 Dagmar Gotthardt
Dagmar Gotthardt Jana Trifinopoulos1
Jana Trifinopoulos1 Veronika Sexl
Veronika Sexl Eva Maria Putz
Eva Maria Putz- 1Department for Biomedical Sciences, Institute of Pharmacology and Toxicology, University of Veterinary Medicine Vienna, Vienna, Austria
- 2St. Anna Children's Cancer Research Institute (CCRI), Vienna, Austria
Natural Killer (NK) cells are cytotoxic lymphocytes of the innate immune system and play a critical role in anti-viral and anti-tumor responses. NK cells develop in the bone marrow from hematopoietic stem cells (HSCs) that differentiate through common lymphoid progenitors (CLPs) to NK lineage-restricted progenitors (NKPs). The orchestrated action of multiple cytokines is crucial for NK cell development and maturation. Many of these cytokines such as IL-2, IL-7, IL-12, IL-15, IL-21, IL-27, and interferons (IFNs) signal via the Janus Kinase / Signal Transducer and Activator of Transcription (JAK/STAT) pathway. We here review the current knowledge about these cytokines and the downstream signaling involved in the development and maturation of conventional NK cells and their close relatives, innate lymphoid cells type 1 (ILC1). We further discuss the role of suppressor of cytokine signaling (SOCS) proteins in NK cells and highlight their potential for therapeutic application.
Introduction
Innate lymphoid cells (ILCs) comprise a variety of cell types with the morphological characteristics of lymphoid cells, but unlike adaptive immune cells, ILCs completely lack rearranged antigen receptors. In analogy to the classification of T cell subsets, ILCs can be sub-divided into three groups according to their dependence on distinct transcription factors and to their cytokine expression repertoire (1). Group 1 ILCs include two major members, conventional NK cells and ILC1s, both of which are characterized by the ability to produce T helper-1 (Th1) cell signature cytokines (e.g., interferon-gamma, IFN-γ) and by their functional and developmental dependence on the transcription factor T-BET. Group 2 cells (ILC2s) produce Th2 cell-type cytokines (e.g., IL-4, IL-5, IL-9, and IL-13) and depend on GATA3, whereas group 3 cells (ILC3s) are potent producers of IL-22 and/or IL-17A and are characterized by RORγt expression (1, 2).
NK cells account for 8–15% of circulating cells in the human blood or 2–6% in mouse blood, and are found throughout the body, in particular in lymphoid organs, lung, liver, uterus and gut (3). Similar to CD8+ cytotoxic T cells, NK cells are important in the defense against tumors and the spread of viral infections by producing pro-inflammatory cytokines such as IFN-γ and TNF-α. However, unlike T cells NK cells do not require prior sensitization and lack antigen-specificity allowing them to patrol and eliminate a broad range of altered and transformed cells. To do so, the activity of NK cells is controlled by a delicate balance of inhibitory and activating receptors, which interact with surface ligands and either prevent or trigger the lysis of a target cell (4, 5). Whereas, NK cells recirculate via blood and lymph vessels and have a license to kill, ILC1s are mostly tissue-resident and show low cytotoxic potential. Besides their common feature of being highly efficient IFN-γ producers, NK cells and ILC1s share many surface markers as well as transcription factors that complicates their discrimination especially under conditions of inflammation or in cancer (6, 7).
In humans, the identification of specific markers for ILC1s remains challenging (6, 8). In mice, surface expression of CD49b and expression of the transcription factor Eomesodermin (EOMES) merge cells under the umbrella of conventional NK cells. In contrast, CD49a expression and the absence of EOMES expression assigns cells to the ILC1 lineage (7, 9). However, to add a layer of complexity it was shown that CD49a expression can be induced on conventional mouse NK cells in vivo upon viral (10) and parasite infection (11) and in the tumor microenvironment (12, 13). Treatment of mouse splenic NK cells with IL-2 and TGF-β induces the expression of ILC1-associated markers, such as CD49a and TRAIL (12). On the other hand, expression of EOMES under the control of the Tbx21 (T-BET) locus induces ILC1s to acquire an NK cell-like phenotype (14).
The high plasticity within group 1 ILCs and the reversible trans-differentiation of group 2 and 3 ILCs into ILC1s (15) complicate the task to dissect the impact of aberrant cytokine signaling or expression of signaling molecules on those cells. It might thus be necessary to re-evaluate some previously published literature on NK cells to determine whether conventional NK cells and/or ILC1s have been analyzed.
NK Cell Development and Maturation
NK cells originate from common lymphoid progenitors (CLPs) in the bone marrow and may traffic to secondary lymphoid tissues, where they undergo terminal maturation and exit to the circulation (16, 17). The α-lymphoid progenitor (α-LP) and the early ILC progenitor (EILP) are the first progenitors with restricted lineage potential for all ILC subsets (18, 19). Downstream of EILPs are NK precursors (NKPs) giving rise to conventional NK cells and common helper-like innate lymphoid precursors (CHILPs), the ancestors of all other ILC subsets including ILC1s (15). The most distinct characteristic of NKPs is the acquisition of CD122 (IL2Rβ) expression, which is pivotal in the transduction of IL-15 signals via JAK1/3 and STAT5. Loss of one of these components unequivocally precludes NK cell development (20–23). This already highlights the central role of the JAK/STAT signaling cascade in NK cell development and maturation.
Human NK cells, classified as CD3−CD56+NKp46+ cells, can be further subdivided based on the expression of the low affinity Fc-receptor CD16 in CD56brightCD16− and CD56dimCD16+ cells. CD56brightCD16− NK cells are more responsive to stimulation by inflammatory cytokines and are thought to be immature precursors of CD56dimCD16+ mature NK cells, which show a higher cytotoxic capacity. The development of human NK cells can be stratified to five stages (16). The final maturation of human NK cells is accompanied by the loss of CD94/NKG2A and CD226 (DNAM1) expression, the acquisition of killer immunoglobulin-like receptors (KIRs) and CD57, and the change in the expression pattern of homing molecules such as CD62L (24, 25). Recently though, several studies have challenged this traditional model and suggested that CD56dimCD16+ and CD56brightCD16− NK cells may arise from separate lineages (26).
Mouse NK cells are defined as CD3−CD49b+NKp46+ cells and in C57BL/6 mice additionally NK1.1+. Their maturation in the periphery is associated with the upregulation of CD11b, CD43, KLRG1, and Ly49 receptors, and the downregulation of CD27 (17). Although the acquisition or loss of these surface markers is happening on a continuous scale, it has become customary to distinguish three subsets of immature (CD27+CD11b−), semi-mature (CD27+CD11b+) and mature (CD27−CD11b+) NK cells (27, 28).
In general, compared to their more immature counterparts, mature NK cells produce less cytokines, show a reduced proliferative capacity, but become more cytotoxic against target cells. However, in the process of terminal differentiation NK cells gradually lose their effector functions as well as the expression of the activating receptor DNAM1 (24, 28).
JAK/STAT Signaling
Most cytokines that influence group 1 ILC development or functions signal via the Janus kinase / signal transducer and activator of transcription (JAK/STAT) pathway (see Figure 1). Depending on the cell type, developmental status and microenvironment, JAK/STAT signaling contributes to the regulation of differentiation, proliferation, migration, survival or cytotoxicity in response to more than 50 cytokines, growth factors and hormones (29–31). Many of these cytokines are crucial for NK cells; their signal transduction and downstream effects are summarized in Figure 2. To allow this enormous complexity, the JAK/STAT signaling cascade transports extracellular signals from the cell membrane to the nucleus via various steps. In the canonical signaling cascade, extracellular binding of a cytokine to its corresponding multimeric receptor leads to conformational changes of the receptor chains. Receptor-associated JAK kinases come into close proximity, and sequentially phosphorylate each other and the intracellular portion of the receptor. This creates docking sites for STAT proteins that are recruited to the receptors and phosphorylated on their tyrosine residues by JAK kinases. STAT phosphorylation provokes detachment from the receptor, the formation of homo- or hetero-dimers with other STAT proteins and nuclear translocation. In the nucleus STATs regulate target gene transcription by binding to promotor or enhancer motifs or other non-coding intra- and intergenic regions (29–31) (see Figure 1). In addition, several non-canonical pathways have been described; these include kinase-independent functions of JAKs, the formation of higher order STAT tetramers or multifactorial complexes with other transcription factors, and pathways building on unphosphorylated STAT proteins (U-STATs) (31, 32). In NK cells, non-canonical functions have so far been described for TYK2, STAT1 and STAT5 (see below).
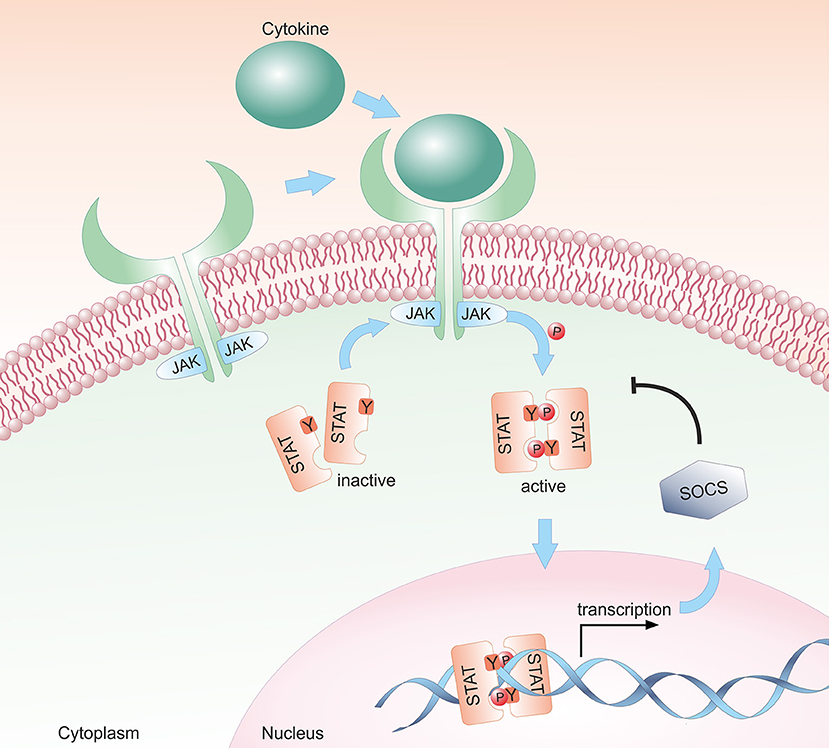
Figure 1. Schematic representation of the canonical JAK/STAT signaling pathway. The JAK/STAT pathway transmits extracellular cytokine signals to the nucleus. Upon binding of a cytokine to its transmembrane receptor, receptor-associated JAKs are activated and phosphorylate STAT proteins. Activated STAT proteins translocate as either homo- or hetero-dimers to the nucleus and modulate target gene transcription. In a negative feedback loop, SOCS proteins are expressed and inhibit the JAK/STAT signaling cascade by suppressing JAK kinase activity, by competing with STAT proteins for binding to the receptor and/or by proteasomal degradation of the proteins.

Figure 2. Schematic overview of crucial cytokines in NK cell biology, their associated JAK and STAT proteins, exemplary target genes and biological effects. One cytokine can lead to the activation of several STAT proteins. STAT proteins predominantly activated by the respective cytokine are depicted in bold font; STAT proteins that have been reported to be activated to a lesser extent are depicted in light font. The details and references for distinct cytokine signaling cascades and the functional responses can be found in the corresponding sections of the main text.
The JAK/STAT pathway is highly conserved among species. Mammals express four members of the JAK family (JAK1-3 and TYK2) and seven STAT proteins (STAT1-4, STAT5A, STAT5B, and STAT6). STAT5A and STAT5B are highly homologous but encoded by distinct genes located on the same chromosome directly adjacent to the Stat3 gene locus indicating that these three genes derived from the duplication of a common primordial gene (33, 34). Although distinct members of the JAK/STAT cascade share high homology, their specific functions vary considerably. Gene-targeted mice have deepened our understanding of distinct roles of individual JAK and STAT proteins (see Figure 3). Deficiency of Jak2 (35) and Stat3 (36) precludes embryonic development, whereas Jak1- (37) and Stat5a/b-deficiencies (38) lead to perinatal lethality. Loss of the other members of the JAK/STAT pathway does not interfere with viability of the animals, but reveals distinct phenotypes including the absence of lymph nodes and/or high sensitivity to infections (39).
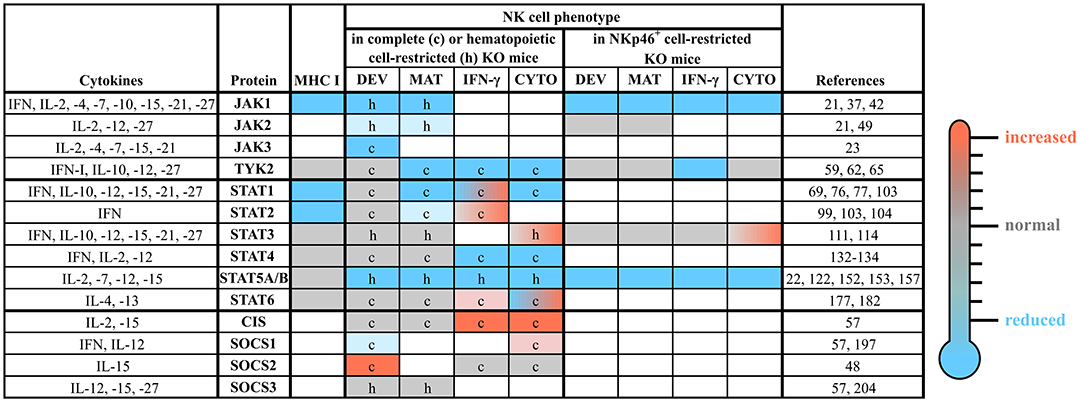
Figure 3. The roles of distinct JAK/STAT and SOCS family members in NK cells. NK cell development, maturation and function are tightly regulated by a plethora of cytokines, which most prominently use the JAK/STAT pathway for their signal transduction. This figure summarizes the available literature about each member of the JAK/STAT signaling pathway and some of their negative regulators (SOCS1-3 and CIS), relevant upstream cytokines and the NK cell phenotypes observed in complete or conditional knockout mice. The individual cells are coded by color: compared to wild-type reduced (blue), unchanged (gray) or increased (red); blank cells indicate not determined yet. c, complete knockout mice; CIS, cytokine induced SH2-containing protein; CYTO, cytotoxicity; DEV, development; h, hematopoietic cell-restricted knockout mice; IFN, interferon; IL, interleukin; IFN-γ, IFN-γ production; JAK, Janus kinase; KO, knockout; MAT, maturation; MHC, major histocompatibility complex; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TYK2, tyrosine kinase 2.
JAK1
JAK1 is involved in the signal transduction of several cytokines crucial for NK cell biology, for instance the IL-2 family cytokines including IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Most importantly, IL-15 represents the major cytokine regulating NK cell development, maturation and function (17, 40). Additionally, as the major component downstream of IFNs and IL-10, JAK1 plays a pivotal role in NK cell biology (41). It should also be mentioned that JAK1 associates with the IL-4 receptor family (transmitting IL-4 and IL-13 signals) and the gp130 receptor family (transmitting, e.g., IL-6, IL-11, IL-27, LIF, and OSM signals) (41).
Given the fundamental role of IL-15 and other IL-2 family cytokines transmitted via the common γ (γc) receptor, it is not surprising that complete loss of Jak1 leads to perinatal lethality in mice, accompanied by a strong reduction in the number of thymocytes and B cells (37). These observations were recently confirmed in adult mice: inducible deletion of Jak1 leads to impaired hematopoietic stem cell homeostasis and a pronounced decrease in immature B220+ NK cells (42).
Using mice with NKp46+ cell-specific deletion of Jak1 uncovered the crucial role of JAK1 in NK cell development and survival (21). Jak1 deficiency reduces the numbers of NK cells and ILC1s in a dose-dependent manner. This indicates that other JAK family members fail to compensate for the loss of JAK1. The consequences of Jak1 deletion within the NK cell compartment exceed the effects seen upon loss of the JAK1 downstream effector STAT5 (21, 22). Different half-lives of JAK1 and STAT5 proteins may contribute to the difference in NK cell frequency. One may also reason that the more pronounced depletion of NK cells results from the combined loss of STAT3 and STAT5-mediated signals in Jak1-deficient animals.
To the best of our knowledge, no reports on JAK1-deficient individuals exist so far, suggesting that like in mice, it might lead to embryonic lethality in humans. A patient harboring a biallelic JAK1 germline mutation leading to a partial loss of kinase activity has been reported. The resulting functional impairment was associated with a mild immunodeficiency, recurrent atypical mycobacterial infections and early onset metastatic bladder carcinoma (43).
JAK2
JAK2 is a critical mediator of growth hormone (GH), erythropoietin and IFN-II signaling and thus plays a pivotal role in hematopoiesis (35, 44). In both NK and T cells, IL-2 signals via JAK1/3 and STAT1/3/5 inducing NK and T cell proliferation and enhancing NK cell cytotoxicity. However, unlike in T cells, in NK cells IL-2 additionally activates JAK2 and STAT4 (45). JAK2 in combination with TYK2 mediates the signal transduction of the IL-12 family members: IL-12 activates STAT4 and to a lesser degree STAT1, STAT3 and STAT5 (46); IL-23 activates mainly STAT3 and STAT4, and IL-27 signals mainly via STAT1 and STAT3 (47). Although it was assumed that IL-15 signals exclusively via JAK1/3, a recent study described an IL-15-mediated JAK2 activation in murine NK cells (48).
Germline deletion of Jak2 results in embryonic lethality at day 12.5 due to impaired hematopoiesis (35). Studies using Jak2-conditional knockout mice uncovered a mild defect in NK cell maturation in the absence of JAK2 in the hematopoietic system (49). In line, treatment of mice with the JAK2 inhibitor BSK805 reduces NK cell numbers due to decreased proliferation and an immature maturation profile resulting in an increased metastatic burden (49). In contrast, deletion of Jak2 in mature NK cells does not impact on NK cell numbers or maturation (21). It is conceivable that JAK2 is crucial for development and maturation of early NK cell progenitor stages in the bone marrow. Alternatively, JAK2-inhibition or deletion may interfere with other cell types to alter NK cells extrinsically by changing the cytokine milieu. JAK2 has been reported to be required for the development of dendritic cells (DCs) (50), which are potent producers of IL-15 and thus indispensable for proper NK cell priming (51). DC-mediated NK cell priming is potentially impaired upon JAK2 inhibition or deletion. Support for this concept stems from the observation that IL-15 treatment overcomes the JAK2 inhibitor-mediated increase of tumor metastasis (49).
JAK3
Together with JAK1, JAK3 transmits signals downstream of the γc cytokines IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 resulting in phosphorylation of STAT1, STAT3, STAT4, STAT5 and STAT6 (41, 52). Jak3−/− mice are immunocompromised, display severe developmental defects in the lymphoid lineage and lack NK, T and B cells (23, 53). The in vivo administration of Tofacitinib, which predominantly inhibits JAK3 and to a lesser degree JAK1 and JAK2, depletes all NK cell subsets in the periphery of rhesus macaques (54). Likewise, human patients harboring JAK3 mutations suffer from severe combined immunodeficiency (SCID) lacking NK and T cells (55). A previous study in mice having a spontaneous Jak3 mutation disclosed the association of an impaired JAK3 signaling with a differentiation block of NK and ILC1s at the pre-NKP and ILCP stage (56). In summary, these findings define a non-redundant role of JAK1 and JAK3 for NK cell development and the differentiation of EILPs.
Interestingly, quantitative mass spectrometry analysis in mature NK cells demonstrated a predominance of JAK1 protein compared to JAK3 (57). It was further proposed that JAK1 dominates JAK3 in the signal transduction of γc cytokines. While loss of the JAK1 kinase function completely abrogates downstream signals, loss of the kinase activity of JAK3 in human cell lines only diminishes STAT5 phosphorylation. It was thus suggested that JAK3 functions by activating JAK1, but does not directly induce STAT5 phosphorylation (58). The details of the molecular interactions in NK cells remain to be determined. It is currently unclear what effect the conditional deletion of Jak3 in mature NK cells will have and how it will affect their proliferation and effector functions.
TYK2
TYK2 associates with the IFN-I (IFNAR1), IL-10Rβ, IL-12Rβ1, and IL13Rα1 receptors and is thus involved in the signal transduction of a large number of cytokines including IFN-I, IL-10, IL-12, IL-23, and IL-27 (46). Despite its broad activity, Tyk2-deficiency does not preclude survival of mice (59, 60).
NK cells derived from Tyk2−/− mice display impaired IL-12-mediated signaling resulting in reduced STAT1, STAT3, and STAT4 activation (61, 62). Although the development of NK cells in the bone marrow is unaltered, the final maturation in the periphery is severely impaired as evidenced by fewer CD27−CD11b+ and KLRG1+ cells in Tyk2-deficient mice. This translates into impaired IFN-γ production and cytolytic responses (62) in line with the involvement of STAT4 in the regulation of IFN-γ and perforin expression, respectively (63, 64). Mice expressing a kinase-inactive version of TYK2 (Tyk2K923E) show a milder defect in NK cell maturation and cytotoxicity compared to Tyk2−/− mice, indicating that TYK2 has kinase-independent functions (62). Using mice with NKp46+ cell-specific deletion of Tyk2 revealed that the impact of TYK2 on NK cell maturation and tumor surveillance is cell-extrinsic and depends on the presence of TYK2 in dendritic cells (65). Accordingly, the defects in NK cell maturation and cytotoxicity related to Tyk2-deficiency are reversed upon treatment with recombinant IL-15/IL-15Rα. However, NK cell-intrinsic TYK2 is required for IL-12-induced IFN-γ production and the defense against Listeria monocytogenes (65).
Tyk2-deficient mice and patients with autosomal recessive TYK2 mutations are susceptible to infections. NK cells from TYK2-deficient patients have an impaired, albeit not completely abrogated, IL-12-mediated IFN-γ production (60). This may explain why their susceptibility to viral infections is less severe when compared to patients harboring a STAT1-deficiency (66). A thorough analysis of peripheral NK cell maturation in the reported TYK2-deficient patients is pending.
STAT1
STAT1 is the predominant transcription factor activated by IFNs, irrespective of the subtype. Whereas, type II IFN (IFN-γ) induces the homo-dimerization of STAT1, type I IFN (IFN-α/β/ε/κ/ω) and type III IFN (IFN-λ 1-3) signaling triggers the formation of the ISGF3 complex consisting of STAT1, STAT2, and IRF9. In addition, STAT1 transmits signals from IL-6, IL-10, IL-12, IL-15, IL-21, IL-27, and IL-35 (31, 67–71).
IFNs play a pivotal role in NK cell maturation, as they provide the necessary signals for IL-15 trans-presentation by DCs (51) and MHC class I expression (37, 72, 73). Stat1-deficient NK cells show profound defects in NK cell maturation, cytokine-induced IFN-γ production, cytolytic capacity and memory formation (69, 74–77). In line, NK cells from Ifnar1-deficient animals display an immature phenotype (78) as well as defects in basal (79) and virus-induced cytotoxicity (80). The maturation defect seen in complete Ifnar1-knockout mice was not recapitulated upon NKp46+ cell-specific Ifnar1 gene deletion (78), suggesting that NK cell-extrinsic factors, such as the presentation of MHC class I and/or IFN-mediated trans-presentation of IL-15, play a crucial role in proper NK cell licensing and maturation. Accordingly, the transfer of Stat1-deficient bone marrow into wild-type mice provided sufficient signals for proper NK cell maturation in vivo (76).
In addition to IFNs, the STAT1/3 activating (70) cytokine IL-21 is known to drive the maturation of mouse and human NK cells (81, 82). Recombinant IL-21 treatment not only increases CD8+ T cell functions (83) but also the cytotoxicity and cytokine production of NK cells by inducing the expression of perforin and IFN-γ, respectively (82, 84). However, mice lacking the IL-21R do not display any defect in NK cell numbers or maturation (85) arguing against a profound effect of IL-21 on NK cell maturation under homeostatic conditions in vivo.
Analogous to IL-21, IL-27 signals mainly via STAT1 and STAT3 (47, 86). Whereas, IL-27 treatment alone does not have a major impact on NK cells, co-stimulation with IL-27 and IL-2, IL-12 or IL-18 leads to enhanced NK cell activation, cytokine production and cytotoxicity (47, 86–88). Accordingly, loss of IL-27R in mice leads to reduced NK cell-mediated IFN-γ production and T-BET expression after influenza virus infection (89). Il27ra-deficient mice are characterized by an unusual maturation profile, represented by fewer mature NK cells in the bone marrow, while more mature NK cells are found in the spleen (89). This phenotype does not reflect the situation in Stat1-deficient mice and may suggest an altered dissemination of NK cells rather than a maturation defect.
However, Stat1-Y701F knock-in mice lacking the tyrosine residue essential for STAT1 translocation and transcriptional activity do mirror the impaired maturation phenotype of NK cells seen in Stat1−/− mice (90). Although the function of STAT1 was considered to depend on the tyrosine phosphorylation, Stat1-Y701F expressing NK cells are more cytotoxic against tumor cells than Stat1−/− NK cells. A novel non-canonical function of STAT1 at the immunological synapse of NK cells regulating tumor surveillance and cytotoxicity may account for that effect (90). Whereas STAT1-Y701 phosphorylation is triggered by cytokine stimulation, non-stimulated primary mouse NK cells display a constitutive CDK8-mediated phosphorylation of STAT1-S727 (69). The introduction of a point-mutation (Stat1-S727A) that prevents the serine phosphorylation event results in NK cells that produce less IFN-γ upon stimulation, and have a mild defect in KLRG1 and NKG2A/C/E expression. Nevertheless, these Stat1-S727A NK cells show enhanced cytotoxicity against tumors in vitro and in vivo, which correlates with increased perforin and granzyme B levels (69), once more highlighting the existence of non-canonical STAT signaling (32).
Like Tyk2-deficient mice, Stat1-deficient mice are highly susceptible to bacterial and viral infections (80, 91–93). Biallelic loss-of-expression or loss-of-function (LOF) STAT1-deficiency in humans is detrimental, with most patients succumbing to lethal infections with mycobacteria or herpes simplex virus 1 (HSV-1) encephalitis before the age of two years (66), which is accompanied by a profound effect on NK cell cytotoxicity (94). Unexpectedly, STAT1 gain-of-function (GOF) mutations are likewise associated with increased susceptibility to infectious diseases, such as chronic mucocutaneous candidiasis, bacterial and viral infections, autoimmune diseases and even cancer (95). STAT1 GOF patients have fewer and highly immature CD56dim NK cells in the periphery showing reduced cytotoxicity, IFN-γ production and cytokine-induced proliferation (96, 97). This defect was partially rescued by treatment with the JAK1/2 inhibitor ruxolitinib (96) and improved the patients' clinical picture (98). The mechanism of how STAT1 GOF mutations result in hyporesponsive NK cells is not fully understood, but it was paralleled by decreased activation of STAT5 (96), which is a master regulator for NK cell functions. These observations indicate that STAT1 signaling needs to be tightly controlled and neither reduced nor excessive pathway activation is beneficial for NK cell maturation and function.
STAT2
STAT2 together with STAT1 and IRF9 are activated in response to IFN-I and IFN-III. This turns STAT2 into a crucial mediator of antiviral defense. Depending on the viral challenge, Stat2−/− mice are more (99, 100) or less (101, 102) susceptible to infection compared to Stat1−/− mice. In the course of lymphocytic choriomeningitis virus (LCMV) infections, STAT1 and STAT2 are both required for optimal viral control, but STAT2 plays a subordinate role compared to STAT1: although both Stat1- and Stat2-deficient NK cells produce increased amounts of IFN-γ early after LCMV infection, this exclusively drives bodyweight loss in the absence of STAT1, but not STAT2 (103). Stat2−/− mice are highly susceptible to MCMV infection and succumb within the first week after infection (100). In line with a crucial role of IFN-I signaling in NK cell expansion and memory formation in the context of MCMV, NK cells from Stat1-, Stat2-, and Irf9-deficient mice are defective in their ability to expand (74, 104). As shown in Stat1−/− and Ifnar1−/− mice, also Stat2−/− NK cells have a defect in NK cell maturation, which could be rescued in bone marrow chimeras (104), again suggesting an NK cell-extrinsic role of IFN-I in NK cell maturation.
STAT2-deficient human patients present a higher incidence of distinct viral infections with astounding variation ranging from asymptomatic adult carriers of the mutation to infants succumbing to viral illness. In particular, fatal prolonged febrile encephalitic illness following measles/mumps/rubella vaccination has been reported in six vaccinated children with a STAT2 deficiency (105–107). Unlike STAT1, STAT2-mediated signaling seems to be dispensable for host defenses against most viral childhood diseases such as respiratory syncytial virus bronchiolitis or HSV-1 as well as infections with intracellular bacteria. This can be partially explained by an unaltered response to IFN-II in STAT2-deficient patients, and the observation that depending on the bacterial infection IFN-I can play adverse roles (108, 109).
Besides its role in antiviral responses, IFN-I has been implicated in anti-tumor immunity. While the contribution of STAT2 for T cell-mediated tumor surveillance has been unequivocally documented (110), the role of STAT2 in NK cell-mediated tumor surveillance is still enigmatic.
STAT3
Cytokines such as IL-2, IL-10, IL-12, IL-15, IL-21, IL-27, and IFN-I induce STAT3-Y705 phosphorylation in NK cells (111, 112). While most of these cytokines positively regulate NK cell maturation and/or activation, IL-10 is classified as immunosuppressive cytokine (113).
Several studies reported constitutive STAT3 phosphorylation of tumor-infiltrating immune cells including NK cells (114, 115). STAT3 phosphorylation is considered to be driven by inflammatory and immunosuppressive cytokines and growth factors produced by both tumor and tumor-infiltrating cells including IL-6, IL-10, or VEGF-A. STAT3 activation in the tumor stroma has been associated with an impaired tumor immune surveillance of both NK and CD8+ T cells (116, 117). High IL-10 levels in the liver also dampen hepatic NK cell responses and restrain the expression of Ly49 receptors (118). In light of recent advances in the discrimination of NK cells and ILC1s, these observations could potentially indicate a specific role of IL-10 in ILC1s, which lack most of the Ly49 receptors (119). This suppressive role is of particular importance in the liver, where IL-10 ensures that liver NK cells/ILC1s remain immune-tolerant, but is undesirable in the context of tumor surveillance (113, 115). Under certain conditions NK cells themselves (120) and the recently described regulatory ILCs have been reported to produce IL-10, which inhibits cytokine-induced IFN-γ production of ILC1s (121).
Studies in mice with constitutive or NKp46+ cell-specific Stat3-deficiency indeed show that STAT3 suppresses NK cell-mediated tumor surveillance in melanoma and leukemia models (111, 114). Loss of Stat3 does not alter classical NK cell maturation but is paralleled by increased expression of the activating receptor and maturation marker DNAM1 as well as increased expression of STAT5 and its downstream targets perforin and granzyme B (111). It is thus attractive to speculate that STAT3 represses STAT5-mediated signaling in wildtype NK cells. As described below, STAT5 represents a master regulator of NK cell function. The fact that IL-15 stimulation induces both STAT3 and STAT5 activation in NK cells (111, 113) endorses the hypothesis that STAT3 is crucial to control IL-15/STAT5-mediated NK-cell cytotoxicity to prevent detrimental hyperactivity. This concept warrants testing of a combined treatment with IL-15 and anti-STAT3 inhibitors in the context of anti-cancer immunotherapy.
Alternatively, STAT3 acts downstream of the cytokine IL-10 (111), which has been shown to transcriptionally induce the tumor promoting factor VEGF-A in NK cells (122). It is thus attractive to speculate that STAT3 activation in NK cells promotes tumor progression by dampening their cytolytic activity and driving tumor angiogenesis.
Besides suppressing cytotoxicity, STAT3 regulates the expression of the activating receptor NKG2D. IL-10 and IL-21 treatment induces NKG2D expression in a STAT3-dependent manner in human and mouse NK cells (71, 123). In line, human NK cells derived from hyper-IgE syndrome patients carrying STAT3 LOF mutations show a pronounced decrease of NKG2D expression (71).
STAT3 GOF mutations in NK cells can be found in patients with chronic lymphoproliferative disorders of NK cells (CLPD-NKs) (124) as well as aggressive NK cell leukemia (125) and extranodal NK/T-cell lymphoma (NKTCL) (126, 127). The identified STAT3 mutations enhance the levels of phosphorylated STAT3 protein and provide a growth advantage to the affected cells. These findings support the concept that STAT3 has an oncogenic potential in NK cells and highlight the importance of tight controls and negative feedback regulators.
STAT4
In contrast to other immune cells such as CD8+ T cells, IL-2 stimulation induces JAK2 and STAT4 activation in NK cells and enhances IL-12 signaling by upregulating the expression of the IL-12R (128, 129). IL-12 is the main driver of STAT4 activation and crucial for IFN-γ production in NK cells and ILC1s (129–131). Under steady-state conditions, Stat4- as well as Il12r-deficient mice harbor an unaltered NK cell repertoire in the periphery. Due to its rather restricted action downstream of IL-12, Stat4-deficiency in mice manifests in reduced IL-12-induced NK cell proliferation, IFN-γ production and cytotoxicity (132, 133). This can be explained by the fact that STAT4 regulates the induction of T-BET, a transcription factor important for NK and ILC1s that induces the transcription of important key players of the cytotoxic machinery, such as IFN-γ, granzyme B and perforin (131, 134). STAT4 and T-BET are also necessary for the generation and maintenance of MCMV-specific memory NK cells (135, 136). In line with the lessons learnt from mice, a heterozygous missense mutation in STAT4 leading to a defect in IL-12-dependent IFN-γ immunity was identified in two patients suffering from acute chronic fungal infections (137).
IL-12 also has a unique and detrimental role in adipose tissue, as diet-induced obesity is associated with IL-12 production and the proliferation and subsequent accumulation of adipose-resident ILC1 and NK cells. IL-12/STAT4 signaling is required for the increased proliferation and IFN-γ production of all group 1 ILC subsets in the adipose tissue driving M1 macrophage polarization and obesity-associated insulin resistance (138).
Apart from IL-12, IFN-I has been reported to induce phosphorylation and dimerization of STAT4 amongst all other STAT proteins (139). NK cells have particularly high basal STAT4 levels pre-bound to IFNAR1 (103). During the early phase of viral infections, STAT4 becomes activated initiating a fast IFN-γ response followed by STAT1 activation, which replaces STAT4 at the IFNAR receptor decreasing the ability to produce IFN-γ (103). These data exemplify how one cytokine activates several STAT molecules enabling a tight regulation of cellular responses.
STAT5A and STAT5B
Of all STAT proteins, STAT5 is the major regulator of NK cell development, maturation, survival and function and is activated by cytokines such as IL-2, IL-7 and IL-15. Compared to IL-15, IL-2, and IL-7 play a minor role in the development and survival of NK cells and ILC1s (140, 141). STAT5 is also implicated in the development, survival and memory formation of CD8+ T cells, which is regulated by IL-2, IL-7, and IL-15 signaling (142, 143). IL-2 plays a crucial role in activating CD8+ T and NK cells against target cells in vivo (144, 145) and it is therefore commonly added to in vitro culture systems. Although mouse NKPs express high levels of CD127 (IL-7Rα) (146), NK cell development and function are unaltered in the absence of IL-7 signaling in mice (147). The only exception are thymic NK cells, whose development depends on IL-7 and the transcription factor GATA3 (148). By contrast, in humans IL-7 controls the survival of immature CD56bright NK cells (149).
Knock-out mice lacking IL-15 or its receptor subunits are devoid of NK cells proving the indispensable role of IL-15 for NK cell development (141, 150, 151). IL-15 trans-presentation by DCs is crucial to prime NK cell maturation and function (51). As STAT5 is a critical transcription factor downstream of IL-15, impaired STAT5 signaling impacts strongly on NK cell viability and function (152, 153).
In general, STAT5 is an umbrella term for two distinct transcription factors: STAT5A and STAT5B sharing 96% sequence homology. Despite largely redundant functions, several non-redundant and tissue-specific roles have been described (154–156). Stat5a/b-deficiency in mice is perinatally lethal due to anemia and hematopoietic failure (38). Early on, STAT5 has been described to be essential for NK cell development, as the first STAT5 knockout mice that express an N-terminally truncated version of Stat5a/b are viable but devoid of peripheral NK cells (153). Single knockout mice for Stat5a or Stat5b verified the impact of STAT5 for NK cell development, maturation and cytoloytic capacity also indicating non-redundant functions of STAT5A and STAT5B. STAT5B is the dominant isoform for NK cells as its deletion has a significantly larger impact than deletion of STAT5A (152, 157). This is explained by a higher abundance of STAT5B over STAT5A transcripts in NK cells (157). Mice expressing only one allele of either Stat5a or Stat5b (Stat5a+/−Stat5b−/− and Stat5a−/−Stat5b+/−) have drastically diminished numbers of NK and ILC1 progenitors, splenic NK cells as well as intestinal and liver NK cells (157). Liver-resident ILC1s and bone marrow NK cells are less sensitive to reduced STAT5 expression levels. STAT5 was also verified as an upstream regulator of the transcription factor T-BET (122, 158) and a recent study showed that both transcription factors co-localize throughout the genome (157).
The cell-intrinsic role of STAT5 in NK cells was studied using mice where Stat5a/b deletion is restricted to NKp46+ cells. This results in a severe reduction of peripheral NK cells (22) as NK cell survival relies on the expression of anti-apoptotic STAT5 target genes such as Mcl1 or Bcl2 (122, 159). The residual NK cells found in the bone marrow of Stat5fl/flNcr1iCreTg mice harbor an immature phenotype and a major developmental block at the NKP stage (22). Enforced expression of the anti-apoptotic factor Bcl-2 rescues survival of Stat5a/b-deficient NK cells, but does not allow proliferation, maturation and reconstitution of effector functions (122). Apart from the central role of STAT5 driving the expression of transcription factors pivotal for NK cell development (ID2, EOMES and T-BET) and regulating the expression of crucial effector molecules (perforin, granzymes and IFN-γ), STAT5 has been reported to suppress the expression of the pro-angiogenic factor VEGF-A in NK cells (122). Further research is necessary to verify if STAT5 directly acts as a transcriptional suppressor or competes with the binding of other activating transcription factors.
Decidual NK cell-derived VEGF-A has a positive impact on neo-angiogenesis and placenta development during pregnancy (160–162). In contrast, the expression of VEGF-A in tumor-infiltrating NK cells promotes tumor formation (122). VEGF-A-secreting tumor-associated NK cells have also been reported in patients and are associated with poor disease outcome (163–165).
In line with observations in mice, patients with a STAT5B LOF mutation harbor significantly reduced NK cell numbers (166–168). STAT5 GOF mutations are found in malignancies of innate and innate-like lymphoid cells (125, 127, 169) and drive tumorigenesis in mouse NKT cells (170).
STATs in general, but STAT5 proteins in particular, are known to form higher order tetramers. A recent study highlighted the importance of STAT5 dimers for NK cell development, while the formation of STAT5 tetramers is a prerequisite for proper NK cell maturation and survival. The authors speculate that interfering with STAT5 tetramer formation could be used therapeutically to restrict the growth of NK cell leukemia and lymphomas (171).
To summarize, STAT5 is a master regulator of NK cells ensuring their development and survival and regulating maturation, proliferation, cytotoxicity and their precarious production of VEGF-A.
STAT6
STAT6 is activated by IL-4 and IL-13 and is involved in Th2 polarization and the development of allergic inflammation (172). Allergies and IL-4 signaling have been suggested to protect from cancer development. Indeed, IL-4 overexpression in combination with phthalic anhydride-induced allergy induction in mice enhances NK cell activity and reduces tumor burden (173). The effect of IL-4 on NK cells is highly controversial, as it was shown that IL-4 treatment of purified NK cells diminishes their cytotoxic capacity (174), while it enhances NK cell cytotoxicity and IFN-γ production when applied in vivo (175). IL-4 synergizes with IL-12 and/or IL-2 to induce IFN-γ production, which was shown to be partially dependent on STAT6 (176). In vitro stimulation of mouse NK cells with a mixture of phorbol 12-myristate 13-acetate (PMA), ionomycin, IL-2, IL-4, and anti-IFN-γ mAb induces IL-5 and IL-13 production in a STAT6-dependent manner (177). Also human NK cells possess the ability to produce IL-5 and IL-13 upon IL-4 stimulation (178, 179). In various allergic diseases, such as asthma and allergic rhinitis, NK cell-derived Th2 cytokine production contributes to eosinophil infiltration and thereby promotes allergic inflammation (180, 181). Although the involvement of STAT6 in this signaling is highly probable, it still awaits formal proof.
It was previously shown that loss of STAT6 in mice does not impact on NK cell development or maturation (177). However, Stat6-deficiency is associated with higher cytotoxic activity of NK cells and increased resistance to ectromelia virus infection (182). The seeming opposing results showing enhanced cytotoxicity of IL-4/STAT6-activated as well as STAT6-deficient NK cells certainly call for a more detailed analysis of the role of STAT6 in NK cells in the context of anti-tumor and anti-viral immunity.
SOCS Proteins
The family of suppressor of cytokine signaling (SOCS) proteins has eight members including SOCS1-7 and CIS which represent important negative regulators of the JAK/STAT signaling pathway (183, 184). SOCS proteins are characterized by a central SH2 domain and an extended SH2 sub-domain, a highly conserved C-terminal SOCS box and a variable N-terminal region. SOCS proteins exert their eponymous function by three means: (i) Via their SH2 domain, SOCS proteins bind to phosphotyrosine residues on cytokine receptors thereby competing with STAT binding and activation. (ii) Via the SOCS box they recruit an E3 ubiquitin ligase complex that leads to proteasomal degradation of signaling molecules including cytokine receptors and JAK kinases. (iii) The N-terminal domain of SOCS1 and SOCS3 contains a kinase inhibitory region serving as pseudo-substrate for JAKs consequently blocking their activity (183, 184).
Immunomodulatory effects of SOCS proteins on NK cells have been reported and suggest them as attractive candidates for immunotherapies (185). SOCS1, 2, 3 and CIS are rapidly induced upon cytokine (57, 186) or GH (187) stimulation. In contrast, little is known about the residual family members SOCS 4-7 that are constitutively expressed in unstimulated cells (185). SOCS4 and SOCS5 are crucial regulators of anti-viral immunity in the context of influenza infection (188, 189). SOCS6 negatively regulates JAK/STAT3 signaling and is epigenetically silenced in NK cell lymphomas (190). Socs7−/− mice suffer from a severe cutaneous disease due to hyperactive mast cells and the increased production of pro-inflammatory cytokines (191).
CIS
The cytokine induced SH2-containing protein (CIS, Cish) is induced by IL-2 and IL-15 and provides a negative feedback loop to inhibit JAK/STAT5-mediated signaling in NK cells (57). CIS interacts with JAK1 to target it for proteasomal degradation and thereby abrogates IL-15-induced signaling. In line with the crucial function of the IL-15/STAT5 axis for NK cell biology, hyperactive IL-15 signaling in Cish−/− mice translates to enhanced NK cell proliferation and cytotoxic function. This ultimately leads to resistance toward experimental metastasis (57) and chemically-induced sarcoma (192). Cish−/− mice react to IL-2 treatment with a further decrease of tumor burden in models that are usually unaffected by IL-2 treatment. Additive effects were also observed when CIS-deficiency was combined with targeted immunotherapies such as BRAF and MEK inhibitors, immune checkpoint blockade antibodies, or IFN-I treatment (192). These data suggest that CIS represents a promising target in immunotherapy especially in combination with other immunomodulatory agents (185).
SOCS1
SOCS1 negatively regulates signaling of IFNs and IL-12 (193, 194) and plays an important role in DC and T cells suppressing antigen-presentation and antitumor immunity (195). Socs1−/− mice die shortly after birth due to severe inflammation and uncontrolled IFN-γ signaling (196). Socs1−/−Ifng−/− double-knockout mice survive until adulthood (196) and IL-12-treated NK cells isolated from these mice display an enhanced capacity to lyse YAC-1 target cells (197). However, Socs1−/−Ifng−/− mice seem to have slightly reduced NK cell numbers in the periphery and hampered NK cell proliferation in response to IL-15 (57). A detailed analysis of the role of SOCS1 in NK cell development, maturation and function is pending.
SOCS2
SOCS2 is closely related to CIS and induced by STAT5-activating cytokines, such as GH and IL-15 in mouse and human NK cells (57, 186, 187). SOCS2 represses NK cell development, as Socs2−/− mice have increased NK cell numbers in bone marrow and spleen while T-, B- and myeloid cell numbers are unaltered (48). The increased NK cell numbers translate into enhanced tumor surveillance. In contrast to the situation in Cish-deficient mice and against the expectations, Socs2-deficiency does not enhance the cytotoxicity or IFN-γ production of NK cells. Intriguingly, the absence of SOCS2 boosts IL-15-induced JAK2/STAT5 activation in NK cells (48), which has commonly been believed to signal via JAK1 and JAK3.
In contrast to murine NK cells, knockdown of SOCS2 has no impact on the IL-15-induced in vitro differentiation of primary human NK cell precursors, but severely diminishes the cytotoxic function of primary NK cells and the human NK cell line NK-92. The reduced cytotoxicity was assigned to impaired degradation and accumulation of the focal adhesion kinase PYK2 (198), which is involved in the formation of the NK/target cell synapse upon killing (199). Although it is clear that SOCS2 interferes with NK cell functions, the distinct roles of SOCS2 in human and mouse NK cells remain enigmatic.
SOCS3
Socs3−/− mice are embryonically lethal due to placental defects (200) and impaired fetal liver hematopoiesis (201). SOCS3 counteracts inflammation by inhibiting a variety of pro-inflammatory signaling pathways (202). Most prominently, SOCS3 inhibits gp130 receptor/STAT3 signaling by direct inhibition and/or ubiquitin-mediated degradation of the receptors or their associated JAK kinases (downstream of IL-6, IL-11, IL-27, OSM and LIF). SOCS3 was also shown to negatively regulate IL-12-induced STAT4 activation by blocking the IL-12Rβ2 subunit via its SH2 domain (203).
In mouse NK cells, SOCS3 is induced upon IL-15 signaling (57) and is a direct target gene of the helix-loop-helix protein ID2 (204). Id2 deletion in NKp46+ cells leads to a complete absence of peripheral NK cells due to impaired IL-15 signaling. NK cell numbers are rescued by additionally deleting Socs3 (204). However, loss of Socs3 alone in the presence of ID2 does not alter the development, maturation or IL-15-induced proliferation of mouse NK cells (57, 204). CRISPR/Cas9-mediated disruption of SOCS3 in human NK cells promotes proliferation and cytotoxicity (205), thereby suggesting that SOCS3 may be a useful target for NK cell-based immunotherapy. However, taking into account the detrimental effect of Socs3-deficiency in mice, any envisaged inhibitor treatment will have to be applied specifically on NK cells to avoid generic adverse side-effects.
Concluding Remarks
The JAK/STAT pathway is evolutionary highly conserved and transmits extracellular signals to the nucleus modulating target gene transcription. Members of this pathway are frequently altered in cancer including malignancies of innate lymphocytes, making them an attractive target for drug development. Several JAK inhibitors are already used for the treatment of rheumatoid arthritis, psoriasis and myelofibrosis, and entered phase 2 and 3 clinical trials for the treatment of other inflammatory diseases and cancer. While the first clinically used inhibitors such as Ruxolitinib or Tofacitinib, proved to target multiple JAK kinases, more specific compounds found their way into clinical trials (206, 207).
We and others have previously shown that treatment with the JAK1/2 inhibitor Ruxolitinib substantially impairs NK cell functions leading to increased susceptibility to viral infections and tumor metastasis (49, 208). In line, in a mouse B cell lymphoma model Ruxolitinib treatment promotes tumor progression by enhancing NK cell-derived VEGF-A expression (122). On the other hand, Ruxolitinib treatment significantly reduces disease burden in the context of CD56+ T-cell large granular lymphocytic (T-LGL) leukemia (170) and restores impaired NK cell functions in patients harboring STAT1 GOF mutations (96).
JAK inhibitor treatment shall be carefully evaluated to identify the complex interplay and potential opposing effects on target and immune cells. While in the context of inflammatory and immune-related diseases JAK inhibitor-induced dampening of NK cell functions may be advantageous, NK cell malfunction in metastatic cancers should be precluded. Besides blocking JAK kinases, considerable effort is undertaken to develop specific STAT inhibitors. This could be of particular interest for the field of immunotherapy, as treatment with STAT3 or STAT6 inhibitors may enhance NK cell cytotoxicity. Finding ways to efficiently improve NK cell functions will promote the use of adoptively transferred NK cells in everyday clinics.
Targeting the negative regulators of the JAK/STAT pathway also holds great promise as novel immunotherapeutic strategy. In particular, CIS was shown to be a checkpoint in NK cell-mediated tumor control making it an attractive candidate for anti-tumor therapy. The expanding knowledge of immune checkpoints and potential drug candidates opens a new avenue for immunotherapy, yet the next challenge is to develop specific and stable compounds suitable for clinical use.
Author Contributions
JT generated the figures. DG, VS, and EP drafted the figures and wrote the manuscript.
Funding
The authors are supported by grants from the Austrian Science Fund (FWF): doc.fund DOC 32-B28 and F6107 awarded to VS, the stand-alone project P32001-B awarded to EP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank our laboratory members and the entire JAK/STAT community for numerous discussions and K. Klein for proofreading of the manuscript and to apologize to all investigators whose research was not cited.
Abbreviations
BRAF, rapidly accelerated fibrosarcoma isoform B; CHILP, common helper-like innate lymphoid precursor; CIS, cytokine inducible SH2-containing protein; Cish, gene coding for CIS protein; CLP, common lymphoid progenitor; CLPD, chronic lymphoproliferative disorder; DC, dendritic cell; DNAM1, DNAX accessory molecule 1 (CD226); EILP, early innate lymphoid cell progenitor; EOMES, Eomesodermin; γc, common gamma chain; GATA3, GATA-binding protein 3; GH, growth hormone; GOF, gain-of-function; HSC, hematopoietic stem cell; HSV-1, herpes simplex virus 1; ID2, inhibitor of DNA binding 2; IFN, interferon; IFN-I, type 1 interferon [e.g., IFN-α (alpha), IFN-β (beta)]; IFN-II, type 2 interferon [IFN-γ (gamma)]; IFN-III, type 3 interferon [IFN-λ (lambda)]; IFNAR, interferon-α/β receptor; IL, interleukin; ILC, innate lymphoid cell; ILCP, innate lymphoid cell precursor; IRF9, interferon regulatory factor 9; ISGF3, interferon-stimulated gene factor 3; JAK, Janus kinase; Klrk1, gene coding for NKG2D protein; LCMV, lymphocytic choriomeningitis virus; LIF, leukemia inhibitory factor; LOF, loss-of-function; mAB, monoclonal antibody; MCMV, mouse cytomegalovirus; MEK, mitogen-activated protein kinase kinase; MHC, major histocompatibility complex; NK, natural killer; NKG2D, natural killer receptor group 2, member D; NKP, NK lineage-restricted progenitor; NKTCL, NK/T-cell lymphoma; OSM, oncostatin M; PMA, phorbol 12-myristate 13-acetate; PYK2, protein tyrosine kinase 2; SCID, severe combined immunodeficiency; SH2, Src homology 2; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; T-BET, T-box expressed in T cells; Tbx21, gene coding for T-BET; TGF-β, transforming growth factor beta; Th1, T-helper cell type 1; Th2, T-helper cell type 2; T-LGL, T-cell large granular lymphocytic; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TYK2, tyrosine kinase 2; RORγt, retinoic acid-related orphan receptor gamma t; U-STAT, unphosphorylated STAT; VEGF-A, vascular endothelial growth factor A.
References
1. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
2. Walker JA, Barlow JL, McKenzie ANJ. Innate lymphoid cells–how did we miss them? Nat Rev Immunol. (2013) 13:75–87. doi: 10.1038/nri3349
3. Caligiuri MA. Human natural killer cells. Blood. (2008) 112:461–9. doi: 10.1182/blood-2007-09-077438
4. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. (2013) 34:182–91. doi: 10.1016/j.it.2013.01.003
5. Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol. (2006) 6:520–31. doi: 10.1038/nri1863
6. Bernink JH, Mjösberg J, Spits H. Human ILC1: to be or not to be. Immunity. (2017) 46:756–7. doi: 10.1016/j.immuni.2017.05.001
7. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
8. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity. (2017) 46:148–61. doi: 10.1016/j.immuni.2016.11.005
9. Roan F, Ziegler SF. Human group 1 innate lymphocytes are negative for surface CD3ε but express CD5. Immunity. (2017) 46:758–9. doi: 10.1016/j.immuni.2017.04.024
10. Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, et al. Molecular definition of the identity and activation of natural killer cells. Nat Immunol. (2012) 13:1000–8. doi: 10.1038/ni.2395
11. Park E, Patel S, Wang Q, Andhey P, Zaitsev K, Porter S, et al. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. Elife. (2019) 8:e47605. doi: 10.7554/eLife.47605
12. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
13. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat Immunol. (2017) 18:995–1003. doi: 10.1038/ni.3809
14. Pikovskaya O, Chaix J, Rothman NJ, Collins A, Chen Y-H, Scipioni AM, et al. Cutting edge: eomesodermin is sufficient to direct type 1 innate lymphocyte development into the conventional NK lineage. J Immunol. (2016) 196:1449–54. doi: 10.4049/jimmunol.1502396
15. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol. (2016) 17:758–764. doi: 10.1038/ni.3482
16. Freud AG, Yu J, Caligiuri MA. Human natural killer cell development in secondary lymphoid tissues. Semin Immunol. (2014) 26:132–7. doi: 10.1016/j.smim.2014.02.008
17. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
18. Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat Immunol. (2015) 16:1044–50. doi: 10.1038/ni.3248
19. Yu X, Wang Y, Deng M, Li Y, Ruhn KA, Zhang CC, et al. The basic leucine zipper transcription factor NFIL3 directs the development of a common innate lymphoid cell precursor. Elife. (2014) 3:3. doi: 10.7554/eLife.04406
20. Gotthardt D, Sexl V. STATs in NK-cells: the good, the bad, and the ugly. Front Immunol. (2017) 7:1–8. doi: 10.3389/fimmu.2016.00694
21. Witalisz-Siepracka A, Klein K, Prinz D, Leidenfrost N, Schabbauer G, Dohnal A, et al. Loss of JAK1 drives innate immune deficiency. Front Immunol. (2019) 10:3108. doi: 10.3389/fimmu.2018.03108
22. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood. (2011) 117:1565–73. doi: 10.1182/blood-2010-06-291633
23. Park SY, Saijo K, Takahashi T, Osawa M, Areas H, Hirayama N, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. (1995) 3:771–82. doi: 10.1016/1074-7613(95)90066-7
24. Stannard KA, Lemoine S, Waterhouse NJ, Vari F, Chatenoud L, Gandhi MK, et al. Human peripheral blood DNAM-1 neg NK cells are a terminally differentiated subset with limited effector functions. Blood Adv. (2019) 3:1681–94. doi: 10.1182/bloodadvances.2018030676
25. Moretta L. Dissecting CD56dim human NK cells. Blood. (2010) 116:3689–91. doi: 10.1182/blood-2010-09-303057
26. Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, et al. Human CD56 bright NK cells: an update. J Immunol. (2016) 196:2923–31. doi: 10.4049/jimmunol.1502570
27. Kim S, Iizuka K, Kang H-SP, Dokun A, French AR, Greco S, et al. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol. (2002) 3:523–8. doi: 10.1038/ni796
28. Goh W, Huntington ND. Regulation of murine natural killer cell development. Front Immunol. (2017) 8:130. doi: 10.3389/fimmu.2017.00130
29. Stabile H, Scarno G, Fionda C, Gismondi A, Santoni A, Gadina M, et al. JAK/STAT signaling in regulation of innate lymphoid cells: the gods before the guardians. Immunol Rev. (2018) 286:148–59. doi: 10.1111/imr.12705
30. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. (2007) 282:20059–63. doi: 10.1074/jbc.R700016200
31. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
32. Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and non-canonical aspects of JAK-STAT signaling: lessons from interferons for cytokine responses. Front Immunol. (2017) 8:29. doi: 10.3389/fimmu.2017.00029
33. Miyoshi K, Cui Y, Riedlinger G, Robinson P, Lehoczky J, Zon L, et al. Structure of the mouse Stat 3/5 locus: evolution from Drosophila to zebrafish to mouse. Genomics. (2001) 71:150–5. doi: 10.1006/geno.2000.6433
34. Wang Y, Levy DE. Comparative evolutionary genomics of the STAT family of transcription factors. JAK-STAT. (2012) 1:23–36. doi: 10.4161/jkst.19418
35. Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. (1998) 93:397–409. doi: 10.1016/S0092-8674(00)81168-X
36. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. (1997) 94:3801–4. doi: 10.1073/pnas.94.8.3801
37. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. (1998) 93:373–83. doi: 10.1016/S0092-8674(00)81166-6
38. Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng C-X, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. (2004) 24:8037–47. doi: 10.1128/MCB.24.18.8037-8047.2004
39. Igaz P, Tóth S, Falus A. Biological and clinical significance of the JAK-STAT pathway; lessons from knockout mice. Inflamm Res. (2001) 50:435–41. doi: 10.1007/PL00000267
40. Rautela J, Huntington ND. IL-15 signaling in NK cell cancer immunotherapy. Curr Opin Immunol. (2017) 44:1–6. doi: 10.1016/j.coi.2016.10.004
41. Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. (2009) 228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x
42. Kleppe M, Spitzer MH, Li S, Hill CE, Dong L, Papalexi E, et al. Jak1 integrates cytokine sensing to regulate hematopoietic stem cell function and stress hematopoiesis. Cell Stem Cell. (2017) 21:489–501.e7. doi: 10.1016/j.stem.2017.08.011
43. Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun. (2016) 7:13992. doi: 10.1038/ncomms13992
44. Park SO, Wamsley HL, Bae K, Hu Z, Li X, Choe S, et al. Conditional deletion of Jak2 reveals an essential role in hematopoiesis throughout mouse ontogeny: implications for Jak2 inhibition in humans. PLoS ONE. (2013) 8:e59675. doi: 10.1371/journal.pone.0059675
45. Wang KS, Ritz J, Frank DA. IL-2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J Immunol. (1999) 162:299–304.
46. Strobl B, Stoiber D, Sexl V, Mueller M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci. (2011) 16:3214–32. doi: 10.2741/3908
47. Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. (2012) 13:722–8. doi: 10.1038/ni.2366
48. Kim WS, Kim MJ, Kim DO, Byun J-E, Huy H, Song HY, et al. Suppressor of cytokine signaling 2 negatively regulates NK cell differentiation by inhibiting JAK2 activity. Sci Rep. (2017) 7:46153. doi: 10.1038/srep46153
49. Bottos A, Gotthardt D, Gill JW, Gattelli A, Frei A, Tzankov A, et al. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun. (2016) 7:12258. doi: 10.1038/ncomms12258
50. Zhong J, Yang P, Muta K, Dong R, Marrero M, Gong F, et al. Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS ONE. (2010) 5:e9593. doi: 10.1371/journal.pone.0009593
51. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. (2007) 26:503–17. doi: 10.1016/j.immuni.2007.03.006
52. Leonard WJ, Lin JX, O'Shea JJ. The γ c family of cytokines: basic biology to therapeutic ramifications. Immunity. (2019) 50:832–50. doi: 10.1016/j.immuni.2019.03.028
53. Nosaka T, Van Deursen JMA, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, et al. Defective lymphoid development in mice lacking Jak3. Science. (1995) 270:800–2. doi: 10.1126/science.270.5237.800
54. Takahashi Y, Byrareddy SN, Albrecht C, Brameier M, Walter L, Mayne AE, et al. In vivo administration of a JAK3 inhibitor during acute SIV infection leads to significant increases in viral load during chronic infection. PLoS Pathog. (2014) 10: doi: 10.1371/journal.ppat.1003929
55. Roberts JL, Lengi A, Brown SM, Chen M, Zhou YJ, O'Shea JJ, et al. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood. (2004) 103:2009–18. doi: 10.1182/blood-2003-06-2104
56. Robinette ML, Cella M, Telliez JB, Ulland TK, Barrow AD, Capuder K, et al. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. (2018) 11:50–60. doi: 10.1038/mi.2017.38
57. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. (2016) 17:816–24. doi: 10.1038/ni.3470
58. Haan C, Rolvering C, Raulf F, Kapp M, Drückes P, Thoma G, et al. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem Biol. (2011) 18:314–23. doi: 10.1016/j.chembiol.2011.01.012
59. Stoiber D, Kovacic B, Schuster C, Schellack C, Karaghiosoff M, Kreibich R, et al. TYK2 is a key regulator of the surveillance of B lymphoid tumors. J Clin Invest. (2004) 114:1650–8. doi: 10.1172/JCI200422315
60. Kreins AY, Ciancanelli MJ, Okada S, Kong X-F, Ramírez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. (2015) 212:1641–62. doi: 10.1084/jem.20140280
61. Prchal-Murphy M, Semper C, Lassnig C, Wallner B, Gausterer C, Teppner-Klymiuk I, et al. TYK2 kinase activity is required for functional type I interferon responses in vivo. PLoS ONE. (2012) 7:e39141. doi: 10.1371/journal.pone.0039141
62. Prchal-Murphy M, Witalisz-Siepracka A, Bednarik KT, Putz EM, Gotthardt D, Meissl K, et al. In vivo tumor surveillance by NK cells requires TYK2 but not TYK2 kinase activity. Oncoimmunology. (2015) 4:e1047579. doi: 10.1080/2162402X.2015.1047579
63. Shimoda K, Tsutsui H, Aoki K, Kato K, Matsuda T, Numata A, et al. Partial impairment of interleukin-12 (IL-12) and IL-18 signaling in Tyk2-deficient mice. Blood. (2002) 99:2094–9. doi: 10.1182/blood.V99.6.2094
64. Schleicher U, Mattner J, Blos M, Schindler H, Rollinghoff M, Karaghiosoff M, et al. Control of Leishmania major in the absence of Tyk2 kinase. Eur J Immunol. (2004) 34:519–29. doi: 10.1002/eji.200324465
65. Simonović N, Witalisz-Siepracka A, Meissl K, Lassnig C, Reichart U, Kolbe T, et al. NK Cells require cell-extrinsic and -intrinsic TYK2 for full functionality in tumor surveillance and antibacterial immunity. J Immunol. (2019) 202:1724–34. doi: 10.4049/jimmunol.1701649
66. Boisson-Dupuis S, Kong X-F, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. (2012) 24:364–78. doi: 10.1016/j.coi.2012.04.011
67. Delgoffe GM, Vignali DAA. STAT heterodimers in immunity. JAK-STAT. (2013) 2:e23060. doi: 10.4161/jkst.23060
68. Zhang C, Zhang J, Niu J, Zhang J, Tian Z. Interleukin-15 improves cytotoxicity of natural killer cells via up-regulating NKG2D and cytotoxic effector molecule expression as well as STAT1 and ERK1/2 phosphorylation. Cytokine. (2008) 42:128–36. doi: 10.1016/j.cyto.2008.01.003
69. Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. (2013) 4:437–44. doi: 10.1016/j.celrep.2013.07.012
70. Wendt K, Wilk E, Buyny S, Schmidt RE, Jacobs R. Interleukin-21 differentially affects human natural killer cell subsets. Immunology. (2007) 122:486–95. doi: 10.1111/j.1365-2567.2007.02675.x
71. Zhu S, Phatarpekar PV, Denman CJ, Senyukov VV, Somanchi SS, Nguyen-Jackson HT, et al. Transcription of the activating receptor NKG2D in natural killer cells is regulated by STAT3 tyrosine phosphorylation. Blood. (2014) 124:403–11. doi: 10.1182/blood-2013-05-499707
72. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. (2009) 28:239–60. doi: 10.1080/08830180902978120
73. Lee C-K. Differential regulation of constitutive major histocompatibility complex class I expression in T and B lymphocytes. J Exp Med. (1999) 190:1451–64. doi: 10.1084/jem.190.10.1451
74. Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, Sun JC. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J Exp Med. (2016) 213:225–33. doi: 10.1084/jem.20150712
75. Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, et al. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell. (2006) 10:77–87. doi: 10.1016/j.ccr.2006.05.025
76. Robbins SH, Tessmer MS, Van Kaer L, Brossay L. Direct effects of T-bet and MHC class I expression, but not STAT1, on peripheral NK cell maturation. Eur J Immunol. (2005) 35:757–65. doi: 10.1002/eji.200425797
77. Lee C-K, Rao DT, Gertner R, Gimeno R, Frey AB, Levy DE. Distinct requirements for IFNs and STAT1 in NK cell function. J Immunol. (2000) 165:3571–7. doi: 10.4049/jimmunol.165.7.3571
78. Mizutani T, Neugebauer N, Putz EM, Moritz N, Simma O, Zebedin-Brandl E, et al. Conditional IFNAR1 ablation reveals distinct requirements of Type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology. (2012) 1:1027–37. doi: 10.4161/onci.21284
79. Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. (2007) 178:7540–9. doi: 10.4049/jimmunol.178.12.7540
80. Müller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science. (1994) 264:1918–21. doi: 10.1126/science.8009221
81. Sivori S, Cantoni C, Parolini S, Marcenaro E, Conte R, Moretta L, et al. IL-21 induces both rapid maturation of human CD34+ cell precursors towards NK cells and acquisition of surface killer Ig-like receptors. Eur J Immunol. (2003) 33:3439–47. doi: 10.1002/eji.200324533
82. Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol. (2006) 176:3840. doi: 10.4049/jimmunol.176.6.3840
83. Sutherland APR, Joller N, Michaud M, Liu SM, Kuchroo VK, Grusby MJ. IL-21 promotes CD8 + CTL activity via the transcription factor T-bet. J Immunol. (2013) 190:3977–84. doi: 10.4049/jimmunol.1201730
84. Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. (2000) 408:57–63. doi: 10.1038/35040504
85. Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. (2002) 16:559–69. doi: 10.1016/S1074-7613(02)00295-9
86. Zwirner NW, Ziblat A. Regulation of NK cell activation and effector functions by the IL-12 family of cytokines: the case of IL-27. Front Immunol. (2017) 8:25. doi: 10.3389/fimmu.2017.00025
87. Ziblat A, Domaica CI, Spallanzani RG, Iraolagoitia XLR, Rossi LE, Avila DE, et al. IL-27 stimulates human NK-cell effector functions and primes NK cells for IL-18 responsiveness. Eur J Immunol. (2015) 45:192–202. doi: 10.1002/eji.201444699
88. Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+T cells. Immunity. (2002) 16:779–90. doi: 10.1016/S1074-7613(02)00324-2
89. Kumar P, Rajasekaran K, Nanbakhsh A, Gorski J, Thakar MS, Malarkannan S. IL-27 promotes NK cell effector functions via Maf-Nrf2 pathway during influenza infection. Sci Rep. (2019) 9:4984. doi: 10.1038/s41598-019-41478-6
90. Putz EM, Majoros A, Gotthardt D, Prchal-Murphy M, Zebedin-Brandl EM, Fux DA, et al. Novel non-canonical role of STAT1 in natural killer cell cytotoxicity. Oncoimmunology. (2016) 5:e1186314. doi: 10.1080/2162402X.2016.1186314
91. Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. (1996) 84:443–50. doi: 10.1016/S0092-8674(00)81289-1
92. Fortin C, Huang X, Yang Y. Both NK cell-intrinsic and -extrinsic STAT1 signaling are required for NK cell response against vaccinia virus. J Immunol. (2013) 191:363–8. doi: 10.4049/jimmunol.1202714
93. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. (1996) 84:431–42. doi: 10.1016/S0092-8674(00)81288-X
94. Vairo D, Tassone L, Tabellini G, Tamassia N, Gasperini S, Bazzoni F, et al. Severe impairment of IFN-γ and IFN-α responses in cells of a patient with a novel STAT1 splicing mutation. Blood. (2011) 118:1806–17. doi: 10.1182/blood-2011-01-330571
95. Toubiana J, Okada S, Hiller J, Oleastro M, Gomez ML, Becerra JCA, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
96. Vargas-Hernández A, Mace EM, Zimmerman O, Zerbe CS, Freeman AF, Rosenzweig S, et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2018) 141:2142–55.e5. doi: 10.1016/j.jaci.2017.08.040
97. Tabellini G, Vairo D, Scomodon O, Tamassia N, Ferraro RM, Patrizi O, et al. Impaired natural killer cell functions in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2017) 140:553–64.e4. doi: 10.1016/j.jaci.2016.10.051
98. Weinacht KG, Charbonnier L-M, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. (2017) 139:1629–40.e2. doi: 10.1016/j.jaci.2016.11.022
99. Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. (2000) 13:795–804. doi: 10.1016/S1074-7613(00)00077-7
100. Le-Trilling VTK, Wohlgemuth K, Rückborn MU, Jagnjic A, Maaßen F, Timmer L, et al. STAT2-dependent immune responses ensure host survival despite the presence of a potent viral antagonist. J Virol. (2018) 92:e00296–18. doi: 10.1128/JVI.00296-18
101. Hofer MJ, Li W, Manders P, Terry R, Lim SL, King NJC, et al. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J Virol. (2012) 86:6932–46. doi: 10.1128/JVI.07147-11
102. Perry ST, Buck MD, Lada SM, Schindler C, Shresta S. STAT2 mediates innate immunity to dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. (2011) 7:e1001297. doi: 10.1371/journal.ppat.1001297
103. Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. (2007) 204:2383–96. doi: 10.1084/jem.20070401
104. Geary CD, Krishna C, Lau CM, Adams NM, Gearty SV, Pritykin Y, et al. Non-redundant ISGF3 components promote nk cell survival in an auto-regulatory manner during viral infection. Cell Rep. (2018) 24:1949–57.e6. doi: 10.1016/j.celrep.2018.07.060
105. Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SMB, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA. (2013) 110:3053–8. doi: 10.1073/pnas.1220098110
106. Shahni R, Cale CM, Anderson G, Osellame LD, Hambleton S, Jacques TS, et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain. (2015) 138:2834–46. doi: 10.1093/brain/awv182
107. Moens L, Van Eyck L, Jochmans D, Mitera T, Frans G, Bossuyt X, et al. A novel kindred with inherited STAT2 deficiency and severe viral illness. J Allergy Clin Immunol. (2017) 139:1995–7.e9. doi: 10.1016/j.jaci.2016.10.033
108. Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe. (2016) 19:760–9. doi: 10.1016/j.chom.2016.05.016
109. Wilson RP, Tursi SA, Rapsinski GJ, Medeiros NJ, Le LS, Kotredes KP, et al. STAT2 dependent type i interferon response promotes dysbiosis and luminal expansion of the enteric pathogen Salmonella Typhimurium. PLoS Pathog. (2019) 15:e1007745. doi: 10.1371/journal.ppat.1007745
110. Yue C, Xu J, Daryl M, Estioko T, Kotredes KP, Lopez-Otalora Y, et al. Host STAT2/type I interferon axis controls tumor growth. Int J Cancer. (2015) 136:117–26. doi: 10.1002/ijc.29004
111. Gotthardt D, Putz EM, Straka E, Kudweis P, Biaggio M, Poli V, et al. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood. (2014) 124:2370–9. doi: 10.1182/blood-2014-03-564450
112. Matsui M, Kishida T, Nakano H, Yoshimoto K, Shin-Ya M, Shimada T, et al. Interleukin-27 activates natural killer cells and suppresses NK-resistant head and neck squamous cell carcinoma through inducing antibody-dependent cellular cytotoxicity. Cancer Res. (2009) 69:2523–30. doi: 10.1158/0008-5472.CAN-08-2793
113. Wu Y, Tian Z, Wei H, Riley E, Rajasekaran K, Goodier MR, et al. Developmental and functional control of natural killer cells by cytokines. Front Immunol. (2017) 8:930. doi: 10.3389/fimmu.2017.00930
114. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. (2005) 11:1314–21. doi: 10.1038/nm1325
115. Cacalano NA. Regulation of natural killer cell function by STAT3. Front Immunol. (2016) 7:128. doi: 10.3389/fimmu.2016.00128
116. Groner B, Lucks P, Borghouts C. The function of Stat3 in tumor cells and their microenvironment. Semin Cell Dev Biol. (2008) 19:341–50. doi: 10.1016/j.semcdb.2008.06.005
117. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. (2007) 7:41–51. doi: 10.1038/nri1995
118. Lassen MG, Lukens JR, Dolina JS, Brown MG, Hahn YS. Intrahepatic IL-10 maintains NKG2A + Ly49 – liver NK cells in a functionally hyporesponsive state. J Immunol. (2010) 184:2693–701. doi: 10.4049/jimmunol.0901362
119. Jiao Y, Huntington ND, Belz GT, Seillet C. Type 1 innate lymphoid cell biology: lessons learnt from natural killer cells. Front Immunol. (2016) 7:426. doi: 10.3389/fimmu.2016.00426
120. Vivier E, Ugolini S. Regulatory natural killer cells: new players in the il-10 anti-inflammatory response. Cell Host Microbe. (2009) 6:493–5. doi: 10.1016/j.chom.2009.12.001
121. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell. (2017) 171:201–16.e18. doi: 10.1016/j.cell.2017.07.027
122. Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov. (2016) 6:414–29. doi: 10.1158/2159-8290.CD-15-0732
123. Takaki R, Hayakawa Y, Nelson A, Sivakumar PV, Hughes S, Smyth MJ, et al. IL-21 enhances tumor rejection through a NKG2D-dependent mechanism. J Immunol. (2005) 175:2167–73. doi: 10.4049/jimmunol.175.4.2167
124. Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Ng KP, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T cell large granular lymphocyte leukemia. Blood. (2012) 120:3048–58. doi: 10.1182/blood-2012-06-435297
125. Dufva O, Kankainen M, Kelkka T, Sekiguchi N, Awad SA, Eldfors S, et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Commun. (2018) 9:1567. doi: 10.1038/s41467-018-03987-2
126. Jiang L, Gu Z-H, Yan Z-X, Zhao X, Xie Y-Y, Zhang Z-G, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. (2015) 47:1061–6. doi: 10.1038/ng.3358
127. Küçük C, Jiang B, Hu X, Zhang WWW, Chan JKC, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun. (2015) 6:6025. doi: 10.1038/ncomms7025
128. Wang KS, Frank DA, Ritz J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood. (2000) 95:3183–90. doi: 10.1182/blood.V95.10.3183.010k36_3183_3190
129. Wang KS, Ritz J, Frank DA. IL-2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J Immunol. (1999) 162:299–304.
130. Weizman O-E, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, et al. ILC1 confer early host protection at initial sites of viral infection. Cell. (2017) 171:795–808.e12. doi: 10.1016/j.cell.2017.09.052
131. Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. (2004) 20:477–94. doi: 10.1016/S1074-7613(04)00076-7
132. Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. (1996) 382:174–7. doi: 10.1038/382174a0
133. Thierfelder WE, Van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. (1996) 382:171–4. doi: 10.1038/382171a0
134. Yamamoto K, Shibata F, Miyasaka N, Miura O. The human perforin gene is a direct target of STAT4 activated by IL-12 in NK cells. Biochem Biophys Res Commun. (2002) 297:1245–52. doi: 10.1016/S0006-291X(02)02378-1
135. Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med. (2012) 209:947–54. doi: 10.1084/jem.20111760
136. Madera S, Geary CD, Lau CM, Pikovskaya O, Reiner SL, Sun JC. Cutting edge: divergent requirement of T-box transcription factors in effector and memory NK cells. J Immunol. (2018) 200:1977–81. doi: 10.4049/jimmunol.1700416
137. Schimke LF, Hibbard J, Martinez-Barricarte R, Khan TA, de Souza Cavalcante R, Borges de Oliveira Junior E, et al. Paracoccidioidomycosis associated with a heterozygous STAT4 mutation and impaired IFN-γ immunity. J Infect Dis. (2017) 216:1623–34. doi: 10.1093/infdis/jix522
138. O'Sullivan TE, Rapp M, Fan X, El Weizman O, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
139. Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
140. Marçais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol. (2013) 4:450. doi: 10.3389/fimmu.2013.00450
141. Ranson T, Vosshenrich CAJ, Corcuff E, Richard O, Müller W, Di Santo JP. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood. (2003) 101:4887–93. doi: 10.1182/blood-2002-11-3392
142. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. (2012) 36:542–50. doi: 10.1016/j.immuni.2012.03.014
143. Mitchell DM, Ravkov E V, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8 + effector and memory T cells. J Immunol. (2010) 184:6719–30. doi: 10.4049/jimmunol.0904089
144. Gasteiger G, Hemmers S, Firth MA, Le Floc'h A, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med. (2013) 210:1065–8. doi: 10.1084/jem.20122462
145. Kamimura D, Bevan MJ. Naive CD8+ T cells differentiate into protective memory-like cells after IL-2-anti-IL-2 complex treatment in vivo. J Exp Med. (2007) 204:1803–12. doi: 10.1084/jem.20070543
146. Carotta S, Pang SHM, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood. (2011) 117:5449–52. doi: 10.1182/blood-2010-11-318956
147. Serafini N, Vosshenrich CAJ, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol. (2015) 15:415–28. doi: 10.1038/nri3855
148. Vosshenrich CAJ, García-Ojeda ME, Samson-Villéger SI, Pasqualetto V, Enault L, Goff OR-L, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol. (2006) 7:1217–24. doi: 10.1038/ni1395
149. Michaud A, Dardari R, Charrier E, Cordeiro P, Herblot S, Duval M. IL-7 enhances survival of human CD56bright NK cells. J Immunother. (2010) 33:382–90. doi: 10.1097/CJI.0b013e3181cd872d
150. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory Cd8 T cell lineages in interleukin 15-deficient mice. J Exp Med. (2000) 191:771–80. doi: 10.1084/jem.191.5.771
151. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. (1998) 9:669–76. doi: 10.1016/S1074-7613(00)80664-0
152. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. (1998) 188:2067–74. doi: 10.1084/jem.188.11.2067
153. Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. (1999) 10:249–59. doi: 10.1016/S1074-7613(00)80025-4
154. Wakao H, Gouilleux F, Groner B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. (1994) 13:2182–91. doi: 10.1002/j.1460-2075.1994.tb06495.x
155. Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. (1997) 11:179–86. doi: 10.1101/gad.11.2.179
156. Hennighausen L, Robinson GW, Wagner KU, Liu X. Developing a mammary gland is a stāt affair. J Mammary Gland Biol Neoplasia. (1997) 2:365–72. doi: 10.1023/A:1026347313096
157. Villarino AV, Sciumè G, Davis FP, Iwata S, Zitti B, Robinson GW, et al. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J Exp Med. (2017) 214: 20150907. doi: 10.1084/jem.20150907
158. Grange M, Verdeil G, Arnoux F, Griffon A, Spicuglia S, Maurizio J, et al. Active STAT5 regulates T-bet and eomesodermin expression in CD8 T cells and imprints a T-bet-dependent Tc1 program with repressed IL-6/TGF-β1 signaling. J Immunol. (2013) 191:3712–24. doi: 10.4049/jimmunol.1300319
159. Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun. (2014) 5:4539. doi: 10.1038/ncomms5539
160. Rätsep MT, Felker AM, Kay VR, Tolusso L, Hofmann AP, Croy BA. Uterine natural killer cells: supervisors of vasculature construction in early decidua basalis. Reproduction. (2015) 149:R91–102. doi: 10.1530/REP-14-0271
161. Lima PDA, Zhang J, Dunk C, Lye SJ, Croy BA. Leukocyte driven-decidual angiogenesis in early pregnancy. Cell Mol Immunol. (2014) 11:522–37. doi: 10.1038/cmi.2014.63
162. Li G, Huang W, Xia Q, Yang K, Liu R, Zhu H, et al. Role of uterine natural killer cells in angiogenesis of human decidua of the first-trimester pregnancy. Sci China Ser C Life Sci. (2008) 51:111–9. doi: 10.1007/s11427-008-0027-7
163. Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst. (2014) 106:dju200. doi: 10.1093/jnci/dju200
164. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. (2018) 9:527. doi: 10.3389/fimmu.2018.00527
165. Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, et al. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. (2015) 6:13835–43. doi: 10.18632/oncotarget.3453
166. Bezrodnik L, Di Giovanni D, Caldirola MS, Azcoiti ME, Torgerson T, Gaillard MI. Long-term follow-up of STAT5B deficiency in three argentinian patients: clinical and immunological features. J Clin Immunol. (2015) 35:264–72. doi: 10.1007/s10875-015-0145-5
167. Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M, et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. (2006) 118:e1584–92. doi: 10.1542/peds.2005-2882
168. Scaglia PA, Martínez AS, Feigerlová E, Bezrodnik L, Gaillard MI, Di Giovanni D, et al. A novel missense mutation in the SH2 domain of the STAT5B gene results in a transcriptionally inactive STAT5b associated with severe IGF-I deficiency, immune dysfunction, and lack of pulmonary disease. J Clin Endocrinol Metab. (2012) 97:E830–9. doi: 10.1210/jc.2011-2554
169. Rajala HLM, Porkka K, MacIejewski JP, Loughran TP, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann Med. (2014) 46:114–22. doi: 10.3109/07853890.2014.882105
170. Klein K, Witalisz-Siepracka A, Maurer B, Prinz D, Heller G, Leidenfrost N, et al. STAT5BN642H drives transformation of NKT cells: a novel mouse model for CD56+ T-LGL leukemia. Leukemia. (2019) 33:2336–40. doi: 10.1038/s41375-019-0471-3
171. Lin J-X, Du N, Li P, Kazemian M, Gebregiorgis T, Spolski R, et al. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat Commun. (2017) 8:1320. doi: 10.1038/s41467-017-01477-5
172. Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. (2011) 50:87. doi: 10.1007/s12026-011-8205-2
173. Son DJ, Jung YY, Park MH, Lee HL, Song MJ, Yoo HS, et al. Activated natural killer cells mediate the suppressive effect of interleukin-4 on tumor development via STAT6 activation in an atopic condition melanoma model. Neoplasia. (2017) 19:537–48. doi: 10.1016/j.neo.2017.02.014
174. Brady J, Carotta S, Thong RPL, Chan CJ, Hayakawa Y, Smyth MJ, et al. The interactions of multiple cytokines control NK cell maturation. J Immunol. (2010) 185:6679–88. doi: 10.4049/jimmunol.0903354
175. Kiniwa T, Enomoto Y, Terazawa N, Omia A, Miyataa N, Ishiwata K, et al. NK cells activated by Interleukin-4 in cooperation with Interleukin-15 exhibit distinctive characteristics. Proc Natl Acad Sci USA. (2016) 113:10139–44. doi: 10.1073/pnas.1600112113
176. Bream JH, Curiel RE, Yu C-R, Egwuagu CE, Grusby MJ, Aune TM, et al. IL-4 synergistically enhances both IL-2- and IL-12-induced IFN-gamma expression in murine NK cells. Blood. (2003) 102:207–14. doi: 10.1182/blood-2002-08-2602
177. Katsumoto T, Kimura M, Yamashita M, Hosokawa H, Hashimoto K, Hasegawa A, et al. STAT6-dependent differentiation and production of IL-5 and IL-13 in murine NK2 cells. J Immunol. (2004) 173:4967–75. doi: 10.4049/jimmunol.173.8.4967
178. Warren HS, Kinnear BF, Phillips JH, Lanier LL. Production of IL-5 by human NK cells and regulation of IL-5 secretion by IL-4, IL-10, and IL-12. J Immunol. (1995) 154:5144–52.
179. Loza MJ, Peters SP, Zangrilli JG, Perussia B. Distinction between IL-13+ and IFN-gamma+ natural killer cells and regulation of their pool size by IL-4. Eur J Immunol. (2002) 32:413–23. doi: 10.1002/1521-4141(200202)32:2<413::AID-IMMU413>3.0.CO;2-X
180. Walker C, Checkel J, Cammisuli S, Leibson PJ, Gleich GJ. IL-5 production by NK cells contributes to eosinophil infiltration in a mouse model of allergic inflammation. J Immunol. (1998) 161:1962–9.
181. Kim JH, Jang YJ. Role of natural killer cells in airway inflammation. Allergy, Asthma Immunol Res. (2018) 10:448–56. doi: 10.4168/aair.2018.10.5.448
182. Mahalingam S, Karupiah G, Takeda K, Akira S, Matthaei KI, Foster PS. Enhanced resistance in STAT6-deficient mice to infection with ectromelia virus. Proc Natl Acad Sci USA. (2001) 98:6812–7. doi: 10.1073/pnas.111151098
183. Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H, Yoshimura A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways. Arterioscler Thromb Vasc Biol. (2011) 31:980–5. doi: 10.1161/ATVBAHA.110.207464
184. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. (2008) 19:414–22. doi: 10.1016/j.semcdb.2008.07.010
185. Keating N, Nicholson SE. SOCS-mediated immunomodulation of natural killer cells. Cytokine. (2018) 118:64–70. doi: 10.1016/j.cyto.2018.03.033
186. Zimmerer JM, Lesinski GB, Kondadasula SV, Karpa VI, Lehman A, RayChaudhury A, et al. IFN-α-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol. (2007) 178:4832–45. doi: 10.4049/jimmunol.178.8.4832
187. Greenhalgh CJ, Alexander WS. Suppressors of cytokine signalling and regulation of growth hormone action. Growth Horm IGF Res. (2004) 14:200–6. doi: 10.1016/j.ghir.2003.12.011
188. Kedzierski L, Linossi EM, Kolesnik TB, Day EB, Bird NL, Kile BT, et al. Suppressor of cytokine signaling 4 (SOCS4) protects against severe cytokine storm and enhances viral clearance during influenza infection. PLoS Pathog. (2014) 10:e1004134. doi: 10.1371/journal.ppat.1004134
189. Kedzierski L, Tate MD, Hsu AC, Kolesnik TB, Linossi EM, Dagley L, et al. Suppressor of cytokine signaling (SOCS)5 ameliorates influenza infection via inhibition of EGFR signaling. Elife. (2017) 6:e20444. doi: 10.7554/eLife.20444
190. Küçük C, Hu X, Jiang B, Klinkebiel D, Geng H, Gong Q, et al. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. (2015) 21:1699–711. doi: 10.1158/1078-0432.CCR-14-1216
191. Knisz J, Banks A, McKeag L, Metcalfe DD, Rothman PB, Brown JM. Loss of SOCS7 in mice results in severe cutaneous disease and increased mast cell activation. Clin Immunol. (2009) 132:277–84. doi: 10.1016/j.clim.2009.04.003
192. Putz EM, Guillerey C, Kos K, Stannard K, Miles K, Delconte RB, et al. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology. (2017) 6:e1267892. doi: 10.1080/2162402X.2016.1267892
193. Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. (2006) 7:33–9. doi: 10.1038/ni1287
194. Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. (1999) 98:597–608. doi: 10.1016/S0092-8674(00)80047-1
195. Shen L, Evel-Kabler K, Strube R, Chen S-Y. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. (2004) 22:1546–53. doi: 10.1038/nbt1035
196. Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. (1998) 95:14395–9. doi: 10.1073/pnas.95.24.14395
197. Eyles JL, Metcalf D, Grusby MJ, Hilton DJ, Starr R. Negative regulation of interleukin-12 signaling by suppressor of cytokine signaling-1. J Biol Chem. (2002) 277:43735–40. doi: 10.1074/jbc.M208586200
198. Lee SH, Yun S, Piao Z, Jeong M, Kim DO, Jung H, et al. Suppressor of cytokine signaling 2 regulates IL-15-primed human NK cell function via control of phosphorylated Pyk2. J Immunol. (2010) 185:917–28. doi: 10.4049/jimmunol.1000784
199. Sancho D, Nieto M, Llano M, Rodríguez-Fernández JL, Tejedor R, Avraham S, et al. The tyrosine kinase PYK-2/RAFTK regulates natural killer (NK) cell cytotoxic response, and is translocated and activated upon specific target cell recognition and killing. J Cell Biol. (2000) 149:1249–61. doi: 10.1083/jcb.149.6.1249
200. Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, et al. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci USA. (2001) 98:9324–9. doi: 10.1073/pnas.161271798
201. Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. (1999) 98:617–27. doi: 10.1016/S0092-8674(00)80049-5
202. Carow B, Rottenberg ME. SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol. (2014) 5:58. doi: 10.3389/fimmu.2014.00058
203. Yamamoto K, Yamaguchi M, Miyasaka N, Miura O. SOCS-3 inhibits IL-12-induced STAT4 activation by binding through its SH2 domain to the STAT4 docking site in the IL-12 receptor β2 subunit. Biochem Biophys Res Commun. (2003) 310:1188–93. doi: 10.1016/j.bbrc.2003.09.140
204. Delconte RB, Shi W, Sathe P, Ushiki T, Seillet C, Minnich M, et al. The helix-loop-helix protein ID2 governs NK cell fate by tuning their sensitivity to interleukin-15. Immunity. (2016) 44:103–15. doi: 10.1016/j.immuni.2015.12.007
205. Kararoudi MN, Elmas E, Lamb M, Chakravarti N, Trikha P, Lee DA. Disruption of SOCS3 promotes the anti-cancer efficacy of primary NK cells. Blood. (2019) 132:4–7. doi: 10.1182/blood-2018-99-116621
206. Fragoulis GE, McInnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology. (2019) 58:i43–54. doi: 10.1093/rheumatology/key276
207. Musumeci F, Greco C, Giacchello I, Fallacara AL, Ibrahim MM, Grossi G, et al. An update on JAK inhibitors. Curr Med Chem. (2018) 26:1806–32. doi: 10.2174/0929867325666180327093502
Keywords: NK cell, ILC1, development, maturation, cytokine, JAK, STAT, SOCS
Citation: Gotthardt D, Trifinopoulos J, Sexl V and Putz EM (2019) JAK/STAT Cytokine Signaling at the Crossroad of NK Cell Development and Maturation. Front. Immunol. 10:2590. doi: 10.3389/fimmu.2019.02590
Received: 14 August 2019; Accepted: 18 October 2019;
Published: 12 November 2019.
Edited by:
Ewa Sitnicka, Lund University, SwedenReviewed by:
Roland Jacobs, Hannover Medical School, GermanyLaurel L. Lenz, University of Colorado Denver School of Medicine, United States
Copyright © 2019 Gotthardt, Trifinopoulos, Sexl and Putz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dagmar Gotthardt, dagmar.gotthardt@vetmeduni.ac.at