Identification and genomic analysis of a novel temperate bacteriophage infecting Labrenzia aggregata isolated from the Mariana Trench
 Bingyu Gu
Bingyu Gu Haowen Wang
Haowen Wang Jiayi Lv3
Jiayi Lv3  Yanfen Zheng
Yanfen Zheng Xiao-Hua Zhang
Xiao-Hua Zhang Yuanchao Zhan
Yuanchao Zhan- 1College of Marine Life Science, Ocean University of China, Qingdao, China
- 2Laboratory of Pathology and Immunology of Aquatic Animals, Laboratory of Pathology and Immunology of Aquatic Animals, Key Laboratory of Mariculture, Ministry of Education (KLMME), Fisheries College, Ocean University of China, Qingdao, China
- 3Haide College, Ocean University of China, Qingdao, China
- 4Marine Agriculture Research Center, Tobacco Research Institute of Chinese Academy of Agricultural Sciences, Qingdao, China
In marine environments, viruses play a pivotal role, yet deep-sea bacteriophages remains largely uncharacterized. The bacterium Labrenzia aggregata RF14, isolated from the Mariana Trench at a depth of 4,000 meters, harbors prophage regions based on a previous study. In this study, we induced a temperate bacteriophage from it using mitomycin C. The bacteriophage exhibited an icosahedral structure with a non-extendable tail and was named vB_LagS-V1. The genome size of it is 39,329 bps with a 59.46% G+C content, encoding 60 putative open reading frames. Genomic and phylogenetic analyses demonstrated that vB_LagS-V1 along with many bacteriophages infecting Hyphomicrobiales, constituted a newly unclassified family, which we designated as Hyphoviridae. Within this novel family, vB_LagS-V1 is distinct with isolated phages and clustered with two uncultured prophages within Labrenzia, forming an unclassified new genus, given a name of Labrenmarinevirus. The codon usage correlation and absence of tRNAs found in vB_LagS-V1, also prevail in some deep-sea bacteriophages, highlighting their adaptations to the deep-sea prokaryotic hosts. Moreover, vB_LagS-V1 encoded two auxiliary metabolic genes, cysteine dioxygenase and phosphoadenosine phosphosulfate reductase, which might help the phage and its host adapt to high hydrostatic pressure in the deep-sea environments. Our study will significantly contribute to the understanding of deep-sea bacteriophages and their interactions with hosts in extreme environments.
1 Introduction
Marine viruses constitute one of the most abundant biological components in the marine environment, and play a critical role in marine ecosystems (Rohwer and Thurber, 2009). They help maintain the balance of microbial communities by infecting and lysing their hosts, thereby driving biogeochemical cycles through “viral shunt” and “viral shuttle” (Wilhelm and Suttle, 1999; Guidi et al., 2016). In addition, viruses can promote gene exchanges and evolution of marine organisms by horizontal gene transfer (Weinbauer, 2004).
Recent global viromic metagenome analyses, such as Tara Ocean, Global Ocean Virome (GOV), and the expanded GOV 2.0, have uncovered a remarkably diverse marine viral ecosystem (Roux et al., 2016; Gregory et al., 2019; Sunagawa et al., 2020). Despite these advances, a significant portion of viruses in these viromic databases remains uncultivated. According to GOV 2.0, a mere 9.8% of the viral population is linked to known isolated viruses, mainly due to the limited sequencing of bacteriophages (Gregory et al., 2019). This underscores a growing necessity for viral isolation efforts to elucidate unexplored metagenomic sequences and identify previously unknown viral populations in the field of viral metagenomics.
The necessity for viral isolation becomes even more evident in the deep sea. In these environments, where native predators are scarce, viruses play a fundamental role in regulating microbial communities and material cycling (Breitbart et al., 2018). Despite their crucial ecological significance, research on deep-sea viruses is notably constrained, with only a limited number of successfully isolated viruses (Cai et al., 2023). In 1968, Johnson and colleagues initiated deep-sea viral exploration by isolating a vibrio phage from sediments at a depth of 3,000 meters (Johnson, 1968). Afterward, two viruses, AmM-1 and PstS-1, were isolated from the Northwest Pacific and Japan Trench. These viruses displayed a mosaic genomic structure, suggesting genetic exchange processes in deep-sea temperate bacteriophages (Yoshida et al., 2015a; Yoshida et al., 2015b). Two Mu-like bacteriophages, vB_ThpS-P1 and vB_PeaS-P1, were also induced from deep-sea Roseobacter (Tang et al., 2017). Interestingly, low temperature induced one filamentous phage, SW1, from the deep-sea bacterium Shewanella piezotolerans WP3 (Wang et al., 2007). This filamentous phage is able to survive under high-pressure and low-temperature conditions, aligning with the characteristics of the deep-sea environment (Jian et al., 2016). Additionally, a siphovirus, vB_HmeY_H4907, was induced, the host of which was isolated from surface sediment in the Mariana Trench at a depth of 8,900 meters (Su et al., 2023). Together, these studies aid in exploring the morphology, infection strategies and phylogenetic relationship, as well as environmental adaptation of deep-sea bacteriophages, advancing our comprehension of these unique ecosystems.
Labrenzia aggregata, a member of order Hyphomicrobiales, class Alphaproteobacteria, is widespread in marine environments and actively contributes to nitrogen and sulfur cycles, playing roles in denitrification, dissimilatory nitrate reduction to ammonium (DNRA) (King, 2003; King and Weber, 2007), thiosulfate oxidation, as well as the biosynthesis and degradation of dimethylsulfoniopropionate (DMSP) (Curson et al., 2017). Recent studies revealed their presence in deep-sea environments, such as the Mariana Trench, due to their ability to survive and grow under high hydrostatic pressure conditions (Priyanka et al., 2014; Zhong et al., 2021). In addition, extensive horizontal gene transfer (HGT) events have been identified in L. aggregata genome, likely mediated by viruses (Zhong et al., 2021).
In our previous study, the L. aggregata RF14 strain was isolated from the Mariana Trench at a depth of 4,000 m and potential prophage sequences were identified in its genome. However, whether this prophage can be induced remains unknown (Zhong et al., 2021). In this study, we successfully induced a prophage from the L. aggregata RF14 strain using mitomycin C, named as vB_LagS-V1. By exploring the physiological features, genomic characteristics, evolutionary position of the phage, we found that vB_LagS-V1 may represent a new genus within a previously uncharacterized family. The adaptation of the phage with host and deep-sea environments were also discussed. The results of this study not only expand our scientific understanding of the biology, genomic information and ecological significance of deep-sea bacteriophages, also provide insight into the mechanisms of their interactions with hosts.
2 Materials and methods
2.1 Culture conditions
The host strain, L. aggregata RF14, was isolated from seawater samples collected at a depth of 4,000 m in the Mariana Trench (11° 23.036′N, 142° 30′E) in April 2016. The seawater sample was spread on marine agar 2216E medium (MA; Becton Dickinson, USA) and cultivated at 28°C. A single colony was carefully selected and purified three times before strain identification (Zhao et al., 2020). The genome of the host strain was also obtained (accession number: CP045617) (Zhong et al., 2021).
To recover L. aggregata RF14, the preserved culture was inoculated onto marine agar 2216E medium. A single colony of the strain was then transferred into marine broth 2216E medium (MB; Becton Dickinson, USA). Subsequently, the culture was further transferred at a 1% inoculation rate for subsequent experiments during the exponential phase (OD600 = 0.2).
2.2 Viral induction assays
Mitomycin C (Sigma-Aldrich, USA) was used to trigger the prophage induction from L. aggregata RF14. After approximately 13.5 hours of cultivation, a final concentration of 1 μg/mL mitomycin C was added and then the culture was kept in the dark for 1 hour (Lorenz et al., 2016; Su et al., 2023). The host cells were washed with fresh medium twice and then kept for cultivation at 28°C. A parallel control group without the addition of mitomycin C was included. Each group was conducted in triplicates.
Samples were collected along a time-series, the values of OD600 and the numbers of viruses were measured. For virus quantification, 1 mL sample were fixed with 1% glutaraldehyde (Sigma-Aldrich, USA) and stored at -80°C before measurement. SYBR Green I (Solarbio, China) was used to stain the virus-like particles as described before (Hewson et al., 2001), followed by measurement using a flow cytometer (CytoFLEX, Beckman Coulter Inc, USA).
2.3 Phage concentration and purification
The induced phage was concentrated using the protocol of Chen et al. with a few modifications (Chen et al., 2006). Briefly, two liters of cultured cells were induced by mitomycin C based on the procedures mentioned above. The lysate was collected approximately 24 hours after induction. Polyethylene glycol 8000 (Sigma-Aldrich, USA) with a final concentration of 100 g/L was added to this lysate and stored at 4°C for 12 - 48 hours for precipitation. The phage concentrate was collected by centrifugation at 15,000xg for 1 hour, then resuspended in Tris-Magnesium buffer (50 mM Tris-HCl, 2.5 mM MgCl2, pH=7.0). The phage concentrate was further purified by iodixanol density gradient ultracentrifugation (OptiPrep™, Sigma-Aldrich, USA). The precipitated phage lysate was centrifuged at 100,000xg at 4°C for 10 hours using an ultracentrifuge (Beckman Coulter Inc, USA). The band of purified phage was extracted and further dialyzed in TM buffer twice and stored at 4°C.
2.4 Transmission electron microscopy observation
The purified and concentrated phage lysate (15-20 µL) was carefully dispensed onto a hydrophilized copper grid for morphological observation one minute for adsorption. Subsequently, the sample was stained by 1% phosphotungstic acid (Sigma-Aldrich, USA) for one minute twice. The grid was air-dried for 3-5 minutes and then examined using a JEM-2100 transmission electron microscope (JEOL Ltd, Japan) at 120 kV.
2.5 Host range test
Another four strains of L. aggregata were selected to test the cross-infectivity of vB_LagS-V1, including LZB033 isolated from surface seawater in the East China Sea, as well as SDL044, ZYF703 and ZYF612 isolated from the Mariana Trench at depth of 4 km, 8 km and 10 km respectively. Through phylogenetic analysis, these four strains were found to have a high evolutionary similarity to L. aggregata RF14, with an evolutionary distance of only 0.2% based on whole-genome phylogenetic analysis (Zhong et al., 2021). For each host strain, one drop of phage lysate was spotted onto a soft-agar overlay plate and incubated for 3 to 4 days at 28°C. The formation of plaques was an indication of cross-infectivity (Rohwer et al., 2000). The original host was also tested in parallel, as a positive control.
2.6 Chloroform sensitivity
The chloroform sensitivity was conducted to assess the presence of lipid in vB_LagS-V1 particles as described before (Yang et al., 2017). Approximately 500 µL of purified phage lysate was mixed with 100 µL of chloroform (Sigma-Aldrich, USA), vigorously shaken for 1 minute, and left to stand for 20 minutes. After centrifuging of 5 minutes at 4,000xg, 10 µL of the upper layer was spotted onto a soft-agar overlay plate, and incubated at 28°C for 3 to 4 days. The persistence of infectivity indicated the phage’s insensitivity to chloroform. The phage without chloroform treatment was also included as a negative control.
2.7 Genome extraction
The DNA of purified bacteriophage was extracted by phenol-chloroform-isopropanol extraction method (Roberts et al., 2004). Briefly, the purified phage particles were treated with a combination of sodium dodecyl sulfate (final concentration 1% w/v, Sigma-Aldrich, USA), proteinase K (final concentration 30 μg/mL, Accurate Biology, China) and EDTA (final concentration 5 μM, Sigma-Aldrich, USA) at 55°C for 3 hours. Then, phage DNA was extracted using phenol/chloroform/isoamyl alcohol (25:24:1) (Sigma-Aldrich, USA), precipitated with isopropanol (Sigma-Aldrich, USA), washed with ethanol (VWR Chemicals, USA), and then resuspended in Tris-EDTA buffer (10 mM Tris-HCl, 1mM EDTA, pH=8.0).
2.8 Sequencing and annotation
The genome of vB_LagS-V1 was sequenced by an Illumina MiSeq PE300 platform (Illumina, San Diego, USA) at Shanghai Meiji Biomedical Technology Company. The raw sequencing data were assembled using the SPAdes algorithm with default settings (Prjibelski et al., 2020). The complete genome was uploaded to the National Center for Biotechnology Information (NCBI) database for a whole-genome BLAST comparison (Pruitt et al., 2007).
Both Prodigal and GeneMark (https://exon.gatech.edu/GeneMark/) were used to predict open reading frames (ORFs) (Besemer, 1999; Hyatt et al., 2010). The final ORFs and their corresponding protein sequences were obtained by integrating the results from both prediction methods. These ORFs protein sequences were compared with known protein sequences using BLASTp against NCBI’s non-redundant viruses databases (E-value ≤ 1e-5 and identity ≥ 30%), providing functional annotation of each ORF (Pruitt et al., 2007). In addition, all ORF protein sequences were queried against the IMG/VR viral protein database v4 using BLASTp (E-value ≤ 1e-5) to recruit as many homologous sequences as possible (Camargo et al., 2023). According to the annotation results, the phages sharing high homology with vB_LagS-V1 were included in whole-genome alignment, which was generated using a custom-made Python script (Supplemental Material) and edited by Adobe Illustrator 2022 software (Adobe, California, USA).
2.9 Phylogenetic analysis
To retrieve the reference phages for phylogenetic analysis, the protein sequences of each ORF of vB_LagS-V1 were compared against both NCBI-nr viruses database and UniProtKB reference proteomes + Swiss-Prot viruses database, using a threshold of E-value ≤ 1e-5 and identity ≥ 30%. Bacteriophages sharing more than two homologous proteins with vB_LagS-V1 were included in our analysis. In addition, uncultivated bacteriophages sharing high homology with vB_LagS-V1 from IMG/VR viral protein database v4 were also incorporated. The complete genomes of these reference phages, as well as the genome of vB_LagS-V1, were uploaded to the VICTOR server (https://ggdc.dsmz.de/victor.php), to investigate the genome-based phylogeny and classification of prokaryotic viruses (Meier-Kolthoff and Göker, 2017). All pairwise comparisons of the nucleotide sequences were conducted using the Genome-BLAST Distance Phylogeny (GBDP) method (Meier-Kolthoff et al., 2013), applying settings recommended for prokaryotic viruses (Meier-Kolthoff and Göker, 2017). Taxon boundaries at the species, genus, and family levels were estimated with the OPTSIL program (Göker et al., 2009), with the recommended clustering thresholds (Meier-Kolthoff and Göker, 2017) and an F value (fraction of links required for cluster fusion) of 0.5 (Meier-Kolthoff et al., 2014). The average nucleotide identity (ANI) values and the corresponding phylogenetic tree were analyzed using OrthoANI (Lee et al., 2016).
To construct a phylogenetic tree for major capsid protein, the protein sequences which share homology with vB_LagS-V1 major capsid protein in NCBI-nr and UniProtKB reference proteomes + Swiss-Prot database (E-value ≤ 1e-5 and identity ≥ 30%), were retrieved. IQ-TREE (version 2.2.0.3) with the best-fitting model was employed to build a maximum-likelihood phylogenetic tree with 1,000 bootstrap (Nguyen et al., 2015; Kalyaanamoorthy et al., 2017). The resulting phylogenetic tree was visualized using the ChiPlot website (Xie et al., 2023).
All NCBI and UniProtKB reference proteomes + Swiss-Prot database were based on versions prior to December 2023.
2.10 Codon usage analysis
tRNAscan-SE was used to detect tRNA sequences within vB_LagS-V1 (Lowe and Eddy, 1997). CodonW (version 1.4.2, available at https://sourceforge.net/projects/codonw) was utilized to calculate the Relative Synonymous Codon Usage (RSCU) of each amino acid for bacteriophages and their corresponding hosts (Yang et al., 2022). Notably, methionine (encoded by ATG) and tryptophan (encoded by ATG) are encoded by a single codon with a constant RSCU value of 1. These two amino acids were not included in the calculation of codon usage preferences. The correlation and linear regression analysis of RSCU values between bacteriophages and their corresponding hosts were analyzed using GraphPad Prism v9 software (GraphPad, California, USA).
2.11 Ecological distribution
The biogeographic distribution of vB_LagS-V1 was investigated by searching against the publicly available prokaryotic metagenomes (PRJNA412741, PRJNA541485) and viromes (PRJNA668558) from the Mariana Trench (Xue et al., 2020; Gao et al., 2022), to assess the both intracellular and extracellular distribution of vB_LagS-V1, respectively. BBmap (version 38.98) was used to map the raw reads to the genome sequence of vB_LagS-V1 (Bushnell, 2014), with a threshold of 75% coverage and 90% identity (Roux et al., 2017). The relative abundance of this bacteriophage was calculated as the reads per kilobase per million mapped reads (RPKM).
The biogeographical distributions of ORF 44 (cysteine dioxygenase) and ORF 32 (phosphoadenosine phosphosulfate reductase) were also investigated. The amino acid sequences of these two ORFs were uploaded to the Ocean Gene Atlas website (https://tara-oceans.mio.osupytheas.fr/ocean-gene-atlas/) and compared against Tara Ocean and Malaspina Gene Database with a threshold E-value ≤ 1e-5 (Vernette et al., 2022).
3 Result and discussion
3.1 Induction and physiological features of the phage
A prophage region was predicted in the genome of L. aggregata RF14 based on previous genome analysis. To investigate the inducibility of this prophage, mitomycin C was added into L. aggregata RF14 culture during the early stage of the exponential growth stage, approximately 13 hours after inoculation. After addition of mitomycin C, the abundance of viral-like particles significantly increased, which escalated from an initial count of 106/mL to approximately 109-1010/mL, marking a substantial 2-3 order of magnitude increase (Figure 1A). Simultaneously, in comparison to the control group, the growth of L. aggregata RF14 was impeded. The addition of mitomycin C and lysis of bacteriophages led to the arrest of further growth, eventually stabilizing at an OD600 of about 0.2.
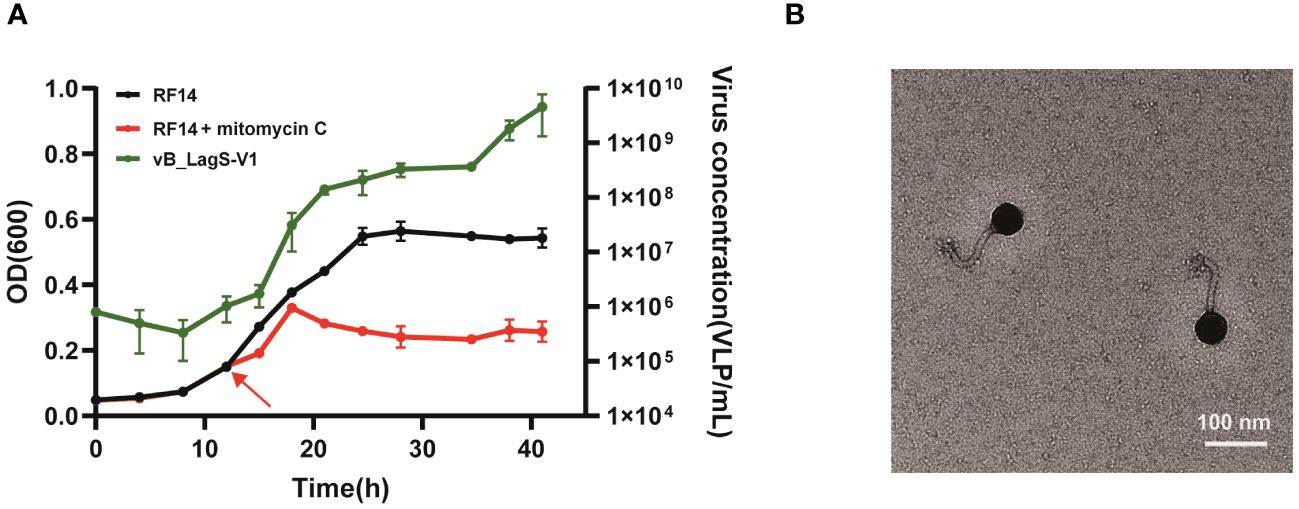
Figure 1 (A) The effect of mitomycin C on the growth of L. aggregata RF14 and the production of prophages. The number of induced prophages was measured by flow cytometry, and the growth of L. aggregata RF14 was monitored through OD600 measurements. The time point at which mitomycin C was added is indicated by an arrow. The presented data represent average values from three independent experiments, and error bars denote standard deviations. (B) Transmission electron microscopy image of the purified bacteriophage induced from the L. aggregata RF14 strain with mitomycin C.
TEM images revealed that the purified induced phage displayed an icosahedral structure along with a non-contractile tail. Measurements indicated a head diameter of about 56 nm and a tail length of approximately 102 nm for this phage (Figure 1B).
The investigation into the host range of the purified phage revealed a limited infectivity spectrum (Supplementary Table S1). Specifically, the phage displayed a relatively narrow host range, exclusively infecting its original host, L. aggregata RF14. Although other four L. aggregata strains included in the analysis are genetical close to L. aggregata RF14, with a mere 0.2% evolutionary distance based on genome comparison, the phage exhibited no infectivity towards these additional strains. Additionally, chloroform treatment did not affect the infectivity of the phage against L. aggregata RF14, suggesting the absence of lipid components in the phage’s structural proteins (Supplementary Figure S1, Supplementary Table S1).
3.2 General genomic features
The genomic sequencing of this induced phage revealed a genome size of 39,329 bps with a G+C content of 59.46%. In total, the phage encodes 60 ORFs. No highly similar phage was identified in the NCBI database through whole-genome BLASTn comparison. Therefore, this temperate phage is considered potentially novel and has been designated as vB_LagS-V1.
Annotation using NCBI-BLASTp revealed that 34 ORFs shared significant similarity with sequences in the non-redundant (nr) viruses protein database (E-value ≤ 1e-5, identity ≥ 30%). Based on the annotation results, the genome of vB_LagS-V1 can be classified into specific functional modules, including packaging, structure, DNA replication and modification, lysis, as well as integration (Figure 2, Supplementary Table S2).
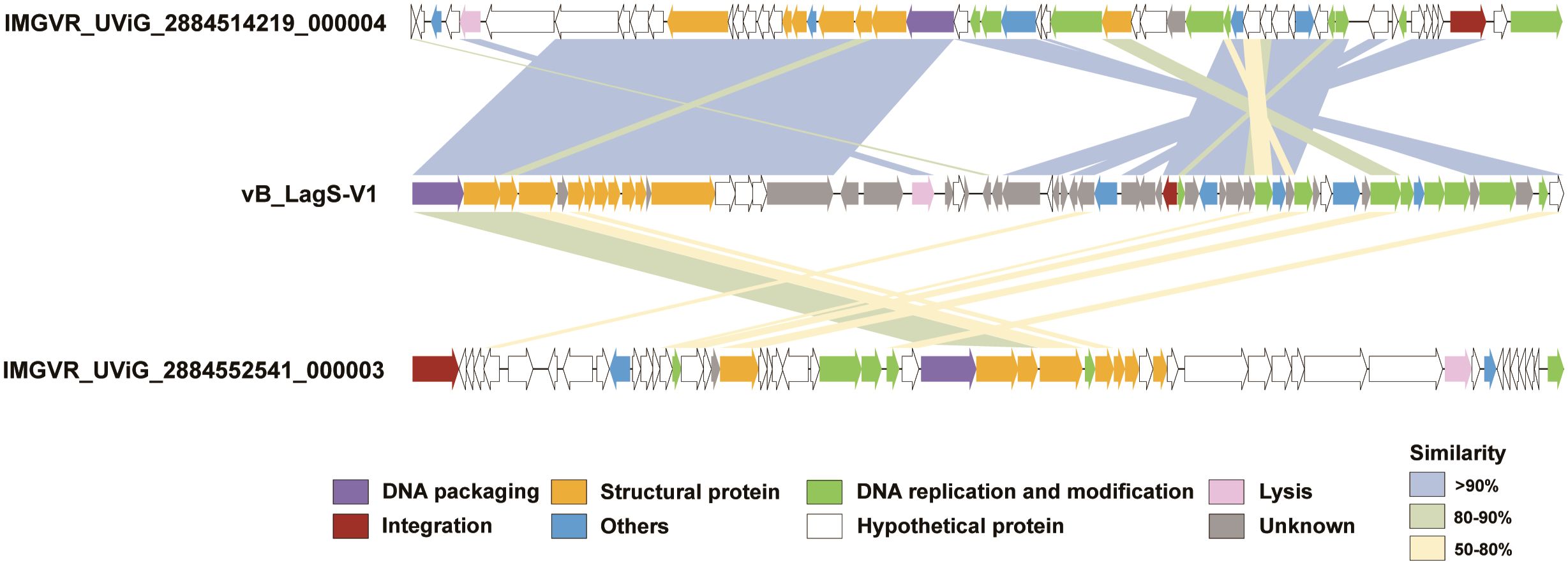
Figure 2 Genome maps of vB_LagS-V1, IMGVR_UViG_2884514219_000004 and IMGVR_UViG_2884552541_000003. ORFs are color-coded according to their predicted functions: purple for DNA packaging; yellow for structural protein; green for DNA replication and modification; pink for lysis; red for integration; blue for others; white for hypothetical protein; and gray for unknown. Similarities between ORFs are also represented by different colored shadings, with light blue shading representing similarities of > 90%, light green shading for similarities between 80 and 90%, and light yellow shading for similarities between 50 and 80%.
Further extending our analysis to the IMG/VR viral protein database, two uncultivated viral genomes, IMGVR_UViG_2884514219_000004 and IMGVR_UViG_2884552541_000003, were found sharing homology with vB_LagS-V1 (Figure 2). IMGVR_UViG_2884514219_000004 and IMGVR_UViG_2884552541_000003 are prophages discovered in Labrenzia sp. THAF187b and Labrenzia sp. THAF82, respectively (accession numbers: NZ_CP045344 and NZ_CP045354). Both strains were obtained from a marine aquarium tank, with one isolated from the surface of a polyethylene microplastic particle, while the other was found within detritus agregates (unpublished). IMGVR_UViG_2884514219_000004 shared 40 proteins with vB_LagS-V1, constituting 68.3% of vB_LagS-V1’s ORFs. High similarity (> 90%) was observed in ORFs related to packaging, DNA replication and modification, as well as structure between our phage and IMGVR_UViG_2884514219_000004. Specifically, almost all structural proteins of vB_LagS-V1 have homologs in IMGVR_UViG_2884514219_000004. For IMGVR_UViG_2884552541_000003, its proteins shared similarity with 31.7% (19/60) of the ORFs in vB_LagS-V1, with most similarities falling within the range of 50-80%.
3.3 vB_LagS-V1 represents a new genus
The lack of known phage isolates similar to vB_LagS-V1 inspired us to further investigate its taxonomic classification and evolution. To improve the accuracy of our taxonomic analysis, only the bacteriophages sharing more than two homologs with vB_LagS-V1 were included in the Genome-BLAST Distance Phylogeny (GBDP) analysis and OPTSIL program. The results indicated that vB_LagS-V1 fell into a previously unclassified family. Most of the phages/prophages in this family were associated with hosts belonging to Hyphomicrobiales with a few exceptions (Figure 3A), suggesting an intimate association between the newly identified viral family and the order Hyphomicrobiales. Hence, we designated this new viral family as Hyphoviridae. In addition, vB_LagS-V1 and the two prophages recruited from the IMG/VR viral protein database, IMGVR_UViG_2884514219_000004 and IMGVR_UViG_2884552541_000003, formed a distinctive clade (Figure 3A), which was consistent with a new viral genus as suggested by OPTSIL program (Figure 3A).
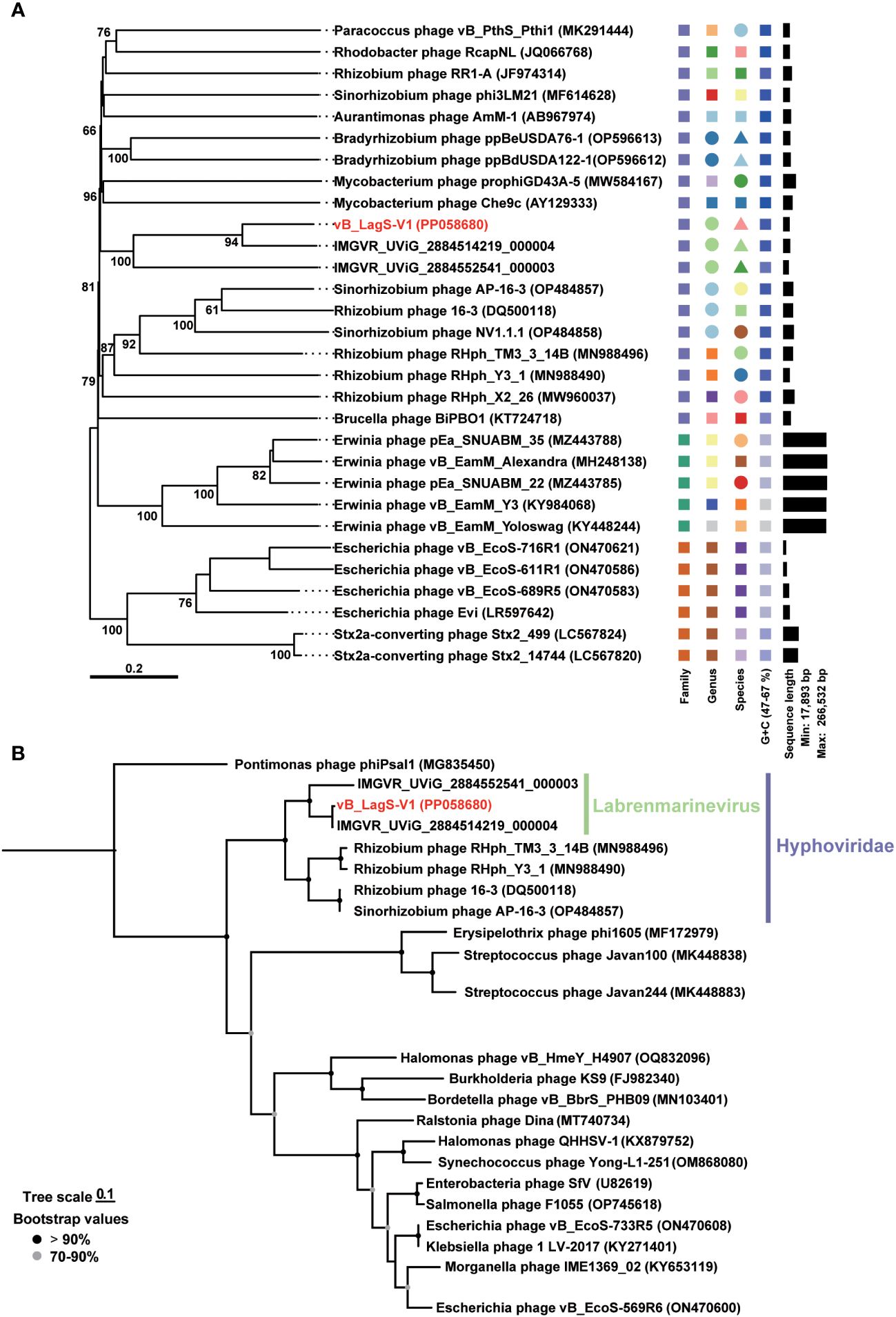
Figure 3 (A) Genome-BLAST Distance Phylogeny (GBDP) tree based on the complete genome sequences of phages. Pseudo-bootstrap support values greater than 50/100 replications are shown. (B) Maximum-likelihood phylogenetic trees based on amino acid sequences of phage DNA major capsid proteins. Bootstrap values are based on 1,000 replicates. Shapes with different colors represent distinct taxonomic units.
This classification was further reinforced by the phylogenetic tree based on the major capsid protein, where the capsid of the members within Hyphoviridae formed a distinct clade and vB_LagS-V1 closely clustered with IMGVR_UViG_2884514219_000004, and IMGVR_UViG_2884552541_000003 (Figure 3B). The ANI analysis also revealed ANI values exceeding 70% among these three entities (Supplementary Figure S2), providing additional support for this genus-level classification (Turner et al., 2021).
In addition to forming a distinct clade, we also recognized that vB_LagS-V1, IMGVR_UViG_2884514219_000004, and IMGVR_UViG_2884552541_000003 were found in varied habitats compared to other members in Hyphoviridae. The hosts of these three phages belong to the Labrenzia and have been isolated from marine environments. Whereas, other members such as Rhizobium phage RHph_TM3_3_14B, Rhizobium phage RHph_Y3_1, Sinorhizobium phage AP-16-3 and Rhizobium phage 16-3 originate from rhizobia isolated from soil environments (Ordogh and Szende, 1961; Santamaría et al., 2022; Kozlova et al., 2023). This divergence in host and corresponding environments highlights a significant ecological distinction of vB_LagS-V1 and its associated new genus. Together, our isolate vB_LagS-V1 can be designated as the representative of the new genus which specifically infects Labrenzia. As an indication of the host specificity of this genus and its association with marine environments, we named this new viral genus as Labrenmarinevirus. Further, we assigned the species name Labrenmarinevirus V1 to vB_LagS-V1.
3.4 A high rRSCU value found in vB_LagS-V1 and other deep-sea phages
The genome of vB_LagS-V1, as analyzed by tRNAscan-SE, lacks tRNA genes, implying the full adaptation of vB_LagS-V1 to the host’s codon usage preferences. To confirm it, the RSCU values of both bacteriophage and the host were calculated separately, followed by a correlation analysis (Chithambaram et al., 2014). A remarkable high rRSCU value of 0.93 between vB_LagS-V1 and the host L. aggregata RF14 was observed (Figure 4A), indicating notable adaptation of vB_LagS-V1 to the host’s translation machinery and proficient utilization of the host’s tRNA pool (Yang et al., 2022).
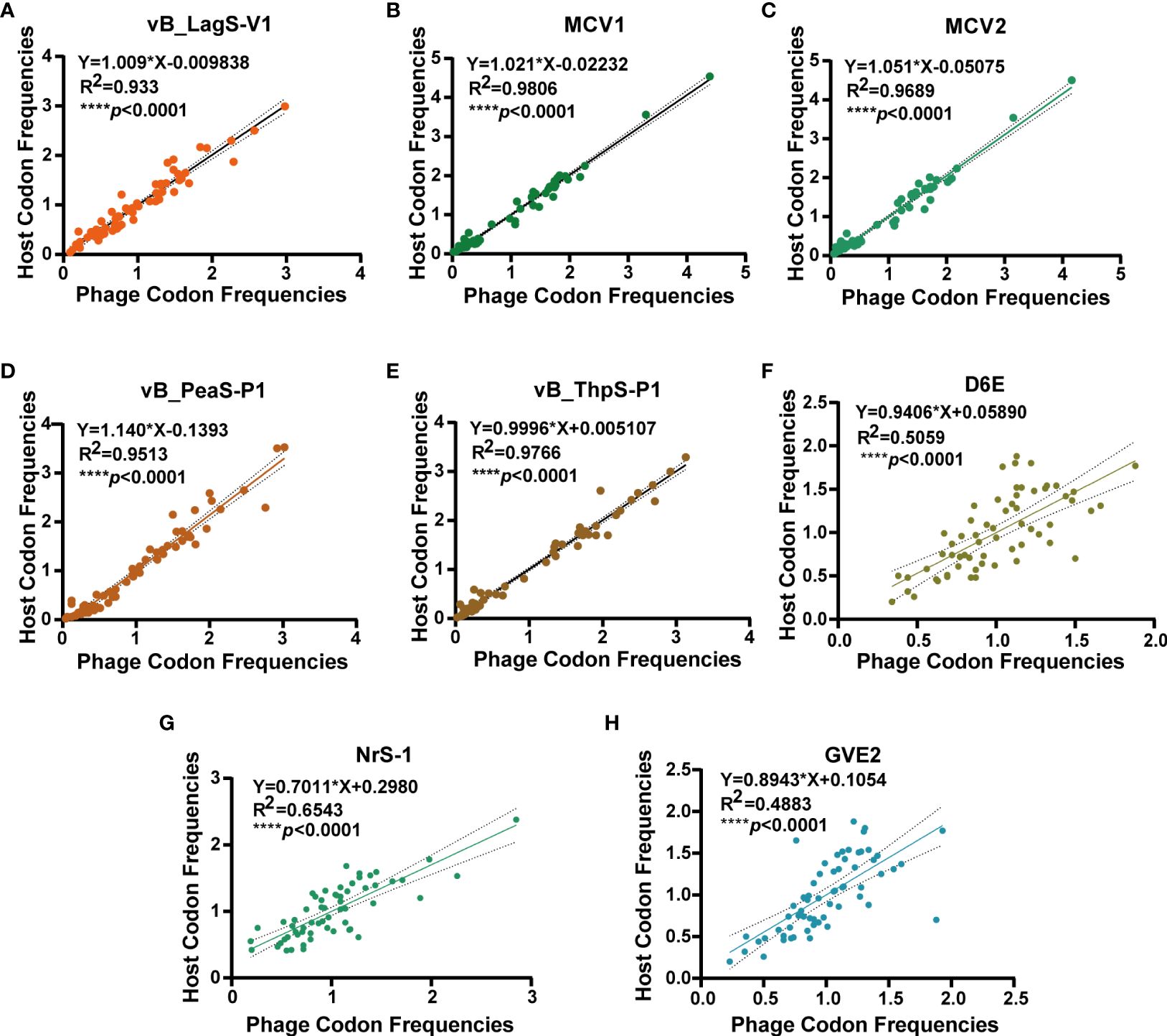
Figure 4 The correlation analysis of RSCU values of 62 codons of vB_LagS-V1 (A), MCV1 (B), MCV2 (C), vB_PeaS-P1 (D), vB_ThpS-P1 (E), D6E (F), NrS-1 (G), GVE2 (H) and their corresponding hosts. The solid line and dashed line represent the best-fit line of linear regression and the 95% confidence interval, respectively.
The absence of tRNA and the high rRSCU value of vB_LagS-V1 inspired an investigation into the prevalence of this observation among other deep-sea phage isolates with available genomes (Supplementary Table S3). Surprisingly, none of these deep-sea phages were found to harbor tRNA genes. Subsequent codon usage analysis revealed a significant correlation (****p < 0.0001) between the RSCU values of these phages and their respective host. Among them, five phages, including vB_LagS-V1, MCV1, MCV2, vB_PeaS-P1, and vB_ThpS-P1, showed relatively higher rRSCU values (Figure 4).
To explain the reason behind the notable alignment on RSCU between these five deep-sea bacteriophages and corresponding hosts, other physiological features of these deep-sea bacteriophages were explored (Supplementary Table S3). These five deep-sea phages with high rRSCU values exhibited a relatively narrow host range. For example, among the five tested L. aggregata strains, vB_LagS-V1 could only infect its original host (Supplementary Table S1). MCV1 and MCV2, which infect Marinitoga bacteria, were unable to infect other potential hosts within the Marinitoga and Thermosipho genera (Mercier et al., 2018). Additionally, vB_PeaS-P1 and vB_ThpS-P1, induced from two deep-sea roseobacters of the genus Salipiger, were also incapable of cross-infecting the two host strains (Tang et al., 2017). Therefore, it is plausible that some deep-sea phages strategically limit their host range to optimize the utilization of resources and energy within a specific or confined spectrum of hosts. This adaptive approach may signify a survival strategy employed by certain deep-sea phages.
The pronounced adaptation to the host and relatively limited host range implies that the co-evolution dynamics between some deep-sea bacteriophages and their hosts might follow the fluctuating selection dynamic (FSD) model. In this model, both bacterial hosts and their phages tend to evolve into highly specialized genotypes (Steven, 1993), in which a virus genotype exclusively infects a specific host genotype (Hamilton, 1980; Decaestecker et al., 2007; Gandon et al., 2008; Lopez Pascua et al., 2014). Many studies have demonstrated that the FSD model is preferred in the unfavorable or nutrient-limited conditions, due to the substantial costs associated with host resistance and virus infectivity (Sasaki, 2000; Agrawal and Lively, 2002). The deep-sea environments, characterized by limited nutrients, high osmotic pressure and perpetual darkness, imposes intensive survival pressure on the resident microbes (Danovaro et al., 2014; Cai et al., 2023). In addition, temperate phages dominate in the deep-sea as indicated by both metagenomic analysis and viral lysogenetic production experiments (Cai et al., 2023). The prevalence of temperate phages reflects the challenges faced by viral population in sustaining the population, probably due to the low abundance and activity of hosts (Breitbart, 2012). Considered the extensive survival pressure they counter, the bacterial hosts and bacteriophages in the deep-sea experience high resistance costs, and therefore they might adhere to the FSD model, as indicated by the observed relatively narrow host range and elevated rRSCU values in our study.
3.5 The genes relevant to host-phage interaction and environmental adaptation
3.5.1 DNA methyltransferases
The vB_LagS-V1 genome encompasses several DNA methyltransferases, including a methyltransferase (ORF 46), a 5’-cytosine methyltransferase (ORF 54), and a DNA adenine methylase (ORF 55). Methyltransferases are typically encoded by bacteriophages to modify their DNA. This modification was widely considered to help the phage to evade host restriction endonucleases, facilitating viral resistance to the host restriction-modification (R-M) systems (Kobayashi, 2001; Wion and Casadesús, 2006; Pouillot et al., 2007). However, when searching for the R-M system in the host genome using rmsFinder, none R-M system was detected (Roberts et al., 2015), leading us to reconsider the reasoning behind the presence of methyltransferases in vB_LagS-V1. Recent analysis revealed that temperate phages are more prone to encoding methyltransferases compared to virulent phages (Kozlova et al., 2023), consistent with the observed prevalence of methyltransferases in the temperate phage, vB_LagS-V1. The over-representation of methyltransferases in temperate phages implies that methyltransferases more likely help the temperate phage to sustain the lysogenic state, and potentially provide protection to the host against infection by other phages (Oliveira et al., 2014; Kozlova et al., 2023).
3.5.2 Adaptation to deep-sea environments
In vB_LagS-V1 genome, a protein associated with cysteine metabolism, cysteine dioxygenase (ORF 44) was found. Cysteine dioxygenase (CDO) serves a crucial role in the regulating cysteine levels and producing essential oxidized cysteine metabolites, including sulfate and taurine (Dominy et al., 2006; Joseph and Maroney, 2007; Stipanuk et al., 2009). Previous studies have shown that taurine, in addition to being utilized by marine prokaryotes as a primary carbon source and energy provider, is also employed by many marine prokaryotes to serve as an osmoprotectant (Clifford et al., 2019). Although the host of vB_LagS-V1 was isolated at a depth of 4,000 m in the Mariana Trench, we did not find additional CDO in the host, except the one encoded by the vB_LagS-V1. Further analysis on the distribution of this protein in the Tara Ocean and Malaspina Gene Database revealed that the homologs of CDO were primarily discovered in the deep-sea region, specifically at depths ranging from 2,000 m to 4,000 m (Figure 5A). Interestingly, the other two members within the genus Labrenmarinevirus, IMGVR_UViG_2884514219_000004 and IMGVR_UViG_2884552541_000003, which were found in a marine aquarium tank, do not contain CDO in their genomes, implying that the presence of CDO might be associated with environments where the phages exist. Although CDO was also found in Marinobacter phage vB_MalS-PS3 isolated from surface seawater (Liu et al., 2021), it is possible that CDO could be a potential AMG for deep-sea phages by synthesizing taurine, which might assist in maintaining the osmotic balance of the host and adapting to the deep-sea environments.
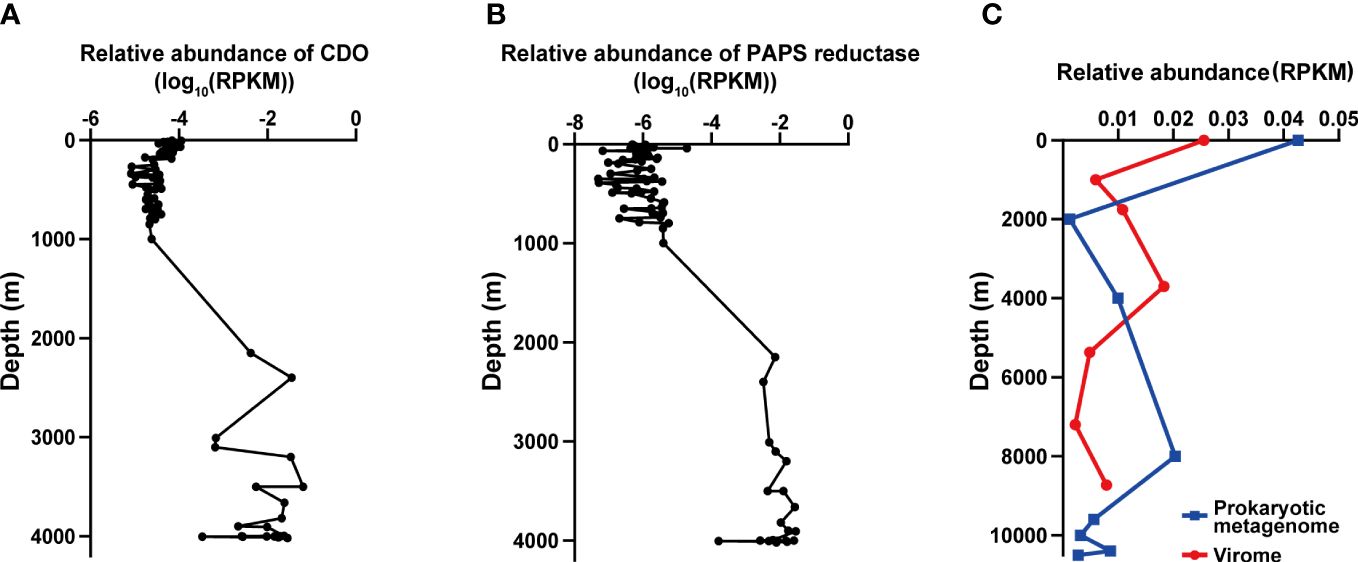
Figure 5 The distribution of vB_LagS-V1 and its genes related to environmental adaptation (A, B). The biogeographical distribution of cysteine dioxygenase (CDO, (A)) and phosphoadenosine phosphosulfate reductase (PAPS reductase, (B)) in the Tara Ocean and Malaspina Gene Database. (C) Biogeographical distribution of vB_LagS-V1 in the Mariana Trench. The blue line represents the prokaryotic metagenome dataset, and the red line represents the viromic metagenome dataset. RPKM represents reads per kilobase per million mapped reads.
The vB_LagS-V1 genome also encodes phosphoadenosine phosphosulfate (PAPS) reductase homolog (ORF 32). Further metagenomic searches revealed that this specific phage PAPS reductase was predominantly found in bathypelagic and abyssopelagic zones, specifically in the range from 2,000 m to 4,000 m (Figure 5B). PAPS reductase is involved in reducing 3′-phosphoadenylylsulfate to phosphoadenosine-phosphate, consequently assimilating sulfates for the biosynthesis of sulfur-containing amino acids, methionine and cysteine (Bick et al., 2000). PAPS reductase has been identified in various aquatic bacteriophages infecting members within Pseudoalteromonas (Sun et al., 2023), SAR11 clade (Du et al., 2021) and archaea (Mizuno et al., 2019). Viral-encoded PAPS reductase has also been found in diverse marine and sediment environments according to metagenomic analysis, including oxygen-deficient water columns (Mara et al., 2020), cold seeps (Li et al., 2021), and deep-sea trenches (Zhao et al., 2022). The viral-encoded PAPS reductase is considered to provide selective advantages to the host by enhancing the biosynthesis of sulfur-containing amino acids (Mara et al., 2020). The host of vB_LagS-V1, L. aggregata RF14, contains the key DMSP biosynthetic gene dsyB, which could utilize the sulfur-containing amino acid methionine to synthesize DMSP (Zheng et al., 2020; Zhong et al., 2021). Bacterial biosynthesis DMSP is believed to protect the bacteria against the osmotic pressure in the deep-sea. Therefore, the vB_LagS-V1 encoded PAPS reductase might contribute to the host’s survival in the high-hydrostatic-pressure environments of the Mariana Trench by providing additional methionine for DMSP biosynthesis.
3.6 Ecological distribution
In our exploration of the biogeographic distribution of vB_LagS-V1, we focused on assessing its relative abundance within its native habitat, the Mariana Trench. Given that vB_LagS-V1 operates as a prophage, initially integrating into the host genome, our analytical framework encompasses both prokaryotic metagenomes (representing intracellular phages, which may manifest as prophages or actively replicating viruses) and viromes (encompassing extracellular phages, which may exist as free-living viruses).
Overall, there was no notable distinction observed in the distribution of vB_LagS-V1 between intracellular and extracellular environments. The abundance of viruses is highest in the surface seawater, gradually decreasing thereafter, but experiencing a subsequent increase in deep seawater (Figure 5C). However, intracellular and extracellular vB_LagS-V1 exhibit different distributions in the deep sea. For intracellular vB_LagS-V1, abundance gradually increases from around 2,000 m, peaking at approximately 8,000 m. In contrast, the extracellular phage reaches its peak abundance in the deep sea around 4,000 m, with lowest abundance in the region below 6,000 m (Figure 5C). Considering that RF14, the lysogenic host of vB_LagS-V1, was isolated from deep-sea environments, it is reasonable to observe a peak of vB_LagS-V1 in the deep-sea prokaryotic metagenome (Zhong et al., 2021). The detection of vB_LagS-V1 in the deep-sea viral community implies the potential induction of vB_LagS-V1 within deep-sea environments, leading to its shift into a lytic cycle. However, the specific factors that could trigger the induction of vB_LagS-V1 in the deep sea remain unclear. Given the constrained presence of mitomycin C in the deep-sea environments, factors such as high pressure, low temperature, or limited nutrient availability, should be taken into consideration (Danovaro et al., 2014).
4 Conclusion
Viruses play a crucial role in deep-sea ecosystems, yet the isolation of deep-sea viruses remains limited, largely due to methodological and cultivation constraints. In this study, we successfully induced a siphovirus, vB_LagS-V1, from the deep-sea bacterium L. aggregata RF14, isolated from a depth of 4,000 meters in the Mariana Trench. Extensive physiological and genomic investigations were undertaken to characterize vB_LagS-V1. Whole-genome alignment results revealed vB_LagS-V1 as a novel bacteriophage. Further phylogenetic analysis found that it formed a new family, Hyphoviridae, with many phages infecting the order Hyphomicrobiales. Moreover, vB_LagS-V1 could be assigned as the representative of the newly classified genus, Labrenmarinevirus, within this new family along with two uncultured prophages within Labrenzia. High regression correlation of codon usage frequencies and a relatively limited host range were identified in vB_LagS-V1, also discovered in some deep-sea phages, suggesting a unique interaction between some deep-sea phages and their hosts. In addition, the presence of CDO and PAPS reductase in the genome of vB_LagS-V1 might help itself and its host adapt to the high-pressure conditions of the deep-sea environments. The distribution of vB_LagS-V1 in the Mariana Trench revealed its prevalence in both prokaryotic metagenomes and viromes. Our in-depth exploration of this new phage enriched the diversity of deep-sea viruses and provided a profound understanding of their adaptation mechanisms to the host and the challenging deep-sea environments.
Data availability statement
The datasets presented in this study can be found in online repositories. The genome of vB_LagS-V1 is available from NCBI GenBank database under accession number PP058680. Other data generated or analyzed during this study were included in the published article and Supplementary Information files.
Author contributions
HW: Conceptualization, Writing – review & editing, Writing – original draft, Visualization, Validation, Software, Methodology, Investigation, Formal analysis, Data curation. BG: Writing – review & editing, Validation, Software, Investigation, Formal analysis, Methodology, Data curation, Conceptualization. JL: Writing – review & editing, Methodology, Investigation, Data curation. YFZ: Writing – review & editing, Project administration, Methodology, Data curation, Conceptualization. X-HZ: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization. YCZ: Writing – review & editing, Validation, Supervision, Resources, Project administration, Methodology, Funding acquisition, Data curation, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (42276108), the Young Scientists Fund of Natural Science Foundation of Shandong Province, China (ZR2022QD052), and the Marine S & T Fund of Shandong Province for Qingdao Marine Science and Technology Center (2022QNLM030004).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1375684/full#supplementary-material
Supplementary Figure 1 | The infectivity of the induced phage on the host. The induced phage is still able to infect the original host L. aggregata RF14 and form clear plaque (A). The addition of chloroform does not affect this infectivity (B).
Supplementary Figure 2 | Whole-genome phylogeny based on the ANI of vB_LagS-V1 and phylogenetically closely related phages in the family, Hyphoviridae.
References
Agrawal A., Lively C. M. (2002). Infection genetics: gene-for-gene versus matching- alleles models and all points in between. Evolutionary Ecol. Res. 4, 79–90.
Besemer J. (1999). Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 27, 3911–3920. doi: 10.1093/nar/27.19.3911
Bick J. A., Dennis J. J., Zylstra G. J., Nowack J., Leustek T. (2000). Identification of a new class of 5′-adenylylsulfate (APS) reductases from sulfate-assimilating bacteria. J. Bacteriol 182, 135–142. doi: 10.1128/JB.182.1.135-142.2000
Breitbart M. (2012). Marine viruses: Truth or dare. Annu. Rev. Mar. Sci. 4, 425–448. doi: 10.1146/annurev-marine-120709-142805
Breitbart M., Bonnain C., Malki K., Sawaya N. A. (2018). Phage puppet masters of the marine microbial realm. Nat. Microbiol. 3, 754–766. doi: 10.1038/s41564-018-0166-y
Bushnell B. (2014). BBMap: a fast, accurate, splice-aware aligner (Berkeley, CA (United States: Lawrence Berkeley National Lab. (LBNL).
Cai L., Weinbauer M. G., Xie L., Zhang R. (2023). The smallest in the deepest: the enigmatic role of viruses in the deep biosphere. Natl. Sci. Rev. 10, nwad009. doi: 10.1093/nsr/nwad009
Camargo A. P., Nayfach S., Chen I.-M. A., Palaniappan K., Ratner A., Chu K., et al. (2023). IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 51, D733–D743. doi: 10.1093/nar/gkac1037
Chen F., Wang K., Stewart J., Belas R. (2006). Induction of multiple prophages from a marine bacterium: A genomic approach. Appl. Environ. Microbiol. 72, 4995–5001. doi: 10.1128/AEM.00056-06
Chithambaram S., Prabhakaran R., Xia X. (2014). Differential Codon Adaptation between dsDNA and ssDNA Phages in Escherichia coli. Mol. Biol. Evol. 31, 1606–1617. doi: 10.1093/molbev/msu087
Clifford E. L., Varela M. M., De Corte D., Bode A., Ortiz V., Herndl G. J., et al. (2019). Taurine is a major carbon and energy source for marine prokaryotes in the North Atlantic Ocean off the Iberian Peninsula. Microb. Ecol. 78, 299–312. doi: 10.1007/s00248-019-01320-y
Curson A. R. J., Liu J., Bermejo Martínez A., Green R. T., Chan Y., Carrión O., et al. (2017). Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat. Microbiol. 2, 17009. doi: 10.1038/nmicrobiol.2017.9
Danovaro R., Snelgrove P. V. R., Tyler P. (2014). Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 29, 465–475. doi: 10.1016/j.tree.2014.06.002
Decaestecker E., Gaba S., Raeymaekers J. A. M., Stoks R., Van Kerckhoven L., Ebert D., et al. (2007). Host–parasite ‘Red Queen’ dynamics archived in pond sediment. Nature 450, 870–873. doi: 10.1038/nature06291
Dominy J. E., Simmons C. R., Karplus P. A., Gehring A. M., Stipanuk M. H. (2006). Identification and characterization of bacterial cysteine dioxygenases: A new route of cysteine degradation for eubacteria. J. Bacteriol 188, 5561–5569. doi: 10.1128/JB.00291-06
Du S., Qin F., Zhang Z., Tian Z., Yang M., Liu X., et al. (2021). Genomic diversity, life strategies and ecology of marine HTVC010P-type pelagiphages. Microbial Genomics 7. doi: 10.1099/mgen.0.000596
Gandon S., Buckling A., Decaestecker E., Day T. (2008). Host–parasite coevolution and patterns of adaptation across time and space. J. Evolutionary Biol. 21, 1861–1866. doi: 10.1111/j.1420-9101.2008.01598.x
Gao C., Liang Y., Jiang Y., Paez-Espino D., Han M., Gu C., et al. (2022). Virioplankton assemblages from challenger deep, the deepest place in the oceans. iScience 25, 104680. doi: 10.1016/j.isci.2022.104680
Göker M., García-Blázquez G., Voglmayr H., Tellería M. T., Martín M. P. (2009). Molecular taxonomy of phytopathogenic fungi: A case study in peronospora. PLoS One 4, e6319. doi: 10.1371/journal.pone.0006319
Gregory A. C., Zayed A. A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., et al. (2019). Marine DNA viral macro-and microdiversity from pole to pole. Cell 177, 1109–1123. doi: 10.1016/j.cell.2019.03.040
Guidi L., Chaffron S., Bittner L., Eveillard D., Larhlimi A., Roux S., et al. (2016). Plankton networks driving carbon export in the oligotrophic ocean. Nature 532, 465–470. doi: 10.1038/nature16942
Hewson I., O’Neil J. M., Fuhrman J. A., Dennison W. C. (2001). Virus-like particle distribution and abundance in sediments and overlying waters along eutrophication gradients in two subtropical estuaries. Limnology Oceanography 46, 1734–1746. doi: 10.4319/lo.2001.46.7.1734
Hyatt D., Chen G.-L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Jian H., Xiong L., Xu G., Xiao X. (2016). Filamentous phage SW1 is active and influences the transcriptome of the host at high-pressure and low-temperature. Environ. Microbiol. Rep. 8, 358–362. doi: 10.1111/1758-2229.12388
Johnson R. M. (1968). Characteristics of a marine vibrio-bacteriophage system. J. Arizona Acad. Sci. 5, 28. doi: 10.2307/40022820
Joseph C. A., Maroney M. J. (2007). Cysteine dioxygenase: structure and mechanism. Chem. Commun. 32, 3338–3349. doi: 10.1039/b702158e
Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., Von Haeseler A., Jermiin L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
King G. M. (2003). Molecular and culture-based analyses of aerobic carbon monoxide oxidizer diversity. Appl. Environ. Microbiol. 69, 7257–7265. doi: 10.1128/AEM.69.12.7257-7265.2003
King G. M., Weber C. F. (2007). Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat. Rev. Microbiol. 5, 107–118. doi: 10.1038/nrmicro1595
Kobayashi I. (2001). Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29, 3742–3756. doi: 10.1093/nar/29.18.3742
Kozlova A. P., Saksaganskaia A. S., Afonin A. M., Muntyan V. S., Vladimirova M. E., Dzyubenko E. A., et al. (2023). A temperate sinorhizobium phage, AP-16-3, closely related to phage 16-3: Mosaic genome and prophage analysis. Viruses 15 1701. doi: 10.3390/v15081701
Lee I., Ouk Kim Y., Park S.-C., Chun J. (2016). OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Systematic Evolutionary Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Li Z., Pan D., Wei G., Pi W., Zhang C., Wang J.-H., et al. (2021). Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. ISME J. 15, 2366–2378. doi: 10.1038/s41396-021-00932-y
Liu Y., Zheng K., Liu B., Liang Y., You S., Zhang W., et al. (2021). Characterization and genomic analysis of Marinobacter phage vb_mals-ps3, representing a new lambda-like temperate siphoviral genus infecting algae-associated bacteria. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.726074
Lopez Pascua L., Hall A. R., Best A., Morgan A. D., Boots M., Buckling A. (2014). Higher resources decrease fluctuating selection during host–parasite coevolution. Ecol. Lett. 17, 1380–1388. doi: 10.1111/ele.12337
Lorenz N., Reiger M., Toro-Nahuelpan M., Brachmann A., Poettinger L., Plener L., et al. (2016). Identification and initial characterization of prophages in Vibrio campbellii. PLoS One 11, e0156010. doi: 10.1371/journal.pone.0156010
Lowe T. M., Eddy S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Mara P., Vik D., Pachiadaki M. G., Suter E. A., Poulos B., Taylor G. T., et al. (2020). Viral elements and their potential influence on microbial processes along the permanently stratified Cariaco Basin redoxcline. ISME J. 14, 3079–3092. doi: 10.1038/s41396-020-00739-3
Meier-Kolthoff J. P., Auch A. F., Klenk H.-P., Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff J. P., Göker M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Meier-Kolthoff J. P., Hahnke R. L., Petersen J., Scheuner C., Michael V., Fiebig A., et al. (2014). Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 9, 2. doi: 10.1186/1944-3277-9-2
Mercier C., Lossouarn J., Nesbø C. L., Haverkamp T. H. A., Baudoux A. C., Jebbar M., et al. (2018). Two viruses, MCV1 and MCV2, which infect Marinitoga bacteria isolated from deep-sea hydrothermal vents: functional and genomic analysis. Environ. Microbiol. 20, 577–587. doi: 10.1111/1462-2920.13967
Mizuno C. M., Prajapati B., Lucas-Staat S., Sime-Ngando T., Forterre P., Bamford D. H., et al. (2019). Novel haloarchaeal viruses from Lake Retba infecting Haloferax and Halorubrum species. Environ. Microbiol. 21, 2129–2147. doi: 10.1111/1462-2920.14604
Nguyen L.-T., Schmidt H. A., Von Haeseler A., Minh B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Oliveira P. H., Touchon M., Rocha E. P. C. (2014). The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 42, 10618–10631. doi: 10.1093/nar/gku734
Ordogh F., Szende K. (1961). Temperate bacteriophages isolated from rhizobium meliloti. Acta Microbiol. Acad. Sci. Hung. 7, 65–71.
Pouillot F., Fayolle C., Carniel E. (2007). A putative DNA adenine methyltransferase is involved in Yersinia pseudotuberculosis pathogenicity. Microbiology 153, 2426–2434. doi: 10.1099/mic.0.2007/005736-0
Priyanka P., Arun A. B., Rekha P. D. (2014). Sulfated exopolysaccharide produced by Labrenzia sp. PRIM-30, characterization and prospective applications. Int. J. Biol. Macromolecules 69, 290–295. doi: 10.1016/j.ijbiomac.2014.05.054
Prjibelski A., Antipov D., Meleshko D., Lapidus A., Korobeynikov A. (2020). Using SPAdes de novo assembler. CP Bioinf. 70, e102. doi: 10.1002/cpbi.102
Pruitt K. D., Tatusova T., Maglott D. R. (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65. doi: 10.1093/nar/gkl842
Roberts M. D., Martin N. L., Kropinski A. M. (2004). The genome and proteome of coliphage T1. Virology 318, 245–266. doi: 10.1016/j.virol.2003.09.020
Roberts R. J., Vincze T., Posfai J., Macelis D. (2015). REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. doi: 10.1093/nar/gku1046
Rohwer F., Segall A., Steward G., Seguritan V., Breitbart M., Wolven F., et al. (2000). The complete genomic sequence of the marine phage Roseophage SIO1 shares homology with nonmarine phages. Limnology Oceanography 45, 408–418. doi: 10.4319/lo.2000.45.2.0408
Rohwer F., Thurber R. V. (2009). Viruses manipulate the marine environment. Nature 459, 207–212. doi: 10.1038/nature08060
Roux S., Brum J. R., Dutilh B. E., Sunagawa S., Duhaime M. B., Loy A., et al. (2016). Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537, 689–693. doi: 10.1038/nature19366
Roux S., Emerson J. B., Eloe-Fadrosh E. A., Sullivan M. B. (2017). Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 5, e3817. doi: 10.7717/peerj.3817
Santamaría R. I., Bustos P., Van Cauwenberghe J., González V. (2022). Hidden diversity of double-stranded DNA phages in symbiotic Rhizobium species. Phil. Trans. R. Soc B 377, 20200468. doi: 10.1098/rstb.2020.0468
Sasaki A. (2000). Host-parasite coevolution in a multilocus gene-for-gene system. Proc. R. Soc Lond. B 267, 2183–2188. doi: 10.1098/rspb.2000.1267
Steven A. F. (1993). Specificity versus detectable polymorphism in host–parasite genetics. Proc. R. Soc Lond. B 254, 191–197. doi: 10.1098/rspb.1993.0145
Stipanuk M. H., Ueki I., Dominy J. E., Simmons C. R., Hirschberger L. L. (2009). Cysteine dioxygenase: a robust system for regulation of cellular cysteine levels. Amino Acids 37, 55. doi: 10.1007/s00726-008-0202-y
Su Y., Zhang W., Liang Y., Wang H., Liu Y., Zheng K., et al. (2023). Identification and genomic analysis of temperate Halomonas bacteriophage vB_HmeY_H4907 from the surface sediment of the Mariana Trench at a depth of 8,900 m. Microbiol. Spectr. 11(5), e01912–e01923. doi: 10.1128/spectrum.01912-23
Sun J., Zhang X., Liang Y., Zheng K., Kennedy F., Han M., et al. (2023). Abundance and ecological footprint of Pseudoalteromonas phage vB_PhoS_XC in the Ulva prolifera green tide. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1201434
Sunagawa S., Acinas S. G., Bork P., Bowler C., Eveillard D., Gorsky G., et al. (2020). Tara Oceans: towards global ocean ecosystems biology. Nat. Rev. Microbiol. 18, 428–445. doi: 10.1038/s41579-020-0364-5
Tang K., Lin D., Zheng Q., Liu K., Yang Y., Han Y., et al. (2017). Genomic, proteomic and bioinformatic analysis of two temperate phages in Roseobacter clade bacteria isolated from the deep-sea water. BMC Genomics 18, 485. doi: 10.1186/s12864-017-3886-0
Turner D., Kropinski A. M., Adriaenssens E. M. (2021). A roadmap for genome-based phage taxonomy. Viruses 13, 506. doi: 10.3390/v13030506
Vernette C., Lecubin J., Sánchez P., Coordinators T. O., Acinas S. G., Babin M., et al. (2022). The Ocean Gene Atlas v2.0: online exploration of the biogeography and phylogeny of plankton genes. Nucleic Acids Res. 50, W516–W526. doi: 10.1093/nar/gkac420
Wang F., Wang F., Li Q., Xiao X. (2007). A Novel Filamentous Phage from the Deep-Sea Bacterium Shewanella piezotolerans WP3 Is Induced at Low Temperature. J. Bacteriol 189, 7151–7153. doi: 10.1128/JB.00569-07
Weinbauer M. G. (2004). Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 28, 127–181. doi: 10.1016/j.femsre.2003.08.001
Wilhelm S. W., Suttle C. A. (1999). Viruses and nutrient cycles in the sea: viruses play critical roles in the structure and function of aquatic food webs. Bioscience 49, 781–788. doi: 10.2307/1313569
Wion D., Casadesús J. (2006). N6-methyl-adenine: an epigenetic signal for DNA–protein interactions. Nat. Rev. Microbiol. 4, 183–192. doi: 10.1038/nrmicro1350
Xie J., Chen Y., Cai G., Cai R., Hu Z., Wang H. (2023). Tree Visualization By One Table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592. doi: 10.1093/nar/gkad359
Xue C.-X., Liu J., Lea-Smith D. J., Rowley G., Lin H., Zheng Y., et al. (2020). Insights into the vertical stratification of microbial ecological roles across the deepest seawater column on Earth. Microorganisms 8, 1309. doi: 10.3390/microorganisms8091309
Yang Y., Cai L., Ma R., Xu Y., Tong Y., Huang Y., et al. (2017). A novel roseosiphophage isolated from the oligotrophic South China Sea. Viruses 9, 109. doi: 10.3390/v9050109
Yang Y., Ma R., Yu C., Ye J., Chen X., Wang L., et al. (2022). A novel alteromonas phage lineage with a broad host range and small burst size. Microbiol. Spectr. 10, e01499–e01422. doi: 10.1128/spectrum.01499-22
Yoshida M., Yoshida-Takashima Y., Nunoura T., Takai K. (2015a). Genomic characterization of a temperate phage of the psychrotolerant deep-sea bacterium Aurantimonas sp. Extremophiles 19, 49–58. doi: 10.1007/s00792-014-0702-5
Yoshida M., Yoshida-Takashima Y., Nunoura T., Takai K. (2015b). Identification and genomic analysis of temperate Pseudomonas bacteriophage PstS-1 from the Japan trench at a depth of 7000 m. Res. Microbiol. 166, 668–676. doi: 10.1016/j.resmic.2015.05.001
Zhao J., Jing H., Wang Z., Wang L., Jian H., Zhang R., et al. (2022). Novel viral communities potentially assisting in carbon, nitrogen, and sulfur metabolism in the upper slope sediments of mariana trench. mSystems 7, e01358–e01321. doi: 10.1128/msystems.01358-21
Zhao X., Liu J., Zhou S., Zheng Y., Wu Y., Kogure K., et al. (2020). Diversity of culturable heterotrophic bacteria from the Mariana Trench and their ability to degrade macromolecules. Mar. Life Sci. Technol. 2, 181–193. doi: 10.1007/s42995-020-00027-1
Zheng Y., Wang J., Zhou S., Zhang Y., Liu J., Xue C.-X., et al. (2020). Bacteria are important dimethylsulfoniopropionate producers in marine aphotic and high-pressure environments. Nat. Commun. 11, 4658. doi: 10.1038/s41467-020-18434-4
Zhong H., Sun H., Liu R., Zhan Y., Huang X., Ju F., et al. (2021). Comparative genomic analysis of labrenzia aggregata (Alphaproteobacteria) strains isolated from the mariana trench: Insights into the metabolic potentials and biogeochemical functions. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.770370
Keywords: bacteriophage induction, deep-sea, genomic analysis, Labrenzia aggregata, Labrenmarinevirus
Citation: Gu B, Wang H, Lv J, Zheng Y, Zhang X-H and Zhan Y (2024) Identification and genomic analysis of a novel temperate bacteriophage infecting Labrenzia aggregata isolated from the Mariana Trench. Front. Mar. Sci. 11:1375684. doi: 10.3389/fmars.2024.1375684
Received: 24 January 2024; Accepted: 06 March 2024;
Published: 26 March 2024.
Edited by:
Wen-Jun Li, Sun Yat-sen University, ChinaReviewed by:
Janina Rahlff, Friedrich Schiller University Jena, GermanyMarike Palmer, University of Nevada, Las Vegas, United States
Copyright © 2024 Gu, Wang, Lv, Zheng, Zhang and Zhan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanchao Zhan, zhanyuanchao@ouc.edu.cn
†These authors have contributed equally to this work and share first authorship