Iron dissolution from Patagonian dust in the Southern Ocean: under present and future conditions
 Clément Demasy1*
Clément Demasy1*  Marie Boye1
Marie Boye1  Barry Lai2 Pierre Burckel1
Barry Lai2 Pierre Burckel1  Yan Feng3 Rémi Losno1 Stephan Borensztajn1
Yan Feng3 Rémi Losno1 Stephan Borensztajn1  Pascale Besson1
Pascale Besson1- 1Université Paris Cité, Institut de Physique du Globe de Paris, CNRS, Paris, France
- 2X-Ray Science Division, Advanced Photon Source, Argonne National Laboratory, Lemont, IL, United States
- 3Environmental Science Division, Argonne National Laboratory, Argonne, IL, United States
Although the input of desert dust as a key source of trace metals in the Southern Ocean (SO) has been previously studied, the dissolution process of metals in surface waters, particularly iron (Fe), remain poorly understood. Given the crucial role of Fe in primary production and the biological carbon pump in the SO, we focused on experimental estimations of Fe dissolution from Patagonian dust, the primary natural dust source in the SO. Our study considered both current and projected future conditions, encompassing sea-surface warming, acidification, increased photosynthetically active radiation, and doubled dust inputs. Through controlled laboratory experiments using filtered SO seawater, conducted over 7 days, we assessed changes in particulate Fe (pFe) concentrations, Fe redox speciation (Fe(II)/Fe(III)), and in the mineralogy of Fe-bearing dust in abiotic condition. The predominant minerals in the dust included quartz and aluminosilicates, with silicon (Si), aluminum (Al), and Fe as the major elements. No significant alterations in the mineralogy and the elemental composition of the dust were recorded during the dissolution experiments, neither under present nor under projected future conditions. The particulate Fe(II)/Fe(III) ratio remained consistently at 0.25 during the experiments, unaffected by changed conditions. Consequently, changes in environmental conditions in the SO would therefore not significantly alter the mineralogy and redox speciation of pFe in the Patagonian dust. On the contrary, pFe exhibited a dissolution rate of 3.8% and 1.6% per day under present and future conditions, respectively. The environmental changes anticipated for 2100 in the SO will likely to result in a decrease in the dissolution rate of pFe. Thus, even though a doubling of dust input by 2100 is anticipated, it will unlikely provide significantly more dissolved Fe (dFe) in seawater in the SO. Consequently, the future intensification of Patagonian dust inputs may not alleviate the Fe limitation in the SO.
Highlights
● The mineralogy of Patagonian dust was dominated by quartz and plagioclase, and the mineralogy of iron (Fe) was dominated by heterosite and Fe(III) sulfate hydrate. Fe(II) redox state was present at ~20% in the dust.
● The mineralogy and redox speciation of particulate Fe in the dust remained stable during the 7-day dissolution experiments, unaffected by the present or future conditions (temperature, PAR, pH, dust concentration).
● The Fe dissolution rate of Patagonian dust was 3.8% per day under present conditions and approximatively half of the rate (1.8% per day) under future conditions.
● Even though a doubling of dust input by 2100 is anticipated, it will unlikely provide significantly more dissolved Fe in seawater in the Southern Ocean.
1 Introduction
Desertification linked to a decrease in precipitation is an emerging issue on Earth in the context of the climate change. Although since 1980, there has been no real trend of global increase in desert areas, Intergovernmental Panel on Climate Change (IPCC) projections for 2100 (RCP8.5) indicate a tendency to a 25% expansion of existing desert regions except for Antarctica (Pörtner et al., 2022). Deserts are the major source of aerosols in the Earth’s atmosphere compared to anthropogenic activities, fire and volcanic sources (Prospero et al., 2002; Hamilton et al., 2022). The desertic dusts are originated from the Earth’s crust and they are dispersed across the atmosphere and the oceans via extensive air-mass circulations (Mahowald et al., 2005). These small particles (< 10 µm) mobilized by the winds have impacts on the climate and human health (Griffin and Kellogg, 2004; Miller et al., 2014). Upon settling on surface waters, these particles undergo dissolution, releasing crustal elements to the oceanic regions farthest from the continents. Thus, the dusts can provide macronutrients and nutritive metals such as iron (Fe), which are essential to the metabolism of microalgae (Lohan and Tagliabue, 2018). The phytoplankton, at the base of the biological carbon pump, indeed needs micro- and macro-nutrients to grow and capture carbon dioxide (CO2) via photosynthesis, while the synthetized organic carbon can be exported at depth (Tréguer et al., 2018). The storage of carbon on the seafloor allows to decrease the greenhouse effect in the atmosphere (Raven and Falkowski, 1999) and ensures a negative feedback on the climate change. Therefore, dusts have a pivotal role by supplying nutrients to phytoplankton, particularly in nutrient-limited oceanic regions.
In the Southern hemisphere, the Patagonian desert is the main source of dust to the Southern Ocean (SO), representing 58% of the total atmospheric deposit at these high latitudes (Gassó and Stein, 2007; Li et al., 2008). Since the pre-industrial revolution, global dust deposition may have doubled, with projections in South America indicating an increasing contribution, partly driven by increased droughts and wind intensity (Zickfeld et al., 2007; Mahowald et al., 2010; Gassó and Torres, 2019; Pörtner et al., 2022). Patagonian dust is transported across the Atlantic and Indian sectors of the SO (Li et al., 2008). South of the Polar Front (> 50°S), surface waters are characterized as High Nutrient-Low Chlorophyll (HNLC), meaning that they are rich in macronutrients but primary production remains low. The deficit of Fe inputs significantly restricts the growth of phytoplankton in these waters (Martin, 1990), compounded by the low inputs of other micronutrients such as manganese (Browning et al., 2021). In this HNLC area, the low net primary production is proportional to the extremely low input of Fe that dissolves from the aerosols (Cassar et al., 2007). Furthermore, dust input into the SO has the potential to modify global climate by providing Fe and other essential limiting micronutrients, as evidenced by connections between increased dust inputs during Glacial periods and the emergence of the deep glaciations that characterize the past 1 Ma of Earth’s history (Martínez-Garcia et al., 2011). In the context of the nowadays climate change, the intensification of Patagonian dust input therefore deserves heightened consideration. Understanding and anticipating the inputs of dissolved and bioavailable micronutrients by the Patagonian dust are critical to accurately project the evolution of the efficiency of the biological carbon pump in the future SO and its feedbacks on atmospheric CO2.
The mineralogy and redox speciation of dust-borne Fe could have played a major role in its bioavailability for the Southern ecosystem during glacial periods (Shoenfelt et al., 2018). The ability of Fe-oxides to release Fe into seawater is closely related with their crystalline structure (Bonneville et al., 2009; Bligh and Waite, 2011), the specific surface area (Roden, 2003) and the aggregation process (Raiswell, 2011). Thus, the nature of Fe in the dust can profoundly impact its dissolution rate in seawater, thereby influencing phytoplankton growth, productivity, and the overall functioning of the biological carbon pump in the SO (Deppeler and Davidson, 2017). Patagonian dust exhibits a distinct chemical composition, dominated by quartz, and crystallinity (grain size<5 μm) (Qu, 2016) compared to other desertic dusts like those from Sahara, characterized by a relatively high presence of illite and a central size class typically ranging between 10-20 μm (Molinaroli, 1996; Guieu et al., 2002; Laity, 2008). Moreover, the SO, where these particles settle, also features specific physical (cold temperature, mixed layer depth of 50-90 m) and biogeochemical (HNLC area, dominance of diatoms south of the Polar Front) characteristics compared to other oceans (De Broyer et al., 2014). As a result, any alterations in the mineralogy and redox speciation of Patagonian Fe-dust in the future SO would be indicative of this region unique characteristics, but such variations have not been investigated yet.
While previous research has focused on the solubility and speciation of dust-borne elements under varying atmospheric pH conditions (Shi et al., 2012; Hamilton et al., 2022), relatively few studies have specifically investigated the behavior of aerosols in seawater (Meskhidze et al., 2019), with even fewer focusing on the speciation of Fe within dust particles (Schroth et al., 2009). For instance, it has been shown that Fe speciation in aerosols can differ depending on the aerosol source, with soils in arid regions dominated by Fe-oxides, glacial flour by primary and secondary ferrous silicates, and oil fly ash by ferric sulphate salts, corresponding, respectively, to Fe solubility of less than 1%, 2-3%, and 77-81% in the ocean (Schroth et al., 2009). To date, no comprehensive study focused on the Fe mineralogy and redox speciation in Patagonian dust. Additionally, only a very few studies have explored aerosol dissolution in seawater. Previous research predominantly simulated atmospheric conditions, incorporating diverse dust origins and experimental variables such as temperature, irradiation, and pH (Shi et al., 2012), illustrating the pivotal role of environmental factors in dust dissolution. In seawater, dissolution experiments were conducted using natural Cape Verde dust and anthropogenic aerosols to investigate the dissolution of cobalt (Co) and zinc (Zn) (Thuróczy et al., 2010), as well as the Fe dissolution in Mediterranean waters using Saharan dust (Wagener et al., 2008). These studies revealed different dissolution rates of metals from dust in seawater, contingent on the dust source and quantity added, although only a subset of different dusts and trace elements were considered. Moreover, while a singular dissolution experiment involving Patagonian dust in seawater has been conducted, it primarily examined the dissolution of macronutrients (P, Si, N) (Paparazzo et al., 2018), indicating rapid dissolution upon contact with seawater. Given the key role that Patagonian dust can play in supplying Fe and supporting the biological carbon pump in the SO, it is essential to conduct new dissolution experiments considering the release of Fe.
To understand the processes of dissolution, it is imperative to investigate the mineralogical structure and redox speciation of the dust at the submicrometric scale. Synchrotron, owing to its power and precision, serves as an invaluable tool in investigating mineral physics. Previous synchrotron studies were performed on aerosols (Oakes et al., 2012; Longo et al., 2016) or marine particles (Campbell, 2013; Ingall et al., 2013; Toner et al., 2016). Atmospheric aerosols and marine particles revealed variations in mineralogical composition and Fe redox speciation typical of the origin and environmental processes undergone by the particle. In contrast, no study tracked the changes in Fe mineralogy and redox speciation during aerosol dissolution in seawater using synchrotron analyses.
In addition to the future intensification of dust deposition, the SO will experience other environmental changes, such as warming of 3°C and acidification of -0.3 pH units of the surface waters, and increased photosynthetically active radiation (PAR) up to 55 μmol quanta m−2 s−1 (Boyd et al., 2016). Since these changes could have an impact on the dissolution of metals from dust, dissolution experiments simulating future SO conditions should also be considered (Breitbarth et al., 2010).
Therefore, this study aimed to assess the dissolution of pFe from Patagonian dust in seawater of the SO in the present and future environmental conditions. Particulate Fe dissolution was complemented by an analysis of associated processes within particulate Patagonian dust, including alterations in mineralogy, elemental composition, and Fe redox speciation. Controlled dissolution experiments were performed simulating the current and future-2100 conditions in terms of the key-selected variables (dust input, temperature, pH, PAR) over a period of one week. Dissolution rates were quantified based on the measurement of pFe using ICP-QMS, enabling estimation of the proportion of Fe released from the particulate phase. The underlying dissolution processes were investigated at the scales of the dust bulk (XRD analyses) and the single particle (synchrotron analyses). This approach allowed a detailed understanding of how the mineralogical composition and redox speciation of pFe were impacted by future conditions, and how they can affect dissolution processes. A significant contribution of this study was its pioneering estimate of Fe released by Patagonian dust, in addition to elucidating the nature of pFe over time, including mineralogical composition and redox speciation. This study provided the first estimations of the concentrations of Fe released by Patagonian dust, which may be readily available to phytoplankton in the present and future SO.
2 Material and methods
2.1 Production of Patagonian dust
The dust was generated from a Patagonian soil sampled in December 2011 at Lago Argentino at 50.29°S-71.81°W. This soil has an average elemental composition consistent with other areas of Patagonia (Qu, 2016), and represents one of the two primary reported dust sources in Patagonia (at 48.65°S-69°W and 50.6°S-70°W; Gassó and Stein, 2007). Therefore, Patagonian dust generated from this soil is a representative proxy of dust emission from South America and deposited in the SO. A quantity of 200 g of soil was sampled from the 0-2 cm surface layer and stored in polyethylene bags. Then, the soil was sifted using a 0.84 mm sieve (Qu, 2016). In the laboratory, the SyGavib system (aerosol generation system by vibration) developed by (Qu et al., 2020) was employed to generate the dust from this sieved soil. This system employs a loudspeaker to induce vibrations and an air-pump system to facilitate the upward movement of the generated aerosols, mimicking natural wind processes. According to Stokes law, particles smaller than 10 µm were transported to the top of the cylinder. Notably, atmospheric dust emitted from deserts consists of particles below 10 µm, with the major fraction lower than 5 µm (Sow et al., 2009). The system generates a bimodal aerosol distribution centered around 2-5 µm and 0.3-0.5 µm, corresponding to the distribution observed by Sow et al. (2009). The generated aerosol was then collected onto a cleaned Polyethersulfone (PES) filter (0.2 µm pore size from Supor®). Then, suprapur methanol was used to extract dust from the filter, rather than distilled water in order to prevent initiating the dissolution phase with acidic distilled water (pH ~5.8) and the formation of aggregates of dust particles. Furthermore, Patagonian dust from steppes contain a very low concentration of organic matter (< 1%) (González Polo et al., 2015), enabling the use of methanol without altering the dust composition. Dust generated was then gently crushed with an agate mortar to eliminate any large particles, thereby preventing sedimentation during the experiment.
2.2 Sampling of the seawater
The seawater used for the dissolution experiments was sampled from the High-Nutrient Low-Chlorophyll area in the Indian sector of the SO at 56.50°S-63.00°E in January 2020 aboard the French R.V. Marion Dufresne II from the surface (approx. 15 m depth) using a clean Teflon diaphragm pump with a flow rate of 15 L min-1 and a 30 m PTFE pipe following ultra-clean trace metal clean sampling procedures (Cutter et al., 2017). Seawater flowed directly from the pipe under a laminar flow fume hood (HEPA 14 filter) in the vessel’s laboratory. The seawater was filtered on-line using a cellulose acetate filter capsule (Sartorius®) featuring an inlet porosity of 0.45 µm and an outlet porosity of 0.2 µm. The filtered seawater was subsequently stored in 10 and 20 L acid cleaned polycarbonate (PC) containers (Nalgene®) at room temperature (21°C) and in the dark. These incubation bottles underwent 48 h in DECON (2.5%), 1 week in HCl (10%) and filled with 2% distilled HCl until use.
2.3 Incubation of dust in seawater: set-up of the experimental conditions
To simulate the surface water conditions of the SO, temperature and light (Table 1) were regulated using a thermostatic cabinet (Aqualytic®) fitted with light emitting diodes (LED) covered by a 201 full convert tungsten blue (CTB) filter (Lee filter®). The artificial light diffusion was set-up according to a daytime cycle from 4:00 am to 8:00 pm for the light and from 8:00 pm to 4:00 am for the night, resulting in a total light exposure of 16 h per day. Photosynthetic Active Radiation (PAR) was monitored in the incubator using a sensor MODEL MQ-510 Apogee® instruments placed in a PC bottle filled with Milli-Q water. The PAR was set at ~30 µmol quanta m-² s-1 in the present conditions, and at ~55 µmol quanta m-² in the future conditions. Adjustment of PAR was achieved by the number of lit LEDs.
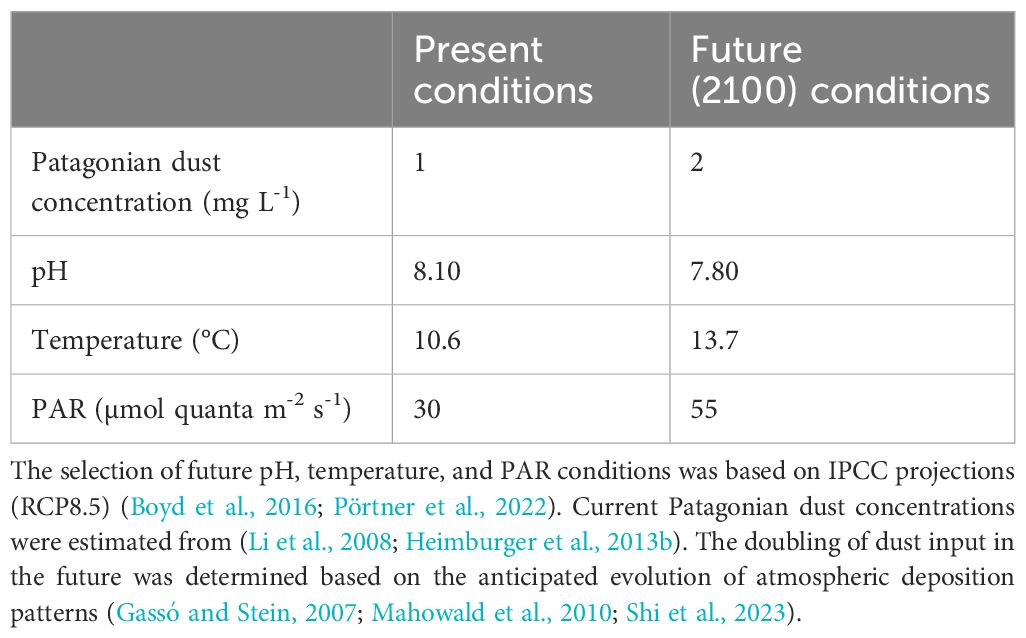
Table 1 Experimental conditions of the dissolution experiments representing the present and future surface waters in the SO.
The pH conditions were regulated by gas exchange from saturation of the overhanging atmosphere of the seawater bottle with CO2 mixture. This approach was specifically designed to emulate the natural acidification of the ocean, without causing any disruption to the carbonate system or altering the alkalinity (Riebesell et al., 2011). To initiate the experiments, the air space of the 10 L bottles containing seawater were saturated with pure CO2 (Alphagas 1 purity®) for 1 min. Subsequently, the bottles were placed on a 3D rotating plate in order to stir the water and enhance the exchange between the air and the water for 15 min. A gas mixture containing 800 ppm of CO2 (Alphagas 1 purity®) was then injected into the bottle air space until the target pH value of 7.80, representing the future conditions, was reached. Continuous pH monitoring was carried out throughout the experiment, and additional injections of the 800 ppm CO2 gas mixture were made to adjust the pH if necessary.
Dry dust deposition was chosen over wet deposition for its prevalence in atmospheric inputs from South America (Li et al., 2008), and in order to mimic the anticipated intensification of winds and aridity, which are expected to increase dry deposition (Pörtner et al., 2022). The mass of dust addition was deliberately overestimated by 1 mg L-1, exceeding a concentration of 9 ng L-1 d-1 or 282 ng L-1 m-1, as measured in the deposition around Kerguelen Island (Heimburger et al., 2012). This overestimation was necessary to ensure accurate measurement of the dust’s weight and to be able to detect the change in pFe concentration over the dissolution. Moreover, a lower mass can increase the heterogeneity in the composition of the inoculated dusts. A microbalance (Mettler Toledo MX5®), periodically calibrated, was used to weigh 10 mg and 20 mg of the dust. This resulted in a final concentration in the medium equivalent to 1 mg L-1 in the present conditions and to 2 mg L-1 in the future conditions. Prior to the addition to seawater, the dust was mixed in a small volume of filtered seawater in an ultra-clean tube, which was placed on a vortex (vortex genie 2 Scientific Industries®) to ensure effective homogenization. Then, the dust-enriched solution was transferred to a final volume of 10 L of filtered seawater. These large volume bottles were vigorously agitated after the dust enrichment and then transferred to an incubator to set the physical conditions.
Throughout the incubation, a set of physico-chemical parameters was sampled upon dust addition (Supplementary Information, Table 1), at the exact time of dust addition, then every 15 minutes during the first hour, every 30 minutes during the second hour, and subsequently every 24 hours over 7 days. This detailed monitoring approach aimed to capture any rapid dissolution processes that might occur during the initial contact of the dust with seawater (Wagener et al., 2008).
2.4 Observation using scanning electronic microscopy (SEM)
A 100 ml volume of seawater was filtered using a PC filter holder (Nalgene®) with a PC filter (porosity of 0.2 µm, diameter of 47 mm, Nuclepore™). The filters were then dried and stored in the dark at room temperature. Prior to microscopic examination, the filters underwent gold metallization. Observations of the dust during the dissolution experiments were conducted using a high-resolution focused ion beam-scanning electron microscope (FEG-FIB-SEM) Auriga 40 from Zeiss®, fitted with a Zeiss Gemini column (Liu et al., 2016). The instrument provides sub-nanometer scale resolution for detailed analysis of the dust particles.
2.5 Analyses of particulate trace metals
The novelty of this study was to monitor the dissolution of Fe by monitoring pFe. The particulate fraction is defined as the size fraction greater than 0.2 μm. Particulate trace metals were analyzed on ultra-clean Supor® PES filters (0.2 µm, 47 mm) after filtration of 250 mL of seawater sample using PC filter holders (Nalgene®), previously cleaned using trace metal clean procedure (Planquette and Sherrell, 2012). The filter holder was connected to a clean air pump (0.5 L min-1), enabling rapid filtration of 1 L of sample within a few minutes. All these manipulations were carried out within a laminar flow fume hood in a clean room (ISO 5). Following filtration, the filters were carefully placed in ultra-clean petri dishes, and then dried under the laminar fume hood. The petri dishes were wrapped with parafilm and stored at -20°C until analyses.
The digestion of the filter was performed in perfluoroalkoxy alkanes (PFA) vials (Savillex®) using the method adapted from Planquette and Sherrell (2012). The filter was positioned on the inner wall of the vial to prevent the complete digestion of the filter by soaking it directly in acid. A 3 mL solution of 8.0 M distilled HNO3-/2.9 M distilled HF was poured at the bottom of the vial and the solution was left to reflux overnight at 130°C on a PFA coating hot plate inside a controlled atmosphere evaporation station (Elemental Scientific®). After cooling, the vial was opened, and the filter was carefully removed from the vial wall. The acid was evaporated at 110°C until complete dryness. Subsequently, the residue was remobilized in 1 mL of a 50% distilled HNO3- solution at 70°C for over an hour. Once cooled, 14 mL of MQ water was added to the solution, resulting in a homogenized solution of particulate trace metals for Fe analysis using inductively coupled plasma quadrupole mass spectrometry (ICP-QMS).
Elemental concentrations were assessed using an Agilent 7900 quadrupole ICP-MS. Fe and Al were measured using a collision-reaction cell with helium gas (5 mL min-1) to remove polyatomic interferences. A scandium internal-standard was injected after inline mixing with the samples to correct for signal drift and matrix effects. A set of multi-element calibration standards was analyzed to confirm and model (through simple linear regression) the linear relationship between signal and concentration. The model was then used to convert measured sample counts to concentrations. Reported uncertainties were calculated using error propagation equations and considering the combination of standard deviation on consecutive signal integrations (n = 3), internal-standard ratio and blank subtraction. The non-linear term (internal-standard ratio) was linearized using a first-order Taylor series expansion to simplify error propagation.
Three measurement sessions on different days were necessary to measure all samples presented in this study. Across these three sessions, the background equivalent concentrations (BEC) of the ICP-MS was below 0.5 ppb for both Al and Fe. Chemistry blanks averaged at 0.15 ± 0.1 ppb (1SD) and 3.2 ± 2 ppb (1SD). These values are well below the lowest concentrations of the samples measured, which were above 400 ppb for both Al and Fe. The impact of the BEC and contaminations on our experiments is therefore neglectable. Repeatability was assessed both within a measurement session and across the different sessions, by measuring some standards, dry dust sample (PB1) and Hawaiian Volcano Observatory Basalt (BHVO1), (Supplementary Information, Table 2) twice within a session (beginning and end) and across sessions. It is calculated as the standard deviation of repeated measurements divided by the mean and expressed in percent. Repeatability results are good as we observe less than 2.5% variability (1SD) within a session and less than 3.5% variability (1SD) across sessions for both Al and Fe. Finally, accuracy was assessed by estimating the deviation of the concentration of the two solid standards from their reference values. It is found to be of -5.1% and 9.8% for Al and -8.9% and 12.8% for Fe, in standards PB1 and BHVO1 respectively. These results are acceptable and can be explained by the relative standard deviation of the measured concentrations of digestion replicates which is between 10% and 15%. These standard deviations are likely related to the accuracy of the scale and the potential loss of material while weighing 1 mg of standards. The absence of systematic accuracy offset further comforts us in the quality of the calibration model. As the experiment could not be replicated, the uncertainty was calculated based on repeated measurements from the ICP-QMS.
2.6 X-Ray diffraction analyses
The X-ray diffraction (XRD) analysis method was employed to identify mineral phases, crystal sizes, and morphologies. The detection was performed using a diffractometer (PANALYTICAL X’PERT PRO) fitted with a Co tube and a X’celerator detector. The analyses were done with the Patagonian soil and dust that were polycrystalline powder, characterized by a condensed and ordered state with fine granulometry, ensuring accurate analysis with wider peaks. The powder was deposited onto a steel sample holder and brushed to create a flat surface for analyses. The data acquisition was repeated 3 times and required 20 minutes to prevent detector saturation and a subsequent decrease in peak intensities. The diffracted ray was captured using a PIXcel 3D detector (Panalytical®), converting the photons into an electrical signal. The HighScore Plus (HS+) software was used to compute the mean curve from the three repeated diffractograms. Each peak position in the diffractogram corresponded to a specific element, and the peak intensity was directly proportional to its relative abundance. A comparison of the peaks with the mineralogical reference database “ICSD FIZ Karlsruhe2” enabled the identification of each mineral present in the sample based on its chemical composition and crystallographic structure. Quantitative analyses were performed by comparing the peak areas of the different minerals. This estimation was conducted using the Rietveld method in conjunction with the HighScore Plus (HS+) software (Chung, 1974; Nowak et al., 2018).
2.7 Synchrotron analyses
Another original objective of this study was to investigate the implication of mineralogy and speciation redox of pFe to Fe dissolution using synchrotron analysis. At different incubation times, 500 ml of seawater samples were filtered onto PC filters Nuclepore™ (pore size of 0.2 µm, diameter of 47 mm), using PC filter holders (Nalgene®). The filters were placed in a petri dish and dried under a fume hood. To prevent any oxidation processes, the samples were stored in a desiccator under a nitrogen atmosphere and shielded from light using 280 black foil (Lee filters®). For the analytical procedures, a section of the filter was selected and cut using ethanol-cleaned tweezers and scissors. The selection of the zone to be cut was guided by a brief observation under an optical microscope to identify regions with the presence of particles. The cut section was smaller than 1 cm2. The piece was then positioned on one of the 2 slots of an aluminium (Al) sample stick. During the analysis, the samples were positioned in a helium-filled chamber to limit the absorption of the fluorescence signal by the oxygen molecules. The analyses were performed at the Chicago synchrotron, APS (Advanced Photon Source) on the Beamline 2-ID-D. The detector used was a silicon drift detector (Vortex EM), with a sensitivity area of 50-mm² and a Be 12.5-mm window SII (NanoTechnology, Northridge CA, USA). Two types of analyses were performed on this sector, micro-fluorescence and micro-XANES analyses (McNulty et al., 2003).
For all micro-fluorescence analyses (micro-XRF), the dwell time was set at 100 ms, and the step size was defined as 0.25 µm. The coarse mode analysis utilized a spot size of 100 x 100 µm, which represented approximately 0.0006% of the total air filter. Based on the coarse scan, the size and location of the spot for the fine scans (56 x 56 µm, equivalent to 0.0002% of total air filter) were selected. Two spots of analysis were then acquired per sample. The X-ray was fixed at 7200 eV to enable the collection of elements from Al to Fe, which included Al, silicon (Si), phosphorus (P), sulfur (S), chlorine (Cl), potassium (K), calcium (Ca), titanium (Ti), vanadium (V), chromium (Cr), manganese (Mn), and Fe. In this study, the target elements were Fe, Al, and Si. To ensure accurate calibration of the elemental scans, NBS 1832 and 1833 thin film standards from the National Bureau of Standards (Gaithersburg, MD) were employed. These standards provide known values for each element in terms of weight percentage (% by wt) and X-ray absorption peak (kα and kβ). Quantification of the samples was achieved through proportionality to the certified standard of known percentage by wt. Then, the average value of density by areas (µg cm-²) was calculated according to Equation 1.
The uncertainty in the film mass (film wt in Equation 1) and area (10.06 cm2) was negligible compared to the estimated uncertainty in the certified values and thus, it can be neglected in the Equation 1. Data processing was carried out using the IDL MAPS software (Vogt, 2003), which converts the data into µg cm-² of micro-fluorescence scans. These results enabled to characterize the metal concentration according to the filter surface, along with the ratios between the elements. Areas identified by the software with high metal concentrations were designated as individual particles. This approach facilitated the calculation of elemental concentrations per particle at a microscopic scale. With the assistance of the software, a plot of the particle area was generated, and mean element concentrations were extracted per pixel (0.062 µm²).
Micro-XANES (X-ray Absorption Near Edge Structure) provides valuable insights into the oxidation state of the element studied. The oxidation state was effectively determined by the position of the pre-edge centroid, as opposed to the K-edge centroid, due to the reduced impact of matrix effects at this specific peak (Bajt et al., 1994). The XANES analysis was measured with a 250 µm beam. The zone plate used was 150 µm in diameter, and the focal length was 64 mm at 7.18 eV, corresponding to the Fe peak. Given the relatively high concentrations of dust added in our experiments compared to the ambient seawater, the signal intensities were well above the detection limits. To determine the oxidation state, the pre-edge centroid was initially normalized by subtracting the background of the absorption peak of the pre-edge, resulting in a Gaussian curve. The position of the peak of this Gaussian curve was then utilized in Equation 2 for converting the position of the K-edge XANES centroid spectrum of Fe(II) mineral to % oxidation state (Oakes et al., 2012).
To discern the nature of the Fe compounds present in the Patagonian dust, our analysis involved a comprehensive comparison of the samples using the data of Fe mineral powders (26 Fe mineral standards) provided by Oakes et al. (2012). The characterization of Fe minerals from the XANES spectra was achieved through a combination linear fitting using the Athena software (Ravel and Newville, 2005). It is important to note that each sample was subjected to dual micro-XANES and micro-XRF analyses, targeting different spots within the filters. Therefore, the uncertainty was determined using the standard deviation based on two measurements from two distinct spots on the same filter. This approach ensured a robust and reliable assessment of the Fe compounds, contributing to a more comprehensive understanding of the oxidation state and mineralogical composition of the Patagonian dust.
3 Results and discussion
3.1 Composition, mineralogy and Fe redox speciation of the Patagonian dust
The elemental composition, the size and the crystallinity of the dust can significantly influence the process of Fe dissolution in seawater (Anbeek, 1992; Rubasinghege et al., 2010). Notably, research has demonstrated that despite the specific surface area, Fe from clays exhibits higher solubility (4%) compared to Fe-oxides (< 1%) (Journet et al., 2008; Ito et al., 2021). In our study, X-ray Diffraction (XRD) analyses were conducted on Patagonian soil and dry dust to characterize the dust mineralogy. Our results indicated the prevalence of framework silicates in both the soil and dust (Table 2), commonly found across Patagonia (Desiage et al., 2018). This family of minerals is the most widespread in the Earth’s upper crust as well as in desertic soils, including quartz (SiO2) and plagioclases (albite NaAlSi3O8 or anorthite CaAl2Si2O8), alongside sheet silicates such as muscovite (KAl2(AlSi3O10)) and tectosilicate with barrerite ((Na,K,Ca)2 Al2 Si7 O18 7H2O) (Nesse, 2017). XRD analysis revealed that all identified minerals within the dust exhibited ordered distributions, suggesting relatively low susceptibility to dissolution (Ramseyer et al., 1992). Similar mineralogy were found between Patagonian soil and dust, as previously observed by Qu (2016), indicating that the production of dust from soil preserved the mineralogical diversity. However, the notable change between soil and dust was a loss of plagioclases (Table 2). Moreover, these measurements, while informative, were unable to capture the presence of amorphous compounds that possess higher dissolution capacity.
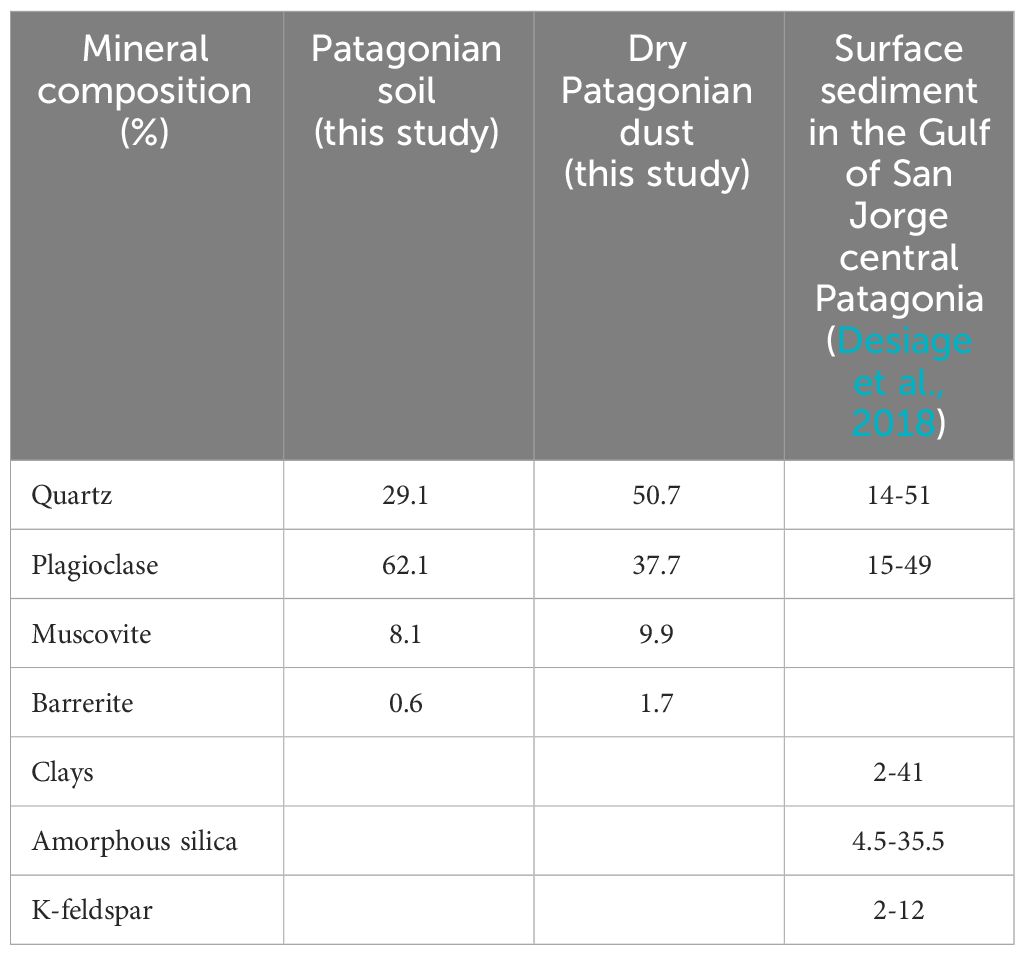
Table 2 X-ray Diffraction (XRD) analyses of Patagonian soil and dry dust (this study) and of surface sediments of the Gulf of San Jorge (Desiage et al., 2018).
The composition of Patagonian dust revealed 7.1% of Al, and 3.9% of Fe, respectively. Furthermore, this finding reinforced the notion that Al and Fe were the dominant metals within the Patagonian dust, as supported by XRD analyses (Table 3). The similar composition of the Patagonian dust used in this study and the average composition of the Earth’s crust (Wedepohl, 1995) suggested that the dust was representative of a desert aerosol. Notably, desert dusts are typically characterized as weathered materials, having undergone the effects of aging, and they are comparatively more resistant to dissolution compared to anthropogenic dust (Desboeufs et al., 2005). Elemental composition analyses of the dust also displayed values ranging in the same records as for river and aeolian samples from Patagonia (Table 3; Gaiero et al., 2003). Furthermore, while Fe was one of the most abundant element in the samples, it was not directly associated with these Si-minerals (Table 2). Notably, in natural dust, Fe-oxide minerals can be difficult to discern among clay minerals (Koffman et al., 2021), because amorphous Fe forms remain undetectable by XRD.

Table 3 Element concentrations in Patagonian dust measured in this study, in Patagonian riverborne and windborne samples, and the average composition of the upper continental crust.
The XRD analysis method was limited in its ability to detect specific Fe minerals in Patagonian dust due to the prevalent abundance of quartz and aluminosilicates compared to other mineral species. Consequently, the XANES spectrum of the samples was employed to conduct a more comprehensive analysis of the dust, particularly regarding Fe minerals, following incubation in seawater. The analysis revealed the presence of several key Fe minerals, primarily heterosite (Fe3 + 0.65 Mn3 + 0.35)PO4 (~77%), Fe(III) sulfate hydrate (~20%), and magnetite (~3%) (Figure 1). Additionally, micro-fluorescence scans were employed to assess the element concentrations of individual particles collected on filters. To enable a comparison between the elemental composition of particles obtained from micro-fluorescence measurements (Figure 1) and those analyzed by ICP-QMS, the average metal concentrations were estimated using the total area of the coarse and fine scans, corresponding to the overall surface of the filter. The initial average Fe concentration estimated on the entire filter surface was 374 nmol L-1, approximately 1.5 times lower than the values measured by ICP-QMS (~550 nmol L-1 Fe). In addition, a notable inconsistency in the estimated Fe concentration was observed among the analyses from different sections of the filter. Given that the surface analyzed by micro-fluorescence was small (100 µm² for coarse scans and 30 µm² for fine scans) relative to the total filter surface (17.34 cm²), the observed disparities among the various sections of the filter likely reflected the inherent heterogeneity in mineral distribution across the filter surface. Furthermore, the Fe/Al elemental ratio measured by ICP-QMS (Fe/Al = 0.27) differed from the ratio obtained in single particles (Fe/Al = 0.03-5.93). However, the X-ray analysis may not fully penetrate the aggregates, thus resulting in a lower Fe concentration estimation by micro-fluorescence compared to ICP-QMS measurements (Spolnik et al., 2004). Additionally, the presence of aggregates counted as a single particle could contribute to measurement inaccuracies. Among the probabilities of occurrence of such aggregates, colloids may be the most likely to create this bias as they are known for their prevalence in seawater and in dust (Wells and Goldberg, 1993; Aguilar-Islas et al., 2010). Moreover, small particles located on the surface of the largest particles could also affect the measurements. The elemental concentrations in each particle were ranked in the order of Si (< 8000 nmol cm−2) > Al (< 2000 nmol cm−2) > Fe (< 1000 nmol cm−2) (Supplementary Information, Table 3), similar to ICP-QMS results (Table 3). The notable presence of Si and Al, detected at the micrometric scale within each particle, further corroborated the findings from the XRD analysis, which indicated a predominance of aluminosilicates in the Patagonian dust (Figure 2).
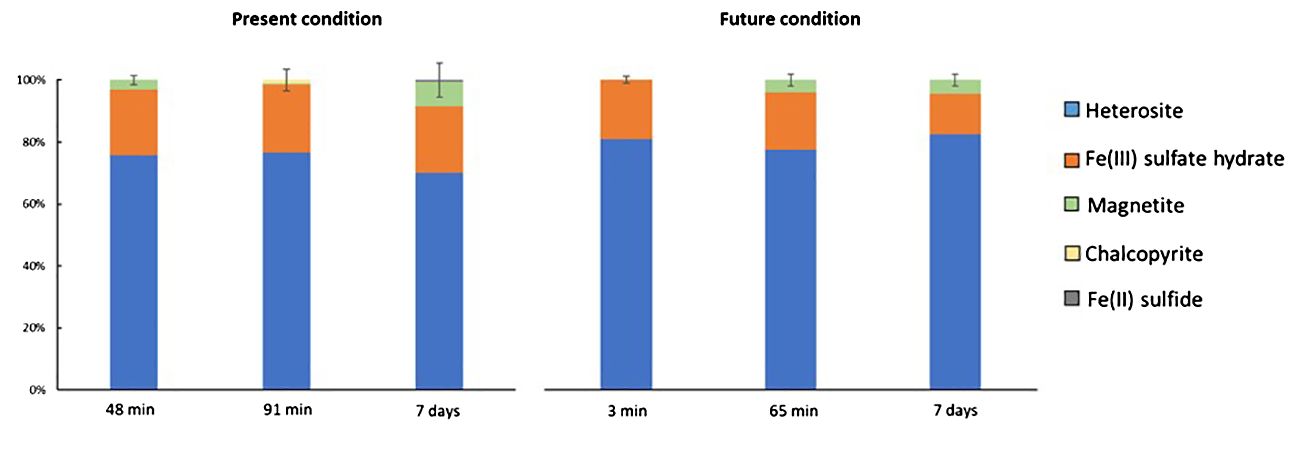
Figure 1 Relative mineral composition of Fe (%) in the dust incubated in seawater obtained with XANES analyses and proceed using Athena software. The standard deviation is computed based on the duplicates conducted at two distinct spots on the same filter.
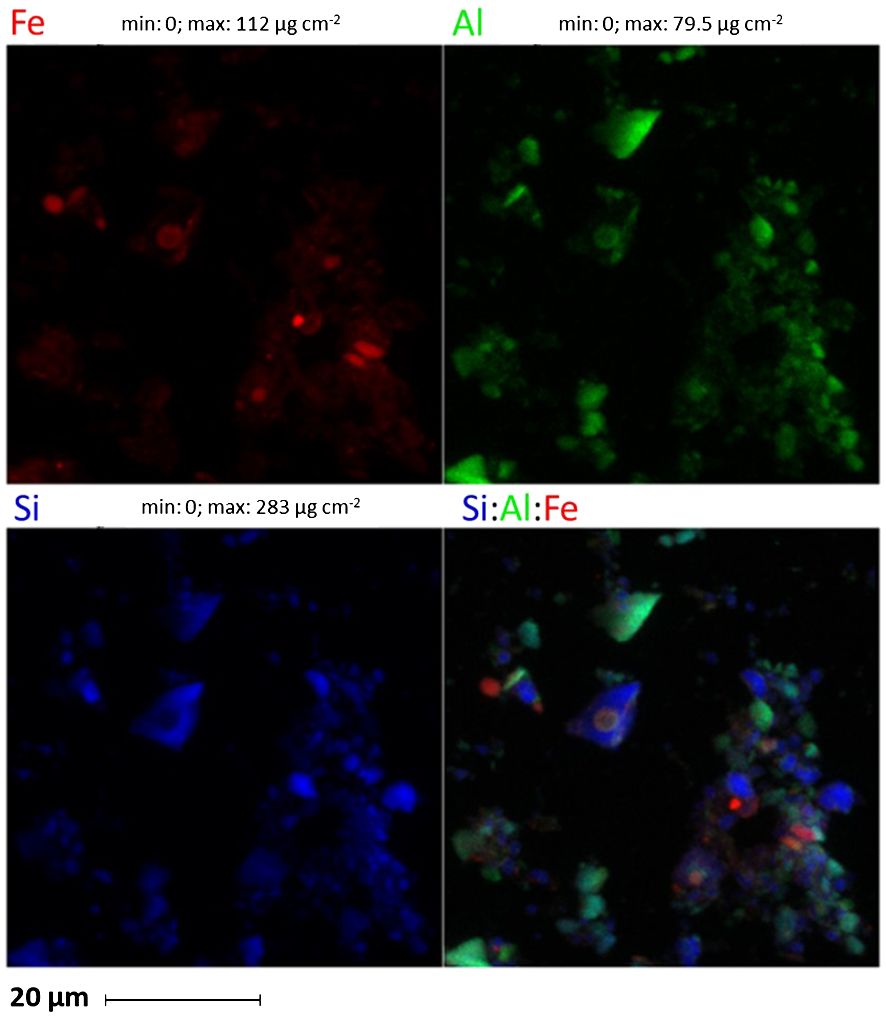
Figure 2 Micro-XRF scan of dust incubated in seawater. The scan shows in detail the colocations of the three major elements of the dust, Fe (in red), Al (in green) and Si (in blue). The scan shows predominance of quartz and aluminosilicates in the Patagonian dust.
Based on the elemental concentrations per particle, elemental ratios were computed to correlate them with the corresponding minerals (Oakes et al., 2012). The Si/Al elemental ratios measured in the incubated dust (Supplementary Information, Table 3) were indicative of quartz and aluminosilicates, with values ranging from 1 to 4 (Oakes et al., 2012). Furthermore, the Fe/Al elemental ratio (Supplementary Information, Table 3) was utilized to identify Fe-oxides, displaying values between 3 and 6 (Oakes et al., 2012). Notably, the Fe-oxides were characterized by a Fe concentration hovering 1000 µg cm-². The relative abundance of these minerals was estimated, with quartz and aluminosilicates accounting for approximately ~57% and Fe-oxides comprising around ~6.9%, aligning closely with the Fe-oxide content reported in Qu (2016) of 6.19%.
In addition to exploring the mineralogy and composition of the incubated dust, investigating the redox speciation of metals within the particles is important in order to understand their dissolution in seawater. However, it has not been thoroughly investigated to date. Tracking the physico-chemical variations of the Fe mixture from its origin to its deposition in oceanic waters presents considerable challenges. Notably, the atmosphere can experience the mixing of dust from diverse sources, which may undergo varying oxidation reactions triggered by light exposure and acidification (Desboeufs et al., 2005; Rubasinghege et al., 2010). Moreover, the meteorology-dependent light radiation can significantly impact the redox speciation and the residence time of dust in the atmosphere, which can span from hours to weeks (Jickells et al., 2005). Oceanic waters can also be the place of mixing for different dust mineralogies and redox states of elements, further complexifying the scenario (Schulz et al., 2012). Thus, it is therefore imperative to define the effects of environmental parameters on the redox speciation of metals in the dust more precisely. This can be achieved through controlled laboratory experiments that allow for a more comprehensive assessment of the underlying processes. To this end, XANES analyses of the filter area were performed to characterize the Fe redox speciation in both dry and incubated dust. The Fe(II)/Fe(III) ratio in Patagonian dust was determined to be 0.25, with ~20 ± 1.42% of Fe(II), which corresponds to the reported atmospheric ranges of 5 to 35% (average of 25%) (Oakes et al., 2012), and with marine aerosols (less than 73%) (Takahama et al., 2008). The ratio of Fe(II) to Fe(III) within particles can serve as a proxy for assessing their crustal origin (low Fe(II)), and of its volcanic and/or anthropogenic sources (up to 98%), in conjunction with their element composition (Oakes et al., 2012; Spolaor et al., 2013), indicating the lithogenic origin of Patagonian dust. The recorded relative abundance of Fe(II) in Patagonian dust (20%) was in the upper range of the crustal signature. Our findings, in line with previous studies (Shi et al., 2012), suggested that the value of Fe(II)/Fe(III) ratio can be used as an effective tracer for discerning the origin of the desertic dust that can persist over time.
3.2 Kinetics of the dissolution of Fe from the Patagonian dust
During the dust dissolution experiments in seawater, there was no indication of a higher dissolution rate of one Fe-mineral relative to the others (Figure 1). In fact, the predominant heterosites, characterized by an orthorhombic dipyramidal system (Fehr et al., 2010) are isostructural with the anhydrous Fe sulfate (FeSO4), even though FeSO4 can manifest itself in various forms, such as monohydrate, tetrahydrate and heptahydrate (Ruggiero et al., 2015). Therefore, given their analogous structural nature, these minerals may exhibit similar behavior in seawater, without any discernible trend of relative dissolution over time. Owing to their ordered structure, the reactivity may be relatively low (Toner et al., 2012), thus indicating a prolonged lifespan for Fe minerals from Patagonian dust in seawater. These Fe-minerals can be indeed classified as minerals with a long reaction time, extending beyond a week (Poulton et al., 2004). For instance, the half-life time of magnetite in seawater at pH 7.5 is 72 days (Poulton et al., 2004). Furthermore, when dust minerals come into contact with seawater, substitutions of elements within the mineral structure might occur, yet these effects have minimal impact on the reactivity of the minerals (Poulton et al., 2004). Moreover, in the two predominant Fe-minerals found in the Patagonian dust (heterosites and Fe(III) sulfate hydrate), the associated elements with Fe were PO4 and SO4, both of which are abundant constituents (PO4 ~2 µmol L-1, SO4 ~2.67 mg L-1) in the seawater of the SO (Brandini et al., 2000; Naik et al., 2006). Thus, these minerals may exhibit reduced solubility owing to the relatively high concentrations of PO4 and SO4 in seawater, thereby slowing down the dissolution process and rendering these minerals less reactive. Additionally, no discernible mineralogical distinctions were observed between the present and projected future conditions (ANOVA test, heterosite: p-value= 0.09, Fe(III) sulfate hydrate: p-value = 0.10, magnetite: p-value = 0.88; Figure 1), implying that changes in temperature, pH, and PAR are unlikely to significantly impact the Fe mineralogy within the dust. Thus, our results regarding the particulate fraction, suggested that the incubated Patagonian dust might be relatively less affected by anticipated environmental changes in the future SO. Crystalline systems, unless disrupted, can retain their constituent elements within their structure, preventing their release into the surrounding environment (Totsche et al., 2018). The environmental changes in the SO did not appear to reach extreme thresholds capable of disrupting the minerals, either during the experimental period of a week or under the future environmental conditions. Nevertheless, biological activities of autotrophs and heterotrophs in ambient seawaters could trigger mechanisms capable of destroying these minerals (Reinl et al., 2022). This includes the production of organic ligands complexing with dFe, thereby competing with Fe scavenging on dust particles (Wuttig et al., 2013), although such processes were not emulated in these abiotic experiments. Additionally, it is important to acknowledge that seasonal fluctuations and daily variations might significantly impact the Fe in Patagonian dust, potentially exceeding the magnitude of changes between the experimental present and future conditions. While Patagonian aerosols could release colloidal and soluble materials, potentially influencing seawater composition, but the reverse influence of seawater on the mineralogy of these dust particles is likely limited.
All samples showed identical XANES spectra, regardless of the dry or incubated dust, the sampling time, or the experimental conditions (pink curve on Figure 3). The consistent Fe(II)/Fe(III) ratio observed at the pre-edge (Figure 3) suggested that environmental changes, such as an increase in PAR that can impact photochemical reactions (Fu et al., 2010), did not affect the redox speciation of Fe in the particles. However, it is possible that the one-week incubation period was insufficient to induce any discernible alterations in the Fe redox speciation (Mayhew et al., 2011). Another explanation is that certain oxidation/reduction processes occurred within a very short time scale, such as during the sample filtration, when Fe(II) might have been rapidly oxidized to Fe(III), making it challenging to capture the initial redox speciation of Fe. Additionally, the time between the incubation and the sample measurement was 3 months, possibly affecting the observed results. It is also possible that the rate of redox conversions between Fe(III) and Fe(II) were too low to be detected within the one-week experimental duration. However, the conversion of particulate Fe(III) to Fe(II) remains elusive, as these processes have been more extensively investigated in the dissolved fraction (Kappler and Newman, 2004). Furthermore, the absence of biological activity in our experiments, especially the absence of biological production of organic ligands and chromophores capable of reducing Fe(III) under light (Barbeau, 2006) may have limited the change in the redox speciation of Fe.
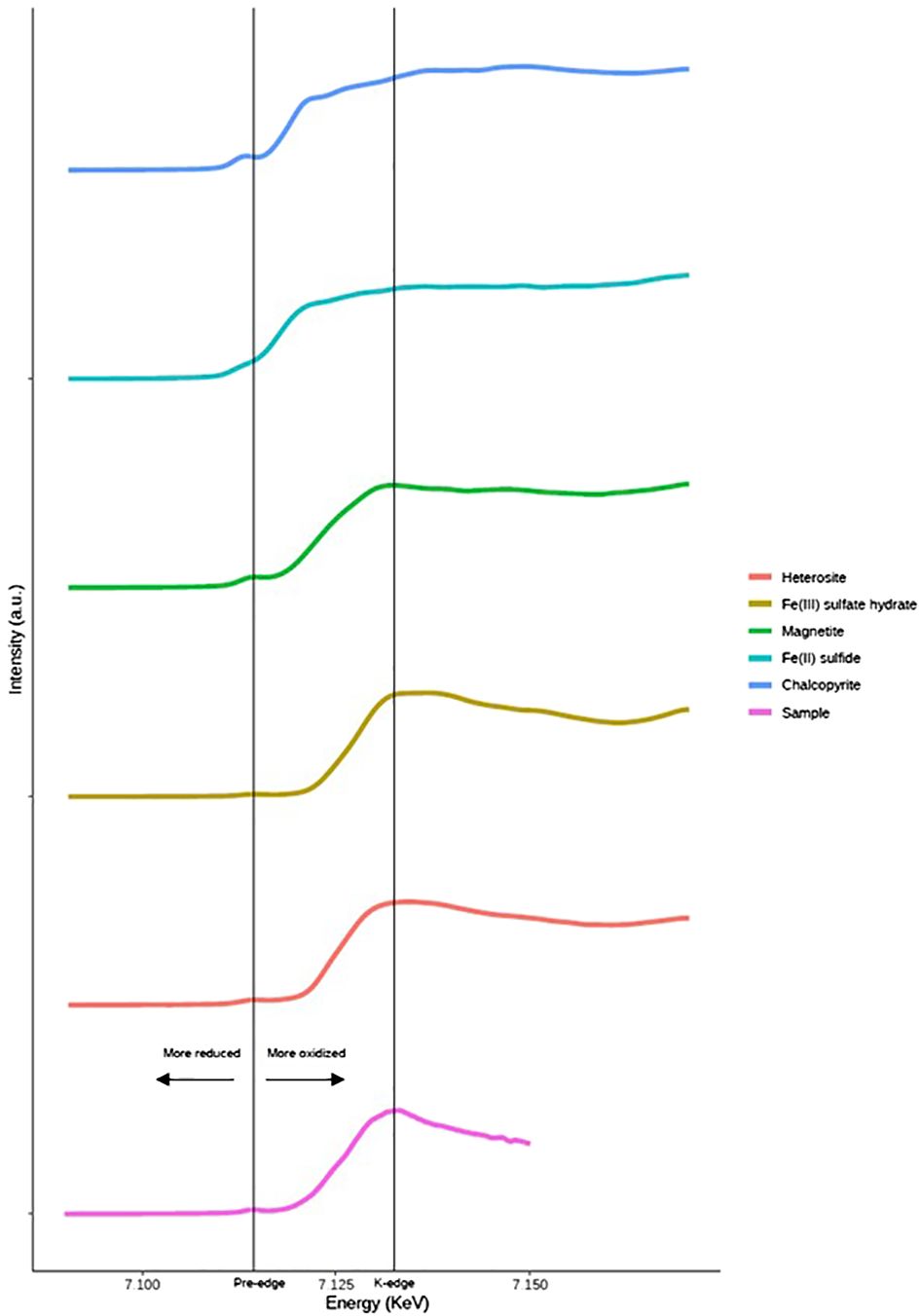
Figure 3 Average of XANES spectra of the all samples (purple) and associated Fe standards selected among 26 references by linear combination fitting.
On the other hand, the dissolution of Fe from incubated Patagonian dust in seawater under present and future conditions was assessed by following the pFe concentration over time of incubation. The particulate trace metals concentrations in the filtered seawater prior to dust addition was below detection limits of ICP-QMS measurements (<1 nmol L-1). Then, the pFe concentration in seawater 3 min after the dust addition (~532 nmol L-1 Fe in current condition) was in the range of the estimated total Fe in the 1 mg L-1 of added dust for the present scenario (~624 nmol Fe mg-1) and 1076 nmol L-1 Fe in future condition. The discrepancy of approximately 92 nmol L-1 might be attributed to uncertainties on the weighted dust and the volume of seawater used for the experiments. Nonetheless, this difference did not affect the interpretation of the dissolution kinetics.
Under the present conditions, pFe increased by ~50 nmol L-1 over the first 1H30, reaching a peak value of 585 nmol L-1 (Figure 4). Then, the concentration decreased, returning to its initial value within another 1H30 period. A third phase of dissolution started after this initial period of 3 hours, characterized by a slow decrease in pFe of 150 nmol L-1 over the course of one week. Under the future condition, the timing and the changes in particulate concentrations were slightly different. Particulate Fe increased by 60 nmol L-1 during the initial 3 hours of the experiment, without an immediate rapid decline. Instead, a gradual decrease in pFe began immediately and showed an amplitude of 200 nmol L-1.
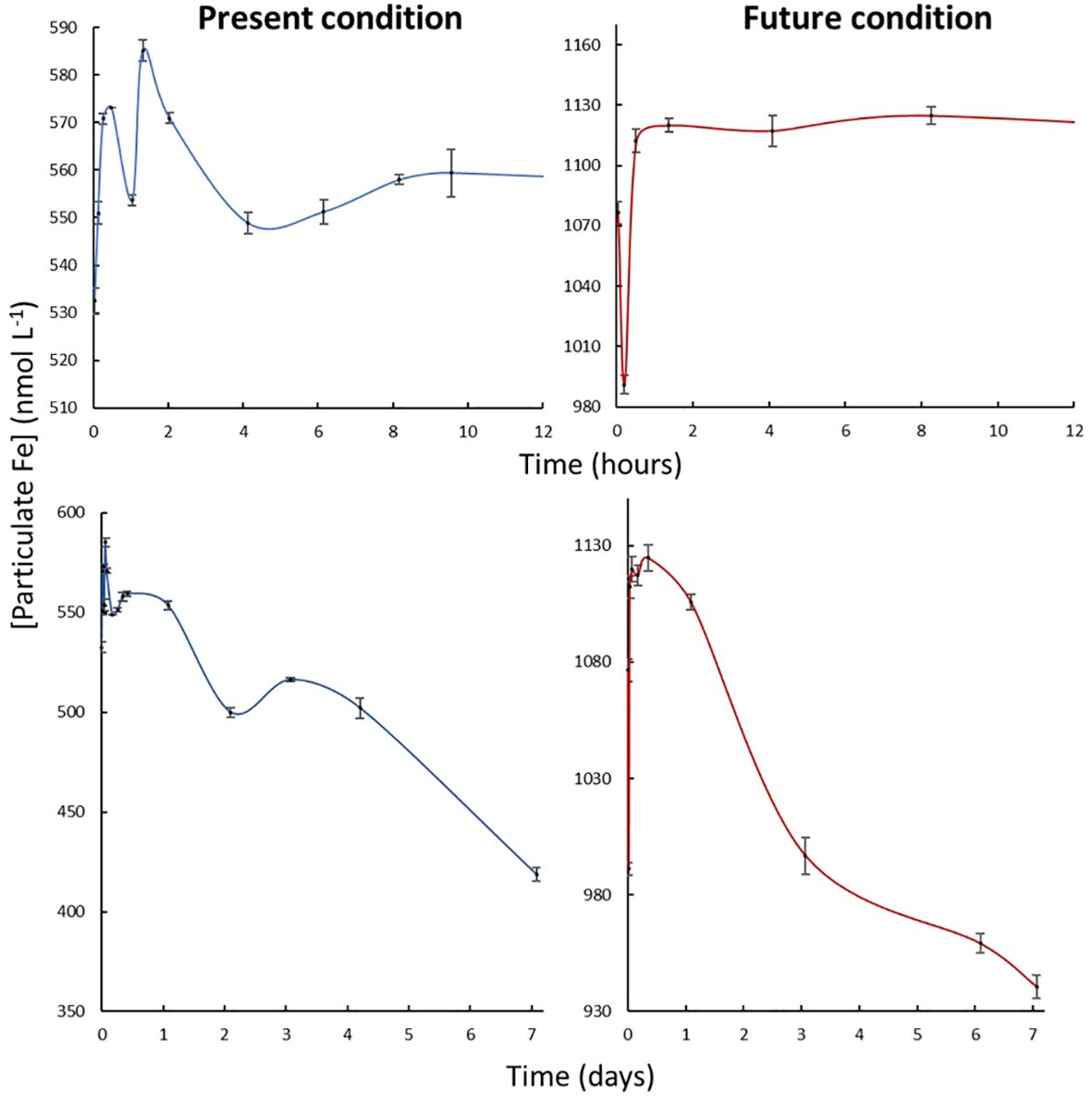
Figure 4 Kinetic of dissolution of pFe from Patagonian dust in seawater at the present and future environmental conditions during the first hours (top panel) and over 7 days (bottom panel). The measured pFe concentration without dust addition corresponds to ~1.3 nmol L-1. Standard deviations are calculated from 5 analysis replicates.
To elucidate the observed pFe dynamics depicted in Figure 4, SEM observations of incubated dust were conducted at different times of incubation and under the two conditions (Figure 5). The SEM analyses revealed a higher incidence of particles with colloids adsorbed to their surfaces, as well as aggregates of particles during the first hours of the kinetics. After one day of incubation, a greater number of dust particles exhibiting a polished surface were identified. No discernible difference was observed in the prevalence of these various particle types and aggregates between the present and future conditions. However, it is important to note that the qualitative assessment of the dust nature using SEM imaging posed challenges in terms of quantitative analysis, and a significant level of uncertainty persisted.
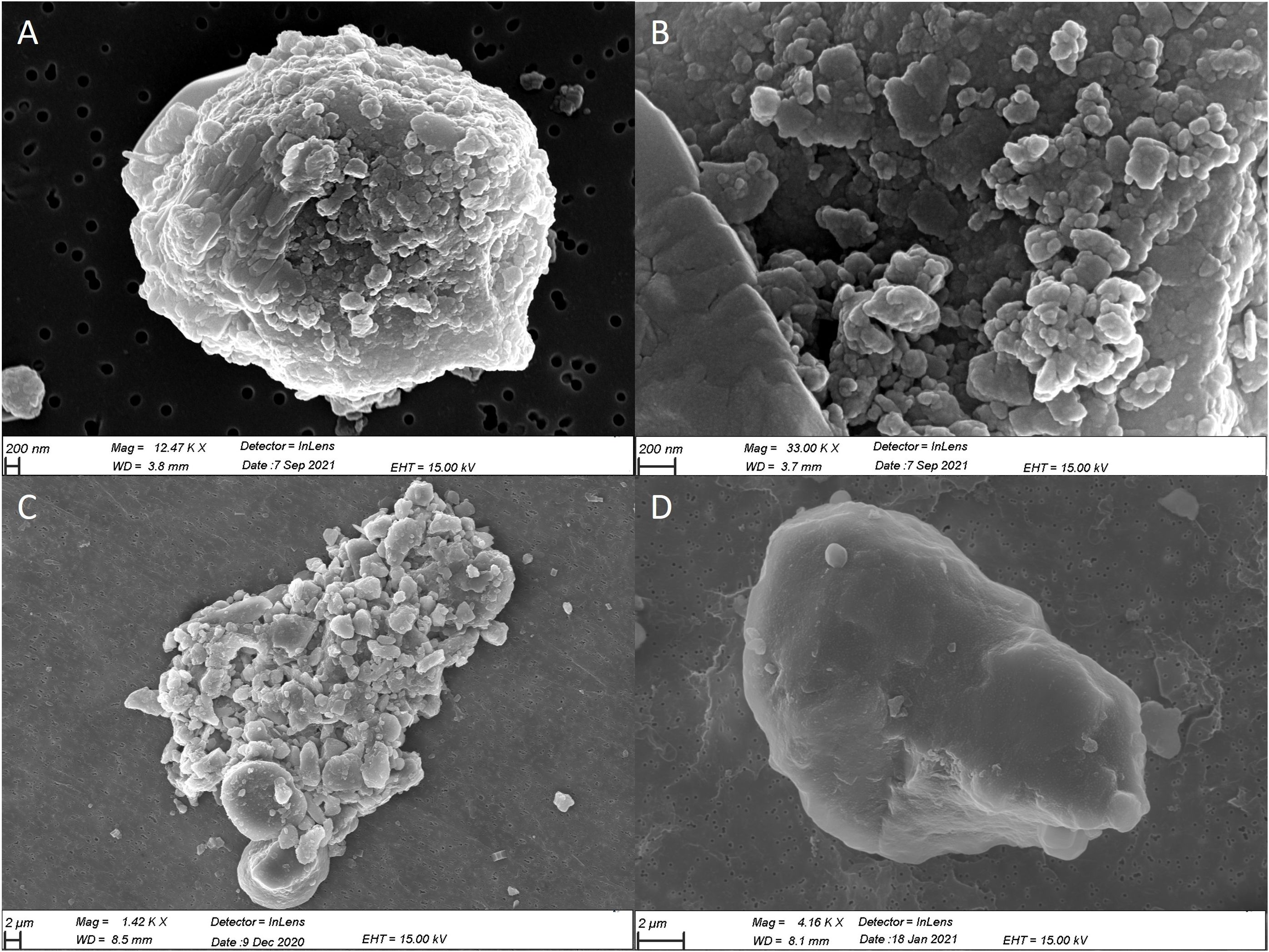
Figure 5 SEM observation of Patagonian dust in current condition; colloids at the surface of the particle (A) at the time of dust addition, (B) an agglomerate of colloids (first few hours), (C) and an aggregate of colloidal and particulate dust, (D) particle with a flat surface after 5 days.
The initial phase of the kinetics was characterized by an unexpected increase in the concentration of the particulate fraction. In an effort to elucidate the behavior of the Patagonian dust in seawater, the aerodynamic model of Shao (2008) was adapted (Figure 6). This model categorizes particle behavior based on size classes, indicating that the finest atmospheric particles (<2 µm) can aggregate as observed in the SEM pictures, a characteristic that corresponds with the distribution of the majority of particles from the Patagonian dust (Sow et al., 2009). For instance, the increase of pFe concentration that has been observed shortly after a dust deposition in a Mediterranean mesocosm coincided with a decrease in dFe, and was attributed to the scavenging of dFe onto the dust particles (Wagener et al., 2010). The formation of Patagonian dust aggregates in seawater may be facilitated under conditions of low ionic strength owing to the bonds with dissolved organic matter (Chekli et al., 2015) or via polymeric effects and Van der Waals forces that neutralize the negative charges on the surface of the particles, resulting in an attractive effect (Winterwerp and van Kesteren, 2004). Thus, the use of seawater fostered aggregate formation, allowing for the slow establishment of equilibrium between the seawater and the metals, an equilibrium that can extend over a week or more (Deguillaume et al., 2005). In addition, labile Fe was unable to coagulate with particulate organic matter, given that seawater was filtered at 0.2 μm, thereby preventing the generation of organic matter in seawater during storage and experimentation. Conversely, the dissolved organic matter originally present in the water might have played a significant role in facilitating dissolution by forming Fe-binding ligands (Bressac and Guieu, 2013). Furthermore, the adsorption of small colloids onto the particles is also a plausible contributing factor as observed in the SEM scans, as the leached dFe from natural aerosols is generally dominated by the colloid fraction (Aguilar-Islas et al., 2010). Indeed, with an addition of about 532 nmol L-1 of pFe, the concentrations were well above the concentration of the Fe solubility limit (~0.2-0.3 nmol L-1) for Fe(III) in seawater (Millero, 1998). Even when compared to a natural input that could be 10 or 100 times lower than the concentration employed in this experiment (Heimburger et al., 2012), the Fe concentration would still exceed the solubility limit. Therefore, this experiment accurately replicates realistic physicochemical processes. Knowing that the dissolved fraction was Fe-saturated by the supply of Patagonian dust, and that no organic ligands were produced in these experiments, most of the released Fe would have been in the form of inorganic complex and colloids (Boye et al., 2005), thereby promoting colloid formation. Consequently, the increase of pFe at the beginning of the kinetics could be linked to the formation of colloids and aggregates, and the adsorption of colloids on the particle surfaces, thus impeding the dissolution of the dust mantle by providing a shelter for the surface area, thereby reducing its contact with water. It is also conceivable that the time step used in this experiment might have hindered the observation of any rapid dissolution processes occurring within the initial minutes of dust incubation (Wagener et al., 2008).
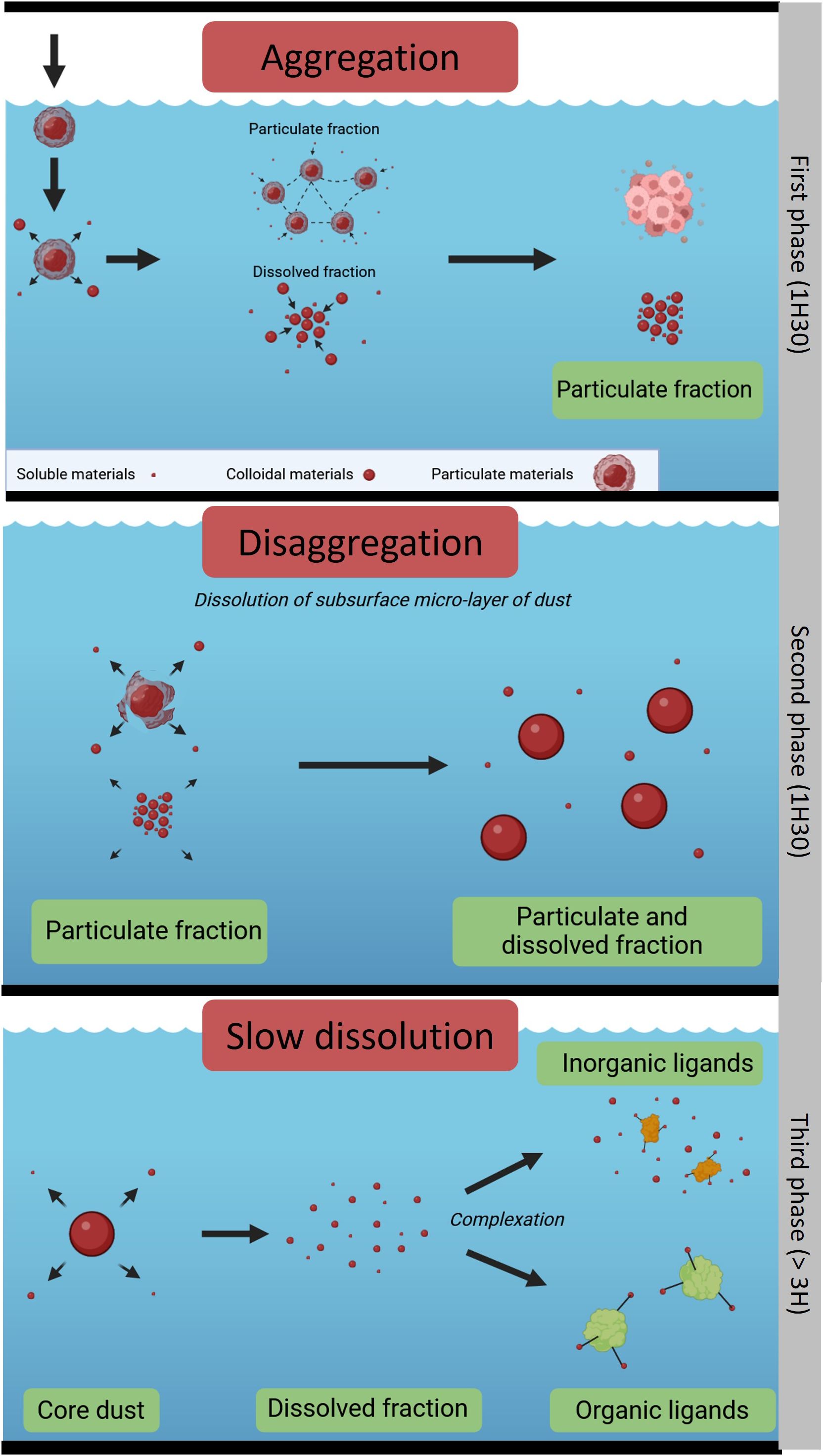
Figure 6 Conceptual schematic of the Patagonian dust dissolution process in seawater (using BioRender) after Shao, 2008.
After 1H30, the concentration rapidly decreased in the particulate phase, exhibiting a temporal and quantitative magnitude comparable to the preceding increase. The rupture of bonds and cohesive forces between colloids aggregates and between colloids and particles could explain such decrease. After extended contact with seawater, the dissolution process would initiate within a thin surface layer of particles and colloids. Here, surface segregation mechanisms (Delage, 1998) might accelerate dissolution at the surface compared to inside of the particles, given that the diffusion of elements is quicker in the surface layers than within the particle volume. Subsequently, as this surface layer dissolves, the cohesive forces between the particles would weaken, leading to the disintegration of the aggregates.
After 3 hours, the dissolution rate decelerated, marking the onset of the third phase of dissolution in Patagonian dust. This pattern of dissolution was akin to the dissolution of Saharan dust in Mediterranean waters, where a slower dissolution phase started after 2.6 hours (Wagener et al., 2008). The deceleration can be attributed to the complete dissolution of the surface thin layer of dust (Desboeufs, 2001), resulting in the collapse of colloid aggregates due to diminished cohesive forces. Subsequently, a new dissolution phase initiated, affecting the core of the particles. This dissolution was much slower than the second phase as it involves the most refractory part of the dust (Desboeufs, 2001).
3.3 Dissolution rates and residence time of Patagonian Fe dust
The dissolution rate of pFe was calculated during the third dissolution phase, following the Equation 3, where PTmti is the pFe concentration at the start of this phase (ti) and PTmtf after 7 days of incubation (tf).
The dissolution rate was subsequently divided by the total quantity of metal present in the dust and converted into a percentage (Equation 4).
Based on these calculations, we estimated the Fe dissolution rate from the Patagonian dust to be around 18.9 nmol L-1 day-1 (3.6% day-1) during the third phase of the dissolution kinetic in the current conditions. The dissolution of Fe can also be affected by the origin of particles and the medium of dissolution. For instance, the observed Fe dissolution rate was lower than those reported for Patagonian dust in Milli-Q water, which reached up to 9% within 15 minutes (Qu, 2016). The dissolution rate of Fe-dust was also lower than those found in Kerguelen Islands, a deposition site for Patagonian dust, where the dissolution rates were measured at 82 ± 18% in rainwater (Heimburger et al., 2013a). The comparatively higher dissolution rates in Milli-Q water and rainwater compared to seawater might be attributed to the acidic conditions. On the other hand, the rates of Patagonian dust dissolution were higher than those recorded for the Saharan aerosol, which exhibited a dissolution rate of approximately ~0.9 nmolFe day-1 (0.02% in 3 days) (Wagener et al., 2008), or 0.05 to 2.2% per day (Bonnet and Guieu, 2004), albeit the Saharan dust containing more Fe (~5%; Mendez et al., 2010) compared to Patagonian dust (~3.94%, this study), and therefore expected to have a higher dissolution rate. In addition, our study used half the amount of dust compared to the study of Bonnet and Guieu (2004), which may not explain the higher dissolution rate observed for Fe from Patagonian dust. Consequently, the discrepancy between our dissolution rate and other studies could primarily be attributed to the nature of the dust (whether it contains Fe in aluminosilicate, Fe-oxide, and amorphous Fe) and the variation in size class distribution. Saharan soils typically are composed of larger grain sizes, averaging around 10-20 µm (Guieu et al., 2002), which could potentially impede the dissolution process. However, the absence of a detailed mineralogical analysis of the dust used for the Saharan aerosol dissolution hampers a comprehensive understanding of the difference dissolution rate of Fe-dust. The methodology employed could also offer insights into these observed differences. Unlike prior investigations, which primarily focused on tracking dissolution rates within the dissolved fraction alone, this study extends its analysis to encompass the particulate fraction. Consequently, our experimental approach may provide a more comprehensive assessment of dissolution dynamics, capturing the transition of Fe from the particulate phase to the dissolved phase. Notably, monitoring dissolution solely within the dissolved fraction overlooks the potential complexation by organic and inorganic ligands, thereby omitting a significant aspect of dissolution behavior.
Conversely, in the future scenario, the dissolution rate was slower with ~19.4 nmol L-1 day-1 (1.8% day-1). The variations in dissolution rates observed between the present and future conditions can be attributed by changes in experimental factors. For instance, during the last glacial maximum, Fe solubility of atmospheric dust in the SO exhibited high variability, ranging from 1 to 42%, with inputs 10 fold higher than modern deposition (Conway et al., 2015). Consequently, Fe dissolution can be impacted by a multitude of physical, chemical, and biological parameters and is likely to evolve under the influence of anthropogenic alterations (Shi et al., 2011). The experimental changes, such as temperature, pH, PAR, and the quantity of added dust, might account for the observed differences in dissolution between the present and future conditions.
For example, the anticipated increase of PAR in the future condition from 30 ± 6 to 55 ± 7 μmol quanta m-2 s-1 (Boyd et al., 2016), is expected to intensify photochemical reactions, thereby rendering trace metals more soluble, and consequently augmenting Fe dissolution. However, photoreduction predominantly occurs at low pH levels of 2.5-5 (Faust and Hoigné, 1990), which was not the case in the future scenario, as the reactions were taking place at the future basic pH of seawater.
Regarding the impact of pH, the common assumption would be that the dissolution should be magnified under more acidic pH conditions due to the increased concentration of H+ ions, leading to a protonation process that weakens the Fe-O bonds on the particle surface, thus causing the release of Fe from the oxidic bulk (Cornell and Schwertmann, 2003). For example, the solubility of Fe in aerosols was shown to increase in Milli-Q water with decreasing pH, from 0.1% at pH 8 to 0.3% at pH 5.5 and to 4.7% at pH 2 (Spokes and Jickells, 1996). Furthermore, the solubility of Fe was found to be lower in the pH range of 7.5 to 9 compared to acidic conditions, and it varied only minimally within this range (Liu and Millero, 2002). Considering the projected shift of the seawater pH from 8.10 to 7.80 by 2100, the pH would not significantly impact the Fe solubility in our experiment. This result contrasts with what was previously suggested by Trimborn et al. (2017) indicating acidification could increase the Fe dissolution in combination with biological activity. A study (Breitbarth et al., 2010) investigating changes in the Fe speciation under varying CO2 treatments, revealed that the increase of dFe in coastal waters was predominantly governed by biological factors compared to acidification, particularly the stability of Fe-ligand complexes.
Next, the dissolution of dust could be amplified with increasing temperature due to the entropy agitation of the particles in the liquid. Nevertheless, the solubility of Fe decreases in seawater with rising temperature (Liu and Millero, 2002). Consequently, the projected +3°C increase in future conditions could be a contributing factor to the observed lower dissolution rate compared to present conditions.
Furthermore, the twofold increase in dust input in the experimental future condition likely led to an increase in aggregate formation, consequently reducing the surface-to-volume ratio of the dust particles. Such surface-to-volume decrease can result in lowering the exchange surface area, lead to a decrease of Fe dissolution, as previously observed with an increase in particle concentration (Biscombe, 2004) and when the dust concentration was lower than 20 mg L-1 (Spokes and Jickells, 1996). Additionally, the introduction of dust could lead to a reduction in dFe due to the adsorption of dFe on the dust particles and the formation of aggregates (Wagener et al., 2010). After deposition, atmospheric Fe exhibited a tendency to scavenge onto particles, particularly in conditions where concentrations of both particles and dissolved organic matter were elevated (Bressac and Guieu, 2013).
In summary (Table 4), the synergistic effect of the combined experimental parameters in the future conditions lead to the observed ~50% reduction in the dissolution rate by the end of the century.
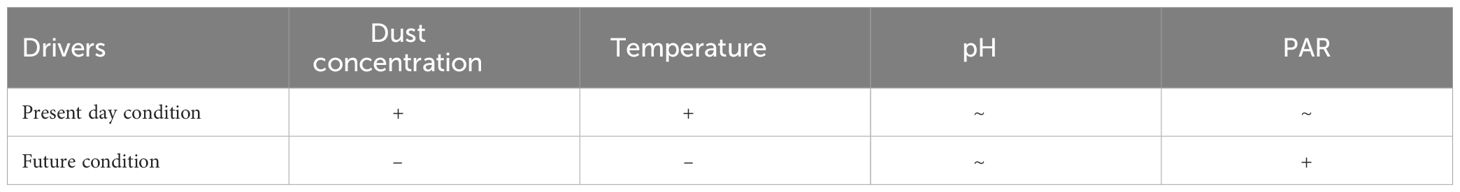
Table 4 Potential drivers that can control the dissolution of Fe from Patagonian dust in present and future seawater of the SO. (+) means increase of the dissolution, (-) means decrease of the dissolution and (~) means no significant impact.
The residence time of pFe in a stratified mixed-layer was estimated using the sinking rate of dust particles in seawater. The sinking rate was calculated using Equation 5 (Omand et al., 2020), which includes wsink (velocity of the particle; m s-1), g (acceleration due to the gravity; m s-2), r (radius of the particle; m), ρparticle (density of the particle; kg m-3), ρseawater (density of seawater; kg m-3), nseawater (kinematic viscosity of seawater; kg m-1 s-1). As the density of the Patagonian dust was not measured in this study, a global average value (~2600 kg m-3) for desert dust particles was adopted (Wagner et al., 2009).
Depending of the particle size, the residence time in the mixed layer can be highly versatile (van der Jagt et al., 2018). Applying the equation yielded an estimated residence time for the largest particles (~10 µm) of 14 days in a 89 m mixed layer (HNLC) and 8 days in a 50 m mixed layer (Polar Frontal Zone) (De Boyer Montégut et al., 2004). However, considering the size distribution of Patagonian dust, which is centered at around 2-5 µm and 0.3-0.5 µm, the calculated residence times were 112 days and 24 years in HNLC and 63 days and 14 years in the PFZ, respectively. Based on the dissolution rates obtained in our study, the Fe content of Patagonian dust would be completely dissolved in 26 days under present conditions and in 55 days under future conditions. Therefore, most of the Fe dissolved from the dust can remain in the mixed-layer depth. Therefore, in future anticipated condition with a doubling of pFe inputs and a 50% reduction in dissolution rate, an equivalent quantity of dFe is expected to be released from dust, potentially becoming bioavailable to phytoplankton. With a dust flux from South America to the SO region of 0.3 mg m-² d-1 (Li et al., 2008), and a mean Fe concentration in Patagonian dust of ~3.94% (this study), the atmospheric deposition flux of Fe from dust would be to around ~0.011 mgFe m-² d-1. Using the observed dissolution rate of 3.6% d-1 in the current conditions, this input would represent a flux of dFe of 0.43 µgFe m-² d-1 (7.62 nmol m-² d-1) in the surface waters of the SO (Equation 6). With an expected doubled input of Patagonian dust in the future conditions, the dissolution rates we recorded was 1.8% d-1, leading to a corresponding flux of dFe in seawater of 0.43 µgFe m-² d-1 (7.6 nmol m-² d-1).
The flux of dFe in the mixed layer found under present and future conditions correspond to 0.09 pmolFe L-1 d-1 in HNLC and 0.15 pmolFe L-1 d-1 in PFZ (Equation 7, in which the mixed layer depth was 89 m in the HNLC area and 50 m in the PFZ). Despite the projected doubling of dust input in the future, the impact of environmental changes was shown to lead to a 2-fold reduction in Fe dissolution rate. As a result, the dFe concentrations are not anticipated to increase in future projections.
The estimated dFe inputs obtained in this study were 0.09 pmol L-1 d-1 in HNLC for both conditions and 0.15 pmolFe L-1 d-1 in PFZ for both conditions.
4 Conclusion
Throughout its journey from source to sink, desert dust from Patagonia undergoes the synergistic effect of various physical and biogeochemical factors, both in the atmosphere and at its contact with the marine environment. This study focused on the dust dynamics after its deposition in seawater.
Initial XRD analyses of the Patagonian dust unveiled the prevalence of silicate minerals, primarily quartz and plagioclase. Further synchrotron analyses highlighted the occurrence of Fe minerals, mainly as heterosites and Fe(III) sulfate hydrates within the dust particles and indicated 20% of Fe(II). The mineralogy and the Fe(II)/Fe(III) ratio were similar in both the dry and incubated dust, with no significant alteration upon submersion in seawater or exposure to future environmental changes.
Despite the absence of mineralogical changes, the examination of pFe indicated that Patagonian dust undergoes dissolution, releasing Fe in seawater. Specifically, the Fe dissolution rates of Patagonian dust in seawater were estimated at 3.8% d-1 in the present conditions and twice less (1.6% d-1) in the future scenario (Figure 7). These dissolution rates resulted in a dFe flux of 0.09 pmol L-1 d-1 in the HNLC region and 0.15 pmol L-1 d-1 in the PFZ. Despite the anticipated doubling of dust input in the future, the slower rate of Fe dissolution negated any significant increase in dFe by 2100, with probably no significant impact to alleviate the Fe limitation in the future SO.
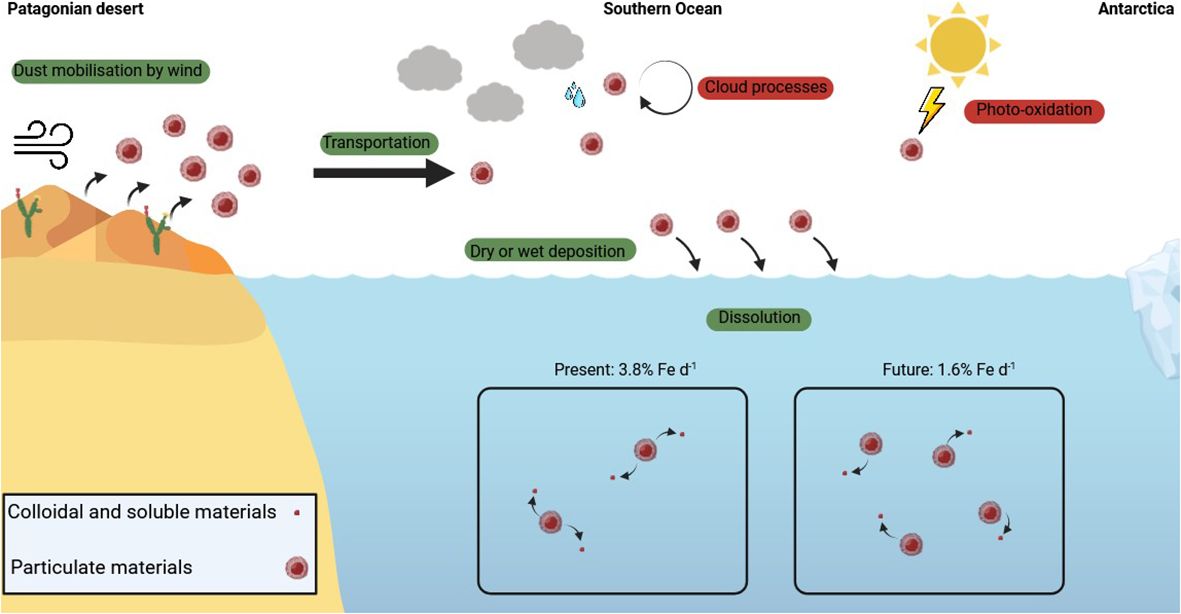
Figure 7 Conceptual scheme of Patagonian dust dissolution under present and future Southern Ocean conditions.
This study underscores the critical role of synchrotron techniques in unrevealing the intricate connections between mineralogy, redox speciation, and Fe-dust dissolution in seawater. Future research should focus on smaller size fractions (<0.2 µm) since nanoparticles exhibit distinct physicochemical properties and significantly heightened reactivity due to their high surface area-to-volume ratio. Another avenue for exploration could be to quantify the labile fraction of potentially bioavailable Fe in dust and compare these findings with the dissolution of pFe in seawater. The labile fraction is generally evaluated by acid treatment (Berger et al., 2008), a process that can yield distinct results from the dissolution of dust in seawater under baseline pH conditions.
The insights garnered from this study offer valuable information for the development of SO biogeochemical models, contributing to a comprehensive understanding of the intricate dynamics underlying Fe dissolution rates from Patagonian dust under present and projected environmental conditions.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
CD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing, Validation, Visualization. MB: Funding acquisition, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Project administration, Validation. BL: Formal analysis, Writing – review & editing. PBu: Data curation, Formal analysis, Writing – review & editing. YF: Conceptualization, Supervision, Writing – review & editing. RL: Formal analysis, Supervision, Writing – review & editing. SB: Formal analysis, Writing – review & editing. PBe: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was conducted in the framework of the SAGAS project (Solubility of Patagonian dust in the future Southern Ocean, PI MB) funded by IDEX-Université Paris Cité. CD benefited from an international mobility grant from the STEP’UP doctoral school of IPGP. The PhD grant of CD was funded by the University Paris Cité. ICP-QMS and SEM analyses were supported by IPGP analytical platform PARI (Plateforme d’analyse haute résolution). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Acknowledgments
The authors thank Ellery Ingall for kindly providing the Fe mineral standards to process the micro-XANES data. The authors thank Sophie Nowak (UPC) for XRD acquisitions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1363088/full#supplementary-material
References
Aguilar-Islas A. M., Wu J., Rember R., Johansen A. M., Shank L. M. (2010). Dissolution of aerosol-derived iron in seawater: Leach solution chemistry, aerosol type, and colloidal iron fraction. Mar. Chem. 120, 25–33. doi: 10.1016/j.marchem.2009.01.011
Anbeek C. (1992). Surface roughness of minerals and implications for dissolution studies. Geochim. Cosmochim. Acta 56, 1461–1469. doi: 10.1016/0016-7037(92)90216-6
Bajt S., Sutton S. R., Delaney J. S. (1994). X-ray microprobe analysis of iron oxidation states in silicates and oxides using X-ray absorption near edge structure (XANES). Geochim. Cosmochim. Acta 58, 5209–5214. doi: 10.1016/0016-7037(94)90305-0
Barbeau K. (2006). Photochemistry of organic iron(III) complexing ligands in oceanic systems. Photochem. Photobiol. 82, 1505–1516. doi: 10.1111/j.1751-1097.2006.tb09806.x
Berger C. J. M., Lippiatt S. M., Lawrence M. G., Bruland K. W. (2008). Application of a chemical leach technique for estimating labile particulate aluminum, iron, and manganese in the Columbia River plume and coastal waters off Oregon and Washington. J. Geophys. Res. Oceans 113, 2007JC004703. doi: 10.1029/2007JC004703
Biscombe A. (2004). Factors influencing the seawater solubility of aerosol associated trace metals. University of Plymouth, Plymouth.
Bligh M. W., Waite T. D. (2011). Formation, reactivity, and aging of ferric oxide particles formed from Fe(II) and Fe(III) sources: Implications for iron bioavailability in the marine environment. Geochim. Cosmochim. Acta 75, 7741–7758. doi: 10.1016/j.gca.2011.10.013
Bonnet S., Guieu C. (2004). Dissolution of atmospheric iron in seawater. Geophys. Res. Lett. 31, L03303. doi: 10.1029/2003GL018423
Bonneville S., Behrends T., Van Cappellen P. (2009). Solubility and dissimilatory reduction kinetics of iron(III) oxyhydroxides: A linear free energy relationship. Geochim. Cosmochim. Acta 73, 5273–5282. doi: 10.1016/j.gca.2009.06.006
Boyd P. W., Dillingham P. W., McGraw C. M., Armstrong E. A., Cornwall C. E., Feng Y.-y., et al. (2016). Physiological responses of a Southern Ocean diatom to complex future ocean conditions. Nat. Clim. Change 6, 207–213. doi: 10.1038/nclimate2811
Boye M., Nishioka J., Croot P. L., Laan P., Timmermans K. R., de Baar H. J. W. (2005). Major deviations of iron complexation during 22 days of a mesoscale iron enrichment in the open Southern Ocean. Mar. Chem. 96, 257–271. doi: 10.1016/j.marchem.2005.02.002
Brandini F. P., Boltovskoy D., Piola A., Kocmur S., Röttgers R., Cesar Abreu P., et al. (2000). Multiannual trends in fronts and distribution of nutrients and chlorophyll in the southwestern Atlantic (30–62°S). Deep Sea Res. Part Oceanogr. Res. Pap. 47, 1015–1033. doi: 10.1016/S0967-0637(99)00075-8
Breitbarth E., Bellerby R. J., Neill C. C., Ardelan M. V., Meyerhöfer M., Zöllner E., et al. (2010). Ocean acidification affects iron speciation during a coastal seawater mesocosm experiment. Biogeosciences 7, 1065–1073. doi: 10.5194/bg-7-1065-2010
Bressac M., Guieu C. (2013). Post-depositional processes: What really happens to new atmospheric iron in the ocean’s surface? Glob. Biogeochem. Cycles 27, 859–870. doi: 10.1002/gbc.20076
Browning T. J., Achterberg E. P., Engel A., Mawji E. (2021). Manganese co-limitation of phytoplankton growth and major nutrient drawdown in the Southern Ocean. Nat. Commun. 12, 884. doi: 10.1038/s41467-021-21122-6
Campbell A. C. (2013). Mineralogy and Chemistry of Marine Particles by Synchrotron X-Ray Spectroscopy, Mössbauer Spectroscopy, and Plasma-Mass Spectrometry. Eds. Hurd D. C., Spencer D. W. (Washington, D. C.: American Geophysical Union), 375–390. doi: 10.1029/GM063p0375
Cassar N., Bender M. L., Barnett B. A., Fan S., Moxim W. J., Levy H., et al. (2007). The southern ocean biological response to aeolian iron deposition. Science 317, 1067–1070. doi: 10.1126/science.1144602
Chekli L., Zhao Y. X., Tijing L. D., Phuntsho S., Donner E., Lombi E., et al. (2015). Aggregation behaviour of engineered nanoparticles in natural waters: Characterising aggregate structure using on-line laser light scattering. J. Hazard. Mater. 284, 190–200. doi: 10.1016/j.jhazmat.2014.11.003
Chung F. H. (1974). Quantitative interpretation of X-ray diffraction patterns of mixtures. I. Matrix-flushing method for quantitative multicomponent analysis. J. Appl. Crystallogr. 7, 519–525. doi: 10.1107/S0021889874010375
Conway T. M., Wolff E. W., Röthlisberger R., Mulvaney R., Elderfield H. E. (2015). Constraints on soluble aerosol iron flux to the Southern Ocean at the Last Glacial Maximum. Nat. Commun. 6, 7850. doi: 10.1038/ncomms8850
Cornell R. M., Schwertmann U. (2003). The iron oxides: structure, properties, reactions, occurrences, and uses. 2nd (Weinheim: Wiley). doi: 10.1002/3527602097
Cutter G., Casciotti K., Croot P., Geibert W., Heimbürger L.-E., Lohan M., et al. (2017). Sampling and Sample-handling Protocols for GEOTRACES Cruises. Version 3, August 2017 (Toulouse, France: GEOTRACES International Project Office). doi: 10.25607/OBP-2
De Boyer Montégut C., Madec G., Fischer A. S., Lazar A., Iudicone D. (2004). Mixed layer depth over the global ocean: An examination of profile data and a profile-based climatology. J. Geophys. Res. Oceans 109, 2004JC002378. doi: 10.1029/2004JC002378
De Broyer C., Koubbi P., Griffiths H. J., Raymond B., Udekem d’Acoz C.d’, Van de Putte A. P., et al. (2014). Biogeographic Atlas of the Southern Ocean (Cambridge: The Scientific Committee on Antarctic Research).
Deguillaume L., Leriche M., Desboeufs K., Mailhot G., George C., Chaumerliac N. (2005). Transition metals in atmospheric liquid phases: sources, reactivity, and sensitive parameters. Chem. Rev. 105, 3388–3431. doi: 10.1021/cr040649c
Delage S. (1998). Cinetique couplees au voisinage des surfaces : segregation, precipitation et dissolution (Paris: Université Paris VI).
Deppeler S. L., Davidson A. T. (2017). Southern ocean phytoplankton in a changing climate. Front. Mar. Sci. 4. doi: 10.3389/fmars.2017.00040
Desboeufs K. (2001). Processus de dissolution des aérosols atmosphériques au sein des gouttes d’eau nuageuses. Université Paris-Diderot - Paris VII, Paris.
Desboeufs K. V., Sofikitis A., Losno R., Colin J. L., Ausset P. (2005). Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter. Chemosphere 58, 195–203. doi: 10.1016/j.chemosphere.2004.02.025
Desiage P.-A., Montero-Serrano J.-C., St-Onge G., Crespi-Abril A., Giarratano E., Gil M., et al. (2018). Quantifying sources and transport pathways of surface sediments in the gulf of San Jorge, Central Patagonia (Argentina). Oceanography 31, 92–103. doi: 10.5670/oceanog.2018.401
Faust B. C., Hoigné J. (1990). Photolysis of Fe (III)-hydroxy complexes as sources of OH radicals in clouds, fog and rain. Atmospheric Environ. Part Gen. Top. 24, 79–89. doi: 10.1016/0960-1686(90)90443-Q
Fehr Th., Hochleitner R., Laumann A., Schmidbauer E., Schneider J. (2010). Mineralogy, Mössbauer spectroscopy and electrical conductivity of heterosite (Fe3+, Mn3+)PO4. Phys. Chem. Miner. 37, 179–189. doi: 10.1007/s00269-009-0322-2
Fu H., Cwiertny D. M., Carmichael G. R., Scherer M. M., Grassian V. H. (2010). Photoreductive dissolution of Fe-containing mineral dust particles in acidic media. J. Geophys. Res. 115, D11304. doi: 10.1029/2009JD012702
Gaiero D. M., Probst J.-L., Depetris P. J., Bidart S. M., Leleyter L. (2003). Iron and other transition metals in Patagonian riverborne and windborne materials: geochemical control and transport to the southern South Atlantic Ocean. Geochim. Cosmochim. Acta 67, 3603–3623. doi: 10.1016/S0016-7037(03)00211-4
Gassó S., Stein A. F. (2007). Does dust from Patagonia reach the sub-Antarctic Atlantic Ocean? Geophys. Res. Lett. 34, L01801. doi: 10.1029/2006GL027693
Gassó S., Torres O. (2019). Temporal characterization of dust activity in the central Patagonia desert (Years 1964–2017). J. Geophys. Res. Atmospheres 124, 3417–3434. doi: 10.1029/2018JD030209
González Polo M., Kowaljow E., Castán E., Sauzet O., Mazzarino M. J. (2015). Persistent effect of organic matter pulse on a sandy soil of semiarid Patagonia. Biol. Fertil. Soils 51, 241–249. doi: 10.1007/s00374-014-0961-4
Griffin D. W., Kellogg C. A. (2004). Dust storms and their impact on ocean and human health: dust in earth’s atmosphere. EcoHealth 1, 284–295. doi: 10.1007/s10393-004-0120-8
Guieu C., Loÿe-Pilot M., Ridame C., Thomas C. (2002). Chemical characterization of the Saharan dust end-member: Some biogeochemical implications for the western Mediterranean Sea. J. Geophys. Res. Atmospheres 107 (D15), ACH-5. doi: 10.1029/2001JD000582
Hamilton D. S., Perron M. M. G., Bond T. C., Bowie A. R., Buchholz R. R., Guieu C., et al. (2022). Earth, wind, fire, and pollution: aerosol nutrient sources and impacts on ocean biogeochemistry. Annu. Rev. Mar. Sci. 14, 303–330. doi: 10.1146/annurev-marine-031921-013612
Heimburger A., Losno R., Triquet S. (2013a). Solubility of iron and other trace elements in rainwater collected on the Kerguelen Islands (South Indian Ocean). Biogeosciences 10, 6617–6628. doi: 10.5194/bg-10-6617-2013
Heimburger A., Losno R., Triquet S., Dulac F., Mahowald N. (2012). Direct measurements of atmospheric iron, cobalt, and aluminum-derived dust deposition at Kerguelen Islands: DUST DEPOSITION AT KERGUELEN ISLANDS. Glob. Biogeochem. Cycles 26 (4). doi: 10.1029/2012GB004301
Heimburger A., Losno R., Triquet S., Nguyen E. B. (2013b). Atmospheric deposition fluxes of 26 elements over the Southern Indian Ocean: Time series on Kerguelen and Crozet Islands. Glob. Biogeochem. Cycles 27, 440–449. doi: 10.1002/gbc.20043
Ingall E. D., Diaz J. M., Longo A. F., Oakes M., Finney L., Vogt S., et al. (2013). Role of biogenic silica in the removal of iron from the Antarctic seas. Nat. Commun. 4, 1981. doi: 10.1038/ncomms2981
Ito A., Ye Y., Baldo C., Shi Z. (2021). Ocean fertilization by pyrogenic aerosol iron. NPJ Clim. Atmospheric Sci. 4, 30. doi: 10.1038/s41612-021-00185-8
Jickells T. D., An Z. S., Andersen K. K., Baker A. R., Bergametti G., Brooks N., et al. (2005). Global iron connections between desert dust, ocean biogeochemistry, and climate. Science 308, 67–71. doi: 10.1126/science.1105959
Journet E., Desboeufs K. V., Caquineau S., Colin J.-L. (2008). Mineralogy as a critical factor of dust iron solubility. Geophys. Res. Lett. 35 (7). doi: 10.1029/2007GL031589
Kappler A., Newman D. K. (2004). Formation of Fe(III)-minerals by Fe(II)-oxidizing photoautotrophic bacteria. Geochim. Cosmochim. Acta 68, 1217–1226. doi: 10.1016/j.gca.2003.09.006
Koffman B. G., Yoder M. F., Methven T., Hanschka L., Sears H. B., Saylor P. L., et al. (2021). Glacial dust surpasses both volcanic ash and desert dust in its iron fertilization potential. Glob. Biogeochem. Cycles 35 (4), e2020GB006821. doi: 10.1029/2020GB006821
Laity J. (2008). Deserts and desert environments, Environmental systems and global change series (Chichester, UK ; Hoboken, NJ: Wiley-Blackwell).
Li F., Ginoux P., Ramaswamy V. (2008). Distribution, transport, and deposition of mineral dust in the Southern Ocean and Antarctica: Contribution of major sources. J. Geophys. Res. 113, D10207. doi: 10.1029/2007JD009190
Liu X., Millero F. J. (2002). The solubility of iron in seawater. Mar. Chem. 77, 43–54. doi: 10.1016/S0304-4203(01)00074-3
Liu Y., King H., van Huis M., Drury M., Plümper O. (2016). Nano-tomography of porous geological materials using focused ion beam-scanning electron microscopy. Minerals 6, 104. doi: 10.3390/min6040104
Lohan M. C., Tagliabue A. (2018). Oceanic micronutrients: trace metals that are essential for marine life. Elements 14, 385–390. doi: 10.2138/gselements.14.6.385
Longo A. F., Feng Y., Lai B., Landing W. M., Shelley R. U., Nenes A., et al. (2016). Influence of atmospheric processes on the solubility and composition of iron in Saharan dust. Environ. Sci. Technol. 50, 6912–6920. doi: 10.1021/acs.est.6b02605
Mahowald N. M., Baker A. R., Bergametti G., Brooks N., Duce R. A., Jickells T. D., et al. (2005). Atmospheric global dust cycle and iron inputs to the ocean: ATMOSPHERIC IRON DEPOSITION. Glob. Biogeochem. Cycles 19 (4). doi: 10.1029/2004GB002402
Mahowald N. M., Kloster S., Engelstaedter S., Moore J. K., Mukhopadhyay S., McConnell J. R., et al. (2010). Observed 20th century desert dust variability: impact on climate and biogeochemistry. Atmospheric Chem. Phys. 10, 10875–10893. doi: 10.5194/acp-10-10875-2010
Martin J. H. (1990). Glacial-interglacial CO2 change the iron hypothesis. Paleoceanography 5, 13. doi: 10.1029/PA005i001p00001
Martínez-Garcia A., Rosell-Melé A., Jaccard S. L., Geibert W., Sigman D. M., Haug G. H. (2011). Southern Ocean dust–climate coupling over the past four million years. Nature 476, 312–315. doi: 10.1038/nature10310
Mayhew L. E., Webb S. M., Templeton A. S. (2011). Microscale Imaging and Identification of Fe Speciation and Distribution during Fluid–Mineral Reactions under Highly Reducing Conditions. Environ. Sci. Technol. 45, 4468–4474. doi: 10.1021/es104292n
McNulty I., Lai B., Maser J., Paterson D. J., Evans P., Heald S. M., et al. (2003). X-ray microscopy at the advanced photon source. Synchrotron Radiat. News 16, 34–42. doi: 10.1080/08940880308603031
Mendez J., Guieu C., Adkins J. (2010). Atmospheric input of manganese and iron to the ocean: Seawater dissolution experiments with Saharan and North American dusts. Mar. Chem. 120, 34–43. doi: 10.1016/j.marchem.2008.08.006
Meskhidze N., Völker C., Al-Abadleh H. A., Barbeau K., Bressac M., Buck C., et al. (2019). Perspective on identifying and characterizing the processes controlling iron speciation and residence time at the atmosphere-ocean interface. Mar. Chem. 217, 103704. doi: 10.1016/j.marchem.2019.103704
Miller R. L., Knippertz P., Pérez García-Pando C., Perlwitz J. P., Tegen I. (2014). “Impact of Dust Radiative Forcing upon Climate,” in Mineral Dust. Eds. Knippertz P., Stuut J.-B. W. (Springer, Netherlands, Dordrecht), 327–357. doi: 10.1007/978-94-017-8978-3_13
Millero F. J. (1998). Solubility of Fe(III) in seawater. Earth Planet. Sci. Lett. 154, 323–329. doi: 10.1016/S0012-821X(97)00179-9
Molinaroli E. (1996). “Mineralogical Characterisation of Saharan Dust with a View to its Final Destination in Mediterranean Sediments,” in The Impact of Desert Dust Across the Mediterranean, Environmental Science and Technology Library. Eds. Guerzoni S., Chester R. (Springer, Netherlands, Dordrecht), 153–162. doi: 10.1007/978-94-017-3354-0_14
Naik S., Noronha R. J., Shirodkar P. V., Gupta R. S. (2006). On the distribution of calcium, magnesium, sulphate and boron in the southwestern Indian Ocean region of the Southern Ocean. Sci. Rep. 1983, 87–94.
Nowak S., Lafon S., Caquineau S., Journet E., Laurent B. (2018). Quantitative study of the mineralogical composition of mineral dust aerosols by X-ray diffraction. Talanta 186, 133–139. doi: 10.1016/j.talanta.2018.03.059
Oakes M., Ingall E. D., Lai B., Shafer M. M., Hays M. D., Liu Z. G., et al. (2012). Iron solubility related to particle sulfur content in source emission and ambient fine particles. Environ. Sci. Technol. 46, 6637–6644. doi: 10.1021/es300701c
Omand M. M., Govindarajan R., He J., Mahadevan A. (2020). Sinking flux of particulate organic matter in the oceans: Sensitivity to particle characteristics. Sci. Rep. 10, 5582. doi: 10.1038/s41598-020-60424-5
Paparazzo F., Crespi-Abril A., Gonçalves R., Barbieri E., Gracia Villalobos L., Solís M., et al. (2018). Patagonian dust as a source of macronutrients in the Southwest Atlantic Ocean. Oceanography 31, 33–39. doi: 10.5670/oceanog.2018.408
Planquette H., Sherrell R. M. (2012). Sampling for particulate trace element determination using water sampling bottles: methodology and comparison to in situ pumps: Particulate trace element sampling. Limnol. Oceanogr. Methods 10, 367–388. doi: 10.4319/lom.2012.10.367
Pörtner H.-O., Roberts D. C., Tignor M., Poloczanska E. S., Mintenbeck K., Alegría A., et al. (2022). IPCC 2022: Climate Change 2022: Impacts, Adaptation and Vulnerability (Camb. UK N. Y. NY USA: Camb. Univ. Press Camb. Univ. Press). doi: 10.1017/9781009325844
Poulton S. W., Krom M. D., Raiswell R. (2004). A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim. Cosmochim. Acta 68, 3703–3715. doi: 10.1016/j.gca.2004.03.012
Prospero J. M., Ginoux P., Torres O., Nicholson S. E., Gill T. E. (2002). Environmental characterization of global sources of atmospheric soil dust identified with the Nimbus 7 total ozone mapping spectrometer (TOMS) absorbing aerosol product. Rev. Geophys. 40, 2–1-2-31. doi: 10.1029/2000RG000095
Qu Z. (2016). Chemical properties of continental aerosol transported over the Southern Ocean: Patagonian and Namibian sources. Université Pierre et Marie Curie, Paris.
Qu Z., Trabelsi A., Losno R., Monna F., Nowak S., Masmoudi M., et al. (2020). A laboratory dust generator applying vibration to soil sample: mineralogical study and compositional analyses. J. Geophys. Res. Atmospheres 125, e2019JD032224. doi: 10.1029/2019JD032224
Raiswell R. (2011). Iceberg-hosted nanoparticulate Fe in the Southern Ocean: Mineralogy, origin, dissolution kinetics and source of bioavailable Fe. Deep Sea Res. Part II Top. Stud. Oceanogr. 58, 1364–1375. doi: 10.1016/j.dsr2.2010.11.011
Ramseyer K., Boles J. R., Lichtner P. C. (1992). Mechanism of plagioclase albitization. J. Sediment. Res. 62, 349–356. doi: 10.1306/D42678FC-2B26-11D7-8648000102C1865D
Ravel B., Newville M. (2005). ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541. doi: 10.1107/S0909049505012719
Raven J. A., Falkowski P. G. (1999). Oceanic sinks for atmospheric CO2. Plant Cell Environ. 22, 741–755. doi: 10.1046/j.1365-3040.1999.00419.x
Reinl K. L., Harris T. D., Elfferich I., Coker A., Zhan Q., De Senerpont Domis L. N., et al. (2022). The role of organic nutrients in structuring freshwater phytoplankton communities in a rapidly changing world. Water Res. 219, 118573. doi: 10.1016/j.watres.2022.118573
Riebesell U., Fabry V. J., Hansson L., Gattuso J.-P. (2011). Guide to best practices for ocean acidification research and data reporting (Luxembourg: Publications Office of the European Union).
Roden E. E. (2003). Fe(III) oxide reactivity toward biological versus chemical reduction. Environ. Sci. Technol. 37, 1319–1324. doi: 10.1021/es026038o
Rubasinghege G., Lentz R. W., Scherer M. M., Grassian V. H. (2010). Simulated atmospheric processing of iron oxyhydroxide minerals at low pH: Roles of particle size and acid anion in iron dissolution. Proc. Natl. Acad. Sci. 107, 6628–6633. doi: 10.1073/pnas.0910809107
Ruggiero M. T., Bardon T., Strlič M., Taday P. F., Korter T. M. (2015). The role of terahertz polariton absorption in the characterization of crystalline iron sulfate hydrates. Phys. Chem. Chem. Phys. 17, 9326–9334. doi: 10.1039/C5CP01195G
Schroth A. W., Crusius J., Sholkovitz E. R., Bostick B. C. (2009). Iron solubility driven by speciation in dust sources to the ocean. Nat. Geosci. 2, 337–340. doi: 10.1038/ngeo501
Schulz M., Prospero J. M., Baker A. R., Dentener F., Ickes L., Liss P. S., et al. (2012). Atmospheric transport and deposition of mineral dust to the ocean: implications for research needs. Environ. Sci. Technol. 46, 10390–10404. doi: 10.1021/es300073u
Shao Y. (2008). Physics and modelling of wind erosion, Atmospheric and oceanographic sciences library (Cambridge: Springer). doi: 10.1007/978-1-4020-8895-7
Shi Z., Krom M. D., Jickells T. D., Bonneville S., Carslaw K. S., Mihalopoulos N., et al. (2012). Impacts on iron solubility in the mineral dust by processes in the source region and the atmosphere: A review. Aeolian Res. 5, 21–42. doi: 10.1016/j.aeolia.2012.03.001
Shi C., Mao R., Gong D.-Y., Kim S.-J., Feng X., Sun Y., et al. (2023). Increased dust transport from Patagonia to eastern Antarctica during 2000–2020. Glob. Planet. Change 227, 104186. doi: 10.1016/j.gloplacha.2023.104186
Shi Z. B., Woodhouse M. T., Carslaw K. S., Krom M. D., Mann G. W., Baker A. R., et al. (2011). Minor effect of physical size sorting on iron solubility of transported mineral dust. Atmospheric Chem. Phys. 11, 8459–8469. doi: 10.5194/acp-11-8459-2011
Shoenfelt E. M., Winckler G., Lamy F., Anderson R. F., Bostick B. C. (2018). Highly bioavailable dust-borne iron delivered to the Southern Ocean during glacial periods. Proc. Natl. Acad. Sci. 115, 11180–11185. doi: 10.1073/pnas.1809755115
Sow M., Alfaro S. C., Rajot J. L., Marticorena B. (2009). Size resolved dust emission fluxes measured in Niger during 3 dust storms of the AMMA experiment. Atmospheric Chem. Phys. 9, 3881–3891. doi: 10.5194/acp-9-3881-2009
Spokes L. J., Jickells T. D. (1996). Factors controlling the solubility of aerosol trace metals in the atmosphere and on mixing into seawater. Aquat. Geochem. 1, 355–374. doi: 10.1007/BF00702739
Spolaor A., Vallelonga P., Cozzi G., Gabrieli J., Varin C., Kehrwald N., et al. (2013). Iron speciation in aerosol dust influences iron bioavailability over glacial-interglacial timescales. Geophys. Res. Lett. 40, 1618–1623. doi: 10.1002/grl.50296
Spolnik Z., Tsuji K., Van Grieken R. (2004). Grazing-exit electron probe x-ray microanalysis of light elements in particles. X-Ray Spectrom. 33, 16–20. doi: 10.1002/xrs.656
Takahama S., Gilardoni S., Russell L. M. (2008). Single-particle oxidation state and morphology of atmospheric iron aerosols. J. Geophys. Res. 113, D22202. doi: 10.1029/2008JD009810
Thuróczy C.-E., Boye M., Losno R. (2010). Dissolution of cobalt and zinc from natural and anthropogenic dusts in seawater. Biogeosciences 7, 1927–1936. doi: 10.5194/bg-7-1927-2010
Toner B. M., Berquó T. S., Michel F. M., Sorensen J. V., Templeton A. S., Edwards K. J. (2012). Mineralogy of iron microbial mats from Loihi seamount. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00118
Toner B. M., German C. R., Dick G. J., Breier J. A. (2016). Deciphering the complex chemistry of deep-ocean particles using complementary synchrotron X-ray microscope and microprobe instruments. Acc. Chem. Res. 49, 128–137. doi: 10.1021/acs.accounts.5b00282
Totsche K. U., Amelung W., Gerzabek M. H., Guggenberger G., Klumpp E., Knief C., et al. (2018). Microaggregates in soils. J. Plant Nutr. Soil Sci. 181, 104–136. doi: 10.1002/jpln.201600451
Tréguer P., Bowler C., Moriceau B., Dutkiewicz S., Gehlen M., Aumont O., et al. (2018). Influence of diatom diversity on the ocean biological carbon pump. Nat. Geosci. 11, 27–37. doi: 10.1038/s41561-017-0028-x
Trimborn S., Brenneis T., Hoppe C., Laglera L., Norman L., Santos-Echeandía J., et al. (2017). Iron sources alter the response of Southern Ocean phytoplankton to ocean acidification. Mar. Ecol. Prog. Ser. 578, 35–50. doi: 10.3354/meps12250
van der Jagt H., Friese C. A., Stuut J.-B. W., Fischer G., Iversen M. H. (2018). Ballasting effects of Saharan dust on the aggregate dynamics in the upwelling region off Cape Blanc (Mauritania) 3 datasets. Limnol. Oceanogr. 63, 1386–1394. doi: 10.1594/PANGAEA.885930
Vogt S. (2003). MAPS : A set of software tools for analysis and visualization of 3D X-ray fluorescence data sets. J. Phys. IV Proc. 104, 635–638. doi: 10.1051/jp4:20030160
Wagener T., Guieu C., Leblond N. (2010). Effects of dust deposition on iron cycle in the surface Mediterranean Sea: results from a mesocosm seeding experiment. Biogeosciences 7, 3769–3781. doi: 10.5194/bg-7-3769-2010
Wagener T., Pulido-Villena E., Guieu C. (2008). Dust iron dissolution in seawater: Results from a one-year time-series in the Mediterranean Sea. Geophys. Res. Lett. 35, L16601. doi: 10.1029/2008GL034581
Wagner F., Bortoli D., Pereira S., Costa M. J., Silva A. M., Weinzierl B., et al. (2009). Properties of dust aerosol particles transported to Portugal from the Sahara desert. Tellus B Chem. Phys. Meteorol. 61, 297. doi: 10.1111/j.1600-0889.2008.00393.x
Wedepohl K. H. (1995). The composition of the continental crust. Geochim. Cosmochim. Acta 59, 1217–1232. doi: 10.1016/0016-7037(95)00038-2
Wells M. L., Goldberg E. D. (1993). Colloid aggregation in seawater. Mar. Chem. 41, 353–358. doi: 10.1016/0304-4203(93)90267-R
Winterwerp J. C., van Kesteren W. G. M. (2004). Introduction to the physics of cohesive sediment in the marine environment (Amsterdam ; Boston: Elsevier). doi: 10.1016/S0070-4571(04)80004-9
Wuttig K., Wagener T., Bressac M., Dammshäuser A., Streu P., Guieu C., et al. (2013). Impacts of dust deposition on dissolved trace metal concentrations (Mn, Al and Fe) during a mesocosm experiment. Biogeosciences 10, 2583–2600. doi: 10.5194/bg-10-2583-2013
Keywords: Patagonian dust, iron, dissolution, Southern Ocean, anthropogenic changes
Citation: Demasy C, Boye M, Lai B, Burckel P, Feng Y, Losno R, Borensztajn S and Besson P (2024) Iron dissolution from Patagonian dust in the Southern Ocean: under present and future conditions. Front. Mar. Sci. 11:1363088. doi: 10.3389/fmars.2024.1363088
Received: 29 December 2023; Accepted: 12 April 2024;
Published: 26 April 2024.
Edited by:
Cecile Guieu, UMR7093 Laboratoire d’océanographie de Villefranche (LOV), FranceReviewed by:
Kathrin Wuttig, University of Tasmania, AustraliaAshwini Kumar, Council of Scientific and Industrial Research (CSIR), India
Copyright © 2024 Demasy, Boye, Lai, Burckel, Feng, Losno, Borensztajn and Besson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clément Demasy, demasy@sun.ac.za