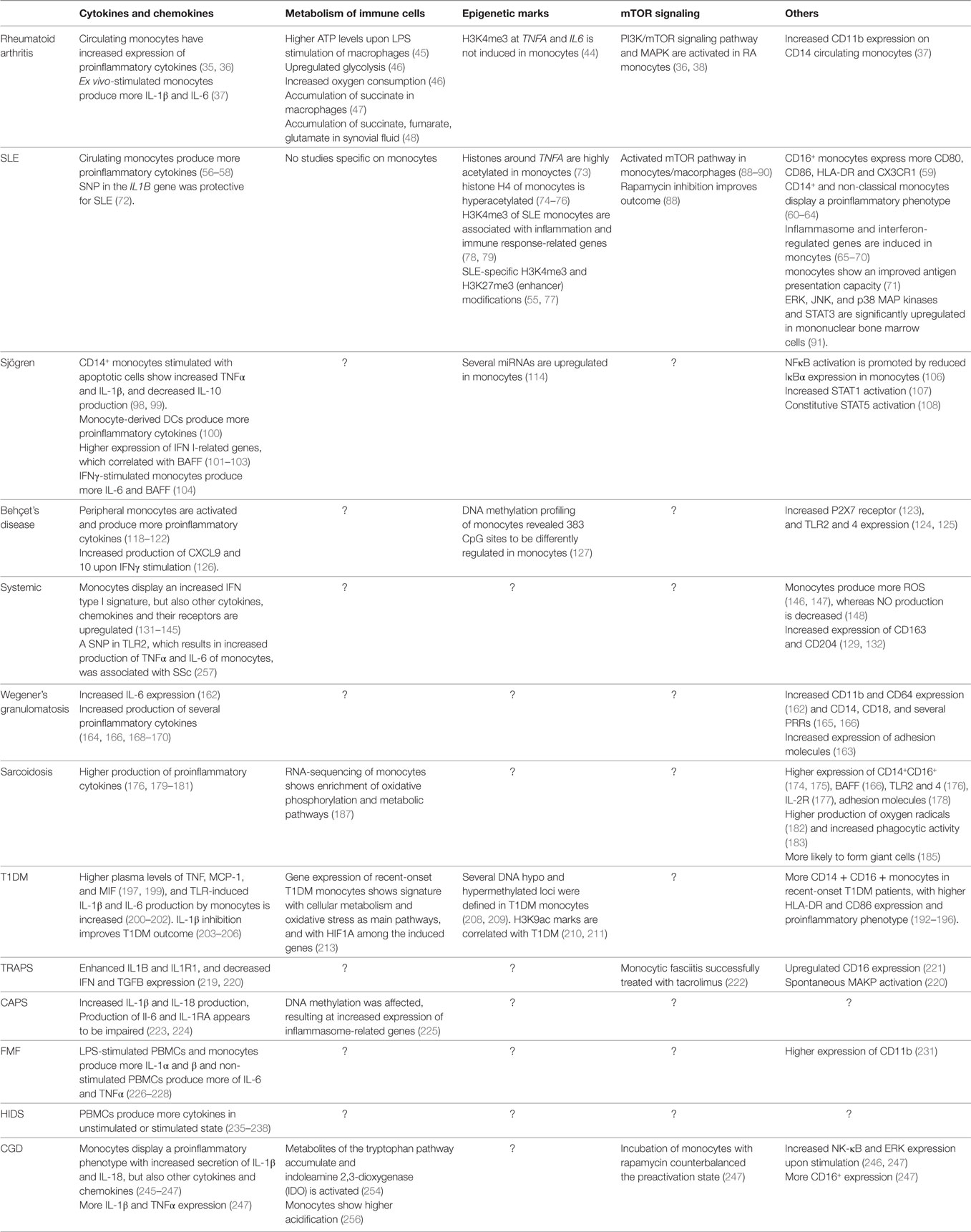
Table 2. Overview of trained immunity-related patterns in autoimmune and autoinflammatory diseases.
By providing a molecular mechanism in the described diseases in terms of trained immunity, we inherently describe potential novel therapies. For certain components of the metabolic pathways and epigenetic pathways described to be important for trained immunity, specific and non-specific inhibitors are already available and new ones are being developed. We have shown before that by inhibiting specific metabolic pathways or by specifically inhibiting certain epigenetic modulating enzymes, the induction of trained immunity can be counteracted (12–15). Hence, by identifying the specific trained immunity pathways that play a role in the induction and progression of disease activity in these autoinflammatory and autoimmune diseases, it is hoped that novel targeted immunotherapies will be developed.
However, all data presented here are circumstantial and do not prove a causal relation between the disease symptoms and monocyte function. Therefore, specifically applied experiments on the role of trained immunity in these diseases are essential to further unravel the role of trained immunity. By elucidating the potential role of trained immunity in these (but supposedly also other) diseases, new steps can be made in better understanding the pathophysiology of these diseases. Even more importantly, this could potentially lead to new approaches for therapeutic intervention in these diseases.
Author Contributions
RA wrote the first draft and LJ and MN made revisions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MN was supported by an ERC Consolidator Grant (#310372) and a Spinoza Grant of the Netherlands Organization for Scientific Research. LJ was supported by a Competitiveness Operational Programme grant of the Romanian Ministry of European Funds (HINT, ID P_37_762; MySMIS 103587).
References
1. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. doi:10.1016/j.chom.2011.04.006
2. Milutinovic B, Kurtz J. Immune memory in invertebrates. Semin Immunol (2016) 28(4):328–42. doi:10.1016/j.smim.2016.05.004
3. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Jacobs C, Xavier RJ, et al. BCG-induced trained immunity in NK cells: role for non-specific protection to infection. Clin Immunol (2014) 155(2):213–9. doi:10.1016/j.clim.2014.10.005
4. O’Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity (2015) 43(4):634–45. doi:10.1016/j.immuni.2015.09.013
5. Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12(2):223–32. doi:10.1016/j.chom.2012.06.006
6. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A (2012) 109(43):17537–42. doi:10.1073/pnas.1202870109
7. Aaby P, Kollmann TR, Benn CS. Nonspecific effects of neonatal and infant vaccination: public-health, immunological and conceptual challenges. Nat Immunol (2014) 15(10):895–9. doi:10.1038/ni.2961
8. Aaby P, Roth A, Ravn H, Napirna BM, Rodrigues A, Lisse IM, et al. Randomized trial of BCG vaccination at birth to low-birth-weight children: beneficial nonspecific effects in the neonatal period? J Infect Dis (2011) 204(2):245–52. doi:10.1093/infdis/jir240
9. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe (2018) 23(1):89–100e5. doi:10.1016/j.chom.2017.12.010
10. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345(6204):1251086. doi:10.1126/science.1251086
11. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science (2016) 352(6284):aaf1098. doi:10.1126/science.aaf1098
12. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi:10.1126/science.1250684
13. Arts RJ, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep (2016) 17(10):2562–71. doi:10.1016/j.celrep.2016.11.011
14. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab (2016) 24(6):807–19. doi:10.1016/j.cmet.2016.10.008
15. Arts RJ, Joosten LA, Netea MG. Immunometabolic circuits in trained immunity. Semin Immunol (2016) 28(5):425–30. doi:10.1016/j.smim.2016.09.002
16. Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol (2012) 227(9):3169–77. doi:10.1002/jcp.24054
17. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell (2018) 172(1–2):135–46e9. doi:10.1016/j.cell.2017.11.025
18. Novakovic B, Habibi E, Wang SY, Arts RJ, Davar R, Megchelenbrink W, et al. beta-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell (2016) 167(5):1354–68e14.
19. Catrina AI, Joshua V, Klareskog L, Malmstrom V. Mechanisms involved in triggering rheumatoid arthritis. Immunol Rev (2016) 269(1):162–74. doi:10.1111/imr.12379
20. Moulton VR, Tsokos GC. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest (2015) 125(6):2220–7. doi:10.1172/JCI78087
21. Bordignon C, Canu A, Dyczko A, Leone S, Monti P. T-cell metabolism as a target to control autoreactive T cells in beta-cell autoimmunity. Curr Diab Rep (2017) 17(5):24. doi:10.1007/s11892-017-0848-5
22. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365(23):2205–19. doi:10.1056/NEJMra1004965
23. Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet (2001) 358(9285):903–11. doi:10.1016/S0140-6736(01)06075-5
24. Theofilopoulos AN, Gonzalez-Quintial R, Lawson BR, Koh YT, Stern ME, Kono DH, et al. Sensors of the innate immune system: their link to rheumatic diseases. Nat Rev Rheumatol (2010) 6(3):146–56. doi:10.1038/nrrheum.2009.278
25. Waldner H. The role of innate immune responses in autoimmune disease development. Autoimmun Rev (2009) 8(5):400–4. doi:10.1016/j.autrev.2008.12.019
26. Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol (2002) 2(8):580–92. doi:10.1038/nri856
27. Toh ML, Bonnefoy JY, Accart N, Cochin S, Pohle S, Haegel H, et al. Bone- and cartilage-protective effects of a monoclonal antibody against colony-stimulating factor 1 receptor in experimental arthritis. Arthritis Rheumatol (2014) 66(11):2989–3000. doi:10.1002/art.38624
28. Horwood NJ, Elliott J, Martin TJ, Gillespie MT. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol (2001) 166(8):4915–21. doi:10.4049/jimmunol.166.8.4915
29. Horwood NJ. Macrophage polarization and bone formation: a review. Clin Rev Allergy Immunol (2016) 51(1):79–86. doi:10.1007/s12016-015-8519-2
30. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol (2014) 5:511. doi:10.3389/fimmu.2014.00511
31. Gerlag DM, Tak PP. Novel approaches for the treatment of rheumatoid arthritis: lessons from the evaluation of synovial biomarkers in clinical trials. Best Pract Res Clin Rheumatol (2008) 22(2):311–23. doi:10.1016/j.berh.2008.02.002
32. Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum (1996) 39(1):115–24. doi:10.1002/art.1780390116
33. Hamilton JA, Tak PP. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum (2009) 60(5):1210–21. doi:10.1002/art.24505
34. Onuora S. Rheumatoid arthritis: anti-TNF agents go head-to-head. Nat Rev Rheumatol (2017) 13(1):2. doi:10.1038/nrrheum.2016.206
35. Stuhlmuller B, Ungethum U, Scholze S, Martinez L, Backhaus M, Kraetsch HG, et al. Identification of known and novel genes in activated monocytes from patients with rheumatoid arthritis. Arthritis Rheum (2000) 43(4):775–90. doi:10.1002/1529-0131(200004)43:4<775::AID-ANR8>3.0.CO;2-7
36. Haupl T, Ostensen M, Grutzkau A, Radbruch A, Burmester GR, Villiger PM. Reactivation of rheumatoid arthritis after pregnancy: increased phagocyte and recurring lymphocyte gene activity. Arthritis Rheum (2008) 58(10):2981–92. doi:10.1002/art.23907
37. Liote F, Boval-Boizard B, Weill D, Kuntz D, Wautier JL. Blood monocyte activation in rheumatoid arthritis: increased monocyte adhesiveness, integrin expression, and cytokine release. Clin Exp Immunol (1996) 106(1):13–9. doi:10.1046/j.1365-2249.1996.d01-820.x
38. Malemud CJ. The PI3K/Akt/PTEN/mTOR pathway: a fruitful target for inducing cell death in rheumatoid arthritis? Future Med Chem (2015) 7(9):1137–47. doi:10.4155/fmc.15.55
39. Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, et al. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum (2010) 62(8):2294–302. doi:10.1002/art.27504
40. Lu Y, Parker N, Kleindl PJ, Cross VA, Wollak K, Westrick E, et al. Antiinflammatory activity of a novel folic acid targeted conjugate of the mTOR inhibitor everolimus. Mol Med (2015) 21:584–96. doi:10.2119/molmed.2015.00040
41. Huber LC, Brock M, Hemmatazad H, Giger OT, Moritz F, Trenkmann M, et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum (2007) 56(4):1087–93. doi:10.1002/art.22512
42. Maciejewska Rodrigues H, Jungel A, Gay RE, Gay S. Innate immunity, epigenetics and autoimmunity in rheumatoid arthritis. Mol Immunol (2009) 47(1):12–8. doi:10.1016/j.molimm.2009.01.010
43. Lin YC, Lin YC, Huang MY, Kuo PL, Wu CC, Lee MS, et al. Tumor necrosis factor-alpha inhibitors suppress CCL2 chemokine in monocytes via epigenetic modification. Mol Immunol (2017) 83:82–91. doi:10.1016/j.molimm.2017.01.009
44. Messemaker TC, Mikkers HMM, Huizinga TW, Toes REM, van der Helm-van Mil AHM, Kurreeman F. Inflammatory genes TNFalpha and IL6 display no signs of increased H3K4me3 in circulating monocytes from untreated rheumatoid arthritis patients. Genes Immun (2017).
45. Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol (2017) 46:112–20. doi:10.1016/j.coi.2017.04.010
46. Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med (2016) 213(3):337–54. doi:10.1084/jem.20150900
47. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med (2016) 213(9):1655–62. doi:10.1084/jem.20160061
48. Kim S, Hwang J, Xuan J, Jung YH, Cha HS, Kim KH. Global metabolite profiling of synovial fluid for the specific diagnosis of rheumatoid arthritis from other inflammatory arthritis. PLoS One (2014) 9(6):e97501. doi:10.1371/journal.pone.0097501
49. Fransen J, Kazemi-Bajestani SM, Bredie SJ, Popa CD. Rheumatoid arthritis disadvantages younger patients for cardiovascular diseases: a meta-analysis. PLoS One (2016) 11(6):e0157360. doi:10.1371/journal.pone.0157360
50. Christ A, Bekkering S, Latz E, Riksen NP. Long-term activation of the innate immune system in atherosclerosis. Semin Immunol (2016) 28(4):384–93. doi:10.1016/j.smim.2016.04.004
51. Crisan TO, Netea MG, Joosten LA. Innate immune memory: implications for host responses to damage-associated molecular patterns. Eur J Immunol (2016) 46(4):817–28. doi:10.1002/eji.201545497
52. Heinhuis B, Koenders MI, van Riel PL, van de Loo FA, Dinarello CA, Netea MG, et al. Tumour necrosis factor alpha-driven IL-32 expression in rheumatoid arthritis synovial tissue amplifies an inflammatory cascade. Ann Rheum Dis (2011) 70(4):660–7. doi:10.1136/ard.2010.139196
53. Pisetsky DS. The role of innate immunity in the induction of autoimmunity. Autoimmun Rev (2008) 8(1):69–72. doi:10.1016/j.autrev.2008.07.028
54. Morell M, Varela N, Maranon C. Myeloid populations in systemic autoimmune diseases. Clin Rev Allergy Immunol (2017) 53(2):198–218. doi:10.1007/s12016-017-8606-7
55. Liu L, Yin X, Wen L, Yang C, Sheng Y, Lin Y, et al. Several critical cell types, tissues, and pathways are implicated in genome-wide association studies for systemic lupus erythematosus. G3 (Bethesda) (2016) 6(6):1503–11. doi:10.1534/g3.116.027326
56. Kavai M, Szegedi G. Immune complex clearance by monocytes and macrophages in systemic lupus erythematosus. Autoimmun Rev (2007) 6(7):497–502. doi:10.1016/j.autrev.2007.01.017
57. Sestak AL, Furnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann Rheum Dis (2011) 70(Suppl 1):i37–43. doi:10.1136/ard.2010.138057
58. Steinbach F, Henke F, Krause B, Thiele B, Burmester GR, Hiepe F. Monocytes from systemic lupus erythematous patients are severely altered in phenotype and lineage flexibility. Ann Rheum Dis (2000) 59(4):283–8. doi:10.1136/ard.59.4.283
59. Zhu H, Hu F, Sun X, Zhang X, Zhu L, Liu X, et al. CD16+ Monocyte subset was enriched and functionally exacerbated in driving T-cell activation and B-cell response in systemic lupus erythematosus. Front Immunol (2016) 7:512. doi:10.3389/fimmu.2016.00512
60. Lyons PA, McKinney EF, Rayner TF, Hatton A, Woffendin HB, Koukoulaki M, et al. Novel expression signatures identified by transcriptional analysis of separated leucocyte subsets in systemic lupus erythematosus and vasculitis. Ann Rheum Dis (2010) 69(6):1208–13. doi:10.1136/ard.2009.108043
61. Dozmorov MG, Dominguez N, Bean K, Macwana SR, Roberts V, Glass E, et al. B-cell and monocyte contribution to systemic lupus erythematosus identified by cell-type-specific differential expression analysis in RNA-seq data. Bioinform Biol Insights (2015) 9(Suppl 3):11–9. doi:10.4137/BBI.S29470
62. Mukherjee R, Kanti Barman P, Kumar Thatoi P, Tripathy R, Kumar Das B, Ravindran B. Non-Classical monocytes display inflammatory features: validation in sepsis and systemic lupus erythematous. Sci Rep (2015) 5:13886. doi:10.1038/srep13886
63. O’Gorman WE, Hsieh EW, Savig ES, Gherardini PF, Hernandez JD, Hansmann L, et al. Single-cell systems-level analysis of human toll-like receptor activation defines a chemokine signature in patients with systemic lupus erythematosus. J Allergy Clin Immunol (2015) 136(5):1326–36. doi:10.1016/j.jaci.2015.04.008
64. Shi L, Zhang Z, Yu AM, Wang W, Wei Z, Akhter E, et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PLoS One (2014) 9(5):e93846. doi:10.1371/journal.pone.0093846
65. Liu J, Berthier CC, Kahlenberg JM. Enhanced inflammasome activity in systemic lupus erythematosus is mediated via type I interferon upregulation of interferon regulatory factor 1. Arthritis Rheumatol (2017) 69(9):1840–9. doi:10.1002/art.40166
66. Lopez P, Rodriguez-Carrio J, Caminal-Montero L, Mozo L, Suarez A. A pathogenic IFNalpha, BLyS and IL-17 axis in systemic lupus erythematosus patients. Sci Rep (2016) 6:20651. doi:10.1038/srep20651
67. Eloranta ML, Ronnblom L. Cause and consequences of the activated type I interferon system in SLE. J Mol Med (Berl) (2016) 94(10):1103–10. doi:10.1007/s00109-016-1421-4
68. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100(5):2610–5. doi:10.1073/pnas.0337679100
69. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197(6):711–23. doi:10.1084/jem.20021553
70. Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum (2004) 50(12):3958–67. doi:10.1002/art.20798
71. Byrne JC, Ni Gabhann J, Lazzari E, Mahony R, Smith S, Stacey K, et al. Genetics of SLE: functional relevance for monocytes/macrophages in disease. Clin Dev Immunol (2012) 2012:582352. doi:10.1155/2012/582352
72. Camargo JF, Correa PA, Castiblanco J, Anaya JM. Interleukin-1beta polymorphisms in Colombian patients with autoimmune rheumatic diseases. Genes Immun (2004) 5(8):609–14. doi:10.1038/sj.gene.6364133
73. Sullivan KE, Suriano A, Dietzmann K, Lin J, Goldman D, Petri MA. The TNFalpha locus is altered in monocytes from patients with systemic lupus erythematosus. Clin Immunol (2007) 123(1):74–81. doi:10.1016/j.clim.2006.12.008
74. Zhang Z, Song L, Maurer K, Petri MA, Sullivan KE. Global H4 acetylation analysis by ChIP-chip in systemic lupus erythematosus monocytes. Genes Immun (2010) 11(2):124–33. doi:10.1038/gene.2009.66
75. Leung YT, Shi L, Maurer K, Song L, Zhang Z, Petri M, et al. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics (2015) 10(3):191–9. doi:10.1080/15592294.2015.1009764
76. Zhang Z, Maurer K, Perin JC, Song L, Sullivan KE. Cytokine-induced monocyte characteristics in SLE. J Biomed Biotechnol (2010) 2010:507475. doi:10.1155/2010/507475
77. Shi L, Zhang Z, Song L, Leung YT, Petri MA, Sullivan KE. Monocyte enhancers are highly altered in systemic lupus erythematosus. Epigenomics (2015) 7(6):921–35. doi:10.2217/epi.15.47
78. Zhang Z, Shi L, Dawany N, Kelsen J, Petri MA, Sullivan KE. H3K4 tri-methylation breadth at transcription start sites impacts the transcriptome of systemic lupus erythematosus. Clin Epigenetics (2016) 8:14. doi:10.1186/s13148-016-0179-4
79. Dai Y, Zhang L, Hu C, Zhang Y. Genome-wide analysis of histone H3 lysine 4 trimethylation by ChIP-chip in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Clin Exp Rheumatol (2010) 28(2):158–68.
80. Oaks Z, Perl A. Metabolic control of the epigenome in systemic lupus erythematosus. Autoimmunity (2014) 47(4):256–64. doi:10.3109/08916934.2013.834495
81. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum (1990) 33(11):1665–73. doi:10.1002/art.1780331109
82. Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, et al. Decreased ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum (2001) 44(2):397–407. doi:10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N
83. Rauen T, Grammatikos AP, Hedrich CM, Floege J, Tenbrock K, Ohl K, et al. cAMP-responsive element modulator alpha (CREMalpha) contributes to decreased Notch-1 expression in T cells from patients with active systemic lupus erythematosus (SLE). J Biol Chem (2012) 287(51):42525–32. doi:10.1074/jbc.M112.425371
84. Zhang Q, Long H, Liao J, Zhao M, Liang G, Wu X, et al. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J Autoimmun (2011) 37(3):180–9. doi:10.1016/j.jaut.2011.09.006
85. Forster N, Gallinat S, Jablonska J, Weiss S, Elsasser HP, Lutz W. p300 Protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J Immunol (2007) 178(11):6941–8. doi:10.4049/jimmunol.178.11.6941
86. Hu N, Long H, Zhao M, Yin H, Lu Q. Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand J Rheumatol (2009) 38(6):464–71. doi:10.3109/03009740902895750
87. Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest (2003) 111(4):539–52. doi:10.1172/JCI16153
88. Oaks Z, Winans T, Huang N, Banki K, Perl A. Activation of the mechanistic target of rapamycin in SLE: explosion of evidence in the last five years. Curr Rheumatol Rep (2016) 18(12):73. doi:10.1007/s11926-016-0622-8
89. Canaud G, Bienaime F, Tabarin F, Bataillon G, Seilhean D, Noel LH, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med (2014) 371(4):303–12. doi:10.1056/NEJMoa1312890
90. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol (2016) 12(3):169–82. doi:10.1038/nrrheum.2015.172
91. Nakou M, Bertsias G, Stagakis I, Centola M, Tassiulas I, Hatziapostolou M, et al. Gene network analysis of bone marrow mononuclear cells reveals activation of multiple kinase pathways in human systemic lupus erythematosus. PLoS One (2010) 5(10):e13351. doi:10.1371/journal.pone.0013351
92. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, et al. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med (2016) 14(1):156. doi:10.1186/s12967-016-0911-z
93. Carvalheiro T, Gomes D, Pinto LA, Ines L, Lopes A, Henriques A, et al. Sera from patients with active systemic lupus erythematosus patients enhance the toll-like receptor 4 response in monocyte subsets. J Inflamm (Lond) (2015) 12:38. doi:10.1186/s12950-015-0083-2
95. Manoussakis MN, Boiu S, Korkolopoulou P, Kapsogeorgou EK, Kavantzas N, Ziakas P, et al. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum (2007) 56(12):3977–88. doi:10.1002/art.23073
96. Zhou D, McNamara NA. Macrophages: important players in primary Sjogren’s syndrome? Expert Rev Clin Immunol (2014) 10(4):513–20. doi:10.1586/1744666X.2014.900441
97. Zhou D, Chen YT, Chen F, Gallup M, Vijmasi T, Bahrami AF, et al. Critical involvement of macrophage infiltration in the development of Sjogren’s syndrome-associated dry eye. Am J Pathol (2012) 181(3):753–60. doi:10.1016/j.ajpath.2012.05.014
98. Hauk V, Fraccaroli L, Grasso E, Eimon A, Ramhorst R, Hubscher O, et al. Monocytes from Sjogren’s syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin Exp Immunol (2014) 177(3):662–70. doi:10.1111/cei.12378
99. Enk C, Oxholm P, Tvede N, Bendtzen K. Blood mononuclear cells in patients with primary Sjogren’s syndrome: production of interleukins, enumeration of interleukin-2 receptors, and DNA synthesis. Scand J Rheumatol Suppl (1986) 61:131–4.
100. Volchenkov R, Brun JG, Jonsson R, Appel S. In vitro suppression of immune responses using monocyte-derived tolerogenic dendritic cells from patients with primary Sjogren’s syndrome. Arthritis Res Ther (2013) 15(5):R114. doi:10.1186/ar4294
101. Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA, et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis (2013) 72(5):728–35. doi:10.1136/annrheumdis-2012-201381
102. Wildenberg ME, van Helden-Meeuwsen CG, van de Merwe JP, Drexhage HA, Versnel MA. Systemic increase in type I interferon activity in Sjogren’s syndrome: a putative role for plasmacytoid dendritic cells. Eur J Immunol (2008) 38(7):2024–33. doi:10.1002/eji.200738008
103. Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann Rheum Dis (2007) 66(5):700–3. doi:10.1136/ard.2006.060772
104. Yoshimoto K, Tanaka M, Kojima M, Setoyama Y, Kameda H, Suzuki K, et al. Regulatory mechanisms for the production of BAFF and IL-6 are impaired in monocytes of patients of primary Sjogren’s syndrome. Arthritis Res Ther (2011) 13(5):R170. doi:10.1186/ar3493
105. Eriksson P, Andersson C, Ekerfelt C, Ernerudh J, Skogh T. Sjogren’s syndrome with myalgia is associated with subnormal secretion of cytokines by peripheral blood mononuclear cells. J Rheumatol (2004) 31(4):729–35.
106. Lisi S, Sisto M, Lofrumento DD, D’Amore M. Altered IkappaBalpha expression promotes NF-kappaB activation in monocytes from primary Sjogren’s syndrome patients. Pathology (2012) 44(6):557–61. doi:10.1097/PAT.0b013e3283580388
107. Pertovaara M, Silvennoinen O, Isomaki P. Cytokine-induced STAT1 activation is increased in patients with primary Sjogren’s syndrome. Clin Immunol (2016) 165:60–7. doi:10.1016/j.clim.2016.03.010
108. Pertovaara M, Silvennoinen O, Isomaki P. STAT-5 is activated constitutively in T cells, B cells and monocytes from patients with primary Sjogren’s syndrome. Clin Exp Immunol (2015) 181(1):29–38. doi:10.1111/cei.12614
109. Lu Q, Renaudineau Y, Cha S, Ilei G, Brooks WH, Selmi C, et al. Epigenetics in autoimmune disorders: highlights of the 10th Sjogren’s syndrome symposium. Autoimmun Rev (2010) 9(9):627–30. doi:10.1016/j.autrev.2010.05.011
110. Le Dantec C, Varin MM, Brooks WH, Pers JO, Youinou P, Renaudineau Y. Epigenetics and Sjogren’s syndrome. Curr Pharm Biotechnol (2012) 13(10):2046–53. doi:10.2174/138920112802273326
111. Peng L, Ma W, Yi F, Yang YJ, Lin W, Chen H, et al. MicroRNA profiling in Chinese patients with primary Sjogren syndrome reveals elevated miRNA-181a in peripheral blood mononuclear cells. J Rheumatol (2014) 41(11):2208–13. doi:10.3899/jrheum.131154
112. Pauley KM, Stewart CM, Gauna AE, Dupre LC, Kuklani R, Chan AL, et al. Altered miR-146a expression in Sjogren’s syndrome and its functional role in innate immunity. Eur J Immunol (2011) 41(7):2029–39. doi:10.1002/eji.201040757
113. Zilahi E, Tarr T, Papp G, Griger Z, Sipka S, Zeher M. Increased microRNA-146a/b, TRAF6 gene and decreased IRAK1 gene expressions in the peripheral mononuclear cells of patients with Sjogren’s syndrome. Immunol Lett (2012) 141(2):165–8. doi:10.1016/j.imlet.2011.09.006
114. Williams AE, Choi K, Chan AL, Lee YJ, Reeves WH, Bubb MR, et al. Sjogren’s syndrome-associated microRNAs in CD14(+) monocytes unveils targeted TGFbeta signaling. Arthritis Res Ther (2016) 18(1):95. doi:10.1186/s13075-016-0987-0
115. Shah M, Edman MC, Janga SR, Shi P, Dhandhukia J, Liu S, et al. A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjogren’s syndrome. J Control Release (2013) 171(3):269–79. doi:10.1016/j.jconrel.2013.07.016
116. Wechsler B, Davatchi F, Mizushima Y, Hamza M, Dilsen N, Kansu E, et al. Criteria for diagnosis of Behcet’s disease. International study group for Behcet’s disease. Lancet (1990) 335(8697):1078–80.
117. Kanekura T, Gushi A, Iwata M, Fukumaru S, Sakamoto R, Kawahara K, et al. Treatment of Behcet’s disease with granulocyte and monocyte adsorption apheresis. J Am Acad Dermatol (2004) 51(2 Suppl):S83–7. doi:10.1016/j.jaad.2003.12.023
118. Slobodin G, Toukan Y, Rosner I, Rozenbaum M, Boulman N, Pavlotzky E, et al. LPS-stimulated production of TNF-alpha by peripheral blood monocytes in patients with Behcet’s disease. Clin Rheumatol (2007) 26(5):764–7. doi:10.1007/s10067-006-0371-6
119. Sahin S, Lawrence R, Direskeneli H, Hamuryudan V, Yazici H, Akoglu T. Monocyte activity in Behcet’s disease. Br J Rheumatol (1996) 35(5):424–9. doi:10.1093/rheumatology/35.5.424
120. Mege JL, Dilsen N, Sanguedolce V, Gul A, Bongrand P, Roux H, et al. Overproduction of monocyte derived tumor necrosis factor alpha, interleukin (IL) 6, IL-8 and increased neutrophil superoxide generation in Behcet’s disease. A comparative study with familial Mediterranean fever and healthy subjects. J Rheumatol (1993) 20(9):1544–9.
121. Nakamura S, Sugita M, Tanaka S, Ohno S. [Enhanced production of in vitro tumor necrosis factor-alpha from monocytes in Behcet’s disease]. Nippon Ganka Gakkai Zasshi (1992) 96(10):1282–5.
122. Morton LT, Situnayake D, Wallace GR. Genetics of Behcet’s disease. Curr Opin Rheumatol (2016) 28(1):39–44. doi:10.1097/BOR.0000000000000234
123. Castrichini M, Lazzerini PE, Gamberucci A, Capecchi PL, Franceschini R, Natale M, et al. The purinergic P2x7 receptor is expressed on monocytes in Behcet’s disease and is modulated by TNF-alpha. Eur J Immunol (2014) 44(1):227–38. doi:10.1002/eji.201343353
124. Neves FS, Carrasco S, Goldenstein-Schainberg C, Goncalves CR, de Mello SB. Neutrophil hyperchemotaxis in Behcet’s disease: a possible role for monocytes orchestrating bacterial-induced innate immune responses. Clin Rheumatol (2009) 28(12):1403–10. doi:10.1007/s10067-009-1261-5
125. Do JE, Kwon SY, Park S, Lee ES. Effects of vitamin D on expression of toll-like receptors of monocytes from patients with Behcet’s disease. Rheumatology (Oxford) (2008) 47(6):840–8. doi:10.1093/rheumatology/ken109
126. Ambrose N, Khan E, Ravindran R, Lightstone L, Abraham S, Botto M, et al. The exaggerated inflammatory response in Behcet’s syndrome: identification of dysfunctional post-transcriptional regulation of the IFN-gamma/CXCL10 IP-10 pathway. Clin Exp Immunol (2015) 181(3):427–33. doi:10.1111/cei.12655
127. Hughes T, Ture-Ozdemir F, Alibaz-Oner F, Coit P, Direskeneli H, Sawalha AH. Epigenome-wide scan identifies a treatment-responsive pattern of altered DNA methylation among cytoskeletal remodeling genes in monocytes and CD4+ T cells from patients with Behcet’s disease. Arthritis Rheumatol (2014) 66(6):1648–58. doi:10.1002/art.38409
128. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med (2009) 360(19):1989–2003. doi:10.1056/NEJMra0806188
129. Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther (2010) 12(4):R128. doi:10.1186/ar3066
130. Kraling BM, Maul GG, Jimenez SA. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology (1995) 63(1):48–56. doi:10.1159/000163933
131. Brkic Z, van Bon L, Cossu M, van Helden-Meeuwsen CG, Vonk MC, Knaapen H, et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis (2016) 75(8):1567–73. doi:10.1136/annrheumdis-2015-207392
132. Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest (2010) 90(6):812–23. doi:10.1038/labinvest.2010.73
133. Duan H, Fleming J, Pritchard DK, Amon LM, Xue J, Arnett HA, et al. Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum (2008) 58(5):1465–74. doi:10.1002/art.23451
134. York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum (2007) 56(3):1010–20. doi:10.1002/art.22382
135. Masuda A, Yasuoka H, Satoh T, Okazaki Y, Yamaguchi Y, Kuwana M. Versican is upregulated in circulating monocytes in patients with systemic sclerosis and amplifies a CCL2-mediated pathogenic loop. Arthritis Res Ther (2013) 15(4):R74. doi:10.1186/ar4251
136. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest (2007) 117(3):557–67. doi:10.1172/JCI31139
137. Truchetet ME, Allanore Y, Montanari E, Chizzolini C, Brembilla NC. Prostaglandin I(2) analogues enhance already exuberant Th17 cell responses in systemic sclerosis. Ann Rheum Dis (2012) 71(12):2044–50. doi:10.1136/annrheumdis-2012-201400
138. Eloranta ML, Franck-Larsson K, Lovgren T, Kalamajski S, Ronnblom A, Rubin K, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis (2010) 69(7):1396–402. doi:10.1136/ard.2009.121400
139. Hasegawa M, Sato S, Takehara K. Augmented production of chemokines (monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1alpha (MIP-1alpha) and MIP-1beta) in patients with systemic sclerosis: MCP-1 and MIP-1alpha may be involved in the development of pulmonary fibrosis. Clin Exp Immunol (1999) 117(1):159–65.
140. Giacomelli R, Cipriani P, Danese C, Pizzuto F, Lattanzio R, Parzanese I, et al. Peripheral blood mononuclear cells of patients with systemic sclerosis produce increased amounts of interleukin 6, but not transforming growth factor beta 1. J Rheumatol (1996) 23(2):291–6.
141. Crestani B, Seta N, De Bandt M, Soler P, Rolland C, Dehoux M, et al. Interleukin 6 secretion by monocytes and alveolar macrophages in systemic sclerosis with lung involvement. Am J Respir Crit Care Med (1994) 149(5):1260–5. doi:10.1164/ajrccm.149.5.8173768
142. Deguchi Y. Spontaneous increase of transforming growth factor beta production by bronchoalveolar mononuclear cells of patients with systemic autoimmune diseases affecting the lung. Ann Rheum Dis (1992) 51(3):362–5. doi:10.1136/ard.51.3.362
143. Umehara H, Kumagai S, Murakami M, Suginoshita T, Tanaka K, Hashida S, et al. Enhanced production of interleukin-1 and tumor necrosis factor alpha by cultured peripheral blood monocytes from patients with scleroderma. Arthritis Rheum (1990) 33(6):893–7. doi:10.1002/art.1780330619
144. Westacott CI, Whicher JT, Hutton CW, Dieppe PA. Increased spontaneous production of interleukin-1 together with inhibitory activity in systemic sclerosis. Clin Sci (Lond) (1988) 75(6):561–7. doi:10.1042/cs0750561
145. Andrews BS, Friou GJ, Berman MA, Sandborg CI, Mirick GR, Cesario TC. Changes in circulating monocytes in patients with progressive systemic sclerosis. J Rheumatol (1987) 14(5):930–5.
146. Allanore Y, Borderie D, Perianin A, Lemarechal H, Ekindjian OG, Kahan A. Nifedipine protects against overproduction of superoxide anion by monocytes from patients with systemic sclerosis. Arthritis Res Ther (2005) 7(1):R93–100. doi:10.1186/ar1614
147. Sambo P, Jannino L, Candela M, Salvi A, Donini M, Dusi S, et al. Monocytes of patients with systemic sclerosis (scleroderma spontaneously release in vitro increased amounts of superoxide anion). J Invest Dermatol (1999) 112(1):78–84. doi:10.1046/j.1523-1747.1999.00476.x
148. Allanore Y, Borderie D, Hilliquin P, Hernvann A, Levacher M, Lemarechal H, et al. Low levels of nitric oxide (NO) in systemic sclerosis: inducible NO synthase production is decreased in cultured peripheral blood monocyte/macrophage cells. Rheumatology (Oxford) (2001) 40(10):1089–96. doi:10.1093/rheumatology/40.10.1089
149. Gunther J, Kill A, Becker MO, Heidecke H, Rademacher J, Siegert E, et al. Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res Ther (2014) 16(2):R65. doi:10.1186/ar4503
150. Yamaguchi Y, Kuwana M. Proangiogenic hematopoietic cells of monocytic origin: roles in vascular regeneration and pathogenic processes of systemic sclerosis. Histol Histopathol (2013) 28(2):175–83. doi:10.14670/HH-28.175
151. Elisa T, Antonio P, Giuseppe P, Alessandro B, Giuseppe A, Federico C, et al. Endothelin receptors expressed by immune cells are involved in modulation of inflammation and in fibrosis: relevance to the pathogenesis of systemic sclerosis. J Immunol Res (2015) 2015:147616. doi:10.1155/2015/147616
152. Ciechomska M, Huigens CA, Hugle T, Stanly T, Gessner A, Griffiths B, et al. Toll-like receptor-mediated, enhanced production of profibrotic TIMP-1 in monocytes from patients with systemic sclerosis: role of serum factors. Ann Rheum Dis (2013) 72(8):1382–9. doi:10.1136/annrheumdis-2012-201958
153. Binai N, O’Reilly S, Griffiths B, van Laar JM, Hugle T. Differentiation potential of CD14+ monocytes into myofibroblasts in patients with systemic sclerosis. PLoS One (2012) 7(3):e33508. doi:10.1371/journal.pone.0033508
154. Yamaguchi Y, Okazaki Y, Seta N, Satoh T, Takahashi K, Ikezawa Z, et al. Enhanced angiogenic potency of monocytic endothelial progenitor cells in patients with systemic sclerosis. Arthritis Res Ther (2010) 12(6):R205. doi:10.1186/ar3180
155. Campioni D, Lo Monaco A, Lanza F, Moretti S, Ferrari L, Fotinidi M, et al. CXCR4 pos circulating progenitor cells coexpressing monocytic and endothelial markers correlating with fibrotic clinical features are present in the peripheral blood of patients affected by systemic sclerosis. Haematologica (2008) 93(8):1233–7. doi:10.3324/haematol.12526
156. Koch AE, Litvak MA, Burrows JC, Polverini PJ. Decreased monocyte-mediated angiogenesis in scleroderma. Clin Immunol Immunopathol (1992) 64(2):153–60. doi:10.1016/0090-1229(92)90193-R
157. Christmann RB, Lafyatis R. The cytokine language of monocytes and macrophages in systemic sclerosis. Arthritis Res Ther (2010) 12(5):146. doi:10.1186/ar3167
158. Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol (2010) 137(1):89–101. doi:10.1016/j.clim.2010.06.017
159. Pantelidis P, McGrath DS, Southcott AM, Black CM, du Bois RM. Tumour necrosis factor-alpha production in fibrosing alveolitis is macrophage subset specific. Respir Res (2001) 2(6):365–72. doi:10.1186/rr87
160. Ciechomska M, O’Reilly S, Przyborski S, Oakley F, Bogunia-Kubik K, van Laar JM. Histone Demethylation and toll-like receptor 8-dependent cross-talk in monocytes promotes transdifferentiation of fibroblasts in systemic sclerosis via Fra-2. Arthritis Rheumatol (2016) 68(6):1493–504. doi:10.1002/art.39602
161. Harper L, Savage CO. Pathogenesis of ANCA-associated systemic vasculitis. J Pathol (2000) 190(3):349–59. doi:10.1002/(SICI)1096-9896(200002)190:3<349::AID-PATH524>3.0.CO;2-A
162. Muller Kobold AC, Kallenberg CG, Tervaert JW. Monocyte activation in patients with Wegener’s granulomatosis. Ann Rheum Dis (1999) 58(4):237–45. doi:10.1136/ard.58.4.237
163. Wikman A, Fagergren A, Gunnar OJS, Lundahl J, Jacobson SH. Monocyte activation and relationship to anti-proteinase 3 in acute vasculitis. Nephrol Dial Transplant (2003) 18(9):1792–9. doi:10.1093/ndt/gfg216
164. Hattar K, van Burck S, Bickenbach A, Grandel U, Maus U, Lohmeyer J, et al. Anti-proteinase 3 antibodies (c-ANCA) prime CD14-dependent leukocyte activation. J Leukoc Biol (2005) 78(4):992–1000. doi:10.1189/jlb.0902442
165. Nowack R, Schwalbe K, Flores-Suarez LF, Yard B, van der Woude FJ. Upregulation of CD14 and CD18 on monocytes In vitro by antineutrophil cytoplasmic autoantibodies. J Am Soc Nephrol (2000) 11(9):1639–46.
166. Uehara A, Sato T, Iwashiro A, Yokota S. PR3-ANCA in Wegener’s granulomatosis prime human mononuclear cells for enhanced activation via TLRs and NOD1/2. Diagn Pathol (2009) 4:23. doi:10.1186/1746-1596-4-23
167. Lamprecht P, Kumanovics G, Mueller A, Csernok E, Komocsi A, Trabandt A, et al. Elevated monocytic IL-12 and TNF-alpha production in Wegener’s granulomatosis is normalized by cyclophosphamide and corticosteroid therapy. Clin Exp Immunol (2002) 128(1):181–6. doi:10.1046/j.1365-2249.2002.01801.x
168. Ralston DR, Marsh CB, Lowe MP, Wewers MD. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. Role of surface proteinase-3, alpha1-antitrypsin, and Fcgamma receptors. J Clin Invest (1997) 100(6):1416–24. doi:10.1172/JCI119662
169. Ludviksson BR, Sneller MC, Chua KS, Talar-Williams C, Langford CA, Ehrhardt RO, et al. Active Wegener’s granulomatosis is associated with HLA-DR+ CD4+ T cells exhibiting an unbalanced Th1-type T cell cytokine pattern: reversal with IL-10. J Immunol (1998) 160(7):3602–9.
170. Casselman BL, Kilgore KS, Miller BF, Warren JS. Antibodies to neutrophil cytoplasmic antigens induce monocyte chemoattractant protein-1 secretion from human monocytes. J Lab Clin Med (1995) 126(5):495–502.
171. Johansson AC, Ohlsson S, Pettersson A, Bengtsson AA, Selga D, Hansson M, et al. Impaired phagocytosis and reactive oxygen species production in phagocytes is associated with systemic vasculitis. Arthritis Res Ther (2016) 18:92. doi:10.1186/s13075-016-0994-1
172. Park J, Lee EB, Song YW. Decreased tumour necrosis factor-alpha production by monocytes of granulomatosis with polyangiitis. Scand J Rheumatol (2014) 43(5):403–8. doi:10.3109/03009742.2014.894568
173. Crawshaw A, Kendrick YR, McMichael AJ, Ho LP. Abnormalities in iNKT cells are associated with impaired ability of monocytes to produce IL-10 and suppress T-cell proliferation in sarcoidosis. Eur J Immunol (2014) 44(7):2165–74. doi:10.1002/eji.201344284
174. Okamoto H, Mizuno K, Horio T. Circulating CD14+ CD16+ monocytes are expanded in sarcoidosis patients. J Dermatol (2003) 30(7):503–9. doi:10.1111/j.1346-8138.2003.tb00424.x
175. Homolka J, Lorenz J, Zuchold HD, Muller-Quernheim J. Evaluation of soluble CD 14 and neopterin as serum parameters of the inflammatory activity of pulmonary sarcoidosis. Clin Investig (1992) 70(10):909–16. doi:10.1007/BF00180437
176. Wiken M, Grunewald J, Eklund A, Wahlstrom J. Higher monocyte expression of TLR2 and TLR4, and enhanced pro-inflammatory synergy of TLR2 with NOD2 stimulation in sarcoidosis. J Clin Immunol (2009) 29(1):78–89. doi:10.1007/s10875-008-9225-0
177. Ina Y, Takada K, Sato T, Yamamoto M, Noda M, Morishita M. Soluble interleukin 2 receptors in patients with sarcoidosis. Possible origin.Chest (1992) 102(4):1128–33. doi:10.1378/chest.102.4.1128
178. Thole AA, Rodrigues CA, Milward G, Negreiros , Porto LC, Carvalho L. Ultrastructural study of expression of adhesion molecules between blood monocytes and alveolar macrophages from patients with pulmonary sarcoidosis. J Submicrosc Cytol Pathol (2001) 33(4):419–24.
179. Sahashi K, Ina Y, Takada K, Sato T, Yamamoto M, Morishita M. Significance of interleukin 6 in patients with sarcoidosis. Chest (1994) 106(1):156–60. doi:10.1378/chest.106.1.156
180. Terao I, Hashimoto S, Horie T. Effect of GM-CSF on TNF-alpha and IL-1-beta production by alveolar macrophages and peripheral blood monocytes from patients with sarcoidosis. Int Arch Allergy Immunol (1993) 102(3):242–8. doi:10.1159/000236532
181. Tercelj M, Stopinsek S, Ihan A, Salobir B, Simcic S, Wraber B, et al. In vitro and in vivo reactivity to fungal cell wall agents in sarcoidosis. Clin Exp Immunol (2011) 166(1):87–93. doi:10.1111/j.1365-2249.2011.04456.x
182. Barth J, Entzian P, Petermann W. Increased release of free oxygen radicals by phagocytosing and nonphagocytosing cells from patients with active pulmonary sarcoidosis as revealed by luminol-dependent chemiluminescence. Klin Wochenschr (1988) 66(7):292–7. doi:10.1007/BF01727514
183. Dubaniewicz A, Typiak M, Wybieralska M, Szadurska M, Nowakowski S, Staniewicz-Panasik A, et al. Changed phagocytic activity and pattern of Fcgamma and complement receptors on blood monocytes in sarcoidosis. Hum Immunol (2012) 73(8):788–94. doi:10.1016/j.humimm.2012.05.005
184. Heron M, Grutters JC, van Velzen-Blad H, Veltkamp M, Claessen AME, van den Bosch JMM. Increased expression of CD16, CD69, and very late antigen-1 on blood monocytes in active sarcoidosis. Chest (2008) 134(5):1001–8. doi:10.1378/chest.08-0443
185. Mizuno K, Okamoto H, Horio T. Heightened ability of monocytes from sarcoidosis patients to form multi-nucleated giant cells in vitro by supernatants of concanavalin A-stimulated mononuclear cells. Clin Exp Immunol (2001) 126(1):151–6. doi:10.1046/j.1365-2249.2001.01655.x
186. Perez RL, Roman J. Fibrin enhances the expression of IL-1 beta by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J Immunol (1995) 154(4):1879–87.
187. Talreja J, Farshi P, Alazizi A, Luca F, Pique-Regi R, Samavati L. RNA-sequencing Identifies Novel Pathways in sarcoidosis monocytes. Sci Rep (2017) 7(1):2720. doi:10.1038/s41598-017-02941-4
188. Liu Y, Li H, Xiao T, Lu Q. Epigenetics in immune-mediated pulmonary diseases. Clin Rev Allergy Immunol (2013) 45(3):314–30. doi:10.1007/s12016-013-8398-3
189. Pociot F, Lernmark A. Genetic risk factors for type 1 diabetes. Lancet (2016) 387(10035):2331–9. doi:10.1016/S0140-6736(16)30582-7
190. Espinoza-Jimenez A, Peon AN, Terrazas LI. Alternatively activated macrophages in types 1 and 2 diabetes. Mediators Inflamm (2012) 2012:815953. doi:10.1155/2012/815953
191. Zacher T, Knerr I, Rascher W, Kalden JR, Wassmuth R. Characterization of monocyte-derived dendritic cells in recent-onset diabetes mellitus type 1. Clin Immunol (2002) 105(1):17–24. doi:10.1006/clim.2002.5265
192. Ren X, Mou W, Su C, Chen X, Zhang H, Cao B, et al. Increase in peripheral blood intermediate monocytes is associated with the development of recent-onset type 1 diabetes mellitus in children. Int J Biol Sci (2017) 13(2):209–18. doi:10.7150/ijbs.15659
193. Ryba-Stanislawowska M, Mysliwska J, Juhas U, Mysliwiec M. Elevated levels of peripheral blood CD14(bright) CD16+ and CD14(dim) CD16+ monocytes may contribute to the development of retinopathy in patients with juvenile onset type 1 diabetes. APMIS (2015) 123(9):793–9. doi:10.1111/apm.12419
194. Brooks-Worrell BM, Iyer D, Coraza I, Hampe CS, Nalini R, Ozer K, et al. Islet-specific T-cell responses and proinflammatory monocytes define subtypes of autoantibody-negative ketosis-prone diabetes. Diabetes Care (2013) 36(12):4098–103. doi:10.2337/dc12-2328
195. Josefsen K, Nielsen H, Lorentzen S, Damsbo P, Buschard K. Circulating monocytes are activated in newly diagnosed type 1 diabetes mellitus patients. Clin Exp Immunol (1994) 98(3):489–93. doi:10.1111/j.1365-2249.1994.tb05517.x
196. Litherland SA, She JX, Schatz D, Fuller K, Hutson AD, Peng RH, et al. Aberrant monocyte prostaglandin synthase 2 (PGS2) expression in type 1 diabetes before and after disease onset. Pediatr Diabetes (2003) 4(1):10–8. doi:10.1034/j.1399-5448.2003.00042.x
197. Mysliwska J, Smardzewski M, Marek-Trzonkowska N, Mysliwiec M, Raczynska K. Expansion of CD14+CD16+ monocytes producing TNF-alpha in complication-free diabetes type 1 juvenile onset patients. Cytokine (2012) 60(1):309–17. doi:10.1016/j.cyto.2012.03.010
198. Harsunen MH, Puff R, D’Orlando O, Giannopoulou E, Lachmann L, Beyerlein A, et al. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm Metab Res (2013) 45(6):467–70. doi:10.1055/s-0032-1331226
199. Ismail NA, Abd El Baky AN, Ragab S, Hamed M, Hashish MA, Shehata A. Monocyte chemoattractant protein 1 and macrophage migration inhibitory factor in children with type 1 diabetes. J Pediatr Endocrinol Metab (2016) 29(6):641–5. doi:10.1515/jpem-2015-0340
200. Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes (2014) 63(7):2538–50. doi:10.2337/db13-1777
201. Wolter TR, Wong R, Sarkar SA, Zipris D. DNA microarray analysis for the identification of innate immune pathways implicated in virus-induced autoimmune diabetes. Clin Immunol (2009) 132(1):103–15. doi:10.1016/j.clim.2009.02.007
202. Alkanani AK, Rewers M, Dong F, Waugh K, Gottlieb PA, Zipris D. Dysregulated toll-like receptor-induced interleukin-1beta and interleukin-6 responses in subjects at risk for the development of type 1 diabetes. Diabetes (2012) 61(10):2525–33. doi:10.2337/db12-0099
203. Hara N, Alkanani AK, Dinarello CA, Zipris D. Modulation of virus-induced innate immunity and type 1 diabetes by IL-1 blockade. Innate Immun (2014) 20(6):574–84. doi:10.1177/1753425913502242
204. Londono P, Komura A, Hara N, Zipris D. Brief dexamethasone treatment during acute infection prevents virus-induced autoimmune diabetes. Clin Immunol (2010) 135(3):401–11. doi:10.1016/j.clim.2010.01.007
205. Hara N, Alkanani AK, Dinarello CA, Zipris D. Histone deacetylase inhibitor suppresses virus-induced proinflammatory responses and type 1 diabetes. J Mol Med (Berl) (2014) 92(1):93–102. doi:10.1007/s00109-013-1078-1
206. Gottlieb PA, Alkanani AK, Michels AW, Lewis EC, Shapiro L, Dinarello CA, et al. alpha1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1beta responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J Clin Endocrinol Metab (2014) 99(8):E1418–26. doi:10.1210/jc.2013-3864
207. Litherland SA. Immunopathogenic interaction of environmental triggers and genetic susceptibility in diabetes: is epigenetics the missing link? Diabetes (2008) 57(12):3184–6. doi:10.2337/db08-1275
208. Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, Schones DE, et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A (2016) 113(21):E3002–11. doi:10.1073/pnas.1603712113
209. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet (2011) 7(9):e1002300. doi:10.1371/journal.pgen.1002300
210. Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes (2014) 63(5):1748–62. doi:10.2337/db13-1251
211. Miao F, Chen Z, Zhang L, Liu Z, Wu X, Yuan YC, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem (2012) 287(20):16335–45. doi:10.1074/jbc.M111.330373
212. Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes (2008) 57(12):3189–98. doi:10.2337/db08-0645
213. Irvine KM, Gallego P, An X, Best SE, Thomas G, Wells C, et al. Peripheral blood monocyte gene expression profile clinically stratifies patients with recent-onset type 1 diabetes. Diabetes (2012) 61(5):1281–90. doi:10.2337/db11-1549
214. Rousseau MC, El-Zein M, Conus F, Legault L, Parent ME. Bacillus Calmette-Guerin (BCG) vaccination in infancy and risk of childhood diabetes. Paediatr Perinat Epidemiol (2016) 30(2):141–8. doi:10.1111/ppe.12263
215. Huppmann M, Baumgarten A, Ziegler AG, Bonifacio E. Neonatal Bacille Calmette-Guerin vaccination and type 1 diabetes. Diabetes Care (2005) 28(5):1204–6. doi:10.2337/diacare.28.5.1204
216. Faustman DL, Wang L, Okubo Y, Burger D, Ban L, Man G, et al. Proof-of-concept, randomized, controlled clinical trial of Bacillus-Calmette-Guerin for treatment of long-term type 1 diabetes. PLoS One (2012) 7(8):e41756. doi:10.1371/journal.pone.0041756
217. Faustman DL. TNF, TNF inducers, and TNFR2 agonists: a new path to type 1 diabetes treatment. Diabetes Metab Res Rev (2018) 34(1). doi:10.1002/dmrr.2941
218. Sag E, Bilginer Y, Ozen S. Autoinflammatory diseases with PERIODIC FEVERS. Curr Rheumatol Rep (2017) 19(7):41. doi:10.1007/s11926-017-0670-8
219. Borghini S, Ferrera D, Prigione I, Fiore M, Ferraris C, Mirisola V, et al. Gene expression profile in TNF receptor-associated periodic syndrome reveals constitutively enhanced pathways and new players in the underlying inflammation. Clin Exp Rheumatol (2016) 34(6 Suppl 102):S121–8.
220. Bachetti T, Ceccherini I. Tumor necrosis factor receptor-associated periodic syndrome as a model linking autophagy and inflammation in protein aggregation diseases. J Mol Med (Berl) (2014) 92(6):583–94. doi:10.1007/s00109-014-1150-5
221. Todd I, Radford PM, Ziegler-Heitbrock L, Ghaemmaghami AM, Powell RJ, Tighe PJ. Elevated CD16 expression by monocytes from patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum (2007) 56(12):4182–8. doi:10.1002/art.23133
222. Ida H, Aramaki T, Arima K, Origuchi T, Kawakami A, Eguchi K. Successful treatment using tacrolimus (FK506) in a patient with TNF receptor-associated periodic syndrome (TRAPS) complicated by monocytic fasciitis. Rheumatology (Oxford) (2006) 45(9):1171–3. doi:10.1093/rheumatology/kel178
223. Carta S, Tassi S, Delfino L, Omenetti A, Raffa S, Torrisi MR, et al. Deficient production of IL-1 receptor antagonist and IL-6 coupled to oxidative stress in cryopyrin-associated periodic syndrome monocytes. Ann Rheum Dis (2012) 71(9):1577–81. doi:10.1136/annrheumdis-2012-201340
224. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol (2016) 17(10):1176–86. doi:10.1038/ni.3538
225. Vento-Tormo R, Alvarez-Errico D, Garcia-Gomez A, Hernandez-Rodriguez J, Bujan S, Basagana M, et al. DNA demethylation of inflammasome-associated genes is enhanced in patients with cryopyrin-associated periodic syndromes. J Allergy Clin Immunol (2017) 139(1):202–11e6. doi:10.1016/j.jaci.2016.05.016
226. Ibrahim JN, Jounblat R, Delwail A, Abou-Ghoch J, Salem N, Chouery E, et al. Ex vivo PBMC cytokine profile in familial Mediterranean fever patients: Involvement of IL-1beta, IL-1alpha and Th17-associated cytokines and decrease of Th1 and Th2 cytokines. Cytokine (2014) 69(2):248–54. doi:10.1016/j.cyto.2014.06.012
227. Schattner A, Lachmi M, Livneh A, Pras M, Hahn T. Tumor necrosis factor in familial Mediterranean fever. Am J Med (1991) 90(4):434–8. doi:10.1016/0002-9343(91)90602-T
228. Davtyan TK, Hakopyan GS, Avetisyan SA, Mkrtchyan NR. Impaired endotoxin tolerance induction in patients with familial Mediterranean fever. Pathobiology (2006) 73(1):26–39. doi:10.1159/000093089
229. Direskeneli H, Ozdogan H, Korkmaz C, Akoglu T, Yazici H. Serum soluble intercellular adhesion molecule 1 and interleukin 8 levels in familial Mediterranean fever. J Rheumatol (1999) 26(9):1983–6.
230. Simsek I, Pay S, Pekel A, Dinc A, Musabak U, Erdem H, et al. Serum proinflammatory cytokines directing T helper 1 polarization in patients with familial Mediterranean fever. Rheumatol Int (2007) 27(9):807–11. doi:10.1007/s00296-006-0301-6
231. Davtyan TK, Harutyunyan VA, Hakobyan GS, Avetisyan SA. Heightened endotoxin susceptibility of monocytes and neutrophils during familial Mediterranean fever. FEMS Immunol Med Microbiol (2008) 52(3):370–8. doi:10.1111/j.1574-695X.2008.00385.x
232. Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet (1999) 22(2):178–81. doi:10.1038/9696
233. Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet (1999) 22(2):175–7. doi:10.1038/9691
234. van der Meer JW, Vossen JM, Radl J, van Nieuwkoop JA, Meyer CJ, Lobatto S, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet (1984) 1(8386):1087–90. doi:10.1016/S0140-6736(84)92505-4
235. Drenth JP, van der Meer JW, Kushner I. Unstimulated peripheral blood mononuclear cells from patients with the hyper-IgD syndrome produce cytokines capable of potent induction of C-reactive protein and serum amyloid A in Hep3B cells. J Immunol (1996) 157(1):400–4.
236. Stoffels M, Jongekrijg J, Remijn T, Kok N, van der Meer JW, Simon A. TLR2/TLR4-dependent exaggerated cytokine production in hyperimmunoglobulinaemia D and periodic fever syndrome. Rheumatology (Oxford) (2015) 54(2):363–8. doi:10.1093/rheumatology/keu341
237. Rigante D, Emmi G, Fastiggi M, Silvestri E, Cantarini L. Macrophage activation syndrome in the course of monogenic autoinflammatory disorders. Clin Rheumatol (2015) 34(8):1333–9. doi:10.1007/s10067-015-2923-0
238. Mulders-Manders CM, Simon A. Hyper-IgD syndrome/mevalonate kinase deficiency: what is new? Semin Immunopathol (2015) 37(4):371–6. doi:10.1007/s00281-015-0492-6
239. Arostegui JI, Anton J, Calvo I, Robles A, Iglesias E, Lopez-Montesinos B, et al. Open-label, phase II study to assess the efficacy and safety of canakinumab treatment in active hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheumatol (2017) 69(i):1679–88. doi:10.1002/art.40146
240. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov (2012) 11(8):633–52. doi:10.1038/nrd3800
241. van der Meer JW, Barza M, Wolff SM, Dinarello CA. A low dose of recombinant interleukin 1 protects granulocytopenic mice from lethal gram-negative infection. Proc Natl Acad Sci U S A (1988) 85(5):1620–3. doi:10.1073/pnas.85.5.1620
242. Van’t Wout JW, Van der Meer JW, Barza M, Dinarello CA. Protection of neutropenic mice from lethal Candida albicans infection by recombinant interleukin 1. Eur J Immunol (1988) 18(7):1143–6. doi:10.1002/eji.1830180728
243. Liese JG, Jendrossek V, Jansson A, Petropoulou T, Kloos S, Gahr M, et al. Chronic granulomatous disease in adults. Lancet (1996) 347(8996):220–3. doi:10.1016/S0140-6736(96)90403-1
244. Rieber N, Hector A, Kuijpers T, Roos D, Hartl D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin Dev Immunol (2012) 2012:252460. doi:10.1155/2012/252460
245. van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A (2010) 107(7):3030–3. doi:10.1073/pnas.0914795107
246. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood (2010) 116(9):1570–3. doi:10.1182/blood-2010-01-264218
247. Gabrion A, Hmitou I, Moshous D, Neven B, Lefevre-Utile A, Diana JS, et al. Mammalian target of rapamycin inhibition counterbalances the inflammatory status of immune cells in patients with chronic granulomatous disease. J Allergy Clin Immunol (2017) 139(5):1641–9e6. doi:10.1016/j.jaci.2016.08.033
248. Selmeczy Z, Szelenyi J, Nemet K, Vizi ES. The inducibility of TNF-alpha production is different in the granulocytic and monocytic differentiated forms of wild type and CGD-mutant PLB-985 cells. Immunol Cell Biol (2003) 81(6):472–9. doi:10.1046/j.1440-1711.2003.01190.x
249. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A (2014) 111(9):3526–31. doi:10.1073/pnas.1322831111
250. van de Veerdonk FL, Dinarello CA. Deficient autophagy unravels the ROS paradox in chronic granulomatous disease. Autophagy (2014) 10(6):1141–2. doi:10.4161/auto.28638
251. Schappi M, Deffert C, Fiette L, Gavazzi G, Herrmann F, Belli D, et al. Branched fungal beta-glucan causes hyperinflammation and necrosis in phagocyte NADPH oxidase-deficient mice. J Pathol (2008) 214(4):434–44. doi:10.1002/path.2298
252. Deffert C, Carnesecchi S, Yuan H, Rougemont AL, Kelkka T, Holmdahl R, et al. Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells. J Pathol (2012) 228(3):341–50. doi:10.1002/path.4061
253. Brown KL, Bylund J, MacDonald KL, Song-Zhao GX, Elliott MR, Falsafi R, et al. ROS-deficient monocytes have aberrant gene expression that correlates with inflammatory disorders of chronic granulomatous disease. Clin Immunol (2008) 129(1):90–102. doi:10.1016/j.clim.2008.06.005
254. De Ravin SS, Zarember KA, Long-Priel D, Chan KC, Fox SD, Gallin JI, et al. Tryptophan/kynurenine metabolism in human leukocytes is independent of superoxide and is fully maintained in chronic granulomatous disease. Blood (2010) 116(10):1755–60. doi:10.1182/blood-2009-07-233734
255. Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, et al. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem (2006) 139(4):655–62. doi:10.1093/jb/mvj072
256. Bernardo J, Brennan L, Brink HF, Ortiz MF, Newburger PE, Simons ER. Chemotactic peptide-induced cytoplasmic pH changes in incubated human monocytes. J Leukoc Biol (1993) 53(6):673–8. doi:10.1002/jlb.53.6.673
257. Broen JC, Bossini-Castillo L, van Bon L, Vonk MC, Knaapen H, Beretta L, et al. A rare polymorphism in the gene for toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum (2012) 64(1):264–71. doi:10.1002/art.33325
Keywords: epigenetics, monocytes, immunometabolism, innate immune memory, rheumatoid arthritis, systemic lupus erythematosus, Wegener’s granulomatosis, hyper Ig-D syndroom
Citation: Arts RJW, Joosten LAB and Netea MG (2018) The Potential Role of Trained Immunity in Autoimmune and Autoinflammatory Disorders. Front. Immunol. 9:298. doi: 10.3389/fimmu.2018.00298
Received: 11 December 2017; Accepted: 02 February 2018;
Published: 20 February 2018
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Min Wu, University of North Dakota, United StatesPaola Italiani, Consiglio Nazionale Delle Ricerche (CNR), Italy
Copyright: © 2018 Arts, Joosten and Netea. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rob J. W. Arts, rob.jw.arts@radboudumc.nl