 Mei Zhang
Mei Zhang Julian A. Kim
Julian A. Kim Alex Yee-Chen Huang
Alex Yee-Chen Huang- 1Department of Biomedical Engineering, Case Western Reserve University, Cleveland, OH, United States
- 2Case Comprehensive Cancer Center, Cleveland, OH, United States
- 3Seidman Cancer Center, University Hospitals, Cleveland, OH, United States
- 4Division of Surgical Oncology, Department of Surgery, Case Western Reserve University School of Medicine, Cleveland, OH, United States
- 5Division of Pediatric Hematology-Oncology, Department of Pediatrics, Case Western Reserve University School of Medicine, Cleveland, OH, United States
Immunotherapy is revolutionizing cancer treatment. Recent clinical success with immune checkpoint inhibitors, chimeric antigen receptor T-cell therapy, and adoptive immune cellular therapies has generated excitement and new hopes for patients and investigators. However, clinically efficacious responses to cancer immunotherapy occur only in a minority of patients. One reason is the tumor microenvironment (TME), which potently inhibits the generation and delivery of optimal antitumor immune responses. As our understanding of TME continues to grow, strategies are being developed to change the TME toward one that augments the emergence of strong antitumor immunity. These strategies include eliminating tumor bulk to provoke the release of tumor antigens, using adjuvants to enhance antigen-presenting cell function, and employ agents that enhance immune cell effector activity. This article reviews the development of β-glucan and β-glucan-based nanoparticles as immune modulators of TME, as well as their potential benefit and future therapeutic applications. Cell-wall β-glucans from natural sources including plant, fungi, and bacteria are molecules that adopt pathogen-associated molecular pattern (PAMP) known to target specific receptors on immune cell subsets. Emerging data suggest that the TME can be actively manipulated by β-glucans and their related nanoparticles. In this review, we discuss the mechanisms of conditioning TME using β-glucan and β-glucan-based nanoparticles, and how this strategy enables future design of optimal combination cancer immunotherapies.
Current Status of Cancer Immunotherapy
Cancer immunotherapy was first reported using Coley’s toxin to provoke patient’s own immune responses (1). Soon after that, many studies have been performed. Preclinical animal studies have identified tumor-specific antigens and the direct killing of tumor cells using T cells specific for these antigens (2, 3). Some of these findings were recapitulated in humans and used to develop cancer immunotherapies based on provoking tumor-antigen-specific T-cell responses (4, 5). However, such immunotherapeutic approaches showed only limited clinical effect and have yet to become clinical standard of care. Subsequently, studies on tumor-induced immune suppressive mechanisms have identified multiple suppressor cell populations and inhibitory molecules, all of which exert critical regulatory roles in tumor generation and progression (6–8). These studies resulted in the development of first cancer immunotherapy agent anti-CTLA-4 monoclonal antibody (mAb), ipilimumab, which blocks the negative co-stimulatory signaling as a result of binding between cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and B7.1 (9). Clinical trial with ipilimumab demonstrated greater durable responses in advanced melanoma patients as compare to conventional therapy; therefore, it was approved by the US Food and Drug administration (FDA) for the treatment of unresectable or metastatic melanoma in 2011 (10). Thereafter, anti-PD-1 mAb, nivolumab, which blocks programmed death-1 (PD-1) binding to PD ligand 1 (PD-L1), was approved for the treatment of unresectable melanoma in 2014 (11). The FDA subsequently expanded the approved use of nivolumab to patients with advanced squamous non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy in 2015 (12). In May 2017, the FDA granted approval to pembrolizumab (anti-PD-1 mAb) for locally advanced or metastatic urothelial carcinoma, the most common type of bladder cancer, which progressed during or after platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant chemotherapy. The FDA also granted accelerated approval to pembrolizumab for patients with locally advanced or metastatic bladder cancer who are not eligible for cisplatin-containing chemotherapy (13).
The clinical successes of ipilimumab and nivolumab have encouraged the development of therapies targeting additional immune checkpoint molecules. As the activation of T cells is controlled by a balance of co-stimulatory molecules that act positively in concert with T-cell receptor (TCR) signaling, and co-inhibitory molecules, including immune checkpoint molecules, which negatively regulate TCR signaling (14), multiple agents are being developed to target co-stimulatory molecules including CD27 (15), CD28 (16), inducible T-cell co-stimulator (17), 4-1BB (18), OX40 (19), and co-inhibitory molecules including (CTLA-4) (20), PD-1 (21), PD-L1 (22), T-cell immunoglobulin and mucin domain-containing molecule-3 (23) and lymphocyte-activation gene 3 (24). These agents aim to repair immune balance at the immune checkpoint in order to promote the activation and generation of tumor-specific effector T cells. Currently, ipilimumab (mAb targeting CTLA-4), nivolumab, pembrolizumab, and pidilizumab (mAbs targeting PD-1), as well as MDX-1105, MPDL3280A, MEDI4736, and MSB0010718C (mAbs targeting PD-L1) are being tested in multiple clinical trials targeting metastatic melanoma, advanced NSCLC metastatic solid tumors, and hematological malignancies with variable clinical outcomes (25–27).
In addition to immunotherapies targeting immune checkpoint molecules, therapies targeting immunosuppressive regulatory T cell (Treg) are also being pursued. The CD25+CD4+FoxP3+ Treg cells are potent suppressors of antigen-specific or non-specific tumor immunity (28), with FoxP3 is a master gene that controls the differentiation of CD25+CD4+ T-cell and its functions (29). FoxP3+ Treg population are heterogeneous and have been classified into three sub-populations: CD45RA+FoxP3low (naïve Treg), CD45RA−FoxP3high (effector Treg) and CD45−FoxP3low (non-Treg). Available data suggest that effector Treg (e-Treg) has the highest suppressive activity among these subpopulations (30). Since CTLA-4 expression is one of the major suppressive mechanisms mediated by Treg, mAb targeting CTLA-4 was developed to inhibit the suppressive activity of CTLA4 on T cell activation (31, 32). Agents targeting GITR, OX40, and CD15S are also being investigated, as these molecules are selectively expressed on e-Treg and have functional roles (33, 34). Some conventional chemotherapeutic drugs such as cyclophosphamide (35), paclitaxel (36), gemcitabine (37), and docetaxel (38) are found to arrest cell cycle of Treg, and are being tested in clinical trials to assess their effect on reducing Treg number and function to improve antitumor immunity in cancer patients.
Another area of immune-modulatory therapy is the targeting of tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs). The presence of TAMs are associated with tumor progression, survival, angiogenesis, and the suppression of antitumor immunity (39), while MDSCs are heterogeneous populations that not only suppress immune activity but also support tumor growth and progression (40). Conventional chemotherapeutic drugs such as gemcitabine, 5-fluorouracil, docetaxel, doxorubicin, paclitaxel, and sunitinib exhibit selective inhibitory effect on MDSCs due to higher proliferative activity of MDSCs as compared to effector T-cells or natural killer (NK) cells (41). In addition, low-dose paclitaxel has been observed to promote MDSC differentiation into dendritic cells (DCs) (42). Doxorubicin exhibits an ability to induce MDSC apoptosis by enhancing the production of ROS (43). Docetaxel can promote MDSC differentiation into M1 macrophages by blocking Stat3 phosphorylation (44). Sunitinib inhibits VEGF and/or c-KIT-mediated signaling pathway in MDSCs (45). Precise molecular mechanisms of these drugs affecting MDSC function remain to be fully investigated.
Other potent immunotherapeutic approaches include chimeric antigen receptor (CAR) T-cell therapy (46), TCR cellular therapy (42), and tumor-associated antigen cancer vaccines (43). The first CAR-T cell therapy has been approved by the FDA in August of 2017. While there is much excitement surrounding CAR-T cell therapy, the broad application of this approach has been limited by the availability of suitable targets in various cancers, cost of making individualized engineered T cells, and the availability of autologous T cells for therapy production, especially in heavily pretreated patients or those with leukemic relapses soon after myeloablative bone marrow transplantation.
In summary, mAbs against immune checkpoint molecules demonstrated superior clinical effects compared with conventional therapy for tumors including advanced melanoma, NSCLC, and renal cancer. However, there is room for greater improvement on the clinical efficacy of this and other aforementioned immunotherapeutic approaches.
Additional Tumor Microenvironment (TME)-Modifying Strategies to Enhance Cancer Immunotherapy
Although ongoing clinical immunotherapies including antibodies targeting immune checkpoint molecules can result in durable responses in some otherwise therapy-refractory cancer patients, only a minority of all cancer patients benefit from these advances. Data suggest that positive responses rely on dynamic enhancing interactions between tumor cells and immune cells within the TME (47). TME plays an important role to either dampen or enhance immune responses in a context-dependent manner. As the understanding of TME increases, strategies are emerging for changing the TME from an immunosuppressive one toward one that supports the enhancement of antitumor immunity.
Current strategies using immune modulators to manipulate TME include the following. (1) The use of immune checkpoint inhibitors as immune modulators to enhance endogenous antitumor responses in TME (48, 49). (2) Targeting regulatory cells in TME. Relevant regulatory cells within TME include Treg, TAMs (M2-type macrophages) and MDSCs. Suppressive mechanisms employed by these cells involve secretion of cytokines (e.g., IL-10 and TGF-β), enzymes (e.g., arginase, NOS, and IDO), and expression of inhibitory receptors (e.g., CTLA-4 and PD-L1). Targeting these regulatory cells and their suppressive mechanisms to alter immunosuppressive nature of TME can be anticipated to enhance immunotherapy (50–59). (3) Modifying the chemokine profile of TME. Cellular composition of tumors is heavily influenced by the chemokine profile in the TME. Various types of leukocytes are attracted in response to specific chemokines. Therefore, manipulation of the chemokine profile can potentiate antitumor activity by altering immune cellular components (60–62). (4) Modulating danger signals: toll-like receptors (TLRs). TLR agonists can trigger broad inflammatory responses that elicit rapid innate immunity and promote activation of the adaptive immune reaction. Oncolytic viruses are highly immunogenic pathogens capable of stimulating TLR, and because they infect or replicate predominantly in tumor cells, much of their activity is localized to tumors (63, 64). (5) Manipulating cytokines in TME. The cytokine content of the microenvironment can tip the balance between immunosuppressive and immune-activating factors within tumors. Many types of immunotherapy benefit from co-administration of cytokines, but delivery of these cytokines is often systemic making it difficult to distinguish between contributions from microenvironment modification and systemic immune modulation. Several studies have directed cytokines specifically to tumors using engineered cytokine-producing T-cells or targeted nanoparticle system. They demonstrate observable changes in the TME and increased the efficacy of additional immunotherapeutic agents (65, 66). (6) The use of virus-like particles (VLPs) as immune modulator of TME. VLPs refer to the spontaneous organization of viral coat proteins into the 3D structure of a particular virus capsid. They are generally in the 20–500 nm size range but lack virus nucleic acid such that they are reported to be non-infectious. It has been reported in a recent study that a VLP system derived from cowpea mosaic virus possess inherent immunogenic properties that stimulate immune responses against tumor cells in preclinical mice models, and protective immunity in the models was associated with leukocyte recruitment, increased antigen-processing capabilities, stimulation of T and B lymphocytes (67). However, VLP-mediated immunomodulation appears to be non-specific and the exact cellular and molecular mechanisms remain to be defined.
The above strategies can be used to modify the TME to support additional immunotherapies (Table 1). However, the majority of these studies were limited to preclinical investigations in mice. Most clinical studies involving combination of immune modulators and immunotherapeutic agent(s) are still in early stages of clinical trials, and the precise mechanisms remains to be established. We believe it is crucial to carefully evaluate immune modulators with respect to the specificity, immune-potentiating capability, and potential toxicity prior to commencing clinical trials.
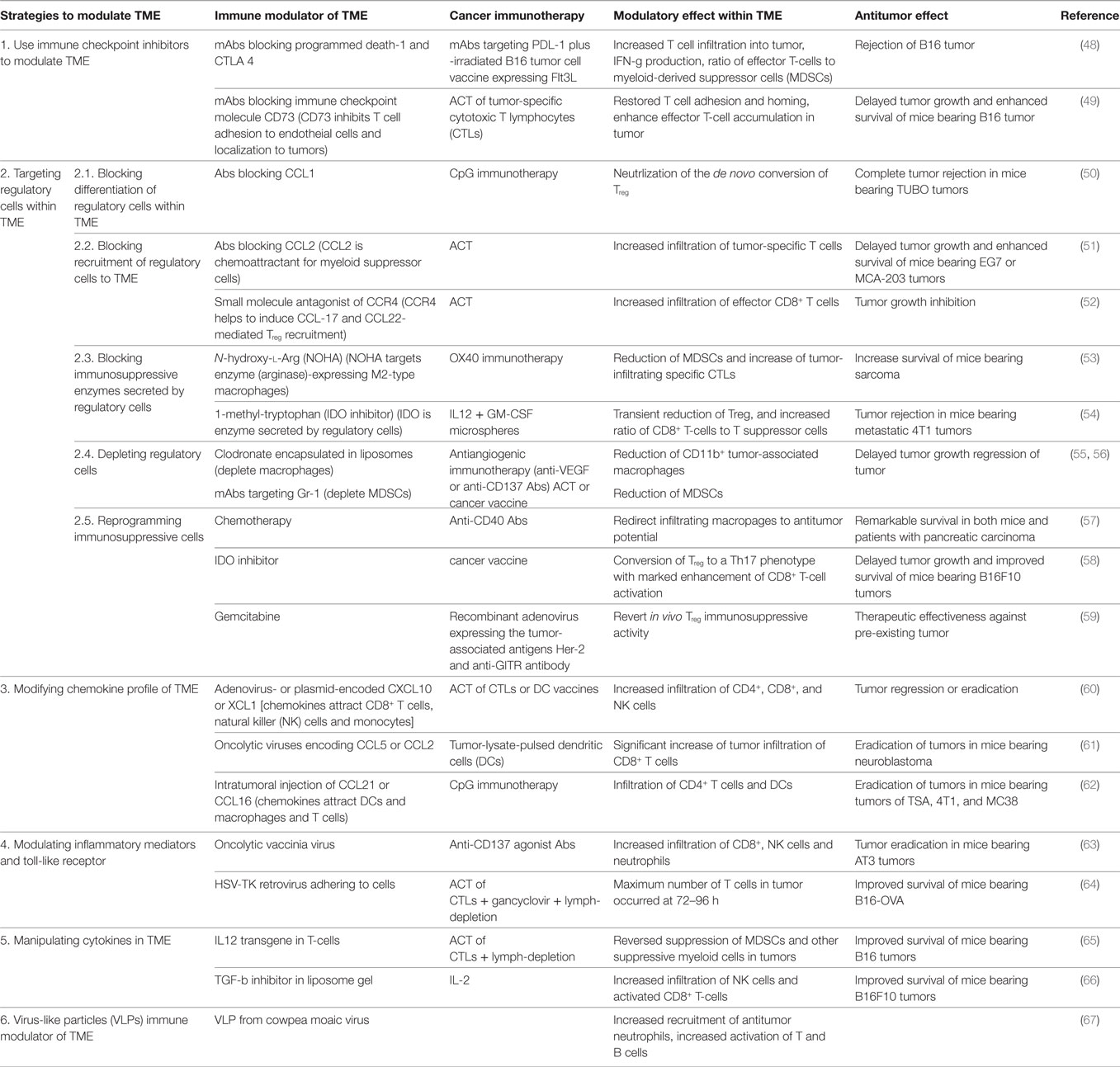
Table 1. Examples of strategies of modulating tumor microenvironment (TME) to enhance cancer immunotherapy.
Our team has focused on the development of β-glucan and β-glucan-based nanoparticles as immune modulators of TME. The β-glucan-based molecules are derived from natural resource and the safety profile has been well demonstrated. These molecules adopt pathogen-associated molecular pattern (PAMP), which has known mechanisms of targeting specific receptors on immune cell subsets. The remainder of this review will discuss the use of β-glucan and β-glucan-based nanoparticles as immune modulators of TME, their specificity, potential benefit, their advantages over other types of immune modulators, and future therapeutic applications. We will also review how β-glucan mediate changes in TME, and how this change enables the design of optimal combination cancer immunotherapies.
β-Glucan and β-Glucan-Based Nanoparticles as Effective Immune Modulator to Enhance Cancer Immunotherapy
Polysaccharides, also known as β-glucans, can be extracted from the cell walls of natural resources such as plant, fungi, and bacteria. They are biomolecules that can adopt pathogen-associated molecular patterns and can modulate host immune responses via priming and/or stimulating innate immune cells such as macrophages, neutrophils, and granulocytes (68). Both in vitro and in vivo studies have suggested that Dectin-1, complement receptor 3 (CR3), and TLR-2/6 are critical receptors mediating such priming and stimulation of innate immune cells by β-glucans (69). Binding of β-glucan on these receptors can trigger immune cells including macrophages, neutrophils, monocytes, NK cells, DCs, as well as T cells (70, 71). Recent preclinical mouse studies have demonstrated that the systemic administration of certain β-glucans could effectively manipulate TME, resulting in significant reduction of primary tumor growth and distant metastases (72). These results suggest that β-glucan molecules are potential immune modulator that can manipulate innate and adaptive immune responses within the TME and improve clinical responses of current cancer immunotherapies. As compared to the protein-, peptide-, virus-, and virus-like-particle-based immune modulators, β-glucan has several advantages: (1) β-glucan is non-immunogenic molecule due to an absence of the protein and peptide components so as not to cause non-specific immune activation; (2) β-glucan has been demonstrated to be non-toxic. A high dose up to 10 mg/kg is well tolerant in vivo with no adverse effect observed; (3) immunomodulatory effect of β-glucan is specific because it mediates immune potentiating activity on immune cells via specific surface receptors (discussed below); (4) β-glucan contains multiple aldehyde and hydroxyl groups, which provide opportunities for structural modification, improved physiological property, and construction of β-glucan-based nanoparticle system with capacity for carrying high payload of immune-modulating agents. We will review below the specific immune potentiating mechanism of β-glucan and the potential clinical benefit, and discuss the therapeutic development of β-glucan and the related nanoparticles in combination with additional cancer immunotherapy for best possible clinical outcome.
Potential Routes of Administration for β-Glucan
The potential route by which external β-glucans enter the blood stream is a very important factor in the eventual development of the β-glucan class of molecules as effective immune modulators. The human body lacks the enzyme(s) capable of digesting β-glucans. It has been well documented that β-glucans are resistant to digestion in the gastrointestinal tract (48) and require the bacterial fermentation process occurring within the large intestine (73). Additional studies over the past 40 years have examined the potential routes by which some β-glucans can enter the bloodstream to exert their biological activities. Rice et al. demonstrated that after oral administration, β-glucans can directly interact with the gastrointestinal mucosa and are then transferred to the general circulation. These studies implicated a process of β-glucan internalization by epithelial cells (possibly M cells), macrophages and DCs, which then rapidly entered the systemic circulation (74). Chan et al. documented uptake and internalization of β-glucans from the gut by macrophages, which then circulated in the blood and released β-glucans throughout the body (71). Sandvik et al. reported a total plasma β-glucan content of ~30 ng following a 14-day oral administration consisting of 5–6 mg β-glucans per day (75). Bioavailability characterization of several soluble and insoluble β-glucans revealed that the ratio of measurable glucan entering the blood to total orally glucan intake ranged from 0.5 to 5% depending on the molecular weight (Mw) and surface charge (74–77). Based upon results of these studies, oral administration is deemed as an effective route for β-glucans to enter the blood stream, and the actual efficiency of β-glucans entering blood circulation varies according to the distributions of their Mw, polydispersity, side chain branching, root mean square radius, and solution conformation.
On the other hand, studies involving systemic administration (i.v. or i.p.) of β-glucans have also been conducted. These results showed that both i.v. and i.p. administration could result in higher blood β-glucans levels with active biological activities. The half-lives of i.v. or i.p. administered β-glucans—typically on the order of hours to 10 h—were dependent on the chemical structure, Mw, side chain branching, soluble and insoluble forms, surface charge, etc. (75, 78).
β-Glucan Sources, Structure, and Active Moiety
β-glucans are polymers composed of glucose. They are cell-wall component of plants and microorganisms such as oat, barley, mushroom, seaweed, some bacteria, and yeast. Oat and barley β-glucans exhibit linear chain structure with large regions of both β-(1–3) and β-(1–4) linkages (hereafter referred to as BG34) (79, 80). Mushroom and fungal β-glucans have β-(1–3) backbone grafted with β-(1–6) side chains of various sizes and numbers and are, therefore, referred to as BG36 (81, 82). β-glucans from different resources and with different structures exhibit different solubility, physiological properties, and biological activities (83). The molecular size and complexity of β-glucans are also important for biological activity because they can influence the interaction of β-glucan with human and murine monocytes and macrophages (84, 85).
Previous studies on yeast β-glucans have suggested that the β-(1–3) glucan fragments may be the active moiety (86). In vitro coculture of primary macrophages with yeast β-glucan resulted in macrophage digestion of glucan into β-(1–3) glucan fragments (Figure 1). The digested fragments can be detected in macrophages as early as 3 days following coculture, but a more complete processing of parent β-glucan requires ~14 days. The fragments were found to prime effector cells such as macrophage, neutrophil, and granulocytes for antitumor efficacy (87). In vivo studies have shown that β-(1–3) glycosidic backbone of yeast glucan could not be digested in stomach so that most glucans enter the proximal small intestine, where the yeast glucans were captured by macrophages and digested into small fragments within macrophages. Glucan fragments could be transported by macrophages to bone marrow and the endothelial reticular system in mice, suggesting that the β-(1–3)-glucan fragments may be the active moiety of yeast glucan (88). However, whether these fragments compose the active moiety of other glucan subtypes remains to be examined. β-glucans with different structures, solubility, Mws, and routes of administration have shown variable immune potencies. Furthermore, the biologically active moieties of other glucan structures may be different from that from yeast and need to be further studied.
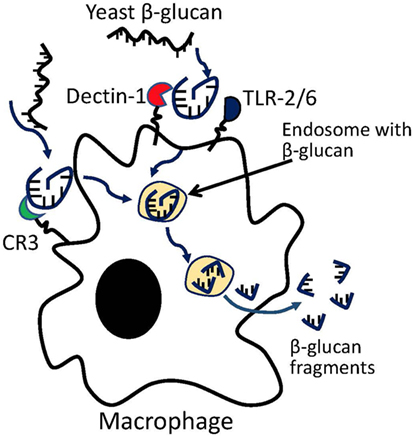
Figure 1. Macrophage processing of yeast β-glucans into small β-glucan fragments.
Mechanisms of β-Glucan-Mediated Antitumor Immune Response and Its Therapeutic Benefits
β-glucans of different resources, structures, and components mediated antitumor immune responses in different manners. For instances, zymosan, an insoluble yeast-derived β-glucan consisting of BG36 glucan and protein complexes, mediates immune responses by increasing the number and function of macrophages as well as activating the complement system (89). Lentinan, a mushroom-derived BG36 glucan with poor water solubility, mediates antitumor immune response by increasing the lymphokine-activated killer cell activity and NK cell activity (90). A particulate β-glucans derived from yeast mediates antitumor immune responses by inducing pro-inflammatory cytokine secretion and stimulating innate immune effector cell activation (70). These observations support the view that β-glucan-based molecules act through different mechanisms to stimulate antitumor immune responses.
In vitro and in vivo preclinical studies on a particulate yeast-derived BG36 glucan-protein complex implicates the dectin-1 pathway as playing a very important role in the glucan-protein complex’s ability to mediate antitumor immune responses. In vitro coculture of macrophages with the particulate yeast-derived BG36 glucan-protein complex has resulted in the activation of the macrophages, which promoted Th1 and cytotoxic T-lymphocyte priming and differentiation. In support of this, glucan-mediated macrophage activation was completely abolished in the presence of anti-dectin-1 blocking Abs. Oral administration of the particulate yeast-derived BG36 glucan-protein complex resulted in downregulating immunosuppressive cells such as Treg and MDSCs, and led to delayed tumor progression in wild-type mice. However, the striking antitumor effects were completely abrogated in dectin-1 knockout (KO) mice using the same treatment schedule with β-glucan (91–94). In addition, MDSCs that exhibited high dectin-1 expression could also be modulated by the particulate yeast-derived BG36 glucan-protein complex. In vitro coculture of MDSCs with the glucan-protein complex has resulted in the reduction of c-jun-molecule-dependent expression of nuclear factor I-A (NFIA) of granulocytic-MDSCs (G-MDSCs). Note that NFIA is an integral transcriptional component of myeloid cells and plays critical role for myeloid differentiation and lineage commitment, and the NFIA knockdown can alter the suppressive function of G-MDSCs, promote the antitumor immune responses, and delay tumor progression in mice (92). These in vitro and in vivo observations suggest that the dectin-1 pathway play a very important role for the particulate yeast-derived β-glucan-protein complex to interact not only with macrophages but also with G-MDSCs to reduce the immunosuppressive function and potentiate the development of antitumor immune responses.
Distinct from particulate yeast-derived BG36 β-glucan-protein complex, a soluble β-glucan fraction derived from yeast was shown to mediate antitumor immune responses through a specific activation of CR3 on leukocytes. CR3 is a αMβ2 integrin family glycoprotein, which is widely expressed on the surface of monocytes, macrophages, granulocytes, NK cells, and subsets of DCs and B cells. One of the critical biological function of CR3 is the CR3-dependent cellular toxicity (CR3-DCC), which is critical for the killing and clearance of microorganisms. Microorganism cell-wall is composed of β-glucan, which can bind to CD11b subunit of CR3 on leukocytes. This binding results in the activation of I-domain on the CD18 subunit of leukocyte CR3. The binding also results in complement activation and iC3b deposition on the surface of microorganisms. The ligation between CD11b-β-glucan-binding and the CD18 (I-domain)-iC3b-binding initiates killing based on a CR3-mediated mechanism, i.e., CR3-DCC, against the microorganisms. In other words, the killing of microorganism by leukocytes (mainly macrophages) requires the ligation of two binding sites within CR3 (Figure 2A) (95–97). Different from microorganisms, mammalian cells including tumor cells do not have β-glucan molecules on the surface. Therefore, although tumor cells could be coated with iC3b molecules, the binding of iC3b to CD18 subunit of CR3 on leukocytes is not sufficient to induce the leukocyte to kill the target (tumor cells) due to a lack of β-glucan-binding to CD11b on leukocytes (Figure 2B).
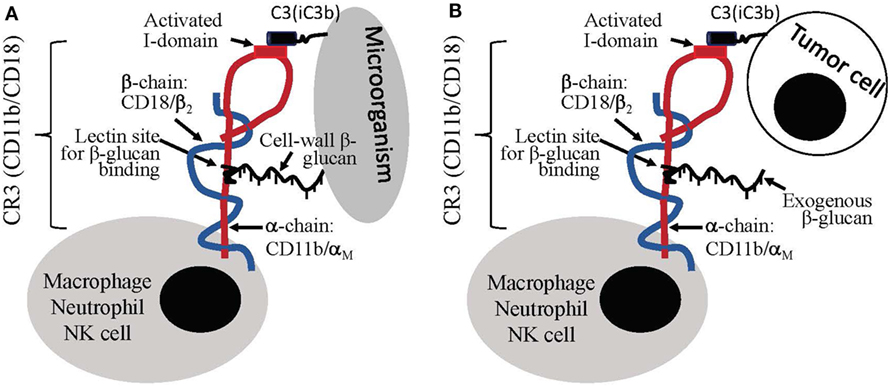
Figure 2. Leukocyte priming/activation induced by β-glucans. (A) Cell-wall β-glucans of microorganism can induce dual binding, i.e., CD11b-β-glucan binding and CD18-iC3b binding, to leukocytes, which stimulate leukocytes for complement receptor 3 (CR3)-depedent cellular cytotoxicity (CR3-DCC). (B) Tumor cells lack of β-glucans do not induce dual binding to leukocytes; however, the introduction of exogenous β-glucan can create dual-binding of leukocyte to iC3b-positive tumor cells to stimulate CR3-DCC for the destruction of the target.
Based upon this knowledge, antitumor therapy using soluble yeast-derived β-glucan was proposed and evaluated. In vitro studies demonstrated that β-glucan co-culture with leukocytes such as macrophage and neutrophil could trigger the binding of soluble β-glucan to CD11b on leukocytes, which resulted in the activation of I-domain of CD18 and its binding to iC3b-positive tumor cells. In response to iC3b-positive tumor cells, β-glucan-primed leukocytes produced cytokines including TNF-α, IFN-γ, IFN-α, and IL-6, which are associated with tumor cell destruction. Interestingly, β-glucan-primed leukocytes did not produce cytotoxic cytokines in response to iC3b-negative cells, suggesting that β-glucan has the potential to mediate specific antitumor effect without undesirable cytotoxicity to bystander cells (98).
To bolster this finding, the CR3-dependent antitumor activity of the soluble yeast-derived β-glucan has been demonstrated in a variety of murine tumor models and human tumor xenografts. The highly purified, low Mw and soluble yeast-derived BG36 β-glucan was intravenously (i.v.) administered to various tumor-bearing mice strains. Tumor regression required the pre-existence of antitumor Abs. In mice with low naturally derived Ab titers, antitumor efficacy of the yeast-derived β-glucan was significantly compromised (99). Indeed, β-glucan therapy completely failed to slow down tumor growth in Ab-deficient SCID mice. However, the β-glucan-mediated antitumor efficacy could be restored by passive immunization with either purified natural Ab in SCID mice or purified antitumor mAbs in mice with low naturally derived antitumor Ab titers (100). β-glucan therapy in combination with antitumor mAbs could enhance both tumor regression and long-term survival as compared to treatment with antitumor mAb or β-glucan therapy alone (101). More importantly, β-glucan-mediated antitumor efficacy was abrogated in either C3-deficient or CR3-deficient mice (102). Altogether, these results showed that soluble yeast-derived BG36 glucan could prime leukocytes via CR3 to induce specific antitumor immune responses via Ab-opsonization and iC3b-coating of tumor cells.
Complement receptor 3-dependent antitumor efficacy has also been demonstrated in tumor-bearing mice treated with soluble BG34 β-glucans derived from barley or oat. Daily oral administration of barley BG34 glucan in combination with an antitumor Ab, mAb14G2a, resulted in significant tumor regression and prolonged survival of neuroblastoma-bearing mice as compared to mice treated with mAb alone. This barley BG34 glucan-mediated rejection was significantly diminished in CR3-deficient mice, suggesting leukocyte CR3 as playing a critical role for soluble barley BG34 glucan-mediated antitumor immune responses (103). In our studies, we observed that oat-derived BG34 β-glucan of a specific Mw range could effectively enhance phagocytosis activity of macrophages via CD11b subunit of CR3. In vitro coculture of fluorescence (FITC)-conjugated BG34 glucans of various Mw with bone marrow-derived macrophages showed that BG34 of Mw less than 500 kD mediated effective uptake by macrophages within 6 h, and these glucan-treated macrophages showed significantly enhanced phagocytosis of latex beads. Macrophage uptake of BG34 was significantly inhibited in the presence of CD11b blocking Ab, but not anti-dectin-1 Ab, suggesting CD11b as playing a very important role for BG34 glucan-mediated activation of macrophages. Coculture of FITC-BG34 glucans with MDSCs showed that BG34 glucan of ~200 kDa could trigger direct binding to CD11b-overexpressing MDSCs (104). Furthermore, daily intraperitoneal administration of oat-derived 200 kDa BG34 glucans resulted in a reversal of TME to a pro-immunogenic one, consisting of M1-type activation of macrophages, activation of DCs, and the induction of cytokines/chemokines such as IFN-γ, TNF-α, CXCL9, CXCL10, PDL-1, and IRF-1 that are associated with T cell infiltration and tumor eradication. The oat-derived BG34 glucan treatment also resulted in a significant increase of activated T cells in the tumor-draining lymph nodes (LNs). BG34-induced alteration in TME and LN immune signatures resulted in regression of established primary and metastasis of B16F10 melanoma model in C57BL/6 mice. Such BG34-induced tumor regression was not observed in the CD11b KO mouse model (105). Results of these studies suggested that soluble BG34 glucans have a great potential to prime immune cells and enable them to mediate innate and adaptive antitumor immune responses.
In addition to the yeast-, oat-, and barley-derived glucans, bacteria-derived BG36 β-glucan, Curdlan, has shown beneficial modulatory effects on adaptive immunity. Coculture of Curdlan with Treg in vitro resulted in the conversion of Treg into Th17 effector cells. Preclinical studies in vivo showed that oral administration of Curdlan could induce both Th1 and Th17 differentiation in mice bearing 4T1 mammary tumors. Curdlan treatment could also improve CD8+ T cell priming via its potent adjuvant effect. The conversion from Treg cells into Th17 effector cells by β-glucan remains to be confirmed in in vivo tumor models. The discovery that β-glucan conversion of Treg into Th17 effector cells may lead to the development of innovative cancer immunotherapies with enhanced efficacy (106).
Purified forms of certain β-glucans from yeast, fungus, bacteria, and plants have demonstrated defined immunomodulatory effects in tumor-bearing hosts. Accumulating studies have provided increasing evidence to support the view that certain species of β-glucans interact with specific immune cell subpopulations to enhance anticancer immunity (76, 107–110). One such mechanism is the effect of β-glucan interactions with DCs and the resulting immune responses. Studies have shown that selective cytokine responses can be produced by stimulating DCs depending on the nature of interacting species of β-glucans (chemical structure, Mw, and surface charge) (111). Systemic administration of fungal β-glucans was shown to stimulate measurable plasma levels of TNF-α, and administration of fungal β-glucans in combination with TLR antagonists resulted in a significant increase of IL-12p70 in plasma, which is a key cytokine involved in initiating Th1-type CD4+ T-cell responses (111). These studies not only support the view that some β-glucans could modulate how host immune system recognizes and responds to antigens but also provide evidence that β-glucan-induced antigen recognition and immune responses could further enhance the adaptive immunity and immune memory.
β-Glucan-Based Nanoparticle Systems
Cancer immunotherapy with nanoparticles is an emerging area with significant promise in oncology. Research into glucan molecules as specific immune modulator has inspired the development of glucan-based multifunctional nanoparticle systems for targeted delivery of glucan combined with therapeutic agents to immune cells, with the anticipation that such approach could amplify glucan-mediated immune activation for more effective antitumor immunotherapy.
The particulate yeast-derived BG36 glucans are hollow, porous 2–4 µm spheres with an outer shell capable of mediating uptake by phagocytic cells via dectin-1 activated pathway. Therefore, high payload of therapeutic agents such as DNA, siRNA, protein/peptide, and small molecules could be encapsulated into the particles using a core-polyplex and layer-by-layer synthetic strategies. Such synthetic particulate yeast BG36-based particle system benefits from a high payload of therapeutics encapsulation and targeted interaction with macrophages. An in situ layer-by-layer synthesis of DNA-caged BG36 particles was shown to not only effectively protect the caged DNA from degradation but also facilitate the systemic delivery of DNA content to macrophages in vivo (112–114).
BG34 glucans are soluble molecules with multiple aldehyde and hydroxyl groups, which provide tremendous opportunity for constructing multifunctional nanoparticle systems. In our studies, we developed technologies to enable BG34 conjugation to the outer shell of carbon nanotubes of 80–100 nm in length and 10–20 nm in diameter. Carbon nanotubes can mediate near infrared trigger imaging and thermal ablation of target cells or tissues; for these reasons, they have been used for image-guided therapy. The elongated shape of nanotube can adapt an ordered alignment within vasculature, leading to fold-increases of blood circulation half-life as compared to spherical nanoparticles of similar sizes. Our BG34-conjugated carbon nanotubes were also conjugated with Fe3O4 nanoparticles to enable the magnetic resonance contrast effect (104). In vitro studies have demonstrated that BG34- and Fe3O4-conjugated carbon nanotubes can mediate excellent MRI-guided imaging of target cells and dose-dependent thermal ablation of PMJ2R macrophages upon near infrared laser irradiation, suggesting the potential of this nanotube system for MRI-guided therapeutic modulation in targeted cells/tissues. The i.v. administration of BG34- and Fe3O4-conjugated carbon nanotubes in mice bearing 4T1 mammary tumors resulted in nanotube accumulation in 4T1 tumors. Pathological analysis of 4T1 tumors showed that tumor-infiltration CD206+F4/80+ TAMs stained positive for iron, suggesting that BG34- and Fe3O4-conjugated carbon nanotubes can efficiently deliver imaging and therapeutic components into macrophage accumulated within TME. Upon accumulation of BG34- and Fe3O4-conjugated carbon nanotubes in TME as observed by MR imaging, three cycles of near infrared laser irradiation were applied directly to tumors resulting in a 24.5% reduction of macrophages in TME and 15% reduction of MDSCs in circulation as compared to untreated mice or mice treated with non-targeted carbon nanotubes. More importantly, the nanotube-mediated reduction in macrophages and MDSCs was associated with a significant reduction of lung metastasis (104).
Clinical Trials of β-Glucan for Cancer Immunotherapy
Since β-glucan therapy has achieved great success in preclinical animal models, many efforts are now underway to determine its clinical therapeutic efficacy. Currently, there are multiple β-glucan-based clinical trials in cancer immunotherapy (summarized in Table 2), many of which utilize β-glucan in conjunction with Abs or tumor vaccines.
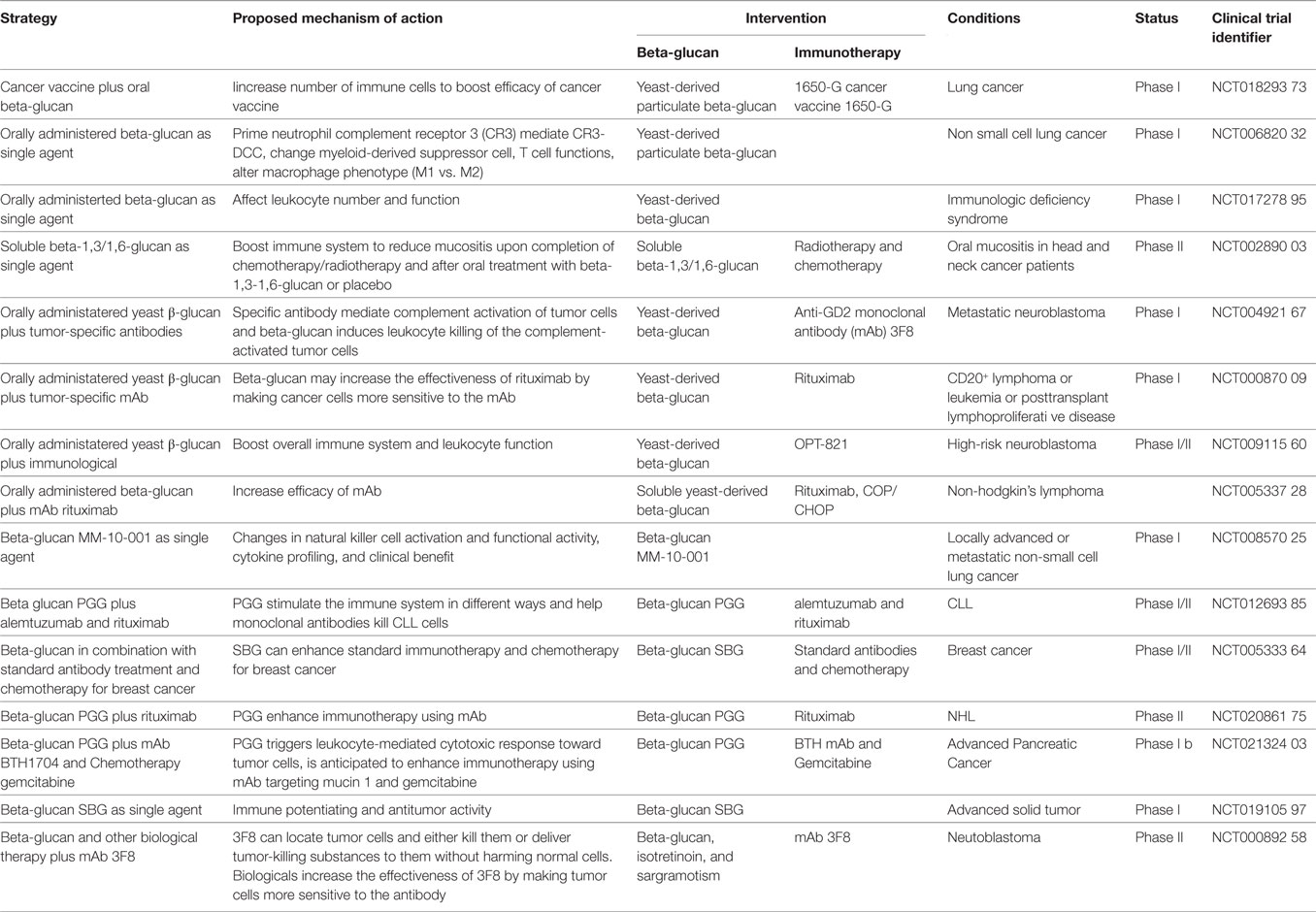
Table 2. Clinical trials of β-glucans.
Clinical experience to-date suggests that combination cancer immunotherapy is the future of cancer therapies for many cancers. Combined immunotherapy utilizing β-glucan and antitumor Abs represents one approach of breaking tumor-induced tolerance to enhance cancer immunotherapy. Combination β-glucan plus Abs therapy offers several unique advantages over other immunotherapeutic approaches. First, this approach uses humanized tumor-specific mAbs to target tumors and sensitize tumor cells for β-glucan treatment and, therefore, does not rely on the patients’ own immune responses. Second, in addition to mAbs listed in Table 2, any antitumor mAbs capable of activating the complement system can be used in combination with β-glucan. Recently, a combination therapy using β-glucan and mAbs targeting immune checkpoint molecules such as PD-1 and PD-L1 has been investigated in preclinical models with promising antitumor efficacy, and is anticipated to be translated into a phase I clinical trial (115, 116). Third, the clinical usage of checkpoint blockade Abs continues to grow and will be incorporated into the standard of care for multiple cancer types. Increases in the number of immune-modulating Abs offer more opportunities to design versatile combinations with β-glucan therapy. Fourth, this approach can also be used in synergy with most tumor vaccines as long as the tumor vaccine can elicit antitumor humoral responses and the Abs can bind to tumor cells to activate the complement system. For the tumor vaccines that elicit potent cytotoxic T cell responses along with humoral responses, combining therapy with β-glucan can add additional efficacy of innate and adaptive immune responses due to a potential role of β-glucan as an immune adjuvant.
Future Perspectives
Although β-glucan appears to stimulate antitumor immune responses via direct interaction with macrophages, it may induce interaction with other type of immune cells. For example, in addition to stimulating macrophages, particulate yeast-derived BG36 β-glucan could also stimulate DCs to secrete pro-inflammatory cytokines including IL-12, TNF-α, and IL-6. The glucan-induced production of IL-12 and TNF-α was not altered in dectin-1 KO DCs, suggesting that glucan-mediated activation of DCs were not dectin-1-dependent (117). Another example was bacteria-derived BG36 glucan, Curdlan, which could directly convert Treg into Th17 effector cells in vitro. It remains to be seen whether and how β-glucan-type molecules could affect phenotype and function of a variety of immune cells, and whether and how β-glucan-mediated immune stimulation involves other cellular receptors and pathways.
β-glucans with different structures appear to stimulate antitumor responses in completely different manners, and these detailed molecular mechanisms need to be further investigated. In vivo administration of oat-derived BG34 glucan could stimulate production of pro-inflammatory cytokines in TME, and the protective immunity was associated with a significant reduction in tumor-infiltrated granulocytes (105). In contrast, in vivo administration of soluble yeast-derived BG36 glucan did not induce any pro-inflammatory cytokines in TME, and the protective immunity was associated with a significant increase of tumor-infiltrating neutrophils that are primed for CR3-DCC (98). These results suggest that β-glucans from different sources with different glycosidic linkages, Mws, solubility, and administrative routes exhibit different mechanisms of actions. Identifying the structural-function relationship of β-glucan will not only enhance our understanding of β-glucans as immune modulator to stimulate innate and adaptive antitumor immune responses but also help to perform well-designed clinical trials to assess antitumor efficacy of β-glucans and related biomolecules.
A recent study showed that CD11b could differentially regulate TLR4-induced signaling pathways in DCs but not in macrophages (118). This study demonstrated CD11b as a key mediator of the adjuvant effect of LPS in DCs, but not in macrophages. Furthermore, the CD11b dependency is not the result of a GM-CSF-mediated cell-programming effects but involves endosomal TRIP-dependent pathways in DCs. These observations suggested that CD11b-mediated effects are likely to be restricted to a specific stage in the macrophage vs. DC lineage differentiation process. Since multiple studies have demonstrated specific binding of BG34 (105) and BG36 (91) with CD11b, these observations provided strong rationale for investigating whether these glucans can specifically affect subtypes of CD11b+ myeloid cells or a particular stage of the CD11b+ macrophage vs. DC lineage differentiation process. Identification of β-glucans capable of promoting this specific differentiation pathway may lead to novel clinical applications in cancer and other immune-mediated diseases.
New emerging data regarding the modulatory effect of β-glucan on regulatory cells such as Treg, TAMs, and MDSCs are of great importance (92, 98, 119). Currently, the main challenge of cancer immunotherapy is to inhibit the tumor-induced suppressive mechanisms and establish a TME, which is favorable for developing therapeutically efficacious antitumor immune responses. It is becoming clear that Treg, TAMs, and MDSCs are major contributors to the immune inhibitory signals limiting the adaptive antitumor immunity. Identification of the molecular mechanisms and inhibitory effects of β-glucan on these regulatory cell subsets is a critical step to develop specific immune modulator for targeted TME modulation to enhance cancer immunotherapy.
Careful selection of β-glucans is essential. In this regard, oat-derived BG34 β-glucan offers several advantages for developing high-quality clinical investigations to enhance cancer immunotherapy. First, BG34 can be highly purified to remove all possible peptide and protein complex contaminants, while BG36 glucans isolated and purified from yeast or mushroom always have undesirable peptide or protein conjugates (~5–30% protein) (120). Second, oat BG34 glucans are derived from cell-wall of plant instead of microorganism (fungi or bacteria). This allows the preparation of BG34 glucans completely free of endotoxins. Third, BG34 glucans exhibit linear and flexible chain structure in solution, allowing for isolating BG34 glucans into fractions with same chemical structures with specific Mws (106). Since the interaction of β-glucan-type molecule with murine and human leukocytes is greatly affected by glucan-associated impurities, endotoxin contaminants, and broad Mw distribution, the abovementioned features of BG34 glucans make them excellent candidates for therapeutic development.
Finally, despite potential therapeutic efficacies of combining β-glucan and various immune-modulating Abs, challenges remain ahead of achieving success with this approach. The anti-inflammatory TME milieu and the overexpression of membrane complement regulatory proteins can limit tumor-infiltrating immune cell subsets as a result of β-glucan priming. Over-expression of CD55 on SKOV-3 tumors can significantly impair complement activation and C5a release within the tumors (121), which also limits the β-glucan-primed immune cells infiltration into tumors. Inefficient iC3b deposition on tumor cells and C5a release within tumor tissue may make combination approach with β-glucan and Abs less efficacious. Therefore, additional strategies such as the use of exogenously administered pro-inflammatory cytokines or agents to induce inflammatory cytokine productions in TME may be added to overcome these obstacles. A cocktail of tumor-directed Abs targeting multiple tumor-associated antigens can also be used as a potential means to amplify complement activation and deposition of iC3b onto tumor cells to improve β-glucan-mediated therapeutic efficacy.
Author Contributions
MZ is first and corresponding author. JK and AH are co-corresponding authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is supported by the following funding sources: VeloSano Cancer Research Award (MZ and JK), NIH R21CA218790 (AH), NIH R03CA219725 (AH), St. Baldrick’s Foundation (AYA), Harrington Discovery Institute (AH), Keira Kilbane Cancer Discovery Fund (AH), Errol’s Cancer Discovery Fund (AH), Steven G. AYA Cancer Research Fund (AH), the Theresia G. and Stuart F. Kline Family Foundation (AH), The Risman Family Foundation (AH), and Chuck and Char Fowler Family Foundation AYA Initiative (AH).
References
1. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J (2006) 26:154–8.
2. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest (2015) 125(10):3981–91. doi:10.1172/JCI82416
3. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi:10.1182/blood-2008-12-195792
4. Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol (2016) 28(1):22–7. doi:10.1016/j.smim.2015.11.002
5. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
6. Stewart TJ, Smyth MJ. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Metastasis Rev (2011) 30(1):125–40. doi:10.1007/s10555-011-9280-5
7. Beavis PA, Slaney CY, Kershaw MH, Neeson PJ, Darcy PK. Enhancing the efficacy of adoptive cellular therapy by targeting tumor-induced immunosuppression. Immunotherapy (2015) 7(5):499–512. doi:10.2217/imt.15.16
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480(7378):480–9. doi:10.1038/nature10673
9. Suzuki S, Ishida T, Yoshikawa K, Ueda R. Current status of immunotherapy. Jpn J Clin Oncol (2016) 46(3):191–203. doi:10.1093/jjco/hyv201
10. Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol (2013) 94(1):41–53. doi:10.1189/jlb.1212631
11. Butler T, Maravent S, Boisselle J, Valdes J, Fellner C. A review of 2014 cancer drug approvals, with a look at 2015 and beyond. P T (2015) 40(3):191–205.
12. Ascierto PA, Marincola FM. The year of anti-PD-1/PD-L1s against melanoma and beyond. EBioMedicine (2015) 2(2):92–3. doi:10.1016/j.ebiom.2015.01.011
13. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol (2017). 14(11):655–668. doi:10.1038/nrclinonc.2017.88
14. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
15. Camerini D, Walz G, Loenen WA, Borst J, Seed B. The T cell activation antigen CD27 is a member of the nerve growth factor/tumor necrosis factor receptor gene family. J Immunol (1991) 147(9):3165–9.
16. Aruffo A, Seed B. Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proc Natl Acad Sci U S A (1987) 84(23):8573–7. doi:10.1073/pnas.84.23.8573
17. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi:10.1038/16717
18. Kwon BS, Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci U S A (1989) 86(6):1963–7. doi:10.1073/pnas.86.6.1963
19. Latza U, Dürkop H, Schnittger S, Ringeling J, Eitelbach F, Hummel M, et al. The human OX40 homolog: cDNA structure, expression and chromosomal assignment of the ACT35 antigen. Eur J Immunol (1994) 24(3):677–83. doi:10.1002/eji.1830240329
20. Dariavach P, Mattéi MG, Golstein P, Lefranc MP. Human Ig superfamily CTLA-4 gene: chromosomal localization and identity of protein sequence between murine and human CTLA-4 cytoplasmic domains. Eur J Immunol (1988) 18(12):1901–5. doi:10.1002/eji.1830181206
21. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95.
22. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest (2015) 125(9):3384–91. doi:10.1172/JCI80011
23. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi:10.1038/415536a
24. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171(5):1393–405. doi:10.1084/jem.171.5.1393
25. Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun (2013) 13:5.
26. Kim JW, Eder JP. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology (Williston Park) (2014) 28(Suppl 3):15–28.
27. Mahoney KM, Freeman GJ, McDermott DF. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin Ther (2015) 37(4):764–82. doi:10.1016/j.clinthera.2015.02.018
28. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol (1999) 163(10):5211–8.
29. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
30. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi:10.1016/j.immuni.2009.03.019
31. Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity (2002) 16(1):23–35. doi:10.1016/S1074-7613(01)00259-X
32. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi:10.1038/nri2343
33. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer (2010) 127(4):759–67. doi:10.1002/ijc.25429
34. Miyara M, Chader D, Sage E, Sugiyama D, Nishikawa H, Bouvry D, et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci U S A (2015) 112(23):7225–30. doi:10.1073/pnas.1508224112
35. Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res (2012) 72(14):3439–44. doi:10.1158/0008-5472.CAN-11-3912
36. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol (2008) 129(2):219–29. doi:10.1016/j.clim.2008.07.013
37. Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, et al. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int J Cancer (2013) 133(1):98–107. doi:10.1002/ijc.27990
38. Chu Y, Wang LX, Yang G, Ross HJ, Urba WJ, Prell R, et al. Efficacy of GM-CSF-producing tumor vaccine after docetaxel chemotherapy in mice bearing established Lewis lung carcinoma. J Immunother (2006) 29(4):367–80. doi:10.1097/01.cji.0000199198.43587.ba
39. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41(1):49–61. doi:10.1016/j.immuni.2014.06.010
40. Dilek N, Vuillefroy de Silly R, Blancho G, Vanhove B. Myeloid-derived suppressor cells: mechanisms of action and recent advances in their role in transplant tolerance. Front Immunol (2012) 3:208. doi:10.3389/fimmu.2012.00208
41. Alizadeh D, Larmonier N. Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res (2014) 74(10):2663–8. doi:10.1158/0008-5472.CAN-14-0301
42. Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol (2012) 9(3):292–300. doi:10.3109/1547691X.2011.642418
43. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res (2014) 74(1):104–18. doi:10.1158/0008-5472.CAN-13-1545
44. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16(18):4583–94. doi:10.1158/1078-0432.CCR-10-0733
45. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res (2009) 69(6):2506–13. doi:10.1158/0008-5472.CAN-08-4323
46. Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol (2016) 13(6):370–83. doi:10.1038/nrclinonc.2016.36
47. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi:10.1038/nm.3394
48. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A (2010) 107(9):4275–80. doi:10.1073/pnas.0915174107
49. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest (2011) 121(6):2371–82. doi:10.1172/JCI45559
50. Morales JK, Kmieciak M, Graham L, Feldmesser M, Bear HD, Manjili MH. Adoptive transfer of HER2/neu-specific T cells expanded with alternating gamma chain cytokines mediate tumor regression when combined with the depletion of myeloid-derived suppressor cells. Cancer Immunol Immunother (2009) 58(6):941–53. doi:10.1007/s00262-008-0609-z
51. Hoelzinger DB, Smith SE, Mirza N, Dominguez AL, Manrique SZ, Lustgarten J. Blockade of CCL1 inhibits T regulatory cell suppressive function enhancing tumor immunity without affecting T effector responses. J Immunol (2010) 184(12):6833–42. doi:10.4049/jimmunol.0904084
52. Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood (2011) 118(18):4853–62. doi:10.1182/blood-2011-01-329656
53. Gu T, Rowswell-Turner RB, Kilinc MO, Egilmez NK. Central role of IFNgamma-indoleamine 2,3-dioxygenase axis in regulation of interleukin-12-mediated antitumor immunity. Cancer Res (2010) 70(1):129–38. doi:10.1158/0008-5472.CAN-09-3170
54. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi:10.1084/jem.20101956
55. Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer (2006) 95(3):272–81. doi:10.1038/sj.bjc.6603240
56. Ko HJ, Kim YJ, Kim YS, Chang WS, Ko SY, Chang SY, et al. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res (2007) 67(15):7477–86. doi:10.1158/0008-5472.CAN-06-4639
57. Alderson KL, Luangrath M, Elsenheimer MM, Gillies SD, Navid F, Rakhmilevich AL, et al. Enhancement of the anti-melanoma response of Hu14.18K322A by alphaCD40 + CpG. Cancer Immunol Immunother (2013) 62(4):665–75. doi:10.1007/s00262-012-1372-8
58. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood (2009) 113(24):6102–11. doi:10.1182/blood-2008-12-195354
59. Gough MJ, Killeen N, Weinberg AD. Targeting macrophages in the tumour environment to enhance the efficacy of alphaOX40 therapy. Immunology (2012) 136(4):437–47. doi:10.1111/j.1365-2567.2012.03600.x
60. Li J, O’Malley M, Urban J, Sampath P, Guo ZS, Kalinski P, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther (2011) 19(4):650–7. doi:10.1038/mt.2010.312
61. Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res (2005) 65(8):3437–46. doi:10.1158/0008-5472.CAN-04-4262
62. Huang H, Xiang J. Synergistic effect of lymphotactin and interferon gamma-inducible protein-10 transgene expression in T-cell localization and adoptive T-cell therapy of tumors. Int J Cancer (2004) 109(6):817–25. doi:10.1002/ijc.20043
63. John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, et al. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res (2012) 72(7):1651–60. doi:10.1158/0008-5472.CAN-11-2788
64. Cole C, Qiao J, Kottke T, Diaz RM, Ahmed A, Sanchez-Perez L, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med (2005) 11(10):1073–81. doi:10.1038/nm1297
65. Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, et al. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater (2012) 11(10):895–905. doi:10.1038/nmat3355
66. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther (2011) 19(4):751–9. doi:10.1038/mt.2010.313
67. Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol (2016) 11(3):295–303. doi:10.1038/nnano.2015.292
68. Muta T. Molecular basis for invertebrate innate immune recognition of (1 – >3)-beta-D-glucan as a pathogen-associated molecular pattern. Curr Pharm Des (2006) 12(32):4155–61. doi:10.2174/138161206778743529
69. Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and toll-like receptor 2. J Exp Med (2003) 197(9):1107–17. doi:10.1084/jem.20021787
70. Qi C, Cai Y, Gunn L, Ding C, Li B, Kloecker G, et al. Differential pathways regulating innate and adaptive antitumor immune responses by particulate and soluble yeast-derived beta-glucans. Blood (2011) 117(25):6825–36. doi:10.1182/blood-2011-02-339812
71. Chan GC, Chan WK, Sze DM. The effects of beta-glucan on human immune and cancer cells. J Hematol Oncol (2009) 2:25. doi:10.1186/1756-8722-2-25
72. Zhang M, Yan L, Kim JA. Modulating mammary tumor growth, metastasis and immunosuppression by siRNA-induced MIF reduction in tumor microenvironment. Cancer Gene Ther (2015) 22(10):463–74. doi:10.1038/cgt.2015.42
73. Lattimer JM, Haub MD. Effects of dietary fiber and its components on metabolic health. Nutrients (2010) 2(12):1266–89. doi:10.3390/nu2121266
74. Rice PJ, Adams EL, Ozment-Skelton T, Gonzalez AJ, Goldman MP, Lockhart BE, et al. Oral delivery and gastrointestinal absorption of soluble glucans stimulate increased resistance to infectious challenge. J Pharmacol Exp Ther (2005) 314(3):1079–86. doi:10.1124/jpet.105.085415
75. Sandvik A, Wang YY, Morton HC, Aasen AO, Wang JE, Johansen FE. Oral and systemic administration of beta-glucan protects against lipopolysaccharide-induced shock and organ injury in rats. Clin Exp Immunol (2007) 148(1):168–77. doi:10.1111/j.1365-2249.2006.03320.x
76. Stier H, Ebbeskotte V, Gruenwald J. Immune-modulatory effects of dietary Yeast Beta-1,3/1,6-D-glucan. Nutr J (2014) 13:38. doi:10.1186/1475-2891-13-38
77. Tomita M, Miwa M, Ouchi S, Oda T, Aketagawa J, Goto Y, et al. Nonlinear intestinal absorption of (1 – >3)-beta-D-glucan caused by absorptive and secretory transporting system. Biol Pharm Bull (2009) 32(7):1295–7. doi:10.1248/bpb.32.1295
78. Rice PJ, Lockhart BE, Barker LA, Adams EL, Ensley HE, Williams DL. Pharmacokinetics of fungal (1-3)-beta-D-glucans following intravenous administration in rats. Int Immunopharmacol (2004) 4(9):1209–15. doi:10.1016/j.intimp.2004.05.013
79. Daou C, Zhang H. Oat beta-glucan: its role in health promotion and prevention of diseases. Compr Rev Food Sci Food Saf (2012) 11:355–65. doi:10.1111/j.1541-4337.2012.00189.x
80. Vannucci L, Krizan J, Sima P, Stakheev D, Caja F, Rajsiglova L, et al. Immunostimulatory properties and antitumor activities of glucans. Int J Oncol (2013) 43:357–64. doi:10.3892/ijo.2013.1974
81. Maeda YY, Watanabe ST, Chihara C, Rokutanda M. Denaturation and renaturation of a beta-1,6;1,3-glucan, lentinan, associated with expression of T-cell-mediated responses. Cancer Res (1988) 48(3):671–5.
82. Samuelsen AB, Schrezenmeir J, Knutsen SH. Effects of orally administered yeast-derived beta-glucans: a review. Mol Nutr Food Res (2014) 58(1):183–93. doi:10.1002/mnfr.201300338
83. Bohn JA, BeMiller JN. (1–3)-β-D-glucans as biological response modifiers: a review of structure-functional activity relationships. Carbohydr Polym (1995) 28:3–14. doi:10.1016/0144-8617(95)00076-3
84. Nisini R, Torosantucci A, Romagnoli G, Chiani P, Donati S, Gagliardi MC, et al. beta-glucan of Candida albicans cell wall causes the subversion of human monocyte differentiation into dendritic cells. J Leukoc Biol (2007) 82(5):1136–42. doi:10.1189/jlb.0307160
85. Zhang M, Kim JA. Effect of molecular size and modification pattern on the internalization of water soluble beta-(1 – > 3)-(1 – > 4)-glucan by primary murine macrophages. Int J Biochem Cell Biol (2012) 44(6):914–27. doi:10.1016/j.biocel.2012.02.018
86. Hong F, Yan J, Baran JT, Allendorf DJ, Hansen RD, Ostroff GR, et al. Mechanism by which orally administered beta-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol (2004) 173(2):797–806. doi:10.4049/jimmunol.173.2.797
87. Yan J, Vetvicka V, Xia Y, Hanikýrová M, Mayadas TN, Ross GD. Critical role of Kupffer cell CR3 (CD11b/CD18) in the clearance of IgM-opsonized erythrocytes or soluble beta-glucan. Immunopharmacology (2000) 46(1):39–54. doi:10.1016/S0162-3109(99)00157-5
88. Li B, Cramer D, Wagner S, Hansen R, King C, Kakar S, et al. Yeast glucan particles activate murine resident macrophages to secrete proinflammatory cytokines via MyD88- and Syk kinase-dependent pathways. Clin Immunol (2007) 124(2):170–81. doi:10.1016/j.clim.2007.05.002
89. Ezekowitz RA, Sim RB, Hill M, Gordon S. Local opsonization by secreted macrophage complement components. Role of receptors for complement in uptake of zymosan. J Exp Med (1984) 159(1):244–60. doi:10.1084/jem.159.1.244
90. Tani M, Tanimura H, Yamaue H, Tsunoda T, Iwahashi M, Noguchi K, et al. Augmentation of lymphokine-activated killer cell activity by lentinan. Anticancer Res (1993) 13(5C):1773–6.
91. Bose N, Chan AS, Guerrero F, Maristany CM, Qiu X, Walsh RM, et al. Binding of soluble yeast beta-glucan to human neutrophils and monocytes is complement-dependent. Front Immunol (2013) 4:230. doi:10.3389/fimmu.2013.00230
92. Baldwin KT, Carbajal KS, Segal BM, Giger RJ, et al. Neuroinflammation triggered by beta-glucan/dectin-1 signaling enables CNS axon regeneration. Proc Natl Acad Sci U S A (2015) 112(8):2581–6. doi:10.1073/pnas.1423221112
93. Ross GD, Cain JA, Myones BL, Newman SL, Lachmann PJ. Specificity of membrane complement receptor type three (CR3) for beta-glucans. Complement (1987) 4(2):61–74. doi:10.1159/000463010
94. Hong F, Hansen RD, Yan J, Allendorf DJ, Baran JT, Ostroff GR, et al. Beta-glucan functions as an adjuvant for monoclonal antibody immunotherapy by recruiting tumoricidal granulocytes as killer cells. Cancer Res (2003) 63(24):9023–31.
95. Thornton BP, Vĕtvicka V, Pitman M, Goldman RC, Ross GD. Analysis of the sugar specificity and molecular location of the beta-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18). J Immunol (1996) 156(3):1235–46.
96. Vĕtvicka V, Hanikýrová M, Vĕtvicková J, Ross GD. Regulation of CR3 (CD11b/CD18)-dependent natural killer (NK) cell cytotoxicity by tumour target cell MHC class I molecules. Clin Exp Immunol (1999) 115(2):229–35. doi:10.1046/j.1365-2249.1999.00800.x
97. Ross GD, Vetvicka V, Yan J, Xia Y, Vetvicková J. Therapeutic intervention with complement and beta-glucan in cancer. Immunopharmacology (1999) 42(1–3):61–74. doi:10.1016/S0162-3109(99)00013-2
98. Russell DG, Wright SD. Complement receptor type 3 (CR3) binds to an Arg-Gly-Asp-containing region of the major surface glycoprotein, gp63, of Leishmania promastigotes. J Exp Med (1988) 168(1):279–92. doi:10.1084/jem.168.1.279
99. Cain JA, Newman SL, Ross GD. Role of complement receptor type three and serum opsonins in the neutrophil response to yeast. Complement (1987) 4(2):75–86. doi:10.1159/000463011
100. Vetvicka V, Thornton BP, Ross GD. Soluble beta-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J Clin Invest (1996) 98(1):50–61. doi:10.1172/JCI118777
101. Yan J, Vetvicka V, Xia Y, Coxon A, Carroll MC, Mayadas TN, et al. Beta-glucan, a “specific” biologic response modifier that uses antibodies to target tumors for cytotoxic recognition by leukocyte complement receptor type 3 (CD11b/CD18). J Immunol (1999) 163(6):3045–52.
102. Zen K, Liu Y, Cairo D, Parkos CA. CD11b/CD18-dependent interactions of neutrophils with intestinal epithelium are mediated by fucosylated proteoglycans. J Immunol (2002) 169(9):5270–8. doi:10.4049/jimmunol.169.9.5270
103. Modak S, Koehne G, Vickers A, O’Reilly RJ, Cheung NK, et al. Rituximab therapy of lymphoma is enhanced by orally administered (1 – >3),(1 – >4)-D-beta-glucan. Leuk Res (2005) 29(6):679–83. doi:10.1016/j.leukres.2004.10.008
104. Yan L, Gao Y, Pierce R, Dai L, Kim J, Zhang M. Development of Y-shaped peptide for constructing nanoparticle systems targeting tumor-associated macrophages in vitro and in vivo. Mater Res Express (2014) 1:25007. doi:10.1088/2053-1591/1/2/025007
105. Zhang M, Chun L, Sandoval V, Graor H, Myers J, Nthale J, et al. Systemic administration of -glucan of 200 kDa modulates melanoma microenvironment and suppresses metastatic cancer. Oncoimmunology (2017) 7(2):e1387347. doi:10.1080/2162402X.2017.1387347
106. Osorio F, LeibundGut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, et al. DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol (2008) 38(12):3274–81. doi:10.1002/eji.200838950
107. Ali MF, Driscoll CB, Walters PR, Limper AH, Carmona EM, et al. Beta-glucan-activated human B lymphocytes participate in innate immune responses by releasing proinflammatory cytokines and stimulating neutrophil chemotaxis. J Immunol (2015) 195(11):5318–26. doi:10.4049/jimmunol.1500559
108. Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK, et al. Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res (2014) 20(5):1375–82. doi:10.1158/1078-0432.CCR-13-1012
109. Vetvicka V, Richter J, Svozil V, Rajnohová Dobiášová L, Král V, et al. Placebo-driven clinical trials of yeast-derived beta-(1-3) glucan in children with chronic respiratory problems. Ann Transl Med (2013) 1(3):26.
110. Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature (2011) 472(7344):471–5. doi:10.1038/nature10071
111. Huang H, Ostroff GR, Lee CK, Wang JP, Specht CA, Levitz SM. Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and toll-like receptor agonists. Infect Immun (2009) 77(5):1774–81. doi:10.1128/IAI.00086-09
112. Sugikawa K, Numata M, Kaneko K, Sada K, Shinkai S. Alternate layer-by-layer adsorption of single- and double-walled carbon nanotubes wrapped by functionalized beta-1,3-glucan polysaccharides. Langmuir (2008) 24(23):13270–5. doi:10.1021/la802211q
113. Soto E, Kim YS, Lee J, Kornfeld H, Ostroff G. Glucan particle encapsulated rifampicin for targeted delivery to macrophages. Polymers (2010) 2:681–9. doi:10.3390/polym2040681
114. Soto ER, Caras AC, Kut LC, Castle MK, Ostroff GR. Glucan particles for macrophage targeted delivery of nanoparticles. J Drug Deliv (2012) 2012:143524. doi:10.1155/2012/143524
115. Qiu X, Chan A, Jonas A, Kangas T, Ottoson N, Graff J, et al. Imprime PGG, a yeast beta-glucan PAMP elicits a coordinated immune response in combination with anti-PD1 antibody. J Immunother Cancer (2016) 196:16. doi:10.1186/2051-1426-2-S3-P191
116. Koido S, Ohkusa T, Homma S, Namiki Y, Takakura K, Saito K, et al. (2013). Immunotherapy for colorectal cancer. World J Gastroenterol 19(46):8531–42.
117. Hernanz-Falcón P, Joffre O, Williams DL, Reis e Sousa C. Internalization of Dectin-1 terminates induction of inflammatory responses. Eur J Immunol (2009) 39(2):507–13. doi:10.1002/eji.200838687
118. Ling GS, Bennett J, Woollard KJ, Szajna M, Fossati-Jimack L, Taylor PR, et al. Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat Commun (2014) 5:3039. doi:10.1038/ncomms4039
119. Municio C, Alvarez Y, Montero O, Hugo E, Rodríguez M, Domingo E, et al. The response of human macrophages to beta-glucans depends on the inflammatory milieu. PLoS One (2013) 8(4):e62016. doi:10.1371/journal.pone.0062016
120. Kollár R, Reinhold BB, Petráková E, Yeh HJ, Ashwell G, Drgonová J, et al. Architecture of the yeast cell wall. Beta(1 – >6)-glucan interconnects mannoprotein, beta(1 – >)3-glucan, and chitin. J Biol Chem (1997) 272(28):17762–75. doi:10.1074/jbc.272.28.17762
121. Li B, Allendorf DJ, Hansen R, Marroquin J, Cramer DE, Harris CL, et al. Combined yeast {beta}-glucan and antitumor monoclonal antibody therapy requires C5a-mediated neutrophil chemotaxis via regulation of decay-accelerating factor CD55. Cancer Res (2007) 67(15):7421–30. doi:10.1158/0008-5472.CAN-07-1465
Keywords: cancer immunotherapy, tumor microenvironment, immune modulator, beta-glucan, beta-glucan-based nanoparticle
Citation: Zhang M, Kim JA and Huang AYC (2018) Optimizing Tumor Microenvironment for Cancer Immunotherapy: β-Glucan-Based Nanoparticles. Front. Immunol. 9:341. doi: 10.3389/fimmu.2018.00341
Received: 15 October 2017; Accepted: 06 February 2018;
Published: 26 February 2018
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Carlos Alfaro, Universidad de Navarra, SpainDmitry Bulavin, Institute of Research on Cancer and Aging in Nice, France
Christopher Gregory, University of Edinburgh, United Kingdom
Copyright: © 2018 Zhang, Kim and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mei Zhang, mxz128@case.edu;
Julian Kim, julian.kim@uhhospitals.org;
Alex Y. Huang, alex.y.huang@case.edu, alex.huang@uhhospitals.org