 Andrew J. Olive
Andrew J. Olive Christopher M. Sassetti
Christopher M. Sassetti- 1Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, United States
- 2Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA, United States
Our understanding of the host response to infections has historically focused on “resistance” mechanisms that directly control pathogen replication. However, both pathogen effectors and antimicrobial immune pathways have the capacity to damage host tissue, and the ability to tolerate these insults can also be critical for host survival. These “tolerance” mechanisms may be equally as important as resistance to prevent disease in the context of a persistent infection, such as tuberculosis, when resistance mechanisms are ineffective and the pathogen persists in the tissue for long periods. Host tolerance encompasses a wide range of strategies, many of which involve regulation of the inflammatory response. Here we will examine general strategies used by macrophages and T cells to promote tolerance in the context of tuberculosis, and focus on pathways, such as regulation of inflammasome activation, that are emerging as common mediators of tolerance.
Introduction
The ultimate goal of the host response to bacterial pathogens is to survive the infection. Much of the research to understand protective immunity has historically had a singular focus on antimicrobial resistance mechanisms that directly control bacterial replication. In general, these “resistance” mechanisms act by poisoning the pathogen, disrupting the pathogen's niche, or sequestering nutrients in an attempt to restrict growth and spread (1, 2). Classic resistance pathways include the antimicrobial peptide production from epithelial surfaces and the microbicidal functions of phagocytes which are augmented by antigen-specific lymphocyte responses. More recently it has become clear that in addition to these resistance strategies, the host also relies on distinct mechanisms that allow it to withstand infections independently of controlling bacterial growth (3, 4). These “tolerance” mechanisms represent host pathways that modulate diverse aspects of physiology. Both the local control of inflammatory tissue damage and repair, as well as systemic responses such as anorexia, and fever have been shown to promote host survival in a number of infection contexts (5, 6). For many self-resolving infections, resistance mechanisms may be sufficient to restrict bacterial replication and minimize pathology (2). However, some pathogens, like Mycobacterium tuberculosis (Mtb), are able to resist many of the resistance mechanisms of the host and persist for long periods (7). In these situations, tolerance pathways are critical for preventing the progressive pathology elicited by the persistent presence of the pathogen. Tolerance responses ensure that the locally infected tissues continue to function and that the overall health of the host is maintained (3, 8).
While potential therapies that promote host resistance have received a great deal of interest, promoting tolerance pathways that decrease morbidity and/or mortality in the face of an ongoing chronic infection could represent an equally appealing avenue for intervention (9, 10). In this review, we will discuss the host response to Mtb infections from the viewpoint of host tolerance. While tolerance encompasses a potentially large array of host functions, we will consider known and emerging mechanisms that limit lung damage and discuss how distinct cell populations like macrophages and T cells contribute to tolerance by controlling cytokine production and metabolic functions. Ultimately, understanding host tolerance mechanisms will define new pathways of protective immunity to tuberculosis (TB), and could identify new therapeutic strategies.
Tuberculosis Pathogenesis
Mtb infections are transmitted by aerosol (7, 11). Following inhalation of contaminated droplets, Mtb is engulfed by alveolar macrophages, where the pathogen replicates and evades the innate antimicrobial mechanisms of this cell (7, 11, 12). After the activation of host adaptive immune responses, bacterial growth is slowed or halted. While evidence from non-human primates (NHP) and human autopsy studies indicate that some infectious foci can be sterilized, the pathogen is able to persist in the face of this adaptive response for long periods. In some individuals, this infection produces the chronic inflammatory disease called, tuberculosis (TB). While any organ in the body can be affected, pulmonary disease promotes transmission of the pathogen, beginning a new infectious cycle.
For most individuals, chronic infection with Mtb does not produce symptomatic disease (7, 13). However, a subset of individuals (5–10%) will progress to develop TB after a period of asymptomatic infection that generally lasts for less than 2 years, but can extend for decades in rare cases (14, 15). What drives the heterogeneity of disease progression is not entirely known and is likely a combination of host and bacterial genetic diversity, as well as environmental factors (3, 8, 16). Several distinct aspects of TB pathogenesis could be affected by host tolerance pathways. Most obviously, the risk of developing disease is likely to depend on host tolerance. Most infected individuals never develop symptoms, and the ability to harbor this immunogenic pathogen for long-periods without suffering from progressive pathology likely depends on the ability to control inflammation(10, 17, 18). In fact, the phenomenon of “latent TB infection” (LTBI) could be considered one of the clearer examples of pathogen tolerance in humans. Patients that are cured of TB by antibiotic therapy suffer from reduced respiratory function, indicating that even after bacteria are eradicated, local tissue damage persists (19–21). In fact, multiple rounds of infection and antibiotic therapy are associated with increased erosion of lung function (21). This effect is not simply additive, as rabbits exposed to 5 sequential low dose infections developed significantly more severe cavitary disease than animals exposed to a single large dose of Mtb (22). Thus, tolerance mechanisms that control local tissue damage could determine long-term outcome and are influenced by environmental factors such as the frequency of infection. Manifestations of Mtb other than pulmonary disease may be even more dependent on host tolerance mechanisms that control inflammation (23). For example, meningeal Mtb infection is associated with very high mortality, which is related to the expression of inflammatory cytokines (24, 25). Similarly, TB immune reconstitution inflammatory syndrome (TB-IRIS) is a condition that occurs in HIV/Mtb co-infected individuals soon after starting antiretroviral therapy (26). This syndrome still results in almost 40% mortality, and is associated with failed regulation of inflammatory cascades (27–30).
The mechanisms that control TB tolerance are complex because interactions between multiple cell types influence disease progression. Following infection and activation of the host immunity, infected cells are walled off in large structures termed a granuloma (7, 16). Granulomas are thought to be required for the host to tolerate Mtb infections, yet their development and progression throughout infection may also drive Mtb survival and transmission. Bacterial barcoding and PET-CT studies in non-human primates have shown individual granuloma that are formed from single founder bacteria can have very distinct fates, some contain the pathogen and while others progressively develop into the large cavities that typify pulmonary TB disease (16, 31, 32). As a result, individual lesions are variable in their disease trajectories and transmission potential suggesting complicated dynamics determine the outcome of each lesion (31, 32). Beyond granuloma development, influx of leukocytes such as neutrophils and the expression of proteases such as matrix metalloproteinases (MMPs) can reduce host tolerance by irreversibly damaging tissue (33, 34). As the role of MMPs and neutrophils in modulating immunopathology to Mtb have been reviewed elsewhere, we will focus on how macrophages and T cells modulate host tolerance to determine the outcome of Mtb infections (35, 36).
Macrophages and Tolerance
Macrophages are an important intracellular niche for Mtb to replicate yet they can also restrict Mtb growth in an activation dependent manner (12). The balance between Mtb replication and control is determined by a diverse array of resistance pathways, including those activated by interferon-γ (IFNγ), granulocyte-macropahge colony stimulating factor (GM-CSF) and interleukin-1β (IL-1β) (37–39). Due to their direct interactions with Mtb, macrophages are also central regulators of host tolerance. Several lines of evidence suggest that tolerance mechanisms modulated by macrophages may play a significant role in determining disease progression and controlling the outcome to Mtb disease.
Nitric Oxide
One compelling case for the role of tolerance in macrophages during chronic Mtb infections is that of inducible nitric oxide synthase (Nos2). For years, it was generally presumed that the protective function of Nos2 could be attributed to the direct antimicrobial activity of nitric oxide (NO) (40). In support of this hypothesis was data that showed that Nos2 deficient mice are extremely susceptible to Mtb infection (40, 41). These animals die within 2 months of infection with 10–100 fold more bacteria in lungs than wild type animals as well as a massive infiltration of tissue-damaging neutrophils. Recent evidence however suggests that the situation is more complex. Mtb expresses a number of defense mechanisms that protect the pathogen from the antimicrobial effects of NO, and recent evidence suggests that the role of Nos2 in regulating inflammatory pathways and host tolerance play a dominant role in protection (Figure 1) (42–45).
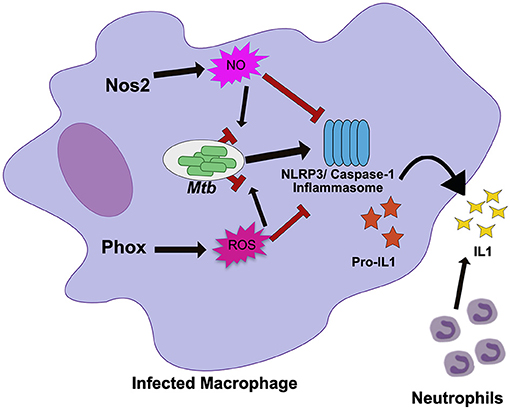
Figure 1. Nos2 and Phox control tolerance during Mtb infection by negatively regulating inflammasome activation. During Mtb infection in macrophages, Nos2 and Phox produce NO and ROS respectively. While these molecules are antimicrobial against many pathogens, Mtb is mostly resistant. Persistent Mtb then activates the NLRP3 inflammasome to produce active IL1β. Prolonged inflammasome activation leads to increased IL1β secretion and neutrophil recruitment that damages the lungs. In order to tolerate persistent infections with Mtb, the NO and ROS produced by macrophages also suppresses inflammasome activation to limit the damage caused by recurring neutrophil recruitment. NO directly nitrosylates NLRP3 while the mechanisms of ROS inhibition remain unknown.
Disentangling tolerance pathways in vivo is a significant challenge due to the interlinked nature of bacterial load and tissue damage; higher bacterial burdens can lead to more inflammation and tissue damage, while higher tissue damage and inflammation may create an environment that drives more bacterial replication (8). The role of each host effector in controlling resistance or tolerance pathways may also be timing and context dependent (46). It is also likely that many pathways control both resistance and tolerance during persistent infection (47). Because of this, distinct in vivo models that control either inflammation or bacterial replication are required to break down the mechanisms of a particular “protective” gene like Nos2. When these models were applied to Nos2, it became clear in Nos2 deficient animals succumb to Mtb infection through hyperinflammatory disease, even when bacterial load is controlled using a conditionally-replicating strain of the pathogen (43, 44). Subsequent mechanistic studies determined that NO nitrosylates the inflammasome component NLRP3, which inhibits the production of bioactive IL-1β and prevents persistent neutrophil recruitment (44). Similarly, Nos2 has also been shown to dampen the inflammatory response by limiting the activation of NF-κβ (48).
Nos2 serves as an important example of the need to understand the mechanisms by which individual immune effectors protect against TB disease progression. While a modest role for Nos2 in modulating Mtb replication in macrophages remains possible, the recent evidence strongly suggests the predominant role of NO production in mice during Mtb is to control tolerance by dampening inflammatory pathways.
NADPH Phagocyte Oxidase
Many immune mediators have similarly pleiotropic effects as Nos2, raising the possibility that other well-characterized pathways may also play unanticipated roles in regulating tolerance. The NADPH Phagocyte Oxidase Complex (Phox) provides another example. This system is required to produce a burst of reactive oxygen species (ROS) that intoxicate the intracellular bacteria. The importance of Phox in protecting the host during Mtb infections is generally considered minimal because Phox deficient animals show no long-term defects in controlling Mtb growth and Mtb is equipped with many strategies to resist ROS-mediated killing (41, 49–51). However, human studies suggest that mutations in Phox, which leads to the condition known as chronic granulomatous disease, are associated with higher susceptibility to mycobacterial infections including TB (52, 53). In other disease contexts Phox deficiencies have been found cause inflammatory disease, particularly those related to IL-1β activation (54). Recent work shows that Phox is also critical for tolerance to Mtb infection (Figure 1) (55). Phox-deficient mice have no deficiency in bacterial control, yet Phox-deficient animals accumulate high numbers of neutrophils in an IL-1β dependent fashion, leading to exacerbated disease (55). Similar to the role of Nos2, the ROS produced by Phox control tolerance by inhibiting the activation of the NLRP3 inflammasome which reduces IL-1β production and limits neutrophil influx to the infected lung. The fact that the important tolerance-regulating functions for both Nos2 and Phox were overlooked for some time, suggests that tolerance-regulating roles may still be found for additional host response pathways.
The similar ability of Nos2 and Phox to control inflammasome activation suggests that preventing persistent IL-1β production is a common strategy used by the host to tolerate persistent infections. In support of this, human studies have found that altered IL-1β expression modulates TB disease severity (56). IL-1β alleles that enhance IL-1β expression are associated with increased risk of developing TB disease, more severe pulmonary disease, and poor treatment outcome (56). In addition, inflammasome activation is associated with the development of TB-IRIS and TB meningitis (57, 58). Two recent studies suggest that expression and activation of inflammasome components including NLRP3 and the high expression of IL-1β in plasma and the nervous system are signatures of failed tolerance during antiretroviral treatment and a major risk factor to developing fatal disease (57, 58). The repeated association with inflammasome activity, IL-1β production and more severe TB-related pathology suggests that this pathway could serve as a therapeutic target, particularly for the severe inflammatory syndromes with poor outcomes.
Lysosomal Function and Autophagy
Proper maintenance of cellular organelles is important to tolerate Mtb infections (59–62). Loss of critical homeostatic pathways can lead to cellular dysfunction and misregulation of inflammatory cytokines during Mtb disease. Mycobacterium infections of zebrafish with mutations in cathepsins leads to loss of granuloma integrity and reduced survival due to improper breakdown in lysosomal contents (59). In humans, this mutation is phenocopied in individuals who smoke tobacco. Mtb infected macrophages from smokers accumulate particulates in their lysosomes, inhibiting their function and likely altering tolerance. It is well known that previous smoking history can increase the risk of developing TB disease by over two-fold and it is possible that alterations to lysosomal function are a key aspect to these patients TB susceptibility (63).
Autophagy is another key pathway that maintains the integrity of organelles and regulates a variety of important immune-related processes (64). Recently, the role of autophagy in antimicrobial resistance during Mtb has been questioned but the importance of Atg5 in tolerance is undeniable (61, 65, 66). Mice with mutations in most autophagy genes control Mtb disease normally (61). However, Atg5-/- mice show a unique susceptibility to TB disease. Infection of Atg5 mice leads to a hyperinflammatory disease state with massive neutrophil migration to the pulmonary tissue and rapid mortality (61). Depletion of neutrophils alone in infected Atg5 deficient mice can reverse the susceptibility and allow long term survival arguing against an inherent defect in antimicrobial control. Exactly how Atg5 controls the inflammatory response, or why loss of Atg5 and not other autophagy components drives neutrophil-mediated disease remains to be understood. But it is clear that altering macrophage homeostasis directly modulates tolerance to Mtb.
Macrophage Metabolism
Recent evidence suggests that macrophage metabolic pathways and byproducts can modulate the inflammatory pathways both locally and systemically (1, 2). Similarly, Mtb infections are influenced by systemic metabolic dysfunction such as diabetes, which can alter the activation state of macrophages at the site of infection (67). Evidence for how essential local and systemic metabolic networks influence host tolerance to Mtb is beginning to emerge.
Central regulators of host cell metabolism are intimately linked with control of inflammatory circuits (68). These pathways, including mammalian target of rapamycin (mTOR), silent mating type information regulation 2 homologs (Sirtuins), and adenosine monophosphate-activated protein kinase (AMPK), are known to regulate cellular functions such as autophagy, NF-kb signaling, and central metabolism. Importantly, many of these networks are disrupted during Mtb infection suggesting that they could play a role in regulating the inflammatory milieu that is activated during Mtb infection and likely influence host tolerance (69). Because FDA approved modulators of these metabolic networks are available, they represent appealing targets for host directed therapies that may enhance tolerance during Mtb infections and improve clinical outcomes (70).
Sirtuin 1 (SIRT1), a known regulator of host stress responses, is downregulated during Mtb infection (71). In order to understand how the loss of SIRT1 function impacts Mtb disease, Singhal and colleagues treated infected macrophages and animals with a known small molecule SIRT1 activator (Figure 2) (71). While activation of SIRT1 resulted in a modest reduction in bacterial growth in vitro and in vivo, it led to dramatic changes in the inflammatory profile of infected macrophages and immunopathology in mice, indicating that activation of SIRT1 promotes host tolerance during Mtb infection. Interestingly, SIRT1 activation during Mtb results in similar outcomes to treatment with the AMPK activator metformin, a common treatment for diabetes (72). During Mtb infection, metformin treatment leads to subtle decreases in bacterial burden but larger decreases in inflammatory cytokines and tissue damage (Figure 2). A retrospective study of diabetic TB patients indicates that metformin may improve outcomes. SIRT1 can also influence AMPK signaling, suggesting that the SIRT1/AMPK signaling axis may be a critical regulator of tolerance during Mtb infection. It is also intriguing that diabetes treatments such as metformin, are so effective against treating Mtb disease. Diabetes increases Mtb risk in humans (73, 74). In a mouse model of hyperglycemia, there was a profound effect on neutrophil accumulation during Mtb infection which worsened disease outcome (75). Thus, while it likely the complex effect of diabetes on immunity could include resistance defects, in the mouse model tolerance defects appear to dominate.
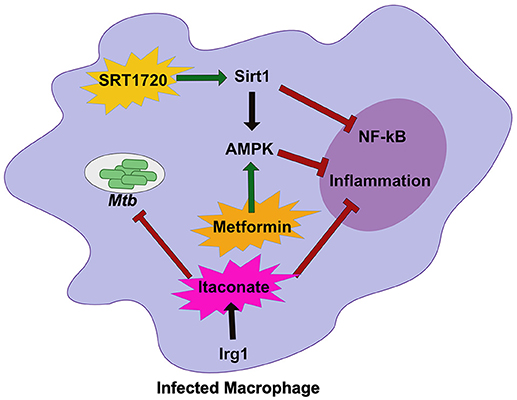
Figure 2. Host metabolic networks modulate tolerance to Mtb infections. Distinct metabolic networks control the inflammatory response during Mtb infection. Small molecule activation of Sirt1 with SRT1720 inhibits NF-κβ signaling and activates AMPK and promote tolerance to Mtb. This is similar to treatment with the diabetes drug Metformin that activates AMPK to inhibit inflammation and allow the host to better tolerate persistent Mtb infections. An alternative metabolic network activated by Irg1, produces the metabolite Itaconate. Itaconate can directly restrict Mtb replication, but in vivo robustly controls tolerance by modulating the inflammatory response to persistent infection. Together these metabolic networks directly and indirectly control tolerance to Mtb infection.
Another important metabolic pathway that modulates tolerance to Mtb is mediated by the mitochondrial enzyme immune responsive gene 1 (Irg1) (76). Irg1 produces the metabolite itaconate that recently was shown to dampen cytokine production and reduce damaging ROS during Mtb infection (Figure 2) (77). Loss of Irg1 in vivo leads to rapid mortality that is driven by hyper-inflammation and neutrophil-mediated disease. Itaconate alone is sufficient to reverse the increase in pro-inflammatory gene expression in infected Irg1 deficient macrophages suggesting this metabolite is a robust regulator of tolerance to Mtb. While itaconate can directly inhibit bacterial growth, in vivo studies indicate that its immunomodulatory function may play a dominant role (76, 77). Future studies will need to carefully dissect the role of Irg1 in both controlling resistance and tolerance to fully understand its pleiotropic functions during Mtb infection.
T Cells and Tolerance
T cells are critical for resistance to Mtb (7, 78). In addition, it is clear that Th1 cells that produce IFNγ promote tolerance by activating the production of NO and by directly inhibiting the recruitment of neutrophils (44, 79). This profound effect on Mtb protection suggested that more robust activation of Th1 cells would lead to improved disease outcomes. In reality, the situation is much more complicated and recent evidence suggests that activating enhanced Th1 responses to Mtb leads to increased susceptibility through failed tolerance.
IFNγ. The cytokine IFNγ is produced by activated T cells during Mtb infection and is essential for protection of the host. During chronic infections, the levels of IFNγ produced by individual T cells can wain due to persistent antigen and T cell exhaustion (80). Targeting inhibitory receptors on T cells might drive enhanced cytokine responses and lead to more robust Mtb control. As a proof of principle of this concept mice lacking the T cell inhibitory receptor PD1 were infected with Mtb (81). Surprisingly, rather controlling Mtb infection better, PD1 deficient animals had decreased tolerance that was characterized by increased susceptibility and immunopathology. This counterintuitive result suggested that more robust T cell responses might be detrimental to long term Mtb protection. What is driving the decrease in tolerance in these animals? One recent study began to examine the mechanisms modulating the tolerance defect in PD1 deficient mice and showed that increased IFNγ production is responsible (82). When PD1 deficient T cells no longer make IFNγ, the defect in tolerance is reversed. In addition, CD4+ T cells that produce more IFNγ on a per cell basis do not control Mtb growth more effectively in the lungs, but rather cause tissue damage and more rapid mortality. Similarly, T cells with mutations in the Calcium channel ORAI1 activating protein Stim1 are unable to undergo apoptosis following infection leading to a significant increase in IFNγ in the lungs (83). This increase in T cell survival and IFNγ makes infected mice susceptible to infection by decreasing tolerance. Therefore, during Mtb infection pushing the expression of IFNγ beyond a protective threshold leads to failed tolerance.
T cell Metabolism. While it is possible that results with PD1 are an outlier additional evidence suggests that other alterations to T cell activation may have deleterious effects on tolerating Mtb. One recent study found an important role for Cyclophilin D in modulating tolerance to Mtb in a T cell dependent manner (84). CyclophilinD (CypD) is a mitochondrial protein that modulates cell death mechanisms such as necrosis (85). Inhibition of CypD in macrophages prevents necrosis and limits Mtb replication (86, 87). On this basis, Divangahi and colleagues infected CypD mice, and found that they were highly susceptible to infection (84). However, these mice succumbed to disease with identical burdens of bacteria compared to wild type animals suggesting loss of CypD decreases tolerance to persistent Mtb infections. Importantly, the defect in tolerance was not related to differences in cell death and control of Mtb replication. Instead CypD was found to regulate a metabolic switch between oxidative phosphorylation and glycolysis in T cells. In the absence of CypD, T cells produced more ROS that drove glycolytic flux, leading to enhanced activation and cytokine production. This critical change in the central metabolism of T cells dramatically reduced the tolerance CypD animals to persistent Mtb infection.
Taken together the findings that increasing T cell numbers and enhancing their function in the lungs of Mtb infected animals reduces tolerance is compelling. We can no longer pursue the development of therapeutics or vaccines that simply drive more activated T cells and more IFNγ production without considering the very real possibility of deleterious effects. Mammalian hosts have clearly evolved an important balance between antimicrobial resistance strategies and tolerance mechanisms to survive persistent infections that must be more adequately evaluated in our research as we pursue more effective Mtb treatment strategies.
Outlook
The studies discussed above suggest a critical role for the regulation of inflammatory cascades in tolerance to persistent Mtb infection, and highlights a number of well-studied pathways in this process. It seems clear that macrophages integrate metabolic and innate immune signals with those derived from T cells to control the extent of inflammatory tissue damage. While these pathways are important determinants of disease progression, they likely represent a small fraction of the mechanisms that contribute to tolerance. Our current understanding of TB tolerance is focused largely on immunological factors with an already appreciated protective role in the mouse model of TB. However, in simpler model systems, it is clear that a wide variety of functions involved in tissue repair, systemic metabolism, and energy utilization also play an important role. Furthermore, it is clear that bacterial factors interact with the immune system to regulate tolerance, and a number of Mtb genes have been found to alter immunopathology without affecting bacterial fitness (88, 89). Developing models for TB where these diverse tolerance pathways can be observed and dissected represents a major challenge for the future.
While our understanding of tolerance generally lags far behind our knowledge of resistance mechanisms, the examples described above highlight the importance of continued research. While antibiotics are generally effective for uncomplicated Mtb infections, several particularly serious and/or long-term sequelae of Mtb infection can be attributed to defects in tolerance. These complications include acute failures of tolerance, such as meningitis and TB-IRIS, as well as the long-term tissue damage and decreased lung function that generally follows infection. Understanding the processes involved in damage and repair will likely produce more effective therapies.
Author Contributions
AO and CS conceived of and wrote the article together.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, MB, and handling editor declared their shared affiliation at the time of the review.
Acknowledgments
We thank the Sassetti lab for helpful discussions. This work was funded by the Arnold and Mabel Beckman Postdoctoral Fellowship (AO), and NIH Grant AI132130 (CS).
References
1. Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol. (2012) 10:525–37. doi: 10.1038/nrmicro2836
2. Olive AJ, Sassetti CM. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol. (2016) 14:221–34. doi: 10.1038/nrmicro.2016.12
3. Ayres JS, Schneider DS. Tolerance of infections. Annu Rev Immunol. (2012) 30:271–94. doi: 10.1146/annurev-immunol-020711-075030
4. Meunier I, Kaufmann E, Downey J, Divangahi M. Unravelling the networks dictating host resistance versus tolerance during pulmonary infections. Cell Tissue Res. (2017) 367:525–36. doi: 10.1007/s00441-017-2572-5
5. Louie A, Song KH, Hotson A, Thomas Tate A, Schneider DS. How many parameters does it take to describe disease tolerance? PLoS Biol. (2016) 14:e1002435. doi: 10.1371/journal.pbio.1002435
6. Schieber AM, Ayres JS. Thermoregulation as a disease tolerance defense strategy. Pathog Dis. (2016) 74:ftw106. doi: 10.1093/femspd/ftw106
7. Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol. (2014) 12:289–99. doi: 10.1038/nrmicro3230
8. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335:936–41. doi: 10.1126/science.1214935
9. Kolloli A, Subbian S. Host-directed therapeutic strategies for tuberculosis. Front Med. (2017) 4:171. doi: 10.3389/fmed.2017.00171
10. Ndlovu H, Marakalala MJ. Granulomas and inflammation: host-directed therapies for tuberculosis. Front Immunol. (2016) 7:434. doi: 10.3389/fimmu.2016.00434
11. Huang L, Russell DG. Protective immunity against tuberculosis: what does it look like and how do we find it? Curr Opin Immunol. (2017) 48:44–50. doi: 10.1016/j.coi.2017.08.001
12. VanderVen BC, Huang L, Rohde KH, Russell DG. The minimal unit of infection: Mycobacterium tuberculosis in the macrophage. Microbiol Spectr. (2016) 4. doi: 10.1128/microbiolspec.TBTB2-0025-2016
13. Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol. (2017) 17:691–702. doi: 10.1038/nri.2017.69
14. Behr MA, Edelstein PH, Ramakrishnan L. Revisiting the timetable of tuberculosis. BMJ (2018) 362:k2738 doi: 10.1136/bmj.k2738
15. Lillebaek T, Dirksen A, Vynnycky E, Baess I, Thomsen VO, Andersen AB. Stability of DNA patterns and evidence of Mycobacterium tuberculosis reactivation occurring decades after the initial infection. J Infect Dis. (2003) 188:1032–9. doi: 10.1086/378240
16. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med (2014) 20:75–9. doi: 10.1038/nm.3412
17. Dorhoi A, Reece ST, Kaufmann SH. For better or for worse: the immune response against Mycobacterium tuberculosis balances pathology and protection. Immunol Rev (2011) 240:235–51. doi: 10.1111/j.1600-065X.2010.00994.x
18. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell (2012) 148:434–46. doi: 10.1016/j.cell.2011.12.023
19. Lee SW, Kim YS, Kim DS, Oh YM, Lee SD. The risk of obstructive lung disease by previous pulmonary tuberculosis in a country with intermediate burden of tuberculosis. J Korean Med Sci. (2011) 26:268–73. doi: 10.3346/jkms.2011.26.2.268
20. Pasipanodya JG, Miller TL, Vecino M, Munguia G, Garmon R, Bae S, et al. Pulmonary impairment after tuberculosis. Chest (2007) 131:1817–24. doi: 10.1378/chest.06-2949
21. Plit ML, Anderson R, Van Rensburg CE, Page-Shipp L, Blott JA, Fresen JL, et al. Influence of antimicrobial chemotherapy on spirometric parameters and pro-inflammatory indices in severe pulmonary tuberculosis. Eur Respir J. (1998) 12:351–6. doi: 10.1183/09031936.98.12020351
22. Urbanowski ME, Ihms EA, Bigelow K, Kubler A, Elkington PT, Bishai WR. Repetitive aerosol exposure promotes cavitary tuberculosis and enables screening for targeted inhibitors of extensive lung destruction. J Infect Dis. (2018) 218:53–63. doi: 10.1093/infdis/jiy127
23. Kritsaneepaiboon S, Andres MM, Tatco VR, Lim CCQ, Concepcion NDP. Extrapulmonary involvement in pediatric tuberculosis. Pediatr Radiol. (2017) 47:1249–59. doi: 10.1007/s00247-017-3867-0
24. Duque-Silva A, Robsky K, Flood J, Barry PM. Risk factors for central nervous system tuberculosis. Pediatrics (2015) 136:e1276–1284. doi: 10.1542/peds.2014-3958
25. Ong CW, Pabisiak PJ, Brilha S, Singh P, Roncaroli F, Elkington PT, et al. Complex regulation of neutrophil-derived MMP-9 secretion in central nervous system tuberculosis. J Neuroinflammation (2017) 14:31. doi: 10.1186/s12974-017-0801-1
26. Meintjes G, Rabie H, Wilkinson RJ, Cotton MF. Tuberculosis-associated immune reconstitution inflammatory syndrome and unmasking of tuberculosis by antiretroviral therapy. Clin Chest Med. (2009) 30, 797–810, x doi: 10.1016/j.ccm.2009.08.013
27. Andrade BB, Singh A, Narendran G, Schechter ME, Nayak K, Subramanian S, et al. Mycobacterial antigen driven activation of CD14++CD16- monocytes is a predictor of tuberculosis-associated immune reconstitution inflammatory syndrome. PLoS Pathog. (2014) 10:e1004433. doi: 10.1371/journal.ppat.1004433
28. Nakiwala JK, Walker NF, Diedrich CR, Worodria W, Meintjes G, Wilkinson RJ, et al. Neutrophil activation and enhanced release of granule products in HIV-TB immune reconstitution inflammatory syndrome. J Acquir Immune Defic Syndr. (2018) 77:221–9. doi: 10.1097/QAI.0000000000001582
29. Tadokera R, Meintjes GA, Wilkinson KA, Skolimowska KH, Walker N, Friedland JS, et al. Matrix metalloproteinases and tissue damage in HIV-tuberculosis immune reconstitution inflammatory syndrome. Eur J Immunol. (2014) 44:127–36. doi: 10.1002/eji.201343593
30. Walker NF, Wilkinson KA, Meintjes G, Tezera LB, Goliath R, Peyper JM, et al. Matrix degradation in human immunodeficiency virus Type 1-associated tuberculosis and tuberculosis immune reconstitution inflammatory syndrome: a prospective observational study. Clin Infect Dis. (2017) 65:121–32. doi: 10.1093/cid/cix231
31. Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog. (2015) 11:e1004603. doi: 10.1371/journal.ppat.1004603
32. Martin CJ, Cadena AM, Leung VW, Lin PL, Maiello P, Hicks N, et al. Digitally barcoding mycobacterium tuberculosis reveals in vivo infection dynamics in the macaque model of tuberculosis. MBio (2017) 8: e00312–17. doi: 10.1128/mBio.00312-17
33. Dorhoi A, Yeremeev V, Nouailles G, Weiner JIII, Jorg S, Heinemann E, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol. (2014) 44:2380–93. doi: 10.1002/eji.201344219
34. Ong CW, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-derived MMP-8 drives ampk-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog. (2015) 11:e1004917. doi: 10.1371/journal.ppat.1004917
35. Elkington PT, Ugarte-Gil CA, Friedland JS. Matrix metalloproteinases in tuberculosis. Eur Respir J. (2011) 38:456–64. doi: 10.1183/09031936.00015411
36. Lyadova IV. Neutrophils in tuberculosis: heterogeneity shapes the way? Mediators Inflamm. (2017) 2017:8619307. doi: 10.1155/2017/8619307
37. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. (1993) 178:2243–7. doi: 10.1084/jem.178.6.2243
38. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35:1023–34. doi: 10.1016/j.immuni.2011.12.002
39. Rothchild AC, Jayaraman P, Nunes-Alves C, Behar SM. iNKT cell production of GM-CSF controls Mycobacterium tuberculosis. PLoS Pathog. (2014) 10:e1003805. doi: 10.1371/journal.ppat.1003805
40. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. (1997) 94:5243–8. doi: 10.1073/pnas.94.10.5243
41. Jung YJ, LaCourse R, Ryan L, North RJ. Virulent but not avirulent Mycobacterium tuberculosis can evade the growth inhibitory action of a T helper 1-dependent, nitric oxide Synthase 2-independent defense in mice. J Exp Med. (2002) 196:991–8. doi: 10.1084/jem.20021186
42. Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science (2003) 302:1963–6. doi: 10.1126/science.1091176
43. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol. (2017) 2:17072. doi: 10.1038/nmicrobiol.2017.72
44. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol. (2013) 14:52–60. doi: 10.1038/ni.2474
45. Samanovic MI, Tu S, Novak O, Iyer LM, McAllister FE, Aravind L, et al. Proteasomal control of cytokinin synthesis protects Mycobacterium tuberculosis against nitric oxide. Mol Cell. (2015) 57:984–94. doi: 10.1016/j.molcel.2015.01.024
46. Jeney V, Ramos S, Bergman ML, Bechmann I, Tischer J, Ferreira A, et al. Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep. (2014) 8:126–36. doi: 10.1016/j.celrep.2014.05.054
47. Soares MP, Teixeira L, Moita LF. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol. (2017) 17:83–96. doi: 10.1038/nri.2016.136
48. Braverman J, Stanley SA. Nitric oxide modulates macrophage responses to Mycobacterium tuberculosis infection through activation of HIF-1alpha and repression of NF-kappaB. J Immunol. (2017) 199:1805–16. doi: 10.4049/jimmunol.1700515
49. Cooper AM, Segal BH, Frank AA, Holland SM, Orme IM. Transient loss of resistance to pulmonary tuberculosis in p47(phox-/-) mice. Infect Immun. (2000) 68:1231–4. doi: 10.1128/IAI.68.3.1231-1234.2000
50. Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, et al. The oxidative stress network of Mycobacterium tuberculosis reveals coordination between radical detoxification systems. Cell Host Microbe. (2015) 17:829–37. doi: 10.1016/j.chom.2015.05.008
51. Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in Mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol. (2004) 52:1291–302. doi: 10.1111/j.1365-2958.2004.04078.x
52. Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous Mycobacterial disease. Nat Immunol. (2011) 12:213–21. doi: 10.1038/ni.1992
53. Deffert C, Cachat J, Krause KH. Phagocyte NADPH oxidase, chronic granulomatous disease and Mycobacterial infections. Cell Microbiol. (2014) 16:1168–78. doi: 10.1111/cmi.12322
54. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. (2014) 111:3526–31. doi: 10.1073/pnas.1322831111
55. Olive AJ, Smith CM, Kiritsy MC, Sassetti CM. The phagocyte oxidase controls tolerance to Mycobacterium tuberculosis infection. J Immunol. (2018). doi: 10.4049/jimmunol.1800202
56. Zhang G, Zhou B, Li S, Yue J, Yang H, Wen Y, Zhan S, Wang W, Liao M, Zhang M, et al. Allele-specific induction of IL-1beta expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog. (2014) 10:e1004426. doi: 10.1371/journal.ppat.1004426
57. Marais S, Lai RPJ, Wilkinson KA, Meintjes G, O'Garra A, Wilkinson RJ. Inflammasome activation underlying central nervous system deterioration in HIV-associated tuberculosis. J Infect Dis. (2017) 215:677–86. doi: 10.1093/infdis/jiw561
58. Tan HY, Yong YK, Shankar EM, Paukovics G, Ellegard R, Larsson M, et al. Aberrant inflammasome activation characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. J Immunol. (2016) 196:4052–63. doi: 10.4049/jimmunol.1502203
59. Berg RD, Levitte S, O'Sullivan MP, O'Leary SM, Cambier CJ, Cameron J, et al. Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell (2016) 165:139–52. doi: 10.1016/j.cell.2016.02.034
60. Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al. (2018). Galectins control mTOR in response to endomembrane damage. Mol Cell. 70:120–35 e128. doi: 10.1016/j.molcel.2018.03.009
61. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature (2015) 528:565–9. doi: 10.1038/nature16451
62. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature (2013) 501:512–6. doi: 10.1038/nature12566
63. Yen YF, Yen MY, Lin YS, Lin YP, Shih HC, Li LH, et al. Smoking increases risk of recurrence after successful anti-tuberculosis treatment: a population-based study. Int J Tuberc Lung Dis. (2014) 18:492–8. doi: 10.5588/ijtld.13.0694
64. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy (2018) 14:243–51. doi: 10.1080/15548627.2017.1402992
65. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA. (2012) 109:E3168–3176. doi: 10.1073/pnas.1210500109
66. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
67. Kumar Nathella P, Babu S. Influence of diabetes mellitus on immunity to human tuberculosis. Immunology (2017) 152:13–24. doi: 10.1111/imm.12762
68. Domblides C, Lartigue L, Faustin B. Metabolic stress in the immune function of T cells macrophages and dendritic cells. Cells (2018) 7:E68. doi: 10.3390/cells7070068
69. Stutz MD, Clark MP, Doerflinger M, Pellegrini M. Mycobacterium tuberculosis: rewiring host cell signaling to promote infection. J Leukoc Biol. (2018) 103:259–68. doi: 10.1002/JLB.4MR0717-277R
70. Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. (2015) 7:127–39. doi: 10.15252/emmm.201404137
71. Cheng CY, Gutierrez NM, Marzuki MB, Lu X, Foreman TW, Paleja B, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. (2017) 2:eaaj1789. doi: 10.1126/sciimmunol.aaj1789
72. Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, et al. (2014). Metformin as adjunct antituberculosis therapy. Sci Transl Med. 6:263ra159. doi: 10.1126/scitranslmed.3009885
73. Joshi N, Caputo GM, Weitekamp MR, Karchmer AW. Infections in patients with diabetes mellitus. N Engl J Med. (1999) 341:1906–12. doi: 10.1056/NEJM199912163412507
74. Ponce-De-Leon A, Garcia-Garcia Md Mde L, Garcia-Sancho MC, Gomez-Perez FJ, Valdespino-Gomez JL, Olaiz-Fernandez G, et al. Tuberculosis and diabetes in southern Mexico. Diabetes Care (2004) 27:1584–90. doi: 10.2337/diacare.27.7.1584
75. Martens GW, Arikan MC, Lee J, Ren F, Greiner D, Kornfeld H. Tuberculosis susceptibility of diabetic mice. Am J Respir Cell Mol Biol. (2007) 37:518–24. doi: 10.1165/rcmb.2006-0478OC
76. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA. (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
77. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection J Exp Med. (2018) 215:1035–45. doi: 10.1084/jem.20180118
78. Sakai S, Mayer-Barber KD, Barber DL. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr Opin Immunol. (2014) 29:137–42. doi: 10.1016/j.coi.2014.06.003
79. Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. (2011) 208:2251–62. doi: 10.1084/jem.20110919
80. Jayaraman P, Jacques MK, Zhu C, Steblenko KM, Stowell BL, Madi A, et al. TIM3 mediates T cell exhaustion during Mycobacterium tuberculosis Infection. PLoS Pathog. (2016) 12:e1005490. doi: 10.1371/journal.ppat.1005490
81. Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. (2011) 186:1598–607. doi: 10.4049/jimmunol.1003304
82. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog. (2016) 12:e1005667. doi: 10.1371/journal.ppat.1005667
83. Desvignes L, Weidinger C, Shaw P, Vaeth M, Ribierre T, Liu M, et al. STIM1 controls T cell-mediated immune regulation and inflammation in chronic infection. J Clin Invest. (2015) 125:2347–62. doi: 10.1172/JCI80273
84. Tzelepis F, Blagih J, Khan N, Gillard J, Mendonca L, Roy DG, et al. Mitochondrial cyclophilin D regulates T cell metabolic responses and disease tolerance to tuberculosis. Sci Immunol. (2018) 3: eaar4135. doi: 10.1126/sciimmunol.aar4135
85. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature (2005) 434:658–62. doi: 10.1038/nature03434
86. Gan H, He X, Duan L, Mirabile-Levens E, Kornfeld H, Remold HG. Enhancement of antimycobacterial activity of macrophages by stabilization of inner mitochondrial membrane potential. J Infect Dis. (2005) 191:1292–300. doi: 10.1086/428906
87. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell (2013) 153:521–34. doi: 10.1016/j.cell.2013.03.022
88. Martinot AJ, Farrow M, Bai L, Layre E, Cheng TY, Tsai JH, et al. Mycobacterial metabolic syndrome: LprG and Rv1410 regulate triacylglyceride levels, growth rate and virulence in Mycobacterium tuberculosis. PLoS Pathog. (2016) 12:e1005351. doi: 10.1371/journal.ppat.1005351
Keywords: Mycobaterium tuberculosis, tolerance, inflammasome, immunometabolism, persistent infections
Citation: Olive AJ and Sassetti CM (2018) Tolerating the Unwelcome Guest; How the Host Withstands Persistent Mycobacterium tuberculosis. Front. Immunol. 9:2094. doi: 10.3389/fimmu.2018.02094
Received: 24 July 2018; Accepted: 24 August 2018;
Published: 12 September 2018.
Edited by:
Maziar Divangahi, McGill University, CanadaCopyright © 2018 Olive and Sassetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew J. Olive, oliveand@msu.edu
Christopher M. Sassetti, christopher.sassetti@umassmed.edu