A critical role for IFN-β signaling for IFN-κ induction in keratinocytes
 Bin Xu1
Bin Xu1  Jon Musai1
Jon Musai1  Yee Sun Tan1 Grace A. Hile2
Yee Sun Tan1 Grace A. Hile2  William R. Swindell3
William R. Swindell3  Benjamin Klein1 J. Tingting Qin4
Benjamin Klein1 J. Tingting Qin4  Mrinal K. Sarkar2
Mrinal K. Sarkar2  Johann E. Gudjonsson2
Johann E. Gudjonsson2  J. Michelle Kahlenberg1*
J. Michelle Kahlenberg1*
- 1Division of Rheumatology, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, United States
- 2Department of Dermatology, University of Michigan, Ann Arbor, MI, United States
- 3Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 4Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, MI, United States
Background/Purpose: Cutaneous lupus erythematosus (CLE) affects up to 70% of patients with systemic lupus erythematosus (SLE), and type I interferons (IFNs) are important promoters of SLE and CLE. Our previous work identified IFN-kappa (IFN-κ), a keratinocyte-produced type I IFN, as upregulated in non-lesional and lesional lupus skin and as a critical regulator for enhanced UVB-mediated cell death in SLE keratinocytes. Importantly, the molecular mechanisms governing regulation of IFN-κ expression have been relatively unexplored. Thus, this study sought to identify critical regulators of IFN-κ and identified a novel role for IFN-beta (IFN-β).
Methods: Human N/TERT keratinocytes were treated with the RNA mimic poly (I:C) or 50 mJ/cm2 ultraviolet B (UVB), followed by mRNA expression quantification by RT-qPCR in the presence or absence neutralizing antibody to the type I IFN receptor (IFNAR). IFNB and STAT1 knockout (KO) keratinocytes were generated using CRISPR/Cas9.
Results: Time courses of poly(I:C) and UVB treatment revealed a differential expression of IFNB, which was upregulated between 3 and 6 h and IFNK, which was upregulated 24 h after stimulation. Intriguingly, only IFNK expression was substantially abrogated by neutralizing antibodies to IFNAR, suggesting that IFNK upregulation required type I IFN signaling for induction. Indeed, deletion of IFNB abrogated IFNK expression. Further exploration confirmed a role for type I IFN-triggered STAT1 activation.
Conclusion: Collectively, our work describes a novel mechanistic paradigm in keratinocytes in which initial IFN-κ induction in response to poly(I:C) and UVB is IFNβ1-dependent, thus describing IFNK as both an IFN gene and an interferon-stimulated gene.
Introduction
Type I interferons comprise 13 subtypes including IFN-α, IFN-β, IFN-ω, IFN-ε and IFN-κ. The dysregulation of type I interferon (IFN) production has been shown to be a critical step in the pathogenesis of many autoimmune diseases with systemic lupus erythematosus (SLE) exhibiting the highest and most pronounced production (1). Type I interferon plays a key role in systemic lupus erythematosus (SLE) pathogenesis through modulation of innate immune responses and activation of the adaptive immune system (2, 3). Importantly, numerous cellular sources of interferon have been identified in SLE that include immune cells and epithelial cell populations. In the skin, an organ frequently inflamed and damaged in SLE patients, keratinocytes are an important contributor to the cutaneous IFN signature in SLE patients (4–6). IFN kappa (IFN-κ) is increased in non-lesional keratinocytes of SLE patients even before disease onset (6) and enhances immune cell activation, cytokine production, and ultraviolet (UV) light sensitivity (4, 5). Thus, understanding the regulation of IFN-κ is a critical undertaking to better investigate the biology of IFNs in the skin and how to target them.
In the epidermis, type I IFN production can be activated via several routes. Sensing of microbial products by pattern-recognition receptors (PRRs) will induce type I IFN production through activation of the interferon regulatory factor (IRF) family of transcription factors, of which IRF3 is the primary regulator of IFN production in non-plasmacytoid dendritic cell populations (7–9). TLR3 stimulation by poly(I:C) generates a robust TRIF-mediated activation of TBK1 which activates IRF3 to induce transcription. Cytoplasmic sensing of nucleic acids through sensors such as RIG-I and cGAS also result in IRF3 activation and upregulation of IFN genes (10). Ultraviolet light, an important trigger for SLE-associated skin disease, also results in cGAS/STING activation and subsequent type I IFN upregulation (11), but the precise signaling mechanisms that lead to IFN production are less clear.
The effects of type I IFNs are mediated through the type I IFN receptor (IFNAR). Canonical IFNAR signaling activates the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway by phosphorylation. JAK1 and TYK2 bind to the cytoplasmic tails of IFNARI and II and phosphorylate STAT1 and STAT2, which dimerize with IRF9 to form the transcriptionally active ISGF3 complexes. ISGF3 binds to interferon-stimulated response elements (ISREs) to upregulate type I IFN response genes (12, 13). Type I IFN signaling can also upregulate the expression of IFN genes as well. For example, treatment of keratinocytes by IFNβ1 induces IFNK expression (14).
In this study, we investigated the production of type I IFNs in the epidermis, an important source of interferons in the skin. We found that in keratinocytes, IFNB and IFNK are differentially regulated in response to poly(I:C) and UVB. Indeed, IFNB is the early IFN produced and IFNK is induced later as an IFN-regulated gene, entirely dependent on IFN-β and STAT1 signaling for its production. Thus, our study provides insight into the sequential and IFN-β-dependent upregulation of downstream keratinocyte IFN responses, including the upregulation of IFNK.
Materials and methods
Cell culture
The immortalized N/TERT keratinocyte cell line was used with the kind permission of Dr James G Rheinwald (15). N/TERTs were grown in Keratinocyte-SFM medium (ThermoFisher #17005–042) supplemented with 30 µg/ml bovine pituitary extract, 0.2 ng/ml epidermal growth factor and 0.3 mM calcium chloride (16) and passaged at ∼60% confluency to avoid differentiation. For treatment with Poly(I:C) and UVB, keratinocyte cultures were grown to 80% confluence and then treated with Poly(I:C) at 10 ug/ml, or irradiated in PBS with 50 mJ/cm2 UVB (310 nm) via a UV-2 irradiator (Tyler Industries, Alberta, Canada) followed by media replacement and harvest at indicated time points. Primary keratinocytes were isolated and cultured from 6 mm punch biopsies as we have previously reported (4, 5).
Generation of knockout (Ko) keratinocytes by CRISPR/cas9
The generation of knock-out (KO) cell lines using non-homologous end joining (NHEJ) via CRISPR/Cas9 and baricitinib treatment to permit efficacious transfection was described previously (4, 17) and was completed with the assistance of the Functional Analytics CRISPR Core of the University of Michigan Skin Biology and Diseases Resource Center. Briefly, for the IFNB1 KO, the following oligonucleotides were used for annealing: IFNB1PCRF1: TGCTCTGGCACAACAGGTAG, IFNB1PCRR1: AGTCTCATTCCAGCCAGTGC. For the Mock−/− line, a sgRNA was designed for the tubulin alpha pseudogene using the following oligonucleotides: GTATTCCGTGGGTGAACGGG. The annealed oligonucleotides were inserted into the cloning vector pSpCas9 (BB)−2A-GFP (PX458) (Addgene # 48138) following the Ran et al. (18) protocol. Ligated plasmids were transformed into competent Escherichia coli (ThermoFisher Catalog #C737303) and then plated on LB-agar. Twelve colonies from each of the groups were selected and cultured in LB medium, and plasmids were purified using Qiagen mini-prep kit (Cat #27106), and then the proper insertion of sgRNA target sequences were verified by Sanger sequencing. Purified plasmid was transfected into an immortalized keratinocyte line (N/TERTs) using the TransfeX transfection kit (ATCC, Cat# ACS4005). Single cells positive for green fluorescent protein (GFP) were sorted into 96-well plates using a MoFlo Astrios #1 cell sorter and grown up to ∼50% confluence. Cells from 96-well plates were transferred into 12-well plates and grown to 50% confluence. DNA was extracted and PCR-amplified using specific primers. Homozygous or heterozygous IFNB1 mutations were verified by Sanger sequencing of the PCR product. For validation of findings, a total of four independent CRISPR/Cas9 KO mutants were generated for IFNB1. Similar experimental design and method was used to generate the STAT1 KO by CRISPR/Cas9 as described previously (19).
RNA isolation, reverse transcription, and RT-qPCR
Total RNA was isolated with an RNeasy Mini kit (Catalog no. 74,104; Qiagen, Germantown, MD) according to the manufacturer's instructions. One microgram of total RNA was reverse transcribed using the iScript™ cDNA Synthesis Kit (Bio-Rad, Catalog# 1708891), and RT-qPCR was performed on a 7900HT Fast Real-time PCR system (Applied Biosystems) as described previously (4). All primer sequences are listed in Supplementary Table S1.
Cell lysis and immunoblotting
Total cellular extracts were prepared in HEPES buffer containing protease inhibitor mixture (Catalog no. 11836170001; Sigma-Aldrich) as described previously (20). Cell lysates were then gently resuspended and incubated at 4°C with gentle rocking for 40 min to 1 h, followed by microcentrifugation at 13,000 rpm for 10 min at 4°C. The supernatants were transferred to new tubes, and protein concentrations were determined by Bradford method using Bio-Rad Protein Assay Dye (Cat# 5000006) and serial dilution of BSA as standard. Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes, and immunoblotting was performed using antibodies to IFNB1 (Cat#73671), P-STAT1 (Tyr701, Cat#9167), STAT1 (Cat#14995) and β-actin (Cat#4967) from Cell Signaling Technology. The primary and secondary antibodies were diluted with signal enhancer HIKAR solution 1 and solution 2, respectively, as described previously (20). Detection by enhanced chemiluminescence was carried out with a SuperSignal West Dura kit (Thermo Fisher Scientific, Catalog #34075), followed by imaging on Omega Lum C (Aplegen). The ratio of phosphorylated to total protein was quantified using Image J.
Reagents
Poly(I:C) was purchased from Tocris Bioscience™ (Cat# 42-871-0). IFN-beta Mouse anti-Human, Clone: 76,703, was from R&D Systems™ (Cat#MAB814100). Cell supernatants were collected after 1 to 24 h post Poly(I:C). IFN-κ levels in keratinocyte supernatants were measured via ELISA, according to the manufacturer's protocol (#MBS936153, MyBiosource, San Diego, CA). IFN-β level was determined using Human IFN-β ELISA Kit (from PBL Assay Science, Catalog# 41410).
Bioinformatic analysis
In silico analysis was used to identify IFNK-correlated genes and motifs enriched in the IFNK and IFNB1 upstream sequences. IFNK-correlated genes were identified by evaluating gene expression across 118 healthy control KC cell line or primary cell microarray samples using methods previously described (21) The 118 samples had been generated using the same commercial microarray platform (Affymetrix Human Genome Plus 2.0 array) and were compiled from 13 Gene Expression Omnibus series submissions (GSE7216, GSE18590, GSE21364, GSE21567, GSE27186, GSE30355, GSE32685, GSE33495, GSE33536, GSE34528, GSE36222, GSE36287 and GSE37637). Microarray samples were generated using RNA from cultured KCs following various forms of treatment (e.g., genetic mutations, siRNA knockdown, cytokines) see Supplementary Table S2.
13,007 genes were identified with detectable expression (p < 0.05, Signed rank test) in at least 5% (≥6/118) of the 118 microarray samples. Of these 13,007 genes, expression of 7,856 was positively correlated with IFNK (rs > 0). Spearman rank correlation estimates varied continuously among these 7,856 genes. To determine an appropriate threshold, a graphical approach was used to identify a critical point in the decay of correlations among the 7,856 genes (Supplementary Figure S1A). This defined a set of 570 genes with IFNK-correlated expression (rs ≥ 0.60). Enrichment of biological processes among the set of 570 IFN-correlated genes (Supplementary Figure S1E) was evaluated using a conditional hypergeometric test (22).
The upstream sequences of IFNK and IFNB1 were further inspected to identify matches to 2,935 binding sites known to interact with transcription factors or unconventional DNA binding proteins (23). The analysis was repeated with respect to regions 1 kb, 2 kb, 5 kb, and 10 kb upstream of both genes. The total number of motif occurrences in the IFNK1 and IFNB1 upstream regions was also evaluated for each of the 2,935 binding sites and Fisher's exact test was used to assess whether the number of occurrences differed significantly between IFNK1 and IFNB1 (Supplementary Tables S3–S6). The consensus motif sequence is shown for each motif along with the International Union of Pure and Applied Chemistry (IUPAC) nucleotide code.
Statistical analysis
For statistical calculations, we used GraphPad Prism version 7. For most studies, we compared mean values for experimental variables between groups using the Student's unpaired 2-sided t-test for normally distributed variables, and 1-way ANOVA for multiple comparisons. In all experiments, p less than 0.05 was considered statistically significant.
Results
IFNB is induced earlier than IFNK in response to poly(I:C) and UVB treatment
IFN-κ is a critical IFN in cutaneous lupus pathology via promotion of enhanced IFN responses and photosensitivity (4). To study the regulation of IFN-κ, we first examined a time course of the upregulation of type I IFNs in the N/TERT human keratinocyte line. When cells were stimulated with poly(I:C), a mimic RNA activator of RIG-I, MDA5, and TLR3 pathways, IFNB1 transcription upregulation was robust and rapid, peaking at 3 h after treatment and dropping close to baseline by 12 h (Figure 1A). This is consistent with other cell types in which poly(I:C) induces a rapid upregulation of IFNB1 in an IRF-3 dependent manner (24). Surprisingly, IFNK upregulation was much slower, with increased transcription starting around 12 h after stimulation and peak expression noted at 24 h after poly(I:C) stimulation (Figure 1B). Activation of IFNA transcription (as measured by a poly-specific primer for all alpha subtypes) was less robust and also delayed until 24 h after stimulation (Figure 1C). A time course of protein production of IFN-β and IFN-κ also followed a similar timeline (Figures 1D,E). These data suggest that regulation of IFNK occurs through an alternate, slower mechanism from IFNB, which is rapidly upregulated following treatment with poly(I:C).
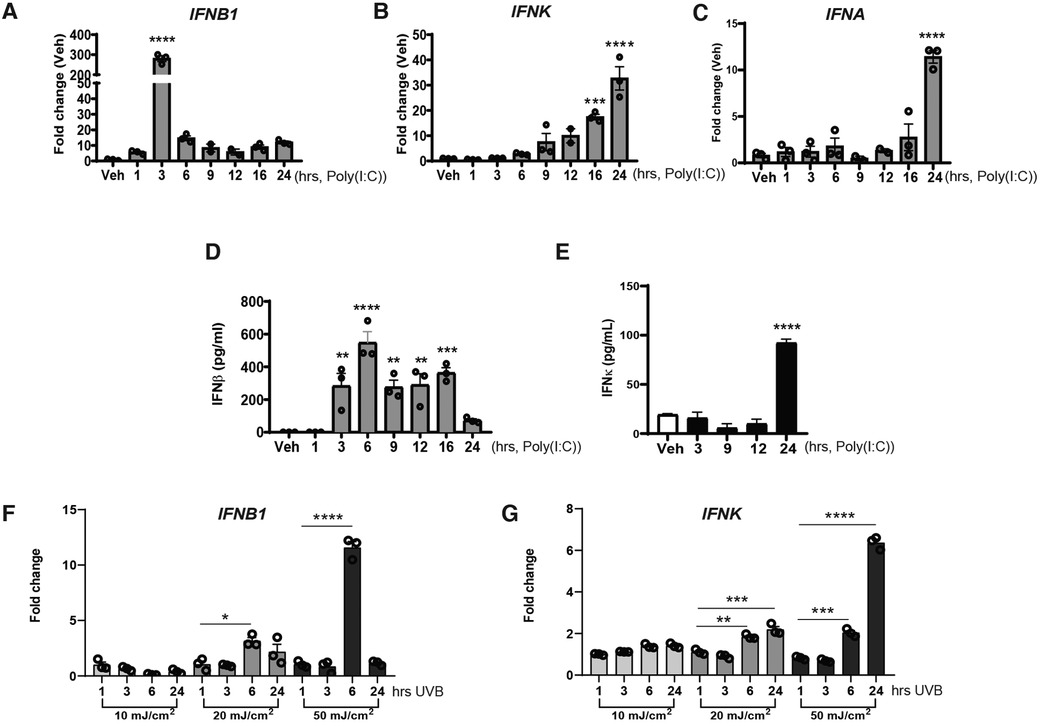
Figure 1. IFNB1 and IFNK are sequentially upregulated in response of Poly(I:C) treatment or exposure to UV radiation. (A–C) N/TERT keratinocytes were treated with Poly(I:C) (10 μg/ml), RNAs were isolated at the different time-points indicated followed by RT-qPCR analysis for mRNA expression. (D,E) IFNβ or IFNκ production in the supernatant was measure via ELISA at the time-points post-Poly(I:C). (F,G) RNA was isolated, and RT-qPCR was performed for gene expression of IFNB1 or IFNK at the time-point indicated after exposure N/TERTs to UVB radiation. (A–G) Data points represent average of triplicates for three separate experiments except for E where it was repeated twice in triplicate and are shown with SEM were normalized to β-actin expression as fold change to vehicle without treatment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 3. Comparison to vehicle was analyzed by 1-way ANOVA for multiple comparisons.
UV radiation is an important stimulus for type I IFN production in keratinocytes and is also an important trigger for SLE-related skin disease. We thus next examined whether the kinetics of IFNB and IFNK regulation were similar in keratinocytes after UVB exposure. N/TERT keratinocytes were treated with increasing doses of UVB followed by RNA harvest at the indicated time points. Similar to poly(I:C), IFNB1 transcription started early after 20 or 50 mJ/cm2 stimulation but upregulation of IFNK did not peak until 24 h after treatment (Figures 1F,G). Similar results for timing of IFNB and IFNK expression in response to poly(I:C) and UV light were obtained in human primary keratinocytes (Supplementary Figure S1).
Induction of IFNK is through type I IFN signaling
The delay in IFNK upregulation suggested that a second signal from the initial stimulus was needed for its upregulation. Given that IFN genes can be activated by other IFNs, we first tested whether signaling through the IFNAR receptor was required. We pretreated the N/TERTs with a blocking IFNAR antibody or control IgG one hour before and during poly(I:C) stimulation. RNA was harvested for analysis at 3 and 24 h. As shown in Figure 2A, anti-IFNAR antibody treatment strongly blocked IFNK upregulation but had much smaller (but significant) effects on overall IFNB1 expression. We then tested to see whether IFN-β treatment of N/TERTs could indeed result in IFNK upregulation. As shown in Figure 2C, IFNK expression was increased after 2 h of IFN-β stimulation, similar to the upregulation of known interferon stimulated genes (ISGs) IRF7 and MX1 (Figure 2C). We then tested whether neutralization of IFN-β in N/TERT cultures blocked the upregulation of IFNK. Indeed, use of IFN-β antibody decreased the upregulation of both IFNK and IRF7 expression after Poly(I:C) (Figure 2D). Together, these data suggest that the induction of IFNK may be through IFNB signaling.
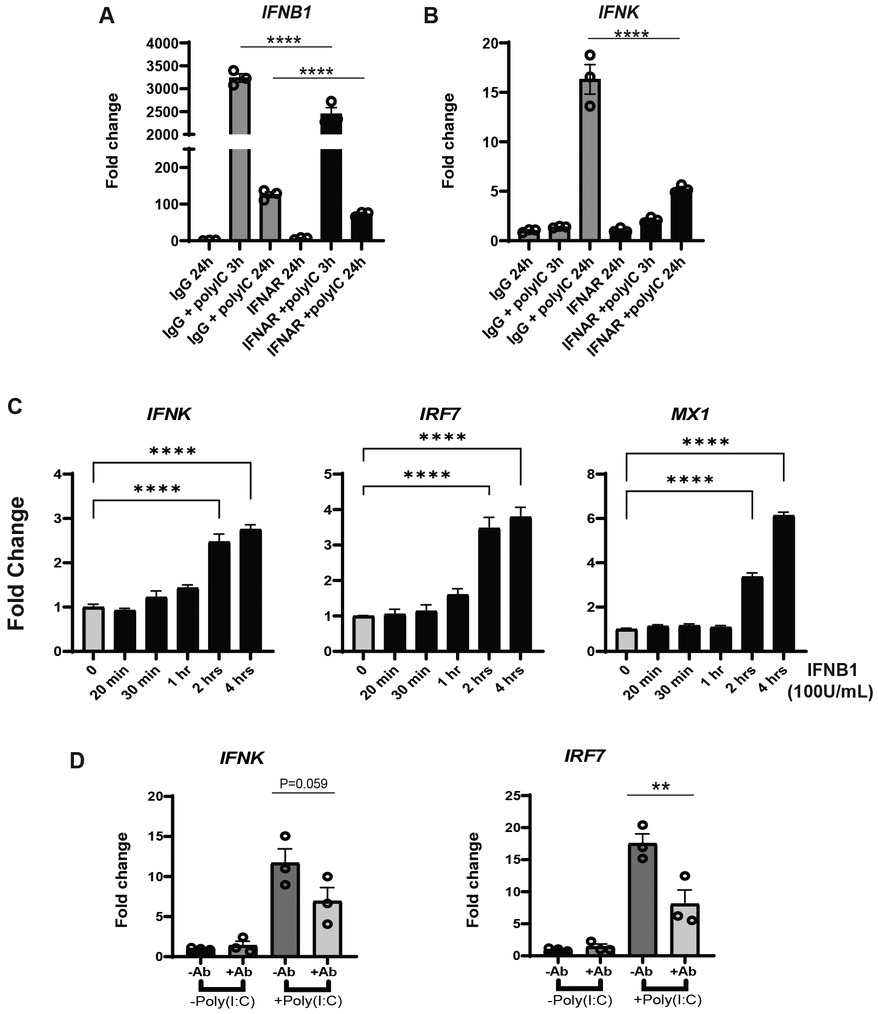
Figure 2. IFNK upregulation is through IFNAR signaling. (A,B) N/TERT keratinocytes were pretreated with (a) neutralizing IFNAR antibody or isotype IgG, followed by the time course treatment with Poly(I:C) (10 μg/ml) with either isotype IgG or neutralizing IFNAR antibody. RNA was isolated at time point indicated followed by the RT-qPCR analysis for gene expression of IFNB1 and IFNK, respectively. (C) N/TERT keratinocytes were treated with 100 U/ml IFNβ and RNA was isolated at indicated time points followed by RT-qPCR analysis for gene expression of IFNK, IRF7 and MX1; (D) N/TERT keratinocytes were pretreated with a neutralizing anti IFNβ antibody (+Ab) or isotype IgG (−Ab), followed by Poly(I:C) induction for 24 h. Then, RNA was isolated, and RT-qPCR were performed for IFNK and IRF7. Data shown with SEM were normalized to β-actin expression as fold change to vehicle without treatment. Datapoints represent average of triplicate for 3 separate experiments except for (C), where assay was repeated twice. Comparisons to vehicle were analyzed by 1-way ANOVA for multiple comparisons: **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 3.
Loss of IFNB1 abrogates the induction of IFNK and decreases the expression of interferon-stimulated-genes after poly(I:C) and UVB
We next took a genetic approach to examine the relationship between IFNB and IFNK. To this end, we generated a knockout line in N/TERTs for IFNB1 and compared it to a line in which a non-coding region was targeted by CRISPR (“mock”). Both lines were generated with the use of baricitinib to avoid selection of low IFNK expression as previously demonstrated (17). As shown in Figure 3A, the basal expression of IFNB1 is very low, but was detectable at 3 and 6 h when cells were induced by Poly(I:C). No expression of IFNB was detected even with stimulation in four knockout lines of IFNB (B1KO #13, #14, #17, and #18), which validated the IFNB1 gene knockout (Figure 3A and Supplementary Figure S2). We then compared Mock−/− and two IFNB1 KO lines to investigate the effect of loss of IFNB1 on IFNK and ISG expression. Strikingly, deletion of IFNB1 resulted in a complete inhibition of IFNK upregulation after poly(I:C) and after UVB treatment (Figures 3B,E, respectively). This also resulted in a significant reduction in the expression of downstream IFN genes such as MX1, IRF7 and ISG15 (Figures 3C,D,F–H). These data suggest that in response to stimuli such as UVB and poly(I:C), IFN-β production is rapid and required for further downstream IFN production. Indeed in this context, IFNK behaves like an ISG.
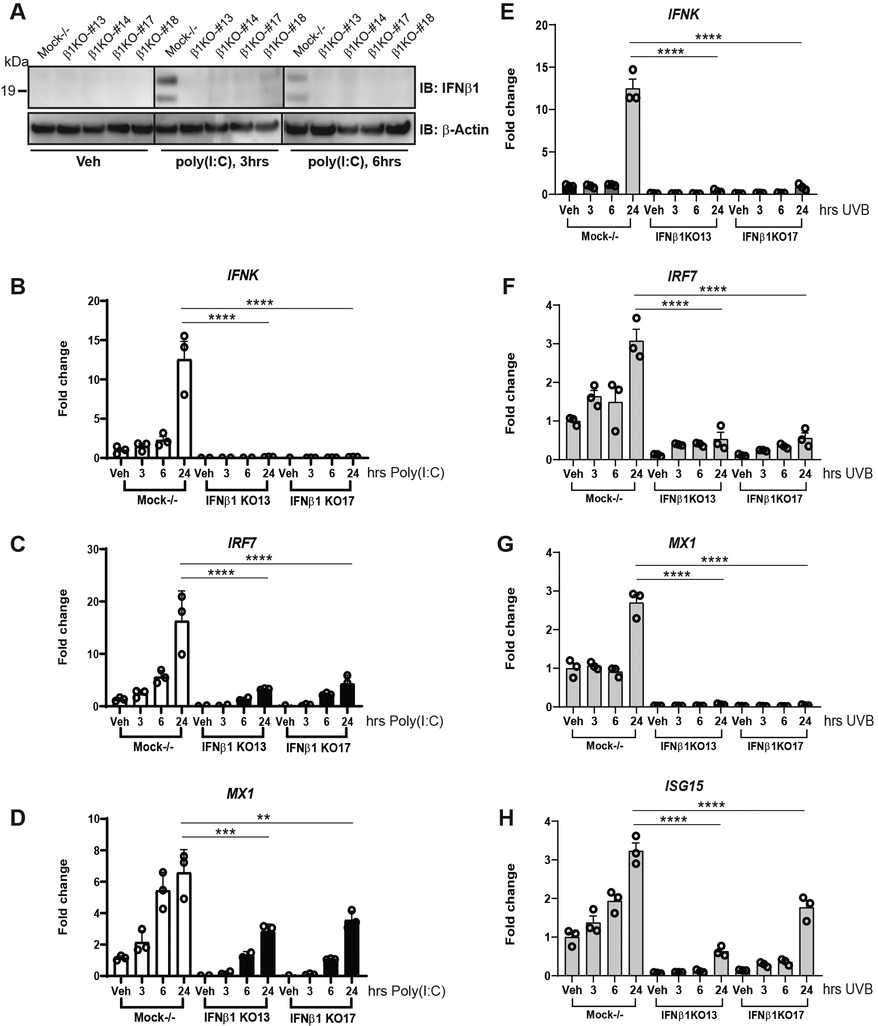
Figure 3. IFNB1 knockout (IFNB1KO) abrogates IFNK expression and decreases ISG induction. (A) IFNβ protein expression in response to Poly(I:C) (10 μg/ml) in Mock−/− control or IFNB1KO N/TERT cells was determined by immunoblot against IFNβ, β-actin as loading control. (B–H) RNA was isolated followed by RT-qPCR was performed for analysis of gene expression at the time point indicated in response to Poly(I:C) (B–D), or exposure to UVB radiation (50 mJ/cm2) (E–H). Data for three separate experiments completed in triplicate shown with SEM were normalized to β-actin expression as fold change to vehicle without treatment. Significance compared to vehicle was analyzed by 1-way ANOVA for multiple comparisons, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 3.
IFNB1 is also required to fully upregulate IFNλ
The type III interferon, IFNλ, is important for epidermal defense against viruses. It is produced by, and acts on, keratinocytes. IFNλ is also produced in the skin in established SLE, and it has structural resemblance to IL-10, but similar signaling and downstream cellular effects to type I interferons (3, 25). We thus also assessed IFNL1 and IFNL3 (we do not detect IFNL2 or IFNL4 in N/TERTs). Our data identified a similar pattern of IFNL3 and IFNB1 regulation where both are early stimulated genes after poly(I:C) treatment. In contrast, IFNL1 behaved like IFNK where expression was seen 24-h post treatment (Figures 4A,B). Similarly, expression of IFNL1 was entirely dependent on IFNB1 whereas only the later expression of IFNL3 had any dependence on IFNB1. Taken together, these data suggest that IFN-β signaling may be also required for IFN-λ1 but not early IFN-λ3 production in keratinocytes.
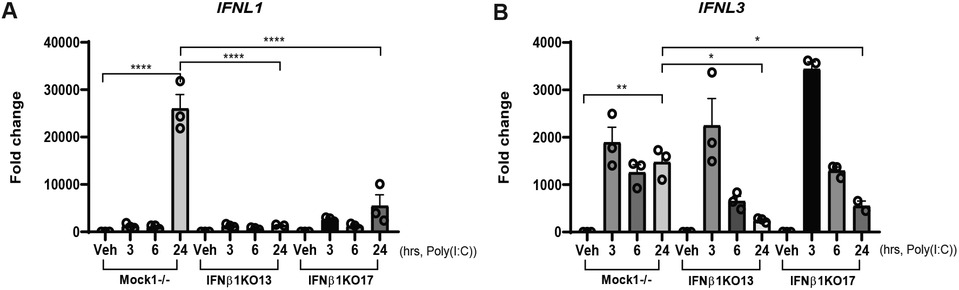
Figure 4. IFNL induction is also regulated by IFNβ. Mock−/− control or IFNB1KO N/TERT keratinocytes were treated with Poly(I:C) (10 μg/ml), RNA was isolated at the indicated time-points followed by RT-qPCR analysis for expression of (A) IFNL1 and (B) IFNL3. Data shown were normalized to β-actin expression as the fold change to vehicle without treatment +/− SEM. These results are representative of two independent experiments performed in triplicate. Significance compared to vehicle was analyzed by 1-way ANOVA for multiple comparisons, *p < 0.05, **p < 0.01, ****p < 0.0001.
IFNK-co-expressed genes and IFNB1/IFNK upstream motifs
Given the difference in time courses of IFNB and IFNK upregulation as well as the essential role of IFN-β in the production of IFN-κ, we next analyzed publicly available microarray data (n = 118 samples, see Supplementary Table S2) generated from cultured keratinocytes and identified 7,856 genes positively correlated with IFNK expression and with detectable expression in at least 5% of microarray samples (Supplementary Figure S3). We defined a set of 570 IFNK-co-expressed genes and identified several overrepresented IFN-related biological processing GO terms (Supplementary Figures S1D,E). Indeed, the most closely correlated genes were known ISGs such as MX1 and 2, IFI44, and ISG15. These data support IFNK as an ISG. We then examined the promoters of IFNB1 and IFNK to determine whether regulatory mechanisms for each IFN's activation would be apparent. For this, we used a screen of 2,935 binding sites known to interact with transcription factors or unconventional DNA binding proteins (23) and focused in on interferon regulatory factor (IRF) binding sites, which are important for both interferon production (especially IRF3) and ISG induction (especially IRF9). As shown in Supplementary Figure S1F, both IFNB and IFNK promoters exhibited binding sites for IRF9 (isgf3f motif), consistent with both genes being activated by signaling through the type I IFN receptor via activated STAT1/2/IRF9 complex formation. However, when we analyzed for binding sites for IRF3, which drives IFN production downstream of poly(I:C) and UV, there were 3 predicted sites for IFNB1 and no predicted sites for IFNK at −5 kb. Further away, at −10 kb total, 3 and 2 were predicted, respectively. While we cannot rule out overlap between these predicted sites, these data suggest that IFNB1 upregulation is more expedient downstream of activation by STING-driven IRF3 activation as there are IRF3 binding sites near the transcription start site whereas IFNK does not have similar availability for IRF3 binding.
IFNAR signaling is required for IFNK production
Type I interferons bind to a shared cell surface receptor, the type I interferon receptor (IFNAR). IFNs binding to the IFNAR initiate activation of Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2), leading to phosphorylation, dimerization and nuclear translocation of STAT proteins. We thus investigated the activation of type I IFN signaling after poly(I:C) and UV stimulation in the presence or absence of IFNB. Strikingly, the deletion of IFNB resulted in decreased activation of STAT1 after poly(I:C) (Figures 5A–D). Less robust activation of STAT1 was noted after UVB at the 6 h time point shown (and at 3 h not shown) (Figures 5E–H). Loss of IFNB resulted in a reduction in total STAT1 expression (Figures 5A,C,E,G and Supplementary Figure S4). We then examined IFN and ISG expression in STAT1 KO KCs. Consistent with our hypothesis, deletion of STAT1 decreased production of IFNK and the ISGs IRF7, ISG15, and MX1 for both poly(I:C) and UVB treatment (Figures 6A,B). However, expression of IFNB1 was not significantly affected by deletion of STAT1 and may have been enhanced, especially with poly(I:C) treatment. These data suggest that IFNB is produced upstream of STAT1 activation, likely through IRF3-dependent means, while IFNK transcription is downstream of IFN-β signaling and relies on IFNAR and STAT1 (Figure 7).
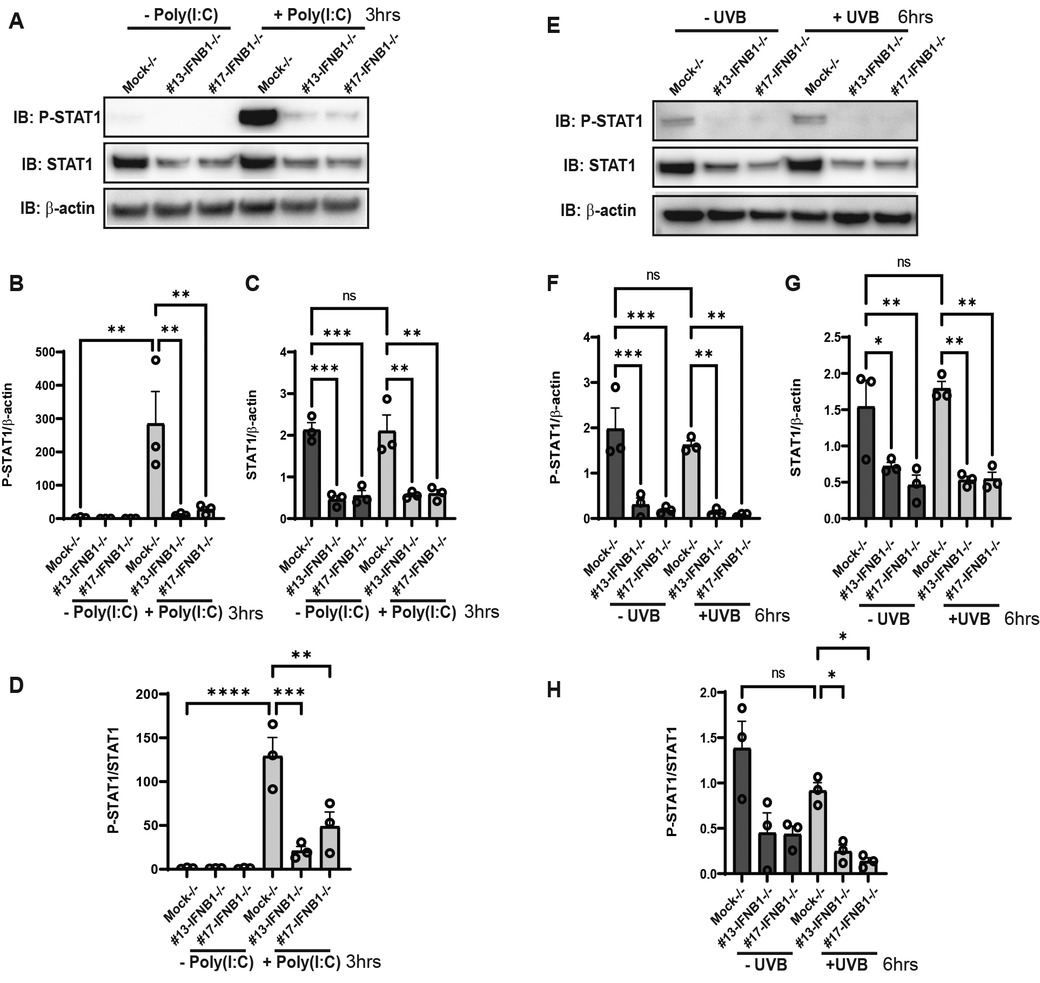
Figure 5. STAT1 activation is decreased in IFNBKO KCs. (A,E) N/TERT keratinocytes were treated with either Poly(I:C) (10 μg/ml) or exposure to UVB radiation (50 mJ/cm2) and were harvested at the time point indicated. Protein expression was then determined by immunoblot using antibodies against P-STAT1, total STAT1, or β-actin as a loading control. (B–D,F–H) Quantification of each band was performed using ImageJ. n = 3 in triplicate. Significance was analyzed by 1-way ANOVA for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.01, ****p < 0.0001, n = 3.
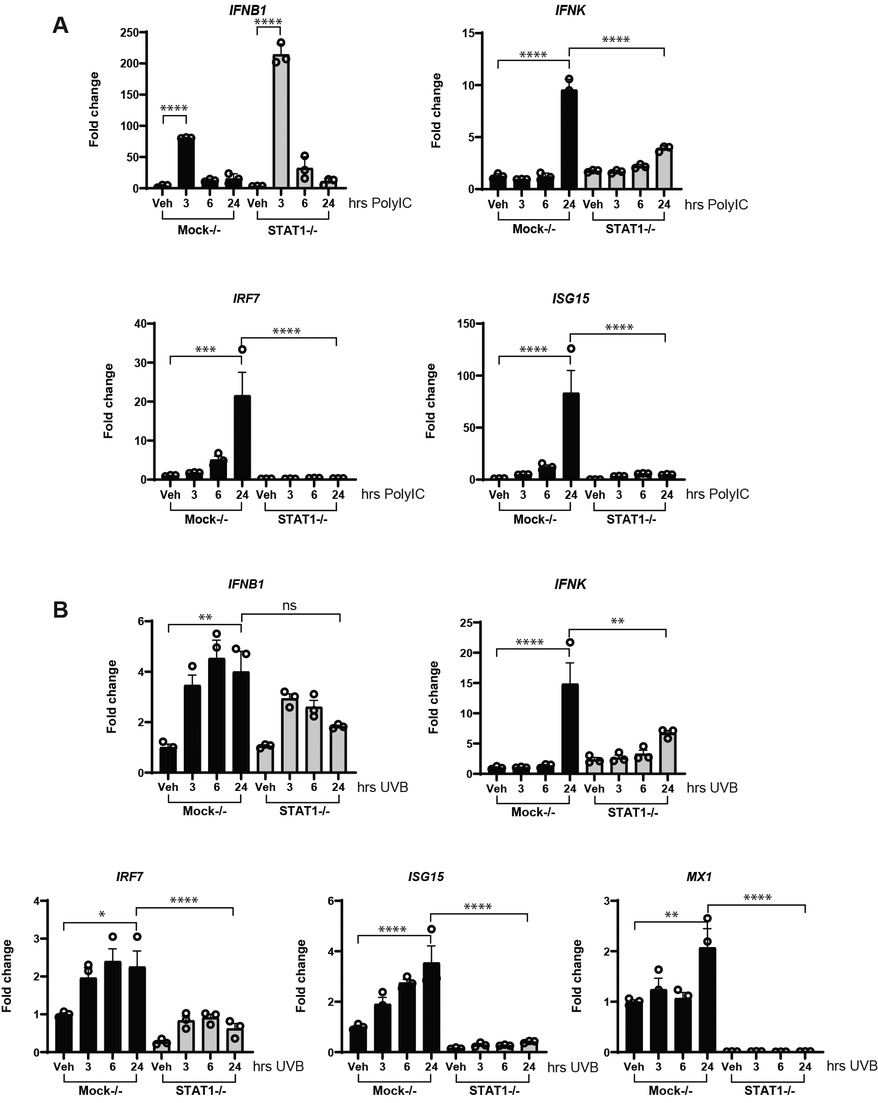
Figure 6. STAT1 is required for upregulation of IFNK and ISG. Mock−/− or STAT1−/− N/TERT keratinocytes were induced with Poly(I:C) (10 μg/ml) in (A), or exposed to UVB radiation (50 mJ/cm2) in (B), followed by RNA isolation and RT-qPCR for gene expressions at the indicated time points. Data were normalized to β-actin expression as fold change to vehicle without treatment. n = 3 in triplicate for each assay. Significance compared to vehicle was analyzed by 1-way ANOVA for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.01, ****p < 0.0001, n = 3.
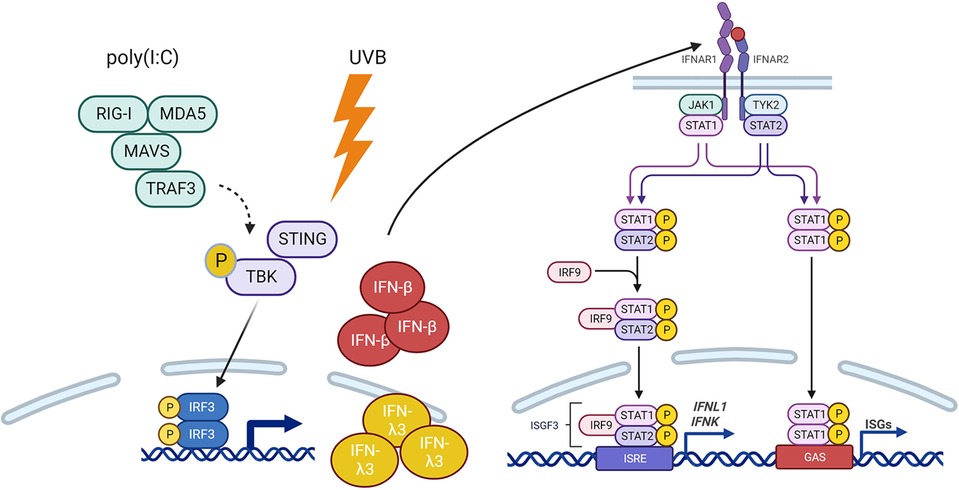
Figure 7. Model of IFNβ-STAT1 signaling for production of IFNK. IFNβ is required for IFNAR-mediated P-STAT1-driven production of IFNK, IFNL1 and ISGs in response to Poly(I:C) and UV treatment.
Discussion
Type I interferons have a crucial role in the pathogenesis of systemic lupus erythematosus (SLE) and other autoimmune diseases and are critical regulators of viral defense (26). Almost every cell type produces type I IFNs (leukocytes, fibroblasts, and endothelial cells), but the primary producers of IFNs may be context- and disease-dependent. While IFN-κ has been found to be chronically upregulated in autoimmune skin diseases such as cutaneous lupus and dermatomyositis (4, 5, 27), the reasons for this remain unclear. Here, we have identified a critical dependence on IFNB for IFNK upregulation. Indeed, IFNK behaves as an IFN response gene after triggers that activate IFNB1 such as poly(I:C) and UV light. These findings are in line with a primary role for IFNB in other stromal cell populations in response to triggers such as poly(I:C) and lipopolysaccharide (28).
The transcriptional regulation of type I interferon gene expression involves the combinatorial action of distinct classes of sequence-specific transcription factors including IRF3, IRF7, NF-κB, and activator protein 1 (AP1,) each of which are activated through upstream kinases activated in response to viral infection (29, 30). Activation of the interferon-β (IFN-β) gene requires assembly of an enhanceosome containing ATF-2/c-Jun, IRF-3/IRF-7, and NFκB (31, 32), IFNA requires IRF7, and IFNL is regulated by IRF3, IRF7, p50/p65 (31, 33, 34). A recent report identified an enhancer ∼5 kb upstream of the IFNK gene driving its expression in keratinocytes. The enhancer consists of binding sites for the transcription factors jun-B, SMAD3/4, AP-2α/γ, and p63, of which the latter two are key regulators of keratinocyte biology (34). Our analysis of the promoter also confirms that there are few IRF3 binding sites near the IFNK transcriptional start site, supporting alternative regulation from classic IRF3 binding as is seen with IFNB.
While our data highlight an important role for IFNB1 in initiation of IFN responses, we note that IFNB1 production is rapid and transient whereas IFNK is upregulated in a delayed fashion and may be responsible for more chronic IFN responses. Indeed, we have previously published that tonic keratinocyte IFN signatures are entirely dependent on IFNK expression (4). This then raises the question as to why IFNK responses are so elevated in autoimmune skin diseases such as SLE. Transcriptional repression of IFNK in keratinocytes can be induced via hypermethylation of the IFNK promoter. Viruses, especially papilloma viruses, utilize this mechanism to downregulate the antiviral responses driven by IFN-κ (35, 36). Other signaling pathways also negatively regulate IFNK expression. Epidermal growth factor receptor (EGFR) signaling is important to minimize IFN signaling in keratinocytes (37). In addition, tonic repression of IFNK in keratinocytes occurs through Mitogen Activated Protein Kinase Kinases (MEK 1 and 2) as addition of MEK inhibitors induce IFNK expression and upregulation of IFN-response genes in an IFN-κ- and STAT-1 dependent manner (38, 39). Further investigation of whether dysfunction of these negative regulatory pathways contribute to high IFN-κ in SLE skin is warranted.
In the skin in established SLE there is also production of type III interferons (IFNλ), which have structural resemblance to IL-10, but similar signaling and downstream cellular effects to type I interferons (25). Indeed, in mice, deletion of the receptor for IFN-λ results in reduced skin inflammation in the MRL/lpr model (40). Intriguingly, we show that loss of IFNB1 in keratinocytes dramatically downregulates the induction of IFNL1, similar to IFNK. IFNL3, however, retains an early and robust upregulation without IFNB present, suggesting its regulation may be more akin to IFNB1. The interplay of type I and type III IFNs in the epidermis deserve additional study.
In sum, our work supports a mechanistic paradigm in keratinocytes in which drivers of IRF3 activation rapidly induce IFNB and IFNL3 and that downstream upregulation of IFNK requires IFN-β driven STAT1 activation. Thus, IFNK appears to be an IFN that is an ISG dependent on IFN-β. Mechanisms that result in chronic upregulation of IFNK in lupus skin when IFNB is not detectible require further study and may be important novel targets for therapy.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
BX: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. JM: Data curation, Formal Analysis, Investigation, Writing – review & editing. YT: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – review & editing. GH: Formal Analysis, Investigation, Methodology, Writing – review & editing. WS: Data curation, Formal Analysis, Investigation, Methodology, Writing – review & editing. BK: Methodology, Resources, Writing – review & editing. JQ: Formal Analysis, Investigation, Writing – review & editing. MS: Methodology, Resources, Writing – review & editing. JG: Resources, Supervision, Writing – review & editing. JK: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Bethesda, MD) of the National Institutes of Health under award numbers R01-AR071384 and K24AR076975 (JMK) and by the German Research Foundation (BK). YST and JMK also received a pilot grant for this work from the U-M Skin Biology and Diseases Resource Center under award P30AR075043 (JEG PI).
Conflict of interest
JK has received grant support from Q32 Bio, Celgene/Bristol-Myers Squibb, Ventus Therapeutics, Rome Therapeutics, and Janssen. JK has served on advisory boards for AstraZeneca, Bristol-Myers Squibb, Eli Lilly, EMD serrano, Exo Therapeutics, Gilead, GlaxoSmithKline, Aurinia Pharmaceuticals, Rome Therapeutics, and Ventus Therapeutics. JG has received support from Eli Lilly, Janssen, BMS, Sanofi, Prometheus, Almirall, Kyowa-Kirin, Novartis, AnaptysBio, Boehringer Ingelheim, Regeneron, Abbvie, and Galderma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/flupu.2024.1359714/full#supplementary-material
References
1. Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. (2017) 214(5):1547–55. doi: 10.1084/jem.20161451
2. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14(1):36–49. doi: 10.1038/nri3581
3. Psarras A, Wittmann M, Vital EM. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat Rev Rheumatol. (2022) 18(10):575–90. doi: 10.1038/s41584-022-00826-z
4. Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. (2018) 77(11):1653–64. doi: 10.1136/annrheumdis-2018-213197
5. Stannard JN, Reed TJ, Myers E, Lowe L, Sarkar MK, Xing X, et al. Lupus skin is primed for IL-6 inflammatory responses through a keratinocyte-mediated autocrine type I interferon loop. J Invest Dermatol. (2017) 137(1):115–22. doi: 10.1016/j.jid.2016.09.008
6. Psarras A, Alase A, Antanaviciute A, Carr IM, Md Yusof MY, Wittmann M, et al. Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nat Commun. (2020) 11(1):6149. doi: 10.1038/s41467-020-19918-z
7. Iwasaki A. A virological view of innate immune recognition. Annu Rev Microbiol. (2012) 66:177–96. doi: 10.1146/annurev-micro-092611-150203
8. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. (2013) 38(5):870–80. doi: 10.1016/j.immuni.2013.05.004
9. Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol. (2019) 10:325. doi: 10.3389/fimmu.2019.00325
10. Yu Y, Xue X, Tang W, Su L, Zhang L, Zhang Y. Cytosolic DNA‒mediated STING-dependent inflammation contributes to the progression of psoriasis. J Invest Dermatol. (2022) 142(3 Pt B):898–906.e4. doi: 10.1016/j.jid.2021.08.430
11. Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, et al. The early local and systemic type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci Rep. (2020) 10(1):7908. doi: 10.1038/s41598-020-64865-w
12. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. (2002) 3(9):651–62. doi: 10.1038/nrm909
13. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity. (2012) 36(4):503–14. doi: 10.1016/j.immuni.2012.03.013
14. LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem. (2001) 276(43):39765–71. doi: 10.1074/jbc.M102502200
15. Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. (2000) 20(4):1436–47. doi: 10.1128/MCB.20.4.1436-1447.2000
16. Swindell WR, Beamer MA, Sarkar MK, Loftus S, Fullmer J, Xing X, et al. RNA-seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared MyD88-dependent gene signature. Front Immunol. (2018) 9:80. doi: 10.3389/fimmu.2018.00080
17. Sarkar MK, Uppala R, Zeng C, Billi AC, Tsoi LC, Kidder A, et al. Keratinocytes sense and eliminate CRISPR DNA through STING/IFN-kappa activation and APOBEC3G induction. J Clin Invest. (2023) 133(9):e159393. doi: 10.1172/JCI159393
18. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. (2013) 8(11):2281–308. doi: 10.1038/nprot.2013.143
19. Kwock JT, Handfield C, Suwanpradid J, Hoang P, McFadden MJ, Labagnara KF, et al. IL-27 signaling activates skin cells to induce innate antiviral proteins and protects against Zika virus infection. Sci Adv. (2020) 6(14):eaay3245. doi: 10.1126/sciadv.aay3245
20. Xu B, O'Donnell M, O'Donnell J, Yu J, Zhang Y, Sartor MA, et al. Adipogenic differentiation of thyroid cancer cells through the Pax8-PPARgamma fusion protein is regulated by thyroid transcription factor 1 (TTF-1). J Biol Chem. (2016) 291(37):19274–86. doi: 10.1074/jbc.M116.740324
21. Swindell WR, Stuart PE, Sarkar MK, Voorhees JJ, Elder JT, Johnston A, et al. Cellular dissection of psoriasis for transcriptome analyses and the post-GWAS era. BMC Med Genomics. (2014) 7:27. doi: 10.1186/1755-8794-7-27
22. Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. (2007) 23(2):257–8. doi: 10.1093/bioinformatics/btl567
23. Swindell WR, Sarkar MK, Stuart PE, Voorhees JJ, Elder JT, Johnston A, et al. Psoriasis drug development and GWAS interpretation through in silico analysis of transcription factor binding sites. Clin Transl Med. (2015) 4:13. doi: 10.1186/s40169-015-0054-5
24. Csumita M, Csermely A, Horvath A, Nagy G, Monori F, Göczi L, et al. Specific enhancer selection by IRF3, IRF5 and IRF9 is determined by ISRE half-sites, 5′ and 3′ flanking bases, collaborating transcription factors and the chromatin environment in a combinatorial fashion. Nucleic Acids Res. (2019) 48(2):589–604. doi: 10.1093/nar/gkz1112
25. Alase AA, El-Sherbiny YM, Vital EM, Tobin DJ, Turner NA, Wittmann M. IFNlambda stimulates MxA production in human dermal fibroblasts via a MAPK-dependent STAT1-independent mechanism. J Invest Dermatol. (2015) 135(12):2935–43. doi: 10.1038/jid.2015.317
26. Ronnblom L, Alm GV. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol. (2001) 22(8):427–31. doi: 10.1016/S1471-4906(01)01955-X
27. Tsoi LC, Gharaee-Kermani M, Berthier CC, Nault T, Hile GA, Estadt SN, et al. IL18-containing 5-gene signature distinguishes histologically identical dermatomyositis and lupus erythematosus skin lesions. JCI Insight. (2020) 5(16):e139558. doi: 10.1172/jci.insight.139558
28. Nistler R, Sharma A, Meeth K, Huard C, Loreth C, Kalbasi A, et al. TLR Stimulation produces IFN-beta as the primary driver of IFN signaling in nonlymphoid primary human cells. Immunohorizons. (2020) 4(6):332–8. doi: 10.4049/immunohorizons.1800054
29. Levy DE, Marie IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol. (2011) 1(6):476–86. doi: 10.1016/j.coviro.2011.11.001
30. You F, Wang P, Yang L, Yang G, Zhao YO, Qian F, et al. ELF4 Is critical for induction of type I interferon and the host antiviral response. Nat Immunol. (2013) 14(12):1237–46. doi: 10.1038/ni.2756
31. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. (2005) 434(7034):772–7. doi: 10.1038/nature03464
32. Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. (2007) 129(6):1111–23. doi: 10.1016/j.cell.2007.05.019
33. Iversen MB, Paludan SR. Mechanisms of type III interferon expression. J Interferon Cytokine Res. (2010) 30(8):573–8. doi: 10.1089/jir.2010.0063
34. Klein K, Habiger C, Iftner T, Stubenrauch F. A TGF-beta- and p63-responsive enhancer regulates IFN-kappa expression in human keratinocytes. J Immunol. (2020) 204(7):1825–35. doi: 10.4049/jimmunol.1901178
35. Habiger C, Jager G, Walter M, Iftner T, Stubenrauch F. Interferon kappa inhibits human papillomavirus 31 transcription by inducing Sp100 proteins. J Virol. (2016) 90(2):694–704. doi: 10.1128/JVI.02137-15
36. Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, et al. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-kappa transcription in keratinocytes. PLoS One. (2014) 9(3):e91473. doi: 10.1371/journal.pone.0091473
37. Lulli D, Carbone ML, Pastore S. Epidermal growth factor receptor inhibitors trigger a type I interferon response in human skin. Oncotarget. (2016) 7(30):47777–93. doi: 10.18632/oncotarget.10013
38. Hile GA, Gudjonsson JE, Kahlenberg JM. The influence of interferon on healthy and diseased skin. Cytokine. (2020) 132:154605. doi: 10.1016/j.cyto.2018.11.022
39. Lulli D, Carbone ML, Pastore S. The MEK inhibitors trametinib and cobimetinib induce a type I interferon response in human keratinocytes. Int J Mol Sci. (2017) 18(10):2227. doi: 10.3390/ijms18102227
Keywords: interferon, keratinocyte, lupus, ultraviolet B, STAT1, poly(I:C)
Citation: Xu B, Musai J, Tan YS, Hile GA, Swindell WR, Klein B, Qin JT, Sarkar MK, Gudjonsson JE and Kahlenberg JM (2024) A critical role for IFN-β signaling for IFN-κ induction in keratinocytes. Front. Lupus 2:1359714. doi: 10.3389/flupu.2024.1359714
Received: 21 December 2023; Accepted: 8 March 2024;
Published: 19 March 2024.
Edited by:
Antony Psarras, University of Oxford, United KingdomReviewed by:
Tammy (Tamara) Nowling, Medical University of South Carolina, United StatesJillian M. Richmond, University of Massachusetts Medical School, United States
© 2024 Xu, Musai, Tan, Hile, Swindell, Klein, Qin, Sarkar, Gudjonsson and Kahlenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Michelle Kahlenberg mkahlenb@umich.edu