The respiratory microbiota of three cohabiting beluga whales (Delphinapterus leucas) under human care
 Susan A. Smith1,2*
Susan A. Smith1,2*  Destiny V. Ropati1,2
Destiny V. Ropati1,2  Luciana F. Santoferrara3
Luciana F. Santoferrara3  Tracy A. Romano1,2
Tracy A. Romano1,2  George B. McManus2
George B. McManus2- 1Research Department, Sea Research Foundation, Inc. d/b/a Mystic Aquarium, Mystic, CT, United States
- 2Department of Marine Sciences, University of Connecticut, Groton, CT, United States
- 3Department of Biology, Hofstra University, Hempstead, NY, United States
We sampled the respiratory mucus from voluntary blowhole exhalations (“blow”) of three healthy beluga whales (Delphinapterus leucas) under professional human care. Blow samples were collected from three resident belugas, one adult male (M1) and two adult females (F1, F2), with voluntary behaviors via non-invasive methods over three days in July 2021 (four days for M1). Samples were weighed and examined microscopically for the enumeration of eukaryotic and prokaryotic microbes, and then were used to evaluate carbon substrate use and taxonomic diversity of prokaryotic communities in the host respiratory sytem. Microscopical observations and 18S rRNA gene sequencing indicated the presence of eukaryotic microbiota, the ciliate genera Planilamina and Kyaroikeus in all three individuals. Exposure of samples to different metabolic carbon substrates indicated significant differences in the number of carbon sources usable by the prokaryotic communities of different whales (range: 11-25 sources), as well as a signficantly decreased diversity of carbon sources used by the community in the habitat water (5 sources). Sequencing of the hypervariable V4 region of the 16S rRNA gene revealed 19 amplicon sequence variants (ASVs) that were present in all whale samples. The oldest female D. leucas (F2) had the lowest overall diversity, and was significantly different from M1 and F1 in taxon composition, including an anomalously low ratio of Baccillota: Bacteroidota (0.01) compared to the other whales. In comparisons of microbial community composition, M1 had a significantly higher diversity than F1 and F2. These results suggest that attention should be given to regular microbiome sampling, and indicate a need for the pairing of microbiome and clinical data for animals in aquaria. Overall, these data contribute to the growing database on the core respiratory microbiota in cohabiting cetaceans under professional human care, indicate the utility of non-invasive sampling, and help characterize a baseline for healthy D. leucas.
Introduction
The respiratory microbiome in humans and model organisms is widely-recognized as a vital physiological component in the protection against pathogens and the maintainence of a dynamic immune system (Lombardo, 2008; Fraune and Bosch, 2010). Disruptions in the natural microbiome community (dysbiosis) can indicate symptomatic response to immune system dysregulation, disease manifestation, nutritional deficiencies, medicinal interactions, as well as psychological and physical stress from external or environmental factors (O’Dwyer et al., 2016; Shukla et al., 2017). However, limited analyses exist for marine mammals. In cetacea, respiratory illness is a primary cause of mortality (Venn-Watson et al., 2012), and contributes to the vulnerability of wild and endangered populations (Waltzek et al., 2012). However, blowhole exhalate (blow) sampling can provide a unique window into the health of the respiratory system, including identification of pathogens, illness, and dysbiosis caused by external stressors (Lombardo, 2008; Fraune and Bosch, 2010; Lima et al., 2012; Nelson et al., 2015) using non-invasive methods. Regular monitoring of the blow microbiome can serve as a warning sign in the detection of early dysbiosis, indicating the presence of an active or ongoing health issue (e.g. infection, external-stress). For cetaceans under professional human care, the non-invasive sampling of blow microbiota also allows for the continuous collection of biological data, which can inform and supplement clinical health monitoring, including the identification of dysbiosis, the effect of antibiotic treatments, and testing for infectious pathogens. Further, as opposed to more canonical health assessments accomplished via blood draw, the respiratory blow of cetaceans can be collected using entirely non-invasive methods, which has application for monitoring health in wild populations. The limited microbiome data available for toothed whales has provided a foundational context, including the identification of respiratory pathogens and the presence of microbial taxa which suggests a core microbial community (Apprill, 2017). However, more data is needed to examine the host-microbe relationship between cohabitating animals.
In this study, we used a non-invasive sampling procedure to collect blow from the beluga whale, Delphinapterus leucas. Access to three resident D. leucas individuals, in combination with specific learned voluntary behaviors for blow sample collection, allowed the opportunity to collect and analyze the metabolic and taxonomic composition of the associated microbial community. To characterize the microbiome, we used three methods. Clone libraries of 16S and 18S ribosomal gene sequences provide a comprehensive phylogenetic assessment of which taxonomic groups are present, including relative abundances. Traditional fluorescence microscopic observation and enumeration of prokaryotic and eukaryotic microbes present in the microbiome provided information on the magnitude of microbial populations, but minimal information on diversity. Both genetic and microscopic analyses are “culture independent” methods that avoid post-sampling changes in the microbial assemblage. The third method we used required incubation of blow samples with single carbon source substrates (Garland and Mills, 1991). This method allowed us to evaluate metabolic diversity of the microbiome community.
These data serve as a valuable examination in the viability of respiratory microbiome testing on D. leucas using non-invasive means. Use of these methods would be ideal to track changes in the health of wild populations, including the detection of pathogens and correlations with existing clinical health assessments (Acevedo-Whitehouse et al., 2010; Thompson et al., 2022). Further, the tested application of these non-invasive sampling methods is particularly critical in the study of endangered populations of D. leucas, which are in immediate need of health monitoring and less-invasive sampling methods.
Materials and methods
Sampling
We collected the exhaled breath condensate (blow) from the voluntary exhalations of three healthy beluga whales under professional human care at the Mystic Aquarium in Mystic, Connecticut. Individuals included M1, a 19 year old adult male, and two adult females, F1 and F2, 38 and 39 years old, respectively. Samples from three dates (7/1/21, 7/14/21, and 7/21/21) were collected from all three whales, with an additional sample collected from M1 on 8/2/21, and used to extract and sequence DNA from the blow microbiome. The whales have been cohabiting in a circulating saltwater system that is disinfected using ozone. Regular clinical health assessments by on-site veterinarians (e.g. blood cell counts, ultrasound, daily visual inspection, etc.) confirmed that all three whales remained stable and healthy throughout the sampling period.
Blow was collected on an inverted, pre-weighed, sterile plastic petri dish via established voluntary positive reinforcement behaviors. Briefly, each animal responded to learned visual cues developed over long-term training from an animal husbandry specialist that signaled the whales to position their head above the water surface, and then exhale on cue. After an initial exhalation to clear seawater, three consecutive exhalations were collected on one petri dish, and two dishes were used at each sampling event. Two mL of filter-sterilized seawater was added to prevent desiccation. The dish was then covered and sealed with parafilm, and transported to the laboratory in an insulated cooler. The amount of mucus per sample was estimated by reweighing the dishes. In addition to the blow samples, 100 mL of water from the whale habitat (“Habitat”) was filtered onto a 0.2 μm glass fiber filter (GFF), cut into pieces using sterilized forceps, and then preserved in a 2.0 mL tube containing lysis buffer (100 mM NaCl, 0.5% SDS, Tris−EDTA at pH 8) and stored at −20°C until DNA extraction.
Bacterial counting
Samples for bacterial counts were preserved with formaldehyde (0.5% final concentration v/v). One mL of preserved sample was stained with the fluorochrome DAPI, collected on a black-stained polycarbonate membrane filter (0.2 μm pore-size), and counted with a fluorescence microscope under ultraviolet illumination. A minimum of 200 bacterial cells were counted per sample. Bacterial abundance is reported as cells per mL of the diluted sample (blow sample plus the added 2 mL filtered seawater).
Prokaryotic carbon metabolism
To assess the carbon substrates used by prokaryotes living in the blowhole and in the habitat water, we used EcoPlates from Biolog, Inc. (Hayward, CA, USA; catalog number 1506). These 96-well plates contain 31 different carbon sources plus a blank, in triplicate. After sample incubation, the presence of species capable of using a given substrate as sole carbon source is indicated by color development of a redox-sensitive tetrazolium dye. Samples of blow or aquarium water were added to each well (150 μl) and diluted with 300 μl of phosphate-buffered saline to avoid calcium precipitation (Pierce et al., 2014). We standardized experimental incubations to three days at 20˚C and measured color development at t=0 and t=3d using a BioTek ELx808 microplate reader, with absorbance at 590 nm. Each well was blanked by its own t=0 value. Wells were considered positive for a given carbon source if they had absorbance values greater than 0.25. The whales and aquarium water were assayed in triplicate (i.e. one 96-well plate for each sample). For the purpose of comparison, we focused on the number of different carbon substrates that could be metabolized in each sample.
DNA extraction, sequencing, and bioinformatics
Frozen habitat water samples collected on GFF were thawed, 15uL of proteinase K was added, and then tubes were incubated in a 56°C water bath overnight. Filters were then processed using the Quick-DNA™ Fecal/Soil Microbe Microprep Kit (Zymo Research; model D6012) using the manufacturer’s protocol for fecal samples. For blow samples, the Qiagen Dneasy Blood and Tissue Kit was used for DNA extraction, following manufacturer’s instructions for tissue samples, except for an additional overnight incubation period in lysis buffer to improve the efficiency of DNA extraction. Briefly, one mL of lysis buffer (for 40 mL: 2mL 10% SDS, 8mL EDTA 0.5 M pH 8, 0.4 mL Tris HCL 1M pH8 + 29.6 mL ddH2O), 1mL of AL buffer, and 5 μL of proteinase K were added to 1 mL of blow sample and digested overnight as above. Extracted DNA concentration was quantified using Qubit high sensitivity DNA assays (ThermoFisher; model Q32851) and through gel visualization. PCR amplification with V4 16S rRNA primers for Bacteria and Archaea (Walters et al., 2016) was completed using DreamTaq polymerase, with PCR amplification conditions as follows: 95°C for 3 min; 35 cycles of 94°C for 45 s, 50°C for 60 s, and 72°C for 90 s; 72°C for 10 min, and then a 10°C hold. For the examination of ciliate presence, PCR amplification of the 18S rRNA gene was achieved using the “Anti-metazoan” primer set (i.e. biased against the amplification of metazoan 18S rRNA; del Campo et al., 2019) 574F (forward: 5’-CGGTAAYTCCAGCTCYV-3’; Hugerth et al., 2014) and UNonMet_DB (reverse: 5’-CTTTAARTTTCASYCTTGCG-3’; Bass and del Campo, 2020) and the following conditions: 95°C for 3 min; 35 cycles of 98°C for 10 s, 51.1°C for 30 s, and 72°C for 1min; with a final 72°C for 10 min. Products were then sent for sequencing with the Illumina MiSeq instrument at the Microbial Analysis, Resources and Services (MARS) facility at the University of Connecticut. Sequences were demultiplexed and quality-filtered in BaseSpace (Illumina).
Subsequent processing was done in QIIME 2 (Bolyen et al., 2019), including primer removal and additional quality filtering. Denoising, dereplication and chimera removal were completed with DADA2 (Callahan et al., 2016). For taxonomic classification, a Naïve Bayes classifier (Pedregosa et al., 2011) pre-trained on a V4-trimmed version of the 16S SILVA database (v.132) (Quast et al., 2012) was used for prokaryote sequences, and a Naïve Bayes classifier pre-trained on pr2 (v. 5.0.1) (Guillou et al., 2013) was used in the classification of sequences from Anti-metazoan primer sequencing. Statistical analyses regarding alpha-diversity indices, multivariate analyses, ANOSIM, and taxonomic visualizations were completed post rarefaction at 8,000 reads using QIIME 2. Eukaryotic sequence analyses regarding taxa identity, query cover and percent identity in regards to ciliates were compared to existing sequences on GenBank using blastn.
Results
Blow observations and weight
After correcting for the 2 mL of filtered seawater added to prevent the blow from drying, the amount of material collected per plate (3 blows) ranged from 0.23 to 0.56 g, (Table 1), with M1 consistently producing the greatest amount of mucus. We observed dysteriid ciliates of the genera Kyaroikeus and Planilamina in the blow (Figures 1A–C; Supplementary Video SV1) in M1 and F1, but not in F2 or in a concentrated volume of habitat water.
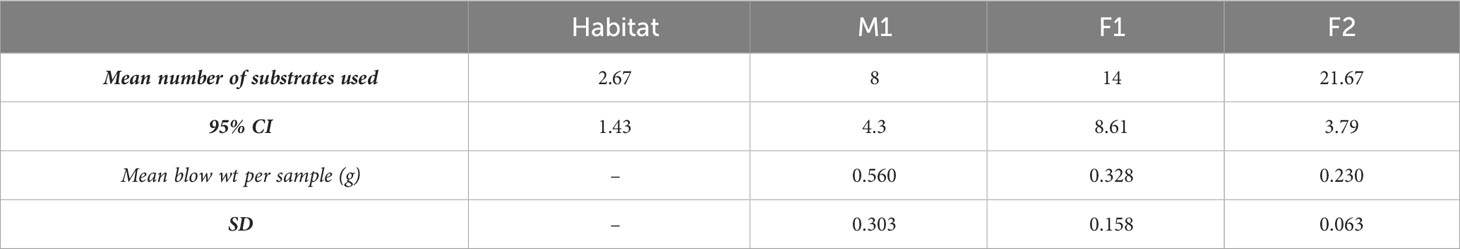
Table 1 Carbon substrate use and blow weight.
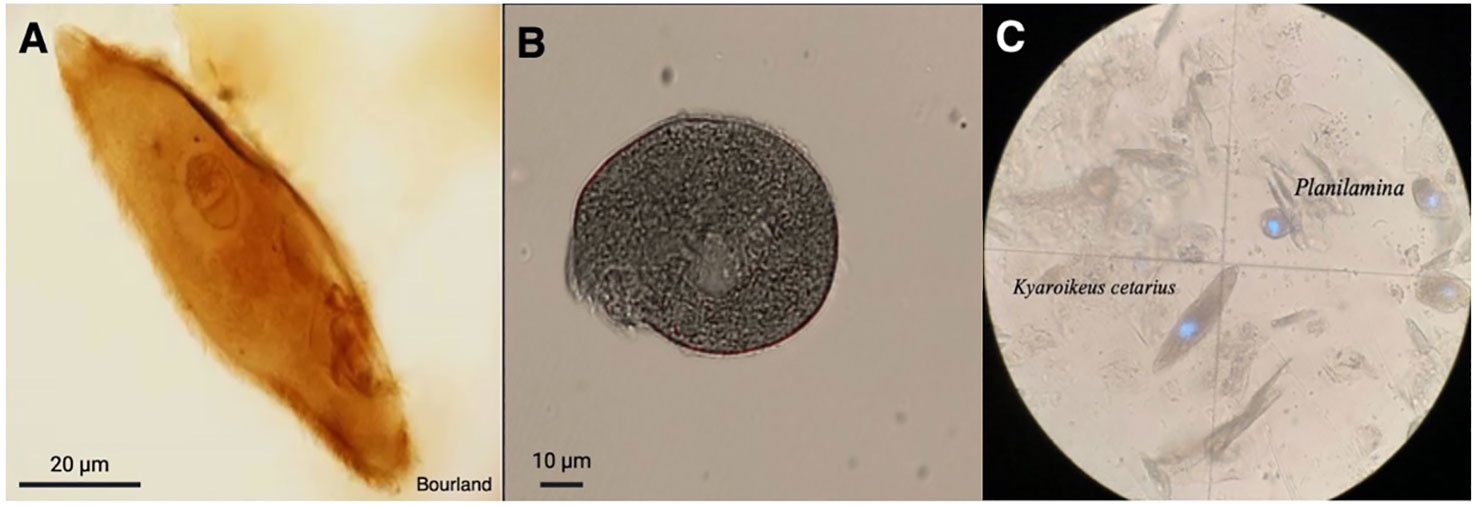
Figure 1 Ciliates observed in respiratory exhalate of Delphinapterus leucas. (A) Kyaroikeus post-silver impregnation using protargol stain; (B) Planilamina under bright field illumination; (C) Kyaroikeus and Planilamina surrounding mucosal exudate collected via blow exhalate on petri dish from M1, imaged in dark field.
Bacterial enumeration
Bacterial concentrations ranged between about 107 and 108 per mL of diluted blow (includes sample plus 2 mL filtered seawater). M1 had the highest levels of bacteria on four of the six sampling dates, while F2 had the lowest.
Carbon substrates
The prokaryotic microbiome community showed growth on various sole carbon source media in the EcoPlates, with the whale assemblage consistently using a higher number of carbon substrates than that of the habitat water (Table 1). Of all 31 carbon sources tested, the total number used, summed across all 3 replicates, was 5 (aquarium water), 11 (M1), 20 (F1), and 25 (F2). Differences were found among the whales (one-way ANOVA; P<0.001), with F2 having the highest carbon source richness (average 22 substrates; n=3), and M1 having the lowest (8 substrates) (Table 1).
Taxonomic diversity
Eukaryotic Microbiota. Although the intended targets for Anti-metazoan 18S rRNA primers were non-metazoan eukaryotes, many sequences were classified as unintended bacterial targets. For remaining sequences, the majority of eukaryotes matched ciliates (family Dysteriidae), dinoflagellates (family Thoracosphaeraceae), diatoms (class Bacillariophyceae), and green algae (family Chlorodendraceae) (Supplementary Table S1). No ciliate sequences were identified in the Habitat, which was instead dominated mostly by dinoflagellates (59.85% relative frequency), diatoms (27.10%), and green algae (4.24%). While the primers were intended to amplify only non-metazoan targets, some sequences in the Habitat were found to match the class Insecta (0.27%). For all whale samples, most sequences were bacteria (avg. relative frequencies for M1: 78%, F21: 87%, F2:94%), while the remainder was dominated by ciliates (M1: 22%, F1: 12%, F2: 6%), although some individuals were found to also have dinoflagellates (F1: 1%), and green algae (M1: 0.2%, F1:0.6%). All Dysteriidae ciliate sequences matched Planilamina ovata under the accession MN830169 or Kyaroikeus paracetarius under the accession number MN830168, and were present in all blow samples from all whales.
Prokaryotic microbiota
Among the prokaryotic 16S rRNA gene sequences found in M1, F1, and F2, 11 ASVs comprised what we consider to be the core prokaryotic microbiota for these three animals (i.e. ASVs shared by all three individuals in 100% of samples for each group) (Figures 2, 3; Supplementary Table S2). In addition, 8 ASVs were also found in Habitat water samples (i.e. 19 ASVs found in all three whales, 8 of which were found in the habitat water and may be of environmental origin). A total of 39 ASVs were found to be unique to M1 and F1samples (Figure 3; Supplementary Table S2), while only 4 ASVs were unique between M1 and F2, and only a single ASV was reported as unique between F1 and F2. For an ASV to be considered it had to be present in all samples for each group, but samples were not pooled due to the possibility for high individual variability within groups. In all three whales, we found a core of three primary phyla at high relative frequency, Proteobacteria, (M1: avg. 37.1± 3.6; F1: avg. 53.9 ± 1.51; F2: avg. 76.7 ± 1.32) Bacteroidota (synonym Bacteroidetes) (M1: avg. 24.2 ± 5.29; F1: avg. 19.3 ± 2.85; F2: avg. 21.3 ± 1.6), and Campylobacterota (M1: avg. 14.35 ± 4.14; F1: avg. 9.6 ± 1.75; F2: avg. 1.23 ± 0.82) (Figure 2). In addition, whales M1 and F1 shared a high relative frequency of the five phyla Actinobacteriota, Patescibacteria, Spirochaetota, Fusobacteriota and Bacillota (Figure 2). Although present in F2, the relative frequency of Bacillota was substantially reduced (avg. 0.17%; range 0-0.4%) in comparison to all other samples from M1 and F1 (avg. 8.8%; range 5.6-12.4%) (Figure 2). Conversely, Bacteroidota remained similar for all three whales, resulting in substantial differences in the Bacillota to Bacteroidota ratio (Bl:B). Expressed via ratio of relative frequency for each phylum averaged for each whale, the Bacillota to Bacteroidota ratio (Bl:B) was 0.52 (12.6: 24.2) in M1, 0.52 (12.6: 24.2) in F1, and 0.01 (0.17: 21.3) in F2. The habitat water samples contained seven phyla that were not observed in the whale samples: WPS-2 (Eremiobacterota), Planctomycetota, Cyanobacteria, Bdellovibrionota, Dependentiae, Crenarchaeota, and MBNT15 (candidate phylum) (Figure 2). Further, habitat water was found to contain the only member of the Archaea identified in this study, which were represented solely by the family Nitrosopumilaceae (genus Candidatus Nitrosotenuis, 0.2% relative frequency). Habitat samples also contained a relatively high frequency (36.5%) of the class Alphaproteobacteria, compared to an average of 0.18% (0-0.5%) in the whale samples (Supplementary Figure S1). There were no variations in the presence/absence of phyla found within each whale microbiome between sampling days; the same prokaryotic communities were present for each whale at all time points.
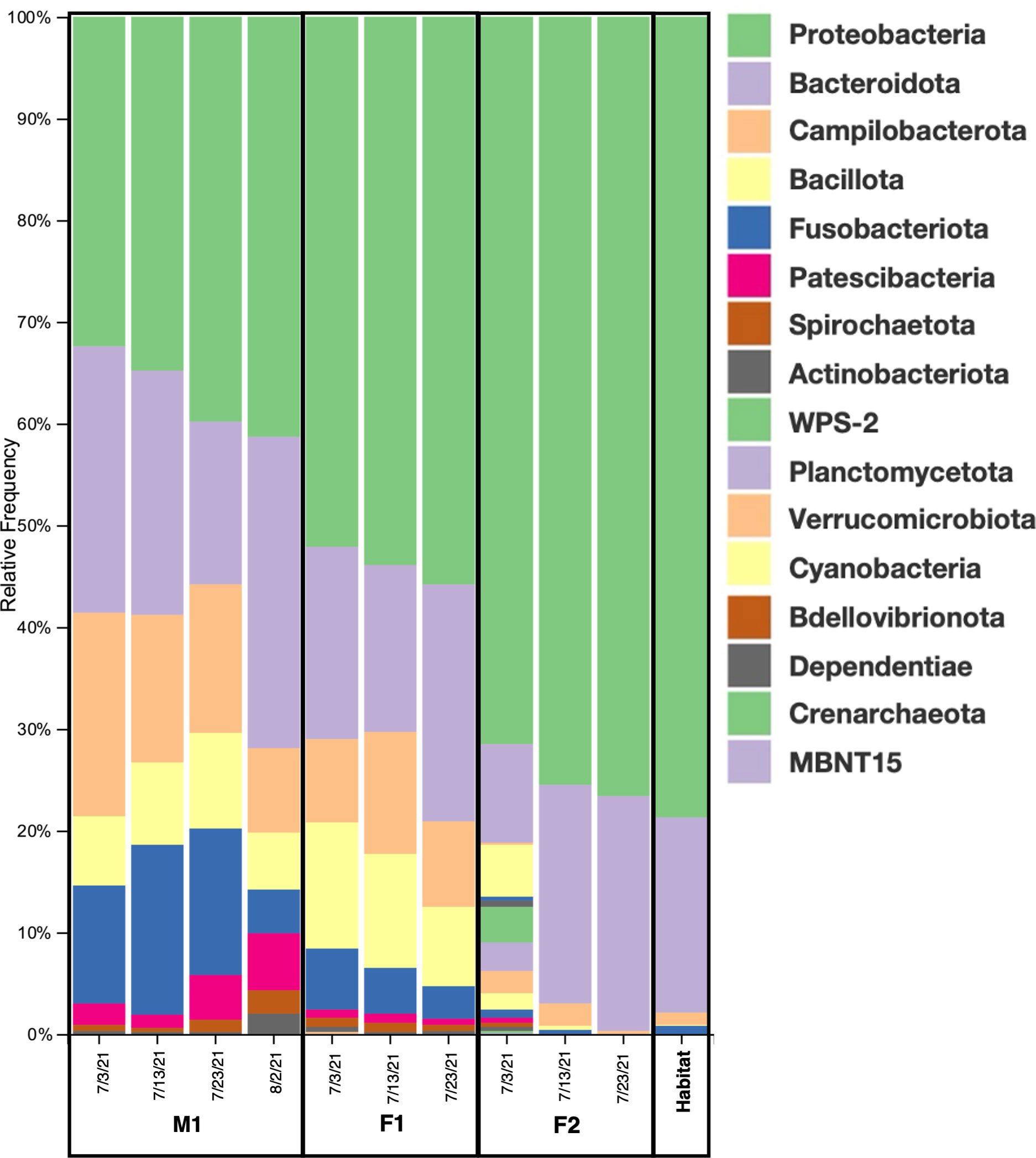
Figure 2 Taxonomic diversity. Relative frequencies of the bacterial taxa present in the blow microbiome of all three animals and habitat water on all sampling dates (M1=male 1; F1= female 1; F2= female 2). Phylum level: 17 total bacterial phyla were detected among all samples.
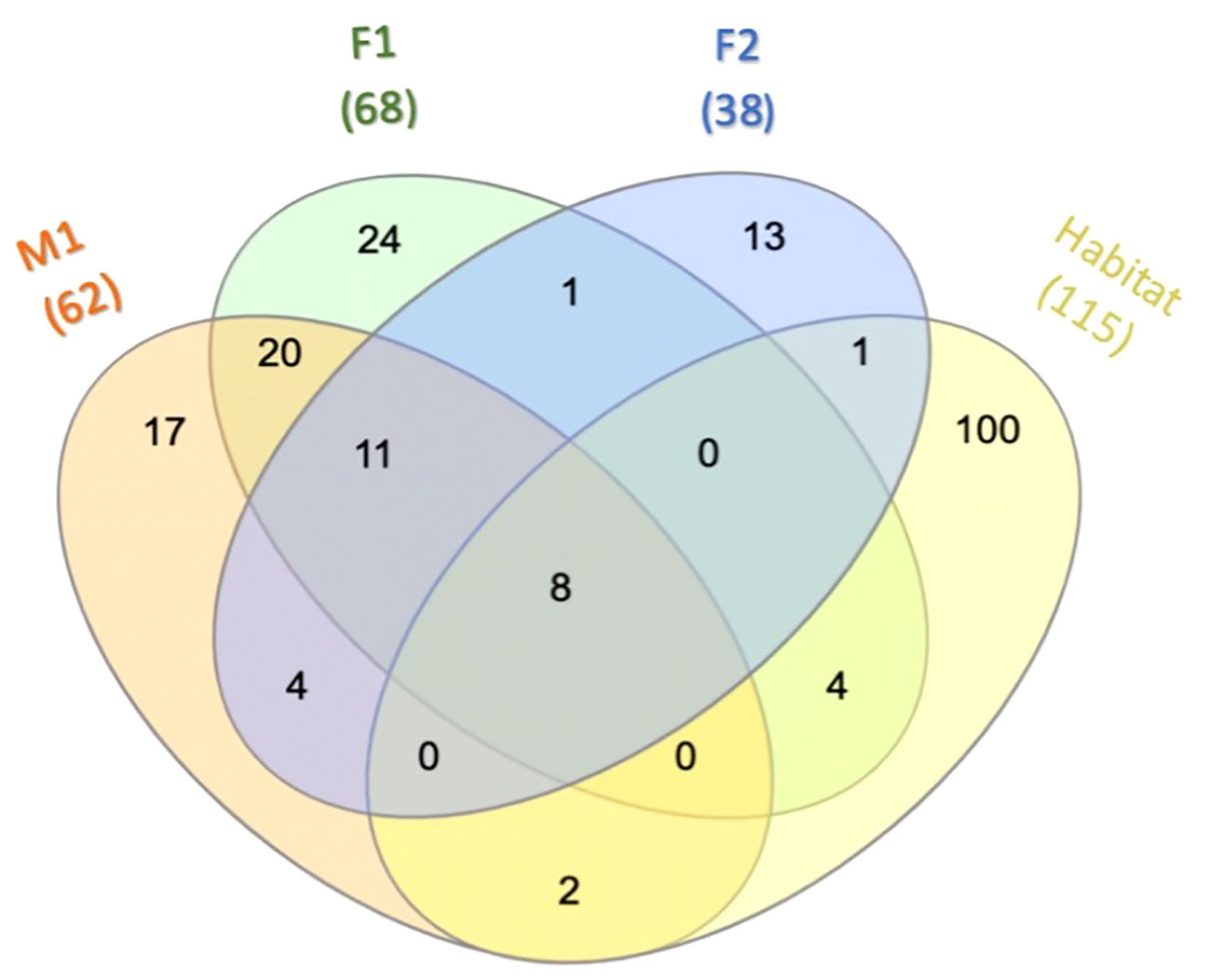
Figure 3 Core microbiota. Venn diagram depicting ASVs among M1, F1, F2, and Habitat that are present in 100% of the samples for each overlapping group. For additional details on ASVs see Supplementary Table S2.
Significant differences were found in alpha diversity, measured by ASV richness, between F2 and M1 (Kruskal-Wallis; p-value 0.034), and F2 and F1 (p-value 0.049) (Figure 4; Supplementary Tables S3-S5), with whale F2 maintaining the lowest diversity among all samples (Figure 4). Nearly significant differences were found in alpha diversity among all four groups (i.e. M1, F1, F2, Habitat) (Kruskal-Wallis for all groups; p-value 0.0579) (Supplementary Table S6). Alpha rarefaction analyses indicated appropriate sampling depth was achieved post-rarefaction via Shannon index analysis (Supplementary Figures S2A, C), but Faith’s PD (phylogenetic diversity) index suggested sampling depth may not have been adequate for the full sequencing of the habitat water microbiome (Supplementary Figure S2B).
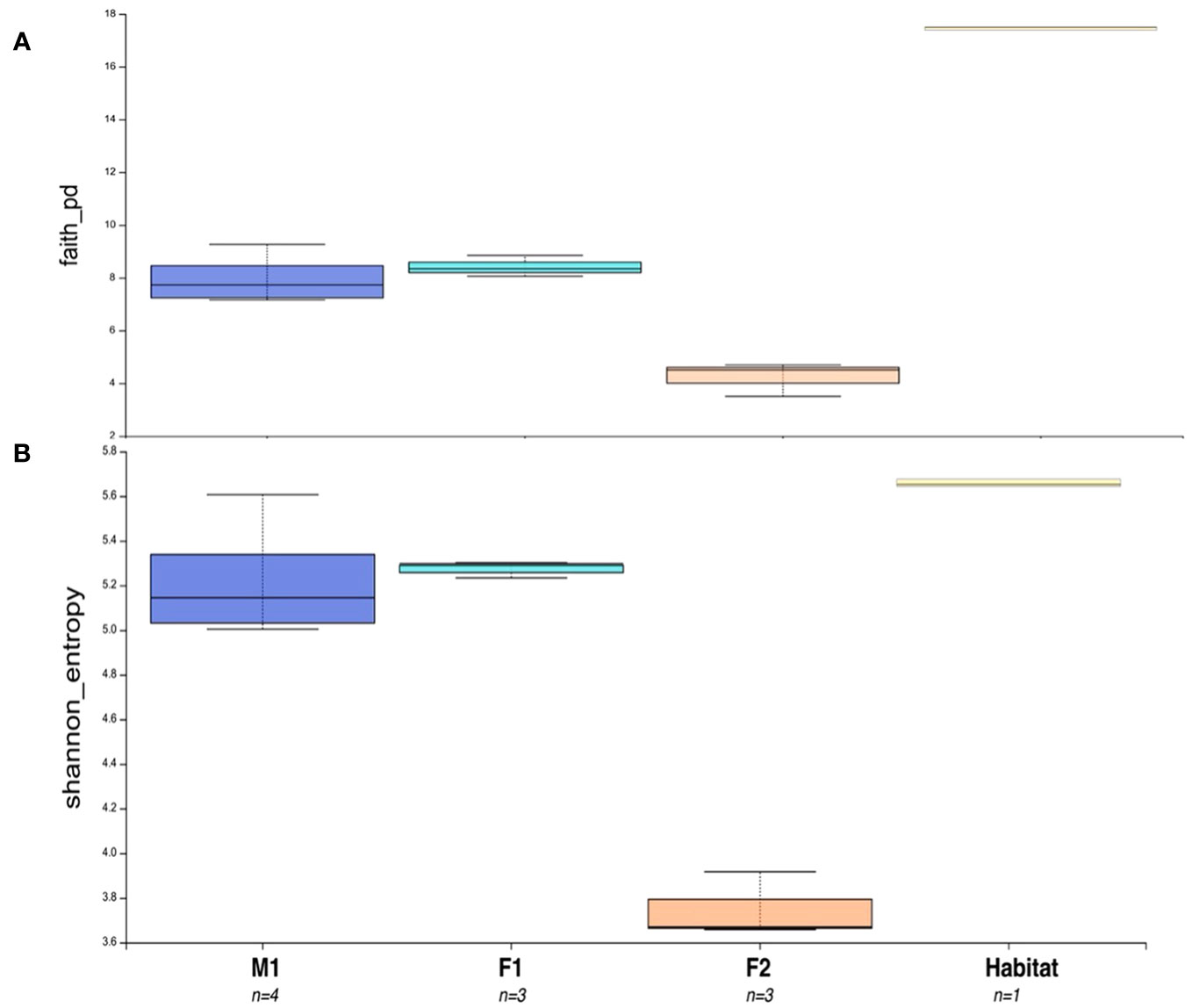
Figure 4 Alpha Diversity Boxplot comparisons between three animals and habitat water. (A) Faith’s Phylogenetic Diversity (PD) Test representing differences in PD between M1, F1, F2, and Habitat microbiome samples. (B) Shannon Entropy for all sample groups. Number of samples for each group = n. Boxes represent the interquartile range (IQR) between the first and third quartiles (25th and 75th percentiles, respectively), and the horizontal line inside the box defines the median. Whiskers represent the lowest and highest values within 1.5 times the IQR from the first and third quartiles, respectively.
Principal Component Analyses (PCA) indicated a clustering among samples for each whale, as well as compositional differences between whales and habitat water (based on Bray-Curtis values) (Figure 5). ANOSIM comparisons via pairwise Bray-Curtis distances indicated significant differences between M1 and F1 (p-value = 0.029) as well as M1 and F2 (p value 0.035) (Figures 4, 6; Supplementary Table S3).
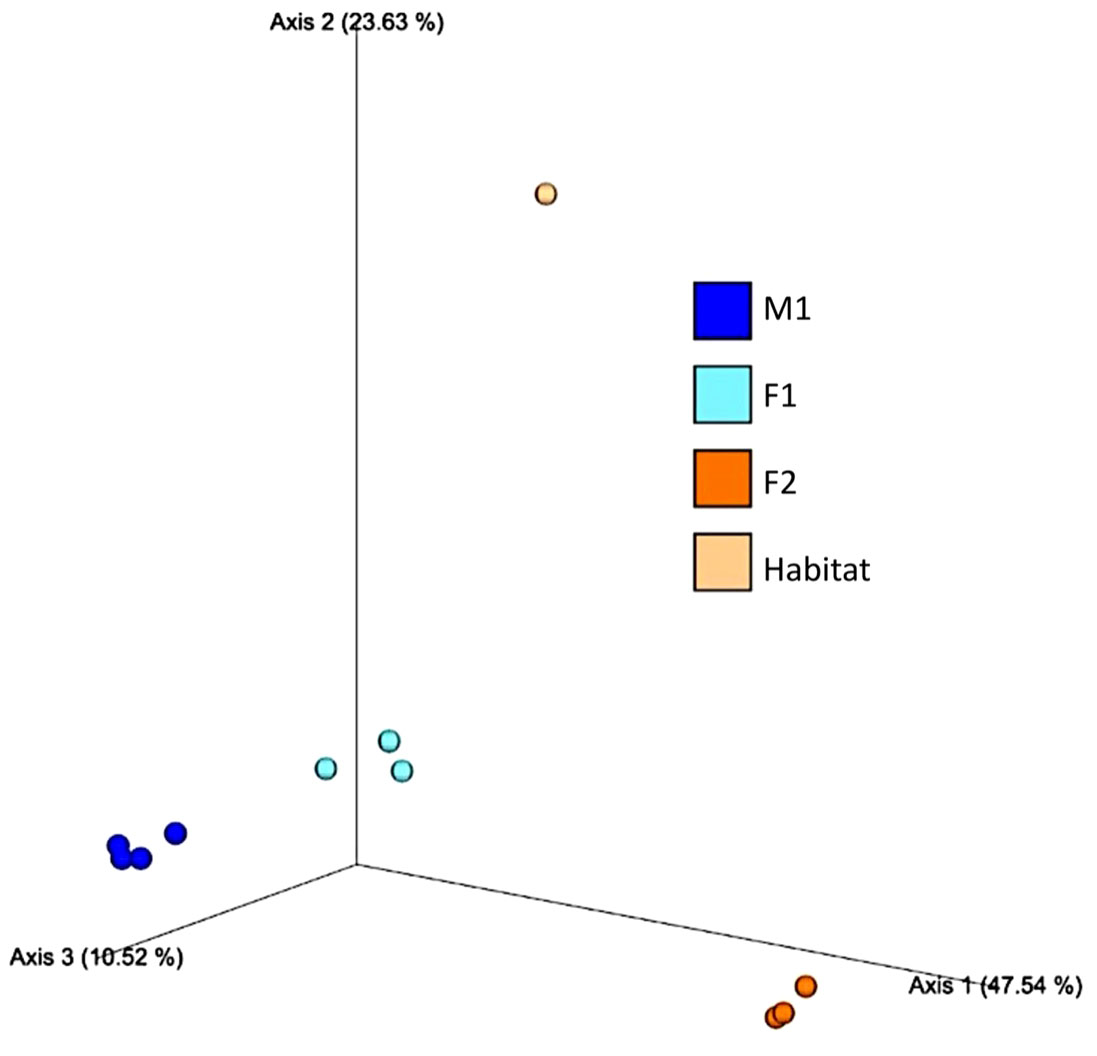
Figure 5 Principal Component Analysis (PCA) based on Bray-Curtis distances among whale blow (M1, F1 and F2) and Habitat water samples.
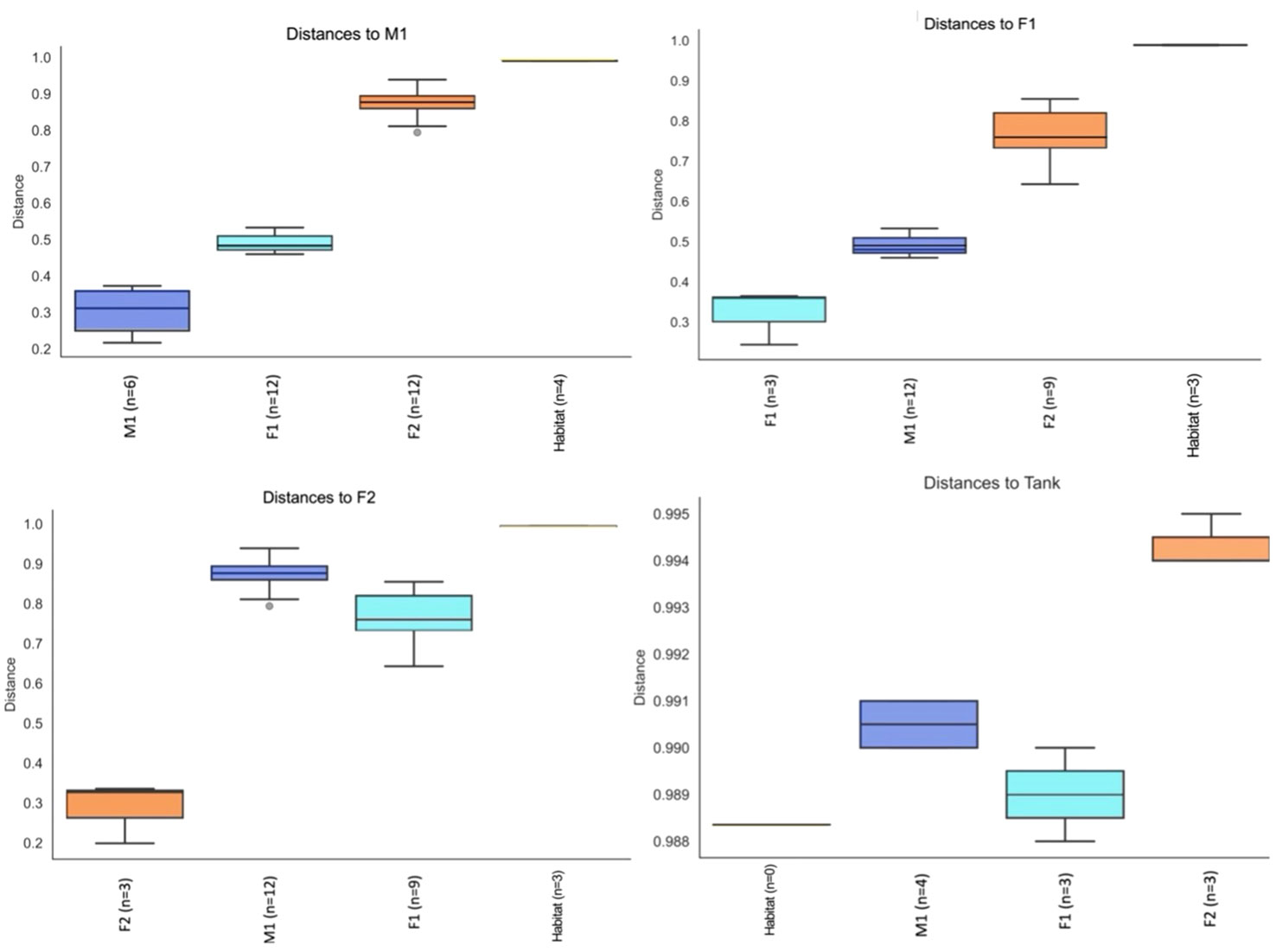
Figure 6 Pairwise Group Significance Comparisons (ANOSIM) between M1, F1, F2, and Habitat microbiome samples (n=number of pairwise comparisons).
Taxa of Interest
There were 11 ASVs found in all whale samples (for all M1, F1, and F2 samples) that were not present in the Habitat water. These were represented by the genera Ganjinia, Suttonella, Porphyromonas, Psychrobacter, Buytrivibrio, Shewanella, Marinifilum, Paracoccus, and genera within the Weeksellaceae and Cardiobacteriaceae families. There was a higher relative frequency of the genus Vibrio (order Vibrionales, family Vibrionaceae) in F2, with an average of 16.1% relative frequency (12.3- 20.5%), as compared to all other samples (average 1.39%; 0.01-3.54%) (Supplementary Table S2). Also found in F2 was a high relative frequency of ASVs affiliated to the species Shewanella putrefaciens (avg. 30.8%; 22.3-40.7%), as compared to all other samples (avg. 2.6%; 0.1-13.3%). Two species within the genus Myroides, Myroides sp. (avg. 7.47%; 4.8-11.6%) and M. odoratimimus (avg. 6.5%; 4-10%), were found exclusively in F2. In the two whales frequently found to be harboring ciliate symbionts in their blow microbiota (M1 and F1), there was a high relative frequency (avg. 9.5%; 3-16.7%) of the genus Oceanivirga, when compared to F2 and Habitat, which had a relative frequency range of 0-0.7%. Whales M1 and F1 also had a high relative frequency of the family Arcobacteraceae (avg. 14.4% and 6.8%, respectively), as opposed to the rest of samples, which had an average of 0.52% (0.1-1.6%).
Discussion
Respiratory illness (e.g. pneumonia) is a primary cause of mortality in marine mammals (Venn-Watson et al., 2012), contributing to the vulnerability of wild populations (Waltzek et al., 2012). In cetaceans, blowhole microbiome sampling can not only identify the presence of infections, but also enables researchers to track physiological changes, especially those reflected in the respiratory system. However, the inherent difficulty in accessing these animals has led to a dearth of baseline information on their respiratory microbiota. Unique access to beluga whales, Delphinapterus leucas, which were trained to carry out behaviors that facilitate biological sampling, enabled us to use respiratory exhalate (blow) to quantify the respiratory microbiome.
Although microbiome studies generally exclude eukaryotic taxa (e.g. Apprill et al., 2017), the presence and sustained high concentrations of the ciliate genera Kyaroikeus and Planilamina in blow samples reflect a need to expand protist microbiome studies. Further, the limited research to date has led to suggestions that these ciliates are pathogenic or parasitic (Sniezek et al., 1995; Ma et al., 2006), whereas our continued observation of these protists in healthy animals suggests that they may instead be commensal or mutualistic members of a normal D. leucas respiratory microbiome.
Among the ASVs unique to all whale samples (i.e. not present in Habitat samples), the genera Ganjinia, Suttonella, Porphyromonas, Weeksellaceae, Porphyromonas, and genera within the Cardiobacteriaceae family had all previously been identified in the oral, respiratory, or mucosal microbiomes of various wild cetaceans while the genera Shewanella and Paracoccus had only been associated with skin samples, and Buytrivibrio solely with the gut biome of wild pygmy sperm whales (see Supplementary Table S7). Interestingly, Paracoccus sp. had only been reported in the skin of killer whales and dolphins under human care by Chiarello et al., 2017, who suggested that the presence of these microbes in cetaceans might indicate a zoonotic transfer from human husbandry specialists to the animals, since Paracoccus sp. is a primary member of the human skin microbiome (Cosseau et al., 2016). While these data are only indicative of a small sample size, and reflect animals under professional human care in a circulating seawater system, our results are similar to published work on cetaceans (Apprill et al., 2017; Apprill et al., 2020; Van Cise et al., 2020), suggesting the existence of “core taxa,” or conserved compositional taxonomy, as we observed in the respiratory exhalate of all three whales, which shared 19 core ASVs in their blow microbiome regardless of sampling date.
While a reduced sample size prevents these data from providing a representative microbiome community for D. leucas, microbiome analyses among cohabiting individuals can be useful in the study of respiratory microbiota by establishing a baseline for healthy animals, which can be used comparatively if dysbiosis occurs (e.g. due to antibiotics, change in immune health, chronic external stress, viral infection, etc.). Individual microbiome communities were also apparent between whales, with significant differences among M1, F1 and F2, including a substantial diversity reduction in F2. Veterinary assessment of all three animals determined the whales to be clinically healthy. F2 was being administered the gastroprotectants sucralfate and omeprazole. Omeprazole is a proton pump inhibitor (PPI), commonly used to decrease stomach acid production (Mishiro et al., 2018). Human studies have identified that PPIs can cause severe dysbiosis with only weeks of administration, which can lead to a sustained disruption to the microbial composition in esophageal microbiomes (Castellani et al., 2017; Mishiro et al., 2018). Further, similar studies have shown that regular doses can substantially alter the relative ratios of microbiome taxa, including an alteration in the Bacillota to Bacteroidota ratio (Naito et al., 2018). In human and non-human terrestrial animals, this ratio is regularly used as a reliable marker in which a high number (i.e. Bl:B) is correlated to the storage of fat (implicated with obesity diagnoses in humans), while a low number is generally linked to inflammatory bowel disease (DeGruttola et al., 2016; Hufnagl et al., 2020). In F2, we saw a substantially reduced Bacillota community in comparison to all other samples from M1 and F1 (Figure 2), while Bacteroidota remained similar for all three whales (Figure 2). This reflected a Bl:B ratio of 0.01 for F2, in comparison to 0.52 and 1.61, for M1 and F1, respectively. These data indicate a need for regular microbiome monitoring, and may warrant an investigation into probiotics or a change in diet for animals requiring PPIs. On the other hand, F2 also had ASVs unique to F2 including the genera Peptostreptococcaceae and Alcaligenes, both of which had previously only been identified in the gut microbiome of marine mammals (Rothenberg et al., 2021; Bai et al., 2022). The presence of these gut-dominant genera in the blow exhalate of F2 may suggest stomach material or acid reflux in this animal, possibly warranting the need for gastroprotectants.
Identifying and quantifying sustained presence of pathogen-associated taxa is complicated; many animals regularly coexist with pathogenic microbiota without showing clinical signs of an affected health assessment (Nelson et al., 2015; Palmer et al., 2020). In some marine mammals, the conserved presence of disease-associated microbes like some Vibrio species and Helicobacter pylori among different populations can exist as a background “pathogen core community” (Apprill et al., 2017; Godoy-Vitorino et al., 2017; Raverty et al., 2017; Li et al., 2019) that is found in the majority of healthy populations (Nelson et al., 2015; Apprill et al., 2017; Ochoa et al., 2018). The frequency of Vibrio sequences identified in F2 may warrant investigation, but may also be an indirect result of the reduced diversity caused by other factors (e.g. medicine, dietary change, etc.). Similarly, M1 and F1 had a higher frequency of the genus Oceanivirga, within which the species O. salmonicida is believed to be a pathogen of Atlantic salmon (Salmo salar) and has been found in oral microbiomes of several marine mammals, including dolphins, humpback whales, and seals (Eisenberg et al., 2016; Palmer et al., 2020). However, both Vibrio and Oceanivirga were identified in all samples, including Habitat water samples, suggesting they may be part of an ambient environmental background biome. This serves as an example where microbiome studies can supplement and inform clinical health assessments, including in decisions regarding quarantine procedures triggered by pathogen presence/absence tests, which may be unnecesary if there is a consistent background of a specific pathogenic taxon in clinically-healthy animals or their environment. Further work including multiple samples taken from various microbiome regions (e.g. oral, skin, fecal, etc.) over time would greatly enhance this dataset and constrain implications regarding core taxa, within-group variation, and pathogenic potential.
The microscopic observations showed prokaryotic cell concentrations about an order of magnitude higher than typical ocean values of c. 106 cells l-1 and the presence of commensal ciliates in two of the whales. Whale F2 had the lowest bacterial concentrations and we did not observe ciliates in her blow during this sampling period. This is surprising, since the ciliates would presumably crop bacterial populations via grazing, so we might expect higher numbers in F2. On the other hand, F2 had the highest metabolic diversity in her microbiome, 1.5-2x higher than the other two whales and about eight-fold higher than that of the habitat water. We know of no other respiratory microbiome studies that looked at metabolic diversity, but there have been observations on changes, for example, in fish gut microbiome metabolic types related to salinity gradients and diet (Mouchet et al., 2012). Probably the most striking result of the metabolic diversity observations is the large difference between the whale respiratory tract and the habitat water. Given the well-known dominance of unculturable bacteria in the ocean, it is perhaps not surprising that the technique we used, which requires incubation over several days, would yield lower diversity in the aquatic habitat. Apparently, the whale respiratory tract is much more similar to a rich culture-medium environment and hence allows for higher diversity of culturable metabolic types to exist there.
The cetacean respiratory microbiome is an important yet understudied system that allows for the identification of infection, disease, and dysbiosis. Respiratory microbiota can maintain host health, support immune system function, and serve as both an indicator and source of illness. In cetaceans, blow microbiome sampling can identify physiological changes in individuals under professional human care, and serve as a non-invasive health index for wild populations. The sampling methods explored in this study represent the utility of non-invasive means to inform clinicians on the health of cetaceans under professional human care, and serve as an ideal method to monitor wild and endangered populations.
Data availability statement
The data presented in the study are deposited in the Sequence Read Archive repository (SRA; https://www.ncbi.nlm.nih.gov/sra) under the Bioproject accession number PRJNA1026968 (BioSample accession numbers: SAMN37769875, SAMN37769876, SAMN37769877, SAMN37769878, SAMN37769879, SAMN37769880, SAMN37769881, SAMN37769882, SAMN37769883, SAMN37769884). In addition, these data are publicly available on FigShare at https://doi.org/10.6084/m9.figshare.24328462 (Smith et al., 2023).
Ethics statement
The animal study was approved by the Mystic Aquarium Institutional Animal Care and Use Committee (IACUC) (protocol # 18018). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SS, DR, TR, and GM conceived and designed study. SS and LS analyzed data. All authors contributed to writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by a National Science Foundation (NSF) Research Experiences for Undergraduates (REU) grant to Mystic Aquarium (Award # 1950480) and the University of Connecticut (Award # 1950415), and NSF Award # OCE-1924527.
Acknowledgments
We thank the staff of Mystic Aquarium for assistance with sampling, W. Bourland (Boise State University) for the protargol staining shown in Figure 1, and R. Cole for encouraging us to sample the whales for ciliates. This constitutes scientific contribution #365 from the Sea Research Foundation, Inc.
Conflict of interest
Authors SS, DR and TR were employed by Sea Research Foundation, Inc. d/b/a Mystic Aquarium.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1168623/full#supplementary-material
References
Acevedo-Whitehouse K., Rocha‐Gosselin A., Gendron D. (2010). A novel non-invasive tool for disease surveillance of free-ranging whales and its relevance to conservation programs. Anim. Conserv. 13, 217–225. doi: 10.1111/j.1469-1795.2009.00326.x
Apprill A. (2017). Marine animal microbiomes: toward understanding host–microbiome interactions in a changing ocean. Front. Mar. Sci. 4, 222. doi: 10.3389/fmars.2017.00222
Apprill A., Miller C. A., Moore M. J., Durban J. W., Fearnbach H., Barrett-Lennard L. G. (2017). Extensive core microbiome in drone-captured whale blow supports a framework for health monitoring. MSystems 2 (5), e00119–e00117. doi: 10.1128/mSystems.00119-17
Apprill A., Miller C. A., Van Cise A. M., U'Ren J. M., Leslie M. S., Weber L., et al. (2020). Marine mammal skin microbiotas are influenced by host phylogeny. R. Soc. Open Sci. 7 (5), 192046. doi: 10.1098/rsos.192046
Bai S., Zhang P., Zhang X., Yang Z., Li S. (2022). Gut microbial characterization of melon-headed whales (Peponocephala electra) stranded in China. Microorganisms 10 (3), 572.
Bass D., del Campo J. (2020). Microeukaryotes in animal and plant microbiomes: Ecologies of disease? Eur. J. Protistol. 76, e125719. doi: 10.1016/j.ejop.2020.125719
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37 (8), 852–857. doi: 10.1038/s41587-019-0209-9
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Castellani C., Singer G., Kashofer K., Huber-Zeyringer A., Flucher C., Kaiser M., et al. (2017). The influence of proton pump inhibitors on the fecal microbiome of infants with gastroesophageal reflux-a prospective longitudinal interventional study. Front. Cell Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00444
Chiarello M., Villéger S., Bouvier C., Auguet J. C., Bouvier T. (2017). Captive bottlenose dolphins and killer whales harbor a species-specific skin microbiota that varies among individuals. Sci. Rep. 7 (1), 15269. doi: 10.3389/fevo.2023.1197323
Cosseau C., Romano-Bertrand S., Duplan H., Lucas O., Ingrassia I., Pigasse C., et al. (2016). Proteobacteria from the human skin microbiota: species-level diversity and hypotheses. One Health 2, 33–41.
DeGruttola A. K., Low D., Mizoguchi A., Mizoguchi E. (2016). Current understanding of dysbiosis in disease in human and animal models. Inflammation Bowel Dis. 22 (5), 1137–1150. doi: 10.1097/MIB.0000000000000750
del Campo J., Pons M., Herranz M., Wakeman K. C., Del Valle J., Vermeij M. J. A., et al. (2019). Validation of a universal set of primers to study animal-associated microeukaryotic communities. Environ. Microbiol. 21, 3855–3861. doi: 10.1111/1462-2920.14733
Eisenberg T., Kämpfer P., Ewers C., Semmler T., Glaeser S. P., Collins E., et al. (2016). Oceanivirga salmonicida gen. nov., sp. nov., a member of the Leptotrichiaceae isolated from Atlantic salmon (Salmo salar). Int. J. Systematic Evolutionary Microbiol. 66, 2429–2437. doi: 10.1099/ijsem.0.001050
Fraune S., Bosch T. C. (2010). Why bacteria matter in animal development and evolution. Bioessays 32 (7), 571–580. doi: 10.1002/bies.200900192
Garland J. L., Mills A. L. (1991). Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl. Environ. Microbiol. 57, 2351–2359. doi: 10.1128/aem.57.8.2351-2359.1991
Godoy-Vitorino F., Rodriguez-Hilario A., Alves A. L., Gonçalves F., Cabrera-Colon B., Mesquita C. S., et al. (2017). The microbiome of a striped dolphin (Stenella coeruleoalba) stranded in Portugal. Res. Microbiol. 168 (1), 85–93. doi: 10.1016/j.resmic.2016.08.004
Guillou L., Bachar D., Audic S., Bass D., Berney C., Bittner L., et al. (2013). The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Hufnagl K., Pali-Schöll I., Roth-Walter F., Jensen-Jarolim E. (2020). Dysbiosis of the gut and lung microbiome has a role in asthma. Semin. Immunopathol. 1), 75–93. doi: 10.1007/s00281-019-00775-y
Hugerth L. W., Muller E. E., Hu Y. O., Lebrun L. A., Roume H., Lundin D., et al. (2014). Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PloS One 9 (4), e95567. doi: 10.1371/journal.pone.0095567
Li C., Tan X., Bai J., Xu Q., Liu S., Guo W., et al. (2019). A survey of the sperm whale (Physeter catodon) commensal microbiome. PeerJ 7, e7257. doi: 10.7717/peerj.7257
Lima N., Rogers T., Acevedo-Whitehouse K., Brown M. V. (2012). Temporal stability and species specificity in bacteria associated with the bottlenose dolphins respiratory system. Environ. Microbiol. Rep. 4, 89–96. doi: 10.1111/j.1758-2229.2011.00306.x
Lombardo M. (2008). Access to mutualistic endosymbiotic microbes: an underappreciated benefit of group living. Behav. Ecol. Sociobiol. 62, 479–497. doi: 10.1007/s00265-007-0428-9
Ma H., Overstreet R. M., Sniezek J. H., Solangi M., Coats W. D. (2006). Two new species of symbiotic ciliates from the respiratory tract of cetaceans with establishment of the new genus Planilamina n. gen.(Dysteriida, Kyaroikeidae). J. Eukaryotic Microbiol. 53 (6), 407–419. doi: 10.1111/j.1550-7408.2006.00124.x
Mishiro T., Oka K., Kuroki Y., Takahashi M., Tatsumi K., Saitoh T., et al. (2018). Oral microbiome alterations of healthy volunteers with proton pump inhibitor. J. Gastroenterol. Hepatol. 33, 1059–1066. doi: 10.1111/jgh.14040
Mouchet M. A., Bouvier C., Bouvier T., Troussellier M., Escalas A., Mouillot D. (2012). Genetic difference but functional similarity among fish gut bacterial communities through molecular and biochemical fingerprints. FEMS Microbiol. Ecol. 79 (3), 568-580.
Naito Y., Kashiwagi K., Takagi T., Andoh A., Inoue R. (2018). Intestinal dysbiosis secondary to proton-pump inhibitor use. Digestion 97, 195–204. doi: 10.1159/000481813
Nelson T. M., Apprill A., Mann J., Rogers T. L., Brown M. V. (2015). The marine mammal microbiome: current knowledge and future directions. Microbiol. Aust. 36 (1), 8–13. doi: 10.1071/MA15004
Ochoa J. L., Sanchez L. M., Koo B. M., Doherty J. S., Rajendram M., Huang K. C., et al. (2018). Marine mammal microbiota yields novel antibiotic with potent activity against Clostridium difficile. ACS Infect. Dis. 4 (1), 59–67. doi: 10.1021/acsinfecdis.7b00105
O’Dwyer D. N., Dickson R. P., Moore B. B. (2016). The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J. Immunol. 196 (12), 4839–4847. doi: 10.4049/jimmunol.1600279
Palmer R., Fleming G. T., Glaeser S., Semmler T., Flamm A., Ewers C., et al. (2020). Marine mammals are natural hosts of Oceanivirga salmonicida, a bacterial pathogen of Atlantic salmon. Dis. Aquat. Organisms 139, 161–174. doi: 10.3354/dao03478
Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830.
Pierce M. L., Ward J. E., Dobbs F. C. (2014). False positives in Biolog EcoPlatesTM and MT2 MicroPlatesTM caused by calcium. J. Microbiological Methods 97, 20–24. doi: 10.1016/j.mimet.2013.12.002
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (D1), D590–D596. doi: 10.1093/nar/gks1219
Raverty S. A., Rhodes L. D., Zabek E., Eshghi A., Cameron C. E., Hanson M. B., et al. (2017). Respiratory microbiome of endangered southern resident killer whales and microbiota of surrounding sea surface microlayer in the Eastern North Pacific. Sci. Rep. 7 (1), 394. doi: 10.1038/s41598-017-00457-5
Rothenberg S. E., Sweitzer D. N., Rackerby B. R., Couch C. E., Cohen L. A., Broughton H. M., et al (2021). Fecal methylmercury correlates with gut microbiota taxa in Pacific walruses (Odobenus rosmarus divergens). Front. Microbiol. 12, 648685.
Shukla S. D., Budden K. F., Neal R., Hansbro P. M. (2017). Microbiome effects on immunity, health and disease in the lung. Clin. Trans. Immunol. 6 (3), e133. doi: 10.1038/cti.2017.6
Smith Susan (2003). The respiratory microbiota of three cohabiting beluga whales (Delphinapterus leucas) under human care. FigShare. Dataset. doi.10.6084/m9.figshare.24328462.v1
Sniezek J. H., Coats D. W., Small E. B. (1995). Kyaroikeus cetarius ng, n. sp.: a parasitic ciliate from the respiratory tract of odonticete cetacea. J. Eukaryotic Microbiol. 42 (3), 260–268.
Thompson L. A., Goertz C. E., Quakenbush L. T., Burek Huntington K., Suydam R. S., Stimmelmayr R., et al. (2022). Serological detection of marine origin Brucella exposure in two Alaska beluga stocks. Animals 12 (15), 1932. doi: 10.3390/ani12151932
Van Cise A. M., Wade P. R., Goertz C. E., Burek-Huntington K., Parsons K. M., Clauss T., et al. (2020). Skin microbiome of beluga whales: spatial, temporal, and health-related dynamics. Anim. Microbiome 2 (1), 1–16. doi: 10.1186/s42523-020-00057-1
Venn-Watson S., Daniels R., Smith C. (2012). Thirty year retrospective evaluation of pneumonia in a bottlenose dolphin Tursiops truncatus population. Dis. Aquat. Organ. 99, 237–242. doi: 10.3354/dao02471
Walters W., Hyde E. R., Berg-Lyons D., Ackermann G., Humphrey G., Parada A. (2016). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems 1 (1), e00009–e00015. doi: 10.1128/mSystems.00009-15
Keywords: microbiome, Delphinapterus leucas, microbiota, ciliate, blowhole, blowhole microbiota, marine mammal
Citation: Smith SA, Ropati DV, Santoferrara LF, Romano TA and McManus GB (2023) The respiratory microbiota of three cohabiting beluga whales (Delphinapterus leucas) under human care. Front. Mar. Sci. 10:1168623. doi: 10.3389/fmars.2023.1168623
Received: 17 February 2023; Accepted: 23 October 2023;
Published: 14 December 2023.
Edited by:
Yunyun Zhuang, Ocean University of China, ChinaReviewed by:
Wang Minxiao, Chinese Academy of Sciences (CAS), ChinaJinsong Zheng, Chinese Academy of Sciences (CAS), China
Jianwei Chen, Beijing Genomics Institute (BGI), China
Copyright © 2023 Smith, Ropati, Santoferrara, Romano and McManus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan A. Smith, susansmith@mysticaquarium.org; susan.smith@uconn.edu