The mussel larvae microbiome changes in response to a temperature rise
 You-Ting Zhu1,2†
You-Ting Zhu1,2†  Xiao Liang1,2†
Xiao Liang1,2†  Tian-Tian Liu1,2† Deborah M. Power1,3
Tian-Tian Liu1,2† Deborah M. Power1,3  Yi-Feng Li1,2*
Yi-Feng Li1,2*  Jin-Long Yang1,2*
Jin-Long Yang1,2*- 1International Research Center for Marine Biosciences, Ministry of Science and Technology, Shanghai Ocean University, Shanghai, China
- 2China-Portugal Belt and Road Joint Laboratory on Space & Sea Technology Advanced Research, Shanghai, China
- 3Centro de Ciências do Mar (CCMAR), Comparative Endocrinology and Integrative Biology, Universidade do Algarve, Faro, Portugal
Ocean warming caused by global climate change influences the function, diversity, and community dynamics of commensal microorganisms, including the hemolymph and the gut microbiota in mussels. However, the microbiota in hard-shelled mussel (Mytilus coruscus) larvae and the effect of temperature on the microbial community structure have yet to be studied. Herein, we investigated the core microbiota of M. coruscus larvae and the impact of acute (4 h) and gradual (4 days) exposure to a rise in seawater temperature from 21 to 25 °C. Eleven core genera were identified in M. coruscus larvae by 16S rDNA gene sequencing: Alteromonas, Brevundimonas, Delftia, Microbacterium, Neptuniibacter, Neptunomonas, Pseudoalteromonas, Rhodococcus, Stenotrophomonas, Tenacibaculum, and Thalassotalea. The microbiota of larvae in the short exposure treatment was similar to the control. However, the abundance of Delftia, Neptunomonas, Pseudoalteromonadaceae, Rhodococcus, and Stenotrophomonas decreased significantly in the long-exposure larvae. In contrast, at the genus level, the abundance of Tenacibaculum increased significantly. Diversity and multivariate analyses confirmed that the microbiota patterns were linked to seawater warming over the long term. Microbiota diversity did not change significantly, regardless of whether the seawater temperature increased quickly or slowly; however, we observed a significant increase in the microbiota species abundance at higher temperatures. Among the altered bacterial genera, Delftia, Neptunomonas, and Rhodococcus function in the degradation of organic compounds; Pseudoalteromonas is closely associated with mussel attachment and metamorphosis, and Tenacibaculum is an opportunistic pathogen that can cause marine mollusk death. The results suggest that marine heat waves caused by climate change may reduce the ability of symbiotic bacteria to degrade environmental toxins, will affect mussel larvae metamorphosis, and increase the abundance of opportunistic pathogens, thereby increasing the risk of disease and death of mussel larvae.
1 Introduction
Multicellular life originated in a microorganism-dominated environment, and interactions between animals and their microbiomes have markedly influenced animal evolution (Nyholm and Graf, 2012). The most studied microbial community, the animal gut microbiome, performs various vital functions, including immune response stimulation, defense against pathogen colonization, essential nutrient provision, and digestive support (Nyholm and Graf, 2012; McFall-Ngai et al., 2013). Research has shown that the composition of host-microbial communities is dictated chiefly by the phylogeny of the host and their environment. Within an individual host, microbes might be unevenly distributed (Cárdenas et al., 2014; Pantos et al., 2015; Brooks et al., 2016; Carrier and Reitzel, 2018). Moreover, environmental factors can change the microbiota composition, significantly affecting the host’s development, homeostasis, survival, and metabolism (Tremaroli and Bäckhed, 2012; McFall-Ngai et al., 2013).
Recent advances in sequencing technology have enabled the investigation of microbiota from marine invertebrates, such as mollusks, corals, sponges, tunicates, echinoderms, polychaetes, and crustaceans (Gobet et al., 2018; Dubé et al., 2019). Such research has revealed that marine invertebrates are hosts to varied core microbial communities that are phylogenetically different from the microbes in the surrounding aquatic environment, and such microbial associations are often host-specific, irrespective of geography (Moitinho-Silva et al., 2014; Reveillaud et al., 2014; Lokmer et al., 2016a; Zhang et al., 2016; Brener-Raffalli et al., 2018). Several biological factors, such as host physiology and genetics (Jaenike, 2012; Wegner et al., 2013; Amato et al., 2019), host health (Cárdenas et al., 2012; Sweet and Bulling, 2017), diet (Carrier et al., 2018), life stage (Trabal et al., 2012; Lema et al., 2014), host-tissue differentiation (Meisterhans et al., 2016; Høj et al., 2018; Dubé et al., 2019), stressors (e.g., antibiotic stress, infection, temperature, starvation, and translocation) (Green and Barnes, 2010; Wegner et al., 2013; Lokmer and Wegner, 2015; Lokmer et al., 2016a, b), and season (Pierce et al., 2016; Gobet et al., 2018) can also contribute to specific variation in microbial communities or shift the microbiome.
In marine ecosystems, microbial community dynamics, function, and diversity are affected by global climate change (Webster et al., 2007), causing shifts in host-microbe interactions which could result in a shift toward pathogen-dominated microbes rather than beneficial bacteria (Lafferty et al., 2004). Temperature changes can influence the composition of hemolymph and gut microbiomes of many invertebrates (Olafsen et al., 1993; Lokmer and Wegner, 2015; Li et al., 2018), and exceeding a species’ thermal tolerance limit is likely to have specific adverse effects on bivalves (Xiao et al., 2005; Li et al., 2018; Li et al., 2019a, b). For example, adult mussels (M. coruscus and Mytilus galloprovincialis) microbiomes and susceptibility to bacterial infections were modified in response to increased water temperature (Li et al., 2018).
In bivalves, the larval microbiota comprises mainly bacteria acquired from the surrounding seawater, with the prevalence of certain groups of bacteria, such as Vibrionaceae, which are presumed to multiply within the gut (Prieur, 1976), and the larval microbiota might be more susceptible to changes in the environment. However, adult mollusks can retain their core microbial communities by selective digestion and multiplication of the bacteria from surrounding seawater (Kueh and Chan, 1985).
The hard-shelled mussel (M. coruscus) is a commercially important species in the Eastern China Sea, and its range covers the coastal waters of the East Asia temperate zones. Our previous study showed that high water temperatures promoted the growth of opportunistic bacteria, such as Arcobacter and Bacteroides, in the gut of M. coruscus, which suggests increased host susceptibility to pathogens at higher temperatures (Li et al., 2018). In addition, increased temperatures reduced the M. coruscus hemolymph microbiome diversity, which could reduce the bacterial elimination function against Vibrio infection in the hemolymph (Li et al., 2019a). However, the life cycle of M. coruscus comprises planktonic larval and benthic adult phases, and the effects of temperature on the larval microbial community are little understood.
The present study investigated the effect of increasing seawater temperature from the perspective of a short-term increase typical of marine heat waves (Field et al., 2014) on the M. coruscus larval microbiota structure and diversity. 16S rRNA gene sequencing was used to provide insight into the likely effects of global warming on the survival of marine bivalve larvae.
2 Materials and methods
2.1 Biological material
Adult M. coruscus were acquired from Gouqi Island, China (30°72’N, 122°77’E) and transported to the laboratory on the same day. The mussels were cleaned and placed in a 10 L polycarbonate tank filled with seawater filtered through a 1.2 μm pore size acetate-fiber membrane (FSW). The water temperature was maintained at 21°C to reflect the collection site’s average winter temperature. The mussels received a daily mixed diet comprising Isochrysis zhanjiangensis and Platymonas helgolandica.
Mussel spawning was induced using a previously described method (Yang et al., 2008) with modifications. In brief, after overnight storage on ice, mussels were placed in 10 L polycarbonate tanks at 21°C until spawning began, when they were transferred immediately to 2 L beakers containing FSW. Egg and sperm FSW suspensions were collected and mixed gently to achieve fertilization, followed by incubation for 20 min. Excess sperm was washed off with FSW using a 20 μm mesh size nylon plankton net, and the fertilized eggs were incubated in a 2 L beaker with FSW at 18°C. Two days later, the swimming larvae were fed daily with 5×104 cells/mL of I. zhanjiangensis and P. helgolandica and cultured at 21°C.
2.2 Experimental setup and sampling collection
The Institutional Animal Care and Use Committee (IACUC) of Shanghai Ocean University approved the animal handling procedures and experimentation.
Pediveliger larvae (35 days post fertilization, larva at this stage can metamorphose into a juvenile) were randomly and evenly distributed in three randomized triplicate groups. The groups were subjected to three temperature treatments terminating at the same time: the larvae were placed directly in seawater at 25°C for 4 hours group Mc25.S); 4 days at 25°C (group Mc25.L), by raising the water one degree a day; and 21°C as control (group Mc21). Four degrees of warming represents the differential temperature rise predicted for 2100. The global mean temperature will have increased by 3.2–4.6 °C (Rogelj et al., 2016), and the increase in seawater temperature over a short time during marine heat waves (Field et al., 2014).
During the trial, the larvae were fed microalgae as usual. At the end of the experiment, 50 µg of larva were combined into a sample (every treatment group has three replicates), frozen, and stored at −80 °C for microbiome analysis.
2.3 Extraction of DNA and PCR amplification
According to the manufacturer’s instructions, a FastDNATM Spin Kit for Soil (MP Biomedicals, Santa Anna, CA, USA) was used to extract genomic DNA from the larval samples. The purity and concentration of the DNA were measured on a Nanodrop One instrument (Thermo Fisher Scientific, Waltham, MA, USA). Sterile, filtered (0.2 μm) water was added to standardize the DNA concentration to 5 ng/μl for all samples.
The universal bacterial primers, 338F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) with a 12 bp barcode (synthesized by Invitrogen, Carlsbad, CA, USA) were used to amplify the bacterial 16S rRNA gene V3-V4 region. The 50 µl PCR reactions comprised 25 μl of 2× Premix Taq (Takara Biotechnology, Dalian, China), one μl of each primer (10 M), three μl of DNA (20 ng/μl) template, and 20 μl of distilled water. The reactions were run on a BioRadS1000 instrument (Bio-Rad, Hercules, CA, USA) with the following thermocycling conditions: initialization for 5 min at 94°C; 30 cycles of 30 s of denaturation at 94°C, 30 s of annealing at 52°C, and 30 s extension at 72°C; followed by a 10 min final elongation at 72°C.
2.4 Illumina HiSeq sequencing
Gel electrophoresis in 1% agarose was used to detect the PCR amplicons. GeneTools (version 4.03.05.0, SynGene, New Delhi, India) was used to assess the density of the PCR bands. The amplicons showing bright leading bands were mixed at equidensity ratios. An EZNA Gel Extraction Kit (Omega Bio-tek, Norcross, GA, USA) was used to purify mixed PCR products. A NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA) was used to generate the sequencing libraries with index codes according to the manufacturer’s protocol. A Qubit@ 2.0 Fluorometer (Thermo Fisher Scientific) and an Agilent Bioanalyzer 2100 system (Agilent Technologies, Waldbron, Germany) were used to assess the quality of the libraries. The Illumina Hiseq 2500 platform (Illumina Inc., San Diego, CA, USA) was used to sequence the libraries, generating 250 bp paired-end reads. The sequencing was carried out by Guangdong Magigene Biotechnology Co. Ltd (Guangzhou, China).
2.5 Bioinformatics and statistics
The Trimmomatic V0.33 (Bolger et al., 2014) was used to filter the raw paired-end reads to generate high-quality, clean reads. FLASH V1.2.11 (Magoč and Salzberg, 2011) was used to merge the clean reads dependent on the overlap between the paired-end reads. Mothur V1.35.1 (Kozich et al., 2013) used the unique barcodes and primers to assign the sequences to the samples, then, the barcodes and primers were removed to obtain effective clean tags. USEARCH V10 (Edgar, 2010) was used for sequence analysis. Sequences sharing greater than or equal to 97% similarity were assigned to the same operational taxonomic unit (OTU). Each OTU was represented by the most frequently occurring sequence, which was screened and annotated. We used the SILVA (Quast et al., 2013) database to annotate the taxonomic information for each representative sequence (using the default confidence threshold ≥ 0.5). After removing contaminating OTUs, we determined the total number of OTU sequences and the OTU types, respectively. A normalized (subsampled) OTU table was obtained based on the sample with the fewest sequences. Using the OTU table, we calculated the annotation ratio for each classification level to determine the sequence composition of each sample at each classification level. The OTU table was also used to construct a Venn graph, histogram, and heat map displayed using R software (v2.15.3).
The complexity of species diversity in the samples was represented by the Chao1 richness index and Simpson’s and Shannon diversity indices, calculated using QIIME software (v1.9.1) (Bolyen et al., 2019) and displayed using the R software (v2.15.3). The vegan package of R software was used to carry out non-metric multidimensional scaling (NMDS) based on the weighted and unweighted distance matrix. Linear discriminant analysis Effect Size (LEfSe) was used to find the biomarker of each group. The obtained raw sequences were submitted to the NCBI database and received the accession number PRJNA1029461.
GraphPad Prism 9.0 (GraphPad Software, Boston, MA, USA) and one-way ANOVA tests were used to analyze the larval microbiota’s relative abundance and taxonomical composition at the different levels and Alpha Diversity.
2.6 Ethics statement
The Animal Ethics Committee at Shanghai Ocean University approved all the protocols for mussel acclimation and experimentation in the present study (registration number SHOU-DW-2020-032).
3 Results
3.1 Composition of M. coruscus microbiota under different temperature treatments
The 16S rDNA gene sequencing yielded 1,233,013 sequences clustered into 6,830 distinct OTUs (Supplementary Table S1). The larvae hosted 29 unique phyla, 72 classes, and 198 orders of bacteria. The most abundant phylum in the Mc21 group was Proteobacteria (73.1%), followed by Actinobacteria (21.2%), Bacteroidetes (4.5%), and many other unclassified bacteria (1.2%). The most abundant phyla in the Mc25.S group were Proteobacteria (71.0%), Actinobacteria (23.0%), Bacteroidetes (4.6%), and many other unclassified bacteria (1.4%). The most abundant phyla in the Mc25.L group were Proteobacteria (35.7%), Actinobacteria (3.8%), and Bacteroidetes (59.2%), and many other unclassified bacteria (1.2%) (Supplementary Table S2, Supplementary Figure S1).
3.2 M. coruscus larvae microbiota at family’s level
We identified 18 different families with an abundance > 1%, and 235 families with an abundance < 1% were classified as “others” (Figure 1A). Most reads (16.60–31.53% of the OTUs) were obtained from the Mc21 and Mc25.S groups and were assigned to Burkholderiaceae, Nocardiaceae, and Xanthomonadaceae. In contrast, most reads (5.00–54.55% of total OTUs) obtained from the Mc25.L group were assigned to Burkholderiaceae, Flavobacteriaceae, and Rhodobacteraceae (Figure 1A; Supplementary Table S3). The analysis of the relative abundance showed that at the family level, the microbial community structure between the Mc21 and Mc25.S groups was similar but different from the Mc25.L group. The relative abundance of Burkholderiaceae, Nitrincolaceae, Nocardiaceae, and Xanthomonadaceae decreased significantly, and that of Flavobacteriaceae and Rhodobacteraceae increased significantly in the Mc25.L group compared with those in the Mc21 and Mc25.S (P < 0.05) (Figures 1B, C; Supplementary Table S3). Notably, the abundance of Pseudoalteromonadaceae decreased significantly irrespective of whether the larvae were in warmer seawater for 4 hours or 4 days. (P < 0.05) (Figures 1B, C; Supplementary Table S3).
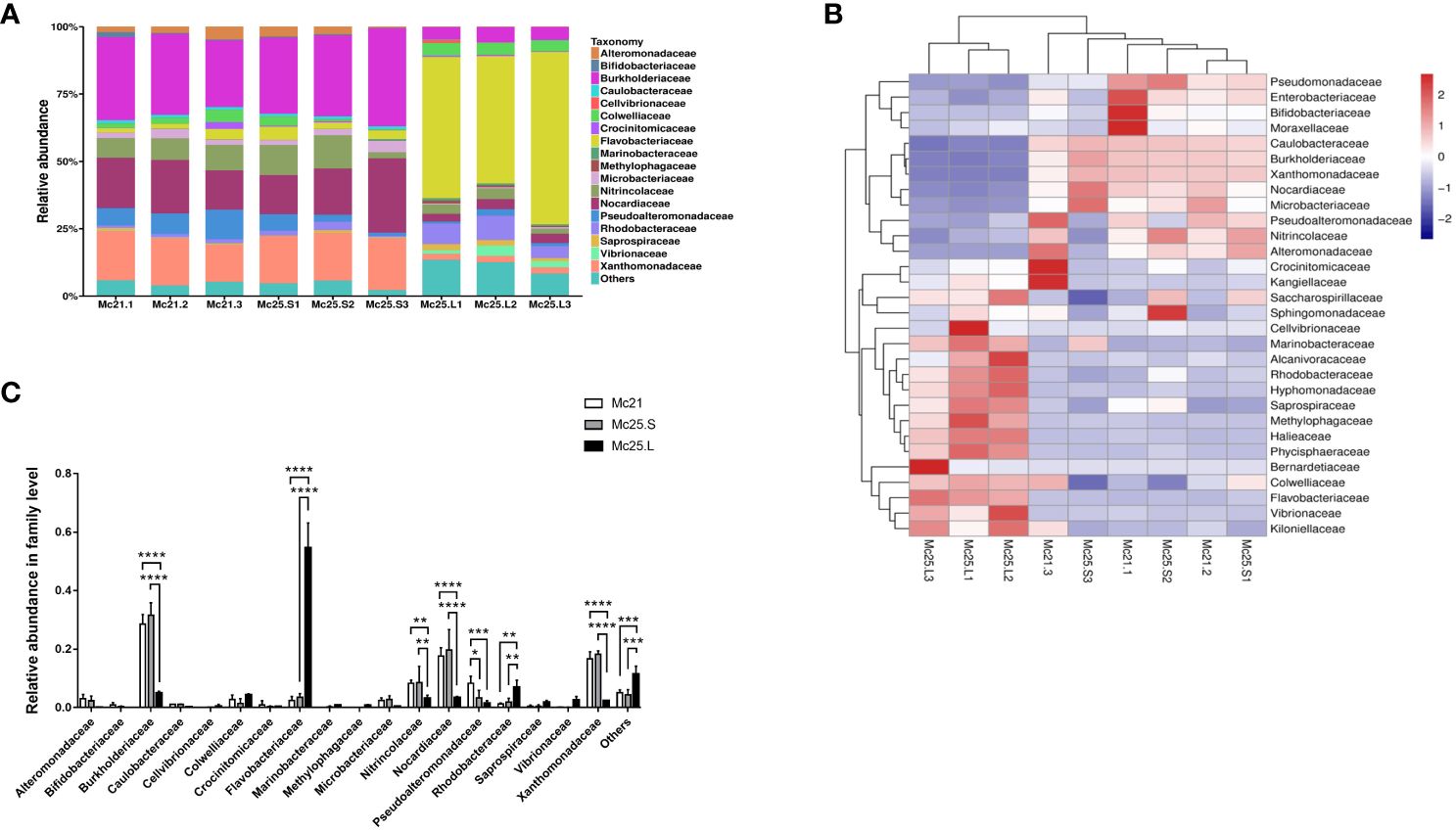
Figure 1 Relative abundance and taxonomical composition of the larval microbiota at family level. The suffix number in the group names indicate individual replicates. (A) Each bar represents the larval bacterial community associated with individual samples. (B) Heatmap showing the top 30 bacterial families (%). Red signifies higher abundance, and blue signifies lower abundance. (C) Comparison of the relative abundance of the 18 main microbial families between the three groups of larvae. P-values mark significant differences in community diversity between the different treatment groups (one-way ANOVA). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.3 M. coruscus larvae microbiota at genera’s level
At the genus level, 21 genera were identified with an abundance > 1%, and 443 genera with an abundance < 1% were classified as “others” (Figure 2A). In the Mc21 and Mc25.S groups, the dominant genera were Alteromonas, Brevundimonas, Delftia, Microbacterium, Neptuniibacter, Neptunomonas, Pseudoalteromonas, Rhodococcus, Stenotrophomonas, Tenacibaculum, and Thalassotalea (average reads per group 1.04–31.40% of the total OTUs) (Figure 2A; Supplementary Table S4). Delftia, Lewinella, Neptuniibacter, Pseudoalteromonas, Rhodococcus, Ruegeria, Stenotrophomonas, Tenacibaculum, Thalassotalea, and Vibrio were the dominant genera in the Mc25.L group (average reads per group 1.09–52.65% of the total OTUs) (Figure 2A; Supplementary Table S4). By comparing the abundance in the different experimental groups, the results showed the similarity of the microbiota between the Mc21 and Mc25.S groups, except for Pseudoalteromonas (P < 0.05) (Figure 2C; Supplementary Table S4). In the Mc25.L group, the larval microbiota had more differences. The abundance of Delftia, Neptunomonas, Pseudoalteromonadaceae, Rhodococcus, and Stenotrophomonas decreased significantly, and Tenacibaculum increased considerably compared with that in the Mc21 and Mc25 groups (P < 0.05) (Figures 2B, C; Supplementary Table S4). These results indicated that rapid warming in a short time does not impact the M. coruscus larval microbial community structure. Conversely, a gradual increase in temperature over four days can change the population levels of certain bacterial species and possibly alter the microbial community composition.
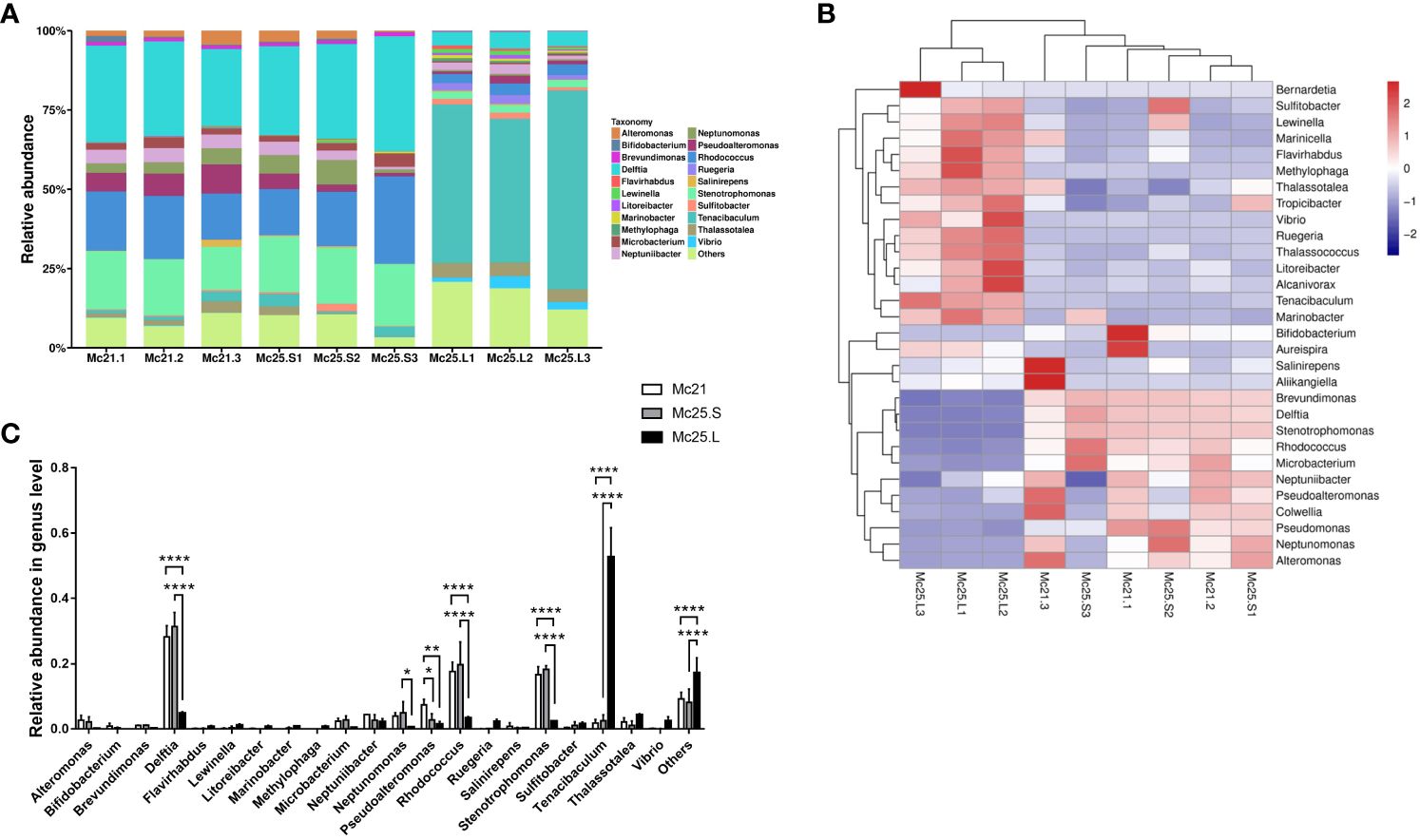
Figure 2 Relative abundance and taxonomical composition of the larval microbiota at the genus level. The suffix number in the group names indicate individual replicates. (A) Each bar represents the larval bacterial community associated with individual samples. (B) Heatmap showing the top 30 bacterial (%). Red signifies higher abundance, and blue signifies lower abundance. (C) Comparison of relative abundance of 21 main microbial genus between the three groups of larvae. P-values mark significant differences in community diversity between different treatment groups (tested using one-way ANOVA). *p < 0.05, **p < 0.01, ****p < 0.0001.
3.4 Composition and temperature-driven fluctuations of the core digestive microbiota
Based on the Venn diagrams, the core microbial communities, i.e., occurring in all treatment groups, included 145 families and 216 genera, representing 57.31% of the total families and 47.68% of the entire genera (Figure 3). Eighteen families and 21 genera with an abundance > 1% belonged to these core families and genera bacterial communities (Supplementary Tables S3, S4). In contrast, unique bacterial communities had an abundance of < 1% in each treatment group (Supplementary Tables S3, S4).
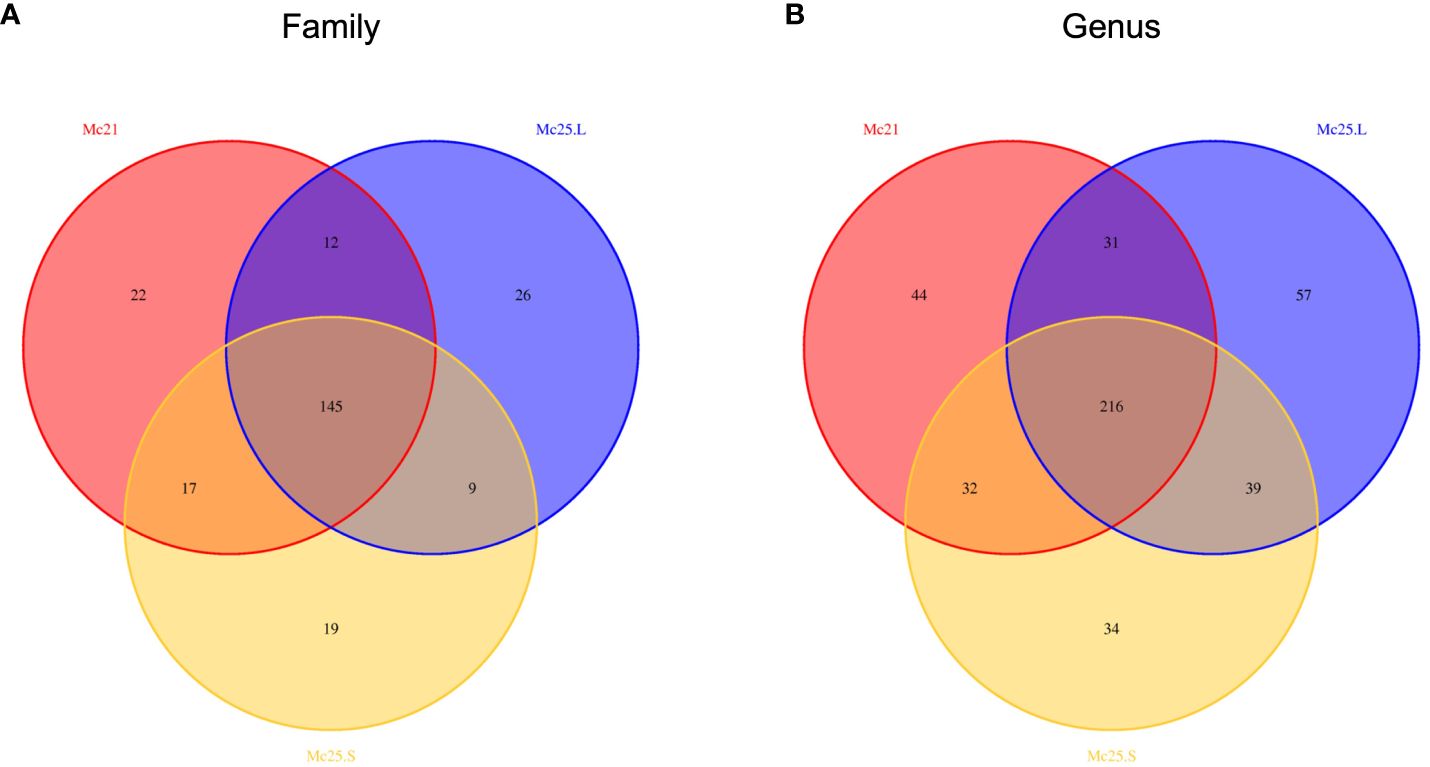
Figure 3 Venn diagrams showing the numbers of microbial OTU composition associated with the three temperature treatment groups in family (A) and genus (B) level. Comparisons are described by numbers of shared or unique OTUs in Mc25.S, Mc25.L, and Mc21 groups.
3.5 Diversity patterns of the microbial community
The microbiota richness and evenness were investigated between the control and the temperature treatment groups. There were no significant differences among the treatment groups in the Shannon and Simpson diversity indices (P > 0.05, Figures 4A, B). However, the Chao1 index of the Mc25.L group was significantly higher than that of the Mc25.S and Mc21 (P < 0.05, Figure 4C), indicating that the gradual increase in temperature caused a significant increment in the microbiota species abundance.
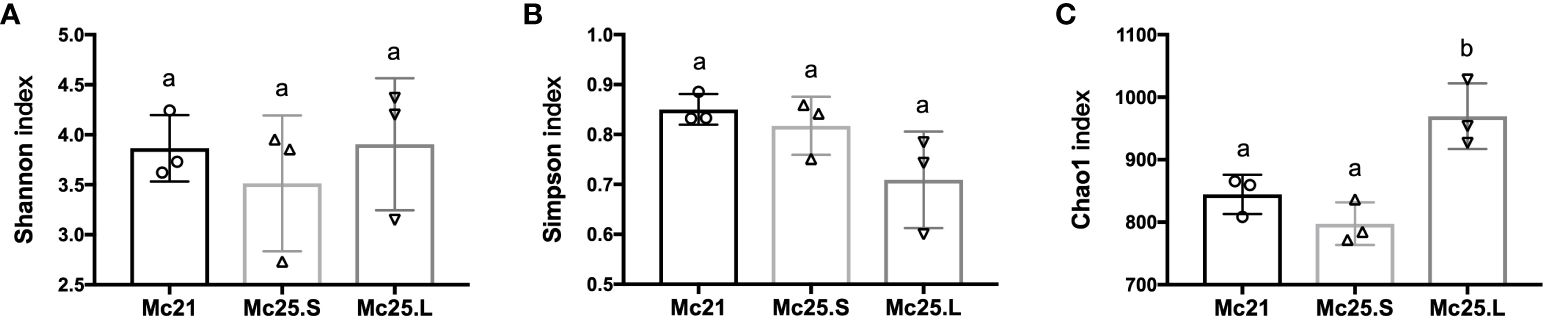
Figure 4 Analysis of microbial diversity. The results of Shannon (A), Simpson (B), and Chao1 (C) indices analysis. Data are presented as the mean ± SE (n = 3). Different low case letters represent significant differences (p < 0.05, one-way ANOVA).
The NMDS analysis was employed as a proxy for temporal stability. It showed that the samples were homogenous in the Mc25.L group, and the larvae microbiota in this group changed compared with the other two groups, which showed some overlap (Figure 5).
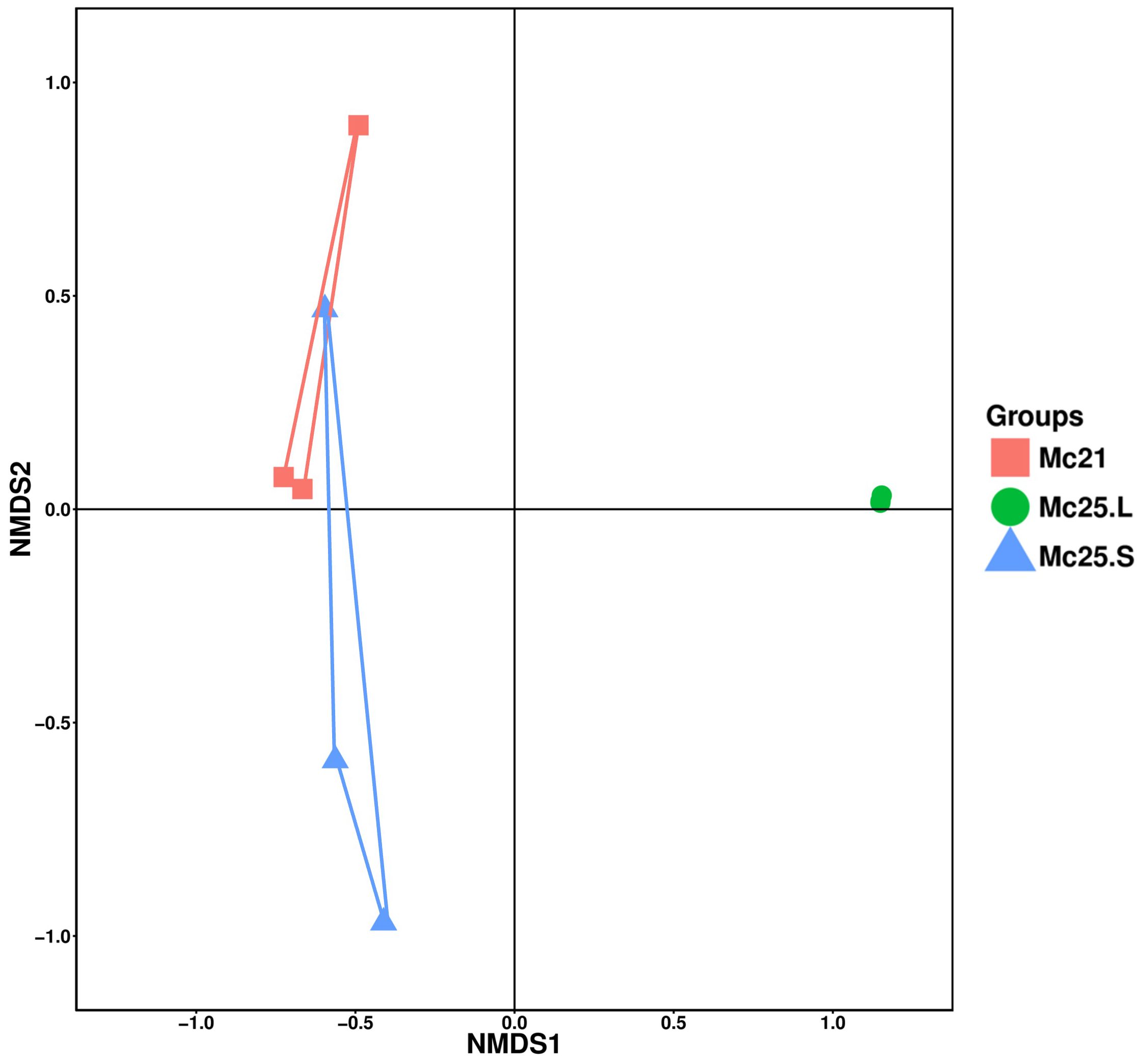
Figure 5 Non-metric multidimensional scaling (NMDS) analysis of the larval bacterial community composition at the OTU-level. Treatment groups are indicated using different colors (Mc21 = red, Mc25.L = green, Mc25.S = blue).
Linear discriminant analysis effect size (LEfSe) analysis was performed to reveal the significant ranking of abundant modules. The linear discriminant analysis (LDA) score and the cladogram showed differences between Mc25.S, Mc25.L, and Mc21 (Figure 6). At the phylum or order level, the biomarkers demonstrating significant differences between the three groups were Caulobacterales in Mc25.S; PB19, Cellvibrionales (genus Halioglobus), Rhodobacterales (genus Litoreibacter and Kiloniella), Caulobacterales (family Hyphomonadaceae), Oceanospirillales (family Alcanivoracaceae), Micavibrionales (family Micavibrionaceae) in Mc25.L; and Alteromonadales (family Pseudoalteromonadaceae and genus Colwellia), Firmicutes (phylum Firmicutes and order Lactobacillales) in Mc21 (Figure 6; Supplementary Table S5).
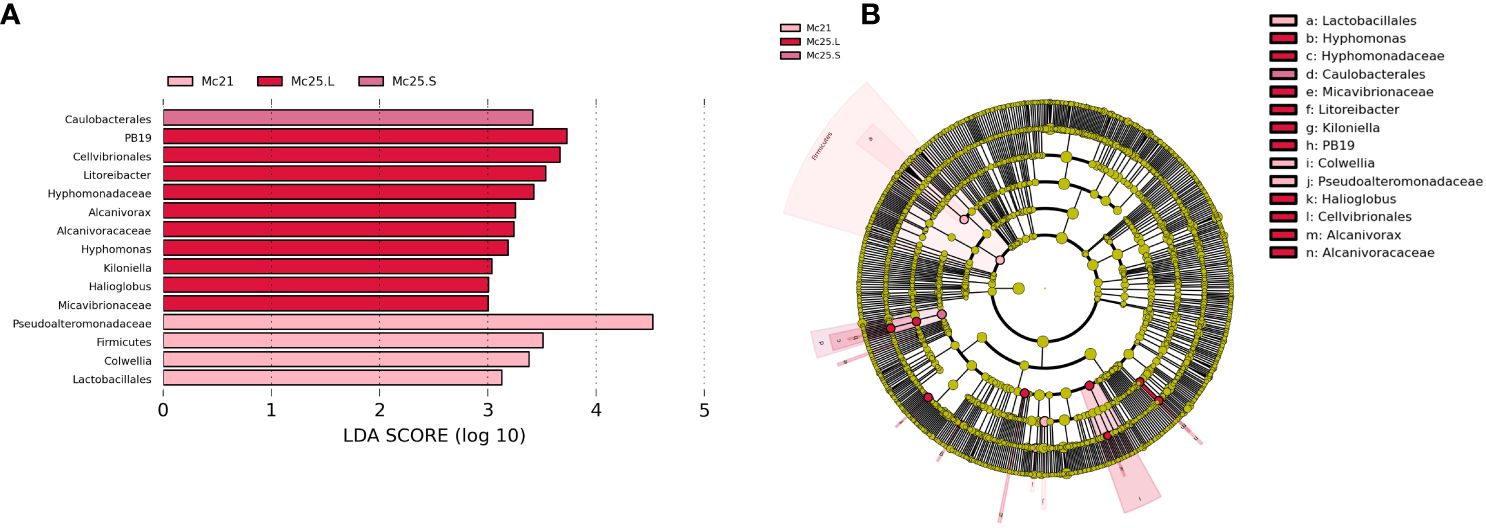
Figure 6 Unique biomarker community composition in larvae. (A) A bar chart of the log-transformed LDA scores for bacterial taxa identified using LEfSe analysis. The threshold used to identify significant taxa was as log-transformed LDA score of 2. LEfSe analysis identified 15 taxa (shown). (B) The phylogenetic relationships for the 15 LEfSe-identified bacterial taxa shown in a cladogram.
4 Discussion
Marine invertebrates host diverse core microbial communities, and we show that this is also the case with the hard-shell mussel larva. However, the microbiome is sensitive to a rise in temperature from 21 to 25°C over 4 days, although it is little affected if the rise is only over 4 hours. In the gradual warming group, temperature impacts some Phyla’s abundance and the community’s richness (Chao1 index). Still, it does not affect the diversity richness and evenness (Shannon and Simpson indices). The little impact over 4 hours may reflect that bacterial growth rates may not be sufficient to make a difference in the number of bacteria in the population.
Marine invertebrates generally have complex life cycles with larval stages, and each of these developmental stages owns different microbiotas, although they also share core microbes (Trabal et al., 2012). The microbiome of the M. coruscus pediveliger in the present study was dominated by Proteobacteria, Actinobacteria, and Bacteroidetes. Adult M. coruscus had fewer than 14 dominant phyla (abundance > 1%), while there were 9 dominant phyla in adult M. galloprovincialis (abundance > 1%), suggesting that the microbiota diversity increases with age (Li et al., 2019a, b). This could be because adult mussels are exposed to environmental microbes over a longer time, and that their more complex bodies offer more niches than larvae for bacteria to colonize. However, there were few dominant phyla in adults of yesso scallop (Patinopecten yessoensis) from Northern China (four phyla) (Lu et al., 2017), sponges Stylissa carteri and Xestospongia testudinaria from the Red Sea (three phyla) (Moitinho-Silva et al., 2014), and Chinese mitten crab (Eriocheir sinensis) from East China (four phyla) (Zhang et al., 2016), which could be indicative of a possible role of the genotype (Wegner et al., 2013), condition (Lokmer and Wegner, 2015), and of neutral processes (Nemergut et al., 2013) in shaping the host microbiome. In addition, at lower taxonomic levels, the untreated pediveliger larvae contained eleven main microbial genera, including Alteromonas, Brevundimonas, Delftia, Microbacterium, Neptuniibacter, Neptunomonas, Pseudoalteromonas, Rhodococcus, Stenotrophomonas, Tenacibaculum, and Thalassotalea. Pseudoalteromonas and Tenacibaculum were the dominant genera in adult M. coruscus, suggesting that these are core bacterial genera throughout this mussel’s life cycle.
In marine poikilotherms, it was proposed that temperature is an important factor that shapes microbial communities by affecting the host and their symbiotic bacteria. Higher temperatures promote the growth of better-adapted microbes and compromise the host defense, leading to a rapid increase in opportunistic pathogens (Malham et al., 2009; Lokmer and Wegner, 2015). In oysters, warmer temperatures led to a higher mortality rate when infected with Vibrio sp., and among the dominant microbiota, the genus Arcobacter, a hemolymph-specific symbiont, decreased in abundance (Lokmer and Wegner, 2015). In contrast, infected oysters harbored more pathogen-containing genera, e.g., Photobacterium and Shewanella (Lokmer and Wegner, 2015). Our previous studies on adult M. coruscus revealed a decreasing abundance of hemolymph Arcobacter when reared in warmer water. In contrast, the abundance of gut Bacteroides, an opportunistic pathogen, increased (Li et al., 2007; Patrick et al., 2009; Li et al., 2018; Li et al., 2019a). The present study is consistent with those observations as indicated by an increase in species richness with higher temperature exposure and a simultaneous decrease in the abundance of the dominant bacteria Delftia, Neptunomonas, Pseudoalteromonadaceae, Rhodococcus, and Stenotrophomonas. The abundance of Tenacibaculum also increased significantly, suggesting that warmer temperatures changed the microbiome’s stability towards more pathogen-dominated communities (Erwin et al., 2012; Lokmer and Wegner, 2015).
Several studies have shown that a healthy bacterial community can help stabilize the microbiome, a requirement for animal health (Green and Barnes, 2010; Lozupone et al., 2012; Lokmer and Wegner, 2015; King et al., 2018; Li et al., 2019a). Among the bacteria that decreased in abundance, Delftia spp. often forms biofilms associated with wastewater treatment and river wetlands (Barrionuevo and Vullo, 2012; Wu et al., 2015) and breaks down or transforms a variety of pollutants, such as acetaminophen (De Gusseme et al., 2011), polycyclic aromatic hydrocarbon (PAH) (Wu et al., 2016), chloroaniline (Zhang et al., 2010), and herbicides (Leibeling et al., 2010). Delftia, Neptunomonas, and Rhodococcus are also in seawater and can degrade PAHs and other organic compounds (Hedlund et al., 1999; Larkin et al., 2005). Interestingly, Neptunomonas has been found in dead clams, although there is no evidence that it is a pathogen (Lee et al., 2012). Members of Pseudoalteromonas have versatile metabolic capacities and can be found in multiple marine habitats where they have vital ecological functions (Ivanova et al., 2014). Moreover, some free species in this genus are associated with mussel metamorphosis and attachment, and it was unclear whether the decline in the genus Pseudoalteromonas would affect larval metamorphosis (Yang et al., 2013; Peng et al., 2020, 2020). Tenacibaculum (family Flavobacteriaceae) is a motile genus and an opportunistic pathogen in fish, which causes an ulcerative disease known as tenacibaculosis (Avendaño-Herrera et al., 2006). Tenacibaculum also causes mortality in mollusk species, including adult Pacific oysters (Burioli et al., 2018). The abundance of Tenacibaculum increased significantly with temperature, supporting the idea that warmer waters could affect mussel larvae by accelerating the proliferation of opportunistic pathogens.
5 Conclusion
In summary, the most striking finding of this study is that the symbiotic bacteria seem to have already been established during the larval stage of the mussel life cycle and that increased seawater temperatures alter the proportion of the symbiotic microbiota. This could reduce the ability of symbiotic bacteria to degrade environmental toxins, regulate metamorphosis in mussel larvae, and increase the abundance of opportunistic pathogens, which together are likely to increase the risk of larval death and reduced larval recruitment in the face of a warmer environment.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/, BioProject: PRJNA1029461.
Ethics statement
The animal study was approved by the Animal Ethics Committee of Shanghai Ocean University (SHOW-DW-2020-032). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Software, Visualization, Writing – original draft. XL: Data curation, Investigation, Writing – original draft, Methodology. T-TL: Data curation, Formal analysis, Investigation, Writing – original draft. DP: Writing – review & editing, Validation. Y-FL: Project administration, Resources, Supervision, Writing – review & editing. J-LY: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key R&D Program of China (No.2022YFE0204600, 2023YFE0115500, 2022YFD2401700), and the National Science Foundation of China (No.32002410, 32172992).
Acknowledgments
This short text acknowledges the contributions of specific colleagues, institutions, or agencies that aided the authors’ efforts.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1367608/full#supplementary-material
References
Amato K. R., Sanders J. G., Song S. J., Nute M., Metcalf J. L., Thompson L. R., et al. (2019). Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 13, 576–587. doi: 10.1038/s41396-018-0175-0
Avendaño-Herrera R., Toranzo A. E., Magariños B. (2006). Tenacibaculosis infection in marine fish caused by Tenacibaculum maritimum: a review. Dis. Aquat Organ 71, 255–266. doi: 10.3354/dao071255
Barrionuevo M. R., Vullo D. L. (2012). Bacterial swimming, swarming and chemotactic response to heavy metal presence: which could be the influence on wastewater biotreatment efficiency? World J. Microbiol. Biotechnol. 28, 2813–2825. doi: 10.1007/s11274-012-1091-5
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brener-Raffalli K., Clerissi C., Vidal-Dupiol J., Adjeroud M., Bonhomme F., Pratlong M., et al. (2018). Thermal regime and host clade, rather than geography, drive Symbiodinium and bacterial assemblages in the scleractinian coral Pocillopora damicornis sensu lato. Microbiome 6, 39. doi: 10.1186/s40168-018-0423-6
Brooks A. W., Kohl K. D., Brucker R. M., Opstal E. J., Bordenstein S. R. (2016). Phylosymbiosis: Relationships and functional effects of microbial communities across host evolutionary history. PloS Biol. 14, e2000225. doi: 10.1371/journal.pbio.2000225
Burioli E. A. V., Varello K., Trancart S., Bozzetta E., Gorla A., Prearo M., et al. (2018). First description of a mortality event in adult Pacific oysters in Italy associated with infection by a Tenacibaculum soleae strain. J. Fish Dis. 41, 215–221. doi: 10.1111/jfd.12698
Cárdenas A., Rodriguez-R L. M., Pizarro V., Cadavid L. F., Arévalo-Ferro C. (2012). Shifts in bacterial communities of two caribbean reef-building coral species affected by white plague disease. ISME J. 6, 502–512. doi: 10.1038/ismej.2011.123
Cárdenas C. A., Bell J. J., Davy S. K., Hoggard M., Taylor M. W. (2014). Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiol. Ecol. 88, 516–527. doi: 10.1111/fem.2014.88.issue-3
Carrier T. J., Reitzel A. M. (2018). Convergent shifts in host-associated microbial communities across environmentally elicited phenotypes. Nat. Commun. 9, 952. doi: 10.1038/s41467-018-03383-w
Carrier T. J., Wolfe K., Lopez K., Gall M., Janies D. A., Byrne M., et al. (2018). Diet-induced shifts in the crown-of-thorns (Acanthaster sp.) larval microbiome. Mar. Biol. 165, 157. doi: 10.1007/s00227-018-3416-x
De Gusseme B., Vanhaecke L., Verstraete W., Boon N. (2011). Degradation of acetaminophen by Delftia tsuruhatensis and Pseudomonas aeruginosa in a membrane bioreactor. Water Res. 45, 1829–1837. doi: 10.1016/j.watres.2010.11.040
Dubé C. E., Ky C. L., Planes S. (2019). Microbiome of the black-lipped pearl oyster Pinctada margaritifera, a multi-tissue description with functional profiling. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01548
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Erwin P. M., Pita L., López-Legentil S., Turon X. (2012). Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Appl. Environ. Microbiol. 78, 7358–7368. doi: 10.1128/AEM.02035-12
Field C. B., Barros V. R., Dokken D. J., Mach K. J., Mastrandrea M. D., Bilir T. E., et al. (2014). “IPCC 2014: Climate Change 2014: Impacts, adaptation, and vulnerability. Part A: Global and sectoral aspects,” in Contribution of working group II to the fifth assessment report of the intergovernmental panel on climate change (Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA), 1132.
Gobet A., Mest L., Perennou M., Dittami S. M., Caralp C., Coulombet C., et al. (2018). Seasonal and algal diet-driven patterns of the digestive microbiota of the European abalone Haliotis tuberculata, a generalist marine herbivore. Microbiome 6, 60. doi: 10.1186/s40168-018-0430-7
Green T. J., Barnes A. C. (2010). Bacterial diversity of the digestive gland of Sydney rock oysters, Saccostrea glomerata infected with the paramyxean parasite, Marteilia sydneyi. J. Appl. Microbiol. 109, 613–622. doi: 10.1111/j.1365-2672.2010.04687.x
Hedlund B. P., Geiselbrecht A. D., Bair T. J., Staley J. T. (1999). Polycyclic aromatic hydrocarbon degradation by a new marine bacterium, Neptunomonas naphthovorans gen. nov., sp. Nov. Appl. Environ. Microbiol. 65, 251–259. doi: 10.1128/AEM.65.1.251-259.1999
Høj L., Levy N., Baillie B. K., Clode P. L., Strohmaier R. C., Siboni N., et al. (2018). Crown-of-thorns sea star Acanthaster cf. solaris has tissue-characteristic microbiomes with potential roles in health and reproduction. Appl. Environ. Microbiol. 84, e00181–e00118. doi: 10.1128/AEM.00181-18
Ivanova E. P., Ng H. J., Webb H. K. (2014). “The Family Pseudoalteromonadaceae,” in The Prokaryotes (Springer, Berlin, Heidelberg). doi: 10.1007/978-3-642-38922-1_229
Jaenike J. (2012). Population genetics of beneficial heritable symbionts. Trends Ecol. Evol. 27, 226–232. doi: 10.1016/j.tree.2011.10.005
King W. L., Jenkins C., Seymour J. R., Labbate M. (2018). Oyster disease in a changing environment: Decrypting the link between pathogen, microbiome and environment. Mar. Environ. Res. 143, 124–140. doi: 10.1016/j.marenvres.2018.11.007
Kozich J. J., Westcott S. L., Baxter N. T., Highlander S. K., Schloss P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kueh C. S., Chan K. Y. (1985). Bacteria in bivalve shellfish with special reference to the oyster. J. Appl. Bacteriol. 59, 41–47. doi: 10.1111/j.1365-2672.1985.tb01773.x
Lafferty K. D., Porter J. W., Ford S. E. (2004). Are diseases increasing in the ocean? Annu. Rev. Ecol. Evol. Syst. 35, 31–54. doi: 10.1146/annurev.ecolsys.35.021103.105704
Larkin M. J., Kulakov L. A., Allen C. C. (2005). Biodegradation and Rhodococcus – masters of catabolic versatility. Curr. Opin. Biotechnol. 16, 282–290. doi: 10.1016/j.copbio.2005.04.007
Lee H. W., Shin N. R., Lee J., Roh S. W., Whon T. W., Bae J. W. (2012). Neptunomonas concharum sp. nov., isolated from a dead ark clam, and emended description of the genus Neptunomonas. Int. J. Syst. Evol. Microbiol. 62, 2657–2661. doi: 10.1099/ijs.0.037473-0
Leibeling S., Schmidt F., Jehmlich N., von Bergen M., Müller R. H., Harms H. (2010). Declining capacity of starving Delftia acidovorans MC1 to degrade phenoxypropionate herbicides correlates with oxidative modification of the initial enzyme. Environ. Sci. Technol. 44, 3793–3799. doi: 10.1021/es903619j
Lema K. A., Bourne D. G., Willis B. L. (2014). Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coral Acropora millepora. Mol. Ecol. 23, 4682–4695. doi: 10.1111/mec.12899
Li Y. F., Chen Y. W., Xu J. K., Ding W. Y., Shao A. Q., Zhu Y. T., et al. (2019a). Temperature elevation and Vibrio cyclitrophicus infection reduce the diversity of haemolymph microbiome of the mussel Mytilus coruscus. Sci. Rep. 9, 16391. doi: 10.1038/s41598-019-52752-y
Li K., Guan W., Wei G., Liu B., Xu J., Zhao L., et al. (2007). Phylogenetic analysis of intestinal bacteria in the Chinese mitten crab (Eriocheir sinensis). J. Appl. Microbiol. 103, 675–682. doi: 10.1111/j.1365-2672.2007.03295.x
Li Y. F., Xu J. K., Chen Y. W., Ding W. Y., Shao A. Q., Liang X., et al. (2019b). Characterization of gut microbiome in the mussel Mytilus galloprovincialis in response to thermal stress. Front. Physiol. 10. doi: 10.3389/fphys.2019.01086
Li Y. F., Yang N., Liang X., Yoshida A., Osatomi K., Power D., et al. (2018). Elevated seawater temperatures decrease microbial diversity in the gut of mytilus coruscus. Front. Physiol. 9. doi: 10.3389/fphys.2018.00839
Lokmer A., Goedknegt M. A., Thieltges D. W., Fiorentino D., Kuenzel S., Baines J. F., et al. (2016a). Spatial and temporal dynamics of pacific oyster hemolymph microbiota across multiple scales. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01367
Lokmer A., Kuenzel S., Baines J. F., Wegner K. M. (2016b). The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ. Microbiol. 18, 970–987. doi: 10.1111/1462-2920.13163
Lokmer A., Wegner K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9, 670–682. doi: 10.1038/ismej.2014.160
Lozupone C. A., Stombaugh J. I., Gordon J. I., Jansson J. K., Knight R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Lu G., Wang F., Yu Z., Lu M., Wang Y., Liu C., et al. (2017). Bacterial communities in gills and intestines of yesso scallop (Patinopecten yessoensis) and its habitat waters in Changhai (Dalian, China). Invertebr Surviv J. 14, 340–351. doi: 10.25431/1824-307x/isj.v14i.340-351
Magoč T., Salzberg S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Malham S. K., Cotter E., O'Keeffe S., Lynch S., Culloty S. C., King J. W., et al. (2009). Summer mortality of the Pacific oyster, Crassostrea gigas, in the Irish Sea: The influence of temperature and nutrients on health and survival. Aquaculture 287, 128–138. doi: 10.1016/j.aquaculture.2008.10.006
McFall-Ngai M., Hadfield M. G., Bosch T. C. G., Carey H. V., Domazet-Lošo T., Douglas A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U S A 110, 3229–3236. doi: 10.1073/pnas.1218525110
Meisterhans G., Raymond N., Girault E., Lambert C., Bourrasseau L., de Montaudouin X., et al. (2016). Structure of Manila clam (Ruditapes philippinarum) microbiota at the organ scale in contrasting sets of individuals. Microb. Ecol. 71, 194–206. doi: 10.1007/s00248-015-0662-z
Moitinho-Silva L., Bayer K., Cannistraci C. V., Giles E. C., Ryu T., Seridi L., et al. (2014). Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 23, 1348–1363. doi: 10.1111/mec.12365
Nemergut D. R., Schmidt S. K., Fukami T., Neill S. P., Bilinski T. M., Stanish L. F., et al. (2013). Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 77, 342. doi: 10.1128/MMBR.00051-12
Nyholm S. V., Graf J. (2012). Knowing your friends: invertebrate innate immunity fosters beneficial bacterial symbioses. Nat. Rev. Microbiol. 10, 815–827. doi: 10.1038/nrmicro2894
Olafsen J. A., Mikkelsen H. V., Giaever H. M., Høvik Hansen G. (1993). Indigenous bacteria in hemolymph and tissues of marine bivalves at low temperatures. Appl. Environ. Microbiol. 59, 1848–1854. doi: 10.1128/aem.59.6.1848-1854.1993
Pantos O., Bongaerts P., Dennis P. G., Tyson G. W., Hoegh-Guldberg O. (2015). Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J. 9, 1916–1927. doi: 10.1038/ismej.2015.3
Patrick S., Houston S., Thacker Z., Blakely G. W. (2009). Mutational analysis of genes implicated in LPS and capsular polysaccharide biosynthesis in the opportunistic pathogen Bacteroides fragilis. Microbiology 155, 1039–1049. doi: 10.1099/mic.0.025361-0
Peng L. H., Liang X., Xu J. K., Dobretsov S., Yang J. L. (2020). Monospecific biofilms of Pseudoalteromonas promote larval settlement and metamorphosis of Mytilus coruscus. Sci. Rep. 10, 2577. doi: 10.1038/s41598-020-59506-1
Pierce M. L., Ward J. E., Holohan B. A., Zhao X., Hicks R. E. (2016). The influence of site and season on the gut and pallial fluid microbial communities of the eastern oyster, Crassostrea virginica (Bivalvia, Ostreidae): community-level physiological profiling and genetic structure. Hydrobiologia 765, 97–113. doi: 10.1007/s10750-015-2405-z
Quast K., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Reveillaud J., Maignien L., Eren A. M., Huber J. A., Apprill A., Sogin M. L., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8, 1198–1209. doi: 10.1038/ismej.2013.227
Rogelj J., den Elzen M., Höhne N., Fransen T., Fekete H., Winkler H., et al. (2016). Paris Agreement climate proposals need a boost to keep warming well below 2 °C. Nature 534, 631–639. doi: 10.1038/nature18307
Sweet M. J., Bulling M. T. (2017). On the importance of the microbiome and pathobiome in coral health and disease. Front. Mar. Sci. 4. doi: 10.3389/fmars.2017.00009
Trabal N., Mazón-Suástegui J. M., Vázquez-Juárez R., Asencio-Valle F., Morales-Bojórquez E., Romero J. (2012). Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microb. Ecol. 64, 555–569. doi: 10.1007/s00248-012-0039-5
Tremaroli V., Bäckhed F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552
Webster N., Hill R., Great Barrier Reef Marine Park Authority. (2007). “Vulnerability of marine microbes on the Great Barrier Reef to climate change,” in Climate Change and the Great Barrier Reef: A Vulnerability Assessment, 97–120. Townsville: The Great Barrier Reef Marine Park Authority Available at: http://hdl.handle.net/11017/538.
Wegner K. M., Volkenborn N., Peter H., Eiler A. (2013). Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol. 13, 252. doi: 10.1186/1471-2180-13-252
Wu W., Huang H., Ling Z., Yu Z., Jiang Y., Liu P., et al. (2016). Genome sequencing reveals mechanisms for heavy metal resistance and polycyclic aromatic hydrocarbon degradation in Delftia lacustris strain LZ-C. Ecotoxicology 25, 234–247. doi: 10.1007/s10646-015-1583-9
Wu Y., Shukal S., Mukherjee M., Cao B. (2015). Involvement in denitrification is beneficial to the biofilm lifestyle of Comamonas testosteroni: a mechanistic study and its environmental implications. Environ. Sci. Technol. 49, 11551–11559. doi: 10.1021/acs.est.5b03381
Xiao J., Ford S. E., Yang H., Zhang G., Zhang F., Guo X. (2005). Studies on mass summer mortality of cultured zhikong scallops (Chlamys farreri Jones et Preston) in China. Aquaculture 250, 602–615. doi: 10.1016/j.aquaculture.2005.05.002
Yang J. L., Satuito C. G., Bao W. Y., Kitamura H. (2008). Induction of metamorphosis of pediveliger larvae of the mussel Mytilus galloprovincialis Lamarck 1819 using neuroactive compounds, KCl, NH4Cl and organic solvents. Biofouling 24, 461–470. doi: 10.1080/08927010802340309
Yang J. L., Shen P. J., Liang X., Li Y. F., Bao W. Y., Li J. L. (2013). Larval settlement and metamorphosis of the mussel Mytilus coruscus in response to monospecific bacterial biofilms. Biofouling 29, 247–259. doi: 10.1080/08927014.2013.764412
Zhang L. L., He D., Chen J. M., Liu Y. (2010). Biodegradation of 2-chloroaniline, 3-chloroaniline, and 4-chloroaniline by a novel strain Delftia tsuruhatensis H1. J. Hazard Mater. 179, 875–882. doi: 10.1016/j.jhazmat.2010.03.086
Keywords: microbe-environment interaction, seawater temperature, larval microbiota, Mytilus coruscus, toxin degradation, opportunistic pathogens
Citation: Zhu Y-T, Liang X, Liu T-T, Power DM, Li Y-F and Yang J-L (2024) The mussel larvae microbiome changes in response to a temperature rise. Front. Mar. Sci. 11:1367608. doi: 10.3389/fmars.2024.1367608
Received: 09 January 2024; Accepted: 12 March 2024;
Published: 08 April 2024.
Edited by:
Yafei Duan, South China Sea Fisheries Research Institute, ChinaReviewed by:
Houguo Xu, Chinese Academy of Fishery Sciences (CAFS), ChinaLijun Ning, South China Agricultural University, China
Copyright © 2024 Zhu, Liang, Liu, Power, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Feng Li, yifengli@shou.edu.cn; Jin-Long Yang, jlyang@shou.edu.cn
†These authors have contributed equally to this work and shared the first authorship