Prokaryotic community structure and key taxa in the Arabian Sea’s oxygen minimum zone
 Ding Li1,2,3†
Ding Li1,2,3†  Liping Wang2†
Liping Wang2†  Fan Jiang1,2,3
Fan Jiang1,2,3  Xiang Zeng2
Xiang Zeng2  Qinzeng Xu4
Qinzeng Xu4  Xuelei Zhang4
Xuelei Zhang4  Qiang Zheng1,3 Zongze Shao2*
Qiang Zheng1,3 Zongze Shao2*- 1State Key Laboratory for Marine Environmental Science, Institute of Marine Microbes and Ecospheres, College of Ocean and Earth Sciences, Xiamen University, Xiamen, China
- 2Key Laboratory of Marine Genetic Resources, Third Institute of Oceanography, Ministry of Natural Resources of PR China, State Key Laboratory Breeding Base of Marine Genetic Resources; Fujian Key Laboratory of Marine Genetic Resources, Xiamen, China
- 3Fujian Key Laboratory of Marine Carbon Sequestration, Xiamen University, Xiamen, China
- 4First Institute of Oceanography, Ministry of Natural Resources, Qingdao, Shandong, China
Microbial communities within oxygen minimum zones (OMZs) play crucial roles in the marine biogeochemical cycling. Arabian Sea (AS) has one of the largest OMZs among the global oceans, however, knowledge about the microbial ecology of the AS OMZ remained limited. In the present study, 44 water samples collected from six stations across the AS, spanning from the deep chlorophyll maximum (DCM) layer to 4000m depth were analyzed. High-throughput sequencing of 16S rRNA genes revealed the structural diversity of bacterial and archaeal communities, influenced primarily by depth and dissolved oxygen (DO) levels. Distinct community compositions were observed across different oxygen gradients, with shifts in the relative abundance of key taxa. Notably, Desulfosarcinaceae, UBA10353, Nitrospina, SUP05, Sva0996_marine_group, Microtrichaceae, and Nitrosopumilus emerged as bioindicator taxa in the AS hypoxic zones. Co-occurrence network analysis identified SAR324, Alteromonadaceae, and Sphingomonadaceae as keystone taxa. The spatial and depth-wise distribution patterns revealed that Desulfosarcinaceae was predominantly found in the hypoxic zones of the Arabian Sea, whereas UBA10353, Nitrospina, SUP05, Microtrichaceae and SAR324 were ubiquitous across AS, Bay of Bengal (BOB), and Eastern Tropical North Pacific (ETNP) OMZs, with OTU-level niche differentiation observed for the latter two. Functional profiling using FAPROTAX predicted higher metabolic potential for nitrogen and sulfur in the OMZ compared to other layers of the AS. Our findings provide valuable insights into the distribution, structure, and diversity of microbial communities in the AS OMZ, highlighting the ecological roles of key taxa in hypoxic environments. The established sequence database offers a foundation for further research into the complex interactions within these microbial ecosystems.
1 Introduction
The oceans boast a captivating ecosystem, replete with diverse habitats like cold seeps, hydrothermal vents, and oxygen minimum zones (OMZs) (Li et al., 2021b; Baumgartner et al., 2022; Chen et al., 2022). The microbial community inhabiting each habitat play a pivotal role in governing the oceans’ biogeochemical cycles, actively engaging in nearly all oceanic biogeochemical processes. Understanding the ecological roles and functions of marine microbes requires thorough investigation of their biodiversity. Till now, extensive research has been conducted on the microbial diversity of the Indian Ocean. AS is a biologically productive tropical basin in Indian Ocean, located between the Horn of Africa and southern Asia, with the Gulf of Oman in the north and the Red Sea through the Gulf of Aden in the west. The northern border between Pakistan and Iran is a world-class transportation hub. Covering an area of 3.86 million square kilometers, with a maximum depth of 5203m and an average depth of 2734m (Corfield et al., 2010).
Ocean models predict that the dissolved oxygen levels in the global ocean will decline by 1% to 7% by 2100 due to a combination of warming-induced decreases in oxygen solubility and reduced deep-sea ventilation (Long et al., 2016). AS, Eastern Tropical North Pacific (ETNP), Eastern Tropical South Pacific (ETSP), and Bay of Bengal (BOB) form the major OMZs of the world ocean (Ulloa and Pantoja, 2009). There are currently three definitions of ocean hypoxia thresholds: hypoxia (< ∼ 63 µM), microbial hypoxia (< ∼ 20 µM) and functional anoxia (< ∼ 0.05 µM), and hypoxia zone throughout the water column is often detected at depths between 200 meters and 1,000 meters (Paulmier and Diana, 2009; Rixen et al., 2020). The total volume of waters characterized by oxygen concentrations below 20 µM in the global ocean estimated to be 15 × 1015 m3, among of which, the northern Indian Ocean accounts for 21%, equivalent to 3.13 × 1015 m3. As a classic hypoxic zone, Arabian Sea hosts the largest proportion of this oxygen-deficient water body, accounting for approximately 2.5 × 1015 m3, while only a small fraction is found in the Bay of Bengal (0.6 × 1015 m3) (Acharya and Panigrahi, 2016). Earlier reports suggested that OMZ regions in the Northern Indian Ocean (AS and BOB combined) made up more than 50% of the global OMZs (Helly and Levin, 2004). The formation of OMZs in the Arabian Sea is linked to inadequate ventilation from the ocean surface to the interiors, along with elevated oxygen consumption due to intense biological productivity. Coinciding with an active denitrification zone, the AS_OMZ is among the largest anoxic regions in the oceans, sometimes DO drops as low as 0.1μM (Gilles-Gonzalez, 2001; Stramma et al., 2008). Understanding the vital role of microbial communities in the Arabian Sea is crucial for unraveling the global biogeochemical and climatic processes mediated by these microbes. Generally, most marine microbes are uncultivable, and with the advent of molecular methods and high-throughput sequencing, methodological advances are more conducive to exploring the biodiversity and biogeography of microorganisms in different sea (Kaeberlein et al., 2002; Puspita et al., 2012; Jin et al., 2022).
In OMZ, element cycling is primarily mediated by special microorganisms. Recurrent reports have highlighted the prevalence of Proteobacteria as the dominant phylum among the microbial communities across the Indian Ocean. Other taxa at class level including the Betaproteobacteria, Deltaproteobacteria, Actinobacteria, Nitrospinia, Planctomycetacia, and SAR406 were also reported, although their relative abundance varied along the depths and locations sampled in the Indian Ocean OMZ (Bandekar et al., 2018; Fernandes et al., 2020; Jin et al., 2022; Li et al., 2022; Rangamaran et al., 2022). Although Proteobacteria predominates in the water column, significant change in the composition of the bacterial community was observed and varied with depth and DO levels in the water and sediment samples in the Arabian Sea (Rangamaran et al., 2022). Previous reports based on clone libraries revealed the vertical partitioning of bacterial communities between the surface and OMZ at the Arabian Sea Time Series (ASTS) location, however no apparent seasonal variation among OMZ bacterial communities (Bandekar et al., 2016, 2018). By comparing the prokaryotic community composition of the ASTS, India’s Idea 2 station (II2) in AS and the BoB Time Series in the BOB, higher relative abundance of bacteria involved in anammox was observed in ASTS and II2 stations (Fernandes et al., 2020). In a recent report, distinct microbial community compositions of OZ and OMZ waters were observed in 131 samples collected from the Andaman Sea and eastern BoB epipelagic waters from 2 to 200 m, and important bioindicator taxa across oxygen gradients were identified (Guo et al., 2022). While several studies have documented the microbial community composition within the Arabian Sea, the majority have concentrated on particular locations or water layers (such as epipelagic waters). A comparative analysis with other significant oxygen minimum zones (OMZs) across the world’s oceans, including the Bay of Bengal (BOB) and the Eastern Tropical North Pacific (ETNP), remains largely absent. Consequently, a thorough understanding of microbial diversity and distribution in the OMZ regions of the Arabian Sea, particularly transitioning from typical hypoxic to non-hypoxic zones, is yet to be fully elucidated.
With the rapid development of molecular methods, high-throughput sequencing technologies have been widely applied in the exploring of biodiversity of microorganisms using targeted sequencing of conserved regions (e.g., 16S or 18S rRNA gene sequencing) or shotgun metagenomics. There are currently some challenges for interpretation of biological information in targeted sequencing approach, due to the limitation such as the primer bias or short sequence amplification. The high costs associated with deep sequencing in shotgun metagenomics also limits full exploitation. Although the information provided by amplicon sequencing of 16S rRNA genes are not enough to interpret the ecological functions of microbial community, it is still an effective and economic approach to characterize the microbial diversity and structure in large number of samples (Lynch and Neufeld, 2015; de Muinck et al., 2017). The composition of environmental bacterial communities are usually complex, so identifying crucial bioindicator taxa would help to understand the community structure and functional differences in specific habitats. In addition to identifying bioindicators, keystone taxa with high degree and tight centrality of microbial interaction networks also play an important role in driving the microbial community structure and maintaining the network stability (Banerjee et al., 2018). Therefore, in order to comprehensively understand the performance of prokaryotes community and their ecological significance in the OMZ, it is necessary to integrate the physicochemical properties, community composition, biomarker taxa, and interaction networks.
In this study, we collected a total of 44 water samples from 6 stations across the typical hypoxic zones in northern Arabian Sea, through transition zones, to the non-hypoxic zones in the southern AS, covering depths from the deep chlorophyll maximum (DCM) layer to 4000m near the bottom. This comprehensive sampling spanned regions with different oxygen gradients to explore and compare the spatial and vertical composition of bacterial and archaeal communities. To achieve this, we employed Illumina sequencing of the 16S rRNA genes from the collected samples. Additionally, we downloaded and integrated publicly available 16S rRNA high-throughput sequencing data from the Bay of Bengal and the eastern tropical North Pacific Ocean for a broader analysis. We identified bioindicators and keystone taxa, analyzing their spatial and depth-wise distribution patterns across the Arabian Sea (AS), Bay of Bengal (Bob), and the eastern tropical North Pacific (ETNP). Furthermore, we investigated the major environmental factors influencing the prokaryotic community structure and predicted potential functional profiles. The sequence database established in this study will contribute to a comprehensive understanding of the distribution and structure of microbial communities and their key members within the AS oxygen minimum zone (OMZ).
2 Materials and methods
2.1 Sample collection and methodology
Seawater samples were collected from 6 stations located the Arabian Sea of northern Indian Ocean during China DY72 cruise onboard the Research Vessel Shen Hai Yi Hao. These stations (Latitude: 10.132553°N-17.326766°N, Longitude: 63.103385°E-68.303971°E, water depth: 3550m-4458m) was sampled from 17th May 2022 to 28th May 2022 (Spring intermonsoon), spanning the north to the south of Arabian Sea. Stations of CTD03, CTD04 and CTD05 located in the northern AS were defined as the core OMZ in AS. Stations CTD02 and CTD06 located in the middle of AS were defined as OMZ transition zones, while CTD08 in the southern AS as a non-hypoxia zone. For most stations, the sampling depths included deep chlorophyll maximum (DCM), oxygen minimum zone (OMZ), 1000 m, 2000 m, and the bottom. In total, 44 samples from 6 stations were collected from AS. These samples were divided into 4 groups based on DO and depth, including euphotic zones (AS_UP), OMZ (AS_OMZ), bathypelagic zone (AS_DOWN) and CK (CTD08). DO levels below 20 μM are defined as the AS_OMZ group. Although the DO values of CTD03-100 was 22.1188 µM, but CTD03-100 was still categorized to AS_OMZ group. DO level higher than 20 μM and depth over or equal to 100m is defined as the AS_UP group. Depth below or equal to 1000m is defined as the AS_DOWN group. The detailed information of sampling sites and depths were provided in Figure 1 and Supplementary Table 1.
Figure 1 The geographic locations of 6 sampling stations in AS. The black solid circles represent the investigation stations in Indian Ocean.
Seawater samples were collected using a Conductivity-Temperature-Depth (CTD) rosette system fitted with 24 Niskin bottle sampler (Seabird Electronics, Washington, NY, United States) mounted with an oxygen sensor (Crescentini et al., 2012). Eight liters’ seawater were filtered through 0.22 μm sterivex filters (Millipore, United States). The filter samples were immediately frozen in liquid nitrogen and stored at −80°C until DNA extraction. The nutrient parameters including silicate, phosphate, nitrate, nitrite and ammonium were measured using Skalar Autoanalyser (Skalar Analytical, Netherlands) following standard methods.
2.2 Total genomic DNA extraction and quality inspection
Genomic DNA was isolated using DNeasy PowerSoil Pro Kit following the manufacturer’s protocol (QIAGEN, Germany, www.qiagen.com/insights). The concentration of the extracted DNA was measured using a Qubit 3.0 Fluorometer (Life Technologies, ThermoFisher Scientific, USA) and the Equalbit 1x dsDNA HS Assay Kit (Vazyme, China, www.vazyme.com). Before sequencing, the quality and concentration of DNA were determined again by 1.0% agarose gel electrophoresis and NanoDrop® ND-2000 spectrophotometer (Thermo Scientific Co., Ltd., USA).
2.3 Amplification of 16S rRNA gene
The primer set 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) was used to amplify the V3-V4 region of bacterial 16S rRNA gene. The primer set 524F10extF (5′- TGYCAGCCGCCGCGGTAA -3′) and Arch958RmodR (5′- YCCGGCGTTGAVTCCAATT -3′) was used for the amplify the V4-V5 region of archaeal 16S rRNA gene (Claesson et al., 2009).
PCR reaction was carried out in a 20 μL reaction volume containing DNA (10 ng), 0.8 μL each of primers (10 μM), 4 μL 5x FastPfu buffer, 0.4 μL FastPfu polymerase, BSA 0.2 μL and 250 mM dNTPs (TransGen, China, www.transgen.com/enzyme/226). PCR cycling conditions maintained were as follows: initial denaturation at 95°C for 3 min, followed by cycle denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 45 s for a total of 27 cycles and a final extension for 10 min at 72°C. For archaeal 16S rRNA gene amplification, PCR cycling conditions maintained 35 cycles.
2.4 Illumina sequencing and bioinformatic analysis
Purified amplicons were pooled in equimolar amounts and paired-end sequenced on an Illumina MiSeq PE300 platform/NovaSeq PE250 platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Raw reads of AS in this study were processed using QIIME 2 pipeline. QIIME 2 was used to do downstream analysis of raw sequence data generated by PE250 (Bokulich et al., 2018; Bolyen et al., 2019). In brief, raw sequence data were demultiplexed using the demux plugin, followed by primer trimming using the cutadapt plugin. Subsequently, sequences underwent quality filtering, denoising, merging, and chimera removal utilizing the DADA2 plugin (Martin, 2011; Callahan et al., 2016). Non-singleton amplicon sequence variants (ASVs) were aligned using MAFFT and then utilized to construct a phylogenetic tree with FastTree2 (Katoh et al., 2002; Price et al., 2009). The bacterial samples were standardized to 12,481 sequences per sample, whereas the archaeal samples were standardized to 7,537 sequences per sample. Alpha diversity metrics, including Sobs (number of species observed), Shannon (microbial diversity in the sample), and Coverage (species coverage), and beta diversity metrics (Jaccard distance) were calculated with Mothur v1.30.1 (Schloss et al., 2009). ASVs were taxonomically classified using the classify-sklearn Naïve Bayes taxonomy classifier against the SILVA Release 138 database, encompassing both bacteria and archaea.
Publicly accessible 16S rRNA gene high-throughput sequencing data were obtained from the National Center for Biotechnology Information (NCBI) for the Bay of Bengal (Bob) and the eastern tropical North Pacific Ocean (ETNP). This dataset encompassed 30 samples from the deep chlorophyll maximum (DCM) and oxygen minimum zone (OMZ) layers of the Bay of Bengal, as well as 66 samples from the DCM, OMZ, and 2000m depth layers of the ETNP (Gu et al., 2022; Li et al., 2022). To enhance our comparative analysis, these datasets were integrated with the 16S rRNA gene sequencing data collected from the Arabian Sea (AS) of this study. Utilizing UPARSE 7.1, the sequences were clustered into operational taxonomic units (OTUs) based on a 97% sequence similarity threshold. For each OTU, the most representative sequence in terms of abundance was chosen, following the methodology established previously (Beman et al., 2021; Gu et al., 2022).
2.5 Diversity and statistical analyses
The similarity matrix was used to perform cluster analysis in Primer 6 (PRIMER-E, Plymouth, UK) using a group-average linking method and non-metric multidimensional scaling analysis (NMDS). Screening of environmental parameters using Variance inflation factor (VIF) analysis. Canonical correspondence analysis (CCA) was performed using Past-3 software to assess the influence of environmental parameters on bacterial and archaeal community structure (https://folk.uio.no/ohammer/past/).
Previous research showed that Molecular Ecological Network Analysis Pipeline (MENAP) can construct ecological association networks named molecular ecological networks (MENs) through Random Matrix Theory (RMT)-based methods to explore the interactions between bacterial and archaeal communities (http://ieg2.ou.edu/MENA) (Deng et al., 2012). In this study, we use the network building process given by the NEMAP creator for analysis and select “Regress Poisson distribution only” to ensure that the type of network belongs to Poisson distribution. The parameters used to construct the network were shown in Supplementary Table 4. In this study, we choose the connectors (zi ≤ 2.5, Pi > 0.62) as the keystone in community (Deng et al., 2012; Berry and Widder, 2014). We used the Kruskal-Wallis rank sum test, FDR multiple testing correction, and the Tukey-kramer Post-hoc test (0.95) to resolve differences between each group of species (Schloss et al., 2009).
Prediction of metabolic pathways based on 16S rRNA amplicon data was performed using FAPROTAX, FAPROTAX is a database that maps prokaryotic taxa to metabolic or other ecologically relevant functions based on representative literature in culture (Deng et al., 2012; Louca et al., 2016).
2.6 Nucleotide accession numbers
The raw sequencing reads have been deposited in the NCBI Sequence Read Archive (SRA) database under the accession number PRJNA993832.
3 Results
3.1 Physico-chemical characteristics
Metabarcoding analysis of 44 water samples from 6 CTD stations in Arabian Sea (AS) was carried out in the present study. Samples were collected from subsurface to deep (bathypelagic) water ranging from 62m to 4000m (Figure 1). All the 44 samples could be divided into 4 groups including AS_UP, AS_OMZ and AS_DOWN, as well as control group (CTD08), which were defined by DO concentrations and depth. The physicochemical parameters of the samples were presented in Supplementary Table 1 and Supplementary Figure 1. In AS, the DO concentrations were almost below the detection threshold in the OMZs ranging from about 150m to 1200m in CTD03, CTD04 and CTD05 stations (Supplementary Figure 1). For CTD02 and CTD06 stations, the DO values were also below 20μM in the OMZs from about 150m to 1000m. However, as a control group, CTD08 located far from the mainland, and the DO values in this water columns were above 62 μM, significantly different from other stations.
3.2 Alpha diversity and beta diversity of microbial communities
In this study, a total of 44 bacterial and 43 archaeal sequence data sets were obtained, respectively. About 1.74 million bacterial sequences and 2.14 million archaeal sequences were retrieved from all the samples, resulting in a total of 11174 bacterial and 1628 archaeal ASVs. The alpha diversity of microbial communities in AS was compared among 44 samples (Supplementary Figures 2, 3). The coverage of microbial communities in each group was almost 1.000, and the rarefaction curves were bent toward the saturation plateau, indicating that the sampling sizes were sufficient to reflect the microbial community diversity in the 44 samples (Supplementary Figures 2, 3).
Alpha diversity analysis further showed that bacteria exhibited the highest richness (Kruskal-Wallis rank sum test, Chao1: 123–646), followed by archaea (Kruskal-Wallis rank sum test, Chao1: 6–134). Bacteria still exhibited the highest diversity (Kruskal-Wallis rank sum test, Shannon, 2.550842–5.18124), followed by archaea (Kruskal-Wallis rank sum test, Shannon, 1.524161–3.996356) (Supplementary Figure 3). The bacterial community diversity showed no significant difference among AS_UP, AS_OMZ and AS_DOWN (p=0.1408). For archaea, increased species richness (p=0.02774) and diversity (p=0.001736) were observed in AS_OMZ, compared to AS_UP and AS_DOWN communities (Supplementary Figure 3).
Non-metric multidimensional scaling (NMDS) analysis was also employed to decipher the community differences at ASV level (Taguchi and Oono, 2005). As illustrated by NMDS for bacteria, the microbial community from AS_UP group was significantly separated from AS_OMZ group along the Y-axis (Figure 2). The archaeal community was structured by DO and depth, and clustered according to different groups (Figure 2). The samples from the AS_UP, AS_OMZ and AS_DOWN clustered individually, indicating the compositions of the archaeal communities in these three groups are distinct (Figure 2).
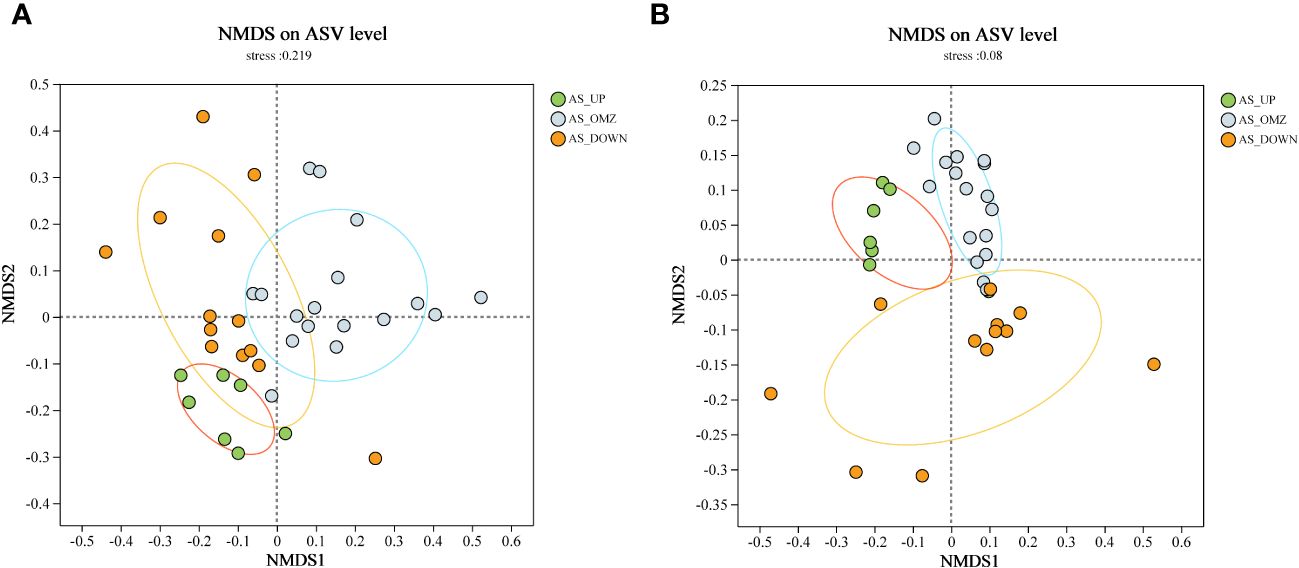
Figure 2 NMDS plot based on Bray-Curtis distance among AS_UP, AS_OMZ and AS_DOWN communities. (A) bacterial communities at ASV level (B) archaeal communities at ASV level.
3.3 Microbial community composition in the water column of Arabian Sea
The composition of the microbial communities of in AS were compared in 44 water samples of 6 stations. For the 3 groups (AS_UP, AS_OMZ and AS_DOWN), 268 shared ASVs of bacteria were identified belonging to 251 shared genera and in respect of the archaea, 109 shared ASVs belonging to 15 shared genera of 6 phyla were identified in 43 samples (Supplementary Figure 4).
Analysis of the bacterial community composition of the 44 samples revealed that Proteobacteria, Actinobacteriota, Bacteroidota, SAR406, Firmicutes, Chloreflexi, Cyanobacteria, SAR324, Nitrospinota were dominant phyla across nearly all samples, although their relative abundance varied among different samples. Proteobacteria (accounting for 27.67%-94.94%) was the most dominant phylum across all samples followed by Actinobacteriota (0.69%-27.64%), Bacteroidota (0.89%-16.69%), Cyanobacteria (0.1%-7.48%), and Marinimicrobia_SAR406 (0%-23.84%) in AS (Figure 3).
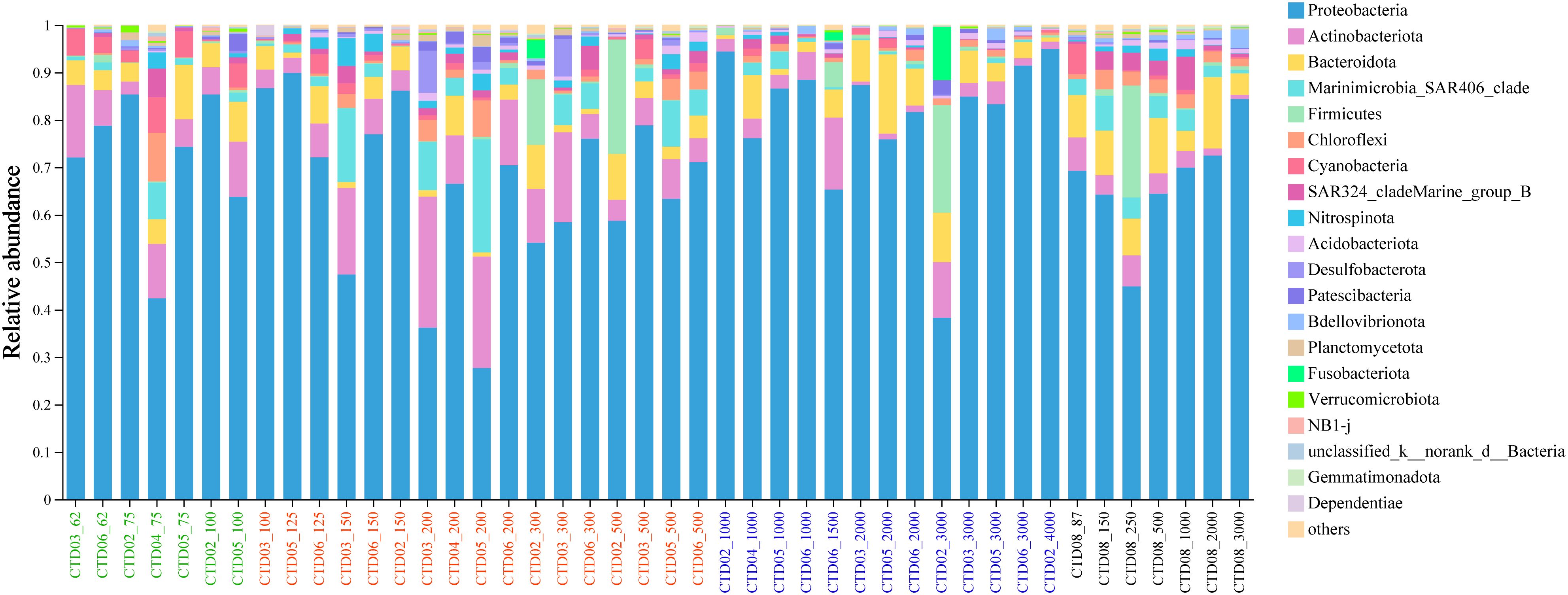
Figure 3 Microbial community composition of AS at phylum level.
At the class level, Alphaproteobacteria (9.23%-52.11%) and Gammaproteobacteria (17.12%-80.05%) was the most dominant classes in AS samples, and the relative abundance of Gammaproteobacteria were higher than that of Alphaproteobacteria. Other predominant class included Acidimicrobiia (0.2%-26.5%), Bacteroidia (0.4%-16.7%), SAR406 (0%-23.8%), Actinobacteria (0%-11.6%), Bacilli (0%-23.4%), Cyanobacteriia (0%-7.5%), Dehalococcoidia (0%-9.3%) and SAR324 (0%-7.0%). The relative abundances of Acidimicrobiia and SAR406 were highest in the OMZs, and lowest in deep sea (AS_DOWN). Besides, Nitrospinia, Desulfobacteria and Vicinamibacteria also exhibited highest relative abundance in the OMZ communities. On the contrary, Cyanobacteriia was more dominant in group AS_UP (2.32%-7.45%), but almost undetectable in group AS_OMZ and AS_DOWN (Supplementary Figure 5).
At the family level, Alteromonadaceae and Sphingomonadaceae were the most dominant family in AS followed by Rhodobacteraceae, Saccharospirillaceae, Flavobacteriaceae, Thalassospiraceae, Microtrichaceae, Moraxellaceae, SAR406_clade, Marinobacteraceae, UBA10353 and Hyphomonadaceae. Alteromonadaceae family dominated in most samples, and exhibited higher relative abundances in AS_UP and AS_DOWN than AS_OMZ. Sphingomonadaceae was also widely distributed in different depths in AS and more abundant in deep sea below 1000 meters (Supplementary Figure 6). Moraxellaceae appeared to increase in abundance with decreasing DO concentrations in AS_OMZ and AS_DOWN, when compared with AS_UP. Microtrichaceae tended to distribute in AS_UP and AS_OMZ, and was most abundant in OMZ communities. Besides, other dominant taxa included UBA10353, Nitrospinaceae, Thioglobaceae, Desulfosarcinaceae were also significantly enriched in AS-OMZ communities. However, Cyanobiaceae, Actinomarinaceae, Legionellaceae, NS9_marine_group, OCS116 and SAR86_clade seemed to mainly distributed in AS_UP, but with low or undectable abundance in AS_OMZ and AS_DOWN (Supplementary Figure 6).
At the genus level, the top 10 abundant genera with high relative abundance included Alteromonas, Acinetobacter, unclassified_f:Alteromonadaceae, Thalassospira, Oleibacter, Sphingobium, Sva0996_marine_group, SAR406_clade, Erythrobacter and Marinobacter (Figure 4). Alteromonas dominated in most of the samples in AS-UP. Acinetobacter and Thalassospira were mainly distributed in OMZ and deep layer in AS. Interestingly, Oleibacter showed high abundance in two samples (CTD05_125m, CTD06_1000m). Sva0996_marine_group, belonging to Acidimicrobiia class, increased relative abundance in AS OMZ layer (Figure 4). Other predominant genus such as Sphingobium, Erythrobacter and Marinobacter showed no significant differences among the three genus in these groups.
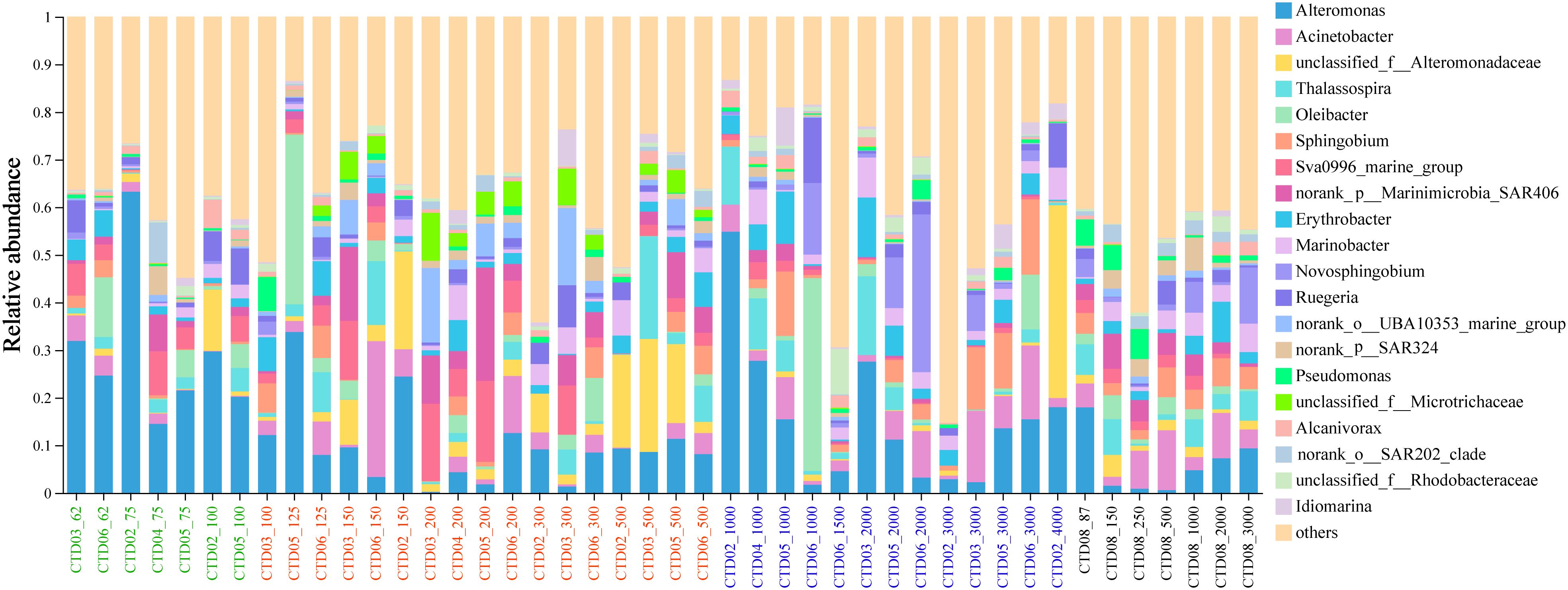
Figure 4 Microbial community composition of AS at genus level.
Among the detected archaea, two classes Nitrososphaeria and Thermoplasmata were found of wide distribution throughout the water column, with the abundance accounting for over 90% in 43 samples. Nitrososphaeria have higher relative abundance below the DCM layers, while Thermoplasmata was more abundant in the AS_UP group (Supplementary Figure 7). At the family level, the three most abundant taxa included Nitrosopumilaceae, norank_o_Marine_Group_II and norank_c_Thermoplasmata in majority of the communities, except for four samples CTD02-500, CTD02-3000, CTD02-4000, CTD06-1500, which were dominated by Nitrososphaeraceae family. At the genus level, Candidatus_Nitrosopelagicus, norank_o_Marine_Group_II and Marine_Group_III dominated in upper water layers above 125m, while Nitrosopumilus, norank_o_Marine_Group_II, Marine_Group_III dominated in deeper water layers (from 150m to bottom). As the most abundant and prevalent taxa in the water column in AS, Nitrosopumilaceae tended to have different ecotypes, among which Candidatus_Nitrosopelagicus distributed in euphotic zones, while Nitrosopumilus genus mainly dominated in meso- and bathy- pelagic water.
3.4 Bioindicator taxa and their distribution patterns in the hypoxic zones of Arabian Sea
Each water layer in AS is distinct with respect to the physical oceanographic features, facilitate or inhibit the growth of specific microorganisms. Research on bioindicator taxa may help understand the connection between microbes and specific habitats. In this study, the microbial community composition was further compared to determine the indicator taxa in the hypoxic zone of Arabian Sea. Kruskal-Wallis H test was used to identify at least 30 bioindicator taxa with significant distribution characteristics that could differentiate AS_UP, AS_OMZ and AS_DOWN communities, as well as hypoxic and non-hypoxic zones communities at the genus or family level (Figure 5). Candidatus_Actinomarina, Prochlorococcus_MIT9313 and unclassified_f_Cyanobiaceae were identified as bioindicator taxa in AS_UP communities, which were exclusively distributed in euphotic zones. Alteromonas was also one of the important indicator taxa and its relative abundance was higher in the AS_UP when compared with AS_OMZ and AS_DOWN group. Although low relative abundance, SAR86_clade, NS2b_marine_group, NS5_marine_group, NS9_marine_group and OCS116_clade were also determined as indicator taxa in AS_UP communities, with significantly higher abundance than in AS_OMZ and AS_DOWN communities. The indicator taxa of the hypoxia zones in AS included unclassified_f_Desulfosarcinaceae, norank_o_UBA10353, Nitrospina, SUP05_cluster, Sva0996_marine_group and unclassified_f_Microtrichaceae, which were significantly enriched and exhibited highest relative abundance in AS_OMZ communities, when compared with AS_UP and AS_DOWN communities. For AS_DOWN, Novosphingobium, Salinicola, Oceanobacter were the indicator taxa of the deep layers (Figure 5).
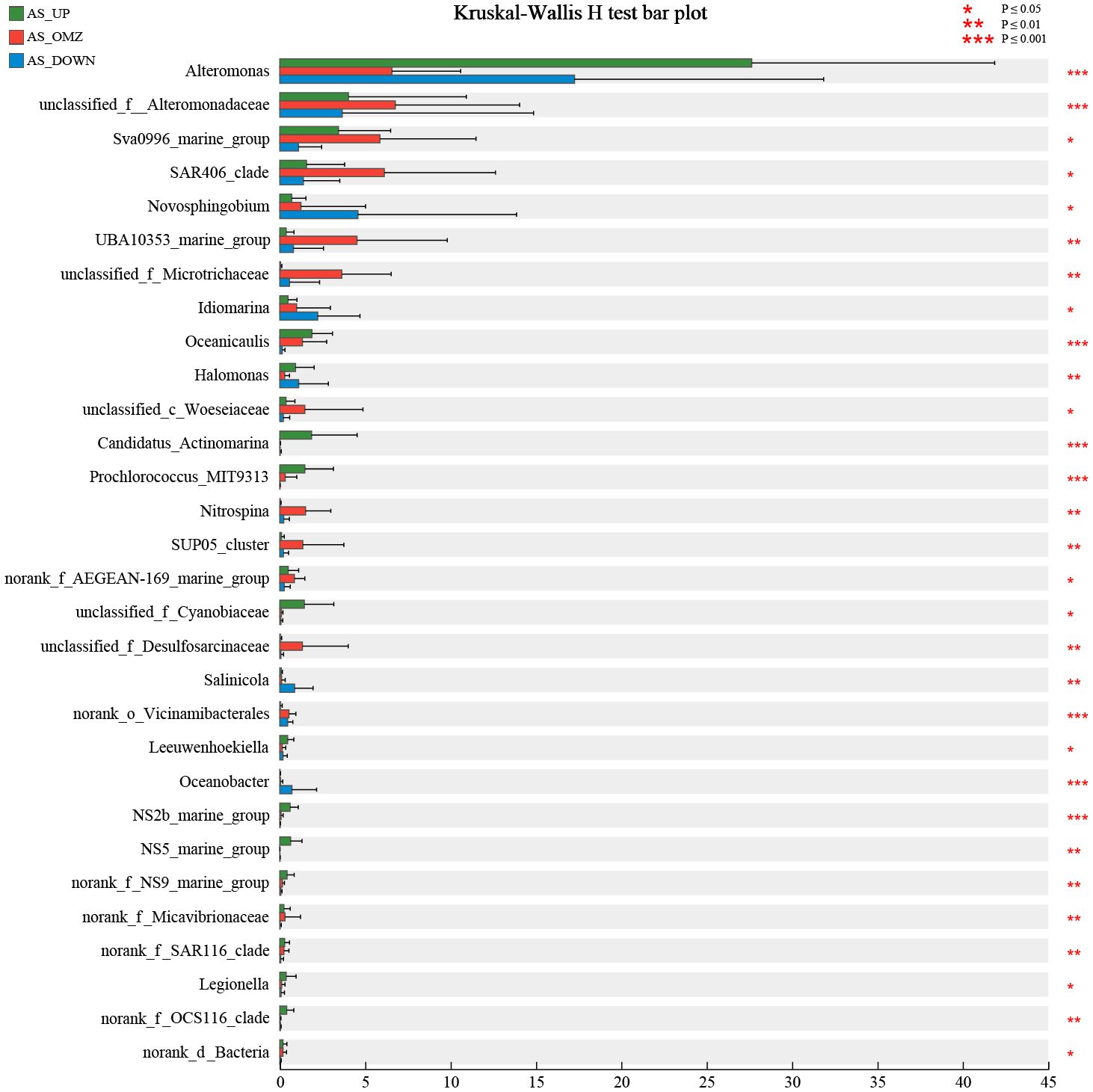
Figure 5 The 30 bioindicator taxa that significantly differentiate AS_UP, AS_OMZ and AS_DOWN communities in AS. *: P ≤ 0.05, **:P ≤ 0.01, ***:P ≤ 0.001.
The spatial and depth-wise distribution patterns of important bioindicator taxa in the hypoxia zones of AS were further analyzed and compared with other OMZs including Bay of Bengal (Bob) and eastern tropical North Pacific Ocean (ETNP) at ASV or OTU levels (Figure 6; Supplementary Figure 8). As one of the most important taxa, Desulfosarcinaceae was almost exclusively predominant in the OMZs in CTD03, CTD04, CTD05 and CTD06 stations of AS. However, it was absent in CTD02 and CTD08 stations, as well as in Bob and ETNP OMZs (Figure 6; Supplementary Figure 8). Only three ASVs were assigned to Desulfosarcinaceae in AS, which belonging to Desulfobacterales of Desulfobacterota phylum, with the most abundant ASV890 belonging to one unclassified taxa, and the other two affiliated with SEEP-SRB1. Similar distribution pattern was observed for another important indicator taxa UBA10353, which was phylogenetically closely related to Arenicellaceae family of Gammaproteobacteria. UBA10353 was also significantly enriched in the OMZ communities in AS. For this taxa, ASV879, ASV678 and ASV666 were the most abundant ASVs, accounting for more than 90%, and were mainly distributed in the hypoxic zone of CTD03, CTD04, CTD05 and CTD06 stations but absent in CTD02 and CTD08. They seemed to increase their relative abundance toward anoxic conditions in AS. Besides, these ASVs (corresponding to OTU11318, OTU11316, OTU11385) were found to be unique to AS OMZ communities when compared with Bob and ETNP OMZs (Figure 6; Supplementary Figure 8). Moreover, Nitrospina was also identified as important bioindicator taxa in OMZs, and predominantly distributed in hypoxic zones of AS. ASVs affiliated with Nitrospina mainly represented by ASV1121 and ASV632. They were almost exclusively dominated in the OMZ communities of CTD03, CTD04, CTD05 and CTD06 except for CTD02 and CTD08, and also enriched in Bob OMZs and ETNP OMZs (as represented by OTU11457 and OTU5252) (Figure 6; Supplementary Figure 8). Likewise, SUP05 group were also significantly enriched in the OMZs of AS except for CTD02 and CTD08 stations, and were identified as important bioindicator taxa. The dominant ASVs represented by ASV635 and ASV4913 were phylogenetically related to Thioglobus genus. This taxa were ubiquitous and abundant not only in AS OMZs but also in Bob and ETNP OMZs (as represented by OTU4763) (Figure 6; Supplementary Figure 8).
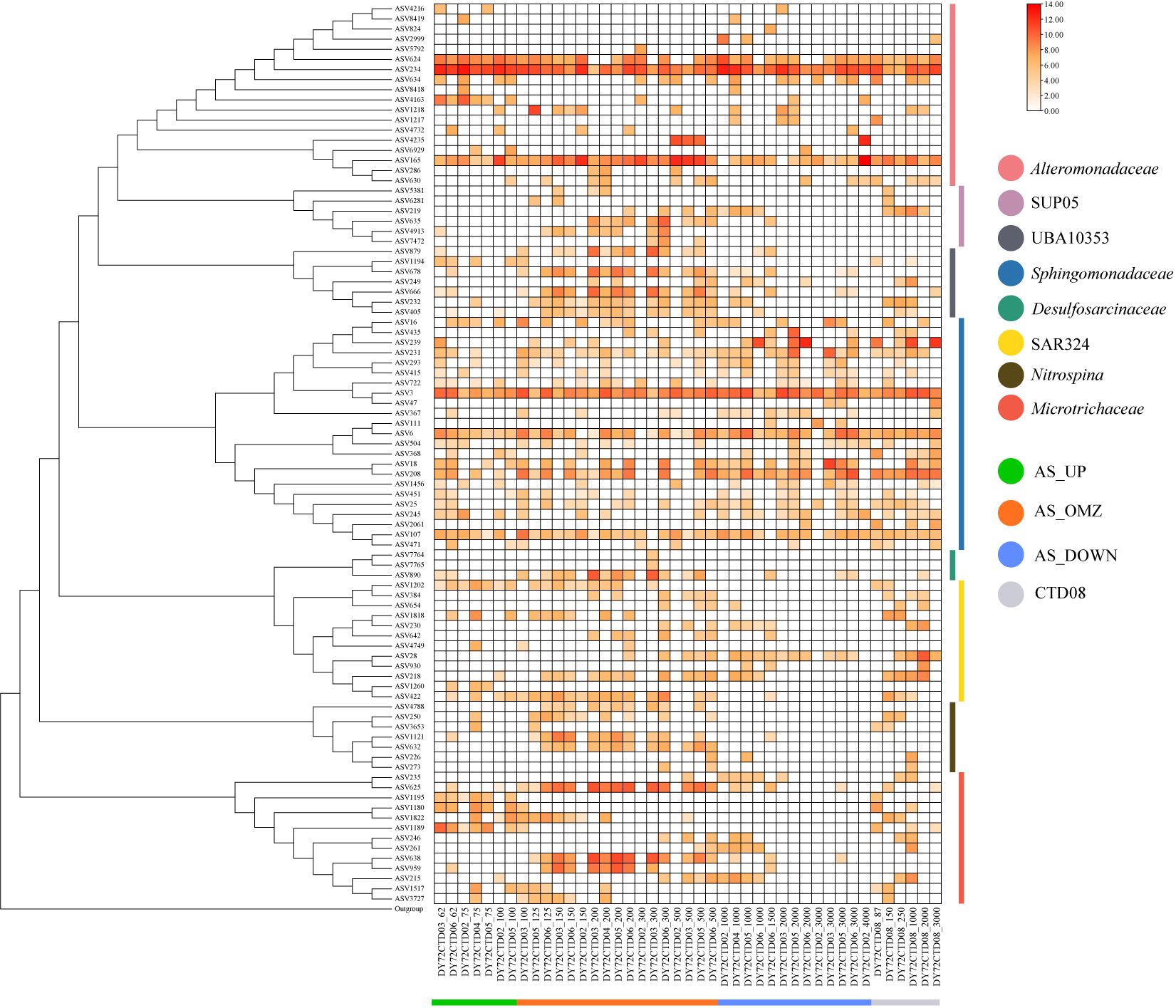
Figure 6 Heatmap displaying the distribution and log-transformed relative abundance of major ASVs.
With regard to Microtrichaceae, it was predominant not only in the OMZs of AS but also in AS_UP. ASVs affiliated with Microtrichaceae formed at least two phylogenetic clades, one belonging to Sva0996_marine_group, and the other belonging to unclassified_f_Microtrichaceae (as represented by ASV625), among which the latter tended to dominated in the mesopelagic water (Figure 6; Supplementary Figure 8). Besides, at least two ecotypes of Sva0996_marine_group were present in AS, one dominated in euphotic zone (as represented by ASV1180 and ASV1189) and the other prevailed in OMZ zones (as represented by ASV638 and ASV959). Similar distribution pattern was also observed in Bob and ETNP OMZs (Figure 6; Supplementary Figure 8).
3.5 Environmental parameters affecting the structure of microbial communities
Prior to Canonical Correspondence Analysis (CCA) to examine the influence of environmental parameters on bacterial and archaeal community structure, Variance Inflation Factor was used to screen the environmental parameters analysis to ensure the accuracy of the CCA analysis (remove Salinity, Silicate, Phosphate and Temperature) (O’brien, 2007).
The results showed that environmental parameters including DO, depth, NO3-, and NO2- exhibited significant influences on bacterial and archaeal communities. DO, depth, and nitrate were the most significant environmental factors. For bacterial communities in AS, the first two ordination axes of CCA accounted for 8.44% of the explained total variance in ASV level (Figure 7, Supplementary Table 2). CCA indicated that among the five different parameters, DO seemed to separate the bacterial communities into ‘oxygen content’ and OMZ communities (Figure 7). For archaeal community in AS, the first two ordination axes of CCA accounted for 16.06% and 6.85% of the explained total variance in ASV level. Similar to bacterial, archaeal samples in AS_OMZ was negatively correlated with DO. The archaeal communities in group AS_DOWN was strongly influenced by depth. Besides, NO2- and NH4+ had a small impact on the archaeal community (Figure 7).
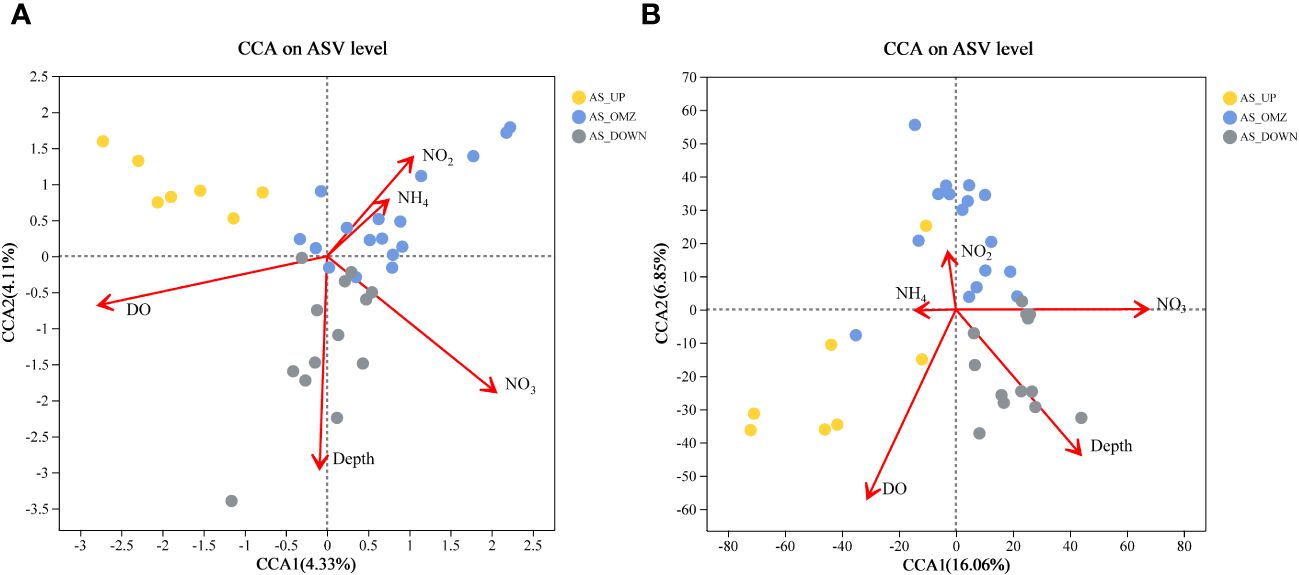
Figure 7 Canonical Correspondence Analysis ordination diagram of bacterial and archaeal communities. (A) bacterial at ASV level in AS (B) archaeal at ASV level in AS.
Spearman correlation heatmap showed the correlation between the bacteria families and environmental parameters (Supplementary Figure 9, Supplementary Table 3). In AS, there was no significant correlation between Altermonadaceae and any environmental parameters. As another important taxon, Sphingomonadaceae, exhibited a significant positive correlation with depth and NO3- in AS. Interestingly, Microtrichaceae were affected by almost all environmental parameters in AS (except for NO3-) (Supplementary Figure 9). In AS, there was no significant correlation of Nitrosopumilaceae with any environmental parameters. However, Marine Group II was significantly positively correlated with DO (Supplementary Figure 10, Supplementary Table 3).
3.6 Co-occurrence patterns of microbial community and functional prediction
Keystone taxa not only maintained the structures and functions of microbial community, but also considerably influence community networks (Berry and Widder, 2014). A co-occurrence network was generated to explore interactions among bacterial and archaeal communities and identify keystone taxa in this study (Supplementary Figure 11, Supplementary Table 4). In the network, each node represents an ASV, with the size of each node proportional to the number of its associations (degree). Connections between nodes represent strong and significant correlations of abundance (|r| > 0.7, p < 0.01). In this study, we choose the connectors (zi ≤ 2.5, Pi > 0.62) as the keystone in community. As a result, ASV84 (Marinobacteraceae), ASV237 (Pseudomonadaceae), ASV264 (Mycobacteriaceae), ASV28 (SAR324_clade), ASV182 (Flavobacteriaceae), ASV16 (Sphingomonadaceae), ASV95 (Rhizobiaceae) and ASV234 (Alteromonadaceae) were identified as keystone of bacterial ASVs. ASV154 (Nitrosopumilaceae) and ASV156 (f_norank_o_norank_c_Thermoplasmata) were identified as keystone of archaeal ASVs.
Through Functional Annotation of Prokaryotic Taxa (FAPROTAX) analysis, various functional process was predicted. Chemoheterotrophy was the most active metabolic process in different water layers in AS, while Photoautotrophy and Photoheterotrophy processes were very active in the up layer and weakened with the increase of water depth. Strong Nitrate_reduction was observed in AS. The potential for denitrification, nitrification, aerobic nitrite oxidation, dark sulfide/sulfur compounds oxidation were predicted and more abundant in the OMZs in AS (Supplementary Figure 12, Supplementary Table 5).
4 Discussion
The OMZs in the ocean have been expanding and intensifying in recent decades. Microbial communities play a crucial role in the formation and function of OMZ, mediating the global biogeochemical and climatic process (Rixen et al., 2020). The present study aimed at elucidating the microbial composition and diversity across different oxygen gradient in the water column of the Arabian Sea, and determination of key bioindicator taxa in the hypoxia zones through 16S rRNA gene high-throughput sequencing.
4.1 Differences in physicochemical parameters and their effect on the prokaryotic community
It is reported that the formation and maintenance of AS OMZ was due to intense seasonal surface blooms, coupled with a land-locked northern boundary and limited northern ventilation (Vallivattathillam et al., 2023). Using switchable trace oxygen (STOX) amperometric microsensors, researchers have identified essentially oxygen-free waters in the nitrite-rich regions of the Arabian Sea, particularly in the Omani Shelf (Jensen et al., 2011). In this study, the variation observed in DO levels along the depth of water column among the sampled locations (except for CTD08) are consistent with results previously described in Arabian Sea OMZs, where the DCM and near bottom depths are well-oxygenated, and the DO values in OMZs are below the defined threshold (<20 μM) (Beal et al., 2013). The thickest OMZs in this study were observed in stations CTD03, CTD04 and CTD05 located in the northern Arabian Sea, ranging from about 100m to 1500m. They were recognized as the core OMZ zone here, with the DO values almost undetectable from about 150m to 1200m. In the OMZ transition zone, for CTD02 and CTD06 stations, their DO values were also below 20 μM in the OMZs from about 150m to 1000m. In contrast, high DO values (above 62 μM) were observed in the water column of CTD08 station located in the southern AS far from the mainland, which might be due to the relatively limited impact of terrestrial organic matter input, indirectly leading to less intense microbial oxygen consumption activity in OMZ (Fernandez-Alamo and Färber-Lorda, 2006). Hence, CTD08 station was considered as non-hypoxic zone, and used as a control group in this study. In AS, no significant difference of the salinity was observed in the horizontal direction of the water column. However, the salinity gradually decreases with increasing water depth, consistent with previous research (Supplementary Figure 1) (Al-Said et al., 2018; Bandekar et al., 2018). The nitrate concentrations were low in DCM layer in this study across the sampled locations, however, they were higher in the OMZs and deep layers. The low surface nitrate concentrations might be due to high primary productivity in AS (Kumar et al., 2001). Nitrite accumulation was observed not only in the surface layers (DCM) but also in the mid and deeper depths in most locations, which might drive nitrification process. High nitrite concentration accompanied with low DO such as at 300m of CTD03 station, was profit for biological processes denitrification, and implied intense denitrification process (Ulloa et al., 2012).
The structure of microbial communities is often influenced by variable ocean environmental conditions with geographical isolation and environmental parameters (Yan et al., 2018). Variations in environmental conditions and competition across the oxygen gradient might account for the difference observed between surface and OMZ bacterial communities (Long et al., 2021). The analysis of relationships between environmental factors with microbial community revealed that dissolved oxygen (DO), depth, nitrate (NO3-), and nitrite (NO2-) were the significant environment factors in this study, which might co-shape microbial communities. DO has been detected as a crucial factor to significantly structure and differentiate the microbial community composition in the oxygenated upper waters and hypoxic OMZs in AS. Similarly, DO was also considered as the important driver responsible for the changes in microbial communities in the hypoxic zone in Gulf of Mexico and the eastern tropical North Pacific Ocean (ETNP) (Beman et al., 2013). Other environmental parameters including depth and nitrate exerted significant impacts on the structure of bacterial and archaeal communities in AS.
4.2 Bacterial community composition and important taxa – key members in OMZ
The alpha diversity index results revealed the highest archaeal richness and diversity in the AS_OMZ and AS_UP communities compared to the AS_DOWN community (Supplementary Figure 3). While the AS_OMZ appeared to have greater bacterial diversity, no significant difference in richness was found between it and the AS_DOWN. This is consistent with previous findings in the epipelagic waters of the Andaman and the eastern Bay of Bengal (Supplementary Figure 3) (Guo et al., 2022). NMDS analysis revealed that bacterial community of AS_OMZ was significantly different from AS_UP group, while AS_DOWN community seemed to overlap with AS_OMZ and AS_UP. Though AS_DOWN samples were geographically adjacent to the AS_OMZ, the DO levels were closer to AS_UP layers, which might lead to mixed characteristics in bacterial community composition between AS_UP and AS_DOWN. For archaea, the beta diversity analysis showed a clear difference between samples from AS_UP, AS_OMZ and AS_DOWN, confirming dissimilar archaeal community structure in the different depths.
Bacterial community in all samples in this study was found to be dominated by Proteobacteria, Actinobacteria, Bacterioda, SAR406_clade, Firmicutes, Chloreflexi, Cyanobacteria, SAR324 and Nitrospinota, similar to others found in various pelagic OMZ (Sun et al., 2021; Gu et al., 2022). Gammaproteobacteria and Alphaproteobacteria dominated through the water column in AS. The dominance of these phylotypes has been well documented in a variety of pelagic marine zones including the ETSP-OMZ, Northwest Mediterranean Sea, Tropical Western Pacific Ocean and Southern AS (Fuchs et al., 2005; Stevens and Ulloa, 2008; Crespo et al., 2013; Ganesh et al., 2013; Sun et al., 2021). The Bacteroidetes was known to be associated with phytoplankton and abundant at surface waters (Fandino et al., 2001). However, Bacterioda abundance did not significantly decrease with depth in the OMZ of AS in this study.
Alteromonadaceae was one of the most dominant representative of Gammaproteobacteria in AS, and exhibited high relative abundances in the whole water column in AS. However, Alteromonadaceae was previously reported to dominate in the deeper water in ETSP (Stevens and Ulloa, 2008). In this study, ASVs affiliated with the family Alteromonadaceae were primarily phylogenetically related to Alteromonas macleodii and Alteromonas lipolytica, with both species being widely distributed throughout the water column in AS. No obvious niche partition was observed for Alteromonas in this study. OTU2530 affiliated with Alteromonas macleodii predominated not only in the UP, OMZ and DOWN of AS, but also in the water column of Bob and ETNP (Figure 4), irrespective of water depth. Previous reports in OMZ suggest that Alteromonas participate in complex carbon metabolism pathways and co-exist with other key microbial players in the carbon cycle, such as Marinobacter and Prochlorococcus (Gao et al., 2017; Koch et al., 2020; Frühe et al., 2021; Henríquez-Castillo et al., 2022). Alteromonas_macleodii has been intensively studied and well-known to niche specialization. The surface ecotype of Alteromonas_macleodii was found to be globally abundant and recognized for its capability to degrade sugars and amino acids and colonize particulate organic matter with much slower sinking rates. In contrast, the deep ecotype displayed a higher potential of degrading recalcitrant compounds and colonizing relatively large particles that sink rapidly to meso and bathy pelagic depths (Ivars-Martinez et al., 2008). Ecologically they are often associated to nutrient-rich environments such as particulate material, marine snow, or marine animals, accompanied with complex metabolic potential which strongly supports the ability to survive in various environments. Although Alteromonadaceae constituted a minor proportion of the metagenomic data, they significantly contributed to gene transcription across various marine environments, from mesotrophic coastal California waters to the oligotrophic subtropical North Pacific, highlighting their crucial role in sustaining ecosystem functions (Sherwood et al., 2015). The dominance of Alteromonas may partly attributed to the abundance of sinking particulate matter in the northern AS (P Vijayan et al., 2023).
Sphingomonadaceae, Thalassospiraceae and Rhodobacteraceae of Alphaproteobacteria were also significantly contributing to microbial communities, which were mainly represented by genera of Sphingomonas, Thalassospira and Ruegeria, respectively. Sphingomonadaceae are prevalent in various environments and metabolically versatile, which could utilize a wide range of organic compounds as well as many types of refractory contaminants, such as Bisphenol A (BPA) and aromatic hydrocarbon compounds (Kertesz and Kawasaki, 2010; Göker et al., 2017; Oh and Choi, 2019). Previous reports in OMZ suggest that high abundance of Sphingomonadaceae may be driven by high concentrations of phosphate and ammonia (Liu et al., 2015). Rhodobacteraceae is considered as one of the most abundant groups of bacteria in marine ecosystem, comprising photoheterotrophic and chemoheterotrophic species. The diverse metabolic capabilities of Rhodobacteraceae, including the utilization of organic compounds, sulfur oxidation, carbon monoxide oxidation, and dimethylsulfoniopropionate (DMSP) degradation, enable them to thrive in a wide range of environments (Brinkhoff et al., 2008; Elifantz et al., 2013; Pujalte et al., 2014). In addition, the ability of Rhodobacteriaceae (Sagittula genus) from the OMZ region to fix N2 has been demonstrated (Martínez-Pérez et al., 2018).
Several important taxa were determined as indicator taxa of the OMZs in AS in this study including unclassified_f_Desulfosarcinaceae, UBA10353, SUP05_cluster, Nitrospina, Sva0996_marine_group, unclassified_f_Microtrichaceae. Among these, relatively high abundances of UBA10353, SUP05, Nitrospina have also been detected in other OMZs, including Gulf of Alaska, ETSP and ETNP (Beman and Carolan, 2013; Muck et al., 2019; Sun et al., 2019). Desulfosarcinaceae, belonging to Desulfobacterales of the class Desulfobacteria, was determined as an abundant member and key indicator taxa in hypoxic zones of AS. The spatial and depth-wise distribution patterns indicated that Desulfosarcinaceae was unique to AS communities, which almost exclusively inhabited in the OMZs of AS (CTD03, CTD04, CTD05, CTD06), however absent in CTD02 and CTD08 stations as well as in Bob and ETNP OMZs communities (Figure 6; Supplementary Figure 8). ASVs (OTUs) affiliated with Desulfosarcinaceae were closely related with SEEP-SRB1. It was reported that the family Desulfosarcinaceae including genera such as Desulfosarcina, Desulfatitalea, SEEP-SRB1, and Sva0081 sediment group, were sulfate-reducing bacteria (SRB). Members of this group have been identified as strict anaerobes, exhibiting both respiratory and fermentative metabolic pathways. They have been found in anoxic brackish water environments as well as marine habitats, including water-flooded oil fields (Galushko and Rozanova, 1991; Watanabe et al., 2017). Previous evidences also suggested that Desulfobacterales were important H2-scavengers in marine sediments as well as in microbial mat community, and also played important roles in nitrogen cycling of mangrove ecosystem (Burow et al., 2014; Dyksma et al., 2017; Nie et al., 2021). Metabolic pathway prediction based on publicly available genomes of Desulfosarcinaceae also indicated the genetic potential of chemolithotrophy via Wood–Ljungdahl pathway as well as nitrogen fixation in this study. The dominant presence of Desulfosarcinaceae suggested the crucial roles involved in the carbon, nitrogen, sulfur and hydrogen cycles in OMZs of AS. The ecological functions and in situ activities of Desulfosarcinaceae in the hypoxic zone of AS will be further explored based on metagenomics and metatranscriptomes data.
UBA10353 was another abundant taxa and predominated in the OMZ core in AS, which was phylogenetically closely related to Arenicellales order of Gammaproteobacteria (Martínez-Pérez et al., 2022). A similar distribution pattern of this group has also been found in OMZs, including the deeper layers of the western Mediterranean Sea, the OMZ core of Tropical Mexican Pacific, the OMZ and OLZ of the Andaman Sea and eastern BoB epipelagic waters (Pajares et al., 2020; Martínez-Pérez et al., 2022). The spatial distribution patterns analysis based on AS, Bob and ETNP data showed that the dominant taxa of UBA10353 shifted between different sea areas (Figure 6; Supplementary Figure 8). For example, OTU11318, OTU11316 and OTU11385 seemed to be exclusively distributed in the hypoxic zone of AS except for CTD02 and CTD08, and they were absent in Bob and ETNP OMZ communities. OTU5267, OTU2157 and OTU4614 dominated in the OMZ and deep communities of ETNP, but showed low relative abundance in AS and Bob OMZ communities. UBA10353 was speculated to have the potential of mixtrophy lifestyle encoding the genes for the Calvin-Benson-Bassham cycle and heterotrophic metabolism (Martínez-Pérez et al., 2022). However, knowledge about the ecology, genetics, and functions of UBA10353 are still limited.
Nitrospina and SUP05 groups were also significantly enriched in the OMZs of AS in this study and were identified as important bioindicator taxa. In this study, the dominant taxa of Nitrospina, as represented by OTU11457 and OTU5252, were enriched not only in the AS OMZ, but also in Bob OMZ and ETNP OMZ communities (Figure 6; Supplementary Figure 8). Surprisingly, this taxa was also detected in the communities from ETNP 2000m. Nitrospina bacteria were recognized as aerobic nitrite oxidizers, playing a crucial role in nitrification processes in the BSI (Red Sea) and in the OMZ (ETSP) habitats (Ngugi et al., 2016). The differences observed by previous studies between novel Nitrospina OMZ MAGs from cultured representatives and all MAGs from non-OMZ environments suggest niche differentiation of Nitrospina in the ocean (Sun et al., 2019). Similar distribution pattern was also observed for SUP05 group. In AS, BOB and ENTP OMZs, SUP05 was mainly dominated by OTU4763, and showed no obvious ecotype differentiation between different sea areas (Figure 6; Supplementary Figure 8). SUP05 are known chemolithoautotrophs and have the potential for carbon fixation and sulfur oxidation (Mattes et al., 2020). Members of these groups are globally distributed in OMZs, such as in OMZs of the Arabian Sea, ETSP, ETNP, and BoB, which might contribute to the carbon, nitrogen and sulfur cycles in the OMZ (Bryant et al., 2012; Ulloa et al., 2012; Bandekar et al., 2018). Microtrichaceae were abundant not only in the OMZs of AS but also in the upper water column (AS_UP). Similar distribution pattern was also observed in Bob and ETNP (Figure 6; Supplementary Figure 8). Most of the ASVs affiliated with Microtrichaceae formed at least two clades, one belonged to Sva0996_marine_group, which was reported that proliferated on the fouling-release surface, one belonged to unclassified_f_Microtrichaceae. Besides, Sva0996_marine_group exhibited the potential for niche partitioning, one ecotype seemed to colonize in the OMZs and the other preferred to inhabit in the euphotic zones in AS (Papadatou et al., 2021; Guo et al., 2022). In this study, the surface ecotype was represented by ASV1195 and ASV1180, and the deep ecotype was represented by ASV625, ASV638 and ASV959 (Figure 6; Supplementary Figure 8). Microtrichaceae has been enriched in the oxygen limited zones communities in Mexican Pacific upwelling, which have been reported to be involved in nitrification-anammox systems and demonstrate the capability to hydrolyze and metabolize complex organic matter (Wang et al., 2020; Li et al., 2021a; Guo et al., 2022; Pajares et al., 2023).
The SAR324 group is known to inhabit a wide range of environments and is particularly abundant in OMZs and deeper depths, including deep-sea hydrothermal vents (Malfertheiner et al., 2022). Although the relative abundance of SAR324 did not significantly differ in the water column of AS in this study, a distinct depth distribution pattern was observed. Specifically, ASV1818 was detected above 200m, ASV1202 and ASV384 were found specifically above the OMZ, ASV422 was exclusively present within the OMZ, and ASV28 was identified from the OMZ to deeper depths (Figure 6; Supplementary Figure 8). However, higher relative abundance of SAR324 were detected in ETNP compared with AS and Bob. In addition, the niche specialization of SAR324 in ETNP were more obvious. For example, OTU2233 dominated in the surface waters, while OTU2280 prevailed in the OMZ and deep layers. SAR324_clade, newly proposed as its own candidate phylum, is globally relevant in the ocean aphotic zone and prevail throughout the water column from surface waters to the abyss, with abundance maxima correlating with low-oxygen concentrations (Wright et al., 1997; DeLong et al., 2006; Wright et al., 2012; Dick et al., 2013; Hawley et al., 2014; Thrash et al., 2017; Aldunate et al., 2018; Rajpathak et al., 2018; Malfertheiner et al., 2022). SAR324 possesses versatile and extensive metabolic repertoire, encompassing both heterotrophic and autotrophic pathways, and exhibited distinct depth and temporal distributions that clearly differentiated ecotypes (Boeuf et al., 2021; Malfertheiner et al., 2022). SAR324_clade was identified as keystone taxa that maintained the structures and functions of microbial community in AS, similar to that in Andaman Sea and eastern BoB (Guo et al., 2022).
Cyanobacteria, Candidatus_Actinomarina, SAR86_clade, NS2b_marine_group, NS5_marine_group, NS9_marine_group and OCS116_clade were also determined as bioindicator taxa in AS_UP communities, with significantly higher abundance than in AS_OMZ and AS_DOWN communities. The most abundant ASVs of Cyanobacteria in this study were affiliated with Prochlorococcus_MIT9313 and unclassified_f_Cyanobiaceae in AS. Prochlorococcus is primarily abundant in warm oligotrophic waters, particularly in the subtropical gyres of the Indian and western Pacific Ocean (Flombaum et al., 2013). As indicator taxa of AS_UP, Candidatus_Actinomarina was reported as a subclass of exclusively marine Actinobacteria with small cells and genomes (Ghai et al., 2013). SAR86 is an abundant and ubiquitous heterotroph in the surface ocean. In previous researches, at least 5 subgroups of SAR86 were identified based on pan-genome analysis, each of which exhibits distinct geographic distribution patterns correlated with specific environmental parameters (Sabehi et al., 2004; Levin and Angert, 2015; Hoarfrost et al., 2019; López-Pérez et al., 2020).
4.3 Archaeal community composition with depth distribution patterns
Archaea are now recognized to be as crucial as bacteria in driving the biogeochemical cycles within the global ocean (Herndl et al., 2005; Knittel et al., 2005; Leininger et al., 2006; Lipp et al., 2008). The occurrence of archaeal groups in the Eastern Tropical South Pacific OMZ, as well as in northern Chile and other oxygen-deficient ecosystems, indicates the active involvement of archaea within these OMZ environments (Woebken et al., 2007; Quinones et al., 2009; Belmar et al., 2011; Stewart et al., 2012). In contrast to the bacterial domain, less was known about archaeal community structure and diversity in the OMZ of AS, although previous studies had reported on ammonia-oxidizing archaea in AS (Pitcher et al., 2011; Peng et al., 2013).
In this study, depth-dependent distribution patterns were observed within the archaeal community. Crenarchaeota and Thermoplasmatota dominated throughout the water column in AS. Not only abundant in the euphotic zone, the relative abundance of Crenarchaeota tended to increase with depth and also dominated the OMZ and bathypelagic archaeal communities, similar to previous results observed in other marine environment such as in Gulf of Alaska and the Altantic Ocean (Shiozaki et al., 2016; Sintes et al., 2016; Muck et al., 2019; Abdulaziz et al., 2020). ASVs of Crenarchaeota were mostly phylogenetically affiliated with Candidatus_Nitrosopelagicus and Nitrosopumilus of Nitrosopumilaceae family, among which Candidatus_Nitrosopelagicus almost exclusively inhabited in the euphotic zones in AS, while Nitrosopumilus prevailed in the OMZs and deeper layers (Rinke et al., 2013; Sow et al., 2022).
Thermoplasmatota (formerly Marine_Group_II) predominated not only in the water column in AS of this study, but also in ETSP, Atlantic Ocean (Pereira et al., 2019). Marine Group II exhibited higher relative abundance in the euphotic zone than the deeper water samples in AS, which was consistent with the common understanding that MGII reside mostly in the surface ocean, Marine Group II showed unique patterns of organic carbon degradation and their energy requirements may be augmented by light in the photic zone (Zhang et al., 2015). Besides, Marine Group II was also abundant in deep-sea waters in this study of AS. Although Marine Group II exhibited high relative abundance throughout the water column of AS, the taxonomic composition of Marine Group II differed in the surface and deep water samples. It has been previously found that Marine Group II lived heterotrophically and displayed great seasonal and spatial variation and phylogenetic diversity (Zhang et al., 2015). Marine Group III (MGIII) were initially described by Fuhrman and Davis from deep marine plankton samples and generally regarded as low-abundance members of deep mesopelagic and bathypelagic communities (Fuhrman and Davis, 1997; Haro-Moreno et al., 2017). Only a few studies report the presence of MG-III in the photic zone. In this study, MG-III was found throughout the water column in AS, not only in the OMZ and bathypelagic layers, but also in the euphotic zones. However, their physiology and ecological roles in oceanic environments remain poorly understood. Crenarchaeota and Thermoplasmatota exhibit specific dominant taxa in different water layers, indicating niche differentiation, metabolic or functional diversity of closely related taxa at the genus or ASV levels, these findings are in line with earlier research results (Rinke et al., 2018; Sow et al., 2022).
4.4 Function prediction from FAPROTAX
FAPROTAX has been extensively utilized for the microbial functional annotation of the biogeochemical process (Louca et al., 2016). The microbial functional prediction based on the taxonomic information of the 16S rDNA gene revealed a significant variation of carbon, nitrogen and sulfur metabolism potential across different oxygen gradients in the Arabian Sea. Photoautotrophy and photoheterotrophy processes were more active in the up layer and weakened with the increase of water depth. The potential for denitrification, nitrification, aerobic nitrite oxidation, nitrogen respiration, nitrate respiration, dark sulfide/sulfur compounds oxidation were more abundant in the OMZs in AS. However, functions including hydrocarbon degradation and aromatic compound degradation seemed to exhibit higher predicted abundances in deeper layers. Similar results were previously observed in the Andaman Sea and eastern BoB (Guo et al., 2022). Nitrogen cycling are intensively active in anoxic water such as OMZs, wherein denitrification and anammox have been recognized as the major process associated with nitrogen loss in the Arabian Sea (Ward et al., 2009; Jensen et al., 2011). Despite oxygen concentrations within the samples from the OMZ of this study being below 20μM, the predicted abundance of denitrification genes was notably lower than that of nitrification genes. This suggests that nitrification might be a predominant nitrogen cycling process in the OMZ of the AS. High abundance of ammonia oxidation genes abundance could be attributed to the significant presence of Nitrosopumilaceae, while aerobic nitrite oxidation genes in AS OMZ samples could well be associated with the abundant presence of Nitrospina. Sulfur cycling are also important for biogeochemical process in OMZs (Wright et al., 2012; Long et al., 2021). Sulfur metabolism functions such as dark sulfide/sulfur compounds oxidation were predicted to be more active in the OMZ of AS than in the oxygenic water samples. Sulfur cycle associated bacterial taxa might contributed by SUP05, SAR324, SAR406, Desulfovibrionales and so on in the OMZ of AS, which also potentially coupled nitrogen and carbon cycling (Bertagnolli et al., 2017; Callbeck et al., 2018; Boeuf et al., 2021). Although potential microbial populations involved in carbon, nitrogen and sulfur metabolism were predicted here, however, their actual activities in situ were unclear. Functional profiling of AS samples should be interpreted with caution, as they rely on predictive models derived from 16S rRNA sequence data. Further investigation based on metagenome and metatranscriptome data is required to understand the ecological roles of the dominant microbes present in the OMZ regions of AS.
5 Conclusion
In conclusion, this study has shed light on the complex prokaryotic community structures within the Arabian Sea’s oxygen minimum zones (OMZs). Our comprehensive analysis of 44 water samples across various depths and oxygen gradients in the Arabian Sea has revealed a significant correlation between microbial community composition and environmental parameters, particularly dissolved oxygen (DO) and depth. Bacterial communities demonstrated greater richness and diversity compared to archaeal communities throughout the water column in the Arabian Sea. The composition of these microbial communities shifted markedly across the different oxygen gradients and depths. Notably, key bioindicator taxa such as Desulfosarcinaceae, UBA10353, Nitrospina, SUP05, Sva0996_marine_group, and Microtrichaceae were prominent in the core of the OMZ. Additionally, SAR324, Alteromonadaceae, and Sphingomonadaceae were recognized for their pivotal roles as keystone taxa. These taxa exhibited distinct spatial and depth-wise distribution patterns across AS, Bob and ETNP. Notably, Desulfosarcinaceae displayed an exclusive distribution within the hypoxic zones of the Arabian Sea, suggesting a specialized adaptation to anoxic niches. In contrast, UBA10353, Nitrospina, SUP05, Microtrichaceae, and SAR324 exhibited a cosmopolitan distribution, indicating their ubiquity and potential adaptability across different OMZs. The functional profiling of microbial communities, as predicted by FAPROTAX, indicated an enhanced potential for denitrification, nitrification, and sulfur oxidation in the OMZ. However, the actual in situ activities of these microbes remain to be elucidated through future metagenomic and metatranscriptomic studies. Collectively, our findings significantly contribute to the understanding of the distribution, structure, and diversity of microbial communities in the Arabian Sea’s OMZ. This knowledge is crucial for predicting the ecological roles and responses of these communities to changing oceanographic conditions and informs further research into the complex microbial ecology and function within OMZs.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
DL: Writing – review & editing, Writing – original draft, Visualization, Formal analysis, Data curation. LW: Writing – original draft, Writing – review & editing, Formal analysis, Data curation, Funding acquisition. FJ: Data curation, Writing – review & editing. XZ: Resources, Investigation, Writing – review & editing. QX: Resources, Investigation, Writing – review & editing. XLZ: Resources, Investigation, Writing – review & editing. QZ: Supervision, Writing – review & editing. ZS: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research was supported by the grants of the National Natural Science Foundation of China (No.42030412), the Natural Science Foundation of Fujian Province (No.2023J011379), Deep Sea Habitats Discovery Project (DY-XZ-04) and Scientific Research Foundation of Third Institute of Oceanography, MNR (No. 2019021).
Acknowledgments
We thank all participants of the oceanographic cruise “Shen Hai Yi Hao”. We thank Xiaowen Li from ShanghaiTech University and Xiangyu Bai from Wuhan Institute of Biological Products for kind help in related experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1380819/full#supplementary-material
References
Abdulaziz A., Balu Tharakan E. M., Jasmin C., Chandran C., Vipindas P., Narayanane S., et al. (2020). Microbial community shifts along an estuarine to open ocean continuum. Regional Stud. Mar. Sci. 41. doi: 10.1016/j.rsma.2020.101587
Acharya S., Panigrahi M. (2016). Eastward shift and Maintenance of Arabian Sea Oxygen Minimum Zone: Understanding the paradox. Deep Sea Res. Part I Oceanographic Res. Papers 115, 240–252. doi: 10.1016/j.dsr.2016.07.004
Aldunate M., de la Iglesia R., Bertagnolli A., Ulloa O. (2018). Oxygen modulates bacterial community composition in the coastal upwelling waters off central Chile. Deep Sea Res. Part II: Topical Stud. Oceanogr. 156, 68–79. doi: 10.1016/j.dsr2.2018.02.001
Al-Said T., Naqvi S. W. A., al-Yamani F., Goncharov O., Fernandes L. (2018). High total organic carbon in surface waters of the northern Arabian Gulf: Implications for the oxygen minimum zone of the Arabian Sea. Mar. pollut. Bull. 129, 35–42. doi: 10.1016/j.marpolbul.2018.02.013
Bandekar M., Ramaiah N., Jain A., Meena R. M. (2018). Seasonal and depth-wise variations in bacterial and archaeal groups in the Arabian Sea oxygen minimum zone. Deep Sea Res. Part II: Topical Stud. Oceanogr. 156, 4–18. doi: 10.1016/j.dsr2.2017.12.015
Bandekar M., Ramaiah N., Jain A., Murti Meena R. (2016). Distinctly different bacterial communities in surface and oxygen minimum layers in the Arabian Sea. Biogeosciences Discussions 2016, 1–36. doi: 10.5194/bg-2016-147
Banerjee S., Schlaeppi K., van der Heijden M. G. (2018). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Baumgartner R., Hu S., Van Kranendonk M., Verrall M. (2022). Taphonomy of microorganisms and microbial microtextures at sulfidic hydrothermal vents: A case study from the Roman Ruins black smokers, Eastern Manus Basin. Geobiology 20, 479–497. doi: 10.1111/gbi.12490
Beal L., Hormann V., Lumpkin R., Foltz G. (2013). The response of the surface circulation of the Arabian sea to monsoonal forcing. J. Phys. Oceanogr. 43, 2008–2022. doi: 10.1175/JPO-D-13-033.1
Belmar L., Molina V., Ulloa O. (2011). Abundance and phylogenetic identity of archaeoplankton in the permanent oxygen minimum zone of the eastern tropical South Pacific. FEMS Microbiol. Ecol. 78, 314–326. doi: 10.1111/j.1574-6941.2011.01159.x
Beman M., Carolan M. (2013). Deoxygenation alters bacterial diversity and community composition in the ocean’s largest oxygen minimum zone. Nat. Commun. 4, 2705. doi: 10.1038/ncomms3705
Beman M., Shih J., Popp B. (2013). Nitrite oxidation in the upper water column and oxygen minimum zone of the eastern tropical North Pacific Ocean. ISME J. 7, 2705. doi: 10.1038/ismej.2013.96
Beman J. M., Vargas S. M., Vazquez S., Wilson J. M., Yu A., Cairo A., et al. (2021). Biogeochemistry and hydrography shape microbial community assembly and activity in the eastern tropical North Pacific Ocean oxygen minimum zone. Environ. Microbiol. 23, 2765–2781. doi: 10.1111/1462-2920.15215
Berry D., Widder S. (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00219
Bertagnolli A., Padilla C., Glass J., Thamdrup B., Stewart F. (2017). Metabolic potential and in situ activity of marine Marinimicrobia bacteria in an anoxic water column. Environ. Microbiol. 19, 4392–4416. doi: 10.1111/1462-2920.13879
Boeuf D., Eppley J., Mende D., Malmstrom R., Woyke T., DeLong E. (2021). Metapangenomics reveals depth-dependent shifts in metabolic potential for the ubiquitous marine bacterial SAR324 lineage. Microbiome 9, 1–19. doi: 10.1186/s40168-021-01119-5
Bokulich N., Kaehler B., Rideout J. R., Dillon M., Bolyen E., Knight R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17. doi: 10.1186/s40168-018-0470-z
Bolyen E., Rideout J. R., Dillon M., Bokulich N., Abnet C., Al-Ghalith G., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 1. doi: 10.1038/s41587-019-0209-9
Brinkhoff T., Giebel H. A., Simon M. (2008). Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch. Microbiol. 189, 531–539. doi: 10.1007/s00203-008-0353-y
Bryant J., Stewart F., Eppley J., DeLong E. (2012). Microbial community phylogenetic and trait diversity declines with depth in a marine oxygen minimum zone. Ecology 93, 1659–1673. doi: 10.1890/11-1204.1
Burow L., Woebken D., Marshall I., Singer S., Pett-Ridge J., Prufert-Bebout L., et al. (2014). Identification of Desulfobacterales as primary hydrogenotrophs in a complex microbial mat community. Geobiology 12, 221–230. doi: 10.1111/gbi.12080
Callahan B., McMurdie P., Rosen M., Han A., Johnson A. J., Holmes S. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Callbeck C., Lavik G., Ferdelman T., Fuchs B., Gruber-Vodicka H., Hach P., et al. (2018). Oxygen minimum zone cryptic sulfur cycling sustained by offshore transport of key sulfur oxidizing bacteria. Nat. Commun. 9 (1), 1729. doi: 10.1038/s41467-018-04041-x
Chen Q., Tang K., Chen X., Jiao N. (2022). Microbial sulfurization stimulates carbon sequestration in marine oxygen minimum zones. Sci. Bull. 67 (9), 895–898. doi: 10.1016/j.scib.2022.01.028
Claesson M., O’Sullivan O., Wang Q., Nikkilä J., Marchesi J., Smidt H., et al. (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PloS One 4, e6669. doi: 10.1371/journal.pone.0006669
Corfield R., Carmichael S., Bennett J., Akhter S., Fatimi M., Craig T. (2010). Variability in the crustal structure of the West Indian Continental Margin in the Northern Arabian Sea. Petroleum Geosci. - PETROL GEOSCI 16, 257–265. doi: 10.1144/1354-079309-902
Crescentini M., Bennati M., Tartagni M. (2012). Design of integrated and autonomous conductivity–temperature–depth (CTD) sensors. AEU - Int. J. Electron. Commun. 66, 630–635. doi: 10.1016/j.aeue.2012.03.013
Crespo B., Pommier T., Fernández-Gómez B., Pedrós-Alió C. (2013). Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. MicrobiologyOpen 2 (4), 541–552. doi: 10.1002/mbo3.92
DeLong E., Preston C., Mincer T., Rich V., Hallam S., Frigaard N.-U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean’s interior. Sci. (New York N.Y.) 311, 496–503. doi: 10.1126/science.1120250
de Muinck E. J., Trosvik P., Gilfillan G. D., Hov J. R., Sundaram A. Y. (2017). A novel ultra high-throughput 16S rRNA gene amplicon sequencing library preparation method for the Illumina HiSeq platform. Microbiome 5, 1–15. doi: 10.1186/s40168-017-0279-1
Deng Y., Jiang Y.-H., Yang Y., He Z., Luo F., Zhou J. (2012). Molecular ecological network analyses. BMC Bioinf. 13, 113. doi: 10.1186/1471-2105-13-113
Dick G., Anantharaman K., Baker B., Li M., Reed D., Sheik C. (2013). The microbiology of deep-sea hydrothermal vent plumes: ecological and biogeographic linkages to seafloor and water column habitats. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00124
Dyksma S., Pjevac P., Ovanesov K., Mussmann M. (2017). Evidence for H2 consumption by uncultured Desulfobacterales in coastal sediments. Environ. Microbiol. 20 (2), 450–461. doi: 10.1111/1462-2920.13880
Elifantz H., Horn G., Ayon M., Cohen Y., Minz D. (2013). Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in Eastern Mediterranean Coastal Seawater. FEMS Microbiol. Ecol. 85 (2), 348–457. doi: 10.1111/femsec.2013.85.issue-2
Fandino L. B., Riemann L., Steward G., Long R., Azam F. (2001). Variations in bacterial community structure during a dinoflagellate bloom analyzed by DGGE and 16S rDNA sequencing. Aquat. Microbial. Ecol. - AQUAT Microb. Ecol. 23, 119–130. doi: 10.3354/ame023119
Fernandes G., Shenoy B. D., Damare S. (2020). Diversity of bacterial community in the oxygen minimum zones of Arabian sea and bay of Bengal as deduced by illumina sequencing. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.03153
Fernandez-Alamo M., Färber-Lorda J. (2006). Zooplankton and the oceanography of the eastern tropical Pacific: A review. Prog. In Oceanogr. 69, 318–359. doi: 10.1016/j.pocean.2006.03.003
Flombaum P., Gallegos J., Gordillo R., Rincón J., Zabala L., Jiao N., et al. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. United States America 110 (24), 9824–9829. doi: 10.1073/pnas.1307701110
Frühe L., Rubel V., Forster D., Keeley N., Laroche O., Pochon X., et al. (2021). Global trends of benthic bacterial diversity and community composition along organic enrichment gradients of salmon farms. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.637811
Fuchs B. M., Woebken D., Zubkov M., Burkill P., Amann R. (2005). Molecular identification of picoplankton populations in contrasting waters of the Arabian Sea. Aquat. Microb. Ecol. 39. doi: 10.3354/ame039145
Fuhrman J., Davis A. A. (1997). Widespread Archaea and novel Bacteria from the deep sea as shown by 16S rRNA gene sequences. Mar. Ecology-progress Ser. - Mar. ECOL-PROGR Ser. 150, 275–285. doi: 10.3354/meps150275
Galushko A., Rozanova E. P. (1991). Desulfobacterium cetonicum sp. nov.: A sulfate-reducing bacterium which oxidizes fatty acids and ketones. Microbiology 60, 742–746.
Ganesh S., Parris D., Delong E., Stewart F. (2013). Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J. 8 (1), 187–211. doi: 10.1038/ismej.2013.144
Gao Y., Liu Q., Wang M., Zhao G., Jiang Y., Malin G., et al. (2017). Characterization and genome sequence of marine alteromonas gracilis phage PB15 isolated from the yellow sea, China. Curr. Microbiol. 74, 821–826. doi: 10.1007/s00284-017-1251-9
Ghai R., Mizuno C. M., Picazo A., Camacho A., Rodriguez-Valera F. (2013). Metagenomics uncovers a new group of low GC and ultra-small marine Actinobacteria. Sci. Rep. 3, 2471. doi: 10.1038/srep02471
Gilles-Gonzalez M.-A. (2001). Oxygen signal transduction. IUBMB Life 51, 165–173. doi: 10.1080/152165401753544232
Göker T., Aşık R., Yılmaz M., Celik I., Tekiner A. (2017). Sphingomonas paucimobilis: A rare infectious agent found in cerebrospinal fluid. J. Korean Neurosurgical Soc. 60, 481–483. doi: 10.3340/jkns.2014.0102.004
Gu B., Liu J., Cheung S., Ho N. H. E., Tan Y., Xia X. (2022). Insights into prokaryotic community and its potential functions in nitrogen metabolism in the Bay of Bengal, a pronounced oxygen minimum zone. Microbiol. Spectr. 10, e00892–e00821. doi: 10.1128/spectrum.00892-21
Guo R., Ma X., Zhang J., Liu C., Thu C. A., Li H., et al. (2022). Microbial community structures and important taxa across oxygen gradients in the Andaman Sea and eastern Bay of Bengal epipelagic waters. Front. Microbiol. 13, 1041521. doi: 10.3389/fmicb.2022.1041521
Haro-Moreno J., Rodriguez-Valera F., Lopez-Garcia P., Moreira D., Martin-Cuadrado A.-B. (2017). New insights into marine group III Euryarchaeota, from dark to light. ISME J. 11 (5), 1102–1117. doi: 10.1038/ismej.2016.188
Hawley A., Brewer H., Norbeck A., Pasa-Tolic L., Hallam S. (2014). Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci. United States America 111 (31), 11395–11400. doi: 10.1073/pnas.1322132111
Helly J., Levin L. (2004). Global distribution of naturally occurring marine hypoxia on continental margins. Deep Sea Res. Part I: Oceanographic Res. Papers 51, 1159–1168. doi: 10.1016/j.dsr.2004.03.009
Henríquez-Castillo C., Plominsky A., Ramirez-Flandes S., Bertagnolli A., Stewart F., Ulloa O. (2022). Metaomics unveils the contribution of Alteromonas bacteria to carbon cycling in marine oxygen minimum zones. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.993667
Herndl G., Reinthaler T., Teira E., Aken H., Veth C., Wendeberg A., et al. (2005). Contribution of archaea to total prokaryotic production in the deep Atlantic Ocean. Appl. Environ. Microbiol. 71, 2303–2309. doi: 10.1128/AEM.71.5.2303-2309.2005
Hoarfrost A., Nayfach S., Ladau J., Yooseph S., Arnosti C., Dupont C., et al. (2019). Global ecotypes in the ubiquitous marine clade SAR86. ISME J. 14, 1–11. doi: 10.1101/635185
Ivars-Martinez E., Martin-Cuadrado A.-B., D’auria G., Mira A., Ferriera S., Johnson J., et al. (2008). Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2, 1194–1212. doi: 10.1038/ismej.2008.74
Jensen M., Lam P., Revsbech N., Nagel B., Gaye B., Jetten M., et al. (2011). Intensive nitrogen loss over the Omani Shelf due to anammox coupled with dissimilatory nitrite reduction to ammonium. ISME J. 5, 1660–1670. doi: 10.1038/ismej.2011.44
Jin J., Yamamoto R., Takeuchi T., Cui G., Miyauchi E., Hojo N., et al. (2022). High-throughput identification and quantification of single bacterial cells in the microbiota. Nat. Commun. 13, 863. doi: 10.1038/s41467-022-28426-1
Kaeberlein T., Lewis K., Epstein S. S. (2002). Isolating “Uncultivable” Microorganisms in pure culture in a simulated natural environment. Sci. (New York N.Y.) 296, 1127–1129. doi: 10.1126/science.1070633
Katoh K., Misawa K., Kuma K.-i., Miyata T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kertesz M., Kawasaki A. (2010). “Hydrocarbon-degrading sphingomonads: Sphingomonas, sphingobium, novosphingobium, and sphingopyxis,” in Handbook of hydrocarbon and lipid microbiology, ed. Timmis K. N. (Professor) (Berlin, Heidelberg: Springer).
Knittel K., Loesekann T., Boetius A., Kort R., Amann R. (2005). Diversity and distribution of methanotrophic archaea at cold seeps. Appl. Environ. Microbiol. 71, 467–479. doi: 10.1128/AEM.71.1.467-479.2005
Koch H., Germscheid N., Freese H., Noriega-Ortega B., Lücking D., Berger M., et al. (2020). Genomic, metabolic and phenotypic variability shapes ecological differentiation and intraspecies interactions of Alteromonas macleodii. Sci. Rep. 10, 809. doi: 10.1038/s41598-020-57526-5
Kumar S. P., Madhupratap M., Dileepkumar M., Muraleedharan P. M., Souza S. N., Gauns M., et al. (2001). High biological productivity in the central Arabian Sea during the summer monsoon driven by Ekman pumping and lateral advection. Curr. Sci. 81, 1633–1638. doi: 10.1016/S0273-1177(01)00615-9
Leininger S., Urich T., Schloter M., Schwark L., Qi J., Nicol G. W., et al. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442, 806–809. doi: 10.1038/nature04983
Levin P., Angert E. (2015). Small but mighty: cell size and bacteria. Cold Spring Harbor Perspect. Biol. 7 (7), e019216. doi: 10.1101/cshperspect.a019216
Li S., Cui Z., Bao M., Luan X., Teng F., Li S., et al. (2022). Contrasting vertical distribution between prokaryotes and fungi in different water masses on the Ninety-East Ridge, Southern Indian Ocean. J. Oceanol. Limnol., 1–15. doi: 10.1007/s00343-021-1046-5
Li Z., Pan D., Wei G., Pi W., Zhang C., Wang J.-H., et al. (2021b). Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. ISME J. 15, 2366–2378. doi: 10.1038/s41396-021-00932-y
Li J., Zheng L., Ye C., Ni B., Wang X., Liu H. (2021a). Evaluation of an intermittent-aeration constructed wetland for removing residual organics and nutrients from secondary effluent: Performance and microbial analysis. Biores. Technol. 329, 124897. doi: 10.1016/j.biortech.2021.124897
Lipp J., Morono Y., Inagaki F., Hinrichs K.-U. (2008). Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 454, 991–994. doi: 10.1038/nature07174
Liu J., Fu B., Yang H., Zhao M., Zhang X.-H. (2015). Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: the potential impact of hypoxia and nutrients. Front. Microbiol. 6, 122282. doi: 10.3389/fmicb.2015.00064
Long M., Deutsch C., Ito T. (2016). Finding forced trends in oceanic oxygen. Global Biogeochem. Cycles 30, n/a–n/a. doi: 10.1002/2015GB005310
Long A., Jurgensen S., Petchel A., Savoie E., Brum J. (2021). Microbial ecology of oxygen minimum zones amidst ocean deoxygenation. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.748961
López-Pérez M., Haro-Moreno J., Iranzo J., Rodriguez-Valera F. (2020). Genomes of the “ Candidatus actinomarinales” Order: highly streamlined marine epipelagic actinobacteria. mSystems 5, e01041–e01020. doi: 10.1128/mSystems.01041-20
Louca S., Parfrey L., Doebeli M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Lynch M. D., Neufeld J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Malfertheiner L., Martínez-Pérez C., Zhao Z., Herndl G., Baltar F. (2022). Phylogeny and metabolic potential of the candidate phylum SAR324. Biology 11, 599. doi: 10.3390/biology11040599
Martin M. (2011). CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Martínez-Pérez C., Greening C., Bay S., Lappan R., Zhao Z., De Corte D., et al. (2022). Phylogenetically and functionally diverse microorganisms reside under the Ross Ice Shelf. Nat. Commun. 13, 117. doi: 10.1038/s41467-021-27769-5
Martínez-Pérez C., Mohr W., Schwedt A., Dürschlag J., Callbeck C. M., Schunck H., et al. (2018). Metabolic versatility of a novel N2-fixing Alphaproteobacterium isolated from a marine oxygen minimum zone. Environ. Microbiol. 20, 755–768. doi: 10.1111/1462-2920.14008
Mattes T., Ingalls A., Burke S., Morris R. (2020). Metabolic flexibility of SUP05 under low DO growth conditions. Environ. Microbiol. 23 (6), 2823–2833. doi: 10.1111/1462-2920.15226
Muck S., De Corte D., Clifford E., Bayer B., Herndl G., Sintes E. (2019). Niche differentiation of aerobic and anaerobic ammonia oxidizers in a high latitude deep oxygen minimum zone. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02141
Ngugi D. K., Blom J., Stepanauskas R., Stingl U. (2016). Diversification and niche adaptations of Nitrospina-like bacteria in the polyextreme interfaces of Red Sea brines. ISME J. 10, 1383–1399. doi: 10.1038/ismej.2015.214
Nie S., Zhang Z., Mo S., Jinhui L., He S., Liang Z., et al. (2021). Desulfobacterales stimulates nitrate reduction in the mangrove ecosystem of a subtropical gulf. Sci. Total Environ. 769, 144562. doi: 10.1016/j.scitotenv.2020.144562
O’brien R. M. (2007). A caution regarding rules of thumb for variance inflation factors. Qual. quantity 41, 673–690. doi: 10.1007/s11135-006-9018-6
Oh S., Choi D. (2019). Microbial community enhances biodegradation of bisphenol a through selection of Sphingomonadaceae. Microbial. Ecol. 77, 631–639. doi: 10.1007/s00248-018-1263-4
Pajares S., Merino-Ibarra M., Farias L. (2023). Prokaryotic community dynamics and nitrogen-cycling genes in an oxygen-deficient upwelling system during La Niña and El Niño conditions. Environ. Microbiol 80 (3), 519–536. doi: 10.1111/1462-2920.16362
Pajares S., Varona F., Hernández-Becerril D. (2020). Spatial distribution patterns of bacterioplankton in the oxygen minimum zone of the tropical Mexican pacific. Microbial. Ecol. 80 (3), 519–536. doi: 10.1007/s00248-020-01508-7
Papadatou M., Robson S. C., Dobretsov S., Watts J. E. M., Longyear J., Salta M. (2021). Marine biofilms on different fouling control coating types reveal differences in microbial community composition and abundance. MicrobiologyOpen 10, e1231. doi: 10.1002/mbo3.1231
Paulmier A., Diana R. (2009). Oxygen Minimum Zones (OMZs) in the modern ocean. Prog. In Oceanogr. 80, 113–128. doi: 10.1016/j.pocean.2008.08.001
Peng X., Jayakumar A., Ward B. (2013). Community composition of ammonia-oxidizing archaea from surface and anoxic depths of oceanic oxygen minimum zones. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00177
Pereira O., Hochart C., Auguet J.-C., Debroas D., Galand P. (2019). Genomic ecology of Marine Group II, the most common marine planktonic Archaea across the surface ocean. MicrobiologyOpen 8 (9), e00852. doi: 10.1002/mbo3.852
Pitcher A., Villanueva L., Hopmans E. C., Schouten S., Reichart G. J., Sinninghe-Damste J. (2011). Niche segregation of ammonia-oxidizing Archaea and anammox bacteria in the Arabian Sea oxygen minimum zone as determined by a combined intact polar lipid and gene-based approach. ISME J. 5, 1–9. doi: 10.1038/ismej.2011.60
Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: neighbor-joining with profiles instead of a dis-tance matrix. Mol. Biol. Evol. 26 (7), 1641–1650. doi: 10.1093/molbev/msp077
Pujalte M., Lucena T., Ruvira M., Arahal D., Macián M. (2014). “The Family Rhodobacteraceae”, ed. Rosenberg E. F., DeLong S., Lory E., Stackebrandt E., Thompson F. (Berlin, Heidelberg: Springer), 439–512.
Puspita I. D., Kamagata Y., Tanaka M., Asano K., Nakatsu C. (2012). Are uncultivated bacteria really uncultivable? Microbes environments/JSME 27 (4), 356–366. doi: 10.1264/jsme2.ME12092
Quinones R., Levipan H., Urrutia H. (2009). Spatial and temporal variability of planktonic archaeal abundance in the Humboldt Current System off Chile. Deep Sea Res. Part II: Topical Stud. Oceanogr. 56, 1073–1082. doi: 10.1016/j.dsr2.2008.09.012
Rajpathak S. N., Banerjee R., Mishra P. G., Khedkar A. M., Patil Y. M., Joshi S. R., et al. (2018). An exploration of microbial and associated functional diversity in the OMZ and non-OMZ areas in the Bay of Bengal. J. Biosci. 43, 635–648. doi: 10.1007/s12038-018-9781-2
Rangamaran V., Sankara Subramnanian S. H., Sundari K., Gopal D. (2022). Vertical microbial profiling of Arabian sea oxygen minimal zone reveals complex bacterial communities and distinct functional implications. Microbial. Ecol. 85 (2), 357–371. doi: 10.1007/s00248-021-01952-z
Rinke C., Rubino F., Messer L., Youssef N., Parks D., ChuvoChina M., et al. (2018). A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.). ISME J. 13, 1. doi: 10.1038/s41396-018-0282-y
Rinke C., Schwientek P., Sczyrba A., Ivanova N., Anderson I., Cheng J.-F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature. Nature 499 (7469), 431–437. doi: 10.1038/nature12352
Rixen T., Cowie G., Gaye B., Goes J., do Rosário Gomes H., Hood R. R., et al. (2020). Reviews and syntheses: Present, past, and future of the oxygen minimum zone in the northern Indian Ocean. Biogeosciences 17. doi: 10.5194/bg-17-6051-2020
Sabehi G., Béjà O., Suzuki M. T., Preston C. M., DeLong E. F. (2004). Different SAR86 subgroups harbour divergent proteorhodopsins. Environ. Microbiol. 6, 903–910. doi: 10.1111/j.1462-2920.2004.00676.x
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sherwood B. P., Shaffer E. A., Reyes K., Longnecker K., Aluwihare L. I., Azam F. (2015). Metabolic characterization of a model heterotrophic bacterium capable of significant chemical alteration of marine dissolved organic matter. Mar. Chem. 177, 357–365. doi: 10.1016/j.marchem.2015.06.027
Shiozaki T., Ijichi M., Isobe K., Hashihama F., Nakamura K.-i., Ehama M., et al. (2016). Nitrification and its influence on biogeochemical cycles from the equatorial Pacific to the Arctic Ocean. ISME J. 10 (9), 2184–2197. doi: 10.1038/ismej.2016.18
Sintes E., De Corte D., Clifford E., Herndl G. (2016). Geographic distribution of archaeal ammonia oxidizing ecotypes in the Atlantic ocean. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00077
Sow S., Brown M., Clarke L., Bissett A., van de Kamp J., Trull T., et al. (2022). Biogeography of Southern Ocean prokaryotes: a comparison of the Indian and Pacific sectors. Environ. Microbiol. 24 (5), 2449–2466. doi: 10.1111/1462-2920.15906
Stevens H., Ulloa O. (2008). Bacterial diversity in the oxygen minimum zone of the eastern tropical South Pacific. Environ. Microbiol. 10, 1244–1259. doi: 10.1111/j.1462-2920.2007.01539.x
Stewart F., Ulloa O., Delong E. (2012). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14, 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Stramma L., Johnson G., Sprintall J., Mohrholz V. (2008). Expanding oxygen-minimum zones in the tropical oceans. Sci. (New York N.Y.) 320, 655–658. doi: 10.1126/science.1153847
Sun X., Kop L., Lau M., Frank J., Jayakumar A., Lücker S., et al. (2019). Uncultured Nitrospina-like species are major nitrite oxidizing bacteria in oxygen minimum zones. ISME J. 13 (10), 2391–2402. doi: 10.1038/s41396-019-0443-7
Sun Q., Song J., Li X., Yuan H., Wang Q. (2021). The bacterial diversity and community composition altered in the oxygen minimum zone of the Tropical Western Pacific Ocean. J. Oceanol. Limnol. 39 (5), 1690–1704. doi: 10.1007/s00343-021-0370-0
Taguchi Y.h., Oono Y. (2005). Relational patterns of gene expression via non-metric multidimensional scaling analysis. Bioinf. (Oxford England) 21, 730–740. doi: 10.1093/bioinformatics/bti067
Thrash J., Seitz K., Baker B., Temperton B., Gillies L., Rabalais N., et al. (2017). Metabolic roles of uncultivated bacterioplankton lineages in the northern Gulf of Mexico “Dead zone”. mBio 8, e01017–e01017. doi: 10.1128/mBio.01017-17
Ulloa O., Canfield D., DeLong E., Letelier R., Stewart F. (2012). Microbial oceanography of anoxic oxygen minimum zones. Proc. Natl. Acad. Sci. 109, 15996–16003. doi: 10.1073/pnas.1205009109
Ulloa O., Pantoja S. (2009). The oxygen minimum zone of the eastern South Pacific. Deep Sea Res. Part II: Topical Stud. Oceanogr. 56, 987–991. doi: 10.1016/j.dsr2.2008.12.004
Vallivattathillam P., Lachkar Z., Levy M. (2023). Shrinking of the Arabian Sea oxygen minimum zone with climate change projected with a downscaled model. Front. Mar. Sci. 10, 1123739. doi: 10.3389/fmars.2023.1123739
Vijayan A. P., Kurian S., Joseph D., Dixon M., Kankonkar H., Khandeparker R., et al. (2023). Variation of amino acid in the sinking particulates in the northeastern Arabian Sea and the northern Bay of Bengal. Prog. Oceanogr. 219, 103168. doi: 10.1016/j.pocean.2023.103168
Wang B., Wang Z., Wang S., Qiao X., Gong X., Qingteng G., et al. (2020). Recovering partial nitritation in a PN/A system during mainstream wastewater treatment by reviving AOB activity after thoroughly inhibiting AOB and NOB with free nitrous acid. Environ. Int. 139, 105684. doi: 10.1016/j.envint.2020.105684
Ward B., Devol A., Rich J., Chang B. X., Naik H., Pratihary A., et al. (2009). Denitrification as the dominant nitrogen loss process in the Arabian Sea. Nature 461, 78–82. doi: 10.1038/nature08276
Watanabe M., Higashioka Y., Kojima H., Fukui M. (2017). Desulfosarcina widdelii sp. nov. and Desulfosarcina alkanivorans sp. nov., hydrocarbon-degrading sulfate-reducing bacteria isolated from marine sediment and emended description of the genus Desulfosarcina. Int. J. systematic evolutionary Microbiol. 67 (8), 2994–2997. doi: 10.1099/ijsem.0.002062
Woebken D., Fuchs B., Kuypers M., Amann R. (2007). Potential interactions of particle-associated anammox bacteria with bacterial and archaeal partners in the Namibian Upwelling System. Appl. Environ. Microbiol. 73, 4648–4657. doi: 10.1128/AEM.02774-06
Wright J., Konwar K., Hallam S. (2012). Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 10, 381–394. doi: 10.1038/nrmicro2778
Wright T. D., Vergin K. L., Boyd P. W., Giovannoni S. J. (1997). A novel delta-subdivision proteobacterial lineage from the lower ocean surface layer. Appl. Environ. Microbiol. 63, 1441–1448. doi: 10.1128/aem.63.4.1441-1448.1997
Yan W., Zhang R., Jiao N. (2018). A long-standing complex tropical dipole shapes marine microbial biogeography. Appl. Environ. Microbiol. 84, AEM.00614–00618. doi: 10.1128/AEM.00614-18
Keywords: hypoxic zone, OMZ, bioindicators, keystone taxa, Arabian Sea, microbial diversity
Citation: Li D, Wang L, Jiang F, Zeng X, Xu Q, Zhang X, Zheng Q and Shao Z (2024) Prokaryotic community structure and key taxa in the Arabian Sea’s oxygen minimum zone. Front. Mar. Sci. 11:1380819. doi: 10.3389/fmars.2024.1380819
Received: 07 February 2024; Accepted: 02 May 2024;
Published: 21 May 2024.
Edited by:
Cinzia Corinaldesi, Marche Polytechnic University, ItalyReviewed by:
Gabriella Luongo, Marche Polytechnic University, ItalyGenevieve L. Fernandes, Agricultural Research Organization, Israel
Copyright © 2024 Li, Wang, Jiang, Zeng, Xu, Zhang, Zheng and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongze Shao, shaozongze@tio.org.cn
†These authors have contributed equally to this work