 Yixin Zheng1,2,3†
Yixin Zheng1,2,3† Jianan Zhao1,2,3†
Jianan Zhao1,2,3† Yu Shan1,2,3
Yu Shan1,2,3 Shicheng Guo4,5*
Shicheng Guo4,5* Steven J. Schrodi4,5*
Steven J. Schrodi4,5* Dongyi He1,2,3,6*
Dongyi He1,2,3,6*- 1Department of Rheumatology, Shanghai Guanghua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Institute of Arthritis Research in Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
- 4Center for Human Genomics and Precision Medicine, University of Wisconsin-Madison, Madison, WI, United States
- 5Department of Medical Genetics, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 6Arthritis Institute of Integrated Traditional and Western medicine, Shanghai Chinese Medicine Research Institute, Shanghai, China
Rheumatoid arthritis (RA) is a complex autoimmune disease characterized by chronic inflammation that affects synovial tissues of multiple joints. Granzymes (Gzms) are serine proteases that are released into the immune synapse between cytotoxic lymphocytes and target cells. They enter target cells with the help of perforin to induce programmed cell death in inflammatory and tumor cells. Gzms may have a connection with RA. First, increased levels of Gzms have been found in the serum (GzmB), plasma (GzmA, GzmB), synovial fluid (GzmB, GzmM), and synovial tissue (GzmK) of patients with RA. Moreover, Gzms may contribute to inflammation by degrading the extracellular matrix and promoting cytokine release. They are thought to be involved in RA pathogenesis and have the potential to be used as biomarkers for RA diagnosis, although their exact role is yet to be fully elucidated. The purpose of this review was to summarize the current knowledge regarding the possible role of the granzyme family in RA, with the aim of providing a reference for future research on the mechanisms of RA and the development of new therapies.
Introduction
Rheumatoid arthritis (RA) is a common, long-term autoimmune disease that causes chronic inflammation of synovial tissues in multiple joints. This inflammation can damage cartilage and bone and cause disability (1, 2). RA is more common in women than that in men and occurs at any age (1). Approximately 1% of the population is affected by RA, which significantly affects individuals and society (3, 4). Therefore, it is important to develop novel strategies for timely diagnosis and treatment to reduce inflammation and prevent further damage. Genome-wide association studies have linked the immunopathogenesis of RA to HLA-DRB1, a class II major histocompatibility gene (5). Other genes and loci also play a role in the development of RA, including co-stimulatory receptors molecules, cytokine receptor signaling pathways, and activation of the innate immune response (6). Multiple factors, including genetic and epigenetic modifications, immunity, inflammation, microorganisms, metabolism, and other mechanisms, constitute the pathological mechanism responsible for RA in which various immune cells and molecules interact with each other to mediate the autoimmune reaction, eventually causing bone and joint destruction or even disability (7–13).
Despite significant progress in understanding the inflammatory processes involved in RA, the exact mechanism underlying its development and progression is still not fully understood. However, recent studies have suggested that members of the granzyme family play an important role in the immunopathology of RA. Zhang F et al. (14) applied single-cell RNA sequencing, mass cytometry, bulk RNA-sequencing, and flow cytometry to identify the cell populations contributing to joint inflammation in RA. They applied intracellular staining to tissues from RA samples and RNA-seq to sorted CD8 T cells. Intracellular staining of GzmK and GzmB proteins in disaggregated tissue samples from patients with RA revealed that the majority of CD8 T cells in synovial tissue express GzmK. Furthermore, most HLA-DR CD8 T cells express both GzmB and GzmK by intracellular protein staining. Therefore, they defined distinct subsets of CD8 T cells characterized by a GzmK, GzmB phenotype. Defining key cellular subsets and their activation states in the inflamed tissue is a critical step to define new therapeutic targets for RA. Gzms are proteases produced and released by certain immune cells, including cytotoxic T cells (CTLs) and natural killer (NK) cells (15, 16). There are five human Gzms, namely granzyme A (GzmA), granzyme B (GzmB), granzyme H (GzmH), granzyme K (GzmK), and granzyme M (GzmM). Gzms are released into the immune synapse between CLs and target cells, enter target cells with the help of the pore-forming protein perforin, and activate various pro-apoptotic pathways by breaking down intracellular substrates (15, 17). Perforin and granulysin are two pore-forming proteins of cytotoxic granules of human killer cells, and they have significant roles in mediating Gzm responses to infection (18). There’s work showing the role of granulysin as a biomarker and pathogenic factor in RA (19). In addition to playing a role in the process of apoptosis or programmed cell death, Gzms are also involved in the immune response to infection and tissue damage (20, 21). Some studies have shown that Gzms are elevated in the synovial fluid and synovial tissue of patients with RA and may contribute to inflammation and joint damage (22, 23). The known extracellular activities of Gzms suggest a proinflammatory effect in RA. This review aims to summarize the current knowledge on the possible roles of the granzyme family in RA, with the goal of providing a reference for further research into the disease mechanism of RA and the development of targeted therapies.
GzmA-mediated proinflammatory cytokine-induced bone destruction in RA
Considering all types of killer cells, GzmA is the most abundant Gzm as it is widely expressed in both CD8 CTLs and NK cells (24). GzmA is a serine protease secreted by various CLs, such as NK cells (25), natural killer T (NKT) cells (26), CTLs (27), and CD4 CTLs (28, 29). It plays a key role in the cell death pathway by targeting the endoplasmic reticulum-associated oxidative stress response complex called the SET complex. The SET complex contains at least two GzmA substrates, the nucleosome assembly protein SET (also known as 12PP2A), and the DNA binding protein HMG2. GzmA-mediated cleavage of SET cause inhibition of GzmA-activated DNase NM23-H1 and leads to single-stranded DNA damage (25). GzmA has been shown to have a variety of proinflammatory mechanisms. Hildebrand D et al. (30) suggested that GzmA enters target cells independently and functions as a mediator for inflammation via interleukin (IL)-1β cleavage. Wensink AC et al. (31) discovered that treatment of monocytes with GzmA in combination with toll-like receptor-2 (TLR2)- and TLR4-agonists markedly increases the release of proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), IL-6, and IL-8. GzmA also promotes inflammation via extracellular activities, such as extracellular cleavage of urokinase (32), proteinase-activated receptor-1 (PAR-1), and PAR-2 (33–36). It is an important proinflammatory mediator in RA (37, 38), psoriasis (39), and osteoarthritis (40). Additionally, NK cells and CTLs transport GzmA into the cytoplasm of target cells through the perforin–granzyme system, and GzmA can cleave the gasdermin B (GSDMB) protein into GSDMB-N and GSDMB-C (at sites K229/K224), releasing its N-terminal pore-forming active fragment, thereby inducing pyroptosis (41).
Abnormal GzmA expression has been linked to inflammatory reactions (23, 42, 43). GzmA and GzmB levels in the plasma and synovial fluid are significantly increased during active periods of RA compared to those during osteoarthritis (OA) (23). GzmA stimulates peripheral blood mononuclear cells to produce TNF-α, IL-6, and IL-8 (36) and stimulates fibroblasts to produce IL-6 and IL-8 (44). These cytokines are largely expressed in the synovium and are mainly produced by macrophages and fibroblast-like synoviocytes (45–47). Therefore, high levels of GzmA in RA joints can promote synovial inflammation owing to its influence on cytokine production. The abnormal expression of GzmA may be related to its abnormal expression in various immune cells, including T cells, NK cells, and NKT cells. Perforin is a 70 kDa glycoprotein that is responsible for the formation of pores on the membrane of target cells (48) and participates in cytotoxic reactions in target cells (49). CD4+ perforin+ and GzmA+ cells have been observed in RA synovial samples (50, 51). Nanki T et al. (52) used flow cytometry to analyze the expression of GzmA and perforin in peripheral blood CD4+ and CD8+T cells of patients with RA and healthy people. GzmA and perforin were mainly expressed by CX3CR1 CD4+ and CD8+T cells in patients with RA and healthy people, with increased expression in patients with RA. In addition, Aggarwal et al. (53) found elevated levels of GzmA in NK and NKT cells and GzmB in NK cells of venous blood samples in patients with RA. Elevated GzmA and GzmB levels are associated with disease severity, tissue damage, and joint damage in RA. Correlation studies showed that the disease activity score (DAS28) is positively associated with enhanced levels of GzmA-expressing NK and NKT cells, perforin-GzmA dual-positive NK, NKT cells, and GzmB-expressing NK cells. Loetscher P et al. (54) analyzed chemokine-mediated enzyme release from cytotoxic lymphocytes using cloned and freshly isolated human blood NK cells and CD8+ T cells. They found that GzmA from CD8+T and NK cells can be activated by chemokines, suggesting chemokines may be involved in regulating cytotoxicity in lymphocyte. Many chemokines capable of inducing Gzms release have been shown to be upregulated in the synovial tissue of patients with RA (55, 56). Therefore, GzmA overexpression in CD8+T and NK cells may be related to chemokines upregulated in RA. Moreover, GzmA and GzmB degrade ECM proteins in vitro (20). Santiago L et al. (37) evaluated inflammatory arthritis induced by type II collagen in wild-type, GzmA-deficient, and perforin-deficient mice, and found that GzmA is more closely associated with cartilage and bone injury in mouse paws and knees than with inflammatory signs and synovial cells. Proliferating osteoclasts (OCs), which are primary bone-resorbing cells, are hematopoietic in origin and have a monocyte/macrophage lineage. The formation and activation of OCs are tightly regulated by systemic and pericellular factors (57). GzmA activates monocytes and other OC precursors to secrete TNF, thus increasing proinflammatory cytokine-induced bone destruction observed in RA. However, the mechanism involved seems to be complex and may be either direct by promoting OC differentiation or indirect via other inflammatory responses (37).
GzmB-mediated inflammation and ECM degradation in RA
GzmB is a granzyme family member with the strongest apoptotic activity because of its caspase-like ability to cleave substrates at aspartic acid residues, thereby activating procaspases directly and cleaving downstream caspase substrates (58). GzmB is a 32 kDa serine protease that is secreted by NK cells and CTLs (59, 60). When released into the gap between those cells and target cells, GzmB can enter the cytoplasm of target cells in the presence of perforin. Subsequently, apoptosis is induced by cleaving various intracellular substrates (61) associated with DNA maintenance, such as inhibitors of caspase-activated DNase, poly (ADP-ribose) polymerase, DNA-dependent protein kinase, and lamin B (62–65). GzmB can be produced by various immune and non-immune cells, including T and B cell subsets, monocytes/macrophages, mast cells, basophils (66–71), vascular smooth muscle cells, lung cells, keratinocytes, chondrocytes, and various types of cancer cells (70, 72–79). GzmB can also have extracellular functions, including the degradation of ECM components, cytokines, cell receptors, and clotting proteins (21, 22, 80). The potential pathophysiological consequences of their cleavage constitute the basis for envisaging a crucial proinflammatory role for GzmB in the pathogenesis of inflammatory diseases (81). In the extracellular pathway, direct processing of caspase-3 and caspase-7 by GzmB promotes caspase-mediated degradation of hundreds of protein substrates, resulting in rapid apoptosis (82).
Abnormal expression of GzmB has been observed in the synovial tissues of patients with RA (83). Studies have shown that most CD8T cells in the synovial tissues of patients with RA express GzmK and GzmB proteins (14). Although Gzms are expressed by CTL, only a small percentage of granzyme-positive cells in the synovial membrane are CD8+ and CD4+ T cells, with the majority being NK cells (84). Elevated levels of GzmB have been found in blood and synovial fluid of patients with RA, which may be a result of GzmB release from inflamed joints (23, 84). Tripathy A et al. (85) indicated that RA patients express functional P2X4 and P2X7 receptors on peripheral CD8+T cells which when ligate with ATP produce high amounts of GzmB. When the ATP molecules induce purinergic signaling and activate T cells via P2X receptors (86), the excess extracellular ATP acts as a self-adjuvant to generate abnormal immune responses (87) and triggers inflammation (88, 89). In the case of RA, the release of ATP and its downstream binding to the purinergic receptors is a key regulator of the inflammatory activity (90, 91). The CD8+T cells from RA patients released significant amounts of GzmB in comparison to the CD8+T cells from HCs when stimulated with extracellular ATP. Moreover, the CD8+T cells from RA patients were increasingly activated over time and hence released greater concentrations of GzmB. GzmB is a specific activation marker protein for CD8+T cells. It thus implies that the excess extracellular ATP in the plasma of RA patients can activate immune cells rapidly and hence can be afflictive for the patients.
Goldbach-Mansky R et al. (92) explored the diagnostic and prognostic value of serum GzmB in patients with a diverse spectrum of early inflammatory arthritis and found that GzmB concentrations were significantly higher in rheumatoid factor positive (RF +) RA than those in RF-RA. Patients with joint erosions had significantly higher levels of GzmB than those without, indicating the independent value of GzmB in the prediction of erosive disease. GZMB+CD4 and CD8 CTL cells have also been found to be upregulated in the peripheral blood of active patients with RA (93), potentially reflecting an autoimmune response. Elevated levels of GzmB in blood may result from extracellular GzmB not taken up by the receptor during the induction of apoptotic cell death (92).
GzmB is a multifunctional proinflammatory molecule (94). It can process and activate proinflammatory, pro-fibrotic, and senescence mediators belonging to the IL-1 cytokine family (95, 96). GzmB can process IL-1α into potent proinflammatory fragments, enhancing inflammation. It stimulates interstitial collagenase production by fibroblasts and ECM remodeling, thereby regulating both, normal and aberrant tissue repair (96, 97). Among proinflammatory cytokines, IL-1α/β and TNF-α can trigger the intracellular molecular signaling pathway responsible for RA pathogenesis, which activates mesenchymal cells and synoviocytes and recruits innate and adaptive immune system cells. Synoviocytes, in turn, activate various mediators, including TNF-α, IL-1, IL-6, and IL-8, resulting in synovium inflammation, increased angiogenesis, and decreased lymphangiogensis (98). Therefore, GzmB may be involved in the inflammatory response of RA by regulating IL-1α expression. The role of GzmB in bone destruction in RA has also been suggested by other studies. Single nucleotide polymorphisms in the GzmB gene have been found to influence the joint destruction rate of RA (99). H. K. Ronday et al. (100)found that GzmB can degrade proteoglycan components in cartilage and contribute to the destruction of articular cartilage in RA. Additionally, GzmB is a potential biomarker for RA diagnosis, with higher levels of GzmB in serum being correlated with increased disease activity as measured by the DAS28-CRP score (101). In summary, GzmB may contribute to inflammation and joint destruction associated with RA through its proinflammatory and tissue-degrading effects.
While several studies have reported the role of GzmB as a proinflammatory molecule in the progression of RA proinflammatory, Xu et al. (102) found the frequency of GzmB production by regulatory B cells (Bregs) in patients with RA to be significantly reduced compared to that in healthy controls. The expression of IL-21 receptor in B cells in patients with RA was also significantly reduced, which may contribute to the reduction in GzmB-producing Bregs in these patients. Further analysis showed that the number of GzmB-producing Bregs was negatively correlated with erythrocyte sedimentation rate tender joint count, and disease activity score DAS28. The number of GzmB-producing Bregs increased significantly after RA treatment. A reduction in Bregs, especially those that produce IL-10 has been shown to be negatively correlated with disease activity in RA (103). Those cells may help maintain immune balance by inhibiting proinflammatory cytokine production and T cell differentiation (104). Whether GzmB has cell-specific functional differences remains to be determined.
GzmH and GzmB are structurally similar with 71% amino acid identity and belong to a gene cluster located on chromosome 14, which also includes cathepsin G and mast cell chymase. Although they have high sequence homology, these enzymes have distinct enzymatic activities (105). GzmH has not been detected in NKT cells, monocytes, or neutrophils (106). High levels of human GzmH mRNA have been found in the peripheral blood lymphocytes, lungs, spleen, and thymus (107, 108). Hou et al. (109) discovered that GzmH can induce rapid apoptosis in target cells, resulting in mitochondrial damage, nuclear condensation, and DNA breakage. GzmH-induced apoptosis depends on caspase activation and cytochrome c release. To date, no research has been conducted on GzmH in the context of RA. GzmH is predominantly expressed at high levels in NK cells (110), and GzmH mRNA has also been detected in activated human T cells (107, 111). IL-15 has significantly higher levels in the serum and synovial fluid of patients with RA than those with OA and healthy control groups (112), and plays key roles in promoting activation of NK and CD8 T cells (113). Zhang B et al. (114) stimulated NK-92 cells with IL-15, and it was found that IL-15 significantly up-regulated GzmA and GzmB gene expression, but GzmH transcripts were down-regulated. Therefore, higher levels of IL-15 in patients with RA might regulate Gzms expression. However, the specific role of GzmH in RA requires further investigation, and IL-15 may be a potential target to focus on.
GzmK- and GzmM-mediated cytokine-based inflammation in RA
GzmK is a trypsin-like molecule in the granzyme family that is expressed by CTLs, NKT, γδ T cells, and CD56bright+ NK cells (110, 115–117). Besides being a member of the granzyme family, little is known about the function of GzmK (118). In vitro studies have demonstrated that GzmK can induce non-apoptotic cell death through the production of reactive oxygen species (ROS) and mitochondrial dysfunction when combined with perforin (119). Further studies have shown that GzmK activates caspase-independent apoptosis by cleaving the SET complex, leading to SET destruction. This results in unleashing GzmA-activated DNase NM23H1, which translocates to the nucleus and nicks DNA (120). GzmK may also cleave the tumor suppressor p53, thus sensitizing tumor cells for apoptosis induction (121) and process a vasolin-containing protein, thus contributing to endoplasmic reticulum stress and caspase-independent cytotoxicity (122). GzmK inhibits influenza virus replication in mice (123) and has an immunoregulatory function in multiple sclerosis (124).Cooper DM et al. (125) demonstrated GzmK-induced activation of both ERK1/2 and p38 MAP kinase signaling pathways and significantly increased fibroblast proliferation in patients with sepsis and acute lung inflammation. Wensink AC et al. (126) demonstrated that extracellular GzmK potentiates the lipopolysaccharide-induced release of inflammatory cytokines from monocytes and that this effect is independent of the catalytic activity of GzmK.
GzmK levels in synovial tissue samples from patients with RA are higher than the levels in those with OA (127). GzmK may have proinflammatory effects and can activate PAR-1, a family of G protein-coupled receptors that mediate the physiological response to serine proteases (125, 128). PAR-1 is activated by thrombin and trypsin and can induce the production of inflammatory cytokines, such as TNF-α, IL-1, IL-6, and monocyte chemotactic protein 1 (129, 130). CD8T cells primarily release GzmK, whereas CD4 T cells primarily release GzmB (131). In the context of RA synovium inflammation, GzmK can act as a key inflammatory agent, inducing synovial fibroblasts to activate proinflammatory pathways, including IL-6, CCL2, and ROS production. This effect does not require perforin or any other agent to induce internalization of GzmK, indicating that GzmK has a proteolytic target on the surface of these cells (129). The protease activity of GzmK can also promote degradation of the ECM, leading to inflammatory cell infiltration and tissue destruction. Blocking GzmK or cytokines that activate CD8T cells, such as IL-12 or IL-15, may be an effective treatment for RA. Anti-citrullinated protein antibody-negative (ACPA-negative) RA comprises up to one-third of patients with RA, whereas lack of biomarkers in ACPA-negative RA poses a big challenge to early diagnosis (132). Lu J et al. (133) reintegrated across the GSE89408 dataset to evaluate the performance of GzmK in the diagnosis of ACPA-negative RA. The expression levels of GzmK in the ACPA-negative RA group were significantly higher than that in the normal and OA groups, and the area under the curve of GzmK expression level was 0.916, suggesting its potential as a biomarker.
GzmM is a trypsin-folding serine protease found specifically in the granules of NK cells (134). High levels of GzmM protein and mRNA have been detected in NK, NKT, γδT, and CD8+T cells (118). Studies have shown that human GzmM promotes cell death in a manner similar to GzmB, including caspase-3 activation, DNA fragmentation, ROS production, and the mitochondrial release of cytochrome c (135, 136). Cytoskeletal components, such as α-tubulin and ezrin, nucleolar phosphoprotein nucleophosmin, and apoptosis-associated p21-activated protein kinase 2, have been identified as direct GzmM subunits and are cleaved during GzmM-induced cell death and cytotoxic lymphocyte-induced cell death (137, 138). Synovial fluid-derived mononuclear cells show GzmM expression, with the highest expression in CTLs and NK cells. Elevated levels of GzmM in synovial fluid from patients with RA compared to OA controls have been shown to stimulate human fibroblasts to release IL-29, a proinflammatory cytokine, and type III interferon (IFN-λ1), suggesting that GzmM may play a local role in the pathophysiology of RA (139). Further studies are needed to fully understand the specific role of GzmM in RA.
Perspectives and challenges
Gzm-inhibiting serpins are believed to act as a fail-safe mechanism for CLs to avoid self-injury during granule exocytosis (140). In recent years, the prevailing theory has been that although circulating Gzms might not be able to enter cells without a high local perforin concentration to induce cell death, they could proteolyze cell surface receptors or extracellular proteins to cause destruction. Particularly when Gzms present at high concentrations at inflamed sites in the absence of natural inhibitors (24). To date, SERPINB12, SERPINB9, SERPINB4, SERPINB1, and inter-alpha inhibitor proteins, have been identified as intracellular inhibitors of GzmA, GzmB, GzmM, GzmH, and GzmK (141–146). Although physiological inhibitors of Gzms are known, no clinical trials have been reported for their use as treatments. Researchers believe that the development of GzmA inhibitors for the treatment of RA may have beneficial effects compared to other commonly used anti-inflammatory drugs, such as corticosteroids or TNF blockers (37). Some studies have suggested that cyclosporine and zidovudine may be potential target drugs for RA treatment in combination with GzmA (43). Zidovudine was developed as an anti-cancer agent in the 1960s and was later approved by the US FDA as an anti-HIV therapeutic drug in the late 1980s after fast track clinical trials (147). Nowadays, this drug is commonly used in the prevention of perinatal HIV-1 transmission (vertical transmission) that consists of the use of this drug by the mother before and during delivery, and treatment of the newborn (148). New potential inhibitors of GzmB, such as tannic acid, mupirocin, cefpiramide, xenazoic acid, vidarabine and phytonadiol sodium diphosphate, have been identified (149). Mi-Sun Kim.et al (150) developed a novel class of weak small-molecule inhibitors against human GzmB by docking studies employing binding site hot spots and three constraints (hydrogen bonding with Arg226, and hydrophobic interactions for S2 and S4 subsites) based on computational solvent mapping using FTMAP. The most distinctive compounds identified were thiazolidinediones 8 (IC50 = 25 μM) and 9 (IC50 = 28 μM), triazole 6 (IC50 = 44 μM), and diazolidinedione 7 (IC50 = 44 μM). Ikram S et al. (151) identified 12 potential inhibitors of GzmH from two separate databases of small molecules. Currently, there are no Gzms inhibitors that are specifically approved for the treatment of RA. Understanding of the precise mechanisms by which granzymes contribute to the development and progression of RA is limited. Lack of clear evidence demonstrating that targeting granzymes is a viable therapeutic strategy for RA. RA is a complex and heterogeneous disease, and it is not clear whether the same granzyme-mediated mechanisms are involved in all patients with RA. There is currently no single, reliable biomarker to indicate patterns of Gzms in patients with RA. Given the intracellular, extracellular, and proinflammatory effects of Gzms on RA, Gzms and their physiological inhibitors may be potential therapeutic targets for RA treatment. It is worth noting that extracellular vesicles (EVs), as one of the important communication carriers between cells and host, may also be a potential contact media between the Gzms family and RA (152). EVs containing granzyme from NK cells and CTLs require Ca2+-dependent signals to release (153). The EVs from activated NK cells include a variety of Gzms, such as GzmA and GzmB, which have cytotoxic effects on tumor cells (154), inhibit cell proliferation, and promote cell death. They are considered a safe and effective immunosuppressive agent, which may have potential therapeutic significance for RA FLS (154, 155).
Discussion
In this review, we described the physiological function, cellular expression, and potential role of five members of the Gzms family in RA (Table 1). Gzms are involved in the induction of apoptotic cell death. In RA, Gzms demonstrate non-cytotoxic activities that include diverse biological effects, such as stimulation of proinflammatory cytokines and remodeling of extracellular matrices. Considering the extracellular and intracellular functions of Gzms, they have the potential to contribute to the pathogenesis of inflammatory diseases (Figure 1). First, GzmA level is significantly elevated in plasma and synovial fluid and can degrade ECM proteins, potentially contributing to bone destruction in RA. Higher levels of GzmB in serum are correlated with increased disease activity. GzmB can degrade proteoglycan components in cartilage and contribute to the destruction of articular cartilage in RA. A subgroup of B cells, Bregs that express GzmB, may inhibit proinflammatory cytokine production and abnormal autoimmune T cell differentiation in patients with RA. GzmM promotes inflammation mainly by stimulating the release of the proinflammatory cytokine IL-29 and is elevated in RA. GzmK is mainly associated with endothelial cells and fibroblasts, suggesting its role in abnormal angiogenesis and synovial hyperplasia in RA. However, the specific role of GzmH in RA requires further investigation. Our search for the latest clinical trials showed that few clinical inhibitors of Gzms have been identified. While the development of clinical drugs targeting the Gzms family is limited, evidence suggests that targeting these proteins may have potential value for the clinical treatment and management of RA. To further enhance our understanding of Gzms in RA, comprehensive use of molecular biology, cellular immunology, and other technologies is necessary. Notably, Gzms primarily play a biological role in cell perforation and target cells. The multiple potential roles of Gzms in RA may include an abnormal manifestation of uncontrolled or excessive cell death. Additionally, the known extracellular activities of Gzms suggest a proinflammatory effect in RA. Therefore, further research on the association between multiple cell death pathways and RA, and experiments defining Gzm-activated proinflammatory pathways may be a promising direction to determine the significance of Gzms as a proinflammatory mediator in future studies.
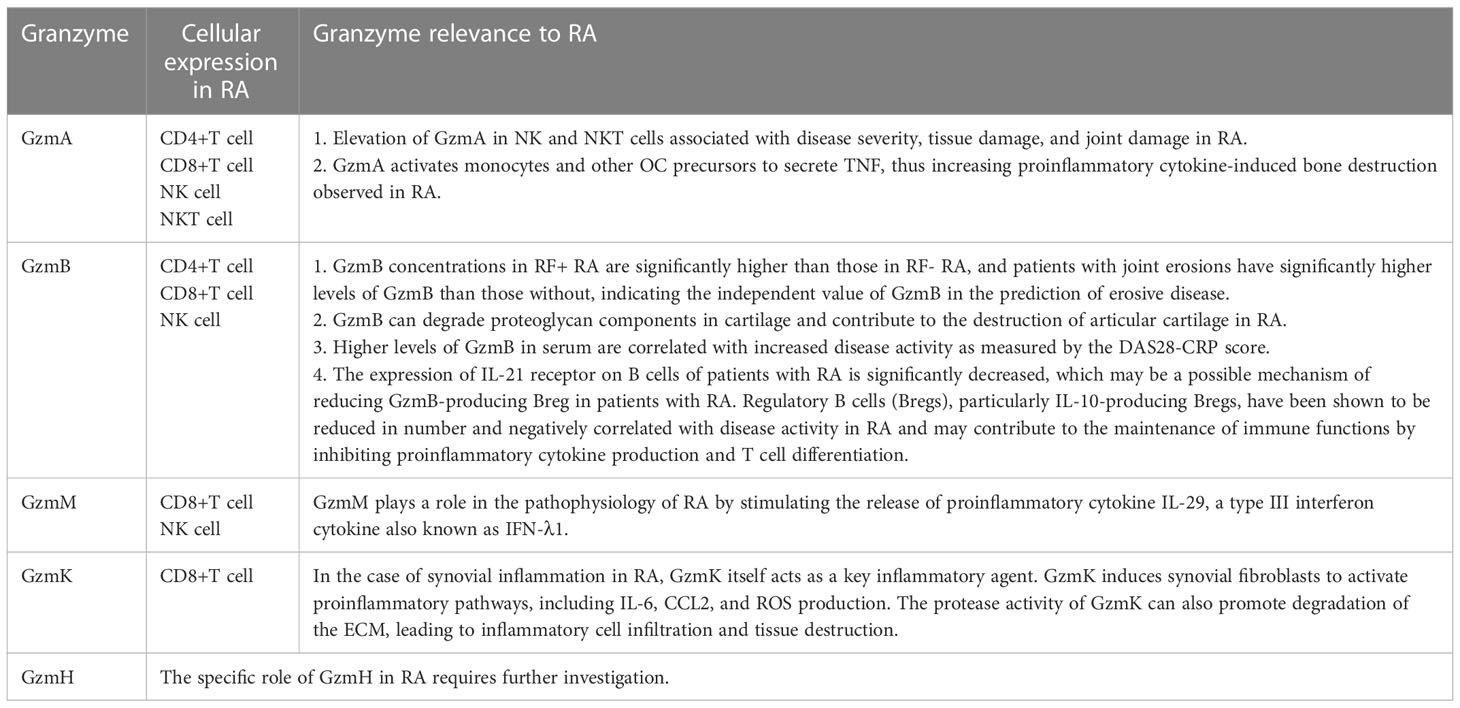
Table 1 The physiological function, cellular expression, and potential role of the Gzms family in RA.
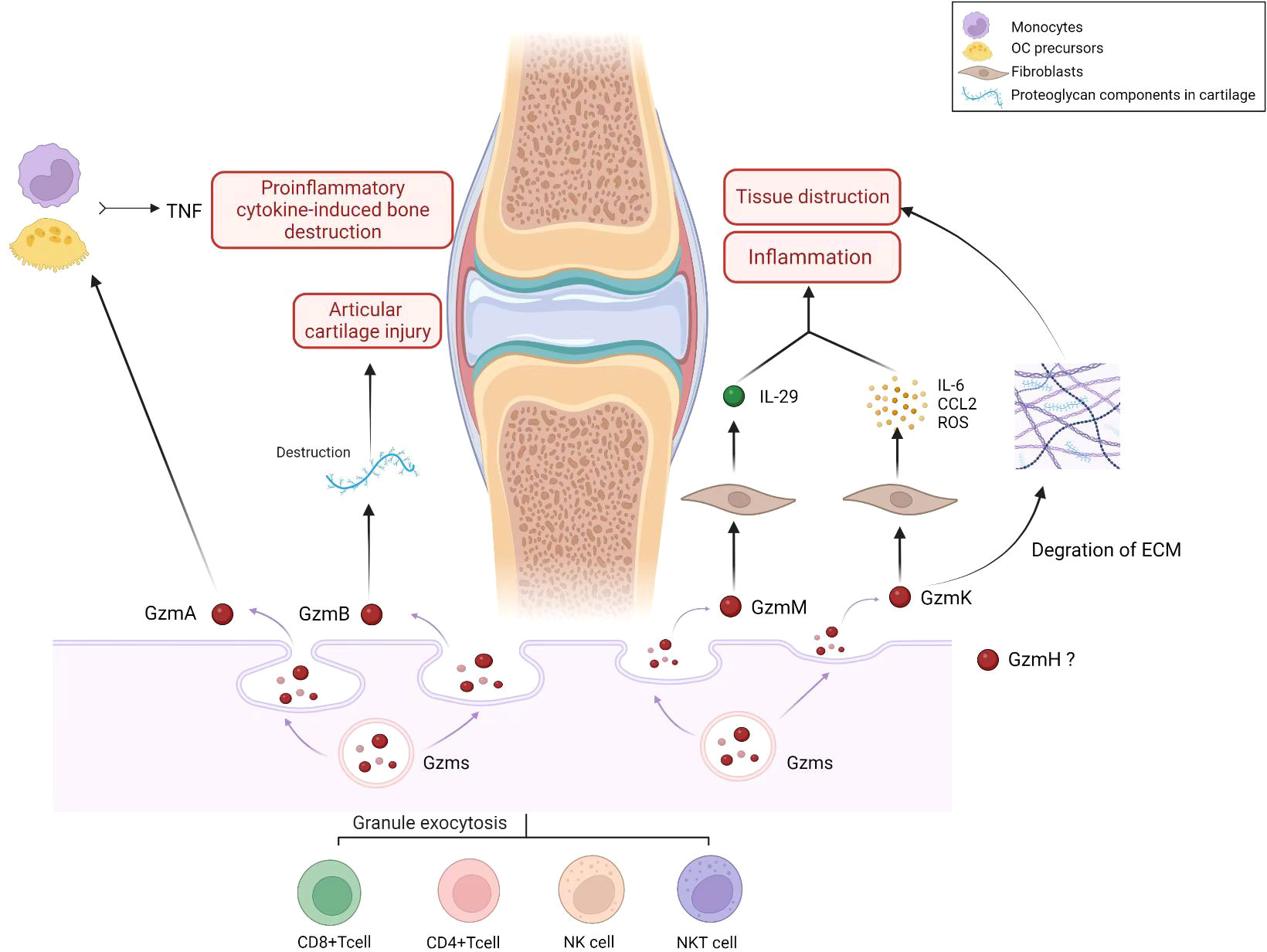
Figure 1 Role of Granzyme family (GzmA, GzmB, GzmH, GzmM, GzmK) in pathology and progression of RA. Gzms are produced and released by immune cells, such as cytotoxic T cells and natural killer cells. They play a role in the ability of the immune system to recognize and eliminate infected or damaged cells. The granzyme family includes several granzyme types, including granzyme A (GzmA), granzyme B (GzmB), granzyme H (GzmH), granzyme M (GzmM), and granzyme K (GzmK). In the context of RA, Gzms may contribute to the pathology and progression of the disease in several ways. RA is an autoimmune disorder characterized by chronic inflammation of the joints that leads to joint damage and deformity. Gzms may contribute to the inflammation and joint damage observed in RA by inducing programmed cell death (apoptosis) in cells within the joint tissue. This can lead to the destruction of joint cartilage and bone, resulting in joint deformity and loss of function. As the figure shows, GzmA activates monocytes and other OC precursors to secrete TNF, thus increasing proinflammatory cytokine-induced bone destruction observed in RA. GzmB can degrade proteoglycan components in cartilage and contribute to the destruction of articular cartilage in RA. GzmM plays a role in the pathophysiology of RA by stimulating the release of proinflammatory cytokine IL-29. GzmK induces synovial fibroblasts to activate proinflammatory pathways, including IL-6, CCL2, and ROS production. The protease activity of GzmK can also promote degradation of the ECM, leading to inflammatory cell infiltration and tissue destruction. (Created with BioRender.com).
Author contributions
YZ and JZ is responsible for the collection, collation, and writing of the original manuscript. YS is responsible for the collection. SG, SS, and DH are responsible for the concept development, revision, and manuscript review. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Funds of China (82074234 and 82071756), National Key Research and Development Project (2018YFC1705200 and 2018YFC1705203), Shanghai Chinese Medicine Development Office, National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology, State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, East China Region-based Chinese and Western Medicine Joint Disease Specialist Alliance, and Shanghai He Dongyi Famous Chinese Medicine Studio Construction Project (SHGZS-202220). Shanghai 13th Five-YearKey Specialized College—Department of Osteoarthritis and Arthritis of Integrated Traditional Chinese and Western Medicine (shslczdzk04801).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RA, Rheumatoid arthritis; CD, Cluster of Differentiation; Gzms, Granzymes; GzmA, Granzyme A; GzmB, Granzyme B; GzmH, Granzyme H; GzmK, Granzyme K; GzmM, Granzyme M; CLs, Cytotoxic lymphocytes; CTLs, Cytotoxic T lymphocytes; NK, Natural killer; NKT, Natural killer T; IL-1b, Interleukin-1b; TLR, Toll-like receptor; TNF, Tumor necrosis factor; RF, Rheumatoid factor; IL-6, Interleukin-6; IL-8, Interleukin-8; IL-1, Interleukin-1; IL-10, Interleukin-10; Il-15, Interleukin-15; IL-29, Interleukin-29; PAR, Proteinase-activated receptor; GSDMB, Gasdermin B; OA, Osteoarthritis; DAS28, Disease activity score; OCs, Proliferation Osteoclasts; ECM, Extracellular matrix; ACPAnegative, Anti-citrullinated protein antibody-negative; EVs, Extracellular vesicles; ROS, Reactive oxygen species.
References
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388(10055):2023–38. doi: 10.1016/s0140-6736(16)30173-8
2. Zhao J, Guo S, Schrodi SJ, He D. Molecular and cellular heterogeneity in rheumatoid arthritis: Mechanisms and clinical implications. Front Immunol (2021) 12:790122. doi: 10.3389/fimmu.2021.790122
3. Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, et al. The global burden of rheumatoid arthritis: Estimates from the global burden of disease 2010 study. Ann Rheum Dis (2014) 73(7):1316–22. doi: 10.1136/annrheumdis-2013-204627
4. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
5. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. (2014) 506(7488):376–81. doi: 10.1038/nature12873
6. Lenz TL, Deutsch AJ, Han B, Hu X, Okada Y, Eyre S, et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat Genet (2015) 47(9):1085–90. doi: 10.1038/ng.3379
7. Zhao J, Wei K, Chang C, Xu L, Jiang P, Guo S, et al. DNA Methylation of T lymphocytes as a therapeutic target: Implications for rheumatoid arthritis etiology. Front Immunol (2022) 13:863703. doi: 10.3389/fimmu.2022.863703
8. Chang C, Xu L, Zhang R, Jin Y, Jiang P, Wei K, et al. MicroRNA-mediated epigenetic regulation of rheumatoid arthritis susceptibility and pathogenesis. Front Immunol (2022) 13:838884. doi: 10.3389/fimmu.2022.838884
9. Wei K, Jiang P, Zhao J, Jin Y, Zhang R, Chang C, et al. Biomarkers to predict DMARDs efficacy and adverse effect in rheumatoid arthritis. Front Immunol (2022) 13:865267. doi: 10.3389/fimmu.2022.865267
10. Zhao J, Wei K, Jiang P, Chang C, Xu L, Xu L, et al. G-Protein-Coupled receptors in rheumatoid arthritis: Recent insights into mechanisms and functional roles. Front Immunol (2022) 13:907733. doi: 10.3389/fimmu.2022.907733
11. Zhao J, Xu L, Chang C, Jiang P, Wei K, Shi Y, et al. Circulating methylation level of HTR2A is associated with inflammation and disease activity in rheumatoid arthritis. Front Immunol (2022) 13:1054451. doi: 10.3389/fimmu.2022.1054451
12. Zhang R, Jin Y, Chang C, Xu L, Bian Y, Shen Y, et al. RNA-Seq and network analysis reveal unique chemokine activity signatures in the synovial tissue of patients with rheumatoid arthritis. Front Med (Lausanne). (2022) 9:799440. doi: 10.3389/fmed.2022.799440
13. Zhao J, Guo S, Schrodi SJ, He D. Absent in melanoma 2 (AIM2) in rheumatoid arthritis: novel molecular insights and implications. Cell Mol Biol Lett (2022) 27(1):108. doi: 10.1186/s11658-022-00402-z
14. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol (2019) 20(7):928–42. doi: 10.1038/s41590-019-0378-1
15. Chowdhury D, Lieberman J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu Rev Immunol (2008) 26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404
16. Garcia-Sanz JA, MacDonald HR, Jenne DE, Tschopp J, Nabholz M. Cell specificity of granzyme gene expression. J Immunol (1990) 145(9):3111–8. doi: 10.4049/jimmunol.145.9.3111
17. Anthony DA, Andrews DM, Watt SV, Trapani JA, Smyth MJ. Functional dissection of the granzyme family cell death and inflammation. Immunol Rev (2010) 235(1):73–92. doi: 10.1111/j.0105-2896.2010.00907.x
18. Dotiwala F, Lieberman J. Granulysin: Killer lymphocyte safeguard against microbes. Curr Opin Immunol (2019) 60:19–29. doi: 10.1016/j.coi.2019.04.013
19. Yu R, Zhang J, Zhuo Y, Hong X, Ye J, Tang S, et al. Identification of diagnostic signatures and immune cell infiltration characteristics in rheumatoid arthritis by integrating bioinformatic analysis and machine-learning strategies. Front Immunol (2021) 12:724934. doi: 10.3389/fimmu.2021.724934
20. Buzza MS, Bird PI. Extracellular granzymes: Current perspectives. Biol Chem (2006) 387(7):827–37. doi: 10.1515/BC.2006.106
21. Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme b in immunity and disease: Challenging the dogma. Lab Invest. (2009) 89(11):1195–220. doi: 10.1038/labinvest.2009.91
22. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular matrix remodeling by human granzyme b via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem (2005) 280(25):23549–58. doi: 10.1074/jbc.M412001200
23. Tak PP, Spaeny-Dekking L, Kraan MC, Breedveld FC, Froelich CJ, Hack CE. The levels of soluble granzyme a and b are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA). Clin Exp Immunol (1999) 116(2):366–70. doi: 10.1046/j.1365-2249.1999.00881.x
24. Lieberman J. Granzyme a activates another way to die. Immunol Rev (2010) 235(1):93–104. doi: 10.1111/j.0105-2896.2010.00902.x
25. Wu CH, Li J, Li L, Sun J, Fabbri M, Wayne AS, et al. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J Extracell Vesicles. (2019) 8(1):1588538. doi: 10.1080/20013078.2019.1588538
26. Gordy LE, Bezbradica JS, Flyak AI, Spencer CT, Dunkle A, Sun J, et al. IL-15 regulates homeostasis and terminal maturation of NKT cells. J Immunol (2011) 187(12):6335–45. doi: 10.4049/jimmunol.1003965
27. Suhrbier A, Fernan A, Burrows SR, Saul A, Moss DJ. BLT esterase activity as an alternative to chromium release in cytotoxic T cell assays. J Immunol Methods (1991) 145(1-2):43–53. doi: 10.1016/0022-1759(91)90309-4.
28. Della-Torre E, Bozzalla-Cassione E, Sciorati C, Ruggiero E, Lanzillotta M, Bonfiglio S, et al. A CD8α- subset of CD4+SLAMF7+ cytotoxic T cells is expanded in patients with IgG4-related disease and decreases following glucocorticoid treatment. Arthritis Rheumatol (2018) 70(7):1133–43. doi: 10.1002/art.40469
29. Muraro E, Merlo A, Martorelli D, Cangemi M, Dalla Santa S, Dolcetti R, et al. Fighting viral infections and virus-driven tumors with cytotoxic CD4(+) T cells. Front Immunol (2017) 8:197. doi: 10.3389/fimmu.2017.00197
30. Hildebrand D, Bode KA, Rieß D, Cerny D, Waldhuber A, Römmler F, et al. Granzyme a produces bioactive IL-1β through a nonapoptotic inflammasome-independent pathway. Cell Rep (2014) 9(3):910–7. doi: 10.1016/j.celrep.2014.10.003
31. Wensink AC, Kok HM, Meeldijk J, Fermie J, Froelich CJ, Hack CE, et al. Granzymes a and K differentially potentiate LPS-induced cytokine response. Cell Death Discovery (2016) 2:16084. doi: 10.1038/cddiscovery.2016.84
32. Brunner G, Simon MM, Kramer MD. Activation of pro-urokinase by the human T cell-associated serine proteinase HuTSP-1. FEBS Lett (1990) 260(1):141–4. doi: 10.1016/0014-5793(90)80087-y
33. Suidan HS, Bouvier J, Schaerer E, Stone SR, Monard D, Tschopp J. Granzyme a released upon stimulation of cytotoxic T lymphocytes activates the thrombin receptor on neuronal cells and astrocytes. Proc Natl Acad Sci USA (1994) 91(17):8112–6. doi: 10.1073/pnas.91.17.8112
34. Suidan HS, Clemetson KJ, Brown-Luedi M, Niclou SP, Clemetson JM, Tschopp J, et al. The serine protease granzyme a does not induce platelet aggregation but inhibits responses triggered by thrombin. Biochem J (1996) 315(Pt 3):939–45. doi: 10.1042/bj3150939.
35. Hansen KK, Sherman PM, Cellars L, Andrade-Gordon P, Pan Z, Baruch A, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci USA (2005) 102(23):8363–8. doi: 10.1073/pnas.0409535102
36. Sower LE, Froelich CJ, Allegretto N, Rose PM, Hanna WD, Klimpel GR. Extracellular activities of human granzyme a. monocyte activation by granzyme a versus alpha-thrombin. J Immunol (1996) 156(7):2585–90. doi: 10.4049/jimmunol.156.7.2585
37. Santiago L, Menaa C, Arias M, Martin P, Jaime-Sanchez P, Metkar S, et al. Granzyme a contributes to inflammatory arthritis in mice through stimulation of osteoclastogenesis. Arthritis Rheumatol (2017) 69(2):320–34. doi: 10.1002/art.39857
38. Chemin K, Gerstner C, Malmström V. Effector functions of CD4+ T cells at the site of local autoimmune inflammation-lessons from rheumatoid arthritis. Front Immunol (2019) 10:353. doi: 10.3389/fimmu.2019.00353
39. Cheuk S, Martini E, Bergh K, Chang D, Rethi B, Ståhle M, et al. Granzyme a potentiates chemokine production in IL-17-stimulated keratinocytes. Exp Dermatol (2017) 26(9):824–7. doi: 10.1111/exd.13284
40. Jaime P, García-Guerrero N, Estella R, Pardo J, García-Álvarez F, Martinez-Lostao L. CD56(+)/CD16(-) natural killer cells expressing the inflammatory protease granzyme a are enriched in synovial fluid from patients with osteoarthritis. Osteoarthritis Cartilage. (2017) 25(10):1708–18. doi: 10.1016/j.joca.2017.06.007
41. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme a from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
42. Spaeny-Dekking EH, Hanna WL, Wolbink AM, Wever PC, Kummer JA, Swaak AJ, et al. Extracellular granzymes a and b in humans: Detection of native species during CTL responses in vitro and in vivo. J Immunol (1998) 160(7):3610–6. doi: 10.4049/jimmunol.160.7.3610
43. Wu B, He Y, Yang D, Liu RX. Identification of hub genes and therapeutic drugs in rheumatoid arthritis patients. Clin Rheumatol (2021) 40(8):3299–309. doi: 10.1007/s10067-021-05650-6
44. Takayama H, Trenn G, Humphrey W Jr., Bluestone JA, Henkart PA, Sitkovsky MV. Antigen receptor-triggered secretion of a trypsin-type esterase from cytotoxic T lymphocytes. J Immunol (1987) 138(2):566–9. doi: 10.4049/jimmunol.138.2.566
45. Chu CQ, Field M, Allard S, Abney E, Feldmann M, Maini RN. Detection of cytokines at the cartilage/pannus junction in patients with rheumatoid arthritis: Implications for the role of cytokines in cartilage destruction and repair. Br J Rheumatol (1992) 31(10):653–61. doi: 10.1093/rheumatology/31.10.653
46. Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheumatol (1997) 40(2):217–25. doi: 10.1002/art.1780400206
47. Rathanaswami P, Hachicha M, Wong WL, Schall TJ, McColl SR. Synergistic effect of interleukin-1 beta and tumor necrosis factor alpha on interleukin-8 gene expression in synovial fibroblasts. evidence that interleukin-8 is the major neutrophil-activating chemokine released in response to monokine activation. Arthritis Rheumatol (1993) 36(9):1295–304. doi: 10.1002/art.1780360914
48. Osińska I, Popko K, Demkow U. Perforin: An important player in immune response. Cent Eur J Immunol (2014) 39(1):109–15. doi: 10.5114/ceji.2014.42135
49. Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol (2006) 6(12):940–52. doi: 10.1038/nri1983
50. Griffiths GM, Alpert S, Lambert E, McGuire J, Weissman IL. Perforin and granzyme a expression identifying cytolytic lymphocytes in rheumatoid arthritis. Proc Natl Acad Sci USA (1992) 89(2):549–53. doi: 10.1073/pnas.89.2.549
51. Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheumatol (1998) 41(12):2108–16. doi: 10.1002/1529-0131(199812)41:12
52. Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, et al. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheumatol (2002) 46(11):2878–83. doi: 10.1002/art.10622
53. Aggarwal A, Sharma A, Bhatnagar A. Role of cytolytic impairment of natural killer and natural killer T-cell populations in rheumatoid arthritis. Clin Rheumatol (2014) 33(8):1067–78. doi: 10.1007/s10067-014-2641-z
54. Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Activation of NK cells by CC chemokines. chemotaxis, Ca2+ mobilization, and enzyme release. J Immunol (1996) 156(1):322–7. doi: 10.4049/jimmunol.156.1.322
55. Ruth JH, Rottman JB, Katschke KJ Jr., Qin S, Wu L, LaRosa G, et al. Selective lymphocyte chemokine receptor expression in the rheumatoid joint. Arthritis Rheumatol (2001) 44(12):2750–60. doi: 10.1002/1529-0131(200112)44:12<2750
56. Haringman JJ, Ludikhuize J, Tak PP. Chemokines in joint disease: The key to inflammation? Ann Rheum Dis (2004) 63(10):1186–94. doi: 10.1136/ard.2004.020529
57. Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev (1999) 20(3):345–57. doi: 10.1210/edrv.20.3.0367
58. Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ (2012) 19(1):28–35. doi: 10.1038/cdd.2011.153
59. Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC. Granzyme b: A natural born killer. Immunol Rev (2003) 193:31–8. doi: 10.1034/j.1600-065x.2003.00044.x
60. Ida H, Utz PJ, Anderson P, Eguchi K. Granzyme b and natural killer (NK) cell death. Mod Rheumatol (2005) 15(5):315–22. doi: 10.1007/s10165-005-0426-6
61. Jans DA, Jans P, Briggs LJ, Sutton V, Trapani JA. Nuclear transport of granzyme b (fragmentin-2). Dependence of perforin in vivo and cytosolic factors in vitro. J Biol Chem (1996) 271(48):30781–9. doi: 10.1074/jbc.271.48.30781
62. Thomas DA, Du C, Xu M, Wang X, Ley TJ. DFF45/ICAD can be directly processed by granzyme b during the induction of apoptosis. Immunity. (2000) 12(6):621–32. doi: 10.1016/s1074-7613(00)80213-7
63. Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme b directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. (1998) 8(4):451–60. doi: 10.1016/s1074-7613(00)80550-6
64. Froelich CJ, Hanna WL, Poirier GG, Duriez PJ, D'Amours D, Salvesen GS, et al. Granzyme b/perforin-mediated apoptosis of jurkat cells results in cleavage of poly(ADP-ribose) polymerase to the 89-kDa apoptotic fragment and less abundant 64-kDa fragment. Biochem Biophys Res Commun (1996) 227(3):658–65. doi: 10.1006/bbrc.1996.1565
65. Zhang D, Beresford PJ, Greenberg AH, Lieberman J. Granzymes a and b directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc Natl Acad Sci USA (2001) 98(10):5746–51. doi: 10.1073/pnas.101329598
66. Hendel A, Hiebert PR, Boivin WA, Williams SJ, Granville DJ. Granzymes in age-related cardiovascular and pulmonary diseases. Cell Death Differ (2010) 17(4):596–606. doi: 10.1038/cdd.2010.5
67. Jin J, Li X, Hu B, Kim C, Cao W, Zhang H, et al. FOXO1 deficiency impairs proteostasis in aged T cells. Sci Adv (2020) 6(17):eaba1808. doi: 10.1126/sciadv.aba1808
68. Bulati M, Buffa S, Martorana A, Candore G, Lio D, Caruso C, et al. Trafficking phenotype and production of granzyme b by double negative b cells (IgG(+)IgD(-)CD27(-)) in the elderly. Exp Gerontol. (2014) 54:123–9. doi: 10.1016/j.exger.2013.12.011
69. Kim WJ, Kim H, Suk K, Lee WH. Macrophages express granzyme b in the lesion areas of atherosclerosis and rheumatoid arthritis. Immunol Lett (2007) 111(1):57–65. doi: 10.1016/j.imlet.2007.05.004
70. Vernooy JH, Moller GM, van Suylen RJ, van Spijk MP, Cloots RH, Hoet PH, et al. Increased granzyme a expression in type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2007) 175(5):464–72. doi: 10.1164/rccm.200602-169OC
71. Pardo J, Wallich R, Ebnet K, Iden S, Zentgraf H, Martin P, et al. Granzyme b is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ (2007) 14(10):1768–79. doi: 10.1038/sj.cdd.4402183
72. Choy JC, McDonald PC, Suarez AC, Hung VH, Wilson JE, McManus BM, et al. Granzyme b in atherosclerosis and transplant vascular disease: Association with cell death and atherosclerotic disease severity. Mod Pathol (2003) 16(5):460–70. doi: 10.1097/01.MP.0000067424.12280.BC
73. Hernandez-Pigeon H, Jean C, Charruyer A, Haure MJ, Titeux M, Tonasso L, et al. Human keratinocytes acquire cellular cytotoxicity under UV-b irradiation. implication of granzyme b and perforin. J Biol Chem (2006) 281(19):13525–32. doi: 10.1074/jbc.M512694200
74. Horiuchi K, Saito S, Sasaki R, Tomatsu T, Toyama Y. Expression of granzyme b in human articular chondrocytes. J Rheumatol (2003) 30(8):1799–810.
75. Hu SX, Wang S, Wang JP, Mills GB, Zhou Y, Xu HJ. Expression of endogenous granzyme b in a subset of human primary breast carcinomas. Br J Cancer. (2003) 89(1):135–9. doi: 10.1038/sj.bjc.6601051
76. D'Eliseo D, Pisu P, Romano C, Tubaro A, De Nunzio C, Morrone S, et al. Granzyme b is expressed in urothelial carcinoma and promotes cancer cell invasion. Int J Cancer (2010) 127(6):1283–94. doi: 10.1002/ijc.25135
77. D'Eliseo D, Manzi L, Merendino N, Velotti F. Docosahexaenoic acid inhibits invasion of human RT112 urinary bladder and PT45 pancreatic carcinoma cells via down-modulation of granzyme b expression. J Nutr Biochem (2012) 23(5):452–7. doi: 10.1016/j.jnutbio.2011.01.010
78. Fang Y, Herrick EJ, Nicholl MB. A possible role for perforin and granzyme b in resveratrol-enhanced radiosensitivity of prostate cancer. J Androl. (2012) 33(4):752–60. doi: 10.2164/jandrol.111.015164
79. D'Eliseo D, Di Rocco G, Loria R, Soddu S, Santoni A, Velotti F. Epitelial-to-mesenchimal transition and invasion are upmodulated by tumor-expressed granzyme b and inhibited by docosahexaenoic acid in human colorectal cancer cells. J Exp Clin Cancer Res (2016) 35:24. doi: 10.1186/s13046-016-0302-6
80. Turner CT, Lim D, Granville DJ. Granzyme b in skin inflammation and disease. Matrix Biol (2019) 75-76:126–40. doi: 10.1016/j.matbio.2017.12.005
81. Hiebert PR, Granville DJ. Granzyme b in injury, inflammation, and repair. Trends Mol Med (2012) 18(12):732–41. doi: 10.1016/j.molmed.2012.09.009
82. Taylor RC, Cullen SP, Martin SJ. Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol Cell Biol (2008) 9(3):231–41. doi: 10.1038/nrm2312
83. Li WC, Bai L, Xu Y, Chen H, Ma R, Hou WB, et al. Identification of differentially expressed genes in synovial tissue of rheumatoid arthritis and osteoarthritis in patients. J Cell Biochem (2019) 120(3):4533–44. doi: 10.1002/jcb.27741
84. Tak PP, Kummer JA, Hack CE, Daha MR, Smeets TJ, Erkelens GW, et al. Granzyme-positive cytotoxic cells are specifically increased in early rheumatoid synovial tissue. Arthritis Rheumatol (1994) 37(12):1735–43. doi: 10.1002/art.1780371205
85. Tripathy A, Padhan P, Swain N, Raghav SK, Gupta B. Increased extracellular ATP in plasma of rheumatoid arthritis patients activates CD8(+)T cells. Arch Med Res (2021) 52(4):423–33. doi: 10.1016/j.arcmed.2020.12.010
86. Ferrero ME. A new approach to the inflammatory/autoimmune diseases. Recent Pat Antiinfect Drug Discovery (2009) 4(2):108–13. doi: 10.2174/157489109788490343
87. Scheckenbach KE, Crespin S, Kwak BR, Chanson M. Connexin channel-dependent signaling pathways in inflammation. J Vasc Res (2011) 48(2):91–103. doi: 10.1159/000316942
88. Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med (2007) 13(8):913–9. doi: 10.1038/nm1617
89. Shen X, Zhao T, Shi P. Plasma ATP increase is a biomarker of hypertension and triggers low-grade inflammation through P2X7 receptor. FASEB J (2019) 33(S1):692.2. doi: 10.1096/fasebj.2019.33.1_supplement.692.2
90. da Silva JLG, Passos DF, Bernardes VM, Leal DBR. ATP and adenosine: Role in the immunopathogenesis of rheumatoid arthritis. Immunol Lett (2019) 214:55–64. doi: 10.1016/j.imlet.2019.08.009
91. Baroja-Mazo A, Pelegrín P. Modulating P2X7 receptor signaling during rheumatoid arthritis: New therapeutic approaches for bisphosphonates. J Osteoporos (2012) 2012:408242. doi: 10.1155/2012/408242
92. Goldbach-Mansky R, Suson S, Wesley R, Hack CE, El-Gabalawy HS, Tak PP. Raised granzyme b levels are associated with erosions in patients with early rheumatoid factor positive rheumatoid arthritis. Ann Rheum Dis (2005) 64(5):715–21. doi: 10.1136/ard.2003.007039
93. Zeng X, Zheng M, Liu T, Bahabayi A, Song S, Alimu X, et al. Cytotoxic T lymphocytes expressing GPR56 are up-regulated in the peripheral blood of patients with active rheumatoid arthritis and reflect disease progression. Immunol Invest. (2022) 51(6):1804–19. doi: 10.1080/08820139.2022.2058403
94. Velotti F, Barchetta I, Cimini FA, Cavallo MG. Granzyme b in inflammatory diseases: Apoptosis, inflammation, extracellular matrix remodeling, epithelial-to-Mesenchymal transition and fibrosis. Front Immunol (2020) 11:587581. doi: 10.3389/fimmu.2020.587581
95. Wensink AC, Hack CE, Bovenschen N. Granzymes regulate proinflammatory cytokine responses. J Immunol (2015) 194(2):491–7. doi: 10.4049/jimmunol.1401214
96. Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr (2006) 83(2):447s–55s. doi: 10.1093/ajcn/83.2.447S
97. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol (2016) 38(4):517–34. doi: 10.1007/s00281-016-0559-z
98. Alam J, Jantan I, Bukhari SNA. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. BioMed Pharmacother (2017) 92:615–33. doi: 10.1016/j.biopha.2017.05.055
99. Knevel R, Krabben A, Wilson AG, Brouwer E, Leijsma MK, Lindqvist E, et al. A genetic variant in granzyme b is associated with progression of joint destruction in rheumatoid arthritis. Arthritis Rheumatol (2013) 65(3):582–9. doi: 10.1002/art.37808
100. Ronday HK, van der Laan WH, Tak PP, de Roos JA, Bank RA, TeKoppele JM, et al. Human granzyme b mediates cartilage proteoglycan degradation and is expressed at the invasive front of the synovium in rheumatoid arthritis. Rheumatol (Oxford). (2001) 40(1):55–61. doi: 10.1093/rheumatology/40.1.55
101. Colombo E, Scarsi M, Piantoni S, Tincani A, Airò P. Serum levels of granzyme b decrease in patients with rheumatoid arthritis responding to abatacept. Clin Exp Rheumatol (2016) 34(1):37–41. doi: 10.1136/annrheumdis-2015-eular.2362
102. Xu L, Liu X, Liu H, Zhu L, Zhu H, Zhang J, et al. Impairment of granzyme b-producing regulatory b cells correlates with exacerbated rheumatoid arthritis. Front Immunol (2017) 8:768. doi: 10.3389/fimmu.2017.00768
103. Daien CI, Gailhac S, Mura T, Audo R, Combe B, Hahne M, et al. Regulatory B10 cells are decreased in patients with rheumatoid arthritis and are inversely correlated with disease activity. Arthritis Rheumatol (2014) 66(8):2037–46. doi: 10.1002/art.38666
104. Rosser EC, Mauri C. Regulatory b cells: Origin, phenotype, and function. Immunity. (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
105. Fellows E, Gil-Parrado S, Jenne DE, Kurschus FC. Natural killer cell-derived human granzyme h induces an alternative, caspase-independent cell-death program. Blood. (2007) 110(2):544–52. doi: 10.1182/blood-2006-10-051649
106. Sedelies KA, Sayers TJ, Edwards KM, Chen W, Pellicci DG, Godfrey DI, et al. Discordant regulation of granzyme h and granzyme b expression in human lymphocytes. J Biol Chem (2004) 279(25):26581–7. doi: 10.1074/jbc.M312481200
107. Connerotte T, Van Pel A, Godelaine D, Tartour E, Schuler-Thurner B, Lucas S, et al. Functions of anti-MAGE T-cells induced in melanoma patients under different vaccination modalities. Cancer Res (2008) 68(10):3931–40. doi: 10.1158/0008-5472.Can-07-5898
108. Przetak MM, Yoast S, Schmidt BF. Cloning of cDNA for human granzyme 3. FEBS Lett (1995) 364(3):268–71. doi: 10.1016/0014-5793(95)00407-z
109. Hou Q, Zhao T, Zhang H, Lu H, Zhang Q, Sun L, et al. Granzyme h induces apoptosis of target tumor cells characterized by DNA fragmentation and bid-dependent mitochondrial damage. Mol Immunol (2008) 45(4):1044–55. doi: 10.1016/j.molimm.2007.07.032
110. Bade B, Boettcher HE, Lohrmann J, Hink-Schauer C, Bratke K, Jenne DE, et al. Differential expression of the granzymes a, K and m and perforin in human peripheral blood lymphocytes. Int Immunol (2005) 17(11):1419–28. doi: 10.1093/intimm/dxh320
111. Haddad P, Jenne D, Tschopp J, Clément MV, Mathieu-Mahul D, Sasportes M. Structure and evolutionary origin of the human granzyme h gene. Int Immunol (1991) 3(1):57–66. doi: 10.1093/intimm/3.1.57
112. Park MK, Her YM, Cho ML, Oh HJ, Park EM, Kwok SK, et al. IL-15 promotes osteoclastogenesis via the PLD pathway in rheumatoid arthritis. Immunol Lett (2011) 139(1-2):42–51. doi: 10.1016/j.imlet.2011.04.013
113. Zhang S, Zhao J, Bai X, Handley M, Shan F. Biological effects of IL-15 on immune cells and its potential for the treatment of cancer. Int Immunopharmacol. (2021) 91:107318. doi: 10.1016/j.intimp.2020.107318
114. Zhang B, Zhang J, Tian Z. Comparison in the effects of IL-2, IL-12, IL-15 and IFNalpha on gene regulation of granzymes of human NK cell line NK-92. Int Immunopharmacol (2008) 8(7):989–96. doi: 10.1016/j.intimp.2008.03.001
115. Bratke K, Kuepper M, Bade B, Virchow JC Jr., Luttmann W. Differential expression of human granzymes a, b, and K in natural killer cells and during CD8+ T cell differentiation in peripheral blood. Eur J Immunol (2005) 35(9):2608–16. doi: 10.1002/eji.200526122
116. Bade B, Lohrmann J, ten Brinke A, Wolbink AM, Wolbink GJ, ten Berge IJ, et al. Detection of soluble human granzyme K in vitro and in vivo. Eur J Immunol (2005) 35(10):2940–8. doi: 10.1002/eji.200526249
117. Bouwman AC, van Daalen KR, Crnko S, Ten Broeke T, Bovenschen N. Intracellular and extracellular roles of granzyme K. Front Immunol (2021) 12:677707. doi: 10.3389/fimmu.2021.677707
118. Bovenschen N, Kummer JA. Orphan granzymes find a home. Immunol Rev (2010) 235(1):117–27. doi: 10.1111/j.0105-2896.2010.00889.x
119. MacDonald G, Shi L, Vande Velde C, Lieberman J, Greenberg AH. Mitochondria-dependent and -independent regulation of granzyme b-induced apoptosis. J Exp Med (1999) 189(1):131–44. doi: 10.1084/jem.189.1.131
120. Zhao T, Zhang H, Guo Y, Zhang Q, Hua G, Lu H, et al. Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ (2007) 14(3):489–99. doi: 10.1038/sj.cdd.4402040
121. Hua G, Wang S, Zhong C, Xue P, Fan Z. Ignition of p53 bomb sensitizes tumor cells to granzyme K-mediated cytolysis. J Immunol (2009) 182(4):2152–9. doi: 10.4049/jimmunol.0802307
122. Guo Y, Chen J, Shi L, Fan Z. Valosin-containing protein cleavage by granzyme K accelerates an endoplasmic reticulum stress leading to caspase-independent cytotoxicity of target tumor cells. J Immunol (2010) 185(9):5348–59. doi: 10.4049/jimmunol.0903792
123. Zhong C, Li C, Wang X, Toyoda T, Gao G, Fan Z. Granzyme K inhibits replication of influenza virus through cleaving the nuclear transport complex importin α1/β dimer of infected host cells. Cell Death Differ (2012) 19(5):882–90. doi: 10.1038/cdd.2011.178
124. Jiang W, Chai NR, Maric D, Bielekova B. Unexpected role for granzyme K in CD56bright NK cell-mediated immunoregulation of multiple sclerosis. J Immunol (2011) 187(2):781–90. doi: 10.4049/jimmunol.1100789
125. Cooper DM, Pechkovsky DV, Hackett TL, Knight DA, Granville DJ. Granzyme K activates protease-activated receptor-1. PLos One (2011) 6(6):e21484. doi: 10.1371/journal.pone.0021484
126. Wensink AC, Kemp V, Fermie J, García Laorden MI, van der Poll T, Hack CE, et al. Granzyme K synergistically potentiates LPS-induced cytokine responses in human monocytes. Proc Natl Acad Sci USA (2014) 111(16):5974–9. doi: 10.1073/pnas.1317347111
127. Long NP, Park S, Anh NH, Min JE, Yoon SJ, Kim HM, et al. Efficacy of integrating a novel 16-gene biomarker panel and intelligence classifiers for differential diagnosis of rheumatoid arthritis and osteoarthritis. J Clin Med (2019) 8(1):50. doi: 10.3390/jcm8010050
128. Willis Fox O, Preston RJS. Molecular basis of protease-activated receptor 1 signaling diversity. J Thromb Haemost. (2020) 18(1):6–16. doi: 10.1111/jth.14643
129. Turner CT, Zeglinski MR, Richardson KC, Zhao H, Shen Y, Papp A, et al. Granzyme K expressed by classically activated macrophages contributes to inflammation and impaired remodeling. J Invest Dermatol (2019) 139(4):930–9. doi: 10.1016/j.jid.2018.09.031
130. Sharma M, Merkulova Y, Raithatha S, Parkinson LG, Shen Y, Cooper D, et al. Extracellular granzyme K mediates endothelial activation through the cleavage of protease-activated receptor-1. FEBS J (2016) 283(9):1734–47. doi: 10.1111/febs.13699
131. Jonsson AH, Zhang F, Dunlap G, Gomez-Rivas E, Watts GFM, Faust HJ, et al. Granzyme k(+) CD8 T cells form a core population in inflamed human tissue. Sci Transl Med (2022) 14(649):eabo0686. doi: 10.1126/scitranslmed.abo0686
132. Li K, Mo W, Wu L, Wu X, Luo C, Xiao X, et al. Novel autoantibodies identified in ACPA-negative rheumatoid arthritis. Ann Rheum Dis (2021) 80(6):739–47. doi: 10.1136/annrheumdis-2020-218460
133. Lu J, Bi Y, Zhu Y, Huipeng S, Duan W, Zhou J. CD3D, GZMK, and KLRB1 are potential markers for early diagnosis of rheumatoid arthritis, especially in anti-citrullinated protein antibody-negative patients. Front Pharmacol (2021) 12:726529. doi: 10.3389/fphar.2021.726529
134. Mahrus S, Kisiel W, Craik CS. Granzyme m is a regulatory protease that inactivates proteinase inhibitor 9, an endogenous inhibitor of granzyme b. J Biol Chem (2004) 279(52):54275–82. doi: 10.1074/jbc.M411482200
135. Lu H, Hou Q, Zhao T, Zhang H, Zhang Q, Wu L, et al. Granzyme m directly cleaves inhibitor of caspase-activated DNase (CAD) to unleash CAD leading to DNA fragmentation. J Immunol (2006) 177(2):1171–8. doi: 10.4049/jimmunol.177.2.1171
136. Hua G, Zhang Q, Fan Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme m-mediated apoptosis. J Biol Chem (2007) 282(28):20553–60. doi: 10.1074/jbc.M703196200
137. Cullen SP, Afonina IS, Donadini R, Lüthi AU, Medema JP, Bird PI, et al. Nucleophosmin is cleaved and inactivated by the cytotoxic granule protease granzyme m during natural killer cell-mediated killing. J Biol Chem (2009) 284(8):5137–47. doi: 10.1074/jbc.M807913200
138. Bovenschen N, de Koning PJ, Quadir R, Broekhuizen R, Damen JM, Froelich CJ, et al. NK cell protease granzyme m targets alpha-tubulin and disorganizes the microtubule network. J Immunol (2008) 180(12):8184–91. doi: 10.4049/jimmunol.180.12.8184
139. Shan L, van den Hoogen LL, Meeldijk J, Kok HM, Jongeneel LH, Boes M, et al. Increased intra-articular granzyme m may trigger local IFN-λ1/IL-29 response in rheumatoid arthritis. Clin Exp Rheumatol (2020) 38(2):220–6. doi: 10.55563/clinexprheumatol/ffb107
140. Sun J, Bird CH, Sutton V, McDonald L, Coughlin PB, De Jong TA, et al. A cytosolic granzyme b inhibitor related to the viral apoptotic regulator cytokine response modifier a is present in cytotoxic lymphocytes. J Biol Chem (1996) 271(44):27802–9. doi: 10.1074/jbc.271.44.27802
141. Grossman WJ, Revell PA, Lu ZH, Johnson H, Bredemeyer AJ, Ley TJ. The orphan granzymes of humans and mice. Curr Opin Immunol (2003) 15(5):544–52. doi: 10.1016/s0952-7915(03)00099-2
142. de Koning PJ, Kummer JA, de Poot SA, Quadir R, Broekhuizen R, McGettrick AF, et al. Intracellular serine protease inhibitor SERPINB4 inhibits granzyme m-induced cell death. PLos One (2011) 6(8):e22645. doi: 10.1371/journal.pone.0022645
143. Bird CH, Sutton VR, Sun J, Hirst CE, Novak A, Kumar S, et al. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme b-mediated apoptosis without perturbing the fas cell death pathway. Mol Cell Biol (1998) 18(11):6387–98. doi: 10.1128/mcb.18.11.6387
144. Wang L, Li Q, Wu L, Liu S, Zhang Y, Yang X, et al. Identification of SERPINB1 as a physiological inhibitor of human granzyme h. J Immunol (2013) 190(3):1319–30. doi: 10.4049/jimmunol.1202542
145. Niehaus JZ, Miedel MT, Good M, Wyatt AN, Pak SC, Silverman GA, et al. SERPINB12 is a slow-binding inhibitor of granzyme a and hepsin. Biochemistry. (2015) 54(45):6756–9. doi: 10.1021/acs.biochem.5b01042
146. Rucevic M, Fast LD, Jay GD, Trespalcios FM, Sucov A, Siryaporn E, et al. Altered levels and molecular forms of granzyme k in plasma from septic patients. Shock. (2007) 27(5):488–93. doi: 10.1097/01.shk.0000246905.24895.e5
147. Fischl MA, Richman DD, Grieco MH, Gottlieb MS, Volberding PA, Laskin OL, et al. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. a double-blind, placebo-controlled trial. N Engl J Med (1987) 317(4):185–91. doi: 10.1056/nejm198707233170401
148. de Vincenzi I. Triple antiretroviral compared with zidovudine and single-dose nevirapine prophylaxis during pregnancy and breastfeeding for prevention of mother-to-child transmission of HIV-1 (Kesho bora study): A randomised controlled trial. Lancet Infect Dis (2011) 11(3):171–80. doi: 10.1016/s1473-3099(10)70288-7
149. Ikram S, Ahmad J, Durdagi S. Screening of FDA approved drugs for finding potential inhibitors against granzyme b as a potent drug-repurposing target. J Mol Graph Model (2020) 95:107462. doi: 10.1016/j.jmgm.2019.107462
150. Kim MS, Buisson LA, Heathcote DA, Hu H, Braddock DC, Barrett AG, et al. Approaches to design non-covalent inhibitors for human granzyme b (hGrB). Org Biomol Chem (2014) 12(44):8952–65. doi: 10.1039/c4ob01874e
151. Ikram S, Ahmad F, Ahmad J, Durdagi S. Screening of small molecule libraries using combined text mining, ligand- and target-driven based approaches for identification of novel granzyme h inhibitors. J Mol Graph Model (2021) 105:107876. doi: 10.1016/j.jmgm.2021.107876
152. Zhang B, Zhao J, Jiang M, Peng D, Dou X, Song Y, et al. The potential role of gut microbial-derived exosomes in metabolic-associated fatty liver disease: Implications for treatment. Front Immunol (2022) 13:893617. doi: 10.3389/fimmu.2022.893617
153. Lettau M, Janssen O. Intra- and extracellular effector vesicles from human T and NK cells: Same-same, but different? Front Immunol (2021) 12:804895. doi: 10.3389/fimmu.2021.804895
154. Jong AY, Wu CH, Li J, Sun J, Fabbri M, Wayne AS, et al. Large-Scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracell Vesicles. (2017) 6(1):1294368. doi: 10.1080/20013078.2017.1294368
Keywords: rheumatoid arthritis, granzymes, apoptosis, inflammation, biomarker
Citation: Zheng Y, Zhao J, Shan Y, Guo S, Schrodi SJ and He D (2023) Role of the granzyme family in rheumatoid arthritis: Current Insights and future perspectives. Front. Immunol. 14:1137918. doi: 10.3389/fimmu.2023.1137918
Received: 05 January 2023; Accepted: 03 February 2023;
Published: 16 February 2023.
Edited by:
Haitao Wang, Center for Cancer Research, National Cancer Institute (NIH), United StatesReviewed by:
Ravichandra Tagirasa, National Cancer Institute at Frederick (NIH), United StatesClaudia Macaubas, Stanford University, United States
Copyright © 2023 Zheng, Zhao, Shan, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Guo, Shicheng.Guo@wisc.edu; Steven J. Schrodi, Schrodi@wisc.edu; Dongyi He, dongyihe@medmail.com.cn
†These authors have contributed equally to this work