Review on the physical chemistry of iodine transformations in the oceans
 George W. Luther III
George W. Luther III- School of Marine Science and Policy, University of Delaware, Lewes, DE, United States
The transformation between iodate (), the thermodynamically stable form of iodine, and iodide (I-), the kinetically stable form of iodine, has received much attention because these species are often dependent on the oxygen concentration, which ranges from saturation to non-detectable in the ocean. As suboxic conditions in the ocean’s major oxygen minimum zones indicate that is minimal or non-detectable, the incorporation of into carbonate minerals has been used as a redox proxy to determine the O2 state of the ocean. Here, I look at the one and two electron transfers between iodine species with a variety of oxidants and reductants to show thermodynamics of these transformations. The to conversion is shown to be the controlling step in the reduction reaction sequence due to thermodynamic considerations. As reduction to is more favorable than reduction to at oceanic pH values, there is no need for nitrate reductase for reduction as other reductants (e.g. Fe2+, Mn2+) and dissimilatory reduction by microbes during organic matter decomposition can affect the transformation. Unfortunately, there is a dearth of information on the kinetics of reductants with ; thus, the thermodynamic calculations suggest avenues for research. Conversely, there is significant information on the kinetics of I- oxidation with various oxygen species. In the environment, I- oxidation is the controlling step for oxidation. The oxidants that can lead to are reactive oxygen species with O3 and •OH being the most potent as well as sedimentary oxidized Mn, which occurs at lower pH than ocean waters. Recent work has shown that iodide oxidizing bacteria can also form . I- oxidation is more facile at the sea surface microlayer and in the atmosphere due to O3.
1 Introduction
The thermodynamically favorable form of iodine in seawater is iodate (). However, iodide (I-) is present in oxic, suboxic and anoxic waters. The one electron transfer reaction of I- with molecular oxygen, 3O2, to form the iodine atom (I•) and superoxide (O2-) is thermodynamically unfavorable as is the reaction of two I- with 3O2 to form I2 and H2O2 (Luther et al., 1995; Luther, 2011). Thus, other oxidants are required to initiate abiotic iodide oxidation, and I- is a known sink for O3. Biotic iodide oxidation has received much interest with one report showing conversion of iodide to iodate (Hughes et al., 2021). Iodate reduction can occur with common reductants (e.g., sulfide, Fe2+), and various organisms that decompose organic matter using iodate as the electron acceptor. Nitrate reductase and dimethyl sulfoxide reductase enzymes from these and planktonic microbes are considered important mediators for biotic iodate reduction (e.g., Hung et al., 2005; Amachi, 2008). Thus, there has been extensive interest in the chemistry of these two iodine species and the possible intermediates that form during their 6-electron redox interconversion ever since the element, iodine, was first discovered as I2 during the study of brown kelp algae of the Laminariales (kelps/seaweeds) by Courtois in the early 1800s (Wong, 1991; Küpper et al., 2008; Küpper et al., 2011).
Possible chemical species that form during the interconversion are given in eqn. (1a, b). The loss of an O atom is equivalent to a two-electron transfer resulting in the reduction of the iodine from +5 in to +3 for (HOIO) to +1 for HOI (IO-) and to -1 for I-. The acid-base species in parentheses are minor species at seawater pH as the pKa values for HOI and HOIO are 10.7 (Ka = 2 x 10-11) and 4.49 (Ka = 3.2 x 10-5), respectively.
Equation 1b shows the interconversion between HOI and I- as HOI undergoes one-electron transfer to I2 followed by another one-electron reduction per I atom to I-.
This work considers the thermodynamics of these transformations during the reduction of , which occurs in anoxic systems, during organic matter decomposition and by phytoplankton, as well as the oxidation of I-, which occurs by the direct oxidation of iodide by iodide oxidizing bacteria, oxidized metals and reactive oxygen species (ROS) that are produced by certain microbes, (macro)algae and abiotic processes including photochemistry. In a previous work (Luther, 2011), the chemistry and thermodynamics of chloride, bromide and iodide oxidation were compared; however, I(+3) species (HOIO, iodous acid, and , iodite) and stepwise iodate reduction were not considered. Here, stepwise reactions of the iodine species in equations 1a and 1b with environmentally important reactants (including transient ROS species) are considered for both the oxidation of I- and the reduction of to affect their interconversion. The kinetics of these stepwise reactions are also considered. Kinetic data for the first step(s) in iodide oxidation are available, but less kinetic information is available for iodate reduction.
2 Methods
2.1 Calculations of aqueous redox potentials from half-reactions
Table 1 gives several equations for redox half-reactions that include the pH dependence for the reaction considered. These are pϵ(pH) relationships based on the balanced chemical equations and the thermodynamics of each chemical species. The basic mathematical approach has been fully developed in standard textbooks (Stumm and Morgan, 1996; Luther, 2016) and used in previous publications (Luther, 2010; Luther, 2011). Aqueous thermodynamic data to calculate the pϵ(pH) or logK(pH) relationships in Table 1 (at 25°C and 1 atm) are from Stumm and Morgan (1996) and other sources (Bard et al., 1985; Stanbury, 1989). The value used for the Gibbs free energy for Fe2+ (-90.53 kJ/mole) is that discussed in Rickard and Luther (2007). Values of the free energy for HOIO and are from Schmitz (2008).
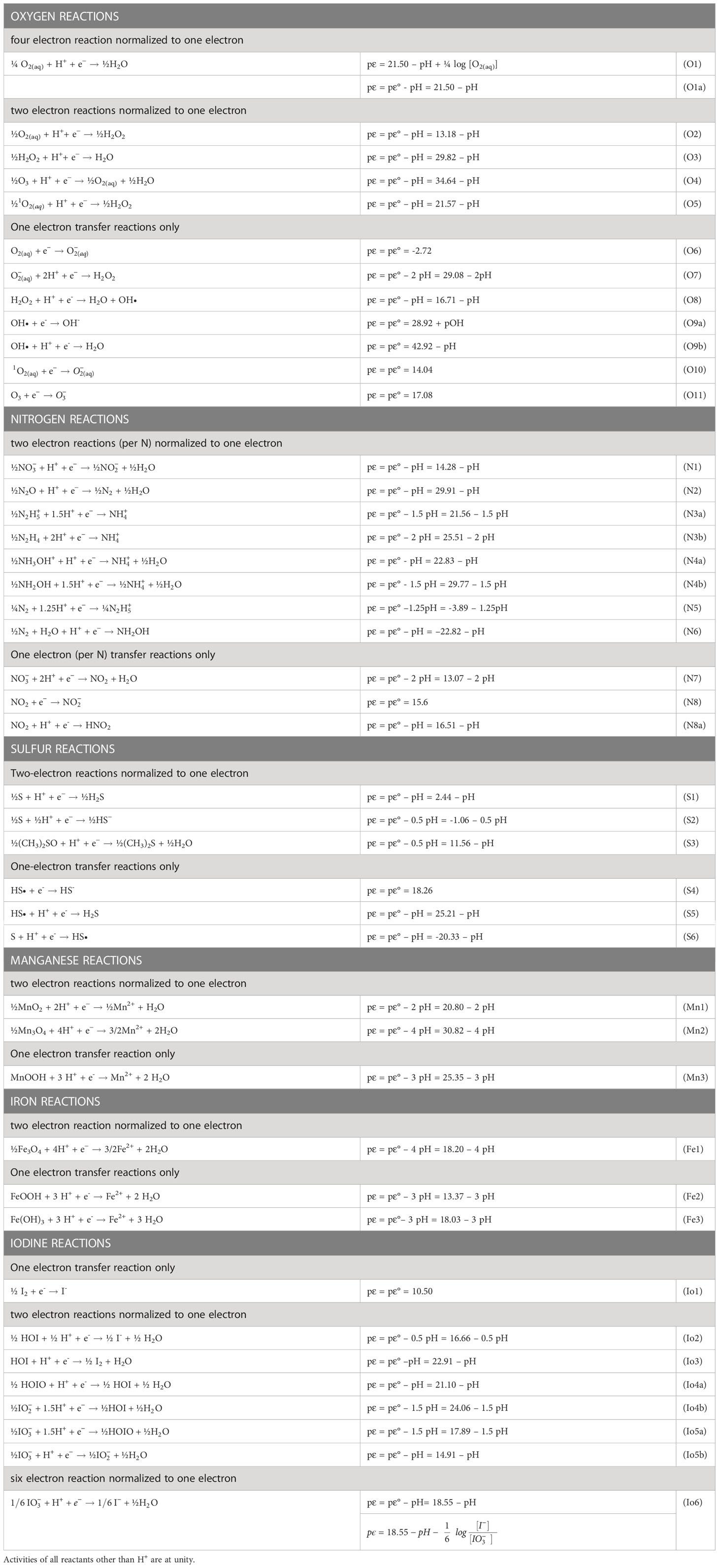
Table 1 Reduction half-reactions for relevant species of oxygen, nitrogen, sulfur, manganese, iron and iodine normalized to one electron.
The calculated pϵ value from each half-reaction is given as a function of pH as in the examples in Table 1, and these half reactions can be used for simple calculations of the pE values of full reactions (see next section). When H+ or OH- is not in a balanced equation for a half-reaction, there is no pH dependence on the half-reaction. The pε calculated is termed pϵ(pH) which provides a log K for each half-reaction at a given pH. Concentration dependence for the other reactants are not considered in the calculation; thus, these are considered standard state calculations. When concentration dependence is considered, the calculated pϵ value can vary as in the following example for the O2/H2O couple (O1 in Table 1).
Using the balanced half reaction and the Gibbs free energy of formation of each species at 25°C, the Gibbs free energy of the reaction and the equilibrium constant are calculated.
The standard state ΔG° for the reaction = - 490.68 kJ/2 moles H2O or 4 moles of electrons. The equilibrium constant () is given in eqn. 2a where {} indicates activity for each chemical species and the activity of H2O is defined as 1.
On expanding, eqn. 2b results.
is for 4 mole of electrons or 21.50 for 1 mole of electrons.
For a one-electron half-reaction, we have
And equation 2b becomes equation 2c
or
From the Nernst Equation, (the standard state value), which on substitution gives equation O1 (see Table 1) where concentration is used for O2(aq).
At ocean surface conditions of 211 µM O2 (211 x 10-6 M; 100% saturation at 25°C and salinity of 35), this expression becomes
and at a pH of 8, pε = 12.58.
At 1µM O2 (10-6 M) which occurs in oxygen minimum zones, this expression becomes
and at a pH of 7.5, pε = 12.50.
At unit activity for all reagents including H+, pε = pε°. At unit activity of all reagents other than the H+, equation O1a results, which is used for many calculations in this paper.
Note that the above equations show a 1.50 log unit change for an O2 concentration range from 1 µM to unity activity (O1a) so the calculations could vary an order of magnitude or more in either direction when concentration dependence is included. However, comparisons can be more easily made when combining different half-reactions at a given pH. This permits an assessment of which combined half-reactions are thermodynamically favorable and thus more likely to occur in a given environmental setting.
2.2 Coupling half-reactions
As an example of coupling two half reactions to determine whether a reaction is favorable, I use the data in Table 1 for the reduction of (Io5b) by (N1) in equation 3.
Equation 4 is used to calculate a complete reaction’s pε or ΔlogKreaction value. All values of ΔlogKreaction > 0 indicate a favorable reaction and all values of ΔlogKreaction< 0 indicate an unfavorable reaction.
At a pH of 7, the pεred values for and are 7.91 and 7.28, respectively. As is the reductant, it is oxidized; thus, the sign for pεred (7.28) is reversed to become pεoxid (-7.28).
For reaction 3, there is no pH dependence as the pH dependence of each half-reaction is similar so cancels.
For this work, Table 1 lists the pϵ(pH) values for Mn, Fe, oxygen, nitrogen, sulfur and iodine species for the relevant iodine redox reactions considered. Dissolved Fe(II) and Mn(II) are primarily hexaaquo species until the pH is > 7, where hydroxo complexes start to become important. As most reactions occur via one and two-electron transfers, the calculations will permit assessment of a thermodynamically unfavorable step along a reaction coordinate of six-electrons as in the reduction of iodate to iodide and the oxidation of iodide to iodate. From surface waters to decomposition zones, seawater pH values range from 8 down to 7; thus, the following discussion will emphasize this pH range.
3 Results and discussion: Iodate reduction
3.1 Iodate and iodide speciation at different seawater oxygen conditions
In the oxic environment, the oxidizing condition of the environment or pε is set by the 4-electron transfer reaction of the O2(aq)/H2O couple [reaction O1 in Table 1]. At a pH of 8, temperature 250C and a salinity of 35, 100% O2(aq) saturation is 211 μM, which gives a pε of 12.58 (Figure 1). As the couple has a pε of 10.56 at is the thermodynamically favored iodine species.
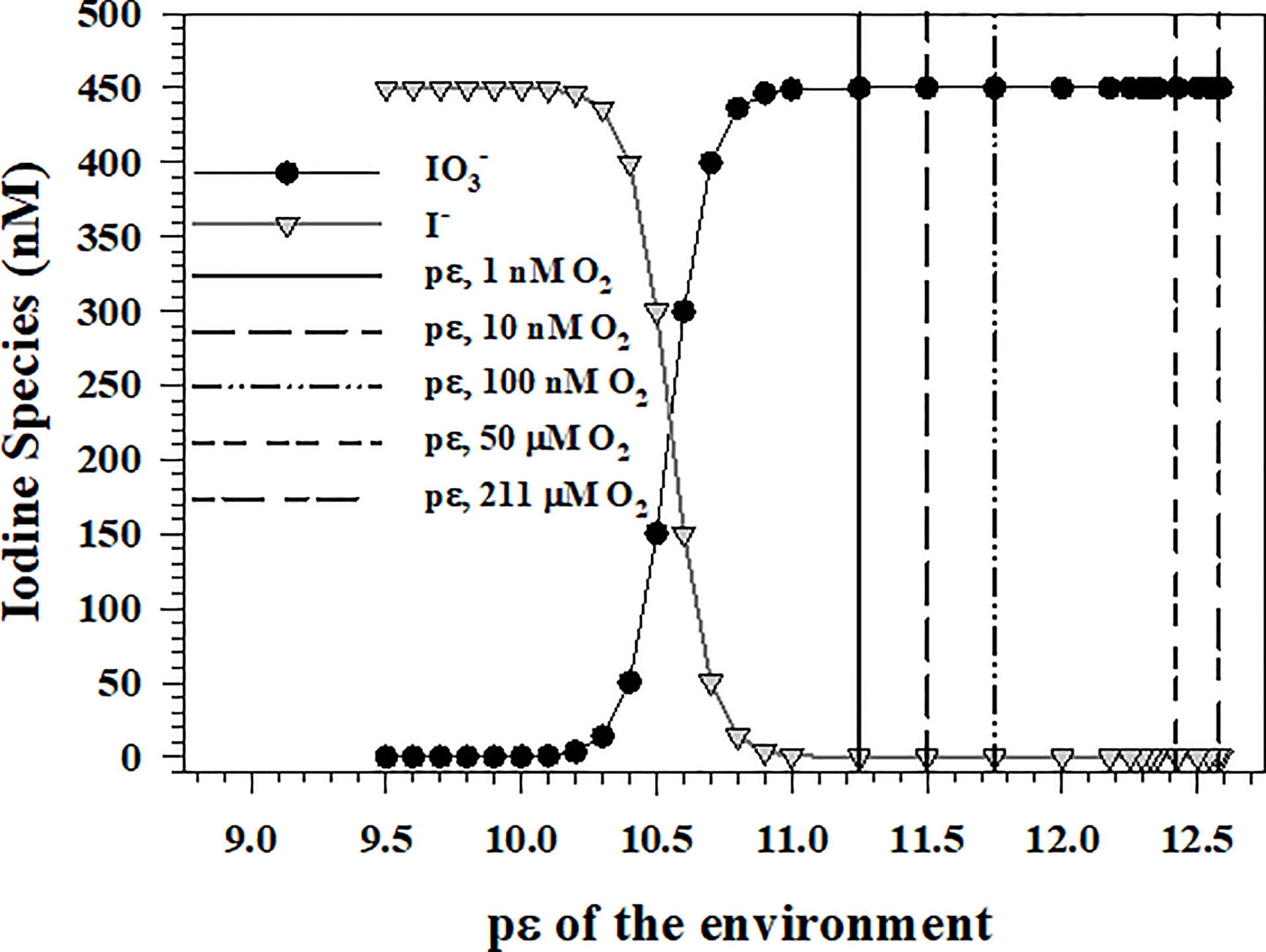
Figure 1 and I- concentrations for a total iodine concentration of 450 nM at different environmental pε values assuming that O2(aq) reduction to H2O sets the pε value of ocean waters.
Entering the pε value for a given [O2(aq)] into equation Io6 allows the determination of the iodide to iodate ratio and the actual concentration of each assuming a total iodine concentration of 450-470 nM (Elderfield and Truesdale, 1980). Figure 1 shows the iodate and iodide concentrations are equivalent at a pε of 10.56. The vertical lines indicate the environmental pε for [O2(aq)] of 1, 10, 100 nM, and 1, 50 and 211 μM. As oxygen minimum zones (OMZ) of the Arabian Sea and the equatorial Pacific Ocean have [O2(aq)] concentrations in the 1-100 nM range (Revsbech et al., 2009; Lehner et al., 2015), calculations show that is the thermodynamically preferred iodine species even at 1 nM O2(aq), which gives a pε of 11.25 for the O2(aq)/H2O couple. However, I- is the dominant iodine species detected in OMZ waters (Wong and Brewer, 1977; Luther and Campbell, 1991; Rue et al., 1997; Farrenkopf and Luther, 2002; Cutter et al., 2018). At [O2(aq)] concentrations ≤ 1 μM, , and Mn2+ concentrations are now similar or higher in concentration and should determine the pε of the water.
As most reactions occur by 1- or 2-electron transfers, Figure 2 shows the redox sequence for two electron transfer redox couples , MnO2/Mn2+ (Mn1) and the one-electron redox couple Fe(OH)3/Fe2+ (Fe3) over a wide range of pH. The redox sequence at pH = 8 is as expected for the N, Mn and Fe systems. As (up to 12 μM) and Mn2+ (up to 8 μM) are formed at OMZ oxic-anoxic transition zones (e.g., Arabian Sea, Black Sea, Equatiorial Pacific, see Lewis and Luther, 2000; Trouwborst et al., 2006; Cutter et al., 2018, respectively) and are in higher concentration than O2(aq), the and MnO2 → Mn2+ (Mn1) couples can be chosen to set the environmental pε. At pH = 8, the to pε is 6.15 and the MnO2 to Mn2+ pε is 4.80. At pH = 7, the to pε is 7.28 and the MnO2 to Mn2+ pε is 6.80. At these pε values, O2(aq) is below 1 nM, and I- is now the thermodynamically favored iodine species when comparing these data with the couple (pε of 10.56 at pH = 8).
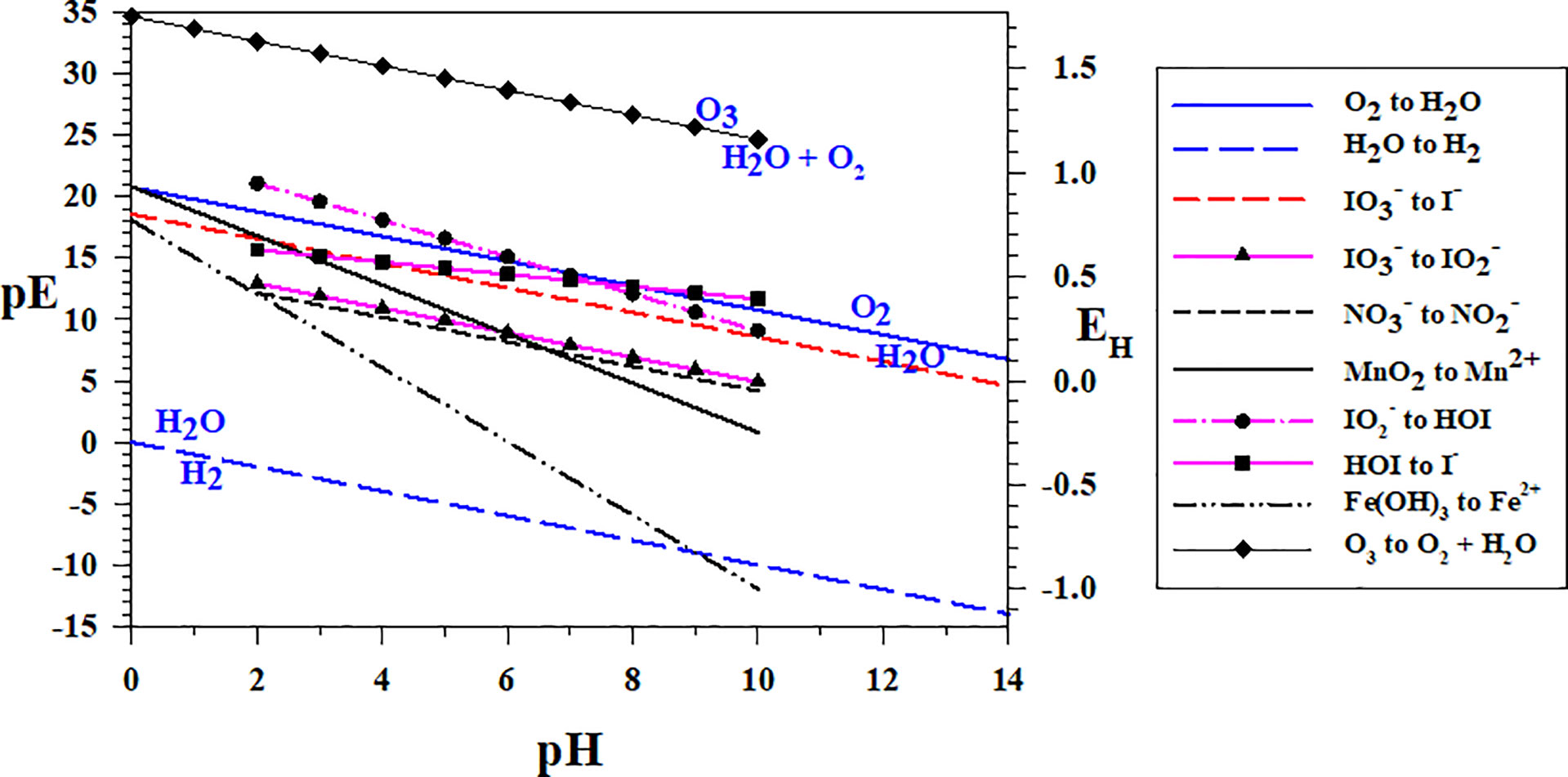
Figure 2 Two electron transfer redox couples for O3 (O4), N (N1), Mn (Mn1) and I (Io2a, Io4b, Io5b), and one electron transfer redox couple for Fe (Fe3). The oxidized species is always above the line and the reduced below the line as in the O2/H2O and the H2O/H2 couples, which dictate the water stability field.
Figure 2 also shows that the couple (Io5b) should be the first step in the reaction sequence of iodate to iodide (eqn. 1a). As the pε of the couple has a more positive pε value than the N, Mn and Fe couples in Figure 2, reduction is more favorable than these couples even though it is very close to the couple. Thus, is predicted to reduce before , and biological activity (e.g., nitrate reductase activity) is not necessary to reduce (see section 3.2). Because of the strong pH dependence for the Mn and Fe couples, they cross the and couples at lower pH, which have similar slopes. Thus, is predicted to reduce to (eqn. 3). Interestingly, Mn2+ and Fe2+ should be poorer reductants than for conversion of to at a pH< 6 and pH< 1, respectively, but are more favorable to reduce than above those pH values (see sections 3.2 and 3.3).
Although the to I- conversion occurs at higher pε, it is a 6-electron transfer (IO6), which is not a facile process. Thus, the intermediates ( and HOI) will dictate the reactivity sequence via a combination of thermodynamic and kinetic considerations.
As shown in Figure 2, reduction to HOI and HOI reduction to I- are also more favorable at higher pε values than the to I- couple. At a pH = 8, the to HOI couple has a pε value of 12.06 corresponding to 2 μM O2 (see Figure 1). Similarly, the HOI to I- couple has a pε value of 12.66 corresponding to 250 μM O2. At a pH of 7, both couples have pε values greater than 13 indicating that, even at O2 saturation, I- is the dominant species predicted when these intermediates form. At a pH = 7.5 (that is found in many OMZ waters), both to HOI and HOI to I- couples have pε values greater than 12.6; also indicating that at O2 saturation, I- is the dominant species predicted. Thus, the intermediates and HOI are not predicted to be stable in marine waters; thus, the conversion of to is a key step. Interestingly, Hardisty et al. (2021) found in situ reduction in the oxycline where [O2(aq)] was 11 μM, but not at [O2(aq)]< 2 μM. Lastly, the O3 to O2 + H2O couple is highly oxidizing indicating that all iodine couples should lead to formation. O3 reactions will be discussed in more detail below (sections 4.2, 4.7).
In the next sections (3.2 – 3.5), the thermodynamics for the conversion of iodate to iodide via the intermediates outlined in equations 1a and 1b by environmental reductants are considered to show what step, if any, in the reduction of iodate to iodide may be unfavorable over a wide range of pH. Iodate reduction is well known in the marine environment (e.g., Wong and Brewer, 1977; Wong et al., 1985; Luther and Campbell, 1991; Rue et al., 1997; Farrenkopf and Luther, 2002; Cutter et al., 2018) and occurs via chemical reductants like sulfide (Zhang and Whitfield, 1986) and via microbes like Shewanella putrfaciens (Farrenkopf et al., 1997) and Shewanella oneidensis (Mok et al., 2018) during dissimilatory reduction coupled with decomposition (oxidation) of organic matter as well as phytoplankton mediated processes (e.g., Chance et al., 2007).
3.2 Iodate reduction by NO2-
Although has not yet been shown to be a reductant for in aqueous lab studies (eqn. 3), HNO2 is a reductant for MnO2 (Luther and Popp, 2002) and Mn(III)-pyrophosphate (Luther et al., 2021). Figure 3A shows the thermodynamic calculations for the stepwise conversion of to I- by reduction ( oxidizes to ). All 2-electron transfer reactions, which involve O atom loss for iodine, are favorable over the pH range. For seawater pH (7-8), the least favorable reaction is the to reaction whereas the to HOI and HOI to I- reactions are more favorable. Thus, the to conversion appears to be the controlling step in the reaction sequence. The 1-electron transfer reaction of HOI to I2 is the most favorable, but the second 1-electron transfer reaction of I2 to I- is only favorable at pH > 4. Thus, reduction of to I- by is predicted via 1-electron or 2-electron transfer reactions at seawater pH values. The data plotted in Figure 2 indicate that once forms there is no thermodynamic barrier to I- formation.
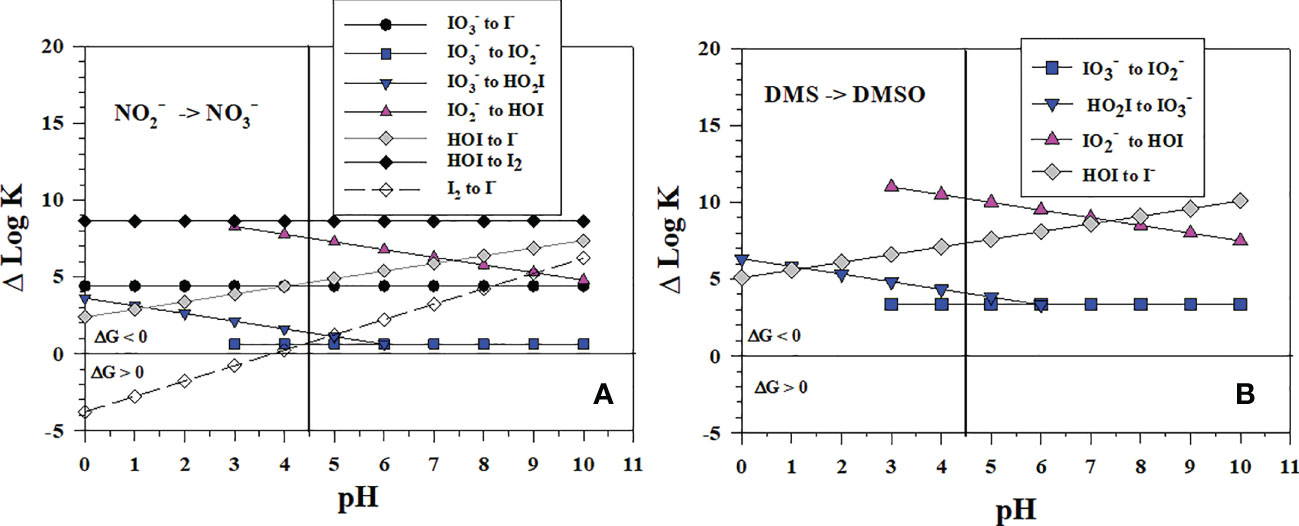
Figure 3 Thermodynamics for (A) the 1-electron and 2-electron reductions of (Io5a, Io5b, Io6), (Io4b), HOI (Io2, Io3), and I2 (Io1) by (N1) and (B) 2-electron reductions of (Io5b), (Io4a, Io4b), HOI (Io2) by DMS (S3). The vertical line represents the pKa value of 4.49 for HO2I. Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
The reaction with has been reported to produce I2 in ice by Kim et al. (2019), but not in solution. The pH in the ice was 3 where HNO2 and H2ONO+ exist and are the likely reductants. Thus, polar areas may be locales for reduction. At seawater pH, the reaction seems to be hindered by kinetics in the transition state as each reactant ( and ) is an anion, which will repel each other.
3.3 Biological iodate reduction
The marine literature has many reports on the uptake of (with or without ) by phytoplankton with the iodine released as I- (e.g., Elderfield and Truesdale, 1980; Wong, 2001; Wong et al., 2002; Chance et al., 2007; Bluhm et al., 2010). As a result of this iodate uptake, reductase was presumed by some researchers to be a key process for reduction to I-. Also, Bluhm et al. (2010) and Carrano et al. (2020) showed that algal senescence enhanced I- release, and Hepach et al. (2020) showed that there is a considerable lag between uptake and I- release due to senescence.
reductase appears to reduce in some phytoplankton (Hung et al., 2005). However, de la Cuesta and Manley (2009) showed that I- can be up taken by phytoplankton, and that different phytoplankton uptake I- whereas other phytoplankton uptake . Thus, there is no need for nitrate reductase for reduction as I- can be up taken by some phytoplankton rather than form from reduction. Moreover, Waite and Truesdale (2003) showed that nitrate reductase was not important for reduction by Isochrysis galbana. The latter study is consistent with the thermodynamics of the reduction to being more favorable than the reduction to .
Furthermore, under anaerobic conditions, dissimilatory reduction occurs without nitrate reductase for the denitrifying bacterium, Pseudomonas stutzeri, (Amachi et al., 2007; Amachi, 2008). Reyes-Umana et al. (2022) and Yamazaki et al. (2020) showed that iodate reductase is in the periplasmic space of Pseudomonas sp SCT. Also, Mok et al. (2018) showed that dissimilatory reduction by Shewanella oneidensis does not involve nitrate reductase. Recently, Shin et al. (2022) showed that Shewanella oneidensis requires extracellular dimethylsulfoxide (DMSO) reductase involving a molybdenum enzyme center for reduction. Guo et al. (2022) studied bacterial genomes in a variety of environments and documented that Shewanella oneidensis are ubiquitous in all fresh and marine waters; they concluded that reduction is a major biogeochemical process. Thus, nitrate reductase (also an O atom transfer reaction) is not a requirement for bacterial reduction to I-.
The interconversion of dimethylsulfoxide with dimethylsulfide during dissimilatory reduction is another 2-electron O-atom transfer reaction. Moreover, the reactions of DMS to reduce HOI, and are thermodynamically favorable (Figure 3B, DMSO reduction is in S3, Table 1 and occurs at a lower pε than and reduction). The reaction of DMS with HOI has been suggested by Müller et al. (2021) to be a sink for DMS based on the rapid reaction of DMS with HOBr.
3.4 Iodate reduction by Mn2+ and Fe2+
Figure 4 shows the thermodynamics for the stepwise conversion of to I- by reduction with Mn2+ and Fe2+. Concentrations of Mn2+ and Fe2+ range from several nM to μM in OMZs (e.g., Trouwborst et al., 2006; Moffett and German, 2020) and in suboxic porewaters (e.g., Oldham et al., 2019; Owings et al., 2021) to mM in waters emanating from hydrothermal vents (e.g., Estes et al., 2022); in these cases, Mn2+ and Fe2+ are normally higher in concentration than the total iodine concentration. For the 2-electron transfer reactions with Mn2+, only the to HOI reaction is favorable over the entire pH range. The to reaction is favorable only at pH > 6 whereas the other reactions are favorable at pH > 3. Thus, the to conversion is the controlling step in the reaction sequence when Mn2+ is the reductant. Using high resolution porewater profiles of I- and Mn2+ obtained by voltammetric microelectrodes, Anschutz et al. (2000) showed that a I- maximum occurred at the depth where upward diffusing Mn(II) was being removed and proposed that I- formed by the reaction of with Mn2+ under suboxic conditions. The reaction has not been investigated in laboratory studies.
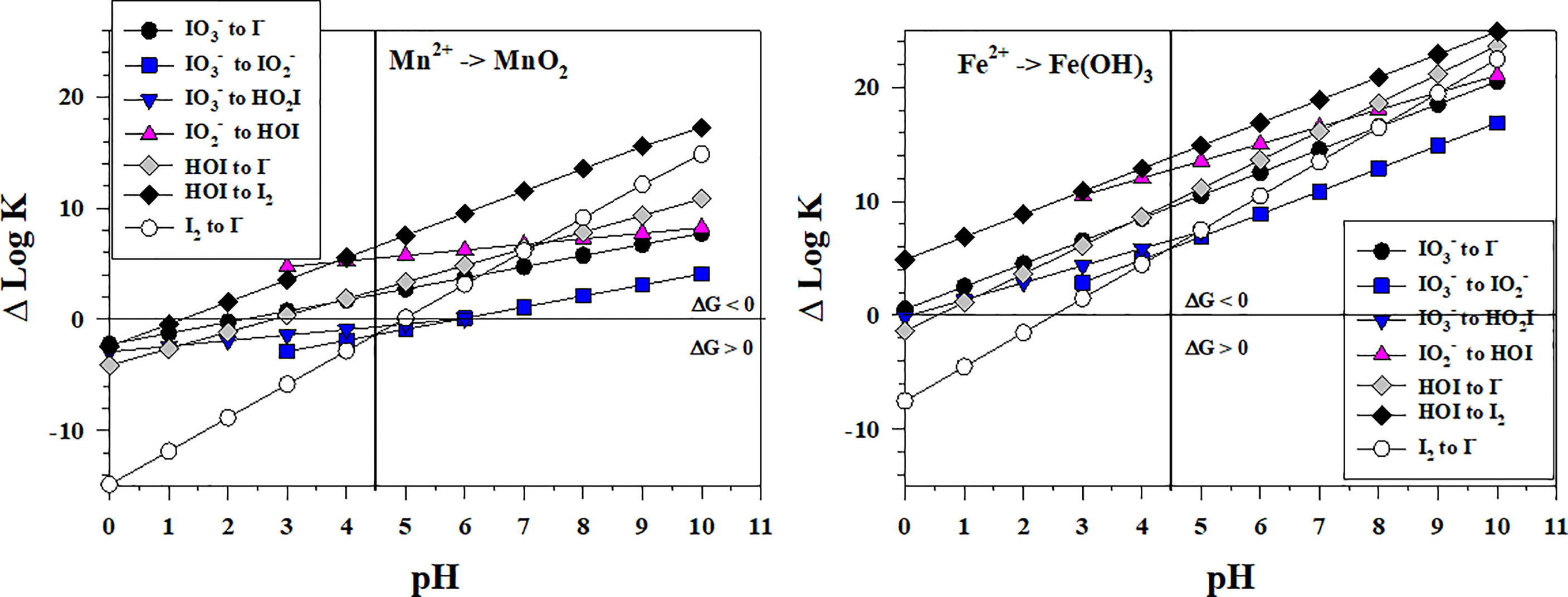
Figure 4 Thermodynamics for the reduction of (Io5a, Io5b, Io6), (Io4b), HOI (Io2, Io3), and I2 (Io1) by Mn2+ (Mn1) and Fe2+ (Fe3). The vertical line represents the pKa value of 4.49 for HO2I. Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
For the reaction sequence with Fe2+, all iodine species reductions are favorable over the entire pH range except for the I2 to I- reaction, which is favorable at pH > 2.5. Thus, there is no thermodynamic inhibition to reduction to I- by Fe2+, and this abiotic reaction at a pH of 7 was reported to be 92% complete after 2 hours using initial concentrations of 2 mM Fe2+ and 0.1 mM (Councell et al., 1997). Because the Fe(OH)3 to Fe2+ couple is a 1-electron transfer, two Fe2+ are required in each step of the sequence. Again, the to conversion is the least favorable and likely controlling step in this reaction sequence.
Comparing Figures 3, 4 indicates that the Mn2+ and reactions with iodine species have a similar range of ΔlogKreaction values whereas the Fe2+ reactions with iodine species are more favorable (higher ΔlogKreaction values).
3.5 Iodate reduction by sulfide
In sulfidic waters and porewaters, does not exist as sulfide reacts readily with it (Zhang and Whitfield, 1986), and S(0) forms as the initial sulfur product. Figure 5 shows the thermodynamics for the stepwise conversion of to and to HOI by sulfide where S(0) forms as an intermediate leading to S8. As the Gibbs free energy of formation for HSOH is unknown, HSOH could not be evaluated as an intermediate, which on continued oxidation would form . The reaction of sulfide with I2 and HOI is well known as the iodometric titration, so calculations were not performed. The only unfavorable iodine reduction reactions are the 1-electron reductions that lead to the formation of the HS radical (HS• or HS rad). The conversion of to HOI is more favorable as it has the larger ΔlogKreaction values. Again, the to conversion is the least favorable and likely controlling step in this reaction sequence.
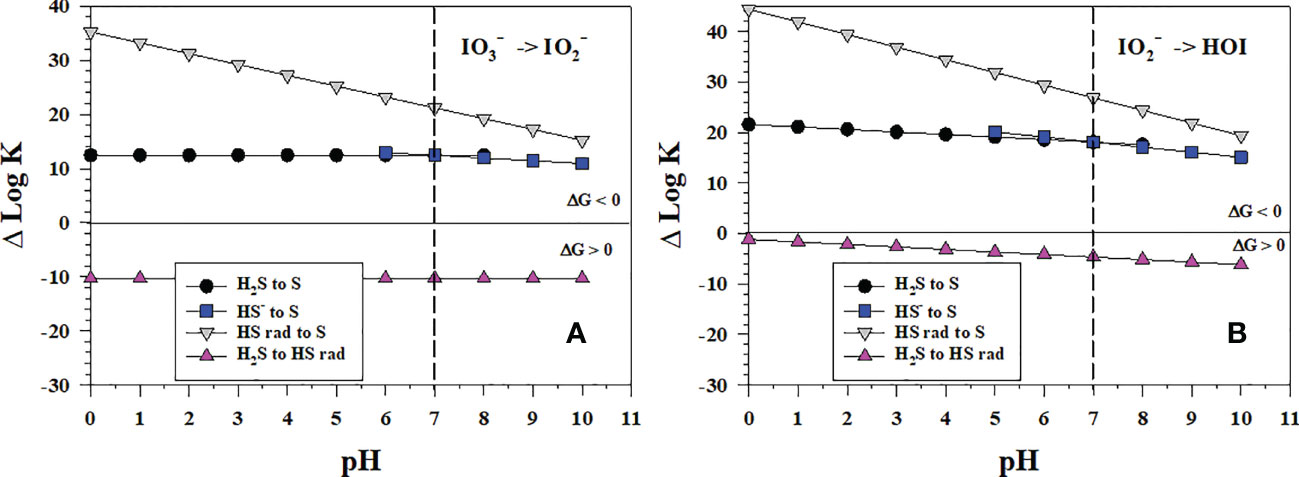
Figure 5 Thermodynamics for the 2-electron transfer reductions of (A) (Io5b) and (B) (Io4b) by sulfide species (S1, S2, S4, S5). The vertical line represents the pKa1 value for H2S. Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
3.6 Iodate reduction by NH4+
Figure 6 shows the thermodynamics for the stepwise conversion of to by where hydrazine (N2H4) and hydroxylamine (NH2OH) as well as their protonated forms could form as the first N intermediates. The thermodynamic calculations for these 2 electron transfers indicate that these reactions are not favorable. However, the reaction of the intermediates, if they could form by other processes, with to form N2 is very favorable. Thus, the to conversion is the controlling step in the reaction sequence with .
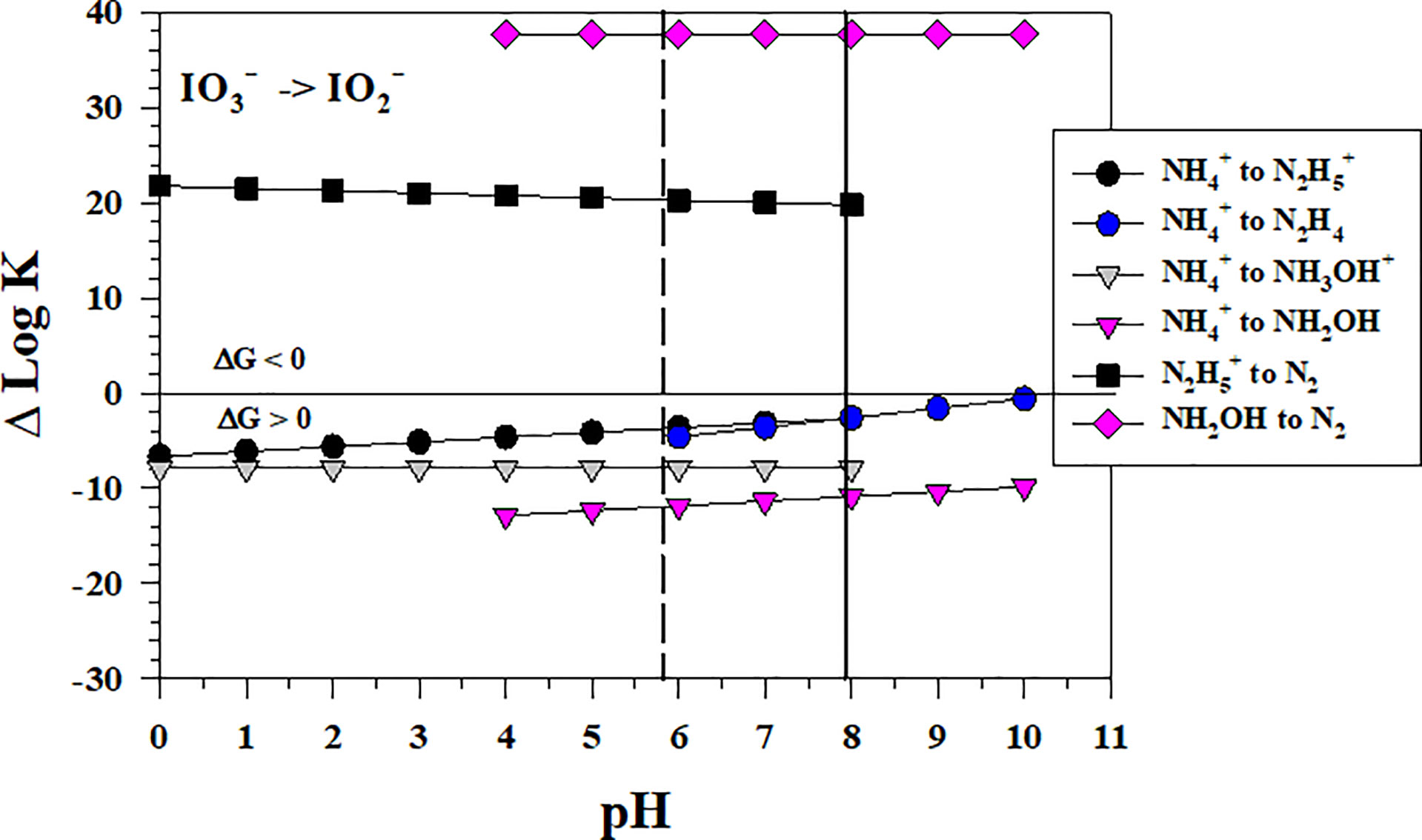
Figure 6 Thermodynamics for the reduction of to (IO5b) by to hydrazine species (N3a, N3b), hydroxylamine species (N4a, N4b) and by (N5) and NH2OH (N6). The vertical lines represent the pKa values for NH3OH+ (5.82) and (7.93), respectively. Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
4 Results and discussion: Iodide oxidation
4.1 Iodide oxidation by NO3-, MnO2 and Fe(OH)3
Figures 3–6 showed the reduction of with reductants. All values of ΔlogKreaction > 0 indicate a favorable reaction and all values of ΔlogKreaction< 0 indicate an unfavorable reaction. These figures can be used to discuss the reverse reaction of I- oxidation with oxidants. For reverse reactions, when a ΔlogKreaction< 0, then I- oxidation is favorable, but when ΔlogKreaction > 0, I- oxidation is unfavorable.
In Figure 3, is not an oxidant for I- (reverse of the and I2 reaction) except for the formation of I2 at a pH< 4.
In Figure 4, MnO2 oxidizes I- to HOI (reverse of the Mn2+ and HOI reaction) at a pH< 3 and I- to I2 (reverse of the Mn2+ and I2 reaction) at a pH< 5. A couple of laboratory studies showed I- oxidation with synthetic birnessite (δ-MnO2). First, Fox et al. (2009) showed that I2 was produced over the pH range 4.50 – 6.25, and that formed in smaller amounts. The kinetics of the reaction were slower at higher pH by 1.5 log units (> 30-fold) and were slower when smaller amounts of MnO2 were added (Table 2). Allard et al. (2009) investigated the same reactants to a pH of 7.5 and found I2 and as products; above pH = 7 the reaction is very slow. Iodate was found mainly in lower pH waters. Both I2 and adsorb to the birnessite surface. Similar results have been found over the pH range 4-6 for Mn(III) solids (Szlamkowicz et al., 2022). These MnOx reactions with I- are much slower that the reactions with reactive oxygen species (Table 2). Nevertheless, these are important as Kennedy and Elderfield (1987a, 1987b) showed that the conversion of iodide to iodate occurred in marine sediments.
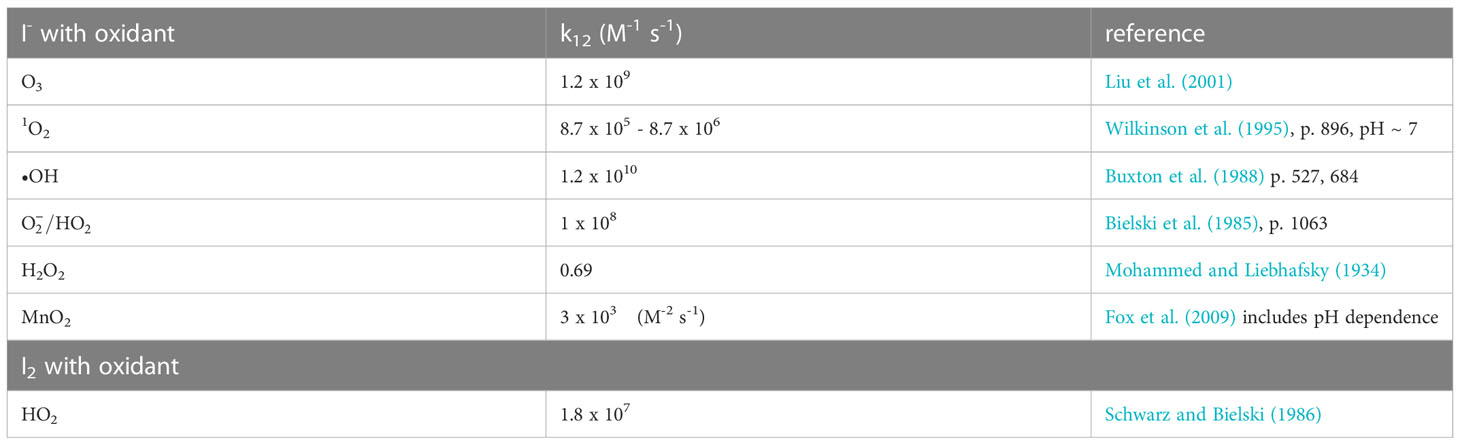
Table 2 Kinetic rate constants for the reaction of oxidants with Iodide and I2.
Figure 4 also shows that I- oxidation by Fe(OH)3 to I2 (reverse of the Fe2+ and I2 reaction) should occur only at a pH< 2.5. The I- to HOI conversion (reverse of the Fe2+ and HOI reaction) is favorable at pH ≤ 0.5.
4.2 Iodide oxidation by oxygen species
Figure 7 shows the thermodynamics of I- oxidation to I2 by oxygen species. Figure 7A shows that the one-electron process for I- oxidation with 3O2 is thermodynamically unfavorable over all pH whereas Figure 7B shows that the two-electron process is favorable at a pH< 3. Figure 7A shows that the successive 1-electron oxidations of I- where superoxide () is reduced to hydrogen peroxide (H2O2), which is reduced to hydroxyl radical (•OH). Only •OH is thermodynamically favorable over the pH range considered. and H2O2 show favorable reactions at pH< 9 and pH< 6, respectively.
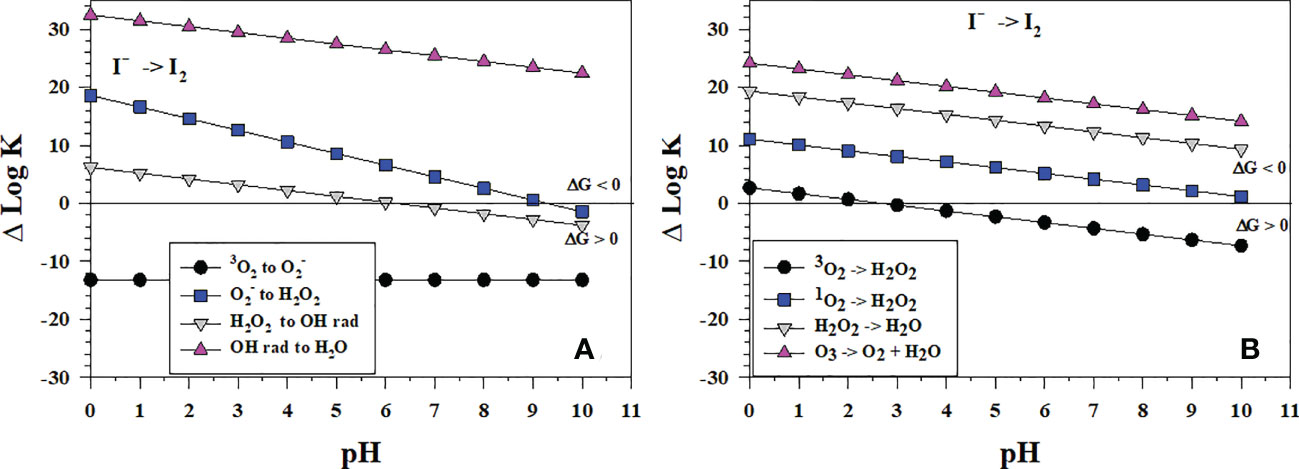
Figure 7 Thermodynamics for I2 formation via the oxidation of I- (Io1) with (A) the successive 1-electron oxidants 3O2 (O6), (O7), H2O2 (O8) and •OH (O9b); (B) the 2-electron oxidants 3O2 (O2), 1O2 (O5), H2O2 (O3), and O3 (O4). Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
By contrast, Figure 7B shows that the reactions of I- with the 2-electron oxidants H2O2, 1O2 and O3 are all thermodynamically favorable. The likely reaction pathway is the loss of 2-electrons to produce I+, which then reacts with I- to form I2. Note that H2O2 reacts to form H2O not •OH in Figure 7B. The 2-electron reaction with O3 (Figure 7B) is more favorable than the 1-electron reaction (Figure 7A).
Wong and Zhang (2008) showed that H2O2 oxidizes I- in artificial seawater from pH 7-9, which is consistent with Figure 7B. However, I- oxidation does not lead to iodate. In fact, I- reforms. They proposed that I2 formed and was reduced back to I-, but they did not provide a mechanism. The reverse reaction of I2 with •OH (ΔlogKreaction< 0 in the plot) is favorable to reform H2O2 and I- at pH > 6 whereas the reverse reaction of H2O2 with I2 to reform and I- is favorable at a pH > 9 (ΔlogKreaction< 0 in the plot). These thermodynamic data indicate that H2O2 can form I2 in a 2-electron transfer (Figure 7B) and then reduce I2 to I- in a 1-electron transfer (Figure 7A).
At seawater pH, superoxide, , can oxidize I- to I2 and the reaction occurs with a rate constant of 108 M-1s-1 (Bielski et al., 1985; Table 2). Because I2 is a good electron acceptor, the subsequent reaction of with I2 leads to (Schwarz and Bielski, 1986). As to be discussed in section 4.3, I2 reacts with organic matter to form organo-iodine compounds. Extracellular is generated by Roseobacter sp. AzwK-3b (Li et al., 2014) and results in the oxidation of Mn2+ to Mn(III,IV) oxides. However, Li et al. (2014) found that also oxidized I-. Considering that extracellular formation is a widespread phenomenon among marine and terrestrial bacteria, this could represent an important first step in the pathway for iodide oxidation in some environments. The Mn oxides formed by Roseobacter sp. AzwK-3b are not the oxidant as MnO2 kinetics is slower (Table 2).
To obtain , further oxidation of I2 to HOI must occur, and •OH is one candidate with a rate constant of 1.2 x 1010 (Buxton et al., 1988; Table 2). Also, O3 has a rate constant of 1.2 x 109 (Liu et al., 2001).
I2 is a prominent intermediate in I- oxidation yet HOI is needed to form . HOI can form directly from I- and I2 oxidation or from hydrolysis of I2 (reverse of eqn. 5), which is fast at basic pH (Wong, 1991). Figure 8A shows that of the successive 1-electron oxidants (starting from O2) for I2 oxidation, only •OH is thermodynamically favorable over all pH to form HOI whereas O3 is favorable at pH > 6, and is favorable at pH< 6. H2O2 as a 1-electron oxidant cannot oxidize I2 to form HOI, but H2O2 can reduce HOI to I2 (reverse of the and I2 reaction). Figure 8B indicates that, as 2-electron oxidants, H2O2 and O3 oxidation can lead to HOI formation. Comparing Δlog K values in Figures 7, 8 indicates that oxidation of I2 to HOI is less favorable than the oxidation of I- to I2.
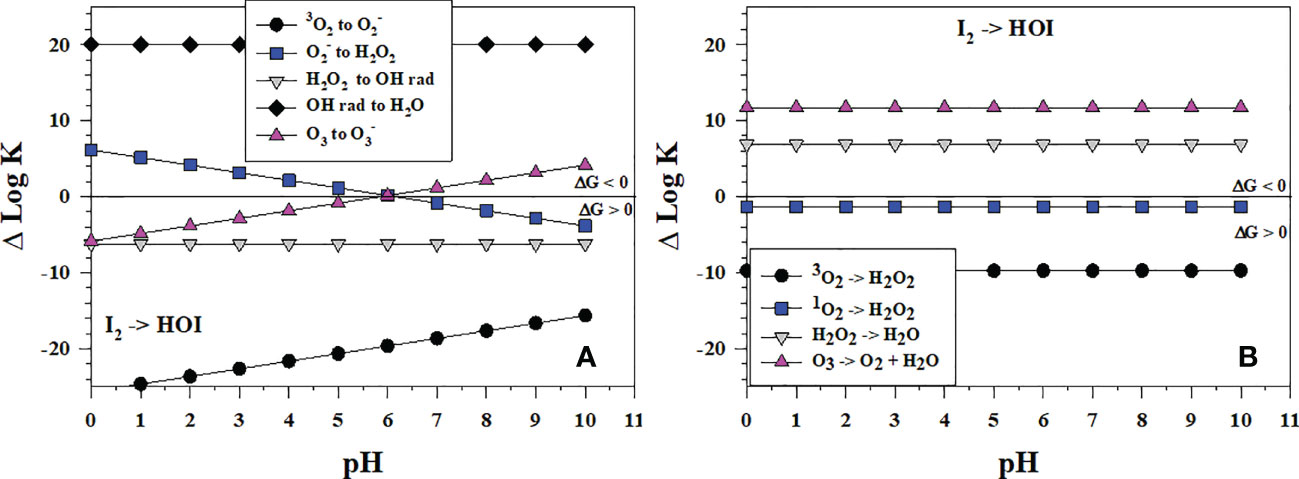
Figure 8 Thermodynamics for the formation of HOI via the oxidation of I2 (Io3) with (A) the successive 1-electron oxidants 3O2 (O6), (O7), H2O2 (O8), •OH (O9b) and O3 (O11); (B) the 2-electron oxidants 3O2 (O2), 1O2 (O5), H2O2 (O3), and O3 (O4). Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
These data also indicate why the comproportionation reaction of HOI with I- to form I2 can occur (eqn. 5, Carpenter et al., 2013).
Although disproportionation of HOI to and I- (eqn. 6) is fast in strongly basic solution, it is not detectable at seawater pH (Wong, 1991).
Figure 9 shows the successive 2-electron oxidation reactions of I-, HOI and with 3O2, 1O2, H2O2 and O3. 3O2 cannot affect the oxidation at any pH. Figure 9 shows that O3 oxidation reactions with I-, HOI and are favorable; thus, O3 can affect the complete oxidation of I- to . Also, the H2O2 oxidation reactions of I-, HOI and are favorable and can lead to formation; however, the kinetics of H2O2 oxidation can be slow. Haloperoxidase enzymes from organisms enhance the kinetics (Butler and Sandy, 2009) as does the reaction of H2O2 with carboxylic acids secreted by microbes to form peroxy carboxylic acids, which in turn oxidize I- to I2 (Li et al., 2012). The reactive oxygen species 1O2 can oxidize I- at pH< 10, oxidize HOI at pH > 5, and over all pH. Thus, 1O2 can be an oxidant of I- to at seawater pH. These data indicate that HOI oxidation leads to formation.
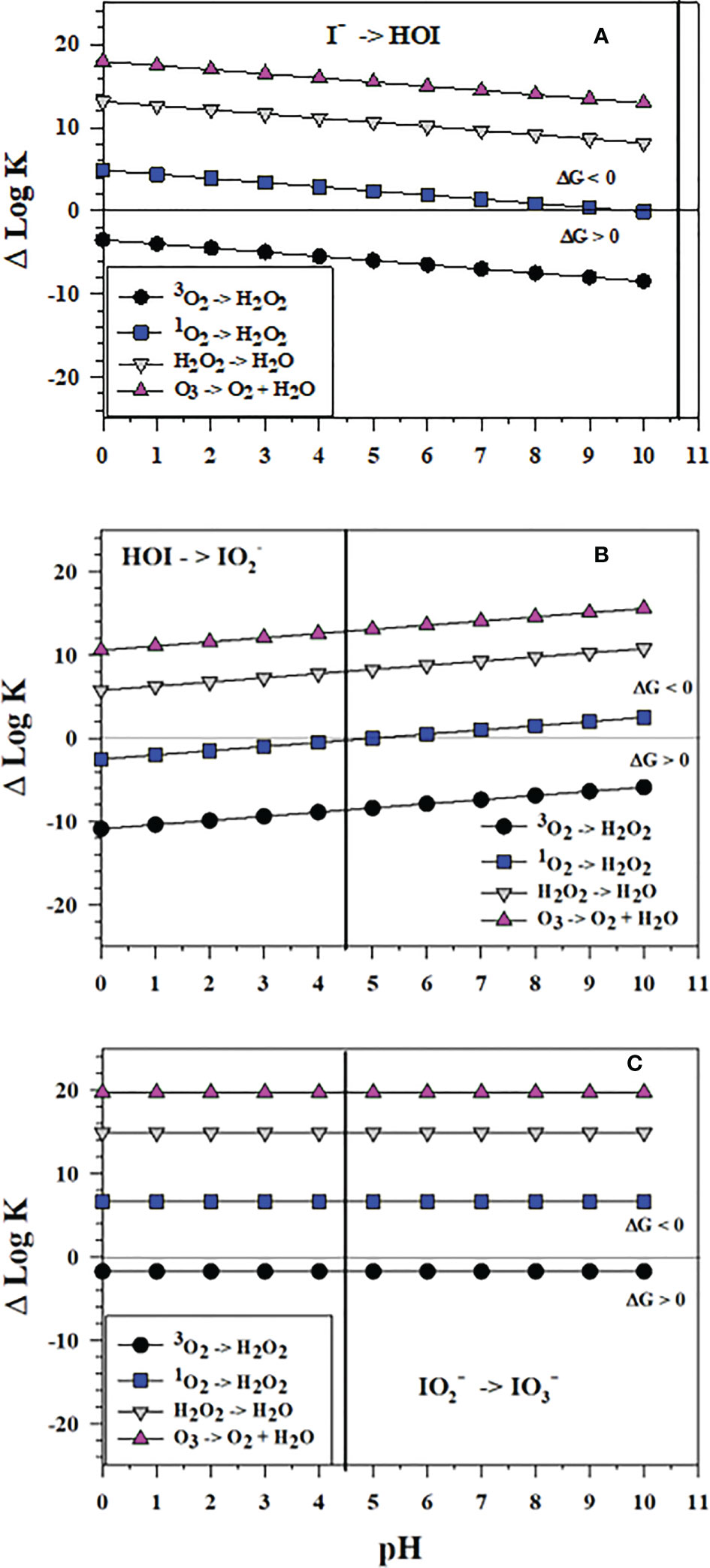
Figure 9 Thermodynamics for the reaction of 3O2 (O2), 1O2 (O5), H2O2 (O3), and O3 (O4) as 2-electron oxidants with (A) I- to form HOI (Io2), (B) HOI to form (Io4b) and (C) to form (Io5b). The vertical lines represent the pKa value of 4.49 for HO2I dissociation to . Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
Interestingly, ΔlogK values in Figure 9A show that the thermodynamics of I- oxidation by the 2-electron oxidants O3 and H2O2 to form HOI is slightly less favorable than I2 formation (Figure 7B). Conversely, thermodynamics of I- oxidation by H2O2 as a 2-electron oxidant to form HOI (Figure 9A) is more favorable than I2 formation (Figure 7A).
As shown in Figure 10, a potentially potent oxidant for I- is N2O, which is an O atom transfer oxidant like O3. However, the N2O concentration in seawater is minor, but the largest reported values are 90 and 250 nmol kg-1 for the OMZs of the Arabian Sea (Freing et al., 2012) and the Eastern Tropical North Pacific (Damgaard et al., 2020), respectively. These values are smaller than the total iodine concentration in seawater. The N2O concentration in the atmosphere is 335 ppbv (August 2022, https://www.n2olevels.org), which is equivalent to 0.0331 Pa or 7.8 nM dissolved in surface seawater (salinity of 35) at 20 0C using the solubility data from Weiss and Price (1980).
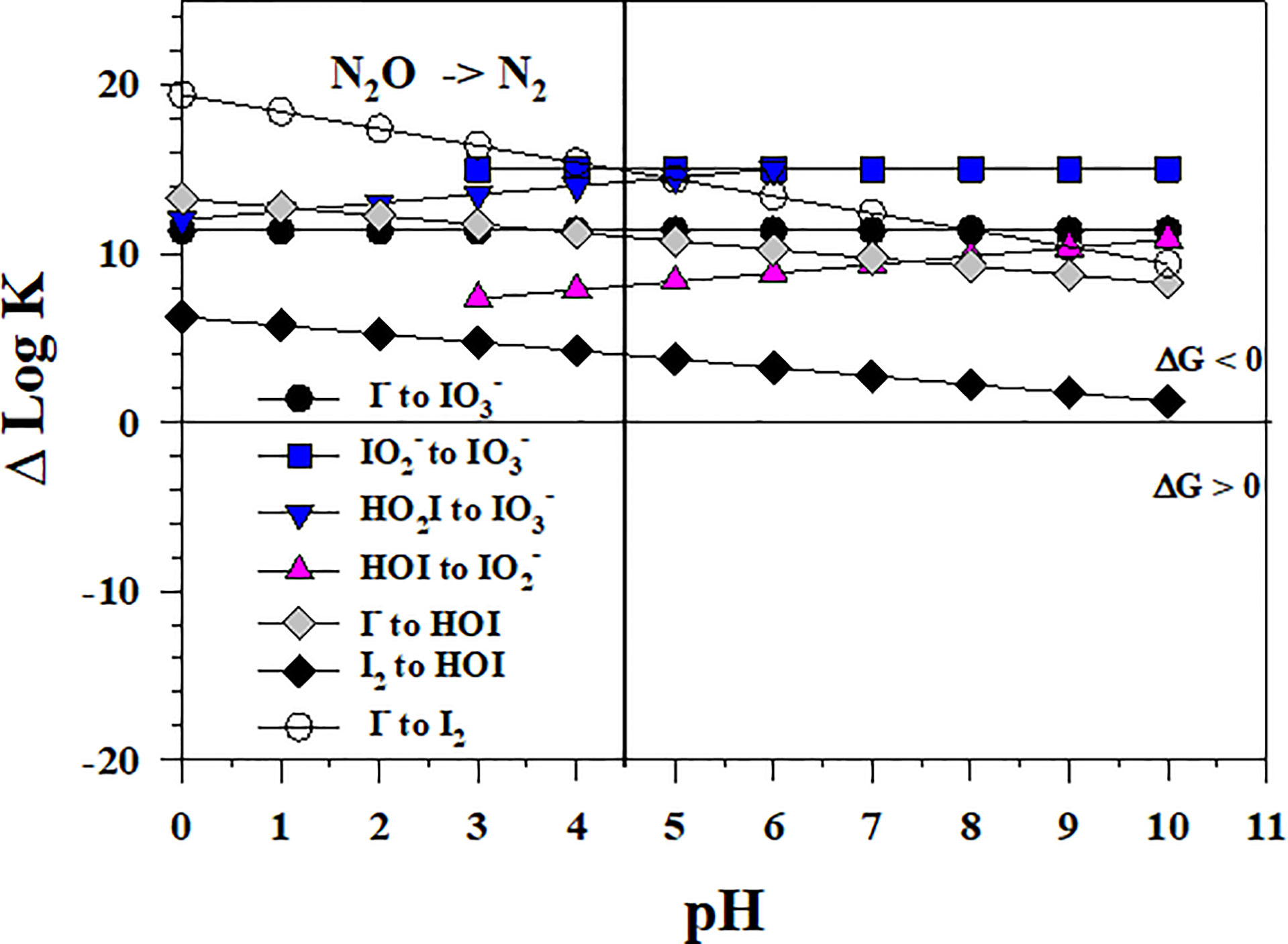
Figure 10 Thermodynamics for the 1- and 2- electron reductions of (Io5a, Io5b, Io6), (Io4b), HOI (Io2, Io3), and I2 (Io1) by N2O (N2). Data above the horizontal line at ΔlogK (ΔlogKreaction) = 0 indicate a favorable reaction and data below the horizontal line indicate an unfavorable reaction.
4.3 ROS in seawater
Reactive oxygen species exist in marine waters, but at low concentrations. O3 penetrates a few micrometers through the water-air interface at surface iodide concentrations (Carpenter et al., 2013). Powers and Miller (2014) showed that solar-induced processes with organic matter in freshwater and seawater are a major source of ROS (as , H2O2, and •OH) with the inventory and production rates for H2O2 in surface seawater being highest of the ROS. Also, Sutherland et al. (2020) report that dark, extracellular production is prolific among marine heterotrophic bacteria, cyanobacteria, and eukaryotes. In surface ocean waters, the concentration of H2O2 ranges from 20 - 80 nM (Yuan and Shiller, 2001), biological production gives a total concentration of ~ 0.07 to 0.30 nM (Sutherland et al., 2020), •OH concentration is ~10-18 M (Mopper and Zhou, 1990), and 1O2 concentration ranges from 10-13 to 10-14 M (Sunday et al., 2020). However, these ROS concentrations are typically smaller than I- concentrations, which range from 10 to 200 nM (Chance et al., 2019). Thus, I- oxidation in seawater samples should be difficult to observe experimentally. Hardisty et al. (2020) tracked the addition of stable isotopes of iodide in sample incubations and report the rate of I- oxidation to be 118–189 nM yr-1, which is similar to rates reported by mass balance approaches (Campos et al., 1996a; Truesdale et al., 2001; Žic and Branica, 2006; Žic et al., 2008). Hardisty et al. (2020) report that the product is likely HOI that results in the formation of organic-iodine compounds (see section 4.6) which on decomposition can release I-.
As the surface concentrations of ROS are smaller than the I- concentration, the question is how does I- get oxidized to in seawater? Microbial processes and the oxidation of I species in the atmosphere by ROS are likely candidates. These are now discussed.
4.4 Iodide oxidation in brown kelp
Brown kelp are the strongest accumulators of iodine as I- among living organisms (up to 100 mM, Küpper et al., 2008). The element iodine was discovered by the formation of I2 during exposure of brown kelp to concentrated sulfuric acid, which oxidized I- to I2. Kelp releases I- on the thallus surface and in the apoplast when undergoing oxidative stress during the partial emersion of the brown kelp forest at low tide; e.g., by exposure to high irradiance, desiccation, and atmospheric O3. Kelp contain vanadium haloperoxidases (Colin et al., 2003; Küpper et al., 2008) that enhance I- oxidation by H2O2. Whereas the nonenzymatic reaction of I- with H2O2 is slow, the reactions with O3, , 1O2, and •OH are very fast (> 108 M-1s-1, Table 2); they are also faster than MnO2 oxidation of I-. Küpper et al. (2008) consider I- as the simplest antioxidant known.
4.5 Iodide oxidation by microbes
Hughes et al. (2021) report that production occurs in cultures of the ammonia-oxidizing bacteria Nitrosomonas sp. and Nitrosococcus oceani supplied with I-, but not in cultures of three different nitrite oxidizing bacteria. Information on the enzymes mediating the oxidation were not studied. Nevertheless, oxidation via nitrification occurs via NH2OH formation which is a 2-electron reaction. Further reaction of NH2OH via metalloenzymes (e.g., Mo and W oxidases that transfer O atoms) leads to (a 4-electron transfer) and . I- likely goes through the intermediates and HOI to form , but these intermediates are reactive and not detectable by present analytical methods unlike .
Consistent with the Hughes et al. (2021) report, Kennedy and Elderfield (1987a, 1987b) showed that the conversion of iodide to iodate occurred in marine sediments. Microbial intervention is likely, but reaction of I- with oxidized Mn is possible depending on the pH.
Amachi and Iino (2022) reviewed the genus Iodidimonas, which was originally found in brines, but was also cultured from seawater enriched with I-. I2 is the first oxidation product. Iodidimonas contains the iodide oxidizing enzyme (IOX), which is an extracellular protein that contains multicopper oxidases. Iodidimonas requires O2, not H2O2, as the electron acceptor. Other oxidants are required to oxidize I2 to with O3 and •OH being the most effective.
4.6 HOI and I2 formation leads to organic iodine
Competing with the inorganic interconversion between iodide and iodate is the formation of organic iodine compounds. Formation of C-I bonds can occur during the reduction of and oxidation of I-. Complete reduction of to I- does not need to occur intercellularly and can lead to HOI and I2 formation as in Figure 4. The first step in I- oxidation also leads to I2 and HOI. I2 is neutral and adds to organic compounds such as olefins, which are not very reactive in seawater, whereas I+ in HOI reacts with α-keto compounds and peptides through keto-enol isomerization (Truesdale and Luther, 1995). Both I2 and HOI lead to volatile and nonvolatile organic-iodine (R-I) compounds with C-I or N-I bonds, and Harvey (1980) showed that N-iodo amides were the main organic iodine components in marine sediments. On decay of organic compounds, the C(N)-I bond breaks leading to I- release, which mimics the senescence pathway outlined by Bluhm et al. (2010) and Hepach et al. (2020). Recently, Ooki et al. (2022) showed that CH3I and CH3CH2I formed in sediments from polar and subpolar seas and was related to increased phytodetritus at the seafloor after the spring bloom.
Allard and Gallard (2013) showed that the oxidation of I- by birnessite in the presence of organic matter also led to CH3I over the pH range 4-5.
As total iodine in surface ocean waters is lower by a few percent compared to deep waters (Wong, 1991), the decomposition of organic-iodine leads to some I- release, which may be oxidized to by ammonia-oxidizing bacteria (Hughes et al., 2021). This is similar to release and oxidation of to from particulate organic matter in deep waters that results in an increase of concentration with depth (recycled element profile). Deep waters contain mainly , so not much I- is released to the deep-water column by in situ water column processes, and most organic-iodine gets to the sediments where it is released as I- (Kennedy and Elderfield, 1987a, Kennedy and Elderfields 1987b; Luther et al., 1995). Kennedy and Elderfield (1987a, 1987b) and Shimmield and Pedersen (1990) report that the molar I/C ratio in planktonic organisms is 10-4 whereas it is typically >10-3 in sediments. Decomposition of sedimentary organic-I releases I- to porewaters and the overlying water column where it can be transported hundreds of kilometers offshore along isopycnal surfaces in OMZs (Farrenkopf and Luther, 2002; Cutter et al., 2018).
4.7 Surface seawater and atmospheric formation of IO3-, and iodine speciation in the atmosphere
There is significant literature showing that coastal and oceanic regions are sources of iodine emissions to the atmosphere, and I note some important aspects of this air-sea connection. I- reacts with O3 to form IO-, which at seawater pH forms HOI. Carpenter et al. (2013) showed that this reaction occurs in the first few micrometers below the air-water interface and that HOI is ten-fold greater than I2 above the sea surface. HOI contributes 75% of the observed iodine oxide aerosol levels over the tropical Atlantic Ocean, and these iodine emissions to the atmosphere have increased 3-fold over the last century due to the increase in anthropogenic O3 (Carpenter et al., 2021). O3 reacts stepwise with this gaseous HOI (IO-) and gaseous to form , which can attach to aerosols.
Formation and release of gaseous I2 from seawater to air permits photochemical breaking of the I-I bond to form gaseous I atoms, •I, which are reactive radicals. Similarly, release of volatile organic-iodine compounds leads to the homolytic cleavage of the C-I bond to form I•. O3 reacts readily with I• to form gaseous IO• in the marine boundary layer (Whalley et al., 2010). Further stepwise oxidation of gaseous IO•/HOI leads to . In laboratory experiments using mass spectrometry detection, Teiwes et al. (2019) showed that hydrated iodide, I(H2O)-, reacts with gaseous O3 to form directly without formation of gaseous HOI or IO-; thus, can form in a two-step reaction sequence in the atmosphere.
Using mass spectrometry to evaluate atmospheric IxOy cluster and (nano)particle formation above seabed macroalgae, Sipilä et al. (2016) showed the stepwise formation of HIO3 via HOI and IO•, which leads to (I2O5)x clusters (x=2-5) containing HIO3 that result in iodine rich aerosol particles. These data on the formation of I2O5 aerosols agree with the exothermic ΔHreaction values of iodine oxide species reacting with O3 and each other calculated using quantum mechanics (Kaltsoyannis and Plane, 2008). Sipilä et al. (2016) also showed that cluster formation increased as a burst at low tide indicating significant I2 release from the macroalgae (and subsequent oxidation) as found by Küpper et al. (2008). Hydration of I2O5 leads to two . In mass spectrometry laboratory studies, Martín et al. (2022) showed that new iodine containing (nano)particles and also form in the presence of and provide ΔHreaction data for the gas phase reactions involved. Experiments using the CERN CLOUD (Cosmics Leaving Outdoor Droplets) chamber documented the formation of HIO3 via iodooxy hypoiodite, IOIO, as an intermediate (Finkenzeller et al., 2022) and the fast growth of HIO3 as (nano)particles (He et al., 2021).
In recent atmospheric campaigns, Koenig et al. (2020) showed that is the main iodine reservoir as it forms on aerosols in the stratosphere with iodine being responsible for 32% of the halogen induced O3 loss. Cuevas et al. (2022) also showed that iodine can dominate (∼73%) the halogen-mediated lower stratospheric ozone loss during summer and early fall, when the heterogeneous reactivation of inorganic chlorine and bromine reservoirs is reduced.
The information in the preceding paragraphs along with the thermodynamic data from Martín et al. (2022), Figure 2 (the half reaction for O3 to O2 and H2O) and Figure 9 predict that should be the dominant species in the atmosphere. Although reduction of is not predicted in an oxidizing atmosphere, analyses of rainwater (Campos et al., 1996b; Truesdale and Jones, 1996; Baker et al., 2001; Hou et al., 2009), aerosols (Gilfedder et al., 2008; Droste et al., 2021) and snow (Gilfedder et al., 2008) in the marine boundary layer indicate that aqueous iodide and iodate coexist. Hou et al. (2009) reviewed wet iodine speciation data and reported that predominates over I- from marine sources/air masses whereas I- predominates from continental air masses.
There are several ways that I- (or reduced I) can form in rainwater and aerosols. The interconversion between and I- at the pH of wet deposition also leads to HOI and I2, which can react with organic material forming C-I bonds that can release I- (section 4.6). This material has been given the term soluble organically bound iodine and can be larger than the sum of the concentrations of and I- in aerosols (Gilfedder et al., 2008; Droste et al., 2021). Soluble organically bound iodine can form from release of natural organic iodine from land and sea (a primary source) or from the reaction of natural organic material with HOI or I2 in the atmosphere (a secondary source). On photolysis of C-I, I• forms and reacts with O3, and on C-I reaction with nucleophiles, I- forms. During a study on the formation of cloud condensation nuclei, Huang et al. (2022) also showed that natural gaseous organic material in the marine boundary layer reacts with in aerosols resulting in gaseous I2, which can be reoxidized to (catalysis) or react to form organic-I compounds. Lastly, Cuevas et al. (2022) reported that photolysis of particles in the stratosphere at a wavelength of about 260 nm can lead to gaseous I• and O2 during transport from the tropics to the Antarctic region. Thus, there are several pathways for reduction of in the atmosphere.
5 Conclusions
The reduction of to I- in solution is a facile process by biotic and abiotic reactions. The intermediates and HOI dictate the reactivity sequence via a combination of thermodynamic and kinetic considerations. The to conversion is the least favorable and likely controlling step in this reaction sequence, but there is no need for nitrate reductase for reduction based on numerous studies. The data from this study indicate that once forms there is no thermodynamic barrier to I- formation. Chemical reduction of all iodine species (not iodide) by sulfide, Fe2+ and Mn2+ are favorable at seawater and sedimentary pH values, but only sulfide has been studied in the laboratory at oceanic pH values. Dissimilatory reduction during organic matter decomposition seems to be a key process as the couple is more favorable than the couple.
However, the oxidation of I- back to via 3O2 has a major thermodynamic barrier in solution, and the disproportionation of HOI at seawater pH values is not measurable. Thus, ROS, oxidized Mn and microbes are important for I- oxidation to due to favorable thermodynamics and kinetics (Table 2). Recent reports of microbial oxidation have not documented the entire six-electron oxidation in a stepwise manner so further work on this topic is necessary. Oxidation of I- by oxidized Mn is a pH dependent reaction and less likely at seawater pH values but could occur in sedimentary environments. The reactions of O3 and •OH with iodine species (not ) are thermodynamically favorable over all pH. However, ROS are not normally in significant concentration in seawater to influence formation. Notable exceptions are for (1) sea surface microlayer, which adsorbs atmospheric O3, and (2) the reaction of Fe2+ with O2 that leads to Fenton chemistry with •OH production. Systems where Fenton chemistry can occur are at/near hydrothermal vents (Shaw et al., 2021), submarine groundwaters (Burns et al., 2010), and sediments or water columns where O2 and Fe2+ concentration profiles overlap including ancient earth (Chan et al., 2016).
I- is a major sink for O3 in the sea surface microlayer and the atmosphere. formation in the atmosphere and redeposition to surface seawater may be major iodine processes with the latter being similar to the deposition of trace metals from wet and dry deposition to the surface ocean (e.g., Chance et al., 2015; Meskhidze et al., 2019). Most atmospheric iodine originates from marine sources where I- oxidation to I2 and homolytic cleavage of C-I bonds occurs; thus, gaseous iodine emissions from the ocean are reduced. forms from these sources during oxidation by O3 in the atmosphere. An estimate of atmospheric deposition of to the ocean surface could be made by using the amount of in rainwater and aerosols that would be returned to the ocean surface, but more information on iodine speciation in rainwater and aerosols is needed as global spatial coverage appears limited. Despite major advances in iodine geochemistry over the last two decades, significant research is still needed on the processes that affect I- oxidation to in the atmosphere, seawater and ocean sediments.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Acknowledgments
The author thanks NSF for funding his group’s research on iodine marine chemistry over his career, and Thomas Church and Timothy Ferdelman for suggesting the author’s initial foray into iodine chemistry. The author thanks the reviewers and guest editor, Rosie Chance, for their comments and constructive suggestions to improve the manuscript.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allard S., Gallard H. (2013). Abiotic formation of methyl iodide on synthetic birnessite: A mechanistic study. Sci. Total Environ. 463–464, 169–175. doi: 10.1016/j.scitotenv.2013.05.079
Allard S., von Gunten U., Sahli E., Nicolau R., Gallard H. (2009). Oxidation of iodide and iodine on birnessite (δ-MnO2) in the pH range 4–8. Water Res. 43, 3417–3426. doi: 10.1016/j.watres.2009.05.018
Amachi S. (2008). Microbial contribution to global iodine cycling: volatilization, accumulation, reduction, oxidation, and sorption of iodine. Microbes Environ. 23 (4), 269–276. doi: 10.1264/jsme2.me08548
Amachi S., Iino T. (2022). The genus Iodidimonas: From its discovery to potential applications. Microorganisms 10, 1661. doi: 10.3390/microorganisms10081661
Amachi S., Kawaguchi N., Muramatsu Y., Tsuchiya S., Watanabe Y., Shinoyama H., et al. (2007). Dissimilatory iodate reduction by marine pseudomonas sp. strain SCT. Appl. Environ. Microbiol. 73, 5725–5730. doi: 10.1128/AEM.00241-07
Anschutz P., Sundby B., Lefrançois L., Luther III, G. W., Mucci A. (2000). Interactions between metal oxides and species of nitrogen and iodine in bioturbated marine sediments, geochim. Cosmochim. Acta 64, 2751–2763. doi: 10.1016/S0016-7037(00)00400-2
Baker A. R., Tunnicliffe C., Jickells T. D. (2001). Iodine speciation and deposition fluxes from the marine atmosphere. J. Geophys. Res. 106, 28743–28749. doi: 10.1029/2000JD000004
Bard A. J., Parsons R., Jordan J. (1985). Standard potentials in aqueous solution. 1st ed (New York: M. Dekker), 834.
Bielski B. H. J., Cabelli D. E., Arudi R. L., Ross A. B. (1985). Reactivity of HO2/O2- radicals in aqueous solution. J. Phys. Chem. Ref. Data 14, 1041–1100. doi: 10.1063/1.555739
Bluhm K., Croot P., Wuttig K., Lochte K. (2010). Transformation of iodate to iodide in marine phytoplankton driven by cell senescence. Aquat. Biol. 11, 1–15. doi: 10.3354/ab00284
Burns J. M., Craig P. S., Shaw T. J., Ferry J. L. (2010). Multivariate examination of Fe(II)/Fe(III) cycling and consequent hydroxyl radical generation. Environ. Sci. Technol. 44, 7226–7231. doi: 10.1021/es903519m
Butler A., Sandy M. (2009). Mechanistic considerations of halogenating enzymes. Nature 460, 848–854. doi: 10.1038/nature08303
Buxton G. V., Greenstock C. L., Helman W. P., Ross A. B. (1988). Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals in aqueous solution. J. Phys. Chem. Ref. Data 17, 513–886. doi: 10.1063/1.555805
Campos M., Farrenkopf A., Jickells T., Luther G. (1996). A comparison of dissolved iodine cycling at the Bermuda Atlantic time-series station and Hawaii ocean time-series station. Deep-Sea Res. II Top. Stud. Oceanogr. 43 (2), 455–466. doi: 10.1016/0967-0645(95)00100-X
Campos M. L. A. M., Nightingale P. D., Jickells T. D. (1996). A comparison of methyl iodide emissions from seawater and wet depositional fluxes of iodine over the southern north Sea. Tellus 48, 106–114. doi: 10.3402/tellusb.v48i1.15830
Carpenter L. J., Chance R. J., Sherwen T., Adams T. J., Ball S. M., Evans M. J., et al. (2021). Marine iodine emissions in a changing world. Proc. R. Soc A 477, 20200824. doi: 10.1098/rspa.2020.0824
Carpenter L., MacDonald S., Shaw M., Kumar R., Saunders R. W., Parthipan R., et al. (2013). Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 6, 108–111. doi: 10.1038/ngeo1687
Carrano M. W., Yarimizu K., Gonzales J. L., Cruz-López R., Edwards M. S., Tymon T. M., et al. (2020). The influence of marine algae on iodine speciation in the coastal ocean. Algae 35 (2), 167–176. doi: 10.4490/algae.2020.35.5.25
Chance R. J., Jickells T. D., Baker A. R. (2015). Atmospheric trace metal concentrations, solubility and deposition fluxes in remote marine air over the south-east Atlantic. Mar. Chem. 177, 45–56. doi: 10.1016/j.marchem.2015.06.028
Chance R., Malin G., Jickells T., Baker A. R. (2007). Reduction of iodate to iodide by cold water diatom cultures. Mar. Chem. 105, 169–180. doi: 10.1016/j.marchem.2006.06.008
Chance R. J., Tinel L., Sherwen T., Baker A. R., Bell T., Brindle J., et al. (2019). Global sea-surface iodide observations 1967–2018. Sci. Data 6, 286. doi: 10.1038/s41597-019-0288-y
Chan C. S., Emerson D., Luther G. (2016). The role of microaerophilic fe-oxidizing micro-organisms in producing banded iron formations. Geobiology 14, 509–528. doi: 10.1111/gbi.12192
Colin C., Leblanc C., Wagner E., Delage L., Leize-Wagner E., van Dorsselaer A., et al. (2003). The brown algal kelp laminaria digitata features distinct bromoperoxidase and iodoperoxidase activities. J. Biol. Chem. 278, 23545–23552. doi: 10.1074/jbc.M300247200
Councell T. B., Landa E. R., Lovely D. R. (1997). Microbial reduction of iodate. Water Air Soil pollut. 100, 99–106. doi: 10.1023/A:1018370423790
Cuevas C. A., Fernandez R. P., Kinnison D. E., Li Q., Lamarques J.-F., Trabelsi T., et al. (2022). The influence of iodine on the Antarctic stratospheric ozone hole. Proc. Natl. Acad. Sci. U.S.A. 119, No. 7, e2110864119. doi: 10.1073/pnas.2110864119
Cutter G. A., Moffett J. G., Nielsdottir M. C., Sanial V. (2018). Multiple oxidation state trace elements in suboxic waters off Peru: in situ redox processes and advective/diffusive horizontal transport. Mar. Chem. 201, 77–89. doi: 10.1016/j.marchem.2018.01.003
Damgaard L. R., Jelly C., Casciotti K., Ward B. B., Revsbech N. P. (2020). Amperometric sensor for nanomolar nitrous oxide analysis. Anal. Chim. Acta 1101, 135–140. doi: 10.1016/j.aca.2019.12.019
de la Cuesta J. L., Manley S. L. (2009). Iodine assimilation by marine diatoms and other phytoplankton in nitrate-replete conditions. Limnology Oceanogr. 54, 1653–1664. doi: 10.4319/lo.2009.54.5.1653
Droste E. S., Baker A. R., Yodle C., Smith A., Ganzeveld L. (2021). Soluble iodine speciation in marine aerosols across the Indian and pacific ocean basins. Front. Mar. Sci. 8, 788105. doi: 10.3389/fmars.2021.788105
Elderfield H., Truesdale V. W. (1980). On the biophilic nature of iodine in seawater. Earth Planet. Sci. Lett. 50, 105–114. doi: 10.1016/0012-821X(80)90122-3
Estes E. R., Berti D., Findlay A. J., Hochella M. F. Jr., Shaw T. J., Yücel M., et al. (2022). Differential behavior of metal sulfides in hydrothermal plumes and diffuse flows. ACS Earth Space Chem. 6, 1429–1442. doi: 10.1021/acsearthspacechem.1c00377
Farrenkopf A. M., Dollhopf M. E., Chadhain S. N., Luther G. W., Nealson K. H. (1997). Reduction of iodate in seawater during Arabian Sea shipboard incubations and in laboratory cultures of the marine bacterium Shewanella putrefaciens strain MR-4. Mar. Chem. 57 (3), 347–354. doi: 10.1016/S0304-4203(97)00039-X
Farrenkopf A. M., Luther G. W. (2002). Iodine chemistry reflects productivity and denitrification in the Arabian Sea: Evidence for flux of dissolved species from sediments of western India into the OMZ. Deep-Sea Res. II Top. Stud. Oceanogr 49 (12), 2303–2318. doi: 10.1016/S0967-0645(02)00038-3
Finkenzeller H., Iyer S., He X. C., Simon M., Koenig T. K., Lee C. F., et al. (2022). The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source. Nat. Chem 15, 129-135. doi: 10.1038/s41557-022-01067-z
Fox P. M., Davis J. A., Luther III, G. W. (2009). The kinetics of iodide oxidation by the manganese oxide mineral birnessite. Geochim. Cosmochim. Acta 73, 2850–2861. doi: 10.1016/j.gca.2009.02.016
Freing A., Wallace D. W. R., Bange H. W. (2012). Global oceanic production of nitrous oxide. Phil. Trans. R. Soc B 367, 1245–1255. doi: 10.1098/rstb.2011.0360
Gilfedder B. S., Lai S. C., Petri M., Biester H., Hoffmann T. (2008). Iodine speciation in rain, snow and aerosols. Atmos. Chem. Phys. 8, 6069–6084. doi: 10.5194/acp-8-6069-2008
Guo J., Jiang J., Peng Z., Zhong Y., Jiang Y., Jiang Z., et al. (2022). Global occurrence of the bacteria with capability for extracellular reduction of iodate. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1070601
Hardisty D. H., Horner T. J., Evans Z. C., Moriyasu R., Babbin A. R., Wankel S. D., et al. (2021). Limited in situ iodate reduction within the Eastern tropical north pacific oxygen deficient zone. Earth Planetary Sci. Lett. 554, 116676. doi: 10.1016/j.chemgeo.2019.119360
Hardisty D. H., Horner T. J., Wankel S. D., Blusztajn J., Nielsen S. G. (2020). Experimental observations of marine iodide oxidation using a novel sparge interface MC-ICP-MS technique. Chem. Geol. 532, 119360. doi: 10.1016/j.chemgeo.2019.119360
Harvey G. R. (1980). A study of the chemistry of iodine and bromine in marine sediments. Mar. Chem. 8, 327–332. doi: 10.1016/0304-4203(80)90021-3
He X.-C., Tham Y. J., Dada L., Wang M., Finkenzeller H., Stolzenberg D., et al. (2021). Role of iodine oxoacids in atmospheric aerosol nucleation. Science 371, 589–595. doi: 10.1126/science.abe0298
Hepach H., Hughes C., Hogg K., Collings S., Chance R. (2020). Senescence as the main driver of iodide release from a diverse range of marine phytoplankton. Biogeosciences 17, 2453–2471. doi: 10.5194/bg-17-2453-2020
Hou X. L., Aldahan A., Nielsen S. P., Possnert G. (2009). Time series of I-129 and I-127 speciation in precipitation from Denmark. Environ. Sci. Technol. 43, 6522–6528. doi: 10.1021/es9012678
Huang R.-J., Hoffmann T., Ovadnevaite J., Laaksonen A., Kokkola H., Xu W., et al. (2022). Heterogeneous iodine-organic chemistry fast-tracks marine new particle formation. Proc. Natl. Acad. Sci. U.S.A. 119, e2201729119. doi: 10.1073/pnas.2201729119
Hughes C., Barton E., Hepach H., Chance R., Pickering M. D., Hogg K., et al. (2021). Oxidation of iodide to iodate by cultures of marine ammonia-oxidising bacteria. Mar. Chem. 234, 104000. doi: 10.1016/j.marchem.2021.104000
Hung C.-C., Wong G. T. F., Dunstan W. M. (2005). Iodate reduction activity in nitrate reductase extracts from marine phytoplankton. Bul. Mar. Sci. 76, 61–72.
Kaltsoyannis N., Plane J. M. C. (2008). Quantum chemical calculations on a selection of iodine-containing species (IO, OIO, INO3, (IO)2, I2O3, I2O4 and I2O5) of importance in the atmosphere. Phys. Chem. Chem. Phys. 10, 1723–1733. doi: 10.1039/b715687c
Kennedy H. A., Elderfield H. (1987a). Iodine diagenesis in pelagic deep-sea sediments. Geochim. Cosmchim. Acta 51, 2489–2504. doi: 10.1016/0016-7037(87)90300-0
Kennedy H. A., Elderfield H. (1987b). Iodine diagenesis in non-pelagic deep-sea sediments. Geochim. Cosmchim. Acta 51, 2505–2514. doi: 10.1016/0016-7037(87)90301-2
Kim K., Ju J., Kim B., Chung H. Y., Vetráková L., Heger D., et al. (2019). Nitrite-induced activation of iodate into molecular iodine in frozen solution. Environ. Sci. Technol. 53, 4892–4900. doi: 10.1021/acs.est.8b06638
Koenig T. K., Baidar S., Campuzano-Jost P., Cuevas C. A., Dix B., Fernandez R. P., et al. (2020). Quantitative detection of iodine in the stratosphere. Proc. Natl. Acad. Sci. U.S.A. 117, 1860–1866. doi: 10.1073/pnas.1916828117
Küpper F. C., Carpenter L. J., McFiggans G. B., Palmer C. J., Waite T. J., Boneberg E.-M., et al. (2008). Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proc. Natl. Acad. Sci. U. S. A. 105, 6954–6958. doi: 10.1073/pnas.0709959105
Küpper F. C., Feiters M. C., Olofsson B., Kaiho T., Yanagida S., Zimmermann M. B., et al. (2011). Commemorating two centuries of iodine research: An interdisciplinary overview of current research. Angewandte Chemie Int. Editions 50, 11598–11620. doi: 10.1002/anie.201100028
Lehner P., Larndorfer C., Garcia-Robledo E., Larsen M., Borisov S. M., Revsbech N.-P., et al. (2015). LUMOS - a sensitive and reliable optode system for measuring dissolved oxygen in the nanomolar range. PloS One 10 (6), e0128125. doi: 10.1371/journal.pone.0128125
Lewis B. L., Luther III, G. W. (2000). Processes controlling the distribution and cycling of manganese in the oxygen minimum zone of the Arabian Sea. Deep Sea Res. 47, 1541–1561. doi: 10.1016/S0967-0645(99)00153-8
Li H.-P., Daniel B., Creeley D., Grandbois R., Zhang S., Xu C., et al. (2014). Superoxide production by a manganese-oxidizing bacterium facilitates iodide oxidation. Appl. Environ. Microbio 80, 2693–2699. doi: 10.1128/AEM.00400-14
Liu Q., Schurter L. M., Muller C. E., Aloisio S., Francisco J. S., Margerum D. W. (2001). Kinetics and mechanisms of aqueous ozone reactions with bromide, sulfite, hydrogen sulfite, iodide, and nitrite ions. Inorg. Chem. 40, 4436–4442. doi: 10.1021/ic000919j
Li H.-P., Yeager C. M., Brinkmeyer R., Zhang S., Ho Y.-F., Xu C., et al. (2012). Bacterial production of organic acids enhances H2O2-dependent iodide oxidation. Environ. Sci. Technol. 46, 4837–4844. doi: 10.1021/es203683v
Luther III, G. W. (2010). The role of one and two electron transfer reactions in forming thermodynamically unstable intermediates as barriers in multi-electron redox reactions. Aquat. Geochem. 16, 395–420. doi: 10.1007/s10498-009-9082-3
Luther III, G. W. (2011). Thermodynamic redox calculations for one and two electron transfer steps: Implications for halide oxidation and halogen environmental cycling. In Aquatic Redox Chemistry editors, Tratnyek P. G., Grundl T. J., Haderlein S. B. (Washington, D.C.: American Chemical Society) 15–35. doi: 10.1021/.bk-2011-1071.ch002
Luther III, G. W. (2016). Inorganic chemistry for geochemistry and environmental sciences: Fundamentals and applications. (Chichester, West Sussex: John Wiley & Sons Ltd.) 456.
Luther G. W., Campbell T. (1991). Iodine speciation in the water column of the black Sea. Deep Sea Res. Part A. Oceanographic Res. Papers 38, S875–S882. doi: 10.1007/978-94-011-2608-3_11
Luther G. W. III, Karolewski J. S., Sutherland K. M., Hansel C. M., Wankel S. D. (2021). The abiotic nitrite oxidation by ligand-bound manganese (III): the chemical mechanism. Aquat. Geochem. 27, 207–220. doi: 10.1007/s10498-021-09396-0
Luther III, G. W., Popp J. I. (2002). Kinetics of the abiotic reduction of polymeric manganese dioxide by nitrite: An anaerobic nitrification reaction. Aquat. Geochem. 8, 15–36. doi: 10.1023/A:1020325604920
Luther III, G. W., Wu J., Cullen J. (1995). “Redox chemistry of iodine in seawater: frontier molecular orbital theory considerations,” in Aquatic chemistry: Interfacial and interspecies processes, vol. 244 . Eds. Huang C. P., O'Melia C. R., Morgan J. J. (Washington, DC: American Chemical Society), 135–155. doi: 10.1021/ba-1995-0244.ch006
Martín J. C. G., Lewis T. R., James A. D., Saiz-Lopez A., Plane J. M. C. (2022). Insights into the chemistry of iodine new particle formation: The role of iodine oxides and the source of iodic acid. J. Am. Chem. Soc 144, 9240–9253. doi: 10.1021/jacs.1c12957
Meskhidze N., Völker C., Al-Abadleh H. A., Barbeau K., Bressac M., Buck C., et al. (2019). Perspective on identifying and characterizing the processes controlling iron speciation and residence time at the atmosphere-ocean interface mar. Chem. 217, 103704. doi: 10.1016/j.marchem.2019.103704
Moffett J. W., German C. R. (2020). Distribution of iron in the Western Indian ocean and the Eastern tropical south pacific: An inter-basin comparison. Chem. Geol. 532, 119334. doi: 10.1016/j.chemgeo.2019.119334
Mohammed A., Liebhafsky H. A. (1934). The kinetics of the reduction of hydrogen peroxide by the halides. J. Amer. Chem. Soc 56, 1680–1685. doi: 10.1021/ja01323a009
Mok J. K., Toporek Y. L., Hyun-Dong S., Lee B. D., Lee M. H., DiChristina T. J. (2018). Iodate reduction by shewanella oneidensis does not involve nitrate reductase. Geomicrobiol. J 35, 570-579. doi: 10.1080/01490451.2018.1430189
Mopper K., Zhou X. (1990). Hydroxyl radical photoproduction in the sea and its potential impact on marine processes. Science 250, 661–664. doi: 10.1126/science.250.4981.661
Müller E., von Gunten U., Bouchet S., Droz B., Winkel L. H. E. (2021). Reaction of DMS and HOBr as a sink for marine DMS and an inhibitor of bromoform formation. Environ. Sci. Technol. 55, 5547–5558. doi: 10.1021/acs.est.0c08189
Oldham V. E., Jones M. R., Siebecker M., Mucci A., Tebo B. M., Luther III, G. W. (2019). The speciation and mobility of Mn and fe in estuarine sediments. Aquat. Geochemistry 25, 3–26. doi: 10.1007/s10498-019-09351-0
Ooki A., Minamikawa K., Meng F., Miyashita N., Hirawake T., Ueno H., et al. (2022). Marine sediment as a likely source of methyl and ethyl iodides in subpolar and polar seas. Commun. Earth Environ. 3, 180. doi: 10.1038/s43247-022-00513-7
Owings S. M., Laurie Bréthous L., Eitel E. M., Fields B. P., Boever A., Beckler J. S., et al. (2021). Differential manganese and iron recycling and transport in continental margin sediments of the northern gulf of Mexico. Mar. Chem. 229, 103908. doi: 10.1016/j.marchem.2020.103908
Powers L. C., Miller W. L. (2014). Blending remote sensing data products to estimate photochemical production of hydrogen peroxide and superoxide in the surface ocean. Environ. Sci. Process. Impacts 16, 792–806. doi: 10.1039/C3EM00617D
Revsbech N. P., Larsen L. H., Gundersen J., Dalsgaard T., Ulloa O., Thamdrup B. (2009). Determination of ultra-low oxygen concentrations in oxygen minimum zones by the STOX sensor. Limnol. Oceanogr. Methods 7, 371–381. doi: 10.4319/lom.2009.7.371
Reyes-Umana V., Henning Z., Lee K., Barnum T. P., Coates J. D. (2022). Genetic and phylogenetic analysis of dissimilatory iodate-reducing bacteria identifies potential niches across the world’s oceans. ISME J. 16, 38–49. doi: 10.1038/s41396-021-01034-5
Rickard D., Luther III, G. W. (2007). Chemistry of iron sulfides. Chem. Rev. 107, 514–562. doi: 10.1021/cr0503658
Rue E. L., Smith G. J., Cutter G. A., Bruland K. W. (1997). The response of trace element redox couples to suboxic conditions in the water column. deep-Sea res. I Oceanogr. Res. Pap. 44 (1), 113–134. doi: 10.1016/S0967-0637(96)00088-X
Schmitz G. (2008). Inorganic reactions of Iodine(III) in acidic solutions and free energy of iodous acid formation. Int. J. Chem. Kinet. 40, 647–652. doi: 10.1002/kin.20344
Schwarz H. A., Bielski B. H. J. (1986). Reactions of HO2 and O2- with iodine and bromine and the I2- and I atom reduction potentials. J. Phys. Chem. 90, 1445–1448. doi: 10.1021/j100398a045
Shaw T. J., Luther G. W. III, Rosas R., Oldham V. E., Coffey N. R., Ferry J. L., et al. (2021). Fe-catalyzed sulfide oxidation in hydrothermal plumes is a source of reactive oxygen species to the ocean. Proc. Natl. Acad. Sci. U.S.A. 118 (40), e2026654118. doi: 10.1073/pnas.2026654118
Shimmield G. B., Pedersen T. F. (1990). The geochemistry of reactive trace metals and halogens in hemipelagic continental margin sediments. Rev. Aquat. Sci. 3, 255–279.
Shin H.-D., Toporek Y., Mok J. K., Maekawa R., Lee B. D., Howard M. H., et al. (2022). Iodate reduction by Shewanella oneidensis requires genes encoding an extracellular dimethylsulfoxide reductase. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.852942
Sipilä M., Sarnela N., Jokinen T., Henschel H., Junninen H, Kontkanen J., et al. (2016). Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3. Nature 537, 532–534. doi: 10.1038/nature19314
Stanbury D. (1989). “Reduction potentials involving inorganic free radicals in aqueous solution,” in Advances in inorganic chemistry, vol. 33 . Ed. Sykes A. G. (New York: Academic Press), 69–138.
Sunday M. O., Takeda K., Sakugawa H. (2020). Singlet oxygen photogeneration in coastal seawater: Prospect of Large-scale modeling in seawater surface and its environmental significance. Environ. Sci. Technol. 54, 6125–6133. doi: 10.1021/acs.est.0c00463
Sutherland K. M., Wankel S. D., Hansel C. M. (2020). Dark biological superoxide production as a significant flux and sink of marine dissolved oxygen. Proc. Natl. Acad. Sci. U.S.A. 117, 3433–3439. doi: 10.1073/pnas.1912313117
Szlamkowicz I. B., Fentress A. J., Longen L. F., Stanberry J. S., Anagnostopoulos V. A. (2022). Transformations and speciation of iodine in the environment as a result of oxidation by manganese minerals. ACS Earth Space Chem. 6, 1948–1956. doi: 10.1021/acsearthspacechem.1c00372
Teiwes R., Elm J., Bilde M., Pedersen H. B. (2019). The reaction of hydrated iodide I(H2O)- with ozone: A new route to IO2- products. Phys. Chem. Chem. Phys. 21, 17546–17554. doi: 10.1039/C9CP01734H
Trouwborst R. E., Clement B. G., Tebo B. M., Glazer B. T., Luther G. W. III (2006). Soluble Mn(III) in suboxic zones. Science 313, 1955–1957. doi: 10.1126/science.1132876
Truesdale V. W., Jones S. D. (1996). The variation of iodate and total iodine in some UK rainwaters during 1980-1981. J. Hydrology 179, 67–86. doi: 10.1016/0022-1694(95)02873-0
Truesdale V. W., Luther III, G. W. (1995). Molecular iodine reduction by natural and model organic substances in seawater. Aquat. Geochem. 1, 89–104. doi: 10.1007/BF01025232
Truesdale V. W., Watts S. F., Rendell A. (2001). On the possibility of iodide oxidation in the near-surface of the black Sea and its implications to iodine in the general ocean. Deep-Sea Res. I Oceanogr. Res. Pap 48 (11), 2397–2412. doi: 10.1016/S0967-0637(01)00021-8
Waite T. J., Truesdale V. W. (2003). Iodate reduction by Isochrysis galbana is relatively insensitive to de-activation of nitrate reductase activity–are phytoplankton really responsible for iodate reduction in seawater? Mar. Chem. 81, 137–148.
Weiss R. F., Price B. A. (1980). Nitrous oxide solubility in water and seawater. Mar Chem. 8, 347–359. doi: 10.1016/0304-4203(80)90024-9
Whalley L. K., Furneaux K. L., Goddard A., Lee J. D., Mahajan A., Oetjen H., et al. (2010). The chemistry of OH and HO2 radicals in the boundary layer over the tropical Atlantic ocean. Atmos. Chem. Phys. 10, 1555–1576. doi: 10.5194/acp-10-1555-2010
Wilkinson F., Helman W. P., Ross A. B. (1995). Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. expanded revised compilation. J. Phys. Chem. Ref. Data 24, 663–1021. doi: 10.1063/1.555965
Wong G. T. F. (2001). Coupling iodine speciation to primary, regenerated or "new" production: A re-evaluation. Deep Sea Res. Part I Oceanographic Res. Papers 48, 1459–1476. doi: 10.1016/S0967-0637(00)00097-2
Wong G. T., Brewer P. G. (1977). The marine chemistry of iodine in anoxic basins. Geochim. Cosmochim. Acta 41 (1), 151–159. doi: 10.1016/0016-7037(77)90195-8
Wong G. T. F., Piumsomboon A. U., Dunstan W. M. (2002). The transformation of iodate to iodide in marine phytoplankton cultures. Mar. Ecol. Prog. Ser. 237, 27–39. doi: 10.3354/meps237027
Wong G. T., Takayanagi K., Todd J. T. (1985). Dissolved iodine in waters overlying and in the orca basin, gulf of Mexico. Mar. Chem. 17, 177–183. doi: 10.1016/0304-4203(85)90072-6
Wong G. T., Zhang L.-S. (2008). The kinetics of the reactions between iodide and hydrogen peroxide in seawater. Mar. Chem. 111, 22–29. doi: 10.1016/j.marchem.2007.04.007
Yamazaki C., Kashiwa S., Horiuchi A., Kasahara Y., Yamamura S., Amachi S. A. (2020). Novel dimethylsulfoxide reductase family of molybdenum enzyme. Environ. Microbiol. 22, 2196–2212. doi: 10.1111/1462-2920.14988
Yuan J., Shiller A. M. (2001). The distribution of hydrogen peroxide in the southern and central Atlantic ocean. Deep Sea Res. Part II Top. Stud. Oceanogr 48, 2947–2970. doi: 10.1016/S0967-0645(01)00026-1
Zhang J.-Z., Whitfield M. (1986). Kinetics of inorganic reactions in seawater. 1. the reduction of iodate by bisulphide. Mar. Chem. 19, 121–137. doi: 10.1016/0304-4203(86)90044-7
Žic V., Branica M. (2006). The distributions of iodate and iodide in rogoznica lake (East Adriatic coast). Estuar. Coast. Shelf Sci. 66 (1), 55–66. doi: 10.1016/j.ecss.2005.07.022
Keywords: iodate, iodide, iodine intermediates, thermodynamics, oxidation, reduction
Citation: Luther GW III (2023) Review on the physical chemistry of iodine transformations in the oceans. Front. Mar. Sci. 10:1085618. doi: 10.3389/fmars.2023.1085618
Received: 31 October 2022; Accepted: 03 January 2023;
Published: 15 February 2023.
Edited by:
Rosie Chance, University of York, United KingdomReviewed by:
Lucy J. Carpenter, University of York, United KingdomAlex Baker, University of East Anglia, United Kingdom
Copyright © 2023 Luther. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George W. Luther III, luther@udel.edu