 Taiga Kuga
Taiga Kuga Asako Chiba
Asako Chiba Goh Murayama
Goh Murayama Sachiko Miyake
Sachiko Miyake- 1Department of Immunology, Juntendo University Faculty of Medicine, Bunkyo-ku, Tokyo, Japan
- 2Department of Internal Medicine and Rheumatology, Juntendo University Faculty of Medicine, Bunkyo-ku, Tokyo, Japan
Type I interferons (IFNs) play crucial roles in the pathogenesis of systemic lupus erythematosus (SLE). Plasmacytoid dendritic cells (pDCs) stimulated by Toll-like receptor (TLR) pathways have been thought to be the major producers of IFNα in patients with SLE. However, the responsiveness of pDCs from SLE patients to stimuli that produce IFNα differs depending on the type of TLR pathway involved. In addition to pDCs, monocytes from SLE patients were found to produce IFNα when responding to the cGAS-STING pathway. Here, we outline the major pathways that induce IFNα production by myeloid cells in SLE, and the possible mechanisms by which IFNα overproduction occurs by these cells. Finally, we discuss the current and future therapeutic strategies to regulate IFNα production in patients with SLE.
Introduction
The involvement of type I interferons (IFNs) in the pathogenesis of systemic lupus erythematosus (SLE) is well established, and recent advances in the treatment of SLE with anifrolumab, a fully human anti-IFN α/β receptor antibody, have highlighted the importance of controlling the disease by inhibiting the type I IFN pathway. The treatment of patients with SLE with anifrolumab improved skin rash, arthritis, overall disease activity, and their health-related quality of life (1–4). Myeloid cells produce type I IFNs including IFNα when nucleic acid receptor pathways are activated. In this review, we describe the pathways and possible mechanisms by which IFNα production occurs in SLE.
Nucleic acid receptor pathways
Type I IFNs, inflammatory cytokines that play a critical role in the self-defense system, are produced when nucleic acid receptors recognize microbial and viral nucleic acids (5, 6). During infection, epithelial cells and fibroblasts at the site of infection produce type I IFNs such as IFNβ, and then plasmacytoid dendritic cells (pDCs) produce large amounts of type I IFNs, particularly IFNα (7, 8). In addition, other myeloid cells such as monocytes, conventional dendritic cells, and macrophages also produce type I IFNs (7, 8). Previous reports suggested the activation of nucleic acid receptor pathways is associated with the overproduction of type I IFNs and the pathogenesis of SLE. In addition to exogenous nucleic acids, self-derived nucleic acids also induce the production of type I IFNs. In SLE, nucleic acids derived from apoptotic cells, those contained in neutrophil extracellular traps (NETs) and mitochondrial DNA released due to mitochondrial stress have been demonstrated to activate nucleic acid receptor pathways (9–11). Immune complexes composed of autoantibodies and nucleic acids have also been shown to induce IFNα by pDCs (12–15). Impaired DNA clearance also leads to the activation of nucleic acid receptor pathways and the production of type I IFNs in SLE. Genetic deficiencies of nucleic acid degradation molecules, such as extracellular DNase-I and TREX1, the most abundant 3’→5’ DNA exonuclease in cells, are associated with a lupus-like syndrome in mice and humans (16–18). Most patients with SLE have reduced DNase I activity (19, 20), which might be explained by anti-DNase antibodies being present in approximately 60% of sera samples from SLE patients (21, 22).
Among the Toll-like receptors (TLRs), TLR4 recognizes pathogen-associated molecular patterns on the cell membrane surface and TLR3/7/8/9 located in endosomes or lysosomes recognize nucleic acids. Upon ligand binding, TLR7/9 induce type I IFN production via the myeloid differentiation factor 88 (MyD88)/TRAF6/IRF7 complex, and TLR3/4 induce type I IFN production by activating interferon regulatory factor 3 (IRF3) through the Toll/IL-1R domain-containing adaptor inducing interferon-β factor (TRIF) pathway in a MyD88-independent manner (8, 23). IFNα production by pDC upon stimulation with immune complexes composed of autoantibodies and nucleic acids was suppressed by blocking FcgRII, and thus these immune complexes appear to be internalized into endosomes where they activate TLR7 and TLR9 pathways (24). pDCs constitutively express high levels of IRF7 and produce large amounts of type I IFNs via TLR7/8/9 (25). Overactivation of the TLR7 pathway caused autoantibody production and lupus-like disease in mice (26–30). TLR7 polymorphisms have been shown to be associated with SLE (31–33). For example, a TLR7 gain-of-function genetic variation (34) and overactive TLR7 pathway as a result of variants in UNC93B1, which binds TLR7 and is essential for TLR7 trafficking to the endosome (35, 36), have provided direct evidence that genetic variants related to TLR7 activation trigger human SLE pathogenesis. Recently, UNC93B1 variants were reported in SLE patients (37, 38), and mice expressing a variant of UNC93B1 developed spontaneous lupus-like disease (37).
Nucleic acid receptors such as cGAS (cGMP-AMP synthase) and the RIG-I-like receptor (RLR) family and melanoma differentiation-associated gene (MDA5) recognize viral nucleic acids and self-derived DNA in the cytoplasm. Upon the recognition of cytoplasmic double-stranded DNA, cGAS synthesizes cyclic GMP-AMP (cGAMP), a second messenger that binds to the transmembrane protein stimulator of interferon gene (STING) located in the endoplasmic reticulum. STING is then transported to the Golgi apparatus where it induces type I IFN production via the TBK1-IRF3 pathway (39). Several lines of evidence suggest the cGAS-STING pathway is activated in SLE. A mutation in TMEM177, which encodes STING, has been associated with SLE-like diseases (40, 41). The cGAS-STING pathway is typically activated in response to DNA derived from pathogenic microbes or viruses, but excess self-DNA, derived from apoptosis-derived membrane vesicles (42), neutrophil extracellular traps (11), and mitochondrial DNA (43) may also contribute to aberrant cGAS-STING activation in SLE. Indeed, cGAS expression is increased in peripheral blood mononuclear cells (PBMCs) from SLE patients (44). Because IFNα impairs mitochondrial metabolism and autophagic degradation leading to the accumulation of mitochondrial DNA in the cytosol, IFNα overproduction in SLE may also contribute to the activation of the cGAS-STING pathway (43). Autoimmune diseases in Trex-1 and DNase II-deficient mice were shown to be dependent on the cGAS-STING pathway (45, 46), and other murine studies have demonstrated the contribution of the cGAS-STING pathway to enhancing type I IFN responses in lupus or lupus-like mouse models (47–49). These findings suggest that activation of the cGAS-STING pathway is deleterious in the pathogenesis of SLE by the induction of type I IFNs. However, cGAS or STING deficiency resulted in exaggerated disease in the MRL/lpr mouse model and pristane-induced lupus model (50). Further studies are needed to understand how the cGAS-STING pathway is involved in the pathogenesis of lupus.
IFNα overproducing myeloid cells in SLE
pDCs are known to be the most potent IFN-I producing cells and thus have been widely proposed to be the dominant source of type I IFN production in SLE. The early ablation of pDCs in BXSB lupus-prone mice prior to disease onset ameliorated lupus nephritis, reduced the tissue expression of IFN-induced genes, and diminished the cascade of IFN-mediated responses, including the reduction of antinuclear antibodies, splenomegaly, and abnormal expansion of T and B cells (51). In humans, the administration of a monoclonal antibody against blood DC antigen 2(BDCA2) expressed on pDCs decreased expression of IFN response genes in the blood, and reduced disease activity of skin disease and arthritis in SLE, suggesting that pDCs may indeed be an important source of type I IFNs in SLE (52–54). The TLR7 and TLR9 pathways induce IFNα production by pDCs. However, pDCs from SLE patients had increased IFNα production when stimulated with a TLR7 agonist, but reduced IFNα production in these cells when stimulated with a TLR9 agonist (Figure 1) (55, 56). Given that most nucleated cells can produce IFN-I during viral infection (57), it is important to consider IFN-I production by other cell subsets. In our study focusing on the cGAS-STING pathway, numbers of IFNα-producing cells among PBMCs from SLE patients were increased when stimulated with 2’3’-cGAMP, a STING-activating ligand (58). IFNα production by pDCs and conventional DCs was also increased in patients with SLE, but the primary IFNα-producing cells among PBMCs were monocytes. In addition, monocytes from SLE patients had higher STING expression and IFN-I expression upon 2’3’-cGAMP stimulation compared with those from healthy controls (Figure 1) (58, 59).
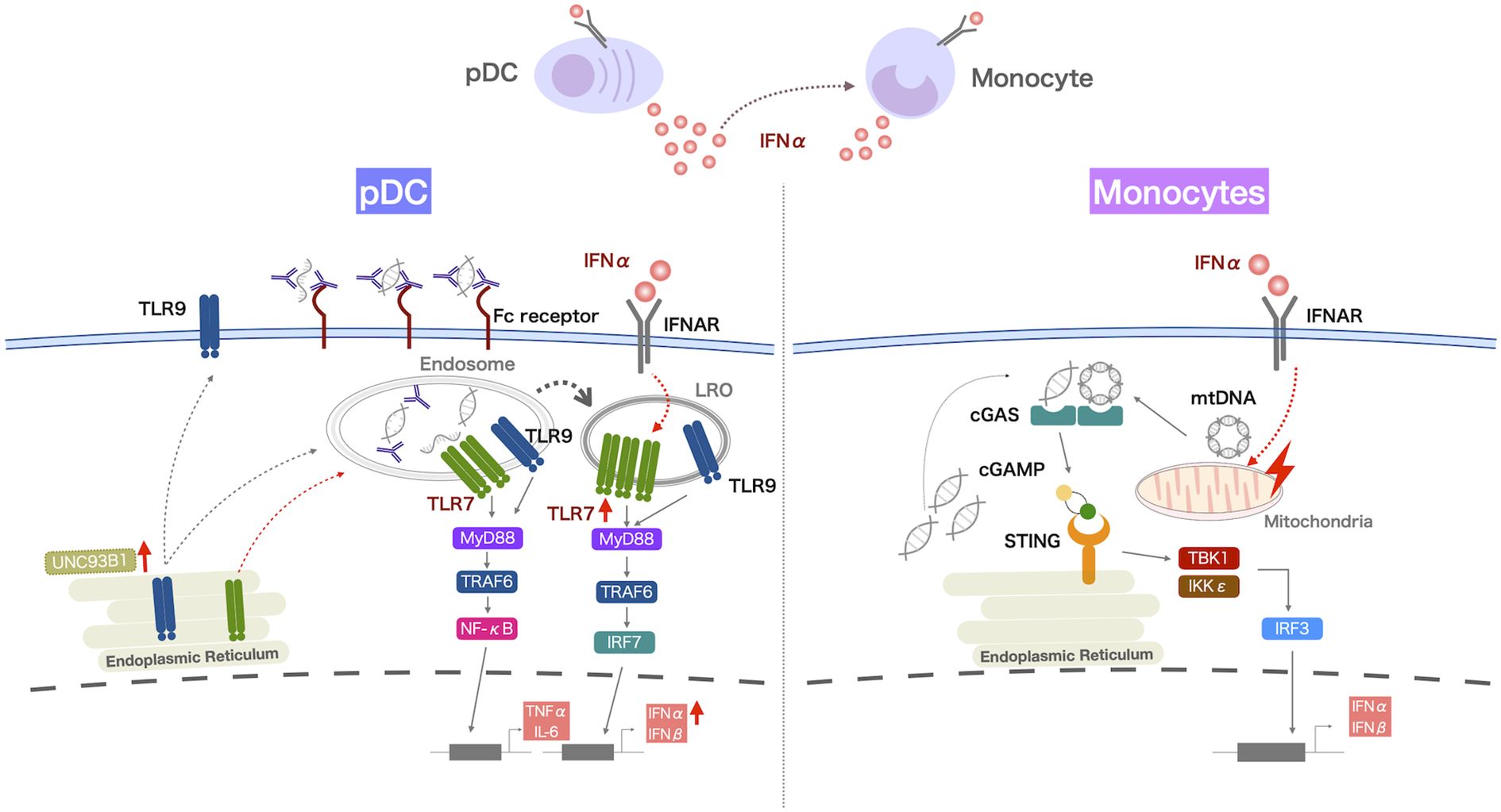
Figure 1. IFNα overproduction by myeloid cells in patients with SLE. IFNα production by pDCs from SLE patients is increased when they are activated via the TLR7 pathway, and the increased retention of TLR7 in lysosome-related organelles (LRO) of pDCs may contribute to this enhanced response. Monocytes from SLE patients produce increased levels of IFNα after activation of the cGAS-STING pathway. IFNα production by pDCs and monocytes correlates with disease activity in SLE, and increased TLR7 responsiveness is observed in pDCs, even in SLE patients with stable disease. These TLR7 and cGAS-STING pathways are enhanced by exposure to IFNα. Thus, IFNα production by pDCs may underlie the pathology of SLE and increased IFNα production by pDCs and monocytes due to the increased nucleic acid load contributes to disease activity.
Increased IFNα production by pDCs and monocytes was positively correlated with disease activity in SLE (55, 56, 58), suggesting that these cells may contribute to the pathogenesis of SLE. Interestingly, the capacity to produce IFNα by pDCs was also increased in patients with stable disease (55). In patients with cutaneous lupus erythematosus, the type I IFN signature was enhanced in keratinocytes from lesional and nonlesional skin, and pDCs were the major subset among myeloid cells in nonlesional skin (60). Thus, IFNα production by pDCs may underlie the pathogenesis of SLE and lupus-like disease. When the cGAS-STING pathway is activated by the accumulation of DNA in the cytosol, monocytes produce IFNα, which can cause disease flares of SLE. Because monocytes are migratory cells, they may produce IFNα at the sites of inflammation. The single-cell RNA-seq analysis of kidney samples from patients with lupus nephritis revealed that non-classical monocytes migrated to the kidney and differentiated into monocyte-derived macrophages, with their characteristics shifting from inflammatory to phagocytic (61). In lupus models, Ly6Clo monocytes, considered non-classical monocytes, were reported to infiltrate into the kidneys of MRL/lpr mice and ABIN1 (Tnip1)-deficient mice (62), as well as the central nervous system of NZB/NZW and FcgRIIB-/- Yaa mice (63). In imiquimod (IMQ)-induced lupus mice, Ly6Chi monocytes were increased in the lymph nodes and Ly6Clo monocytes expressing high levels of TLR7, adhesion molecules, and cytokines such as IL-6, were increased in the peripheral blood and infiltrated into the kidneys (64). Ly6Chi and Ly6Clo monocytes upregulated the expression of proinflammatory cytokine genes when stimulated with a TLR7 agonist, and only Ly6Chi monocytes upregulated IFNα genes when stimulated via the cGAS-STING pathway. It is unknown whether human IFNα-producing monocytes are CD16-negative classical monocytes because activated monocytes downregulate CD16 expression. Studies in mice suggested that Ly6Chi classical monocytes with IFNα-producing capacity migrated to the lymph nodes, whereas Ly6Clo non-classical monocytes infiltrated into the site of inflammation (62, 64). IFN-responsive genes were upregulated in microglia from FcgRIIB-/- Yaa mice and Ly6Clo monocytes present in the central nervous system of FcgRIIB-/- Yaa mice may produce IFNα (63). Pseudotime analysis of the RNA sequencing data of myeloid cells in the blood and skin lesions from patients with cutaneous lupus erythematosus indicated the transition of circulating non-classical monocytes to skin infiltrating CD16+ DCs, which was associated with the expression of type I IFN (60). Thus, circulating monocytes may also become IFNα-producing cells and contribute to disease pathogenesis at the site of inflammation.
Mechanisms of IFNα overproduction by myeloid cells in SLE
IFNα production by pDCs was increased when activated by a TLR7 agonist, and decreased by the TLR9 pathway, but the expression levels of TLR7 and TLR9 were reported to be comparable in pDCs from SLE patients and healthy controls (65). IRF7 activation for type I IFN expression requires TLR localization to lysosomes, and UNC93B1 is important for the trafficking of TLR7 and TLR9. Of note, we observed the increased retention of TLR7 in the lysosomes of pDCs from SLE patients (55). TLR9 competes with TLR7 for trafficking by UNC93B1 (66), and the accelerated localization of TLR7 to lysosomes may suppress TLR9 trafficking by UNC93B1. Exposure to IFNα increased IFNα production by pDCs and this was associated with increased TLR7 localization to lysosome-related organelles (55). Thus, the in vivo exposure of pDCs to IFNα in SLE may enhance their responses to TLR7 stimulation. UNC93B1 variants were shown to cause hyperresponsiveness to TLR7 stimulation, but not to TLR9 stimulation (37, 38). Thus, UNC93B1 variants may also contribute to the increased IFNα production by pDCs stimulated by a TLR7 agonist.
We found that STING expression and its colocalization with TBK1 was increased in monocytes from SLE patients (58). In contrast, monocytes from healthy individuals produced low levels of IFNα upon cGAS-STING activation, although their in vitro exposure to IFNα induced the expression of STING and IFNα (58). IFNα-induced STAT1, a transcription factor known to be activated by cytokines including IFNα, binds directly to the promoter of STING to enhance STING induction (67), and exposure to IFNα induced the colocalization of STING and TBK1 (58), which might be associated with the effect of IFNα on the induction of mitochondrial DNA accumulation in the cytoplasm. Thus, prior exposure to IFNα may accelerate the TLR7 and cGAS-STING pathways. Increased STING expression was accompanied by increased phosphorylated-mTOR? in SLE monocytes, suggesting that increased STING expression is also related to impaired autophagy. Indeed, the treatment of monocytes with rapamycin, an inhibitor of mTOR, decreased STING and IFNα production by SLE monocytes. Thus, impaired autophagy may also contribute to the increased STING and IFNα expression by SLE monocytes (58).
To uncover the cell-intrinsic mechanisms underlying enhanced IFNα production by STING stimulation, we performed the transcriptomic analysis of monocytes from SLE patients and in vitro IFNα-exposed monocytes from healthy individuals, and found that the transcription factor GATA4 was upregulated in monocytes from SLE patients (Figure 2) (68). GATA4 is a key transcription factor that contributes to the production of cytokines including IL-1 and IL-6, via NF-κB in cells with cellular senescence, a phenomenon known as the senescence-associated secretory phenotype (69). Cellular senescence refers to a phenomenon whereby cells stop proliferating due to various stresses and factors that cause DNA damage. GATA4 expression also enhanced IFNα production in monocytic U937 cells (68). GATA4 binds to the enhancer region of the IFIT family. Furthermore, IFIT3 was reported to be upregulated in SLE monocytes, and its inhibition reduced type I IFN production by monocytes activated by the cGAS-STING pathway (59). In addition to GATA4 expression, SLE monocytes exhibited cellular senescence-like features, including high CDKN2A expression and increased senescence-associated β-galactosidase activity (68). The DNA damage response, as measured by γ-H2AX levels, was reported to be increased in monocytes (70), B cells (71), and bone marrow-derived mesenchymal stem cells from SLE patients (72). The in vitro exposure of activated human B cells to IFNα and human carcinoma cell lines to a mixture of IL-1β, IFNγ, and TNFα has been shown to induce DNA damage responses in these cells (71, 73). Thus, IFNα overproduction in SLE may contribute to enhanced DNA damage responses (71). Thus, the accumulation of cytoplasmic DNA, such as micronuclei, due to DNA damage induces cellular senescence and may also activate the cGAS-STING pathway.
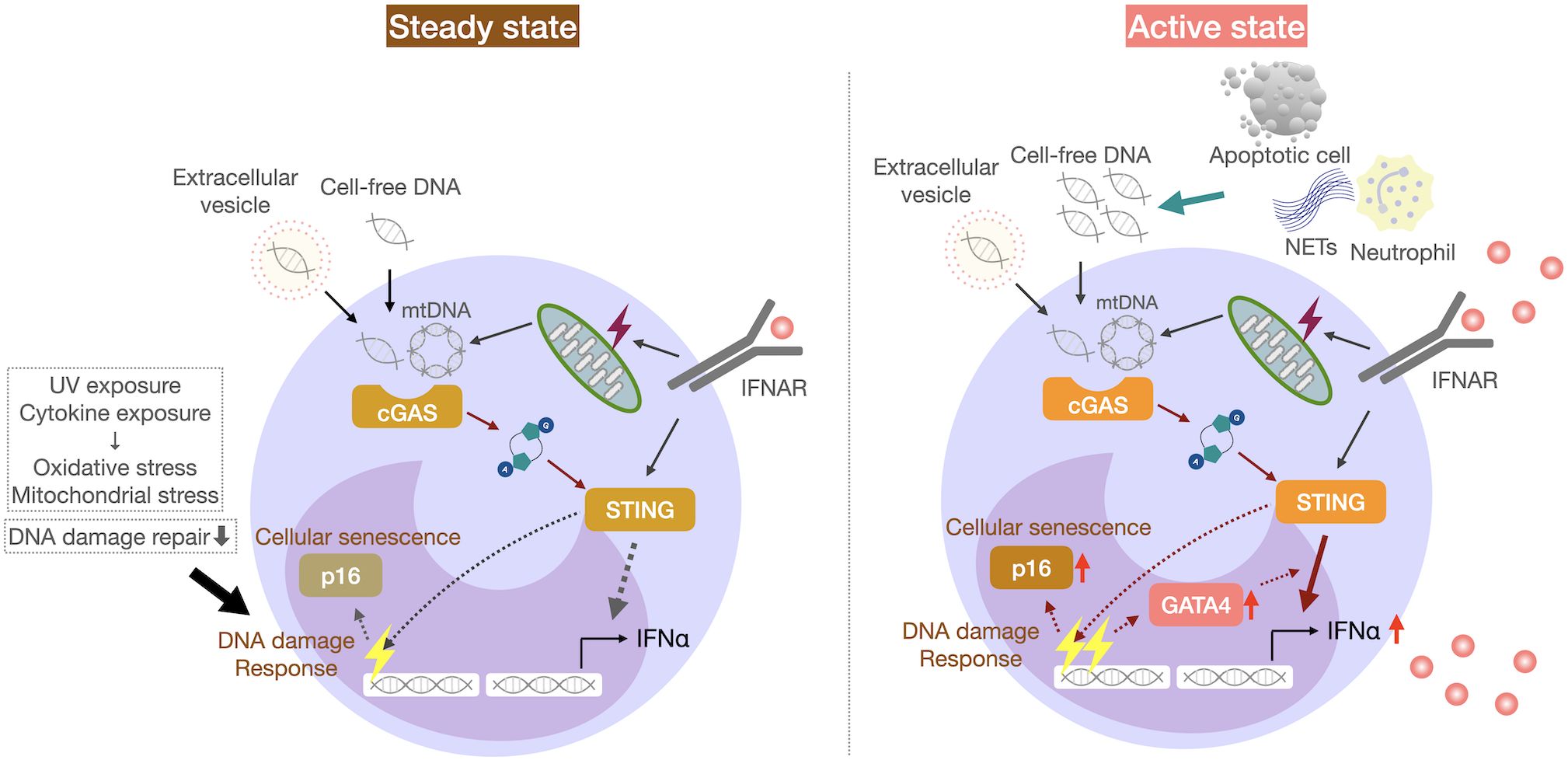
Figure 2. IFNα overproduction by senescent monocytes in patients with SLE. SLE monocytes from SLE patients exhibit cellular senescence-like features, including p16 expression and increased senescence-associated β-galactosidase activity. GATA4 expression increases the responsiveness of the cGAS-STING pathway and IFNα production by monocytes, which may increase the disease activity in SLE.
Therapeutic strategies for SLE by inhibiting IFNα production
Some of the current therapeutic strategies for SLE have been shown to suppress IFNα production. Many lines of research have clearly demonstrated that hydroxychloroquine (HCQ) reduces disease flares in SLE patients (74, 75) by inhibiting the activation of endosomal TLRs, particularly TLR7 and TLR9 (76, 77) and antigen presentation (78), as well as reducing NETs formation (79). Indeed, HCQ suppressed IFNα production by pDCs exposed to CpG-A (80). pDCs isolated from cutaneous lupus erythematosus and SLE patients administered HCQ had significantly lower IFNα production upon TLR7 or TLR9 stimulation compared with patients not receiving HCQ (80, 81). Furthermore, HCQ levels in the blood of patients with cutaneous lupus erythematosus correlated negatively with the IFNα-producing capacity of their pDCs upon TLR9 stimulation, with a weaker correlation observed upon TLR7/8 stimulation (80). HCQ suppresses the formation of cGAMP from cGAS; therefore, HCQ may also inhibit IFNα production via the cGAS-STING pathway (82).
Prior exposure to IFNα increased IFNα production by pDC and monocytes (55, 56, 58). Thus, anifrolumab treatment may reduce IFNα production by these cells. The MUSE study, a trial of anifrolumab, showed that SLE patients with high IFN gene signature (IFNGS) or high disease activity had decreased numbers of lymphocytes, neutrophils, pDCs and monocytes, and the decrease in these cells was reversed after anifrolumab treatment (83). This anifrolumab treatment effect was shown to be associated with the suppression of interferon-inducible chemokines such as interferon gamma-induced protein 10 (IP-10) and IFN-inducible T cell alpha chemoattractant (ITAC). Therefore, anifrolumab treatment may reduce the migration of immune cells, including those capable of producing IFNα, to sites of inflammation.
Plasma dsDNA from SLE patients was shown to activate the cGAS-STING pathway in monocytic cells when using a cell-based reporter system that detected the bioavailability and inducing activity of IFN-I (42, 84). Type I IFN bioavailability was decreased in the plasma of SLE patients treated with double filtration plasmapheresis (DFPP) and this was associated with decreased plasma cDNA levels (84). This suggests that the beneficial effects of DFPP may be related to the removal of nucleic acids in addition to autoantibodies.
Rapamycin, a drug that inhibits T-cell proliferation, has shown efficacy in a phase 1/2 clinical trial for the treatment of SLE (85). Autophagy was suppressed in SLE monocytes, and rapamycin reduced STING expression and IFNα production by these cells, suggesting that the cGAS-STING pathway may be a therapeutic target for the suppression of IFNα production. Indeed, several inhibitors of cGAS or STING are already in development (86). In SLE patients, the source of IFNα may vary between individuals, and TLR7 and pDCs might also be a good therapeutic target. Inhibitors of TLR7/8 such as E6742, afimeoran (NCT04493541), and enpatoran (NCT05540327) are being or have been investigated in clinical trials for SLE. E6742, a selective dual antagonist for TLR7/8, has been investigated in a double-blind phase 1/2 study in SLE, and 57.1% of patients achieved a positive response on the British Isles Lupus Assessment Group-based Composite Lupus Assessment (BICLA), compared with 33.3% in the placebo group (87). SLE patients with active cutaneous lupus erythematosus (CLE) treated with anti-BDCA2 antibody litifilimab (also known as BIIB059) showed an approximately 50% reduction in IFNGS expression in whole blood 24 hours after treatment, and litifilimab treatment was highly effective in normalizing IFN response proteins (myxovirus resistance protein 1 and IFN-induced transmembrane protein 3) in lesional skin and reducing CD45+ immune cell infiltration (52). Phase 2 studies of litifinimab demonstrated the reduction of Cutaneous Lupus Erythematosus Disease Area and Severity Index Activity scores and in the number of swollen and tender joints in SLE (53, 54). The efficacy of litifilimab is being further investigated in phase 3 trials (NCT04895241 and NCT04961567). Therapeutic strategies targeting specific immune cells or nucleic acid receptor pathways may be an option depending on which cell type is the prominent IFNα producing cell in SLE.
Conclusions
Although increased IFNα production by pDCs was observed in SLE patients including those with stable disease, IFNα production by monocytes in SLE patients positively correlated with disease activity. Thus, IFNα overproduction by pDCs may enhance the responsiveness of pDCs and monocytes to nucleic acid receptor pathways, and IFNα overproduction by monocytes may be responsible for the disease activity in SLE. Thus, the suppression of the TLR pathway may be more important for the maintenance of stable disease and the activation of the cGAS-STING pathway may need to be suppressed to inhibit the disease flares in SLE. The future therapeutic targets of nucleic acid receptor pathways and myeloid cells should be decided depending on the disease status in SLE.
Author contributions
TK: Writing – original draft. AC: Writing – original draft, Writing – review & editing. GM: Writing – review & editing. SM: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported in part by a Grant-in-Aid for Special Research in Subsidies for ordinary expenses of private schools from The Promotion and Mutual Aid Corporation for Private Schools of Japan.
Acknowledgments
We thank J. Ludovic Croxford, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KF is currently organizing a Research Topic with the author SM.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. (2018) 5:e000261. doi: 10.1136/lupus-2018-000261
2. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
3. Stull D, O'Quinn S, Williams B, Bean S, Schwetje E, Abreu G, et al. Causal cascade of direct and indirect effects of anifrolumab on patient-reported outcomes: structural equation modelling of two Phase 3 trials. Rheumatol (Oxford England). (2022) 61:4731–40. doi: 10.1093/rheumatology/keac138
4. Merrill JT, Furie R, Werth VP, Khamashta M, Drappa J, Wang L, et al. Anifrolumab effects on rash and arthritis: impact of the type I interferon gene signature in the phase IIb MUSE study in patients with systemic lupus erythematosus. Lupus Sci Med. (2018) 5:e000284. doi: 10.1136/lupus-2018-000284
5. McNab F, Mayer-Barber K, Sher A, Wack A, and O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
6. Seo YJ and Hahm B. Type I interferon modulates the battle of host immune system against viruses. Adv Appl Microbiol. (2010) 73:83–101. doi: 10.1016/S0065-2164(10)73004-5
7. Ali S, Mann-Nüttel R, Schulze A, Richter L, Alferink J, and Scheu S. Sources of type I interferons in infectious immunity: plasmacytoid dendritic cells not always in the driver's seat. Front Immunol. (2019) 10:778. doi: 10.3389/fimmu.2019.00778
8. Porritt RA and Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. (2015) 36:150–60. doi: 10.1016/j.it.2015.02.002
9. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
10. Darrah E and Andrade F. NETs: the missing link between cell death and systemic autoimmune diseases? Front Immunol. (2012) 3:428. doi: 10.3389/fimmu.2012.00428
11. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
12. Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. (2005) 202:1131–9. doi: 10.1084/jem.20050914
13. Lövgren T, Eloranta ML, Båve U, Alm GV, and Rönnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheumatol. (2004) 50:1861–72. doi: 10.1002/art.20254
14. Lövgren T, Eloranta ML, Kastner B, Wahrenraren M, Alm GV, and Rönnblom L. Induction of interferonn. by immune complexes or liposomes containing systemic lupus erythematosus autoantigen– and Sjögren's syndrome autoantigen–associated RNA. Arthritis Rheumatism. (2006) 54:1917–27. doi: 10.1002/art.21893
15. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, and Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. (2005) 115:407–17. doi: 10.1172/JCI23025
16. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, and Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. (2000) 25:177–81. doi: 10.1038/76032
17. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. (2001) 28:313–4. doi: 10.1038/91070
18. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39:1065–7. doi: 10.1038/ng2091
19. Tinazzi E, Puccetti A, Gerli R, Rigo A, Migliorini P, Simeoni S, et al. soluble Fas/FasL levels and cell surface Fas expression in patients with SLE: a possible explanation for the lack of efficacy of hrDNase I treatment. Int Immunol. (2009) 21:237–43. doi: 10.1093/intimm/dxn142
20. Chitrabamrung S, Rubin RL, and Tan EM. Serum deoxyribonuclease I and clinical activity in systemic lupus erythematosus. Rheumatol Int. (1981) 1:55–60. doi: 10.1007/BF00541153
21. Trofimenko AS, Gontar IP, Zborovsky AB, and Paramonova OV. Anti-DNase I antibodies in systemic lupus erythematosus: diagnostic value and share in the enzyme inhibition. Rheumatol Int. (2016) 36:521–9. doi: 10.1007/s00296-016-3437-z
22. Yeh TM, Chang HC, Liang CC, Wu JJ, and Liu MF. Deoxyribonuclease-inhibitory antibodies in systemic lupus erythematosus. J BioMed Sci. (2003) 10:544–51. doi: 10.1007/BF02256116
23. Kawai T and Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
24. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
25. Ngo C, Garrec C, Tomasello E, and Dalod M. The role of plasmacytoid dendritic cells (pDCs) in immunity during viral infections and beyond. Cell Mol Immunol. (2024) 21:1008–35. doi: 10.1038/s41423-024-01167-5
26. Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. (2006) 25:429–40. doi: 10.1016/j.immuni.2006.07.014
27. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, and Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. (2006) 25:417–28. doi: 10.1016/j.immuni.2006.07.013
28. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, and Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Sci (New York NY). (2006) 312:1669–72. doi: 10.1126/science.1124978
29. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. (2007) 27:801–10. doi: 10.1016/j.immuni.2007.09.009
30. Fairhurst AM, Hwang SH, Wang A, Tian XH, Boudreaux C, Zhou XJ, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. (2008) 38:1971–8. doi: 10.1002/eji.200838138
31. dos Santos BP, Valverde JV, Rohr P, Monticielo OA, Brenol JC, Xavier RM, et al. TLR7/8/9 polymorphisms and their associations in systemic lupus erythematosus patients from southern Brazil. Lupus. (2012) 21:302–9. doi: 10.1177/0961203311425522
32. Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. (2010) 107:15838–43. doi: 10.1073/pnas.1001337107
33. Wang CM, Chang SW, Wu YJ, Lin JC, Ho HH, Chou TC, et al. Genetic variations in Toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Sci Rep. (2014) 4:3792. doi: 10.1038/srep03792
34. Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. (2022) 605:349–56. doi: 10.1038/s41586-022-04642-z
35. Kim YM, Brinkmann MM, Paquet ME, and Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. (2008) 452:234–8. doi: 10.1038/nature06726
36. Ishida H, Asami J, Zhang Z, Nishizawa T, Shigematsu H, Ohto U, et al. Cryo-EM structures of Toll-like receptors in complex with UNC93B1. Nat Struct Mol Biol. (2021) 28:173–80. doi: 10.1038/s41594-020-00542-w
37. Al-Azab M, Idiiatullina E, Liu Z, Lin M, Hrovat-Schaale K, Xian H, et al. Genetic variants in UNC93B1 predispose to childhood-onset systemic lupus erythematosus. Nat Immunol. (2024) 25:969–80. doi: 10.1038/s41590-024-01846-5
38. Wolf C, Lim EL, Mokhtari M, Kind B, Odainic A, Lara-Villacanas E, et al. UNC93B1 variants underlie TLR7-dependent autoimmunity. Sci Immunol. (2024) 9:eadi9769. doi: 10.1126/sciimmunol.adi9769
39. Decout A, Katz JD, Venkatraman S, and Ablasser A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
40. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
41. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. (2014) 124:5516–20. doi: 10.1172/JCI79100
42. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1507–15. doi: 10.1136/annrheumdis-2018-212988
43. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNα Impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep. (2018) 25:921–33.e5. doi: 10.1016/j.celrep.2018.09.001
44. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol (Hoboken NJ). (2017) 69:800–7. doi: 10.1002/art.40002
45. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. (2015) 112:E5699–705. doi: 10.1073/pnas.1516465112
46. Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr., Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. (2012) 36:120–31. doi: 10.1016/j.immuni.2011.11.018
47. Thim-Uam A, Prabakaran T, Tansakul M, Makjaroen J, Wongkongkathep P, Chantaravisoot N, et al. STING mediates lupus via the activation of conventional dendritic cell maturation and plasmacytoid dendritic cell differentiation. iScience. (2020) 23:101530. doi: 10.1016/j.isci.2020.101530
48. Martin GR, Henare K, Salazar C, Scheidl-Yee T, Eggen LJ, Tailor PP, et al. Expression of a constitutively active human STING mutant in hematopoietic cells produces an Ifnar1-dependent vasculopathy in mice. Life Sci Alliance. (2019) 2:e201800215. doi: 10.26508/lsa.201800215
49. Liu Y, Carmona-Rivera C, Seto NL, Oliveira CB, Patino-Martinez E, Baumer Y, et al. Role of STING deficiency in amelioration of mouse models of lupus and atherosclerosis. Arthritis Rheumatol. (2024) 77:547–59. doi: 10.1002/art.43062
50. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci U S A. (2015) 112:E710–7. doi: 10.1073/pnas.1420217112
51. Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. (2014) 211:1977–91. doi: 10.1084/jem.20132620
52. Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. (2019) 129:1359–71. doi: 10.1172/JCI124466
53. Furie RA, van Vollenhoven RF, Kalunian K, Navarra S, Romero-Diaz J, Werth VP, et al. Trial of anti-BDCA2 antibody litifilimab for systemic lupus erythematosus. N Engl J Med. (2022) 387:894–904. doi: 10.1056/NEJMoa2118025
54. Werth VP, Furie RA, Romero-Diaz J, Navarra S, Kalunian K, van Vollenhoven RF, et al. Trial of anti-BDCA2 antibody litifilimab for cutaneous lupus erythematosus. N Engl J Med. (2022) 387:321–31. doi: 10.1056/NEJMoa2118024
55. Murayama G, Furusawa N, Chiba A, Yamaji K, Tamura N, and Miyake S. Enhanced IFN-α production is associated with increased TLR7 retention in the lysosomes of palasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. (2017) 19:234. doi: 10.1186/s13075-017-1441-7
56. Sakata K, Nakayamada S, Miyazaki Y, Kubo S, Ishii A, Nakano K, et al. Up-regulation of TLR7-mediated IFN-alpha production by plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Front Immunol. (2018) 9:1957. doi: 10.3389/fimmu.2018.01957
57. Stetson DB and Medzhitov R. Type I interferons in host defense. Immunity. (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
58. Murayama G, Chiba A, Kuga T, Makiyama A, Yamaji K, Tamura N, et al. Inhibition of mTOR suppresses IFNα production and the STING pathway in monocytes from systemic lupus erythematosus patients. Rheumatol (Oxford England). (2020) 59:2992–3002. doi: 10.1093/rheumatology/keaa060
59. Wang J, Dai M, Cui Y, Hou G, Deng J, Gao X, et al. Association of abnormal elevations in IFIT3 with overactive cyclic GMP-AMP synthase/stimulator of interferon genes signaling in human systemic lupus erythematosus monocytes. Arthritis Rheumatol. (2018) 70:2036–45. doi: 10.1002/art.2018.70.issue-12
60. Billi AC, Ma F, Plazyo O, Gharaee-Kermani M, Wasikowski R, Hile GA, et al. Nonlesional lupus skin contributes to inflammatory education of myeloid cells and primes for cutaneous inflammation. Sci Transl Med. (2022) 14:eabn2263. doi: 10.1126/scitranslmed.abn2263
61. Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. (2019) 20:902–14. doi: 10.1038/s41590-019-0398-x
62. Kuriakose J, Redecke V, Guy C, Zhou J, Wu R, Ippagunta SK, et al. Patrolling monocytes promote the pathogenesis of early lupus-like glomerulonephritis. J Clin Invest. (2019) 129:2251–65. doi: 10.1172/JCI125116
63. Nomura A, Noto D, Murayama G, Chiba A, and Miyake S. Unique primed status of microglia under the systemic autoimmune condition of lupus-prone mice. Arthritis Res Ther. (2019) 21:303. doi: 10.1186/s13075-019-2067-8
64. Nomura A, Mizuno M, Noto D, Aoyama A, Kuga T, Murayama G, et al. Different spatial and temporal roles of monocytes and monocyte-derived cells in the pathogenesis of an imiquimod induced lupus model. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.764557
65. Sakata K, Nakayamada S, Miyazaki Y, Kubo S, Ishii A, Nakano K, et al. Up-regulation of TLR7-mediated IFN-α Production by plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.01957
66. Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. (2011) 35:69–81. doi: 10.1016/j.immuni.2011.05.010
67. Ma F, Li B, Yu Y, Iyer SS, Sun M, and Cheng G. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. (2015) 16:202–12. doi: 10.15252/embr.201439366
68. Kuga T, Chiba A, Murayama G, Hosomi K, Nakagawa T, Yahagi Y, et al. Enhanced GATA4 expression in senescent systemic lupus erythematosus monocytes promotes high levels of IFNα production. Front Immunol. (2024) 15:1320444. doi: 10.3389/fimmu.2024.1320444
69. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. (2015) 349:aaa5612. doi: 10.1126/science.aaa5612
70. Stergioti EM, Manolakou T, Sentis G, Samiotaki M, Kapsala N, Fanouriakis A, et al. Transcriptomic and proteomic profiling reveals distinct pathogenic features of peripheral non-classical monocytes in systemic lupus erythematosus. Clin Immunol. (2023) 255:109765. doi: 10.1016/j.clim.2023.109765
71. Manolakou T, Nikolopoulos D, Gkikas D, Filia A, Samiotaki M, Stamatakis G, et al. ATR-mediated DNA damage responses underlie aberrant B cell activity in systemic lupus erythematosus. Sci Adv. (2022) 8:eabo5840. doi: 10.1126/sciadv.abo5840
72. Gu Z, Meng Y, Tao T, Guo G, Tan W, Xia Y, et al. Endoplasmic reticulum stress participates in the progress of senescence of bone marrow-derived mesenchymal stem cells in patients with systemic lupus erythematosus. Cell Tissue Res. (2015) 361:497–508. doi: 10.1007/s00441-015-2131-x
73. Jaiswal M, LaRusso NF, Burgart LJ, and Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. (2000) 60:184–90.
74. The Canadian Hydroxychloroquine Study Group. A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. N Engl J Med. (1991) 324:150–4. doi: 10.1056/NEJM199101173240303
75. Dima A, Jurcut C, Chasset F, Felten R, and Arnaud L. Hydroxychloroquine in systemic lupus erythematosus: overview of current knowledge. Ther Adv Musculoskelet Dis. (2022) 14:1759720X211073001. doi: 10.1177/1759720X211073001
76. Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, and Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol (Baltimore Md: 1950). (2011) 186:4794–804. doi: 10.4049/jimmunol.1000702
77. Cenac C, Ducatez MF, and Guery JC. Hydroxychloroquine inhibits proteolytic processing of endogenous TLR7 protein in human primary plasmacytoid dendritic cells. Eur J Immunol. (2022) 52:54–61. doi: 10.1002/eji.202149361
78. Gies V, Bekaddour N, Dieudonne Y, Guffroy A, Frenger Q, Gros F, et al. Beyond anti-viral effects of chloroquine/hydroxychloroquine. Front Immunol. (2020) 11:1409. doi: 10.3389/fimmu.2020.01409
79. Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. (2014) 66:2532–44. doi: 10.1002/art.38703
80. Gardet A, Pellerin A, McCarl CA, Diwanji R, Wang W, Donaldson D, et al. Effect of in vivo Hydroxychloroquine and ex vivo Anti-BDCA2 mAb Treatment on pDC IFNα Production From Patients Affected With Cutaneous Lupus Erythematosus. Front Immunol. (2019) 10:275. doi: 10.3389/fimmu.2019.00275
81. Sacre K, Criswell LA, and McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. (2012) 14:R155. doi: 10.1186/ar3895
82. Schrezenmeier E and Dorner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol. (2020) 16:155–66. doi: 10.1038/s41584-020-0372-x
83. Casey KA, Guo X, Smith MA, Wang S, Sinibaldi D, Sanjuan MA, et al. Type I interferon receptor blockade with anifrolumab corrects innate and adaptive immune perturbations of SLE. Lupus Sci Med. (2018) 5:e000286. doi: 10.1136/lupus-2018-000286
84. Saito T, Takatsuji R, Murayama G, Yamaji Y, Hagiwara Y, Nishioka Y, et al. Double-filtration plasmapheresis reduces type I interferon bioavailability and inducing activity in systemic lupus erythematosus. Immunol Med. (2024) 47:264–74. doi: 10.1080/25785826.2024.2372918
85. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet. (2018) 391:1186–96. doi: 10.1016/S0140-6736(18)30485-9
86. Decout A, Katz JD, Venkatraman S, and Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
87. Tanaka Y, Kumanogoh A, Atsumi T, Ishii T, Tago F, Aoki M, et al. Safety, pharmacokinetics, biomarker response and efficacy of E6742: a dual antagonist of Toll-like receptors 7 and 8, in a first in patient, randomised, double-blind, phase I/II study in systemic lupus erythematosus. RMD Open. (2024) 10:e004701. doi: 10.1136/rmdopen-2024-004701
Keywords: SLE, pDC, monocyte, interferon, TLR, cGAS-STING, cellular senescence
Citation: Kuga T, Chiba A, Murayama G and Miyake S (2025) Myeloid cells as IFNα producers in systemic lupus erythematosus. Front. Immunol. 16:1562221. doi: 10.3389/fimmu.2025.1562221
Received: 17 January 2025; Accepted: 01 July 2025;
Published: 17 July 2025.
Edited by:
Tianfu Wu, University of Houston, United StatesReviewed by:
Keishi Fujio, The University of Tokyo, JapanUma Sriram, Temple University, United States
Melissa Anne Cunningham, Medical University of South Carolina, United States
Copyright © 2025 Kuga, Chiba, Murayama and Miyake. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asako Chiba, YS1jaGliYUBqdW50ZW5kby5hYy5qcA==; Sachiko Miyake, cy1taXlha2VAanVudGVuZG8uYWMuanA=