 Yanwei Cheng
Yanwei Cheng Tony N. Marion
Tony N. Marion Xue Cao
Xue Cao Wanting Wang1
Wanting Wang1- 1West China Hospital Emergency Department, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, and Collaborative Innovation Center of Biotherapy, Chengdu, China
- 2Disaster Medicine Center, Sichuan University, Chengdu, China
- 3Department of Rheumatology and Immunology, West China Hospital, Sichuan University, Chengdu, China
- 4Department of Microbiology, Immunology, and Biochemistry, The University of Tennessee Health Science Center, Memphis, TN, United States
Sepsis remains a serious and life-threatening condition with high morbidity and mortality due to uncontrolled inflammation together with immunosuppression with few therapeutic options. Macrophages are recognized to play essential roles throughout all phases of sepsis and affect both immune homeostasis and inflammatory processes, and macrophage dysfunction is considered to be one of the major causes for sepsis-induced immunosuppression. Currently, Parkinson disease protein 7 (Park 7) is known to play an important role in regulating the production of reactive oxygen species (ROS) through interaction with p47phox, a subunit of NADPH oxidase. ROS are key mediators in initiating toll-like receptor (TLR) signaling pathways to activate macrophages. Emerging evidence has strongly implicated Park 7 as an antagonist for sepsis-induced immunosuppression, which suggests that Park 7 may be a novel therapeutic target for reversing immunosuppression compromised by sepsis. Here, we review the main characteristics of sepsis-induced immunosuppression caused by macrophages and provide a detailed mechanism for how Park 7 antagonizes sepsis-induced immunosuppression initiated by the macrophage inflammatory response. Finally, we further discuss the most promising approach to develop innovative drugs that target Park 7 in patients whose initial presentation is at the late stage of sepsis.
Introduction
Sepsis is a common clinical disease with high morbidity and mortality. Annually, ~30 million (1) people are affected by sepsis and more than 6–8 million (2) of those affected die. Despite significant advances in treatment, sepsis is still a major clinical problem and remains the leading cause of death in the critically ill patient population (3, 4) with an associated severe cost burden (5). In 2013, sepsis was responsible for more than $23 billion (6) of hospital costs in the USA alone. Thus, sepsis has been described as “the quintessential medical disorder of the twenty-first century.” On 26 May 2017, the World Health Organization listed sepsis as a global health priority by adopting a resolution to improve the prevention, diagnosis and management of this deadly disease (7).
In the recent “sepsis-3” consensus (8), sepsis is defined as a life-threatening, multiorgan dysfunction caused by a dysregulated host response to infection, which is primarily caused by Gram-negative bacteria. However, a global study of 14,000 critically ill patients found that 47% of isolates were Gram-positive, indicating that more patients currently become septic from Gram-positive infections (9). Even after an inciting infection has been resolved, septic patients continue to mount an excessive inflammatory response (10) that leads to tissue damage and organ failure. Key advances have made earlier recognition and treatment of sepsis feasible with the result that some patients can restore immune homeostasis, completely clear infection, and achieve complete recovery (11). Otherwise, patients progress into late stage sepsis and suffer from severe immunosuppression characterized by an impaired activation of the immune response and a hypo-inflammatory response (12), resulting in more difficult recovery and poor long-term outcomes with risk of cognitive and physical impairments, even an increased incidence of delayed death due to the lack of effective treatment for sepsis-induced immunosuppression (13). At present, immunosuppression in septic patients constitutes an important focus of research. Thus far, various interrelated, non-mutually exclusive mechanisms have been proposed to explain sepsis-induced immunosuppression, including cellular apoptosis (14), autophagy (15, 16), regulation by the central nervous system (17, 18), metabolic reprogramming (19), epigenetic regulation (20–22), and endotoxin tolerance (23–25). The immunopathogenesis of sepsis-induced immunosuppression is a very complex process that involves both innate and adaptive immune cells. In fact, it is at least partially caused by the dysfuction of macrophages.
Macrophages and Sepsis-Induced Immunosuppression
Macrophages play essential roles throughout all phases of sepsis with their ubiquitous presence and comprehensive effects on immune homeostasis and inflammatory process. After infection, macrophage is activated through Toll-like receptor (TLR) that recognizes pathogen-associated molecular patterns (PAMPs) of the invading pathogen, such as lipopolysaccharide (LPS) in Gram-negative bacteria and lipoteichoic acid (LTA)/peptidoglycan (PGN) in Gram-positive bacteria (26). In the early stage of sepsis, macrophages undergo M1 differentiation and promote host defense by eliminating invading pathogens or damaged tissues and releasing massive amounts of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-a), interleukin-1 (IL-1), interleukin-6 (IL-6) and interleukin-8 (IL-8) (27). However, macrophages may be excessively activated during the early phase and produce excessive pro-inflammatory cytokines (28), which have been identified as one of the major causes for the high mortality rate in the early stage of sepsis (29). If macrophage-mediated pro-inflammatory responses cannot be adequately regulated, a cytokine storm may emerge (30) with the pro-inflammatory response becoming pathogenic and eventually immunosuppressive in late stage sepsis (31–33). As activated pro-inflammatory macrophages undergo apoptosis and/or polarize to the M2 phenotype that dampens the pro-inflammatory response, they may contribute to immunosuppression. Due to the cytokine storm, a large number of apoptosis-inducing factors are generated and released, including TNF-a, high mobility group box-1 protein (HMGB1) (34), thereby inducing and promoting macrophage apoptosis (35). Previous studies (36, 37) have determined the presence of an excessive level of macrophages apoptosis in human autopsies and animal models of sepsis. However, escaped M1 macrophages from apoptosis convert into M2 macrophages, showing downregulated inflammatory cytokines but upregulated anti-inflammatory cytokines (38). Certain cytokines (i.e., TNF-a, IL-13, IL-4, IL-10 etc.) can stimulate the polarization of macrophages toward M2 phenotype (39–41). Porta et al. (42) found that LPS-tolerant macrophages have the same characteristics as M2 macrophages. When a gram-negative infection persists, long-term accumulation of LPS can reprogram inflammatory responses (43) from activation to suppression leading to decreased production of inflammatory cytokines (44). The affected host may present a LPS-tolerant state, and macrophages also display the phenomenon of LPS-tolerance (45–47). In addition, M2 phenotype macrophages also accelerate T cell apoptosis and suppress Th1 cell responses (48). Collectively, this “dysfunctional” macrophage plays a key role in the pathogenesis of sepsis-induced immunosuppression because their pro-inflammatory cytokine secretions to support effective immune reactivity against primary or secondary pathogens is compromised. Therefore, modulating homeostasis of pro- and anti-inflammatory responses and functional stabilities of macrophages can be of great benefits for sepsis-induced immunosuppression.
Reactive Oxygen Species (ROS) and Macrophages
In addition to its cytotoxic function, reactive oxygen species (ROS) can initiate multiple signal transduction cascades to modulate macrophage function and are critical to the regulation of immune responses against pathogens (49). Previous studies have shown that ROS have an established role in regulating TLR signaling pathways, such as TLR/NF-κB and TLR/MARKs pathways (50–52). In LPS-tolerant macrophages, LPS tolerance blunts the TLR4 signaling, inhibiting the activation of the NF-κB signaling pathway downstream of TLR4, resulting in reduced production of inflammatory cytokines in response to LPS challenge (53–55). ROS can modulate the production of pro-inflammatory cytokines from LPS-tolerant macrophages by activating TLR4/NF-κB and TLR/MARKs pathways (49) mainly by accelerating the phosphorylation of IκBα and MAPK phosphatases (56, 57), respectively. In addition, it has been reported that TLR2-deficient macrophages lacked the response to Gram-positive LTA and PGN (58, 59), which can interact with TLR2, leading to NF-κB activation and induction of proinflammatory mediators in macrophages (59, 60). Rajamani (61) also demonstarted that high glucose mediated ROS could induce TLR-2 activation and downstream NF-κB signaling mediating increased inflammation during diabetic retinopathy. TLR4/NF-κB pathway also plays a central role in the regulation of macrophage polarization (48). M1 macrophage polarization is related to the activation of the TLR4/NF-κB pathway (62), whereas M2 macrophage polarization is associated with the down-regulatation of NF-κB pathway (63). A recent study has confirmed that the p50 subunit of NF-κB inhibits the NF-κB pathway and M1 polarization (42). Kuchler et al. (64) reported that impaired ROS formation contributed to an M2 phenotype shift of macrophages in sepsis by inhibiting NF-κB signaling. Consequently, increased ROS formation may reduce the M2 polarization of macrophages and protect against sepsis-induced immunosuppression.
The TLR4/MARKs pathway is also involved in regulating the LPS/pro-inflammatory cytokines-induced autophagy (65). Autophagy can induce cell death but can also be a cytoprotective process. Deficient autophagy suppresses the immune response in sepsis and increases mortality (15, 16, 66). Macrophage autophagy is considered an important part of the host immune defense, eliminating intracellular pathogens through heterophagy. It has been reported that ROS can influence the MAPK pathways to activate macrophage autophagy. In hepatoma cells, migration inhibitory factor, produced by many cells including macrophages, induced autophagy via ROS generation (67). Likewise, autophagy also participates in regulating functions of macrophages and affects their ability to defend and clear pathogens through activating NF-κB pathway (68) and enhancing phagocytic capacity of macrophages (69). All of this suggests that ROS can activate macrophages to improve bactericidal and autophagy and increase production of pro-inflammatory cytokines, thereby helping to maintain immune homeostasis. Thus, a novel approach to improve ROS production in macrophages may be a useful therapy for sepsis-induced immunosuppression.
Parkinson Disease 7
Parkinson disease 7 (Park 7), also known as DJ-1 (70), is highly conserved in almost all organisms and is ubiquitously expressed in all tissues and organs (71). Park 7 was initially discovered as a novel oncogene product (72) and is considered as a major causal factor for the early onset of Parkinson's disease (73). In the past two decades, Park 7 has been intensely studied in many diseases including cancer (74), neurodegenerative disorderes (75) and stroke (76). Among these diseases, Park 7 not only serves as a reliable predictor of auxiliary diagnosis, but also is a useful therapeutic target. Park 7 is a multi-functional protein with transcriptional regulation, protein chaperone, protease, and antioxidative stress functions (77). At present, increasing evidence has demonstrated that Park 7 plays important functions in protecting neurons (78), astrocytes (79), cardiomyocytes (80, 81), and renal proximal tubule cells (82) against oxidative stress-induced cell injury. In addition, Park 7 played an important role in restoring impaired autophagy and ameliorated phenylephrine-induced cardiac hypertrophy in a repression of cardiac hypertrophy model (83). Oxidative stress is strongly related to inflammation and is thought to be involved in the processes of many diseases, including sepsis (84). Recently, accumulating lines of evidence for Park 7 in activating the inflammatory response through modulating ROS regulating oxidative stress have also been reported (53, 85). As an antioxidant, Park 7 helps to limit to cell and tissue injury in a number of diseases by removing accumulated ROS (82, 86–89). However, studies had shown that Park 7 surprisingly seems to be required for high intracellular ROS production (85, 90). Therefore, Park 7 plays a dual role in buffering cellular ROS levels: functions as a scavenger in high ROS levels, whereas helps ROS production when essential ROS are required. In view of the hypo-inflammation characteristics of sepsis-induced immunosuppression and the critical role of Park 7 in modulating ROS production and initiating an inflammatory response, recently it has been reported that Park 7 can protect against sepsis-induced immunosuppression.
Park 7 Protects Against Sepsis-Induced Immunosuppression (Figure 1)
In a park 7 knock-out (KO) mouse injected with LPS, Liu et al. (53) found that park 7 KO mice present immunosuppression phenotypes similar to the late stage of sepsis but not acute inflammation state, suggesting that park7 KO mice could serve as an animal model of sepsis-induced immunosuppression. In this model, Park 7 absence led to macrophage paralysis that resulted in increased abdominal bacterial burdens, reduced local and systemic inflammation, and impaired pro-inflammatory cytokines induction, eventually leading to high susceptibility to LPS. Neutrophil paralysis, similar to macrophage paralysis described above, was described in experimental studies of patients and sepsis animal models and was associated with decreased production of ROS in neutrophils (91, 92). In a liver fibrosis model, Park 7 deficiency inhibited ROS production in macrophages (93). Similarly, Liu et al. also observed greatly reduced ROS production in macrophages from park 7 KO mice (53). Macrophages with Park 7 deficiency showed downregulation of NF-κB and MAPK signaling pathways downstream of TLR suggesting that Park 7 deficiency can reduce the ROS production to limit TLR signaling and impair the activation of macrophages. Restoration of Park 7 expression with an inducible Park7 transgene restored the production of ROS in Park 7 KO macrophages to subsequently restore TLR signaling, pro-inflammatory cytokine production, bactericidal function, and eventually improve survival of the Park 7 KO mice in the late stage of sepsis. However, immunosuppressive IL-10 was not simultaneously enhanced after restoration of Park7 expression. During the late stage of sepsis, Park 7 may also enhance the macrophage functions by restoring impaired macrophages autophay through increased ROS and TLR/MARK signaling. Macrophage autophagy can affect cell death via complex pathways involving crosstalk with apoptosis, which may also partly attenuate immunosuppression (94). Moreover, Park 7 may contribute to the M1 macrophages polarization and inhibit the M2 macrophages polarization by the increased ROS.
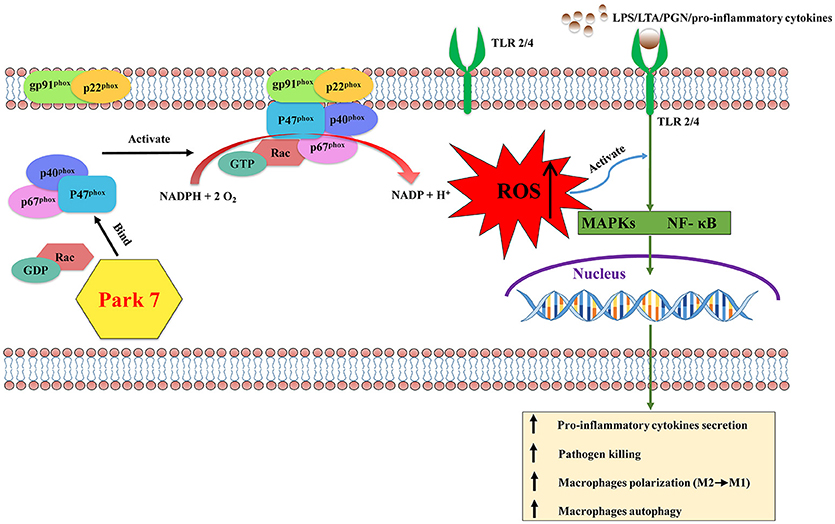
Figure 1. The effects of Park 7 on macrophages in sepsis-induced immunosuppression. During the late stage of sepsis, the activation of macrophages is impaired due to the blunted TLR/NF-κB and/or TLR/MARKs signaling pathways induced with LPS/LTA/PGN/pro-inflammatory cytokines. p47phox, a proenzyme subunit of NADPH oxidase, is key to the assembly process of NADPH oxidase. Park 7 can interact with p47phox and promote its phosphorylation and membrane translocation to form the holoenzyme complex. Subsequently the activation of NADPH oxidase produces ROS, which can activate the MAPKs and NF-κB signaling pathways downstream of TLR signaling, resulting in the activation of macrophages. Activated macrophages protect against sepsis-induced immunosuppression by releasing pro-inflammatory cytokines, killing pathogen, polarizing to M1 phenotype and the enhanced capacity of autophagy. Park 7, Parkinson disease 7; LPS, lipopolysaccharide; LTA, lipoteichoic acid; PGN, peptidoglycan; ROS, reactive oxygen species; TLR, Toll-like receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; MAPKs, mitogen-activated protein kinases.
Although there are many sources of ROS within macrophages, NADPH oxidase-derived ROS are critical in host defense. When macrophages are stimulated by an extracellular stimulus such as hormones, cytokines, and other inflammatory factors, the six proenzyme subunits of NADPH oxidase (95), including p22phox, gp91phox, GTPase Rac, p40phox, p47phox, and p67phox, form the holoenzyme complex that catalyzes the transfer of NADPH electrons to oxygen molecules to produce ROS (96). Key to the assembly process of the holoenzyme complex is p47phox (97, 98). After macrophages are stimulated extracellularly, p47phox, which resides in the cytosol during the resting state (95), is phosphorylated and translocated to the plasma membrane together with the remaining proenzyme subunits and activation of NADPH oxidase (99, 100). Consistent with Liu's study (53), by interacting with p47phox and modulating phosphorylation and membrane translocation of p47phox, Park7 promoted NADPH oxidase assembly and induced the production of ROS in macrophages. This mechanism supports the hypothesis that Park 7-targeted therapy maybe useful in the future in the treatment of sepsis-induced immunosuppression.
Is Park 7 a Potential Target for Drug Treatment in the Future?
In this decade, many reports have shown the therapeutic potency of Park 7 and Park 7-targeting molecules/compounds in treating several neurodegenerative disorders (101–103). Can Park 7 be a potential target for drug treatment for sepsis-induced immunosuppression in the future? Structure-based drug design (SBDD) (104), as a valuable pharmaceutical lead discovery tool, opens up new opportunities for drug design for the patient with sepsis-induced immunosuppression. A typical example is the successful design of many valuable drugs by SBDD based on the crystal structure of Class B G-protein-coupled receptors (105). As noted above, the interaction of Park 7 and p47phox is a decisive factor in activating macrophages to ameliorate sepsis-induced immunosuppression, suggesting that the interaction between Park 7 and p47phox may be an ideal target for drug design. Single crystal structures of Park 7 and p47phox have been determined. Human Park 7 consists of 189 amino acids from N-terminus to C-terminus, which folds into a helix-strand-helix sandwich structure (106). The C-terminal domain (CTD) of Park 7 physically interacts with p47phox in vitro (53). In addition, the C106 and L166 residues in the CTD of Park 7 are important for its functions (107, 108), suggesting the two residues might play a key role in Park 7 interacting with p47phox. However, the details of the interaction depend on the crystal structure of the Park7-p47phox complex. Therefore, determing Park7-p47phox complex structure should be an urgent issue for future research.
With regard to a potential drug treatment based on Park 7 in the future it might be important to discuss three relevant points here. (1) It would be necessary to detect/diagnose the immune status of the patient in sepsis-induced immunosuppression. (2) In line with this it would be crucial to find the right timing to start drug treatment to overcome sepsis-induced immunosuppression. (3) Considering the complexity of the host response during sepsis and the variety of pathophysiological pathways involved, it is unlikely that the current “one-target” and “one-size-fits-all” approach will ever be successful. To date, absolute lymphocyte count and decreased expression of HLA-DR by monocytes seem to be the most robust markers for patient stratification in multicenter clinical trials (109–112). Measurement of soluble mediators such as IL-6, IL-10, and TNF-a can also help detect immune status. However, a convenient, faster detection protocol and other effective drugs are extremely necessary. These are interesting issues that are worth pursuing in the future.
Conclusion
In summary, macrophages, as one of the most important cells of the innate immune system, play an important role in inflammatory and immune processes. In the early stage of sepsis, macrophages usually have a pro-inflammatory phenotype, whereas the excessive inflammatory macrophage response can lead to macrophages apoptosis and change macrophage polarization contributing to the immunosuppression. ROS have the capacity to initiate many TLR signaling pathways and in turn modulate macrophage functions and are produced by the activation of NADPH oxidase. Park 7 has been extensively studied in many diseases and can serve as an effective therapeutic target. For research on sepsis in the late stage, Park7 KO mice can be an ideal model. The interaction of Park7 and p47phox can activate NADPH oxidase and subsequently increase ROS in macrophages to initiate TRL signaling to in turn, reinforce macrophage functions to protect against sepsis–induced immunosuppression. In light of this understanding, the Park 7/p47phox/ROS axis may become an effective therapeutic target for sepsis induced immunosuppression.
Author Contributions
YaC wrote the first draft of this article. XC and WW designed the figure. YC and TM critically revised the manuscript for important intellectual content. All authors approved the final version.
Funding
The present work was supported by the National Natural Science Foundation of China (Grant Nos. 81471836 and 81772037), the Chengdu Science and Technology Huimin Project (Grant No. 2016-HM0M2-00099-SF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current estimates and limitations. Am J Respir Crit Care Med. (2016) 193:259–72. doi: 10.1164/rccm.201504-0781OC
2. Seymour CW, Rea TD, Kahn JM, Walkey AJ, Yealy DM, Angus DC. Severe sepsis in pre-hospital emergency care: analysis of incidence, care, and outcome. Am J Respir Crit Care Med. (2012) 186:1264–71. doi: 10.1164/rccm.201204-0713OC
3. Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. (2007) 35:1244–50. doi: 10.1097/01.ccm.0000261890.41311.e9
4. Melamed A, Sorvillo FJ. The burden of sepsis-associated mortality in the United States from 1999 to 2005: an analysis of multiple-cause-of-death data. Crit Care (2009) 13:R28. doi: 10.1186/cc7733
5. Tiru B, DiNino EK, Orenstein A, Mailloux PT, Pesaturo A, Gupta A, et al. The Economic and Humanistic burden of severe sepsis. Pharmacoeconomics (2015) 33:925–37. doi: 10.1007/s40273-015-0282-y
6. Torio CM, Moore BJ. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013: Statistical Brief #204, in Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. (2006). Rockville, MD: Agency for Healthcare Research and Quality (US).
7. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing sepsis as a global health priority - A WHO resolution. N Engl J Med. (2017) 377:414–7. doi: 10.1056/NEJMp1707170
8. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
9. Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA (2009) 302:2323–9. doi: 10.1001/jama.2009.1754
10. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. (2017) 17:407–20. doi: 10.1038/nri.2017.36
11. Kumar A, Kethireddy S. Emerging concepts in optimizing antimicrobial therapy of septic shock: speed is life but a hammer helps too. Crit Care (2013) 17:104. doi: 10.1186/cc11890
12. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. (2013) 13:260–8. doi: 10.1016/s1473-3099(13)70001-x
13. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304:1787–94. doi: 10.1001/jama.2010.1553
14. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. (1999) 27:1230–51.
15. Lin CW, Lo S, Hsu C, Hsieh CH, Chang YF, Hou BS, et al. T-cell autophagy deficiency increases mortality and suppresses immune responses after sepsis. PLoS ONE (2014) 9:e102066. doi: 10.1371/journal.pone.0102066
16. Oami T, Watanabe E, Hatano M, Sunahara S, Fujimura L, Sakamoto A, et al. Suppression of T cell autophagy results in decreased viability and function of T cells through accelerated apoptosis in a murine sepsis model. Crit Care Med. (2017) 45:e77–85. doi: 10.1097/ccm.0000000000002016
17. Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, et al. Cecal ligation and puncture results in long-term central nervous system myeloid inflammation. PLoS ONE (2016) 11:e0149136. doi: 10.1371/journal.pone.0149136
18. Martelli D, Yao ST, McKinley MJ, McAllen RM. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol. (2014) 592:1677–86. doi: 10.1113/jphysiol.2013.268573
19. Arts RJ, Gresnigt MS, Joosten LA, Netea MG. Cellular metabolism of myeloid cells in sepsis. J Leukoc Biol. (2017) 101:151–164. doi: 10.1189/jlb.4MR0216-066R
20. Cazalis MA, Lepape A, Venet F, Frager F, Mougin B, Vallin H, et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp. (2014) 2:20. doi: 10.1186/s40635-014-0020-3
21. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med. (2011) 208:2581–90. doi: 10.1084/jem.20111354
22. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. (2016) 4:259–71. doi: 10.1016/s2213-2600(16)00046-1
23. Allantaz-Frager F, Turrel-Davin F, Venet F, Monnin C, De Saint Jean A, Barbalat V, et al. Identification of biomarkers of response to IFNg during endotoxin tolerance: application to septic shock. PLoS ONE (2013) 8:e68218. doi: 10.1371/journal.pone.0068218
24. Giza DE, Vasilescu C. [MicroRNA's role in sepsis and endotoxin tolerance. More players on the stage]. Chirurgia (2010) 105:625–30. doi: 10.3109/19401736.2015.1060434
25. Andrades ME, Morina A, Spasic S, Spasojevic I. Bench-to-bedside review: sepsis - from the redox point of view. Crit Care (2011) 15:230. doi: 10.1186/cc10334
26. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol (2011) 30:16–34. doi: 10.3109/08830185.2010.529976
27. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
28. Wang TS, Deng JC. Molecular and cellular aspects of sepsis-induced immunosuppression. J Mol Med. (2008) 86:495–506. doi: 10.1007/s00109-007-0300-4
29. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. (2013) 369:840–51. doi: 10.1056/NEJMra1208623
30. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. (2008) 8:776–87. doi: 10.1038/nri2402
31. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140:871–82. doi: 10.1016/j.cell.2010.02.029
32. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. (2011) 121:985–97. doi: 10.1172/jci44490
33. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. (2011) 12:231–8. doi: 10.1038/ni.1990
34. Zhu XM, Yao YM, Liang HP, Liu F, Dong N, Yu Y, et al. Effect of high mobility group box-1 protein on apoptosis of peritoneal macrophages. Arch Biochem Biophys. (2009) 492:54–61. doi: 10.1016/j.abb.2009.09.016
35. Luan YY, Yao YM, Xiao XZ, Sheng ZY. Insights into the apoptotic death of immune cells in sepsis. J Interferon Cytokine Res. (2015) 35:17–22. doi: 10.1089/jir.2014.0069
36. Rimmele T, Payen D, Cantaluppi V, Marshall J, Gomez H, Gomez A, et al. Immune cell phenotype and function in sepsis. Shock (2016) 45:282–91. doi: 10.1097/shk.0000000000000495
37. Luan YY, Dong N, Xie M, Xiao XZ, Yao YM. The significance and regulatory mechanisms of innate immune cells in the development of sepsis. J Interferon Cytokine Res. (2014) 34:2–15. doi: 10.1089/jir.2013.0042
38. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity (2005) 23:344–6. doi: 10.1016/j.immuni.2005.10.001
39. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
40. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/jci59643
41. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science (2017) 356:513–9. doi: 10.1126/science.aal3535
42. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci USA. (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
43. Seeley JJ, Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol. (2017) 101:107–119. doi: 10.1189/jlb.3MR0316-118RR
44. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
45. Sato S, Takeuchi O, Fujita T, Tomizawa H, Takeda K, Akira S. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. Int Immunol. (2002) 14:783–91. doi: 10.1093/intimm/dxf046
46. Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. (2000) 164:5564–74. doi: 10.4049/jimmunol.164.11.5564
47. Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, et al. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J Immunol. (2003) 170:508–19. doi: 10.4049/jimmunol.170.1.508
48. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci. (2014) 10:520–9. doi: 10.7150/ijbs.8879
49. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472:476–80. doi: 10.1038/nature09973
50. Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol. (2012) 12:168–79. doi: 10.1038/nri3151
51. Gallego C, Golenbock D, Gomez MA, Saravia NG. Toll-like receptors participate in macrophage activation and intracellular control of Leishmania (Viannia) panamensis. Infect Immun. (2011) 79:2871–9. doi: 10.1128/iai.01388-10
52. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell (2005) 120:649–61. doi: 10.1016/j.cell.2004.12.041
53. Liu W, Wu H, Chen L, Wen Y, Kong X, Gao WQ. Park7 interacts with p47(phox) to direct NADPH oxidase-dependent ROS production and protect against sepsis. Cell Res. (2015) 25:691–706. doi: 10.1038/cr.2015.63
54. Hoogendijk AJ, Garcia-Laorden MI, van Vught LA, Wiewel MA, Belkasim-Bohoudi H, Duitman J, et al. Sepsis patients display a reduced capacity to activate nuclear factor-kappaB in multiple cell types. Crit Care Med. (2017) 45:e524–31. doi: 10.1097/ccm.0000000000002294
55. Nomura F, Akashi S, Sakao Y, Sato S, Kawai T, Matsumoto M, et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. (2000) 164:3476–9. doi: 10.4049/jimmunol.164.7.3476
56. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev. (2016) 2016:4350965. doi: 10.1155/2016/4350965
57. Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci Lett. (2015) 584:191–6. doi: 10.1016/j.neulet.2014.10.016
58. Park OJ, Han JY, Baik JE, Jeon JH, Kang SS, Yun CH, et al. Lipoteichoic acid of Enterococcus faecalis induces the expression of chemokines via TLR2 and PAFR signaling pathways. J Leukoc Biol. (2013) 94:1275–84. doi: 10.1189/jlb.1012522
59. Hong SW, Baik JE, Kang SS, Yun CH, Seo DG, Han SH. Lipoteichoic acid of Streptococcus mutans interacts with Toll-like receptor 2 through the lipid moiety for induction of inflammatory mediators in murine macrophages. Mol Immunol. (2014) 57:284–91. doi: 10.1016/j.molimm.2013.10.004
60. Paul-Clark MJ, McMaster SK, Belcher E, Sorrentino R, Anandarajah J, Fleet M, et al. Differential effects of Gram-positive versus Gram-negative bacteria on NOSII and TNFalpha in macrophages: role of TLRs in synergy between the two. Br J Pharmacol. (2006) 148:1067–75. doi: 10.1038/sj.bjp.0706815
61. Rajamani U, Jialal I. Hyperglycemia induces Toll-like receptor-2 and−4 expression and activity in human microvascular retinal endothelial cells: implications for diabetic retinopathy. J Diabetes Res. (2014) 2014:790902. doi: 10.1155/2014/790902
62. Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. (2008) 4:619–26. doi: 10.1038/ncpendmet0976
63. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. (2009) 9:259–70. doi: 10.1038/nri2528
64. Kuchler L, Giegerich AK, Sha LK, Knape T, Wong MS, Schroder K, et al. SYNCRIP-dependent Nox2 mRNA destabilization impairs ROS formation in M2-polarized macrophages. Antioxid Redox Signal. (2014) 21:2483–97. doi: 10.1089/ars.2013.5760
65. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity (2007) 27:135–44. doi: 10.1016/j.immuni.2007.05.022
66. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
67. Chuang YC, Su WH, Lei HY, Lin YS, Liu HS, Chang CP, et al. Macrophage migration inhibitory factor induces autophagy via reactive oxygen species generation. PLoS ONE (2012) 7:e37613. doi: 10.1371/journal.pone.0037613
68. Ren C, Zhang H, Wu TT, Yao YM. Autophagy: a potential therapeutic target for reversing sepsis-induced immunosuppression. Front Immunol. (2017) 8:1832. doi: 10.3389/fimmu.2017.01832
69. Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, et al. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. (2014) 20:432–42. doi: 10.1089/ars.2013.5368
70. Bandyopadhyay S, Cookson MR. Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol Biol. (2004) 4:6. doi: 10.1186/1471-2148-4-6
71. Shen ZY, Sun Q, Xia ZY, Meng QT, Lei SQ, Zhao B, et al. Overexpression of DJ-1 reduces oxidative stress and attenuates hypoxia/reoxygenation injury in NRK-52E cells exposed to high glucose. Int J Mol Med. (2016) 38:729–36. doi: 10.3892/ijmm.2016.2680
72. Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, SM, Iguchi-Ariga et al. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. (1997) 231:509–13. doi: 10.1006/bbrc.1997.6132
73. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (2003) 299:256–9. doi: 10.1126/science.1077209
74. Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson's disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci USA. (2006) 103:15091–6. doi: 10.1073/pnas.0607260103
75. Honbou K, Suzuki NN, Horiuchi M, Niki T, Taira T, Ariga H, et al. The crystal structure of DJ-1, a protein related to male fertility and Parkinson's disease. J Biol Chem. (2003) 278:31380–4. doi: 10.1074/jbc.M305878200
76. Dongworth RK, Mukherjee UA, Hall AR, Astin R, Ong SB, Yao Z, et al. DJ-1 protects against cell death following acute cardiac ischemia-reperfusion injury. Cell Death Dis. (2014) 5:e1082. doi: 10.1038/cddis.2014.41
77. Hijioka M, Inden M, Yanagisawa D, Kitamura Y. DJ-1/PARK7: a new therapeutic target for Neurodegenerative disorders. Biol Pharm Bull. (2017) 40:548–52. doi: 10.1248/bpb.b16-01006
78. Kim W, Kim DW, Jeong HJ, Yoo DY, Jung HY, Nam SM, et al. Tat-DJ-1 protects neurons from ischemic damage in the ventral horn of rabbit spinal cord via increasing antioxidant levels. Neurochem Res. (2014) 39:187–93. doi: 10.1007/s11064-013-1205-y
79. Yanagida T, Tsushima J, Kitamura Y, Takata K, Shibaike T, et al. Oxidative stress induction of DJ-1 protein in reactive astrocytes scavenges free radicals and reduces cell injury. Oxid Med Cell Longev. (2009) 2:36–42. doi: 10.4161/oxim.2.1.7985
80. Yu HH, Xu Q, Chen HP, Wang S, Huang XS, Huang QR, et al. Stable overexpression of DJ-1 protects H9c2 cells against oxidative stress under a hypoxia condition. Cell Biochem Funct. (2013) 31:643–51. doi: 10.1002/cbf.2949
81. Billia F, Hauck L, Grothe D, Konecny F, Rao V, Kim RH, et al. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc Natl Acad Sci USA. (2013) 110:6085–90. doi: 10.1073/pnas.1303444110
82. Cuevas S, Zhang Y, Yang Y, Escano C, Asico L, Jones JE, et al. Role of renal DJ-1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension (2012) 59:446–52. doi: 10.1161/hypertensionaha.111.185744
83. Xue R, Jiang J, Dong B, Tan W, Sun Y, Zhao J, et al. DJ-1 activates autophagy in the repression of cardiac hypertrophy. Arch Biochem Biophys. (2017) 633: 124–32. doi: 10.1016/j.abb.2017.09.012
84. Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. (2011) 107:57–64. doi: 10.1093/bja/aer093
85. Chen L, Luo M, Sun X, Qin J, Yu C, Wen Y, et al. DJ-1 deficiency attenuates expansion of liver progenitor cells through modulating the inflammatory and fibrogenic niches. Cell Death Dis. (2016) 7:e2257. doi: 10.1038/cddis.2016.161
86. Xu X, Martin F, Friedman JS. The familial Parkinson's disease gene DJ-1 (PARK7) is expressed in red cells and plays a role in protection against oxidative damage. Blood Cells Mol Dis. (2010) 45:227–32. doi: 10.1016/j.bcmd.2010.07.014
87. Zhang XL, Yuan YH, Shao QH, Wang ZZ, Zhu CG, Shi JG, et al. DJ-1 regulating PI3K-Nrf2 signaling plays a significant role in bibenzyl compound 20C-mediated neuroprotection against rotenone-induced oxidative insult. Toxicol Lett. (2017) 271:74–83. doi: 10.1016/j.toxlet.2017.02.022
88. Taira T, Saito Y, Niki T, SM Iguchi-Ariga, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. (2004) 5:213–8. doi: 10.1038/sj.embor.7400074
89. Amatullah H, Shan Y, Beauchamp BL, Gali PL, Gupta S, T, Maron-Gutierrez et al. DJ-1/PARK7 impairs bacterial clearance in sepsis. Am J Respir Crit Care Med. (2017) 195:889–905. doi: 10.1164/rccm.201604-0730OC
90. Vasseur S, Afzal S, Tomasini R, Guillaumond F, Tardivel-Lacombe J, Mak TW, et al. Consequences of DJ-1 upregulation following p53 loss and cell transformation. Oncogene (2012) 31:664–70. doi: 10.1038/onc.2011.268
91. Alves-Filho JC, Spiller F, Cunha FQ. Neutrophil paralysis in sepsis. Shock (2010) 34(Suppl. 1):15–21. doi: 10.1097/SHK.0b013e3181e7e61b
92. Kovach MA, Standiford TJ. The function of neutrophils in sepsis. Curr Opin Infect Dis. (2012) 25:321–7. doi: 10.1097/QCO.0b013e3283528c9b
93. Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y, et al. Deficiency of DJ-1 Ameliorates liver Fibrosis through inhibition of Hepatic ROS production and inflammation. Int J Biol Sci. (2016) 12:1225–35. doi: 10.7150/ijbs.15154
94. Qiu P, Liu Y, Zhang J. Review: the role and mechanisms of macrophage autophagy in sepsis. Inflammation (2018) 1–14. doi: 10.1007/s10753-018-0890-8
95. Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell (2003) 113:343–55. doi: 10.1016/S0092-8674(03)00314-3
97. de Mendez I, Homayounpour, N, Leto TL. Specificity of p47phox SH3 domain interactions in NADPH oxidase assembly and activation. Mol Cell Biol. (1997) 17:2177–85.
98. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. (2004) 4:181–9. doi: 10.1038/nri1312
99. El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. (2009) 41:217–25. doi: 10.3858/emm.2009.41.4.058
100. Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, et al. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox−/− and gp91phox−/− mice. J Immunol. (2002) 168:3974–82. doi: 10.4049/jimmunol.168.8.3974
101. Kitamura Y, Watanabe S, Taguchi M, Takagi K, Kawata T, Takahashi-Niki K, et al. Neuroprotective effect of a new DJ-1-binding compound against neurodegeneration in Parkinson's disease and stroke model rats. Mol Neurodegener. (2011) 6:48. doi: 10.1186/1750-1326-6-48
102. Lev N, Barhum Y, Ben-Zur T, Aharony I, Trifonov L, Regev N, et al. A DJ-1 based Peptide attenuates dopaminergic degeneration in mice models of Parkinson's disease via enhancing Nrf2. PLoS ONE (2015) 10:e0127549. doi: 10.1371/journal.pone.0127549
103. Kitamura Y, Inden M, Kimoto Y, Takata K, Yanagisawa D, Hijioka M, et al. Effects of a DJ-1-binding compound on spatial learning and memory impairment in a mouse model of Alzheimer's Disease. J Alzheimers Dis. (2017) 55:67–72. doi: 10.3233/jad-160574
104. Grey JL, Thompson DH. Challenges and opportunities for new protein crystallization strategies in structure-based drug design. Expert Opin Drug Discov. (2010) 5:1039–45. doi: 10.1517/17460441.2010.515583
105. Zhang H, Qiao A, Yang D, Yang L, Dai A, de Graaf C, et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature (2017) 546:259–64. doi: 10.1038/nature22363
106. Wilson MA, Collins JL, Hod Y, Ringe D, Petsko GA. The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc Natl Acad Sci USA. (2003) 100:9256–61. doi: 10.1073/pnas.1133288100
107. Olzmann JA, Brown K, Wilkinson KD, Rees HD, Huai Q, Ke H, et al. Familial Parkinson's disease-associated L166P mutation disrupts DJ-1 protein folding and function. J Biol Chem. (2004) 279:8506–15. doi: 10.1074/jbc.M311017200
108. Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. (2011) 15:111–22. doi: 10.1089/ars.2010.3481
109. Schefold JC. Measurement of monocytic HLA-DR (mHLA-DR) expression in patients with severe sepsis and septic shock: assessment of immune organ failure. Intensive Care Med. (2010) 36:1810–2. doi: 10.1007/s00134-010-1965-7
110. Cheron A, Monneret G, Landelle C, Floccard B, Allaouchiche B. [Low monocytic HLA-DR expression and risk of secondary infection]. Ann Fr Anesth Reanim. (2010) 29:368–76. doi: 10.1016/j.annfar.2010.02.015
111. Monneret G, Venet F. Sepsis-induced immune alterations monitoring by flow cytometry as a promising tool for individualized therapy. Cytometry B Clin Cytom. (2016) 90:376–86. doi: 10.1002/cyto.b.21270
Keywords: Park 7, sepsis-induced immunosuppression, inflammation, macrophages, ROS, p47phox, NADPH, crystal structure
Citation: Cheng Y, Marion TN, Cao X, Wang W and Cao Y (2018) Park 7: A Novel Therapeutic Target for Macrophages in Sepsis-Induced Immunosuppression. Front. Immunol. 9:2632. doi: 10.3389/fimmu.2018.02632
Received: 11 August 2018; Accepted: 25 October 2018;
Published: 13 November 2018.
Edited by:
Lukas Martin, Uniklinik RWTH Aachen, GermanyReviewed by:
Johannes Ehler, University of Rostock, GermanyDaniel Remick, Boston University, United States
Copyright © 2018 Cheng, Marion, Cao, Wang and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Cao, yuyuer@126.com