 Ruirui Zhao
Ruirui Zhao Shanshan Cai1
Shanshan Cai1 Shengyong Xu
Shengyong Xu- 1Fishery College, Zhejiang Ocean University, Zhoushan, China
- 2Zhejiang Marine Fisheries Research Institute, Zhoushan, China
- 3Third Institute of Oceanography, Ministry of Natural Resources, Xiamen, China
The white-spotted bamboo shark (Chiloscyllium plagiosum) is an ecologically and commercially important marine cartilaginous fish in the Indo-Western Pacific. However, the resources of this species are declining in recent years due to habitat degradation and overfishing. Studies on genomics and genetic markers can provide valuable information for sound management and conservation of C. plagiosum. In this study, genome resequencing data of a domesticated C. plagiosum individual were generated for genomic comparison between wild and domesticated samples. Based on 281.17 Gb of clean sequencing data, the genome size of the domesticated sample was estimated as 4.99 Gb, with heterozygosity of 0.51% and repeat ratio of 74.67%, which is about 1 Gb larger than that of the wild sample (3.85 Gb with a repeat ratio of 63.53%). By using a reference-based approach, we assembled a nearly 3.45 Gb genome sequence of the domesticated sample, with the scaffold N50 of 69.45 Mb. Subsequent identification of genome-wide microsatellite markers confirmed the different abundance of repeat elements in wild and domesticated C. plagiosum. Additionally, a total of 8,703,211 single-nucleotide polymorphisms (SNPs) were detected and annotated. The demographic analysis based on identified SNPs revealed a large and constant effective population size of C. plagiosum after the last population expansion (~0.3 million years ago). The genomic data and identified genetic markers in this study can provide fundamental and useful information for further comparative genomics, evolutionary biology, and conservation genetics of C. plagiosum.
Introduction
The white-spotted bamboo shark, Chiloscyllium plagiosum (Orectolobiformes, Hemiscylliidae), is a benthic species that is widely distributed in the Indo-West Pacific (Compagno, 1984) (Figure 1). This species is an oviparous cartilaginous fish with small body sizes (nearly 1 m in length) (Wang et al., 2019). Given the unique skin pattern, abundant nutrition, special immunoglobulins, and key position in marine food webs, the white-spotted bamboo shark is of great economic and ecological value (Baum et al., 2003; Masstor et al., 2014; Zielonka et al., 2014), which make this species as one of the main targets in the fishery and aquarium industries in China. However, due to low fecundity and the combined effects of overfishing and environmental pressures, the white-spotted bamboo shark has been listed as a near-threatened species (IUCN, 2021) in recent years with the continuously declined population resources. Therefore, it is necessary to investigate genome information and the population genetics pattern of this species, which could provide crucial references for effective management and conservation. Previous studies of the white-spotted bamboo shark were mainly focused on physiology, biology, and biomedicine (Lowry and Motta, 2007; Maia and Wilga, 2013; Alexander et al., 2016; Straube et al., 2016), while genetic analyses of this species are relatively limited (Ding et al., 2009). Till now, a handful of molecular markers were reported, mainly focusing on mitogenomes (Fu et al., 2010) and microsatellite loci (Ding et al., 2009). Limited molecular markers may affect population genetics studies of this species, as well as biodiversity and conservation from a microevolution perspective.

Figure 1 Photograph of Chiloscyllium plagiosum (photo by Yuan Li).
The development of next-generation sequencing (NGS) provides a rapid and cost-effective way to uncover genomic information about non-model organisms (Kumar and Kocour, 2017). Based on high-depth genome sequencing data, genome survey analysis is generally implemented to obtain fundamental genomic information (i.e., genome size, GC content, heterozygosity ratio, and repeat ratio), as well as to develop large numbers of molecular markers including mitochondrial genomes, single-nucleotide polymorphisms (SNPs) and microsatellites . For instance, Xu et al. (2020b) performed genome survey analyses of the brown-spotted flathead (Platycephalus sp.1) to estimate the fundamental genomic information of both male and female individuals, suggesting possible male heterogamety. Furthermore, the mitochondrial genome sequence and genome-wide microsatellites were also identified using genome sequencing data, providing valuable genetic markers for this species. Moreover, Yu et al. (2015) discovered 25,140 SNPs of the Pacific white shrimp (Litopenaeus vannamei) for linkage map construction using genome survey sequencing data; these SNP data could be useful in further comparative genomics studies. By using genome survey sequencing data, one can identify and develop thousands of genome-wide molecular markers efficiently and cost-effectively.
Recently, the chromosome-scale genome assembly of the white-spotted bamboo shark was reported (Zhang et al., 2020). This assembled genome sequence, which was obtained from a wild natural individual, was 3.85 Gb in length with a scaffold N50 of 57.92 Mb. Furthermore, a total of 19,595 protein-coding genes were predicted, and 63.53% of the assembly was annotated as repeat contents (Zhang et al., 2020). This genomic and annotation information can be used as references for comparative genomics and evolution studies of the white-spotted bamboo shark. In the present study, an artificially domesticated white-spotted bamboo shark individual was sampled, and subsequent whole-genome sequencing and genome survey analyses were implemented. Genomic comparison of wild and domesticated individuals was performed, which can provide supplemental genomic information for the white-spotted bamboo shark. Moreover, by using the reported chromosome-scale genome assembly and annotation information as references, genome-wide molecular markers including SNPs and microsatellites were also identified based on these genome sequencing data. These genomic data and identified molecular markers in this study should provide valuable information for further population genetics and evolution studies of the white-spotted bamboo shark.
Materials and Methods
Sample Collection, DNA Extraction, and Illumina Sequencing
The female individual used in this study was collected from Haichang Ocean Park, Yantai, China, in October 2020. The sample was naturally dead when collected, and a piece of muscle tissue was sampled and stored in 95% ethanol at −80°C. Total genomic DNA was extracted using a standard phenol-chloroform method.
A 350-bp paired-end genome survey sequencing library was constructed and sequenced using the Illumina NovaSeq6000 platform (Illumina, Inc., San Diego, CA, USA). The library construction and sequencing were performed at Novogene Co., Ltd. (Beijing, China). The Illumina sequencing data were deposited in the Sequence Read Archive (SRA) database under accession number PRJNA823509.
Genome Survey Analyses
After low-quality data were removed, retained clean data were used for k-mer analyses. The formula (Xu et al., 2020a) Genome size = k-mer_num/peak_depth was used to estimate genome size, heterozygosity, and repeat ratio, where k-mer_num is the total number of predicted best k-mer and peak_depth is the expected value of the k-mer depth. K-mer analyses were performed using software GCE v1.0.0 (Liu et al., 2013). The clean reads were assembled into contigs with a k-mer of 41 by applying the de Bruijn graph structure, which was completed in SOAPdenovo v2.01 (Luo et al., 2012). Then, the contig was assembled into a scaffold using the paired-end information. The preliminary assembly was further scaffolded with RagTag v1.1.0 software (Alonge et al., 2021) using the chromosome-scale assembly in Zhang et al. (2020) as a reference. The assembly completeness was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.1 (Simão et al., 2015) against the actinopterygii_odb10 database.
Software MicroSatellite (MISA, http://pgrc.ipk-gatersleben.de/misa/) was used to identify genome-wide microsatellite motifs based on the assembled genome sequences. The search parameters were set for the detection of mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide microsatellite motifs with a minimum of 10, 6, 5, 5, 5, and 5 repeats, respectively.
Single-Nucleotide Polymorphism Detection, Annotation, and Demographic Analysis
The reported genome of C. plagiosum (accession no. GCF_004010195.1) (Zhang et al., 2020) was downloaded from the GenBank database as a reference sequence for genome-wide SNP calling. The clean reads of our genome survey sequencing data were mapped to the genome assembly using BWA software (Li and Durbin, 2009) with default parameters. After the alignment, SNP calling was performed using SAMtools v1.3.1 (Li et al., 2009). SNP filtering was produced using VCFtools (Danecek et al., 2011) with the following parameters: i) the SNP was called in 100% of individuals, ii) the minor allele frequency (MAF) was greater than 10%, iii) only two alleles were present, iv) sites that contained an indel were excluded, v) sites that failed the Hardy–Weinberg equilibrium (HWE) test at p < 0.05 were excluded, vi) only sites with quality value >30 were included, and vii) loci with sequencing depth <50 were eliminated. Only SNPs satisfying the above criteria were kept in the final SNP dataset. Subsequently, the final SNP variants were annotated using SnpEff v4.3t software (Cingolani et al., 2012).
To reconstruct the population history of the white-spotted bamboo shark, demographic analysis was performed based on the generated SNP matrix, using a pairwise sequentially Markovian coalescent (PSMC) model in the PSMC package (Li and Durbin, 2011). The reconstructed population history was plotted using “psmc_plot.pl” script using the substitution rate “-u 1.1e-9” and a generation time of 7 years. The generation time was calculated as g = a + [s/(1 − s)] (Yuan et al., 2018), where s is the expected adult survival rate, which is assumed as 80%, and a is the sexual maturation age, which is 3 years for the white-spotted bamboo shark (Chen et al., 2011). Therefore, the generation time was determined as 7 in the PSMC analysis. The substitution rate of the white-spotted bamboo shark was estimated as 1.6e−10 substitution/site/year (Martin, 1999). However, the unit of substitution rate in PSMC analysis is substitution/site/generation (Li and Durbin, 2011); thus, “-u 1.1e-9” was used in the PSMC analysis. A total of 100 bootstraps were performed in the PSMC analysis to determine the variance in the estimated effective population size (Ne).
Results
Information Estimation
A total of 282.94 Gb raw data were obtained, representing the sequencing depth of more than 50× (Table 1). The Q20 and Q30 values of raw data were evaluated as 95.65% and 90.73%, respectively, showing high sequencing quality. After the low-quality data were filtered, about 281.17 Gb of clean data were retained with an effective rate of 99.56%. The clean data were used for k-mer analysis, which showed that the genome size of our sample was estimated as 4.99 Gb, with heterozygosity and repeat ratio of 0.51% and 74.67%, respectively (Table 2).

Table 1 Statistics of genome sequencing data in this study.

Table 2 Estimation of Chiloscyllium plagiosum genome based on 17-mer statistics.
Microsatellite Identification and Characterization
The genome sequence of our C. plagiosum sample was preliminarily assembled at the contig and scaffold levels (Table 3). The total length of this genome sequence was nearly 3.3 Gb, with the contig N50 of 3,129 bp and the scaffold N50 of 5,412 bp. This assembly was further updated with in silico RagTag software using the reported chromosome-scale genome assembly in Zhang et al. (2020) as a reference, and the final assembly was 3,450,307,416 bp in length with a scaffold N50 of 69.45 Mb and high genomic completeness (Table 3). Based on these two assemblies, a total of 736,562 and 736,565 microsatellites were identified in the preliminary and updated assemblies, respectively (Figure 2A, Table S1). Thus, the microsatellite distribution frequency in our C. plagiosum genome assembly was about 223.13 microsatellites per Mb.
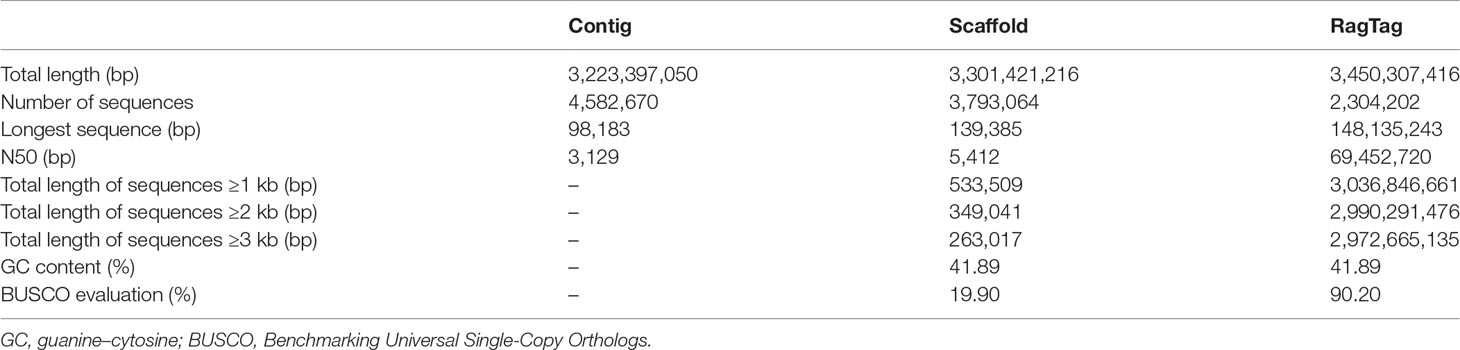
Table 3 Genome assembly statistics in our study.
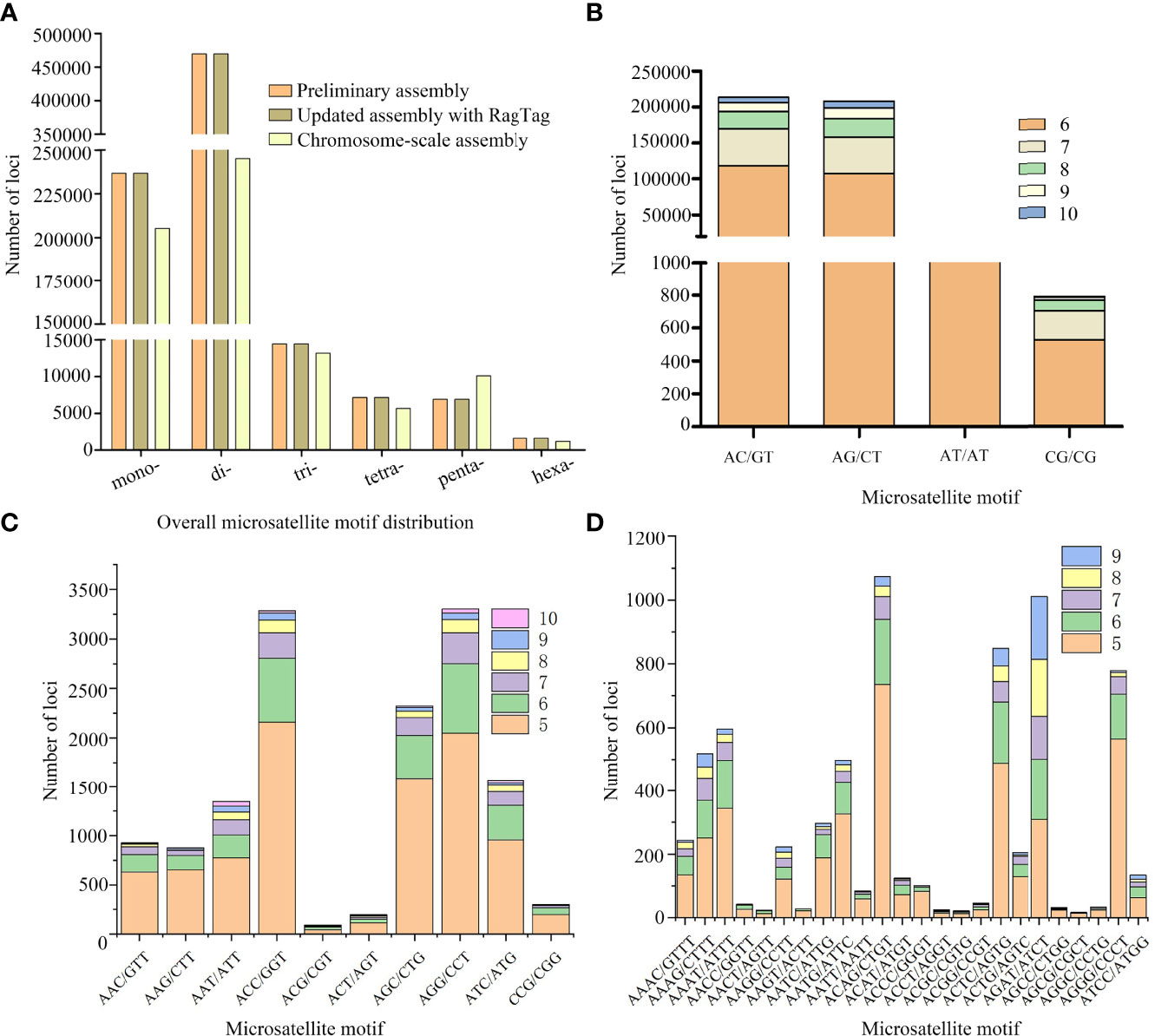
Figure 2 Distribution and frequency of microsatellite loci in Chiloscyllium plagiosum.(A) Frequency of different microsatellite repeat types in three genome assemblies. (B–D) Frequency of dinucleotide, trinucleotide, and tetranucleotide microsatellite motifs in our assembly.
For microsatellite motif types, comparative analyses showed that higher numbers of mono- and di-nucleotide motifs in our assemblies were identified, while the number of pentanucleotide motifs was higher in the chromosome-scale assembly (Figure 2A). In our assemblies, the results showed that dinucleotide motifs were the most frequent (63.76%, 469,630), followed by mononucleotide (32.15%, 236,807) and trinucleotide (1.96%, 14,451) motifs, while the proportions of tetranucleotide (0.97%, 7,161), pentanucleotide (0.94%, 6,899), and hexanucleotide (0.22%, 1,614) repeat were quite low (Figure 2A). Furthermore, the top 5 abundant motifs were AC/GT (31.10%), AG/CT (30.80%), A/T (18.08%), C/G (14.07%), and AT/AT (1.75%), while the total proportion of the remaining 283 motifs was less than 1%. In terms of each motif type, the most abundant motif was AC/GT, AGG/CCT, and ACAG/CTGT in di-, tri-, and tetra-nucleotide motif types, respectively (Figures 2B–D).
Single-Nucleotide Polymorphism Identification, Annotation, and Demographic Analysis
In our study, the initial SNP calling generated a total of 22,535,935 genome-wide SNPs, among which 8,703,211 high-confidence SNPs were finally retained after SNP filtering. These SNPs covered 3,517 Mb of the total assembly length; thus, the SNP density was one SNP per 404 bp. Among these identified SNPs, most were transitions, accounting for 63.74%, while transversions accounted for 36.26%, leading to the transition/transversion ratio of 1.76. The results of SNP annotation (Table 4) revealed that a total of 13,758,432 (39.46%) SNPs mapped to transcripts, 13,586,846 (38.97%) SNPs were present in intron regions, 4,777,409 (13.70%) SNPs were in intergenic regions, and 133,720 (0.38%) SNPs were located in exon regions. For functional effect, 42,164 SNPs were annotated as missense mutations, 306 were nonsense, and 71,715 were silent mutations. For impact effect, most of the identified SNPs were categorized as modifiers (34,731,306, 99.62%), while 130,858 SNPs had “high to moderate impacts” on the coding sequences.

Table 4 SNP annotation information in our study.
Based on these identified SNPs, our PSMC plotting revealed three times of population expansion (ca. 100, 5, and 0.3 million years ago) and two times of population reduction (ca. 40 and 1 million years ago) of the white-spotted bamboo shark (Figure 3). After the last population expansion, the Ne was relatively constant (~1,430,000 individuals), showing a large effective population size of this species.
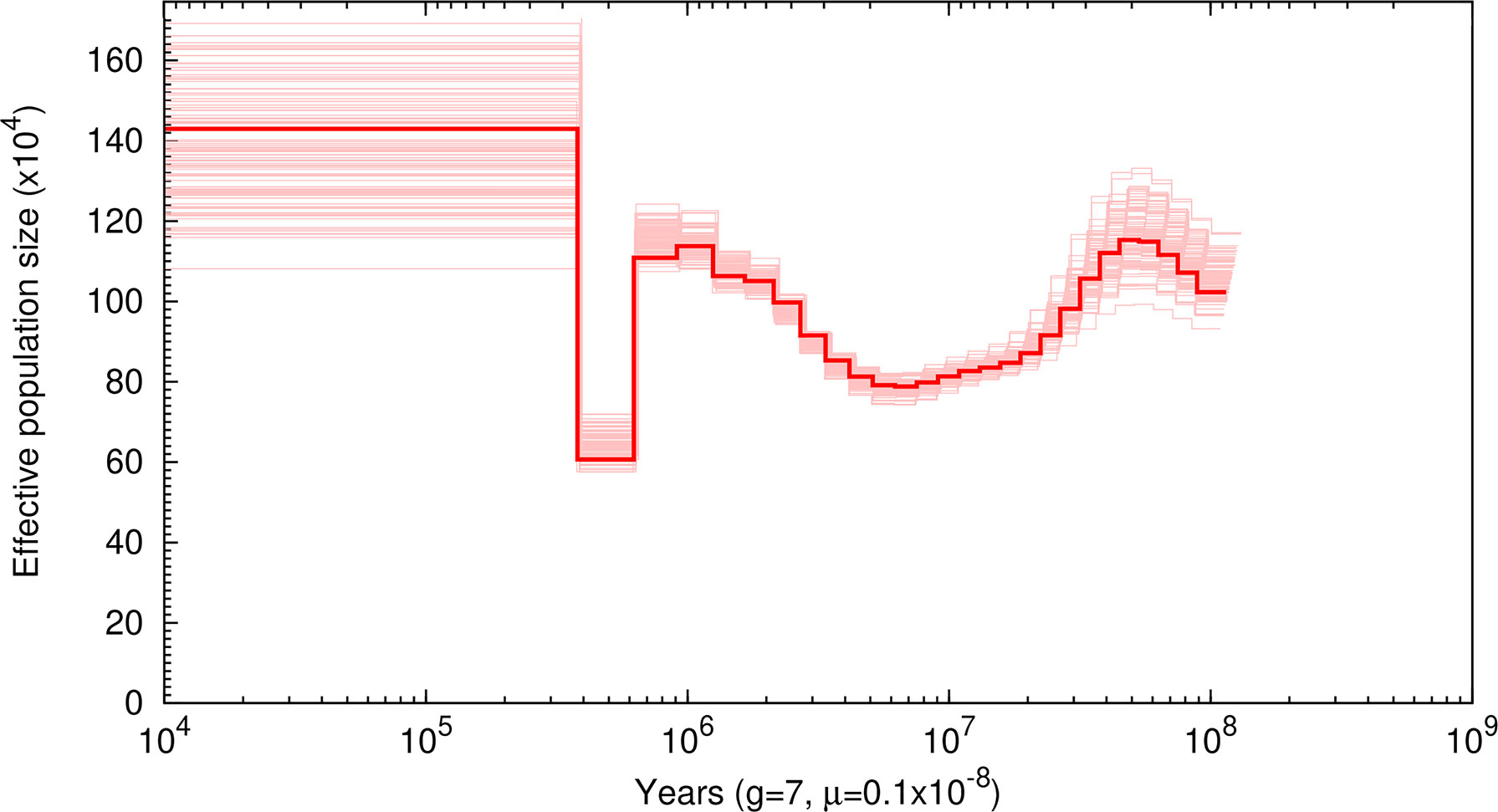
Figure 3 Demographic history of Chiloscyllium plagiosum in this study. Thick lines represent the median, and thin light lines correspond to 100 rounds of bootstrapping.
Discussion
In the present study, the genomic resequencing data of an artificially domesticated white-spotted bamboo shark individual were sequenced, and genomic comparison of wild and domesticated individuals was performed. Furthermore, genome-wide molecular markers including SNPs and microsatellites were also identified. These genomic data and molecular markers should be useful for further conservation genetics and evolutionary studies of the white-spotted bamboo shark.
Genomic Information Comparison
The k-mer analysis showed that the genome size of our sample was estimated as 4.99 Gb, with heterozygosity and repeat ratio of 0.51% and 74.67%, respectively (Table 2). Comparatively, the previously reported genome of C. plagiosum was 3.85 Gb in length with a repeat ratio of 63.53% (Zhang et al., 2020). Our results showed that the domesticated sample had a larger genome size and repeat ratio, and when the repeat sequences were excluded, the revised genome sizes were comparable (domesticated, ~1.26 Gb; wild, ~1.40 Gb). We hypothesized that the difference in genome size and repeat ratio of this species should be due to different (i.e., natural vs. artificial) habitat environments of samples. Similar genomic patterns were also revealed in the silver sillago (Sillago sihama) (Li et al., 2019b; Lin et al., 2020), in which the genome size of the natural sample was 521.63 Mb with a repeat ratio of 13.78%, while the domesticated one had a genome size of about 680 Mb and a repeat ratio of 29.60%. Chalopin et al. (2015) discovered a positive correlation between repeat elements and genome size in 26 vertebrate species, suggesting that repeat sequences largely contribute to the genome size of vertebrates. Furthermore, a positive logarithmic correlation between repeat ratio and genome size in 60 jawed fish species was also revealed in our recent study (unpublished data). This might be also a plausible explanation for the larger genome sizes of chondrichthians and sarcopterygians than those of actinopterygian fish species. Till now, genomic resources of chondrichthians are still limited (Marra et al., 2019), due to large genome size (Venkatesh et al., 2007). Compared to other reported chondrichthian genomes (Pearce et al., 2021), the genome size of the white-spotted bamboo shark was at a medium level, yet the repeat ratio was the highest, even using the genomic information in Zhang et al. (2020). Repetitive sequences can precisely regulate gene expression and are considered one of the main factors controlling structural gene recombination and regulation. Moreover, they are also an important part of the non-coding region of the genome (Li et al., 2019a). As a result, further studies are needed to analyze the functional attributes of repeat sequences, which decipher the evolutionary mechanism of higher repeat ratio in C. plagiosum genome.
Microsatellite Distribution in Chiloscyllium plagiosum Genome
In our study, a total of 736,562 and 736,565 microsatellites were identified in the preliminary and updated assemblies, and the microsatellite distribution frequency in our C. plagiosum genome assembly was about 223.13 microsatellites per Mb. To our knowledge, there is no other study investigating the genome-wide microsatellite distribution of chondrichthians. As a result, we investigated an interspecific comparison of microsatellite distribution frequency between the white-spotted bamboo shark and actinopterygian fish species. Compared to actinopterygian fish species such as the marbled rockfish Sebastiscus marmoratus (314.6/Mb) (Xu et al., 2020a), silver sillago (S. sihama) (257.5/Mb) (Li et al., 2019b), and spotted scat Scatophagus argus (512.1/Mb) (Huang et al., 2019), the microsatellite distribution frequency of C. plagiosum was relatively low. It is generally believed that genomes with a high proportion of repeat sequences contain more microsatellites. However, despite the high repeat ratio, comparatively low levels of microsatellite distribution frequency were detected in our assemblies. The results of repeat sequences annotation in Zhang et al. (2020) showed that most of the repeat elements (91.70%) were transposable elements (TEs) in the C. plagiosum genome, while the proportion of tandem repeat was quite low. This might be a reasonable explanation for the low microsatellite distribution frequency in our study. Furthermore, we also analyzed the microsatellite distribution of the chromosome-scale assembly in Zhang et al. (2020), and a total of 480,452 microsatellites were identified (Figure 2A), representing the frequency of 124.73/Mb. Such a lower frequency of microsatellite distribution confirmed our above inference, which also mirrored the difference in repeat ratio between our assembly and the chromosome-scale assembly in Zhang et al. (2020). In addition, the results of microsatellite motif types showed that dinucleotide and mononucleotide motifs were the dominant types. In general, our results were consistent with the expectation that short motif types (i.e., mono- and di-nucleotide) were generally dominant in fish genomes (e.g., marbled rockfish S. marmoratus (Xu et al., 2020a), silver sillago S. sihama (Li et al., 2019b), and crocodile flathead Cociella crocodilus (Zhao et al., 2021)). These identified microsatellites in our study could provide substantial genetic markers for further population genetics studies of C. plagiosum.
Single-Nucleotide Polymorphism Identification and Demographic Analysis
In our study, a total of 8,703,211 high-confidence SNPs were identified, and the SNP density was one SNP per 404 bp. The SNP density of C. plagiosum was much higher than that of Oreochromis niloticus (one SNP per 9 kb) (Yáñez et al., 2020) and Clupea harengus (ca. one SNP per 1 kb) (Corander et al., 2013) but lower than that of threespine stickleback (ca. one SNP per 100 bp) (Shanfelter et al., 2019), suggesting relatively high levels of genetic variability in the white-spotted bamboo shark genome. Among these identified SNPs, in terms of impact effect, most of the identified SNPs were categorized as modifiers (34,731,306, 99.62%), while 130,858 SNPs had “high to moderate impacts” on the coding sequences. Moderate-impact mutations are single-residue missense variants that do not affect translation, while high-impact mutations are coding sequence changes that may result in a knockout of the affected gene (Hawkins et al., 2016). High-impact mutations generally include frameshifts, “start_lost,” “stop_lost,” “stop_gained,” and loss or gain of splice sites. In this study, a total of 771 SNPs were categorized as high-impact mutations. Despite being lower in number, these high-impact mutations play crucial roles in the genetic and phenotypic evolution of the white-spotted bamboo shark. Additionally, these identified genome-wide SNPs could facilitate future phenotype–genotype association analysis, population genomics, and evolutionary biology, which might provide valuable clues for the effective management and conservation of this species.
For demographic analysis, our PSMC plotting revealed three times of population expansion and two times of population reduction of the white-spotted bamboo shark. Unexpectedly, we failed to detect population reduction during the Last Glacial Maximum (LGM; ~20,000 years ago), a period when most of the marine species in the Northwest Pacific underwent drastic population decline due to wide-range marine regression (Liu et al., 2010). This observed demographic pattern perhaps indicated high levels of adaptability of the white-spotted bamboo shark to survive LGM or perhaps resulted from the well-known difficulties in estimating recent effective population sizes using the PSMC approach (Zhou et al., 2018). More samples from distinct geographic populations should be collected to further confirm the demographic pattern of the white-spotted bamboo shark. Overall, our results provided novel insights into the population history of the white-spotted bamboo shark, which should be useful in fishery resource management and conservation.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
Ethical review and approval was not required for the animal study because the sample was naturally dead when collecting.
Author Contributions
SX and YL conceived the study. SX, RZ, SC, DL, and PL analyzed the data. SX and RZ drafted the manuscript. All authors revised and approved the final manuscript.
Funding
This study was supported by the National Programme on Global Change and Air-Sea Interaction (GASI-02-SCS-YDsum), the Basic Scientific Research Projects of Fujian Provincial Public Service Research Institutes (2020R1101) and Scientific Research Startup Foundation of Zhejiang Ocean University (2018-2019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Mr. Qingjie Zhou for the sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.936681/full#supplementary-material
References
Alexander A. B., Parkinson L. A., Grant K. R., Carlson E., Campbell T. W. (2016). The Hemic Response of White-Spotted Bamboo Sharks (Chiloscyllium Plagiosum) With Inflammatory Disease. Zoo Biol. 35, 251–259. doi: 10.1002/zoo.21280
Alonge M., Lebeigle L., Kirsche M., Aganezov S., Wang X. G., Lippman Z. B., et al. (2021) . Automated Assembly Scaffolding Elevates a New Tomato System for High-Throughput Genome Editing. bioRxiv. doi: 10.1101/2021.11.18.469135
Baum J. K., Myers R. A., Kehler D. G., Worm B., Harley S. J., Doherty P. A. (2003). Collapse and Conservation of Shark Populations in the Northwest Atlantic. Science 299, 389–392. doi: 10.1126/science.1079777
Chalopin D., Naville M., Plard F., Galiana D., Volff J. N. (2015). Comparative Analysis of Transposable Elements Highlights Mobilome Diversity and Evolution in Vertebrates. Genome Biol. Evol. 7, 567–580. doi: 10.1093/gbe/evv005
Chen L., Chen M. R., Yang S. Y. (2011) . The Spousing, Mating and Reproductive Behavior of the White-Spotted Bamboo Shark (Chiloscyllium Plagiosum) From South Fujian Coastal Waters. J. Xiamen Univ. Nat. Sci. 50, 641–644. doi: CNKI:SUN:XDZK.0.2011-03-028
Cingolani P., Platts A., Wang L. L., Coon M., Nguyen T., Wang L., et al. (2012). A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Corander J., Majander K. K., Lu C., Merilä J. (2013). High Degree of Cryptic Population Differentiation in the Baltic Sea Herring Clupea Harengus. Mol. Ecol. 22, 2931–2940. doi: 10.1111/mec.12174
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., Depristo M. A., et al. (2011). The Variant Call Format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Ding S. X., Pan Y., Zeng H. S., Wang J. (2009). Characterization of 12 Polymorphic Microsatellite Loci for the Whitespotted Bamboo Shark (Chiloscyllium Plagiosum Bennett). Mol. Ecol. Resour. 9, 1398–1400. doi: 10.1111/j.1755-0998.2009.02676.x
Fu M. N., Wang J., Ding S. X., Du J. Y., Su S. Q. (2010). Studies on the Genetic Structure and Genetic Subdivision of White Spotted Bamboo Shark, Chiloscyllium Plagiosum, by Analysing Mitochondrial Cyt B Genes. J. Trop. Oceanogr. 29, 86–91. doi: 10.3969/j.issn.1009-5470.2010.06.013
Hawkins C., Caruana J., Schiksnis E., Liu Z. C. (2016). Genome-Scale DNA Variant Analysis and Functional Validation of a SNP Underlying Yellow Fruit Color in Wild Strawberry. Sci. Rep. 6, 29017. doi: 10.1038/srep29017
Huang Y. Q., Jiang D. N., Li M., Mustapha U. F., Tian C. X., Chen H. P., et al. (2019). Genome Survey of Male and Female Spotted Scat (Scatophagus Argus). Animals 9, 1117. doi: 10.3390/ani9121117
Kumar G., Kocour M. (2017). Applications of Next-Generation Sequencing in Fisheries Research: A Review. Fish. Res. 186, 11–22. doi: 10.1016/j.fishres.2016.07.021
Li H., Durbin R. (2009). Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Durbin R. (2011). Inference of Human Population History From Individual Whole-Genome Sequences. Nature 475, 493–496. doi: 10.1038/nature10231
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lin X. H., Huang Y., Jiang D. N., Chen H. P., Deng S. P., Zhang Y. L., et al. (2020). Chromosomal-Level Genome Assembly of Silver Sillago (Sillago Sihama). Genome Biol. Evol. 13, evaa272. doi: 10.1093/GBE/EVAA272
Li G. Q., Song L. X., Jin C. Q., Li M., Gong S. P., Wang Y. F. (2019a). Genome Survey and SSR Analysis of Apocynum Venetum. Bioscience. Rep. 39, BSR20190146. doi: 10.1042/BSR20190146
Li Z. Y., Tian C. X., Huang Y., Lin X. G., Wang Y. R., Jiang D. G., et al. (2019b). A First Insight Into a Draft Genome of Silver Sillago (Sillago Sihama) via Genome Survey Sequencing. Animals 9, 756. doi: 10.3390/ani9100756
Liu J., Gao T., Wu S., Zhang Y. (2010). Pleistocene Isolation in the Northwestern Pacific Marginal Seas and Limited Dispersal in a Marine Fish, Chelon Haematocheilus (Temminck & Schlegel 1845). Mol. Ecol. 16, 275–288. doi: 10.1111/j.1365-294X.2006.03140.x
Liu B. H., Shi Y. J., Yuan J. Y., Hu X. S., Zhang H., Li N., et al. (2013). Estimation of Genomic Characteristics by Analyzing K-Mer Frequency in Denovo Genome Projects. Quant. Biol. 35, 62–67. doi: 10.48550/arXiv.1308.2012
Lowry D., Motta P. J. (2007). Ontogeny of Feeding Behavior and Cranial Morphology in the Whitespotted Bamboo Shark Chiloscyllium Plagiosum. Mar. Biol. 151, 2013–2023. doi: 10.1007/s00227-007-0642-z
Luo R. B., Liu B. H., Xie Y. L., Li Z. Y., Huang W. H., Yuan J. Y., et al. (2012). SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read De Novo Assemble. GigaScience 1, 18. doi: 10.1186/2047-217X-1-18
Maia A., Wilga C. D. (2013). Anatomy and Muscle Activity of the Dorsal Fins in Bamboo Sharks and Spiny Dogfish During Turning Maneuvers. J. Morphol. 274, 1288–1298. doi: 10.1002/jmor.20179
Marra N. J., Stanhope M. J., Jue N. K., Wang M. H., Sun Q., Bitar P. P., et al. (2019). White Shark Genome Reveals Ancient Elasmobranch Adaptations Associated With Wound Healing and the Maintenance of Genome Stability. P. Natl. A. Sci. 116, 4446–4455. doi: 10.1073/pnas.1819778116
Martin A. P. (1999). Substitution Rates of Organelle and Nuclear Genes in Sharks: Implicating Metabolic Rate (Again). Mol. Biol. Evol. 16, 996–1002. doi: 10.1093/oxfordjournals.molbev.a026189
Masstor N. H., Samat A., Nor S. M., Md-Zain B. M. (2014). Molecular Phylogeny of the Bamboo Sharks (Chiloscyllium Spp.). BioMed. Res. Int. 2014, 213896. doi: 10.1155/2014/213896
Pearce J., Fraser M. W., Sequeira A. M., Kaur P. (2021). State of Shark and Ray Genomics in an Era of Extinction. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.744986
Shanfelter A. F., Archambeault S. L., White M. A. (2019). Divergent Fine-Scale Recombination Landscapes Between a Freshwater and Marine Population of Threespine Stickleback Fish. Genome Biol. Evol. 11, 1573–1585. doi: 10.1093/gbe/evz090
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: Assessing Genome Assembly and Annotation Completeness With Single-Copy Orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Straube N., Lampert K. P., Geiger M. F., Weiss J. D., Kirchhauser J. X. (2016). First Record of Second-Generation Facultative Parthenogenesis in a Vertebrate Species, the Whitespotted Bambooshark Chiloscyllium Plagiosum. J. Fish. Biol. 88, 668–675. doi: 10.1111/jfb.12862
Venkatesh B., Kirkness E. F., Loh Y. H., Halpern A. L., Lee A. P., Johnson J., et al. (2007). Survey Sequencing and Comparative Analysis of the Elephant Shark (Callorhinchus Milii) Genome. PLoS Biol. 5, e101. doi: 10.1371/journal.pbio.0050101
Wang J. Q., Li J., Huang L. M. (2019). Biological Characteristics of Striped Bamboo Shark (Chiloscyllium Plagiosum Bennett 1830) in Xiamen Bay. J. Jimei Univ. Nat. Sci. 24, 161–166. doi: 10.19715/j.jmuzr.2019.03.01
Xu S. Y., Song N., Xiao S. J., Gao T. X. (2020a). Whole Genome Survey Analysis and Microsatellite Motif Identification of Sebastiscus Marmoratus. Bioscience. Rep. 40, BSR20192252. doi: 10.1042/BSR20192252
Xu S. Y., Zhang H., Gao T. X. (2020b). Comprehensive Whole Genome Survey Analyses of Male and Female Brownspotted Flathead Fish Platycephalus sp.1. Genomics 112, 4742–4748. doi: 10.1016/j.ygeno.2020.08.030
Yáñez J. M., Yoshida G., Barria A., Palma V. R., Travisany D., Díaz D., et al. (2020). High-Throughput Single Nucleotide Polymorphism (SNP) Discovery and Validation Through Whole-Genome Resequencing in Nile Tilapia (Oreochromis Niloticus). Mar. Biotechnol. 22, 109–117. doi: 10.1007/s10126-019-09935-5
Yuan Z. H., Huang W., Liu S. K., Xu P., Dunham R., Liu Z. J. (2018). Historical Demography of Common Carp Estimated From Individuals Collected From Various Parts of the World Using the Pairwise Sequentially Markovian Coalescent Approach. Genetica 146, 235–241. doi: 10.1007/s10709-017-0006-7
Yu Y., Zhang X. J., Yuan J. B., Li F. H., Chen X. H., Zhao Y. Z., et al. (2015). Genome Survey and High-Density Genetic Map Construction Provide Genomic and Genetic Resources for the Pacific White Shrimp Litopenaeus Vannamei. Sci. Rep. 5, 15612. doi: 10.1038/srep15612
Zhang Y. L., Gao H. Y., Li H. B., Guo J., Ouyang B. J., Wang M. N., et al. (2020). The White-Spotted Bamboo Shark Genome Reveals Chromosome Rearrangements and Fastevolving Immune Genes of Cartilaginous Fish. iScience 23, 101754. doi: 10.1016/j.isci.2020.101754
Zhao R., Lu Z., Cai S., Gao T., Xu S. (2021). Whole Genome Survey and Genetic Markers Development of Crocodile Flathead Cociella Crocodilus. Anim. Genet. 52, 891–895. doi: 10.1111/age.13136
Zhou X., Guang X., Sun D., Xu S., Li M., Seim I., et al. (2018). Population Genomics of Finless Porpoises Reveal an Incipient Cetacean Species Adapted to Freshwater. Nat. Commun. 9, 1276. doi: 10.1038/s41467-018-03722-x
Keywords: genome assembly, Chiloscyllium plagiosum, microsatellite, single nucleotide polymorphism, effective population size
Citation: Zhao R, Cai S, Lu D, Li P, Xu S and Li Y (2022) Genomic Comparison and Genetic Marker Identification of the White-Spotted Bamboo Shark Chiloscyllium plagiosum. Front. Mar. Sci. 9:936681. doi: 10.3389/fmars.2022.936681
Received: 05 May 2022; Accepted: 09 June 2022;
Published: 13 July 2022.
Edited by:
Massimo Milan, University of Padua, ItalyReviewed by:
Li Lian Wong, Universiti Malaysia Terengganu, MalaysiaCarlos Saavedra, Institute of Aquaculture Torre de la Sal (CSIC), Spain
Copyright © 2022 Zhao, Cai, Lu, Li, Xu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengyong Xu, a2V2aW44OTAyMjNAMTYzLmNvbQ==; Yuan Li, bGl5dWFuNDIyQDE2My5jb20=