RNAseq Reveals Sensitive, Concentration-Dependent Transcriptional Markers of Copper in Mytilus californianus Larvae and Adults
 Megan R. Hall
Megan R. Hall James W. Moffett
James W. Moffett Andrew Y. Gracey
Andrew Y. Gracey- Department of Biological Sciences, University of Southern California, Los Angeles, CA, United States
Copper contamination of coastal waters is a long-standing problem in many regions. Copper water quality criteria in marine waters is often determined with bivalve embryo-larval toxicity tests which measure survival and normal development as endpoints. Gene expression data is increasingly incorporated into such assays as a complementary and sensitive marker of contaminant exposure or toxicity. Here, we measured the impacts of copper on Mytilus californianus larval transcriptional profiles, and identified sensitive biomarkers of copper exposure. Key functional categories that were identified among these genes include biomineralization/shell formation, metal binding, and development. Finally, we compared the transcriptional response of larvae to that of adult gill tissue, and show in both datasets that patterns of declining transcript expression occur at lower copper concentrations than those required to induce increases in transcript expression, suggesting that down-regulated genes serve as the most sensitive marker of copper exposure.
Introduction
In recent decades, improvements in coastal water quality have been achieved by regulation of discharges of contaminants from point sources. These regulations have been based in part on toxicity testing of marine species and on documenting the accumulation of contaminants in their tissues (EPA, 1985). Mussels of the genus Mytilus have been regularly used for such testing because they occur in almost every coastal habitat and are filter feeders. As larvae, Mytilus are highly sensitive to their chemical environment, so short-term bivalve embryo-larval toxicity assays are standard to determine the toxicity of metals, effluent, and numerous other chemicals (EPA, 1995; E50 Committee, 2013), and LC50 data arising from laboratory testing of these effects serve as the criteria for setting water quality regulations (Rivera-Duarte et al., 2005). The bay mussel species, Mytilus edulis and M. galloprovincialis, have been most widely used in contaminant testing, but their deployment presents challenges because together with M. trossulus, they are almost impossible to distinguish morphologically and can hybridize with one another (Bierne et al., 2003; Riginos and Cunningham, 2005), making it very difficult to standardize testing protocols without genetic analysis of the animals (Arnold et al., 2009).
Bivalve embryo-larval toxicity assays currently use abnormal development as the most sensitive endpoint, and rarely consider other detrimental effects that may occur at low contaminant concentrations. Over the past two decades, advances in large-scale gene expression analysis have created the potential to provide more sensitive indicators of toxicity, while simultaneously characterizing biochemical and physiological regulation in response to a toxin (Ankley et al., 2006; Calzolai et al., 2007; Schirmer et al., 2010; Hahn, 2011). Transcriptional characterization may also reveal unexpected pathways involved in the toxicity of a chemical (Poynton et al., 2007). Ultimately from these datasets, biomarker genes can be identified that are highly correlated with toxin exposure (Nordberg, 2010), or with some negative outcome at the whole-organism or population level (Ankley et al., 2010; Connon et al., 2010).
Copper is a common environmental pollutant in coastal waters globally (Sadiq, 1992), but Southern California has been a hotspot for copper contamination (Schiff et al., 2003, 2007; Rivera-Duarte et al., 2005). Mytilus mussels are a key component of ecosystems in this region, and thus provide an environmentally relevant model for copper research. The effects of copper on larval Mytilus spp. survival and development are relatively well studied (Johnson, 1988; Hoare et al., 1995a, b; Arnold et al., 2009), yet there has been little investigation into the biochemical effects of copper on this sensitive stage, nor on the potential molecular drivers of developmental abnormality. All investigations of Mytilus spp. global transcriptome responses to toxins have been conducted in adults (Venier et al., 2006; Negri et al., 2013; Varotto et al., 2013; Xu et al., 2016), so we know little about how early life history stages respond to toxins at the transcriptional level, especially at low, sublethal concentrations.
Traditional endpoints of toxicity assays (i.e., developmental abnormality) are often measured in concentration–response experiments, which allow for modeling of the response to a toxin and for consistent determination of biologically relevant concentration thresholds, such as the lowest observed effect concentration (Walker et al., 2000). The transcriptional response to a drug or toxin can also be analyzed in a dose–responsive fashion (Daston, 2008; Ji et al., 2009, 2011). Concentration–responsive transcriptional analysis has been used to some extent in ecotoxicological investigations (Poynton et al., 2008; Brulle et al., 2010; Whitehead et al., 2010), yet most transcriptomic studies assess only a few concentrations, if more than one is even used. Transcriptional dose–response or concentration–response experiments allow for a more complete characterization of the discrete physiological responses to different levels of a toxin (Denslow et al., 2007). More specifically, concentration-specific biomarkers can be identified, which could provide a high-resolution metric of toxin-exposure level in environmental monitoring. Biomarkers that respond to low concentrations of a contaminant can be especially useful for identifying sublethal physiological changes that may not be evident at the whole-organism level.
This work presents an analysis of concentration–responsive transcription in Mytilus californianus larvae and adults after short-term (48 h or 24 h, respectively) exposures to copper. This species was chosen because M. californianus is morphologically distinct from bay mussels, possesses a ribbed shell, and cannot hybridize with other Mytilus species. Reports indicate that incorrect identification of bay mussel species can confound efforts to standardize regulatory criteria and have called for data collected in M. californianus to be added to toxicity databases (Arnold et al., 2009). We aimed to identify low-concentration transcriptional biomarkers of short-term copper exposure, and to characterize functional pathways associated with a range of copper exposure concentrations. We also compared the concentration-dependent copper response of adults and larvae, and identified transcriptional biomarkers of copper exposure and toxicity. The biomarkers identified in this study represent robust indicators of copper exposure that could be incorporated into environmental monitoring and toxicity testing.
Materials and Methods
Preparation of Mussel Larvae
Adult M. californianus were collected from Santa Monica, California and transported to the Wrigley Marine Science Center (WMSC) on Catalina Island, where they were held in a subtidal cage hanging from the dock for 1.5 years prior to spawning. Although M. californianus does not appear to exhibit significant population differences throughout its range (Addison et al., 2008), we further sought to avoid potential population-level sources of genetic variation by collecting all the mussels from the same site in Santa Monica. Animals were collected for two trial spawns during May–June of 2013. During both experiments, 15–20 animals were removed from the subtidal cage, and placed directly on ice for 1 h. To induce spawning, mussels were then rinsed in fresh DI water, scraped clean of any fouling organisms, and transferred to a tank of filtered seawater at 23–25°C. Mussels were sometimes fed a small quantity of Shellfish Diet 1800 (Reed Mariculture) to induce spawning if thermal shock alone was not sufficient.
Once an adult mussel started spawning, it was removed from the main tank, rinsed thoroughly with filtered seawater, and transferred to a separate beaker containing 0.2 μm filtered seawater. Each individual that spawned was held alone in a beaker to prevent cross-contamination of gametes. When spawning was complete, the adult mussels were removed from the beakers, and the appearance of eggs and motility of sperm were examined under low power on a compound microscope. Once eggs had transformed from club-shaped to round, sperm from a single male was added to eggs of a single female to reach an average density of ∼5 sperm per egg. Fertilization was generally completed within ∼30 min and the number of fertilized embryos, evidenced by the formation of a polar body and first division of the zygote, was counted.
Embryo-Larval Toxicity Assay
Copper solutions were prepared in 0.2 μm filtered natural seawater (34 parts per thousand salinity) using a 1 mM stock solution of copper sulfate (CuSO4). Each copper exposure experiment consisted of one control container and nine containers with increasing copper concentrations—2, 3.1, 4, 6, 8, 10, 15, 20, and 25 μg/L Cu. Larval exposure concentrations were selected based on EC50 and LC50 values in the literature (e.g., Martin et al., 1981; Arnold et al., 2009), as well as pilot work conducted by our lab. For the purposes of our experiment, a greater number of exposure concentrations was more important than technical replicates of each concentration. The experiment relied on the notion that more concentrations are important to effectively model concentration response of transcripts, as opposed to fewer concentrations with more replicates, which would be favorable for use of ANOVA or differential expression analysis (“regression” design vs. “ANOVA” design, described by Graney, 1993). As our study primarily sought to model transcriptional dose responses, we opted for higher resolution in the concentration, with counts from each larger container serving as replicates. This is similar to the approach taken in Ji et al. (2009), the study that introduced and first applied the Sigmoidal Dose Response Search program (described in more detail below).
After spiking copper into each container, jars were mixed thoroughly and allowed to equilibrate for 1 h. Embryos were transferred into the 1L treatment containers at a density of 10 per mL (total count of 10,000 per container), and incubated at 20°C with a 16:8 h L:D cycle. After a 48-h incubation, 5 replicate 10 mL samples were taken from each culture and preserved with EtOH for counting and morphological analysis. The 48-h larval exposure time was chosen to match the industry standard for 48-h bivalve toxicity assays (EPA, 1995), which is based on the standard time it takes for bivalve embryos to reach the D-hinge larval stage. Microscope counts of larvae were used to calculate the number of larvae surviving and normally developing in each container. To calculate the proportion of control-normalized survival and normal development, each estimated number was divided by the mean estimated number of larvae surviving in the no-copper control. These normalized proportions were analyzed in the r package ‘drc’ (Ritz et al., 2015), with replicate counts from each concentration serving as replicates for the model. Four-parameter log-logistic curves were fitted to each dataset to calculate 50% lethal concentration (LC50) and 50% normal development effective concentration (EC50) values. The remainder of each culture was collected on a 20 μm sieve, concentrated by centrifugation at 2,000 × g for 4 min, and larval pellets were frozen at −80°C for subsequent RNA extraction.
RNA Sequencing of Larval Samples
A total of 20 samples were processed for RNAseq (9 copper concentrations and 1 control seawater sample × 2 copper exposure experiments). Total RNA was extracted using Trizol (Thermo Fisher) in MaxTract tubes (Qiagen). RNAseq libraries were prepared for next generation sequencing using a modified version of the Illumina TruSeq protocol, and the 20 libraries were multiplexed and sequenced across two lanes on the Illumina HiSeq 2500 platform as 50 bp single end reads. In addition to the libraries prepared for experimental samples, two longer libraries were prepared for transcriptome assembly. The first was prepared using larvae exposed to 8 μg/L Cu for 48 h, and was sequenced on one lane of an Illumina HiSeq 2500 as 150 bp single end reads. The second library was prepared from larvae reared under control conditions for 48 h, processed using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina, and run over 10% of an Illumina NextSeq lane as 75 bp paired-end reads. The resulting raw sequence data is deposited in the Sequence Read Archive (SRA) database on NCBI under BioProject ID PRJNA639354.
Larval Transcriptome Assembly
Reads collected in the two assembly sequencing runs were quality trimmed and vetted for adapter sequence contamination in Trimmomatic v0.33 (Bolger et al., 2014). (ILLUMINACLIP:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36). Paired end reads were merged using fq2fa, part of IDBA v1.1.1(–merge –filter) (Peng et al., 2013). Both sets of reads were then assembled individually with IDBA-tran (maxk 124). For the longer read library (150 bp), the short.sequence.h script in IDBA-tran was edited to run on longer sequences by changing the kMaxShortSequence parameter to 200. Both resulting assemblies were merged into one file with a previously published Expressed Sequence Tag (EST) library (Gracey et al., 2008), and further consolidated by running cd-hit-est v4.6.5 (-c 0.98) (Li and Godzik, 2006; Fu et al., 2012) once, and CAP3 three times (–o 50 –p 98) (Huang and Madan, 1999). Cd-hit-est was then run once more (-c 0.95). Contigs corresponding to mitochondrial and ribosomal RNAs were removed.
Peptides were predicted for the resulting assembly (Supplementary File S1), and the contigs and predicted peptides were annotated using Trinotate v 2.0.2 with default parameters (Haas et al., 2013). Full taxonomic paths for hits from the nt NCBI BLAST database1 were attained by parsing the NCBI taxonomic files. UniProt annotations from Trinotate were filtered to only include hits with E-values lower than 1e-15, and hits to the nr NCBI BLAST database (see text footnote 1) were filtered to only include best hits with E-values lower than 1e-20. All annotations were searched and only those containing the term ‘Metazoan’ in the taxonomic path were retained. Contigs with a ‘Metazoan’ annotation from either the UniProt or nt database were retained. The final assembly consisted of 23,025 contigs with an average length of 1905.38 bp (Supplementary File S2).
Larval RNAseq Mapping
For mapping, RNAseq reads were quality trimmed and contaminating adapter sequence was removed using Trimmomatic v0.33 (Bolger et al., 2014) with default parameter settings. Reads arising from mitochondria were removed by mapping to the M. californianus mitochondrial genome using BBMap v34 (minid = 0.95 ambiguous = all sssr = 1.0) (Bushnell, n.d.). The remaining reads were mapped to our M. californianus transcriptome assembly detailed above with BBMap v34 (minid = 0.95, ambiguous = all, sssr = 1.0, nhtag = t). Read counts were obtained using FeatureCounts v1.5.0-p3 (Liao et al., 2014), counting on the feature level and only allowing for uniquely mapping reads to be counted (-T 8 -O -F). The two samples from the highest copper concentrations (25 μg/L) were removed at this point, as the majority of read count values were 0, and the samples had consisted of very few living larvae. The resulting count table was further processed in EdgeR (Robinson et al., 2010; McCarthy et al., 2012) to generate normalized trimmed mean of M-values (TMM) counts. Methods for larval transcriptome assembly and mapping are summarized in Supplementary Figure S1.
Adult Mussel Copper Exposure and Microarray-Based Gene Expression Analysis
Similar experiments were conducted with adult animals to compare adult and larval transcriptional responses and look at commonalities and differences across these life history stages. For adults, copper exposure was performed at 15°C in individual acid-washed polycarbonate containers containing 1L of seawater that was spiked with either 3, 6, 9, 15, 30, 60, 90 or 120 μg/L copper by dilution of a CuSO4 stock solution. Concentrations were selected to span a range where no toxicity was expected for adult animals (3 ppb is lower than dissolved copper water quality criteria) through to near lethal concentrations for the adult animals (determined based on pilot work by our lab). The concentration range for adult exposure was different than for larvae because for animals of each life history stage, we wanted to elicit a full range of transcriptional responses, from non-toxic exposure through a toxic response that was associated with mortality. Because adult mussels have a much higher tolerance for copper than larval mussels, it was necessary to expose adult animals to a wider range of copper concentrations to elicit a similar response. Importantly, there was overlap in the doses that each life-history stage experienced, so larvae experienced 2, 3.1, 4, 6, 8, 10, 15, 20, and 25 μg/L and adults were exposed to 3, 6, 9, 15, 30, 60, 90 or 120 μg/L. The concentrations were extended to higher levels in the adults to reflect their higher resistance to copper, with the goal of inducing a maximal transcriptional response.
Three individual mussels of identical size (∼4 cm long) were placed in each jar with gentle aeration to maintain saturated levels of dissolved oxygen and to promote water movement. Every 4 h the water in each jar was completely replaced with a liter of freshly prepared copper-spiked seawater to compensate for copper draw-down. Two replicate containers of natural seawater without addition of copper served as controls. The exposure was continued for 24 h, and then the mussels were quickly dissected and samples of gill tissue were flash frozen and archived at −80°C. Adult mussels were exposed for 24 h because there was no standardized incubation time for adult mussel exposures, and out pilot data showed that markers of cell stress (namely HSP70 mRNA induction) were maximally expressed at 24 h of exposure. The 24-h timepoint was also intended to capture an acute response which compares more directly to the goals of the EPA’s standard mussel embryo-larval toxicity assay. Gill was analyzed because in adult mussels, the gill and mantle represent the largest tissues, with gill tissue being deployed previously in many physiological studies because of its central role in feeding and respiration and ease of dissection. More importantly, gill tissue is known to be the key site of dissolved metal uptake in aquatic mollusks (reviewed in Marigómez et al., 2002), and has thus been used regularly in studies of molluscan response to metals. Understanding tissue-specific physiological responses is ideal, but in early larvae gills are still developing (Cannuel et al., 2009), so direct tissue comparison would not be possible.
Total RNA was isolated from the 30 samples of gill tissue using Trizol (Invitrogen) and the resulting RNA was purified further across RNAeasy filter columns (Qiagen). Amplified RNA (aRNA) was prepared for each individual gill sample as previously described (Connor and Gracey, 2011). The 30 fluorescently labeled aRNA samples were hybridized to 40 M. californianus arrays (Connor and Gracey, 2011) using an interwoven loop design which yielded a balanced design in which aliquots of each labeled RNA sample were hybridized to either two or four arrays with fluorescent dye reversal. In total, expression data were generated for six replicate mussels sampled under control conditions, and three replicate mussels for each of the 8 concentrations of copper. The resulting expression data were deposited in the ArrayExpress public database as accession number E-MTAB-810. Genes whose expression varied across the series of samples were identified using analysis of variance (ANOVA) in MAANOVA Version 0.98–7 (Kerr et al., 2000) in a model in which dye was treated as a fixed effect and array was treated as a random effect, testing for differences in expression between mussels sampled after different copper concentration exposure. The p-values were adjusted for false discovery rate (Benjamini and Yekutieli, 2001) and a statistical threshold of p < 0.05 defined a list of genes that were statistically significantly differentially expressed across treatments. These differentially expressed genes were analyzed with respect to copper exposure as described below. The microarray probe sequences were aligned to the RNAseq-based transcriptome assembly using a reciprocal BLASTn search so that sequence annotation across the microarray and RNAseq datasets could be compared. Methods for adult Mytilus microarray printing, hybridization and analysis are summarized in Supplementary Figure S1.
Data Analysis
For both larval and adult data, concentration–responsive gene expression was analyzed with Sigmoidal Dose Response Search (SDRS) v0.04 (Ji et al., 2011) to identify genes with sigmoidal expression, and to parameterize the expression dose–response curve of each gene. For genes with significant (p < 0.05) sigmoidal expression, the program calculates an expression EC50, representing the concentration at which the gene’s expression was induced (or repressed) by 50% of its maximum expression value (Supplementary Figure S2). The low and high dose for the 95% confidence interval of the expression EC50 are also calculated. The lower threshold of the 95% confidence interval for the EC50 value, which is the lowest concentration of significant induction or suppression of the gene, was considered to be the lowest observed transcriptional effect concentration (LOTEC) for genes (Supplementary Figure S2).
For larval expression data, Weighted Gene Co-expression Network Analysis (WGCNA) (Langfelder and Horvath, 2007) was used to create gene co-expression networks and identify modules of genes with similar expression patterns. WGCNA was run using all genes with a summed TMM across all samples > 300, a module-merging threshold of 0.15, and distinct modules being assigned different colors. Functional enrichment of gene groupings obtained with SDRS and WGCNA were conducted using Gene Ontology (GO) (Ashburner et al., 2000) enrichment analysis in the Cytoscape (Shannon et al., 2003) plugin program, BINGO (Maere et al., 2005). Enrichment was calculated using a hypergeometric test for overrepresentation (p < 0.05), with a Benjamini and Hochberg False Discovery rate correction.
Results
Embryo-Larval Development and Survival
The proportion of larvae surviving (survival) and the proportion of larvae reaching a normal D-hinge shape (normal development) in each copper treatment at the end of the 48-h period exhibited significant sigmoidal concentration–responsive patterns in each trial (Figure 1). Based on the modeled curves, the EC50 (corresponding to the effect of copper on normal development) in exposure trial 1 was calculated to be 6.04 μg/L Cu, and the LC50 (corresponding to the effect of copper on larval survival) was 6.5 μg/L Cu. The larvae in trial 2 exhibited greater sensitivity to copper, with an EC50 of 3.78 μg/L, and an LC50 of 3.96 μg/L. Thus, despite differences in the apparent sensitivity of the different larval cultures, LC50 and EC50 values were relatively similar to one another in a given trial.
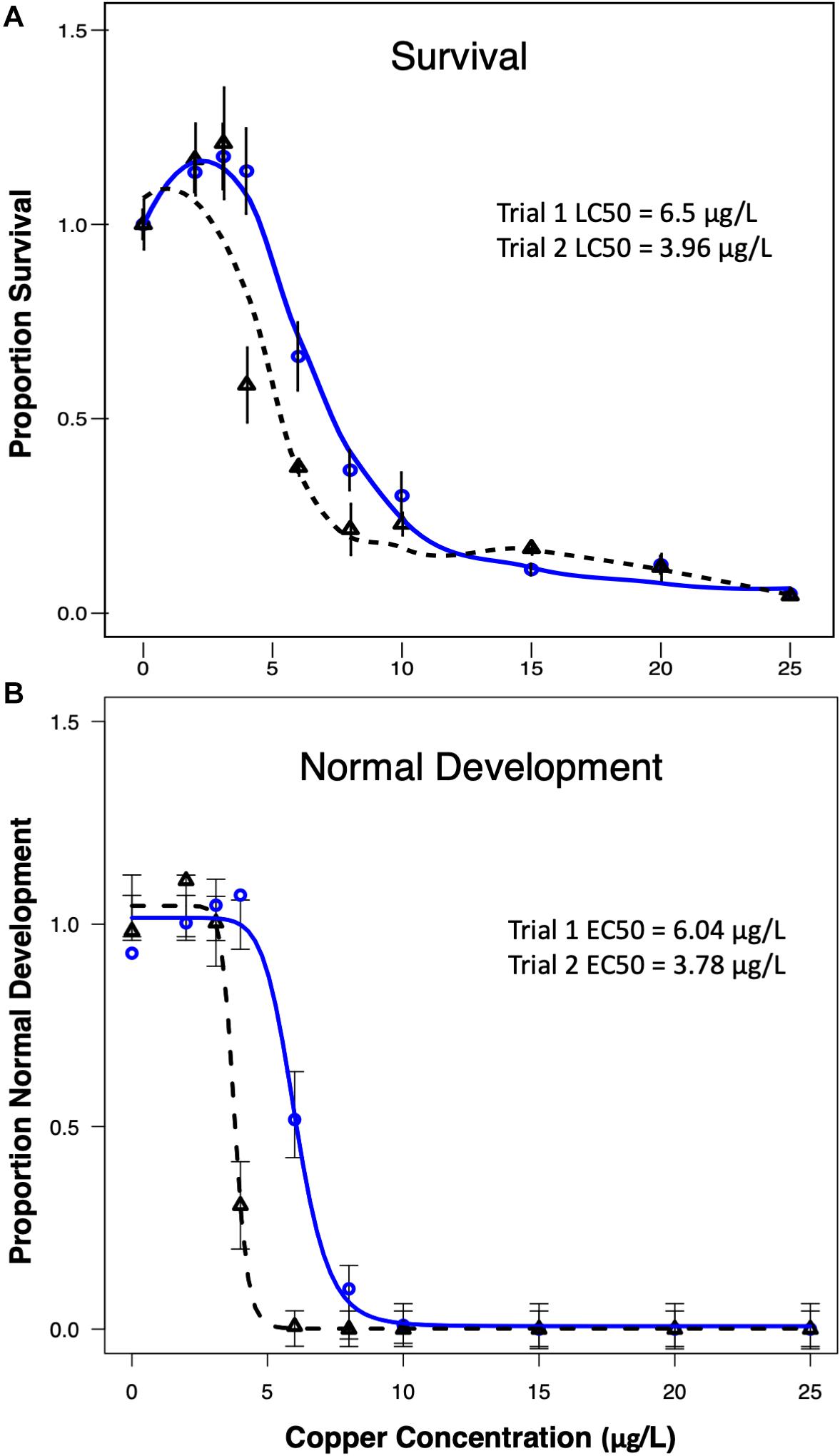
Figure 1. Control-normalized survival (A) and normal development (B) were plotted against copper concentration for trial 1 (blue line) and trial 2 (dashed black line). Data indicate that larvae were slightly more sensitive to copper in trial 2.
The Transcriptional Concentration–Response to Copper in Larvae
In larvae, 1,483 transcripts exhibited sigmoidal expression in at least one trial (Supplementary Figure S3), and 317 transcripts exhibited sigmoidal expression in both trials. Of these common transcripts, 248 were upregulated in response to increasing copper (Figure 2A), and 61 were downregulated (Figure 2B).
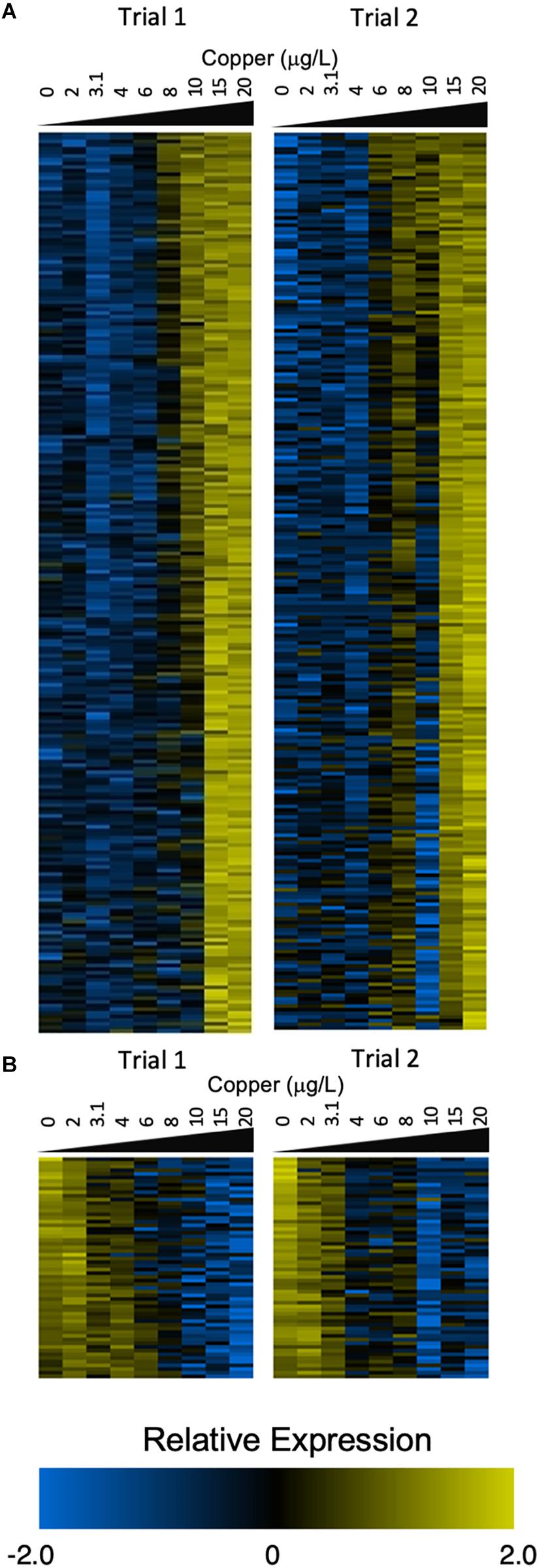
Figure 2. Heatmaps depict expression of sigmoidal genes that were upregulated (A) and downregulated (B) in both trials 1 and 2 (left and right panels). Count values are control-normalized and log2 transformed. Genes were ranked by LOTEC value to create the order shown in the heat maps (A,B).
Calculation of expression EC50 and LOTEC values provided a quantitative measurement of each gene’s responsiveness to copper allowing genes to be organized with respect to their transcriptional sensitivity to copper. Plotting the expression LOTEC and EC50 values with the corresponding phenotypic data showed that nearly all downregulated genes exhibit an expression LOTEC and EC50 that are lower than the EC50 that was calculated based on the gross phenotypic marker of normal development (Figure 3 and Supplementary Figure S4). For example in Trial 1, the mode LOTEC and mode expression EC50 were 3.46 μg/L and 3.29 μg/L, respectively, while the normal development EC50 was 6.04 μg/L Cu, and in Trial 2, the mode LOTEC and mode expression EC50 were of 0.001 μg/L and 2.12 μg/L, respectively, while the normal development EC50 was 3.78 μg/L. In contrast, almost all upregulated genes had expression LOTECs and EC50s that exceeded the normal development EC50 in each respective trial (Figure 3 and Supplementary Figures S4B,D), with a mode LOTEC of 9.63 μg/L and mode expression EC50 of 12.29 μg/L compared to a normal development EC50 of 6.04 μg/L Cu in Trial 1, and a mode LOTEC of 9.63 μg/L and mode expression EC50 of 14.23 μg/L compared to a normal development EC50 of 3.78 μg/L in Trial 2. Thus, overall transcript repression occurs at lower copper concentrations than transcript induction, and in many cases the repression of specific genes can be detected before the deleterious effects of copper exposure are phenotypically evident as abnormal development and mortality. GO enrichment analysis revealed that the genes induced by copper were enriched in functional categories related to the proteasome core complex, proteasome regulation, the ESCRT III complex, ribonucleotide binding, oxidoreductase activity, hydrolase activity, peptidase activity, and copper ion binding. Copper-repressed genes were enriched in just two categories, polysaccharide binding and metabolism.
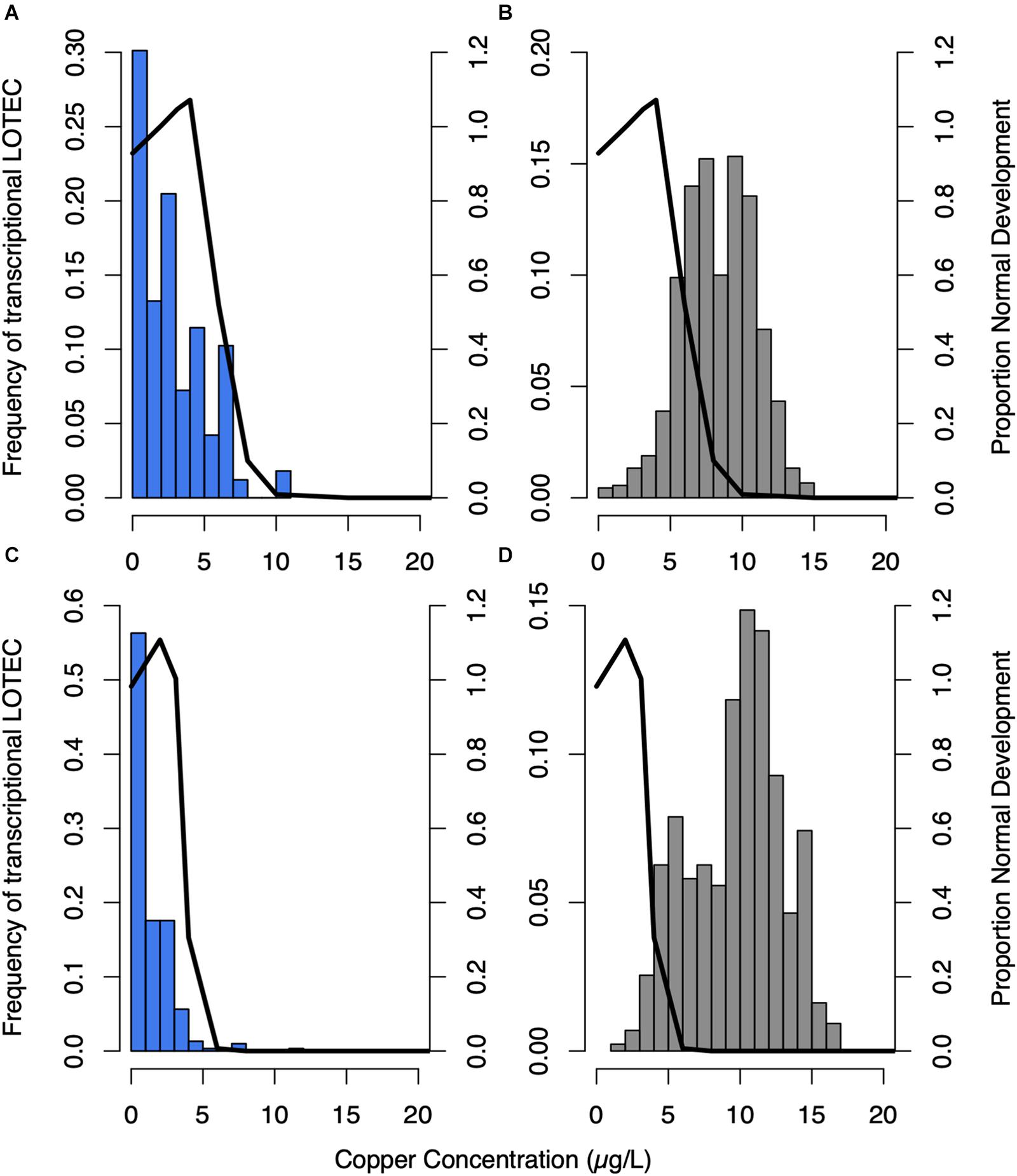
Figure 3. The frequency of sigmoidal genes’ lowest observed transcriptional effect concentration (LOTEC) was plotted as a histogram for downregulated genes in blue - trials 1 (A) and 2 (C), and upregulated genes in gray - trials 1 (B) and 2 (D). These values were compared to normal development (black line) in each trial. LOTEC thresholds are more sensitive indicators of copper-induced expression changes than EC50 values, which is evidenced by the shift toward lower copper concentrations compared to frequency histograms of EC50 values (Supplementary Figure S4).
Modularity of Dose–Responsive Gene Sets
Visual inspection of the data indicated that SDRS did not capture the full-extent of the transcriptional response because a proportion of genes did not show a significant sigmoidal expression response. Clustering co-expressed genes into modules has proven a good way to classify gene sets that respond in the same way to external stimuli, and often share involvement in the same functional pathways (Langfelder and Horvath, 2007). Therefore, WGCNA was used to construct 23 modules of co-expressed genes, of which five modules were significantly correlated (p < 0.05), with the experimental parameter (copper concentration) or the observed phenotypic response (normal development or survival) (Supplementary Figure S5). Three of the modules (colors white, yellow, and steel blue), which collectively comprised 326 transcripts, captured down-regulated expression in response to copper (Supplementary Figure S5). Indeed, 85% (52 transcripts) of the sigmoidally down-regulated genes identified by SDRS were represented in these three modules, indicating that WGCNA effectively captured an expanded set of copper-repressed transcripts. Examination of the expression EC50 and LOTEC values for the SDRS genes associated with each module indicated that the steel blue module contained transcripts that were most sensitive to copper, while transcripts in the yellow module were the least sensitive (Table 1). The most prominent functional category of down-regulated genes was related to development (Table 1 and Supplementary Table S1) and included genes involved in cell differentiation and organogenesis (Supplementary Figure S6A), neural and muscular development (Supplementary Figure S6B), and muscle formation (Supplementary Figure S6E). Another functional grouping was genes associated with biomineralization and shell matrix proteins, which were primarily members of the white and yellow modules (Table 1; Supplementary Table S1, and Supplementary Figure S6C). Finally, genes coding for divalent metal-containing enzymes were well represented in the white module with genes primarily coding for proteins involved in the uptake or transport of zinc, iron, and molybdenum (Table 1, Supplementary Table S1, and Supplementary Figure S6D).
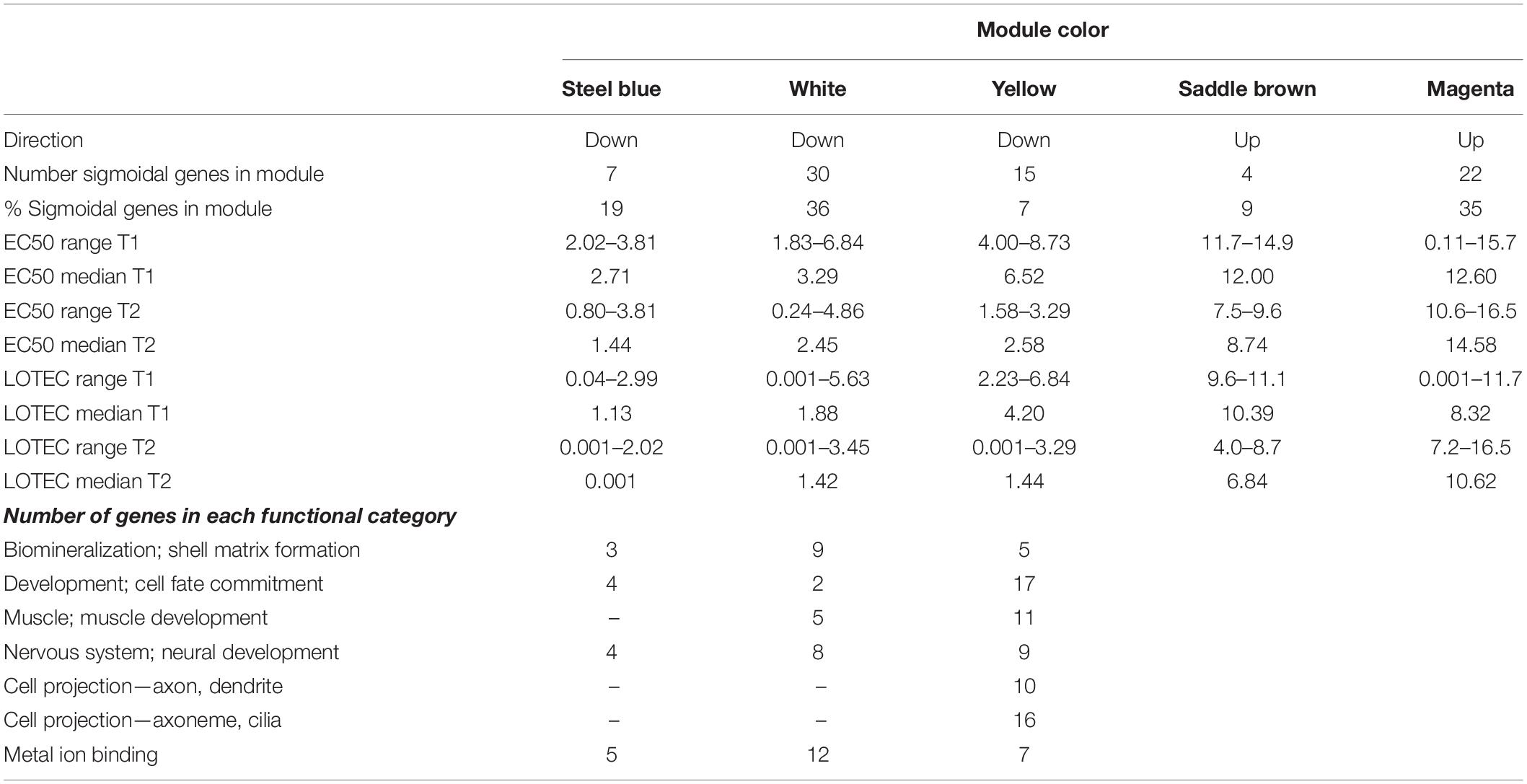
Table 1. SDRS-defined characteristics of three sensitive downregulated modules and two significant upregulated modules, and the number of genes from each down regulated module that fall within notable functional categories that were well-represented among sigmoidal and modular genes.
Weighted Gene Co-expression Network Analysis also revealed two modules of upregulated transcripts (magenta and saddle brown) that were significantly correlated with copper, and significantly anti-correlated with normal development and survival (Supplementary Figure S5). The magenta module had the most significant correlation with copper concentration of any module, and consisted of genes whose induction was first detected at 8–10 μg/L, and whose expression was further elevated at progressively high copper concentrations. The saddle brown module was characterized by substantial upregulation between 8 and 15 μg/L copper. While neither module was enriched for any specific GO terms, the saddle brown module contained numerous cysteine peptidase genes, as well as several additional genes with activity related to superoxide metabolism, lysosome processes, ubiquitination, and autophagy. Cysteine peptidases are proteases that contain an active site cysteine. Cysteine-containing proteins are redox sensitive, and can play a role in modulating metal ions that cause oxidative stress (Giles et al., 2003). The magenta module also contained several genes involved in the oxidoreductase activity or mediating the response to oxidative stress, including DBH-like monooxygenase protein 1 homolog, glutathione S-transferase 1, peroxisome biogenesis factor 10, putative ferric-chelate reductase 1 homolog, and sulfotransferase 1C4. Three subunits of cytochrome P450, an enzyme involved in responding to toxic compounds, were also upregulated as part of the magenta module.
The Transcriptional Concentration–Response to Copper in Adult Gill Tissue
ANOVA was used to identify a subset of 1,012 transcripts that were significantly differentially expressed across the adult gill tissue dataset, and of these 88% exhibited a sigmoidal concentration–response to copper according to SDRS, with 572 induced transcripts and 323 repressed transcripts (Figure 4). As was observed in larvae, transcript repression occurs at lower concentrations of copper than transcript induction in adult gill tissue, with the mode EC50 value for downregulated transcripts in adults at 12.5 μg/L, while the mode EC50 for upregulated transcripts was 22.5 μg/L (Supplementary Figure S7).
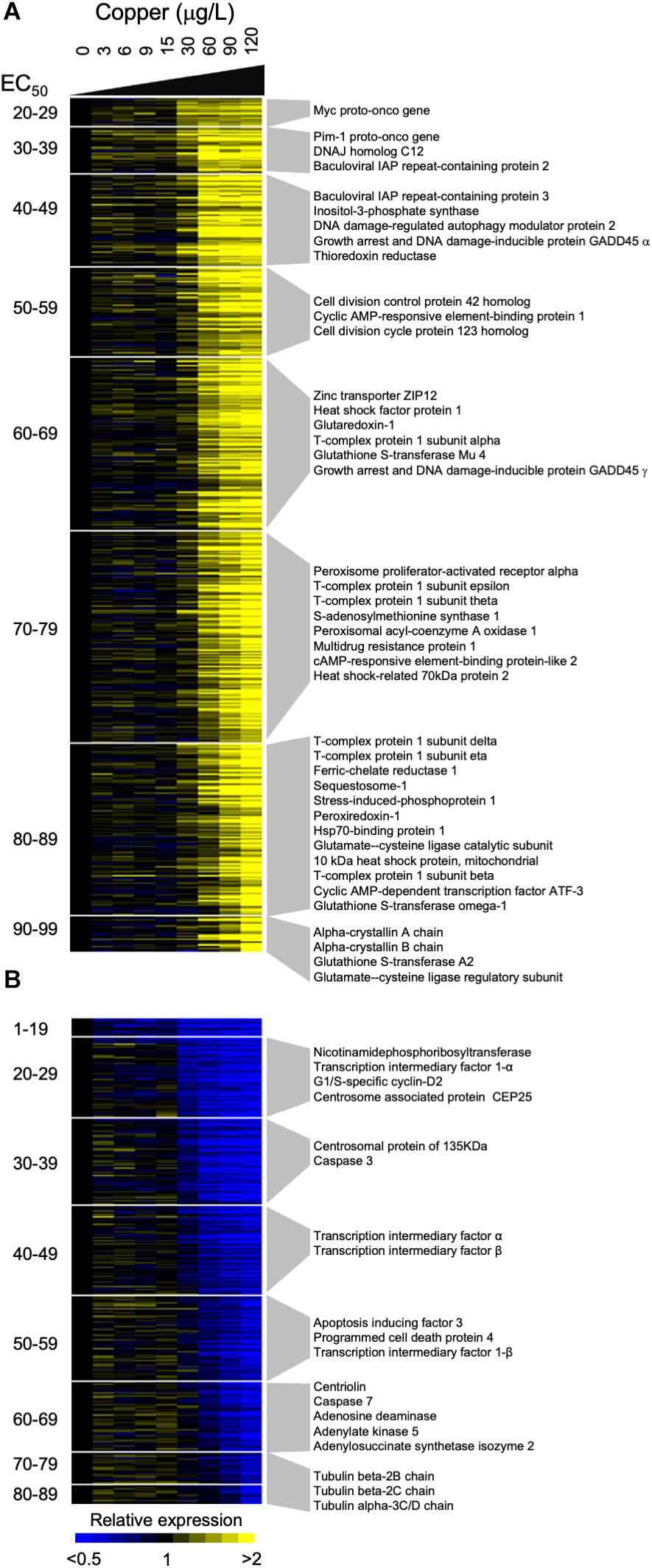
Figure 4. Gene expression dose response profiles detected in gill tissue of adult Mytilus californianus exposed to increasing concentrations of copper. Heatmaps depict induced (A) and repressed (B) genes that exhibited a statistically significant sigmoidal transcriptional dose response. Each row corresponds to a single gene and each column corresponds to a particular concentration of copper. The average expression of each gene at each concentration is represented by a color, with yellow or blue indicating that the gene is up-regulated or down-regulated relative to the expression observed in control mussels. The genes are ranked and grouped according to their observed EC50 value, with more responsive transcripts located at the top of each heatmap. Selected genes whose expression may be pertinent to the response to copper are indicated next to each grouping.
Examination of copper-induced transcripts revealed evidence of a cell stress response, with many of the induced genes being associated with protein folding, apoptosis, and oxidative stress/redox chemistry (Figure 4, Supplementary Table S2, and Supplementary Figure S8). For example, several genes that participate in the detoxification of reactive electrophilic compounds by catalyzing their conjugation to glutathione were induced, including three isoforms of the glutathione-S-transferase family of proteins (A2, Mu4, omega-1), and glutaredoxin 1. Consistent with the induction of enzymes that utilize glutathione, data indicated that expression of both the regulatory and catalytic subunits of glutamate-cysteine ligase, the enzyme which catalyzes the first step in the biosynthesis of glutathione (Krejsa et al., 2010), were induced at high concentrations of copper. Other induced transcripts with roles in the detoxification of electrophilic compounds included thioredoxin reductase 3 which catalyzes the reduction of thioredoxin (Supplementary Figure S8) (Conterato et al., 2013), and peroxiredoxin 1 which controls the level of peroxides (Ishii and Yanagawa, 2007). Also induced were three transcription factors that are activated by changes in levels of cyclic AMP and which are reported to play a pivotal role in the cellular response to oxidative stress, cyclic AMP-dependent transcription factor ATF-3, cAMP-responsive element-binding protein 1, and cAMP-responsive element-binding protein-like 2 (Hai et al., 1999; Lee et al., 2009).
The list of repressed transcripts was less coherent with respect to the functional roles of the members (Supplementary Table S2). At lower concentrations of copper we identified a down-regulation of transcripts that are associated with the cell cycle including Centrosome associated protein CEP25, centrosomal protein of 135 KDa, centriolin and G1/S-specific cyclin-D2 (Figure 4 and Supplementary Figure S8). At higher copper concentrations we detected the reduced expression transcripts associated with adenylate metabolism, such as adenylate kinase and adenosine deaminase, while at the highest concentrations we detected a decline in the expression of a number of subunits of Tubulin.
Larval Versus Adult Gill Transcriptional Response
Sigmoidal Dose Response Search analysis was more effective at capturing the expression dynamics of the adult response that that of the larvae. This likely reflects the toxicity of copper on the different life stages, with adult mussels showing no mortality even after exposure to 120 μg/L whereas exposure of larvae to just 25 μg/L resulted in close to 100% mortality. Thus, while adult mussels were capable of mounting an adaptive transcriptional response throughout the concentration range, the larvae are highly impacted at the high concentrations and die. Only 48 genes were identified as significantly sigmoidal by SDRS in both life history stages (Supplementary Table S3). This included 5 downregulated genes and 43 upregulated genes. The low number of shared genes indicates that the larval and adult responses to copper largely consist of different sets of genes. The genes that were upregulated in both datasets were generally involved in the cell stress response. This list included seven HSP-70 isoforms and HSP-binding proteins, stress-induced phosphoprotein 1 and two subunits of the T-complex chaperonin protein that assists in protein folding (Supplementary Table S3). In both datasets, we detected a particularly robust induction of sequestosome 1, a ubiquitin-binding protein involved in the transfer of ubiquitinated proteins to the proteasome, and a marker of autophagy (Myeku and Figueiredo-Pereira, 2011). The five genes that were commonly downregulated in adults and larvae did not exhibit a consistent functional theme (Supplementary Table S3).
Discussion
The M. californianus Larval Response to Copper
Normal development EC50s observed in this experiment were consistent with EC50s of Mytilus galloprovincialis, Mytilus edulis, and Mytilus californianus from previous studies (Martin et al., 1981; Arnold et al., 2009; Funk, 2014; EPA, 2016). The measured EC50s of 6.04 and 3.78 μg/L copper in the first and second trial, respectively, were higher than the U.S. Environmental Protection Agency recommended chronic water quality criteria of 3.1 μg/L copper, and the trial 1 EC50 was higher than the recommended acute criteria of 4.8 μg/L (EPA, U. S, n.d.). The observed differences in LC50 and EC50 between trials is similar to previous reports showing that mussel larvae-based water quality testing conducted by different entities can yield different results, and that copper tolerance varies between populations (Hoare et al., 1995a). In this study, trials were conducted with progeny from the same population, but from different parental crosses. Bivalves exhibit natural high-standing genetic variation, which can result in a range of phenotypes from different crosses of bivalves from the same population (Curole and Hedgecock, 2007). The differences in toxicity responses to copper reported here are expected to be linked to genetic variation between the parents used in each cross. This is manifest as left or right-shifted differences in copper concentration–response curves in each of the crosses.
Despite differences in the toxicity results of the two trials, the transcriptional response was qualitatively similar between the trials, with progeny from the more sensitive cross exhibiting a similar transcriptional response at lower concentrations than the more copper-resistant cross. Sigmoidal concentration response modeling and co-expression analysis revealed two broad patterns of modulation—transcriptional repression was a marker of exposure to lower copper concentrations, while transcript induction occurred at higher copper concentrations (Figures 2, 3). Generally, transcriptional repression, as measured by the expression LOTEC and EC50 values, occurred at copper concentrations that were below the observed normal development EC50, indicating that repressed genes will probably serve as the most sensitive biomarkers of copper exposure (Figure 3 and Supplementary Figure S3). Indeed, a number of transcripts appear to be repressed at copper concentrations at which toxicity is not apparent in the larvae with respect to development or mortality, so these genes appear to indicate copper exposure, and provide an early warning that copper concentrations are approaching toxic levels. On the other hand, transcript induction was generally initiated at higher copper concentrations, at which toxicity was evident in the larvae. Thus, transcripts that are elevated by copper can serve as markers of toxicity and the induction of a stress response, but they aren’t as useful as predictors of impending stress.
Functional Characterization of Sensitive Larval Biomarkers
Sensitive downregulated transcripts were characterized by several broad functional categories. First, genes involved in development were well-represented among the transcripts that were repressed at low copper concentrations (Table 1, Supplementary Table S1, and Supplementary Figure S6A). Downregulation of development genes has been observed before in two studies of post-larval red abalone and scallops exposed to low copper concentrations (Zapata et al., 2009; Silva-Aciares et al., 2011). Specifically, developmental genes related to the nervous and muscle system, including numerous calcium binding proteins, were notable among downregulated genes (Table 1, Supplementary Table S1, and Supplementary Figures S5B, S6E). While metals such as mercury and lead are well known for inhibiting nervous system function (Weis, 2014), data reflecting the effects of copper on neural development is sparse, although exposure of zebrafish embryos to copper reduced the formation of sensory neuroblasts that are used in swimming (Johnson et al., 2007). Several genes with regulatory roles in muscle development, such as calponin, were also down-regulated in response to copper, which is plausible given that muscle and neuronal development are closely integrated in early larval stages (Dyachuk and Odintsova, 2009). Although developmental pathways are not commonly recognized biochemical targets of copper toxicity in invertebrates, altered expression of these genes is probably not that surprising given that copper exposure results in morphological abnormalities in larva. Fundamental neuronal and muscular developmental pathways are activated early in development, generally during or immediately after the trochophore stage (Voronezhskaya et al., 2008; Dyachuk and Odintsova, 2009), and continue to progress through the early veliger stage at 48 h post-fertilization. Thus, drastic structural malformation and erratic swimming behavior of abnormal larvae could be explained by the systemic down-regulation of developmental programs and neurological function. Here, we have discovered some potential drivers of this phenomenon which precede developmental abnormality, and may be indicative of the early traces of failure. The exact mechanism by which copper causes developmental and neurological malfunction, and whether copper generally affects these genes systematically or targets primarily a few regulators, is still unclear.
A second key functional group that comprised many sensitive, down-regulated biomarkers was biomineralization and shell matrix protein genes. This group included several known and purported shell formation genes (Supplementary Figure S6C): protein PIF (Suzuki et al., 2009), chitin synthase (Schönitzer and Weiss, 2007), several tyrosine and tyrosinase genes (Aguilera et al., 2014), and components of the tyrosine metabolic pathway (Liu et al., 2015). Copper suppression of shell-formation pathways in larvae has not been observed, yet this offers additional potential explanation of the eventual failure of abnormal larvae to develop a normal D-shaped shell in the early veliger stage. The suppression of calcium binding proteins, which was mentioned as a pathway involved in nervous system development, could also be associated with failure in shell development.
Finally, genes coding for divalent metal-containing enzymes were down-regulated across a range of low copper concentrations (Table 1, Supplementary Table S1, and Supplementary Figure S6D). Interestingly, most of these genes code for enzymes that bind to divalent cations other than copper, including Zn2+, Mo2+, and Fe2+. Copper-induced downregulation of iron and zinc binding stress-protein transcripts was observed previously in juvenile abalone (Silva-Aciares et al., 2011). Metals often bind organic ligands without much specificity (Williams, 1981, 1984), and copper is the most likely to replace almost any other divalent metal at a binding site (Irving and Williams, 1953). Copper is already known to replace zinc in some proteins (Viarengo, 1985), so as copper concentration increases relative to other metal ions, enzymes may exhibit even more binding promiscuity. Thus, downregulation of metal-binding enzymes that have become functionally disrupted by erroneous copper binding may be a necessary and protective response mechanism. Similarly, downregulation of metal-binding enzymes may serve as a mechanism to maintain homeostasis by limiting metal requirements and thus limiting metal uptake from the environment.
Metallothioneins in our study showed either no response in adults, or induction and down-regulation, depending on the isoform, in larvae. Previous research also confirms that metallothionein gene expression is not a consistent marker of copper exposure. In zebra mussel larvae, metallothionein exhibited an inconsistent response to copper (Navarro et al., 2011), and adult gill in the same study also showed no response of metallothionein to copper. In our study of larvae, MT 10-III and IV were downregulated in response to copper, while MT 20-IV was upregulated. Previous research shows that MT-10 is a constitutive isoform, while MT-20 is a metal-inducible isoform, and that each isoform exhibits differential copper binding (Vergani et al., 2007). These differences could explain the opposing expression patterns of the two MT isoforms observed in our study.
The Toxic Response Across Life History Stages
We compared adult and larval transcriptional responses to look at commonalities and differences across life history stages. The functional themes associated with the sets of up-regulated genes were somewhat consistent between larvae and adults. Genes involved in oxidative stress, damaged protein turnover, and signaling were up-regulated in both life history stages. As many of these are generic cellular stress response pathways, which are highly conserved across taxa as well (Kültz, 2005), it is not surprising that these pathways are common in larvae and adults. The specific genes in each of these categories were quite different, however (Supplementary Tables S2–S4), which indicates that larvae and adults may be using different means to reach the same end of cellular defense and protein turnover. The copper-induced expression of certain genes such as heat-shock protein 70, sequestosome-1, and glutathione-S-transferase have been reported in other studies of both adult and larval mollusks suggesting that these genes likely represent universal markers of copper stress (Navarro et al., 2011; Negri et al., 2013; e.g., Zapata et al., 2009; Varotto et al., 2013; Xu et al., 2016).
The most striking expression response observed in both developing larvae and a mature adult tissue was that transcript repression began at relatively low copper concentrations, and preceded any evidence of transcript induction. This conserved pattern is consistent with what we might expect from an energetic model of stress tolerance. In such a framework, the earliest phases of stress response involve limitation of growth, development, and reproduction to divert energy to maintenance and homeostasis (Sokolova, 2013). As these processes all require metals to some extent, limitation of these processes could also reduce metal requirements and thus reduce copper uptake from the environment. As stress intensifies, pathways involved in oxidative stress and protein chaperoning are activated to reduce further harm to vital metabolic and respiratory processes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA639354; https://www.ebi.ac.uk/arrayexpress/, accession number E-MTAB-810.
Author Contributions
MH contributed to the experimental design, executed the experiments, analyzed the data, and authored the manuscript. JM contributed to the research idea and experimental design. AG contributed to the research idea and experimental design, data analysis, and authored the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The University of Southern California Provost’s Ph.D. Fellowship provided stipend to MH. The Wrigley Institute for Environmental Studies provided stipend to MH. University of Southern California Sea Grant funded research supplies through a USC Sea Grant award to AG and JM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank University of Southern California Sea Grant, the University of Southern California Office of the Provost, and the University of Southern California Wrigley Institute of Environmental Studies for funding this work. We also thank Chase A. Femrite for conducting copper exposure experiments, RNA extractions, microarray preparation and scanning, and data analysis for adult mussels. Thanks to John F. Heidelberg and Rohan Sachdeva for providing computational support and support in bioinformatic analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.572496/full#supplementary-material
Footnotes
- ^ https://ftp.ncbi.nlm.nih.gov/blast/db/; accessed September 2016.
References
Addison, J. A., Ort, B. S., Mesa, K. A., and Pogson, G. H. (2008). Range-wide genetic homogeneity in the California sea mussel (Mytilus californianus): a comparison of allozymes, nuclear DNA markers, and mitochondrial DNA sequences. Mol. Ecol. 17, 4222–4232. doi: 10.1111/j.1365-294X.2008.03905.x
Aguilera, F., McDougall, C., and Degnan, B. M. (2014). Evolution of the tyrosinase gene family in bivalve molluscs: independent expansion of the mantle gene repertoire. Acta Biomater. 10, 3855–3865. doi: 10.1016/j.actbio.2014.03.031
Ankley, G. T., Bennett, R. S., Erickson, R. J., Hoff, D. J., Hornung, M. W., Johnson, R. D., et al. (2010). Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 29, 730–741. doi: 10.1002/etc.34
Ankley, G. T., Daston, G. P., Degitz, S. J., Denslow, N. D., Hoke, R. A., Kennedy, S. W., et al. (2006). Toxicogenomics in regulatory ecotoxicology. Environ. Sci. Technol. 40, 4055–4065.
Arnold, W. R., Cotsifas, J. S., Smith, D. S., Le Page, S., and Gruenthal, K. M. (2009). A comparison of the copper sensitivity of two economically important saltwater mussel species and a review of previously reported copper toxicity data for mussels: important implications for determining future ambient copper saltwater criteria in the USA. Environ. Toxicol. 24, 618–628. doi: 10.1002/tox.20452
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1665–1668.
Bierne, N., Borsa, P., Daguin, C., Jollivet, D., Viard, F., Bonhomme, F., et al. (2003). Introgression patterns in the mosaic hybrid zone between Mytilus edulis and M. galloprovincialis. Mol. Ecol. 12, 447–461. doi: 10.1046/j.1365-294x.2003.01730.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brulle, F., Morgan, A. J., Cocquerelle, C., and Vandenbulcke, F. (2010). Transcriptomic underpinning of toxicant-mediated physiological function alterations in three terrestrial invertebrate taxa: a review. Environ. Pollut. 158, 2793–2808. doi: 10.1016/j.envpol.2010.06.019
Bushnell, B. (n.d.). BBMap. Available online at: https://sourceforge.net/projects/bbmap/ (accessed April, 2015).
Calzolai, L., Ansorge, W., Calabrese, E., Denslow, N., Part, P., and Lettieri, T. (2007). Transcriptomics and proteomics. Applications to ecotoxicology. Comp. Biochem. Physiol. D Genomics Proteomics 2, 245–249. doi: 10.1016/j.cbd.2007.04.007
Cannuel, R., Beninger, P. G., McCombie, H., and Boudry, P. (2009). Gill Development and its functional and evolutionary implications in the blue mussel Mytilus edulis (Bivalvia: Mytilidae). Biol. Bull. 217, 173–188. doi: 10.1086/BBLv217n2p173
Connon, R. E., Beggel, S., D’Abronzo, L. S., Geist, J. P., Pfeiff, J., Loguinov, A. V., et al. (2010). Linking molecular biomarkers with higher level condition indicators to identify effects of copper exposures on the endangered delta smelt (Hypomesus transpacificus). Environ. Toxicol. Chem. 30, 290–300. doi: 10.1002/etc.400
Connor, K. M., and Gracey, A. Y. (2011). Circadian cycles are the dominant transcriptional rhythm in the intertidal mussel Mytilus californianus. Proc. Natl. Acad. Sci. U.S.A. 108, 16110–16115. doi: 10.1073/pnas.1111076108
Conterato, G. M. M., Bulcão, R. P., Sobieski, R., Moro, A. M., Charão, M. F., de Freitas, F. A., et al. (2013). Blood thioredoxin reductase activity, oxidative stress and hematological parameters in painters and battery workers: relationship with lead and cadmium levels in blood. J. Appl. Toxicol. 33, 142–150. doi: 10.1002/jat.1731
Curole, J. P., and Hedgecock, D. (2007). “Bivalve genomics: complications, challenges, and future perspectives,” in Aquaculture Genome Technologies, ed. Z. Liu (Oxford: Blackwell Publishing Ltd), 525–544. doi: 10.1002/9780470277560.ch29
Daston, G. P. (2008). Gene expression, dose-response, and phenotypic anchoring: applications for toxicogenomics in risk assessment. Toxicol. Sci. 105, 233–234. doi: 10.1093/toxsci/kfn138
Denslow, N. D., Garcia-Reyero, N., and Barber, D. S. (2007). Fish “n” chips: the use of microarrays for aquatic toxicology. Mol. BioSyst. 3, 172–177. doi: 10.1039/B612802P
Dyachuk, V., and Odintsova, N. (2009). Development of the larval muscle system in the mussel Mytilus trossulus (Mollusca, Bivalvia). Dev. Growth Diff. 51, 69–79. doi: 10.1111/j.1440-169X.2008.01081.x
E50 Committee (2013). Standard Guide for Conducting Static Acute Toxicity Tests Starting with Embryos of Four Species of Saltwater Bivalve Molluscs. West Conshohocken, PA: ASTM International, 1–22.
EPA (1985). Guidelines for Deriving Numerical National Water Quality Criteria for the Protection of Aquatic Organisms and Their Uses. Washington, DC: EPA, 1–59.
EPA (1995). Short-term Methods for Estimating the Chronic Toxicity of Effluents and Receiving Waters to West Coast Marine and Estuarine Organisms. Washington, DC: EPA, 1–666.
EPA (2016). Draft Aquatic Life Ambient Estuarine/Marine Water Quality Criteria for Copper - 2016. Washington, DC: EPA.
EPA (n.d.). National Recommended Water Quality Criteria - Aquatic Life Criteria Table. Available online at: https://www.epa.gov/wqc/national-recommended-water-quality-criteria-aquatic-life-criteria-table (accessed February, 2019).
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Funk, C. (2014). The effects of anthropogenic copper on the native marine mussel Mytilus californianus in spud point marina, Bodega Bay, California. Explorat. UC Davis Undergrad. Res. J. 16, 1–13.
Giles, N. M., Watts, A. B., Giles, G. I., Fry, F. H., Littlechild, J. A., and Jacob, C. (2003). Metal and redox modulation of cysteine protein function. Chem. Biol. 10, 677–693. doi: 10.1016/S1074-5521(03)00174-1
Gracey, A. Y., Chaney, M. L., Boomhower, J. P., Tyburczy, W. R., Connor, K., and Somero, G. N. (2008). Rhythms of gene expression in a fluctuating intertidal environment. Curr. Biol. 18, 1501–1507. doi: 10.1016/j.cub.2008.08.049
Graney, R. L. (1993). Aquatic Mesocosm Studies in Ecological Risk Assessment. Boca Raton, FL: CRC Press.
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hahn, M. E. (2011). Mechanistic research in aquatic toxicology: perspectives and future directions. Aquat. Toxicol. 105, 67–71. doi: 10.1016/j.aquatox.2011.06.001
Hai, T., Wolfgang, C. D., Marsee, D. K., Allen, A. E., and Sivaprasad, U. (1999). ATF3 and stress responses. Gene Express. 7, 321–335.
Hoare, K., Beaumont, A. R., and Davenport, J. (1995a). Variation among populations in the resistance of Mytilus edulis embryos to copper:adaptation to pollution? Mar. Ecol. Prog. Ser. 120, 155–161. doi: 10.3354/meps120155
Hoare, K., Davenport, J., and Beaumont, A. R. (1995b). Effects of exposure and previous exposure to copper on growth of veliger larvae and survivorship of Mytilus edulis juveniles. Mar. Ecol. Prog. Ser. 120, 163–168. doi: 10.3354/meps120163
Huang, X., and Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Irving, H., and Williams, R. J. P. (1953). The stability of transition-metal complexes. J. Chem. Soc. 637, 3192–3210. doi: 10.1039/jr9530003192
Ishii, T., and Yanagawa, T. (2007). Stress-induced peroxiredoxins. Sub Cell. Biochem. 44, 375–384. doi: 10.1007/978-1-4020-6051-9_18
Ji, R.-R., de Silva, H., Jin, Y., Bruccoleri, R. E., Cao, J., He, A., et al. (2009). Transcriptional profiling of the dose response: a more powerful approach for characterizing drug activities. PLoS Comp. Biol. 5:e1000512. doi: 10.1371/journal.pcbi.1000512
Ji, R.-R., Siemers, N. O., Lei, M., Schweizer, L., and Bruccoleri, R. E. (2011). SDRS–an algorithm for analyzing large-scale dose-response data. Bioinformatics 27, 2921–2923. doi: 10.1093/bioinformatics/btr489
Johnson, A., Carew, E., and Sloman, K. (2007). The effects of copper on the morphological and functional development of zebrafish embryos. Aquat. Toxicol. 84, 431–438. doi: 10.1016/j.aquatox.2007.07.003
Johnson, D. (1988). Development of Mytilus edulis embryos: a bio-assay for polluted waters. Mar. Ecol. Prog. Ser. 46, 135–138. doi: 10.3354/meps046135
Kerr, M. K., Martin, M., and Churchill, G. A. (2000). Analysis of variance for gene expression microarray data. J. Comput. Biol. 7, 819–837. doi: 10.1089/10665270050514954
Krejsa, C. M., Franklin, C. C., White, C. C., Ledbetter, J. A., Schieven, G. L., and Kavanagh, T. J. (2010). Rapid activation of glutamate cysteine ligase following oxidative stress. J. Biol. Chem. 285, 16116–16124. doi: 10.1074/jbc.M110.116210
Kültz, D. (2005). Molecular and Evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 67, 225–257. doi: 10.1146/annurev.physiol.67.040403.103635
Langfelder, P., and Horvath, S. (2007). Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1:54. doi: 10.1186/1752-0509-1-54
Lee, B., Cao, R., Choi, Y.-S., Cho, H.-Y., Rhee, A. D., Hah, C. K., et al. (2009). The CREB/CRE transcriptional pathway: protection against oxidative stress-mediated neuronal cell death. J. Neurochem. 108, 1251–1265. doi: 10.1111/j.1471-4159.2008.05864.x
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Liu, J., Yang, D., Liu, S., Li, S., Xu, G., Zheng, G., et al. (2015). Microarray: a global analysis of biomineralization-related gene expression profiles during larval development in the pearl oyster, Pinctada fucata. BMC Genomics 16:980. doi: 10.1186/s12864-015-1524-2
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Marigómez, I., Soto, M., Cajaraville, M. P., Angulo, E., and Giamberini, L. (2002). Cellular and subcellular distribution of metals in molluscs. Microsc. Res. Tech. 56, 358–392. doi: 10.1002/jemt.10040
Martin, M., Osborn, K. E., Billig, P., and Glickstein, N. (1981). Toxicities of ten metals to Crassostrea gigas and Mytilus edulis embryos and Cancer magister larvae. Mar. Pollut. Bull. 12, 305–308. doi: 10.1016/0025-326X(81)90081-3
McCarthy, D. J., Chen, Y., and Smyth, G. K. (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. doi: 10.1093/nar/gks042
Myeku, N., and Figueiredo-Pereira, M. E. (2011). Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J. Biol. Chem. 286, 22426–22440. doi: 10.1074/jbc.M110.149252
Navarro, A., Faria, M., Barata, C., and Piña, B. (2011). Transcriptional response of stress genes to metal exposure in zebra mussel larvae and adults. Environ. Pollut. 159, 100–107. doi: 10.1016/j.envpol.2010.09.018
Negri, A., Oliveri, C., Sforzini, S., Mignione, F., Viarengo, A., and Banni, M. (2013). Transcriptional response of the mussel Mytilus galloprovincialis (Lam.) following exposure to heat stress and copper. PLoS One 8:e66802. doi: 10.1371/journal.pone.0066802
Nordberg, G. F. (2010). Biomarkers of exposure, effects and susceptibility in humans and their application in studies of interactions among metals in China. Toxicol. Lett. 192, 45–49. doi: 10.1016/j.toxlet.2009.06.859
Peng, Y., Leung, H. C. M., Yiu, S.-M., Lv, M.-J., Zhu, X.-G., and Chin, F. Y. L. (2013). IDBA-tran: a more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 29, i326–i334. doi: 10.1093/bioinformatics/btt219
Poynton, H. C., Loguinov, A. V., Varshavsky, J. R., Chan, S., Perkins, E. J., and Vulpe, C. D. (2008). Gene expression profiling in Daphnia magna part I: concentration-dependent profiles provide support for the no observed transcriptional effect level. Environ. Sci. Technol. 42, 6250–6256. doi: 10.1021/es8010783
Poynton, H. C., Varshavsky, J. R., Chang, B., Cavigiolio, G., Chan, S., Holman, P. S., et al. (2007). Daphnia magna ecotoxicogenomics provides mechanistic insights into metal toxicity. Environ. Sci. Technol. 41, 1044–1050.
Riginos, C., and Cunningham, C. W. (2005). Local adaptation and species segregation in two mussel (Mytilus edulis x Mytilus trossulus) hybrid zones. Mol. Ecol. 14, 381–400. doi: 10.1111/j.1365-294X.2004.02379.x
Ritz, C., Baty, F., Streibig, J. C., and Gerhard, D. (2015). Dose-response analysis using R. PLoS One 10:e0146021. doi: 10.1371/journal.pone.0146021
Rivera-Duarte, I., Rosen, G., Lapota, D., Chadwick, D. B., Kear-Padilla, L., and Zirino, A. (2005). Copper toxicity to larval stages of three marine invertebrates and copper complexation capacity in San Diego Bay, California. Environ. Sci. Technol. 39, 1542–1546. doi: 10.1021/es040545j
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Schiff, K., Brown, J., Diehl, D., and Greenstein, D. (2007). Extent and magnitude of copper contamination in marinas of the San Diego region, California, USA. Mar. Pollut. Bull. 54, 322–328. doi: 10.1016/j.marpolbul.2006.10.013
Schiff, K. C., Bay, S. M., and Diehl, D. (2003). Stormwater toxicity in chollas creek and San Diego Bay, California. Environ. Monit. Assess. 81, 119–132. doi: 10.1007/978-94-017-0299-7_12
Schirmer, K., Fischer, B. B., Madureira, D. J., and Pillai, S. (2010). Transcriptomics in ecotoxicology. Anal. Bioanal. Chem. 397, 917–923. doi: 10.1007/s00216-010-3662-3
Schönitzer, V., and Weiss, I. M. (2007). The structure of mollusc larval shells formed in the presence of the chitin synthase inhibitor Nikkomycin Z. BMC Struct. Biol. 7:71. doi: 10.1186/1472-6807-7-71
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Silva-Aciares, F., Zapata, M., Tournois, J., Moraga, D., and Riquelme, C. (2011). Identification of genes expressed in juvenile Haliotis rufescens in response to different copper concentrations in the north of Chile under controlled conditions. Mar. Pollut. Bull. 62, 2671–2680. doi: 10.1016/j.marpolbul.2011.09.023
Sokolova, I. M. (2013). Energy-limited tolerance to stress as a conceptual framework to integrate the effects of multiple stressors. Integr. Comp. Biol. 53, 597–608. doi: 10.1093/icb/ict028
Suzuki, M., Saruwatari, K., Kogure, T., Yamamoto, Y., Nishimura, T., Kato, T., et al. (2009). An acidic matrix protein, Pif, is a key macromolecule for nacre formation. Science 325, 1388–1390. doi: 10.1126/science.1173793
Varotto, L., Domeneghetti, S., Rosani, U., Manfrin, C., Cajaraville, M. P., Raccanelli, S., et al. (2013). DNA damage and transcriptional changes in the Gills of Mytilus galloprovincialis exposed to nanomolar doses of combined metal salts (Cd, Cu, Hg). PLoS One 8:e54602. doi: 10.1371/journal.pone.0054602
Venier, P., De Pittà, C., Pallavicini, A., Marsano, F., Varotto, L., Romualdi, C., et al. (2006). Development of mussel mRNA profiling: Can gene expression trends reveal coastal water pollution? Mutat. Res. Fundament. Mol. Mech. Mutagenesis 602, 121–134. doi: 10.1016/j.mrfmmm.2006.08.007
Vergani, L., Grattarola, M., Grasselli, E., Dondero, F., and Viarengo, A. (2007). Molecular characterization and function analysis of MT-10 and MT-20 metallothionein isoforms from Mytilus galloprovincialis. Arch. Biochem. Biophys. 465, 247–253. doi: 10.1016/j.abb.2007.05.023
Viarengo, A. (1985). Biochemical effects of trace metals. Mar. Pollut. Bull. 16, 153–158. doi: 10.1016/0025-326x(85)90006-2
Voronezhskaya, E. E., Nezlin, L. P., Odintsova, N. A., Plummer, J. T., and Croll, R. P. (2008). Neuronal development in larval mussel Mytilus trossulus (Mollusca: Bivalvia). Zoomorphology 127, 97–110. doi: 10.1007/s00435-007-0055-z
Walker, C. H., Sibly, R. M., and Peakall, D. B. (2000). Principles of Ecotoxicology, Second Edition. Boca Raton, FL: CRC Press.
Weis, J. (2014). Delayed behavioral effects of early life toxicant exposures in aquatic biota. Toxics 2, 165–187. doi: 10.3390/toxics2020165
Whitehead, A., Triant, D. A., Champlin, D., and Nacci, D. (2010). Comparative transcriptomics implicates mechanisms of evolved pollution tolerance in a killifish population. Mol. Ecol. 19, 5186–5203. doi: 10.1111/j.1365-294X.2010.04829.x
Williams, R. J. P. (1981). The bakerian lecture, 1981: natural selection of the chemical elements. Proc. R. Soc. B Biol. Sci. 213, 361–397. doi: 10.1098/rspb.1981.0071
Williams, R. J. P. (1984). Zinc: what is its role in biology? Endeavour 8, 65–70. doi: 10.1016/0160-9327(84)90040-1
Xu, M., Jiang, L., Shen, K.-N., Wu, C., He, G., and Hsiao, C.-D. (2016). Transcriptome response to copper heavy metal stress in hard-shelled mussel (Mytilus coruscus). Genomics Data 7, 152–154. doi: 10.1016/j.gdata.2015.12.010
Keywords: RNAseq, Mytilus californianus, mussel larvae, concentration response curve, copper, biomarker of exposure
Citation: Hall MR, Moffett JW and Gracey AY (2020) RNAseq Reveals Sensitive, Concentration-Dependent Transcriptional Markers of Copper in Mytilus californianus Larvae and Adults. Front. Mar. Sci. 7:572496. doi: 10.3389/fmars.2020.572496
Received: 14 June 2020; Accepted: 12 August 2020;
Published: 31 August 2020.
Edited by:
Vanessa F. Fonseca, Center for Marine and Environmental Sciences (MARE), PortugalReviewed by:
Jianmin Zhao, Yantai Institute of Coastal Zone Research (CAS), ChinaHelen Piontkivska, Kent State University, United States
Copyright © 2020 Hall, Moffett and Gracey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew Y. Gracey, gracey@usc.edu