New Viewpoint in Exaggerated Increase of PtiO2 With Normobaric Hyperoxygenation and Reasons to Limit Oxygen Use in Neurotrauma Patients
 Gurgen Harutyunyan
Gurgen Harutyunyan Garnik Harutyunyan
Garnik Harutyunyan Gagik Mkhoyan3
Gagik Mkhoyan3
- 1Emergency, Hospital 9 de Octubre, Valencia, Spain
- 2Faculty of Pharmacy, Universitat De València, Valencia, Spain
- 3Anesthesiology and Intensive Care, Erebouni Medical Center, Yerevan, Armenia
Multiple studies have shown that some of cerebral metabolic abnormalities occurring after traumatic brain injuries (TBI) are not of ischemic origin (1–4). The described pattern is called “metabolic crisis without brain ischemia” (5), and is associated with poor outcome within 6 months after trauma (6–8). Nevertheless, targeting adequate cerebral blood flow (CBF) and oxygen delivery (DO2) remains the cornerstone in the clinical management of TBI, especially in the early phase. Surprisingly, DO2 targeted therapy based on an escalation of dissolved O2, has been shown positive outcome in a few cases only, although it causes a significant increase in O2 pressure in cerebral tissue (PtiO2). High inspired O2 fraction or normobaric hyperoxia (NH) leads to a dramatic elevation of PtiO2 to “arterial” levels of 147 ± 36 mmHg (9). This daily and well-known phenomenon so far does not have definitive explanation and differs from the classical notions: oxyhemoglobin saturation (SO2) must remain nearly 100% over a wide range of O2 partial pressures (PO2) greater than 80 mmHg (10). NH induces a negligible increase in the amount of arterial O2 content and DO2, whereas it is associated with an important augmentation of PtiO2, without significant change in cerebral metabolic rate of O2 consumption (CMRO2) (11–14). Furthermore, studies using positron emission tomography, magnetic resonance imaging (MRI), and near-infrared spectroscopy showed an active O2 extraction fraction (OEF) in the non-necrotic tissue during NH (9, 15–18) with negligible increase of regional SO2 (rSO2) at 2.8 ± 1.82% (9) or without changes in rSO2 in tissue with intact autoregulation (16). The data of OEF at 0.56 ± 0.06 in reversible tissue, which although reduced from viable tissue to infarction (19), discards the possibility of a luxury perfusión in these cases. In general, NH causes an insignificant increase or no in rSO2 with an exaggerated elevation of PtiO2, which is equal to or less than end capillary PO2 (20). Thus, with NH the PO2 of end capillary venous blood reaches to “arterial” levels. Therefore, the exaggerated increase of PtiO2 with a negligible increase of rSO2 is incompatible with the classical sigmoidal form of the oxyhemoglobin dissociation curve (ODC). Hereby, the circulated hypotheses of mitochondrial dysfunction as a contributor (21) to high PtiO2 and the loss of O2 homeostatic mechanisms in the injured tissue during NH (22) requires an alternative explanation based on the conformational change of hemoglobin (Hb) quaternary structure (Max Perutz—the Nobel prize in chemistry 1962). This change produces a significant decrease in Hb–O2 affinity, considerable Hb buffering capacity augmentation, and convert the sigmoidal form of ODC to hyperbola. In this commentary, we explain the mechanisms underlying the exaggerated rise in PtiO2 with NH and consider the factors that limiting oxygen use in the damaged cerebral tissue.
Basic Biochemistry
Hemoglobin plays an important role in the transport of CO2 by the blood. In the brain tissue with a respiratory quotient (RQ) of 1, metabolism of 1 mol of O2 results in 1 mol of CO2. Also, 1 mol of CO2 in the erythrocyte generates 1 mol of protons (H+) by two mechanisms:
(*This reaction is catalyzed by carbonic anhydrase ranging between 104 and 106 reactions per second (23); therefore, the participation of other buffers of blood in CO2 buffering is not discussed).
The H+ uptake by Hb facilitates CO2 transport by stimulating bicarbonate formation: erythrocyte membranes do have a rapid anion exchange protein (capnoporin), which exchanges for Cl− at a ratio of 1:1. The H+ released by carbamate formation is taken up by the Hb as well. Thus, Hb remains the main buffer for protons (i.e., for CO2), but its capacity is biochemically limited (24):
where k = 1, 2, 3 or 4; 0.6 is the Haldane coefficient (the amount of the oxygen-linked H+ binding of Hb).
Thus, the release of 1 mol of O2 will allow the Hb to bind a maximum 0.6 mol of H+ or the Hb buffering capacity is lost in the brain tissue when OEF exceeded 60% (i.e., full saturation of Hb with H+) and CO2 begins to accumulate intensively in the brain tissue, provoking important acidosis. The imbalance of Hb capacities as a H+ buffer and as an O2 transporter in this setting (i.e., Eq. 1) makes the buffering capacity of Hb more important for brain cell survival to maintain a stable pH than its O2 carrier function. With the constant CMRO2, unchanged capillary permeability, temperature, pH, and DO2, the PtiO2 generated depends only on the Hb–O2 affinity, which decreases when Hb is saturated with negative allosteric effectors [2,3-diphosphoglycerate (2,3 DPG), CO2, H+, Cl−] (24).
The Hill coefficient (n) and the ODCs provide a way to assess these effects:
where P50O2 is O2 half-saturation pressure of Hb.
The increase of concentrations of the negative allosteric effectors in erythrocytes shifts the sigmoidal shape of ODC to the right and when Hb saturates with them, the ODC becomes hyperbolic in form (Figure 1). In this condition, Hb converts from the quaternary conformational state R (relax) to a state T (tense), which has a lower Hb–O2 affinity, hyperbolic form of ODC and increases up to six times the buffer capacity of Hb (24).
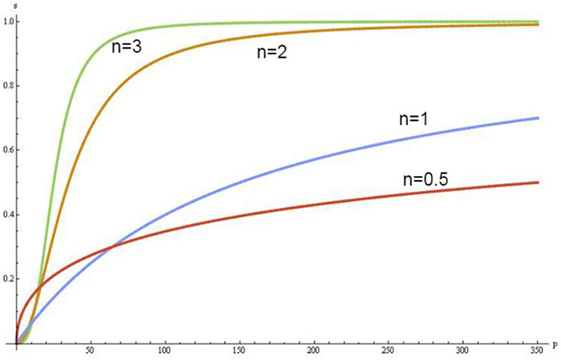
Figure 1. Oxyhemoglobin dissociation curves according to different Hill coefficients.
Physiology and Pathophysiology
In brain tissue (RQ at 1.0), use of 1 mol of O2 will result in the production of more CO2 than in the other tissues (RQ at 0.8). That is, the Hb buffering capacity is depleted with higher SO2 in the brain tissue (i.e., SO2 ≈40%, Eq. 1) than in other tissues. From this critical point, the generated CO2 is transported only in dissolved states and accumulated in the brain tissue causing clinically important acidosis with “normal” values of PtiO2. High levels of CO2 and intracellular acidosis directly or indirectly suppress the mitochondrial respiration: the CMRO2 is inhibited proportionally to decrease the perfusion by compensatory mechanisms [i.e., OEF is unable to raise (25)] trying to counteract the life-threatening intracellular acidosis. This is a blood-buffering capacity (BBC)-dependent mitochondrial suppression while independent of tissue O2 availability for cell aerobic metabolism. The O2 consumption (VO2)/buffer’s delivery inter-independency is shown in Figure 2.
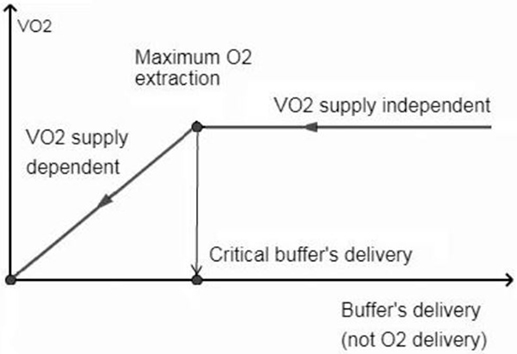
Figure 2. Oxygen consumption (VO2) is independent when perfusion allows hemoglobin not to be fully saturated with protons. From the critical buffer’s delivery or maximum O2 extraction point, the CO2 abruptly accumulates in the brain tissue, causing VO2 dependence on blood flow buffering capacity (not on O2 delivery) to counteract the life threatening acidosis.
Therefore, the accepted term “O2 delivery” does not reflect the pathophysiology of metabolism and has to be replaced by “buffer’s delivery,” which depends on the amount of Hb, the CBF, the total content of CO2 in arterial blood, and the Hb quaternary conformational state at the end of capillaries. The exaggerated increase in PtiO2 by NH in the VO2 supply-dependent phase has no metabolic effect due to the depletion of BBC. Moreover, augmentation in BBC in this phase through the increases of Hb concentration, augmentation of CBF or controlled hyperventilation without a change of Hb conformational state at the end of capillaries, is able to unlock the mitochondrial suppression and raise the CMRO2. Importantly, the high concentrations of 2,3 DPG as a significant negative allosteric effector can reduce the Hill coefficient to one without depleting the buffering capacity of Hb and can convert the ODC to hyperbola. In this case, the NH also causes an exaggerated increase in PtiO2 in relatively healthy tissue by physiological reduction on rCBF through vasoconstriction.
Clinical Interpretation
Many drugs and techniques used commonly during therapy of severe TBI, including mannitol, thiopental, ketorolac, nimodipine, hypothermia, deep sedation, transfusion of red blood cells (RBC), etc., can reduce the PtiO2 in the damaged tissue (26–30). This is because it improves the relationship within BBC and CMRO2 causing a decrease of Hb loading by negative effectors (i.e., increase in Hb–O2 affinity). The effects of medically induced augmentation of cerebral perfusion pressure on cerebral oxygenation are difficult to predict (31, 32). Indeed, the effects depend on which phase (VO2 buffer supply dependent or independent) the rCBF (i.e., BBC) change is made. Coles et al. showed that with hyperventilation, many regions of brain demonstrate an increase in CMRO2 despite the reduction in CBF (33). In fact, the reduction of Hb loading by negative allosteric effectors in the VO2 buffer supply-dependent phase, the hyperventilation rehabilitates the suppressed mitochondrial function by augmentation of BBC. Often uncontrolled hyperventilation causes an important decrease in PtiO2 due to an increase in Hb–O2 affinity. The worst is, when hyperventilation causes end capillary T to R conformational change of Hb with a decrease in BBC, which is dangerous for damaged tissue. Unlike hyperventilation, hypoventilation improves tissue oxygenation (34). Despite this effect, hypoventilation has no clinical use in patients with TBI. The increase in PtiO2 is related not so much to the increase of CBF by vasodilatation as to the increase in Hb loading by the negative allosteric effectors and the decrease in Hb–O2 affinity. Depletion of BBC will cause the decrease of CMRO2. Several studies have shown an association between anemia and poor outcome after TBI (35–37). As expected, the higher Hb levels are associated with improved outcome after subarachnoid hemorrhage (38) and post-cardiac arrest patients (39–41) (i.e., higher BBC maintains the metabolism in the VO2 supply independent phase). On the other hand, the transfusion of RBC is an independent risk factor for poor neurological outcome after 3 and 6 months in TBI patients (42). The stored blood with low values of erythrocyte 2,3-DPG can be used without hesitation when correcting a chronic anemia for instance (43). However, in acute situations with important tissue acidosis, the organism needs to rapidly augment the buffering capacity of Hb by R to T states shift, but re-synthesis of 2,3-DPG is obviously insufficient in stored blood. In such situations, fresh blood or blood with a near normal 2,3-DPG content should be used (43). According to proposed hypothesis, the augmentation of BBC by increasing the Hb concentration is the main role of RBC transfusion in TBI patients. Inconsistent results and contradictory findings have been reported for the effects of NH in brain energy metabolism (8, 10, 19, 44–46). The lactate MR spectroscopy demonstrates that in ischemic core and contralateral striatum, the lactate levels are not affected by NH, whereas, in the region of mismatch, lactate levels changed with changes in O2 delivery (47). The T2*-weighted MRI studies showed positive CBF changes to O2 challenge in the mismatch region, in contrast to negative CBF changes in normal tissue due to O2-induced vasoconstriction (16, 48). Indeed, the increase in BBC through augmentation of rCBF in the VO2 buffer supply dependent phase (i.e., in mismatch region) improves local acidosis, unlock the mitochondrial suppression, and decrease the lactate levels.
Discussion
Martini and colleagues determined that severe TBI management guided by PtiO2 monitoring was associated with a poor neurological outcome and was an inefficient use of hospital resources (49). Of course, recording of “normal” or high PtiO2 values with acceptable high inspired O2 fraction or positive end-expiratory pressures without sufficient BBC may create a false impression of safety and negatively impact clinical decision making. The use of absolute levels of PtiO2 as a marker of ischemia is confusing because a more important BBC deficit happens much earlier. The transition from the concept of “increase in O2 supply” to the concept of “increase of buffer supply” has important clinical implications in the strategy of treatment in neurotrauma patient. In addition, the characteristics of T Hb (i.e., hyperbolic ODC, high buffering capacity) allow us to understand and correctly assess the effects of current treatment and evolution of damaged brain tissue through PtiO2 changes with NH. The explanation of known paradoxes in neurotrauma such as an exaggerated rise in PtiO2 by NH, inexplicable decrease in PtiO2 after many drugs and techniques commonly used, and metabolic crisis without “ischemia” is explained for the first time by the Hb conformational changes and BBC-dependent mitochondrial suppression. As we have seen the metabolic crisis always coincides with ischemia but in the sense of lack of BBC. The hypothesis is structural and revolutionary. Despite the enormous amount of data available in literature, targeted practical observations are needed to confirm (or deny) this hypothesis.
Conclusion
1. The exaggerated increase of PtiO2 by NH occurs in the “T” state of Hb when ODC as a hyperbole and does not always coincide with the mitochondrial dysfunction.
2. The BBC is a limiting factor of O2 consumption by damaged brain tissues.
Author Contributions
GH, as the first author, he developed the hypothesis and prepared the manuscript. All the co-authors helped with their important input to finalize the hypothesis discussed and to write the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chieregato A, Tanfani A, Compagnone C, Turrini C, Sarpieri F, Ravaldini M, et al. Global cerebral blood flow and CPP after severe head injury: a xenon-CT study. Intensive Care Med (2007) 33(5):856–62. doi:10.1007/s00134-007-0604-4
2. Soustiel JF, Sviri GE. Monitoring of cerebral metabolism: non-ischemic impairment of oxidative metabolism following severe traumatic brain injury. Neurol Res (2007) 29(7):654–60. doi:10.1179/016164107X240017
3. Glenn TC, Kelly DF, Boscardin WJ, McArthur DL, Vespa P, Oertel M, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab (2003) 23(10):1239–50. doi:10.1097/01.WCB.0000089833.23606.7F
4. Soustiel JF, Glenn TC, Vespa P, Rinsky B, Hanuscin C, Martin NA. Assessment of cerebral blood flow by means of blood-flow-volume measurement in the internal carotid artery: comparative study with a 133xenon clearance technique. Stroke (2003) 34(8):1876–80. doi:10.1161/01.STR.0000080942.32331.39
5. Vespa P, Bergneider M, Hattori N, Wu HM, Huang SC, Martin NA, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab (2005) 25:763–74. doi:10.1038/sj.jcbfm.9600073
6. Stein NR, McArthur DL, Etchepare M, Vespa PM. Early cerebral metabolic crisis after TBI influences outcome despite adequate hemodynamic resuscitation. Neurocrit Care (2012) 17:49–57. doi:10.1007/s12028-012-9708-y
7. Marcoux J, McArthur DA, Miller C, Glenn TC, Villablanca P, Martin NA, et al. Persistent metabolic crisis as measured by elevated cerebral microdialysis lactate-pyruvate ratio predicts chronic frontal lobe brain atrophy after traumatic brain injury. Crit Care Med (2008) 36:2871–7. doi:10.1097/CCM.0b013e318186a4a0
8. Xu Y, Mc Arthur DL, Alger JR, Etchepare M, Hovda DA, Glenn TC, et al. Early non ischemic oxidative metabolic dysfunction leads to chronic brain atrophy in traumatic brain injury. J Cereb Blood Flow Metab (2010) 30:883–94. doi:10.1038/jcbfm.2009.263
9. McLeod AD, Igielman F, Elwell C, Cope M, Smith M. Measuring cerebral oxygenation during normobaric hyperoxia: a comparison of tissue microprobes, near-infrared spectroscopy, and jugular venous oximetry in head injury. Anesth Analg (2003) 97(3):851–6. doi:10.1213/01.ANE.0000072541.57132.BA
10. Severinghaus JW. Simple, accurate equations for human blood O2 dissociation computations. J Appl Physiol Respir Environ Exerc Physiol (1979) 46:599–602. doi:10.1152/jappl.1979.46.3.599
11. Magnoni S, Ghisoni L, Locatelli M, Caimi M, Colombo A, Valeriani V, et al. Lack of improvement in cerebral metabolism after hyperoxia in severe head injury: a micro-dialysis study. J Neurosurg (2003) 98:952–8. doi:10.3171/jns.2003.98.5.0952
12. Diringer MN. Hyperoxia: good or bad for the injured brain? Curr Opin Crit Care (2008) 14:167–71. doi:10.1097/MCC.0b013e3282f57552
13. Diringer MN, Aiyagari V, Zazulia AR, Videen TO, Powers WJ. Effect of hyperoxia on cerebral metabolic rate for oxygen measured using positron emission tomography in patients with acute severe head injury. J Neurosurg (2007) 106:526–9. doi:10.3171/jns.2007.106.4.526
14. Xu F, Liu P, Pascual JM, Xiao G, Lu H. Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation and oxidative metabolism. J Cereb Blood Flow Metab (2012) 32:1909–18. doi:10.1038/jcbfm.2012.93
15. Ghosh A, Highton D, Kolyva C, Tachtsidis I, Elwell CE, Smith M. Hyperoxia results in increased aerobic metabolism following acute brain injury. J Cereb Blood Flow Metab (2017) 37:2910–20. doi:10.1177/0271678X16679171
16. Sahoo S, Sheshadri V, Sriganesh K, Madhsudana Reddy KR, Radhakrishnan M, Umamaheswara Rao GS. Effect of hyperoxia on cerebral blood flow velocity and regional oxygen saturation in patients operated on for severe traumatic brain injury-the influence of cerebral blood flow autoregulation. World Neurosurg (2017) 98:211–6. doi:10.1016/j.wneu.2016.10.116
17. Shen Q, Huang S, Du F, Duong TQ. Probing ischemic tissue fate with BOLD fMRI of brief oxygen challenge. Brain Res (2011) 1425:132–41. doi:10.1016/j.brainres.2011.09.052
18. Christen T, Bouzat P, Pannetier N, Coquery N, Moisan A, Lemasson B, et al. Tissue oxygen saturation mapping with magnetic resonance imaging. J Cereb Blood Flow Metab (2014) 34(9):1550–7. doi:10.1038/jcbfm.2014.116
19. Powers WJ, Grubb RL Jr, Darriet D, Raichle ME. Cerebral blood flow and cerebral metabolic rate of oxygen requirements for cerebral function and viability in humans. J Cereb Blood Flow Metab (1985) 5:600–8. doi:10.1038/jcbfm.1985.89
20. Gupta AK, Hutchinson PJ, Fryer T, Al-Rawi PG, Parry DA, Minhas PS, et al. Measurement of brain tissue oxygenation performed using positron emission tomography scanning to validate a novel monitoring method. J Neurosurg (2002) 96(2):263–8. doi:10.3171/jns.2002.96.2.0263
21. Johnston AJ, Steiner LA, Gupta AK, Menon DK. Cerebral oxygen vasoreactivity and cerebral tissue oxygen reactivity. Br J Anaesth (2003) 90(6):774–86. doi:10.1093/bja/aeg104
22. Vilalta A, Sahuquillo J, Merino MA, Poca MA, Garnacho A, Martinez–Valverde T, et al. Normobaric hyperoxia in traumatic brain injury. Does brain metabolic state influence the response to hyperoxic challenge? J Neurotrauma (2011) 28:1139–48. doi:10.1089/neu.2010.1720
23. Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther (1997) 74(1):1–20. doi:10.1016/S0163-7258(96)00198-2
25. Gutierrez G, Pohil R. Oxygen consumption is linearly related to O2 supply in critically ill patients. J Crit Care (1986) 1(1):45–53. doi:10.1016/S0883-9441(86)80116-2
26. Steiner T, Pilz J, Schellinger P, Wirtz R, Friederichs V, Aschoff A, et al. Multimodal online monitoring in middle cerebral artery territory stroke. Stroke (2001) 32:2500–6. doi:10.1161/hs1101.097400
27. Gupta AK, Al-Rawi PG, Hutchinson PJ, Kirkpatrick PJ. Effect of hypothermia on brain tissue oxygenation in patients with severe head injury. Br J Anesth (2002) 88:188–92. doi:10.1093/bja/88.2.188
28. Stiefel MF, Heuer GG, Abrahams JM, Bloom S, Smith MJ, Maloney-Wilensky E, et al. The effect of nimodipine on cerebral oxygenation in patients with poor-grade subarachnoid hemorrhage. J Neurosurg (2004) 101:594–9. doi:10.3171/jns.2004.101.4.0594
29. Zygun DA, Nortje J, Hutchinson PJ, Timofeev I, Menon DK, Gupta AK. The effect of red blood cell transfusion on cerebral oxygenation and metabolism after severe traumatic brain injury. Crit Care Med (2009) 37:1074–8. doi:10.1097/CCM.0b013e318194ad22
30. Sakowitz OW, Stover JF, Sarrafzadeh AS, Unterberg AW, Kiening KL. Effects of mannitol bolus administration on intracranial pressure, cerebral extracellular metabolites, and tissue oxygenation in severely head-injured patients. J Trauma (2007) 62:292–8. doi:10.1097/01.ta.0000203560.03937.2d
31. LeRoux PD, Oddo M. Parenchymal brain oxygen monitoring in the neurocritical care unit. Neurosurg Clin N Am (2013) 24:427–39. doi:10.1016/j.nec.2013.03.001
32. Sahuquillo J, Amoros S, Santos A, Poca MA, Panzardo H, Domínguez L, et al. Does an increase in cerebral perfusion pressure always mean a better oxygenated brain? A study in head-injured patients. Brain Edema XI. Acta Neurochirurgica Supplements. (Vol. 76), Springer, Vienna (2000).
33. Coles JP, Fryer TD, Coleman MR, Smielewski P, Gupta AK, Minhas PS, et al. Hyperventilation following head injury: effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med (2007) 35(2):568–78. doi:10.1097/01.CCM.0000254066.37187.88
34. Akça O, Doufas AG, Morioka N, Iscoe S, Fisher J, Sessler DI. Hypercapnia improves tissue oxygenation. Anesthesiology (2002) 97(4):801–6. doi:10.1097/00000542-200210000-00009
35. Salim A, Hadjizacharia P, DuBose J, Brown C, Inaba K, Chan L, et al. Role of anemia in traumatic brain injury. J Am Coll Surg (2008) 207(3):398–406. doi:10.1016/j.jamcollsurg.2008.03.013
36. Sekhon MS, McLean N, Henderson WR, Chittock DR, Griesdale DE. Association of hemoglobin concentration and mortality in critically ill patients with severe traumatic brain injury. Crit Care (2012) 16(4):R128. doi:10.1186/cc1143
37. Steyerberg EW, Mushkudiani N, Perel P, Butcher I, Lu J, McHugh GS, et al. Predicting outcome after traumatic brain injury: development and international validation of prognostic scores based on admission characteristics. PLoS Med (2008) 5(8):165. doi:10.1371/journal.pmed.0050165
38. Naidech AM, Jovanovic B, Wartenberg KE, Parra A, Ostapkovich N, Connolly ES, et al. Higher hemoglobin is associated with improved outcome after subarachnoid hemorrhage. Crit Care Med (2007) 35(10):2383–9. doi:10.1097/01.CCM.0000284516.17580.2C
39. SOS-KANTO Study Group. Relationship between the hemoglobin level at hospital arrival and post–cardiac arrest neurologic outcome. Am J Emerg Med (2012) 30:770–4. doi:10.1016/j.ajem.2011.03.031
40. Wang CH, Huang CH, Chang WT, Tsai MS, Yu PH, Wang AY, et al. Association between hemoglobin levels and clinical outcomes in adult patients after in-hospital cardiac arrest: a retrospective cohort study. Intern Emerg Med (2016) 11:727–36. doi:10.1007/s11739-015-1386-2
41. Johnson NJ, Rosselot B, Perman SM, Dodampahala K, Goyal M, Gaieski DF, et al. The association between hemoglobin concentration and neurologic outcome after cardiac arrest. J Crit Care (2016) 36:218–22. doi:10.1016/j.jcrc.2016.07.012
42. Warner MA, O’Keeffe T, Bhavsar P, Shringer R, Moore C, Harper C, et al. Transfusions and long-term functional outcomes in traumatic brain injury. J Neurosurg (2010) 113(3):539–46. doi:10.3171/2009.12.JNS091337
43. Stan A, Zsigmond E. The restoration in vivo of 2,3-diphosphoglycerate (2,3-DPG) in stored red cells, after transfusion. The levels of red cells 2,3-DPG. Rom J Intern Med (2009) 47(2):173–7.
44. Vidal-Jorge M, Sánchez-Guerrero A, Mur-Bonet G, Castro L, Rădoi A, Riveiro M, et al. Does normobaric hyperoxia cause oxidative stress in the injured brain? A microdialysis study using 8-iso-prostaglandin F2α as a biomarker. J Neurotrauma (2017) 34:2731–42. doi:10.1089/neu.2017.4992
45. Reinert M, Barth A, Rothen HU, Schaller B, Takala J, Seiler RW. Effects of cerebral perfusion pressure and increased fraction of inspired oxygen on brain tissue oxygen, lactate and glucose in patients with severe head injury. Acta Neurochir (Wien) (2003) 145:341–9. doi:10.1007/s00701-003-0027-0
46. Tolias CM, Reinert M, Seiler R, Gilman C, Scharf A, Bullock MR. Normobaric hyperoxia-induced improvement in cerebral metabolism and reduction in intracranial pressure in patients with severe head injury: a prospective historical cohort-matched study. J Neurosurg (2004) 101:435–44. doi:10.3171/jns.2004.101.3.0435
47. Holmes WM, Lopez-Gonzalez MR, Gallagher L, Deuchar GA, Macrae IM, Santosh C, et al. Detection of the ischemic penumbra: direct assessment of metabolic integrity. NMR Biomed (2012) 25:295–304. doi:10.1002/nbm.1748
48. Wu O, Lu J, Mandeville JB, Murata Y, Egi Y, Dai G, et al. Dynamic functional cerebral blood volume responses to normobaric hypoxia in acute ischemic stroke. J Cereb Blood Flow Metab (2012) 32:1800–9. doi:10.1038/jcbfm.2012.87
Keywords: normobaric hyperoxia, PtiO2, metabolism, brain trauma, blood-buffering capacity
Citation: Harutyunyan G, Harutyunyan G and Mkhoyan G (2018) New Viewpoint in Exaggerated Increase of PtiO2 With Normobaric Hyperoxygenation and Reasons to Limit Oxygen Use in Neurotrauma Patients. Front. Med. 5:119. doi: 10.3389/fmed.2018.00119
Received: 13 September 2017; Accepted: 10 April 2018;
Published: 22 May 2018
Edited by:
Samir G. Sakka, Universität Witten/Herdecke, GermanyReviewed by:
Suren Soghomonyan, The Ohio State University, United StatesCopyright: © 2018 Harutyunyan, Harutyunyan and Mkhoyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gurgen Harutyunyan, varsenik@hotmail.es